


Abstract
The novel multitargeted tyrosine kinase inhibitor sunitinib is used as an antiangiogenic agent for the treatment of several types of cancer, including metastatic renal cell carcinoma (RCC). Sunitinib was shown to positively change the immunosuppressive phenotype in RCC patients. To improve its antitumor efficacy, and offer strategies for its combination with other approaches, it is critical to fully elucidate its mechanisms of action. We show that sunitinib induces tumor cell apoptosis and growth arrest in RCC tumor cells, which correlates with signal transducer and activator of transcription 3 (Stat3) activity inhibition. Sunitinib-mediated direct effects on tumor cells occur regardless of von Hippel-Lindau tumor suppressor gene status and hypoxia-inducible transcription factor-2α levels. Reduction of Stat3 activity enhances the antitumor effects of sunitinib, whereas expression of a constitutively activated Stat3 mutant rescues tumor cell death. Intravital multiphoton microscopy data show that sunitinib induces mouse Renca tumor cell apoptosis in vivo before tumor vasculature collapse. Sunitinib also inhibits Stat3 in Renca tumor–associated myeloid-derived suppressor cells (MDSC), down-regulates angiogenic gene expression, and reduces MDSCs and tumor T regulatory cells. These results suggest that Stat3 activity is important for RCC response to sunitinib, and Stat3 inhibition permits the direct proapoptotic activity of sunitinib on tumor cells and positive effects on tumor immunologic microenvironment. [Cancer Res 2009;69(6):2506–13]
Introduction
Sunitinib is a multitargeted tyrosine kinase inhibitor known to selectively inhibit several growth factor receptors, including vascular endothelial growth factor receptor 1 (VEGFR1), 2 (VEGFR2), and 3 (VEGFR3); platelet-derived growth factor receptor α (PDGFRα) and β (PDGFβ); and stem cell factor (1, 2). Recent research has explored the potential antiangiogenic effects of sunitinib, given the critical role of many of these receptor kinases in tumor angiogenesis (1, 2). Sunitinib has been shown in vitro to inhibit the VEGF-induced mitogenic response of human endothelial cells, to abrogate migration of endothelial cells, and to inhibit their ability to form capillary-like tubes (1–3). In vivo, sunitinib treatment reduced tumor microvessel density, inhibited neovascularization in a tumor vascular window model, and prevented metastasis in a lung carcinoma model (4). Based on these observations, sunitinib has been evaluated in the clinic as an antiangiogenic agent in several types of cancer, including gastrointestinal stromal tumors (GIST) and renal cell carcinoma (RCC; refs. 1, 2). It has shown improvements in progression-free when compared with IFN-α therapy in metastatic RCC patients (5) and has become a frontline therapy for this disease (6, 7). However, its clinical effects remain suboptimal (8), suggesting the need to further explore the mechanisms of action of sunitinib, at both the cellular and molecular levels, so as to facilitate its rational and effective combinatorial use.
Although sunitinib has been explored in the clinic mainly for its antiangiogenesis effects, a recent report of tumor biopsy results from a GIST patient treated with sunitinib showed marked tumor response in the form of tumor cell necrosis, which was not associated with reduction in tumor vasculature (9). Other reports also suggested sunitinib effects on glioma cells, although no detailed molecular mechanisms were included (10). Recent clinical studies also indicated that sunitinib treatment reduced the number of myeloid-derived suppressor cells (MDSC; ref. 11), improved Th-1 response, and diminished T regulatory cells (Treg) in RCC patients (8). However, the underlying mechanism(s) mediating sunitinib-induced tumor cell apoptosis in GIST patients and reducing immunosuppressive cells in RCC patients remains unknown.
Signal transducer and activator of transcription 3 (Stat3) is constitutively activated in diverse cancer cell types, including RCC (12, 13). Frequent Stat3 activation in tumor cells is largely due to the fact that Stat3 is a point of convergence for numerous tyrosine kinases, including VEGFR, PDGFR, EGFR, and Src (12, 14). Stat3 is also activated in diverse tumor-associated immune cells, including myeloid cells, promoting tumor immune evasion as well as tumor angiogenesis (15–17). Stat3 mediates tumor immune evasion by inhibiting expression of Th-1 immunostimulatory molecules and by promoting production of immunosuppressive factors (16–18). Ablating Stat3 in tumor myeloid cells increased dendritic cell activation, tumor Treg reduction, and CD8+ T-cell activation (17, 19). Recently, it was documented that Stat3 was critically involved in tumor accumulation of MDSCs, which play an important role in suppressing antitumor immune responses (S100A9; ref. 20).
In the current study, we assessed the direct effects of sunitinib on RCC tumor cells in vitro and in vivo as well as its effects on the tumor immunologic microenvironment.
Materials and Methods
Reagents. Sunitinib (SU11248; Sutent) was purchased from City of Hope Medical Center Pharmacy. Human Stat3 small interfering RNA (siRNA), control siRNA, anti-Stat3 (C-20), anti–Bcl-xL (H-5), anti-survivin (D-8), anti-HDAC1 (H-11), and anti–poly(ADP-ribose) polymerase (PARP) were obtained from Santa Cruz Biotechnology; anti–cyclin E (clone HE12) was from BD Biosciences; anti–cyclin D1 (AB-3) and anti–Bcl-2 were from Calbiochem; anti–β-actin (AC-74) was from Sigma; and anti–phosphorylated Stat3 (p-Stat3; Tyr705), anti–phosphorylated Src (p-Src; Tyr416)/anti-Src, anti–phosphorylated AKT (p-AKT; Ser473)/anti-AKT, anti–phosphorylated extracellular signal-regulated kinase 1/2 (p-ERK1/2; Thr202/Tyr204)/anti-ERK1/2, and anti–phosphorylated Janus-activated kinase 2 (p-JAK2; Tyr1007/1008)/anti-JAK2 were from Cell Signaling Technology, Inc. FITC-conjugated, phycoerythrin-conjugated, or allophycocyanin (APC)–conjugated monoclonal antibodies to mouse CD4, CD8, CD11b, CD11c, Gr1, and phosphotyrosine-Stat3 were from BD Biosciences Pharmingen.
Cells. Human RCC cell lines 786-O, RCC4, and their derivatives, including 786-O-vector (empty pBabe-pure) and 786-O-VHL(HA-VHLwt-pBabe-pure), were generous gifts from Dr. William G. Kaelin (Harvard University, Cambridge, MA; ref. 21). The Renca murine cell line was obtained from Dr. Alfred Chang (University of Michigan Medical Center, Ann Arbor, MI) and was grown in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). For establishing the Stat3C-expressing stable cells, plasmids of pRC/CMV-vector and pRC/CMV-Stat3C-Flag were transected into 786-O cells using Lipofectamine 2000 (Invitrogen) and selected in 2 mg/mL G418 (Invitrogen). Resistant pooled cells were characterized by Western blot and maintained in medium containing 0.5 mg/mL G418.
In vivo experiments. Female BALB/c mice (7–8 wk old) were purchased from The Jackson Laboratory. Animal use procedures were approved by the institutional committee of the Beckman Research Institute at City of Hope Medical Center. For s.c. tumor challenge, female BALB/c mice were implanted s.c. with 2.5 × 106 Renca cells. After tumors reached 5 to 7 mm in diameter, sunitinib or vehicle control was administered orally, once a day, at dose levels of 40, 20, and 10 mg/kg body weight. Tumor growth was monitored every other day.
Flow cytometry. Preparation and staining of single-cell suspensions of spleen, lymph node, or tumor tissues were as described previously (17).
Proliferation assay. Cells were seeded onto 96-well plates (5,000 per well) in DMEM with 5% FBS and allowed to attach overnight. Vehicle (DMSO) or drug was added at different concentrations the following day. After 24 or 48 h of treatment, cell proliferation assays were performed using CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit (Promega) according to the manufacturer's instructions. Absorbance was measured at 490 nm using Mikrotek Laborsysteme. Experiments were done in triplicate and repeated a minimum of three times.
Apoptotic assays. Cells (3 × 105) were seeded in 60-mm dishes in DMEM with 5% FBS and treated the following day with sunitinib (24 or 48 h). Floating and attached cells were then collected and stained with Annexin V-APC and 4′,6-diamidino-2-phenylindole (DAPI; eBioscience) for flow cytometry measurement of apoptosis levels. Cells were analyzed using FlowJo as described above.
Real-time quantitative PCR. Total RNA was extracted by RNeasy kit (Qiagen) and cDNA was synthesized with iScript cDNA Synthesis (Bio-Rad). Primers for human Stat3, survivin, Bcl-xL, and Cdc25A and mouse-specific primers of VEGF and CXCL2 were purchased from SuperArray Bioscience Corp. Human and mouse glyceraldehyde-3-phosphate dehydrogenase primers were used as an endogenous control. Sequence-specific amplification was detected with increased fluorescent signal of SYBR Green (Bio-Rad) with a Chromo4 Real-time PCR Detector (Bio-Rad).
Live cell imaging and immunofluorescence. Murine 3T3/v-Src fibroblasts were transfected with pcDNA5/TO/FRT-Stat3-CFP-YFP (a kind gift of Michael Vogt, Klinikum Aachen, Aachen, Germany) using Lipofectamine 2000 and then treated with sunitinib (20 μmol/L, 2 h). Localization of Stat3-CY in living cells was analyzed using an LSM 510 Meta Inverted microscope (Zeiss). Tumor sections were stained using indirect immunofluorescence as previously described (15).
Intravital multiphoton microscopy. BALB/c mice bearing Renca tumors were anaesthetized with isoflurane/oxygen followed by i.v. injection (via retro-orbital route) with dextran-rhodamine (100 μg; Molecular Probes), Hoechst 33342 (250 μg), and Annexin V-FITC (10 μg; BioVision). Fifteen minutes later, tumor tissues in mice were surgically exposed for intravital multiphoton microscopy (IVMPM) and imaged with an Ultima Multiphoton Microscopy System (Prairie Technologies). Hoechst 33342 (λ = 730 nm) and fluorescein conjugate and rhodamine (λ = 860 nm) excitation wavelengths were used. Band-pass filters optimized for Hoechst 33342 (BP λ = 460/50 nm), fluorescein (BP λ = 525/50 nm), and rhodamine (BP λ = 570–620 nm) were used for detection.
Statistics. A two-sided t test was used to evaluate differences between treated and control groups (***, P < 0.001; **, P < 0.01; and *, P < 0.05).
Results
Sunitinib induces RCC cell apoptosis and growth arrest. To determine whether sunitinib has direct effects on RCC tumor cells, we tested its ability to kill 786-O and RCC4 human RCC cell lines, as well as murine Renca tumor cells. The effects of sunitinib on cell death were dose dependent in all three tumor cell lines (Fig. 1A,, top). In addition to inducing RCC tumor cell apoptosis, sunitinib also inhibited cell proliferation in a dose-dependent manner (Fig. 1A,, bottom). To confirm that sunitinib-induced tumor cell death was associated with apoptosis, we examined the levels of cleaved PARP. Results showed that for concentrations at which sunitinib caused effective tumor cell death, there were corresponding increases in cleaved PARP (Fig. 1B). Western blot analyses were also performed to determine the downstream genes mediating the effects of sunitinib on RCC cells. Sunitinib treatment (24 hours) of 786-O, RCC4, and Renca tumor cells reduced expression of several key antiapoptotic and pro-proliferation genes, including cyclin E, cyclin D1, and survivin (Fig. 1B). Real-time reverse transcription-PCR confirmed sunitinib down-regulation of survivin in 786-O cells (Fig. 1B).
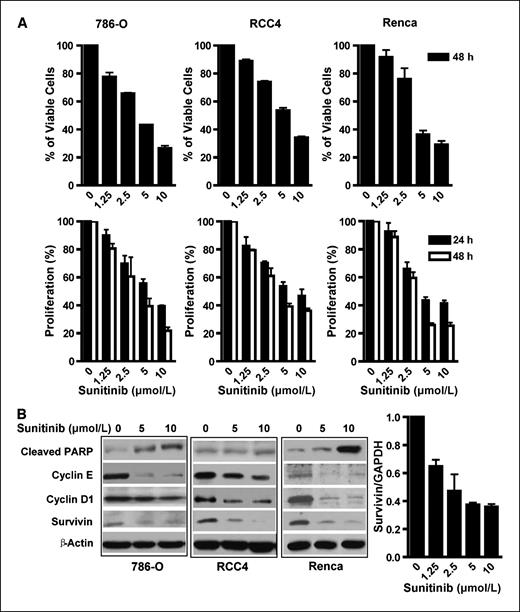
Sunitinib induces apoptosis and inhibits RCC cell proliferation. A, analysis of RCC cell apoptosis and proliferation following treatment of sunitinib. Renca, 786-O, and RCC4 cells were treated (24 and 48 h) at indicated doses, harvested, and stained with Annexin V-APC and DAPI. Top, viable cells represented by Annexin V-APC and DAPI double-negative cells were determined by flow cytometry; bottom, cell proliferation was evaluated by MTS assay following sunitinib treatment. Columns, mean (n = 3, in triplicate); bars, SD. B, sunitinib treatment of RCC cells reduces expression of antiapoptotic and proliferative genes. Western blot analyses of RCC cells treated (24 h) with sunitinib to evaluate protein levels of cleaved PARP, cyclin E, cyclin D1, and survivin. Bottom, real-time PCR was performed to confirm the sunitinib-induced reduction of survivin expression in 786-O tumor cells. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Sunitinib induces apoptosis and inhibits RCC cell proliferation. A, analysis of RCC cell apoptosis and proliferation following treatment of sunitinib. Renca, 786-O, and RCC4 cells were treated (24 and 48 h) at indicated doses, harvested, and stained with Annexin V-APC and DAPI. Top, viable cells represented by Annexin V-APC and DAPI double-negative cells were determined by flow cytometry; bottom, cell proliferation was evaluated by MTS assay following sunitinib treatment. Columns, mean (n = 3, in triplicate); bars, SD. B, sunitinib treatment of RCC cells reduces expression of antiapoptotic and proliferative genes. Western blot analyses of RCC cells treated (24 h) with sunitinib to evaluate protein levels of cleaved PARP, cyclin E, cyclin D1, and survivin. Bottom, real-time PCR was performed to confirm the sunitinib-induced reduction of survivin expression in 786-O tumor cells. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Effects of sunitinib on major oncogenic signaling pathways: the role of Stat3. Because sunitinib is a known tyrosine kinase inhibitor, we examined several major downstream signaling molecules of tyrosine kinases, including AKT, mitogen-activated protein kinase (MAPK), and Stat3. No inhibitory effects were observed on p-AKT and p-ERK1/2 levels in 786-O cells following 2-hour sunitinib treatment (Fig. 2A). However, increased treatment time (24 hours) and higher sunitinib concentration (10 μmol/L) resulted in decreased p-ERK1/2 levels as well as a minor reduction in p-AKT levels. Similar results were observed in RCC4 cells (Fig. 2B). We further assessed the potential effects of sunitinib on JAK2, which is also frequently activated in cancer cells. RCC4 tumor cells contained constitutively activated p-JAK2 (Fig. 2B) and Western blot data indicated that sunitinib had no inhibitory effect on p-JAK2 in these cells. Elevated activated JAK2 (p-JAK2) was not observed in 786-O tumor cells (data not shown).

Effects of sunitinib on major oncogenic signaling pathways in 786-O and RCC4 cells. Sunitinib reduced Stat3 and Src activity, with no dramatic reduction of AKT, MAPK, and JAK signaling in 786-O (A) and RCC4 tumor cells (B). Tumor cells were treated with sunitinib at indicated concentrations for 2 h (left) or 24 h (right). Total cell lysates were prepared and Western blots were performed using relevant antibodies to detect total protein levels, with β-actin used as the loading control. C, sunitinib inhibits activated Src and Stat3, leading to cell apoptosis. 3T3 fibroblasts transformed by v-Src were transfected with Stat3-CY and visualized by confocal microscopy. Top, Stat3-CY localization in cells untreated (left) or treated (2 h) with sunitinib (right); bottom, sunitinib induces apoptosis of 3T3 v-Src cells. The 3T3 v-Src cells were stained for Annexin V-APC (red); Hoechst 33342 was used to visualize nuclei (blue). Scale bar, 10 μm.
Effects of sunitinib on major oncogenic signaling pathways in 786-O and RCC4 cells. Sunitinib reduced Stat3 and Src activity, with no dramatic reduction of AKT, MAPK, and JAK signaling in 786-O (A) and RCC4 tumor cells (B). Tumor cells were treated with sunitinib at indicated concentrations for 2 h (left) or 24 h (right). Total cell lysates were prepared and Western blots were performed using relevant antibodies to detect total protein levels, with β-actin used as the loading control. C, sunitinib inhibits activated Src and Stat3, leading to cell apoptosis. 3T3 fibroblasts transformed by v-Src were transfected with Stat3-CY and visualized by confocal microscopy. Top, Stat3-CY localization in cells untreated (left) or treated (2 h) with sunitinib (right); bottom, sunitinib induces apoptosis of 3T3 v-Src cells. The 3T3 v-Src cells were stained for Annexin V-APC (red); Hoechst 33342 was used to visualize nuclei (blue). Scale bar, 10 μm.
Given that Stat3 is constitutively activated in diverse cancers, including RCC (13), we assessed whether sunitinib-induced tumor cell death was associated with Stat3 inhibition. Although sunitinib had no effects on total Stat3 protein levels in tumor cells, it inhibited p-Stat3 as early as 2 hours after sunitinib treatment, with continued p-Stat3 inhibition at 24 hours after treatment (Fig. 2A and B). The reduction of p-Stat3 correlated well with sunitinib-induced tumor cell death (Fig. 1A,, top) and inhibition of tumor cell proliferation, as shown by 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Fig. 1A , bottom).
Because Src is a tyrosine kinase upstream of Stat3, and frequently overactivated in diverse cancers (12, 22, 23), we examined the effects of sunitinib on Src activity (p-Src). Similar to its effects on p-Stat3, sunitinib effectively and rapidly inhibited Src activity (Fig. 2A and B). To confirm sunitinib inhibitory effects on Src and Stat3, we used 3T3 fibroblasts transformed by v-Src to evaluate the subcellular distribution of Stat3 following sunitinib treatment. Stat3-CFP-YFP (Stat3-CY) was expressed in 3T3/v-Src cells and confocal microscopy was used to analyze Stat3-CY localization in living cells. When v-Src–transformed cells were treated with sunitinib, nuclear Stat3 translocated to the cytoplasm, indicating abrogation of Src-induced Stat3 activation (Fig. 2C), which was accompanied by cell apoptosis (Fig. 2C , bottom). These results suggested that Src tyrosine kinase was also a target of sunitinib in RCC cells.
RCC cell apoptosis and growth arrest induced by sunitinib is independent of VHL status and hypoxia-inducible factor-2α levels. Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene occur frequently in RCC cells, and the lack of a functional VHL is associated with poor prognosis (21, 24). VHL inhibits hypoxia-inducible factor-2α (HIF-2α) expression, thereby reducing its ability to up-regulate VEGF expression, which is critical for RCC progression (24). Recent publications have also indicated an important role of HIF-2α in regulating renal tumor cell oncogenic potential by up-regulating several c-myc downstream genes (25). To test whether VHL status would affect RCC cell response to sunitinib-induced tumor cell apoptosis and proliferation inhibition, we compared the effects of sunitinib on RCC isotype tumor lines with different VHL status and HIF-2α levels (Supplementary Fig. S1).
Stat3 is critical for sunitinib-induced direct antitumor effects in RCC tumor cells. Our initial results (Fig. 1B) showed that sunitinib treatment affected several Stat3 target genes important for tumor cell survival and proliferation. Western blot analyses of RCC cells treated with Stat3 siRNA confirmed that Stat3 inhibition reduced the expression of several known Stat3 downstream genes, such as survivin, cyclin E, Bcl-xL, and Bcl-2 (Fig. 3A). These results suggested that sunitinib-induced RCC tumor cell apoptosis and growth arrest was at least partially mediated by Stat3 inhibition. To test whether combining Stat3 targeting and sunitinib treatment could lead to better antitumor efficacy, RCC tumor cells were transfected with either control siRNA (scrambled) or Stat3 siRNA and treated with increasing doses of sunitinib. Combination treatment leads to small apoptotic effects, resulting in enhanced tumor cell apoptosis as well as inhibition of proliferation (Fig. 3B).

Levels of Stat3 activity affect the direct antitumor effects of sunitinib. A, Stat3 inhibition reduces expression of genes important for survival and proliferation. RCC tumor cells were transfected with Stat3 or control siRNAs and total cell lysates were collected 48 h after transfection. Left, Western blot analyses of lysates with indicated antibodies; right, total RNA was extracted from the same samples and gene expression of Stat3, survivin, Bcl-xL, and Cdc25A was assessed by real-time PCR. B, knockdown of Stat3 enhances the effects of sunitinib on RCC tumor cell apoptosis and growth arrest. 786-O tumor cells transfected with either control or Stat3 siRNA followed by treatment (24 h) with sunitinib at indicated doses. Tumor cells positive for both Annexin V and DAPI, as determined by flow cytometry, were considered apoptotic. Cell proliferation was analyzed by MTS assay. Columns, mean (n = 3, in triplicate); bars, SD. C, overexpression of a constitutively activated Stat3 (Stat3C) rescues RCC cells from apoptosis induced by sunitinib. Pooled 786-O tumor cells containing a control vector, pRC-vector, or the pRC-Stat3C expression vector were treated (24 h) with sunitinib at indicated concentrations. Percentage of dead cells was calculated by dividing the number of trypan blue–stained cells by the total cell number (n = 3, in triplicate). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Levels of Stat3 activity affect the direct antitumor effects of sunitinib. A, Stat3 inhibition reduces expression of genes important for survival and proliferation. RCC tumor cells were transfected with Stat3 or control siRNAs and total cell lysates were collected 48 h after transfection. Left, Western blot analyses of lysates with indicated antibodies; right, total RNA was extracted from the same samples and gene expression of Stat3, survivin, Bcl-xL, and Cdc25A was assessed by real-time PCR. B, knockdown of Stat3 enhances the effects of sunitinib on RCC tumor cell apoptosis and growth arrest. 786-O tumor cells transfected with either control or Stat3 siRNA followed by treatment (24 h) with sunitinib at indicated doses. Tumor cells positive for both Annexin V and DAPI, as determined by flow cytometry, were considered apoptotic. Cell proliferation was analyzed by MTS assay. Columns, mean (n = 3, in triplicate); bars, SD. C, overexpression of a constitutively activated Stat3 (Stat3C) rescues RCC cells from apoptosis induced by sunitinib. Pooled 786-O tumor cells containing a control vector, pRC-vector, or the pRC-Stat3C expression vector were treated (24 h) with sunitinib at indicated concentrations. Percentage of dead cells was calculated by dividing the number of trypan blue–stained cells by the total cell number (n = 3, in triplicate). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To provide direct evidence that Stat3 activity in RCC cells affected sunitinib-induced tumor cell apoptosis, an expression vector encoding a constitutively activated Stat3 mutant, Stat3C (26), and an empty control vector were transfected into RCC cells followed by selection of cells with the transfected vectors. Pooled tumor cells that survived the antibiotic selection, and which were positive for either the control vector or Stat3C-expressing vector, were then treated with sunitinib. We found that expression of the constitutively activated Stat3 mutant rendered RCC cells significantly more resistant to sunitinib-induced tumor cell death (Fig. 3C).
Sunitinib inhibits tumor Stat3 in vivo and induces tumor cell apoptosis before collapse of tumor vasculature. We next assessed whether sunitinib inhibited RCC tumor growth by inducing tumor cell apoptosis through Stat3 inhibition. Western blot analysis confirmed that sunitinib inhibited Stat3 in murine Renca tumor cells in vitro (Fig. 4A,, left). Sunitinib treatment of Renca tumor–bearing mice, with clinically relevant doses (10–40 mg/kg/d via oral administration; refs. 3, 27), resulted in inhibition of tumor growth (Fig. 4A,, right), which was correlated with a reduction in Stat3 activity in tumors (Fig. 4B,, left) and a reduction of cyclin E and survivin protein expression (Fig. 4B,, right). Consistent with that observed in RCC cell lines in vitro, sunitinib inhibited p-Stat3 and p-Src effectively in growing tumors as early as 24 hours after treatment (Fig. 5A,, left). In addition, similar to that seen in RCC tumor cells in vitro, the effects of sunitinib on p-AKT and p-ERK1/2 were not detectable in growing tumors (Fig. 5A , left).

Sunitinib inhibits Renca tumor growth, which corresponds to p-Stat3 reduction. A, left, sunitinib inhibits Stat3 activity in cultured Renca tumor cells. Tumor cells were treated (2 h) with sunitinib at indicated concentrations and cell lysates were then used for Western blot analysis. Right, sunitinib inhibits Renca tumor growth. BALB/c mice were implanted s.c. with Renca cells (2.5 × 106). Sunitinib or vehicle control was administered orally, once daily, at the indicated doses 7 d after tumor challenge. Points, mean (n = 6); bars, SE. P < 0.01. B, sunitinib inhibits p-Stat3 protein level (left) and reduces cyclin E and survivin expression (right) in Renca tumors. Western blot analyses of tumor tissues harvested 10 d after sunitinib treatment using indicated antibodies.
Sunitinib inhibits Renca tumor growth, which corresponds to p-Stat3 reduction. A, left, sunitinib inhibits Stat3 activity in cultured Renca tumor cells. Tumor cells were treated (2 h) with sunitinib at indicated concentrations and cell lysates were then used for Western blot analysis. Right, sunitinib inhibits Renca tumor growth. BALB/c mice were implanted s.c. with Renca cells (2.5 × 106). Sunitinib or vehicle control was administered orally, once daily, at the indicated doses 7 d after tumor challenge. Points, mean (n = 6); bars, SE. P < 0.01. B, sunitinib inhibits p-Stat3 protein level (left) and reduces cyclin E and survivin expression (right) in Renca tumors. Western blot analyses of tumor tissues harvested 10 d after sunitinib treatment using indicated antibodies.
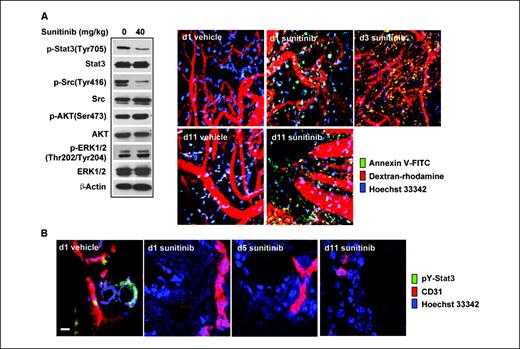
Sunitinib inhibits tumor Stat3 activity in vivo, leading to tumor cell apoptosis, which occurs before tumor vessel collapse. A, BALB/c mice were challenged with Renca tumor cells and then treated with sunitinib 7 d later. On indicated days after first sunitinib treatment, mice were given retroorbital injections of FITC-labeled Annexin V (green, to detect apoptotic cells), dextran-rhodamine (red, to stain vessels), and Hoechst 33342 (blue, to stain nuclei). IVMPM analysis was performed, 15 min after injection, on tumors grown in mice treated with vehicle or sunitinib. Western blot analysis of Renca tumor tissues harvested 1 d after sunitinib treatment shows its effects on p-Stat3 and other indicated molecules. B, frozen tumor sections of vehicle- and sunitinib-treated tumors (same as in A) were stained for phosphotyrosine-Stat3 (pY-Stat3; green), CD31/PECAM-1 (red), and Hoechst 33342 (blue) and analyzed by confocal laser scanning microscopy. Scale bar, 10 μm.
Sunitinib inhibits tumor Stat3 activity in vivo, leading to tumor cell apoptosis, which occurs before tumor vessel collapse. A, BALB/c mice were challenged with Renca tumor cells and then treated with sunitinib 7 d later. On indicated days after first sunitinib treatment, mice were given retroorbital injections of FITC-labeled Annexin V (green, to detect apoptotic cells), dextran-rhodamine (red, to stain vessels), and Hoechst 33342 (blue, to stain nuclei). IVMPM analysis was performed, 15 min after injection, on tumors grown in mice treated with vehicle or sunitinib. Western blot analysis of Renca tumor tissues harvested 1 d after sunitinib treatment shows its effects on p-Stat3 and other indicated molecules. B, frozen tumor sections of vehicle- and sunitinib-treated tumors (same as in A) were stained for phosphotyrosine-Stat3 (pY-Stat3; green), CD31/PECAM-1 (red), and Hoechst 33342 (blue) and analyzed by confocal laser scanning microscopy. Scale bar, 10 μm.
IVMPM was then used at different time points after treatment to determine if sunitinib was able to directly induce tumor cell apoptosis in vivo; to this aim, FITC-labeled Annexin V was injected retro-orbitally shortly before microscopy assay to capture in vivo tumor cell apoptosis. Dextran-rhodamine (red) and Hoechst (blue) dyes were also given retro-orbitally to label blood vessels and cell nuclei, respectively. Our data suggested that sunitinib induced tumor cell apoptosis in vivo as early as 1 day after treatment, which occurred in the presence of apparently intact tumor vessels (Fig. 5A,, right). There seemed to be more apoptosis in the tumor on days 3 and 11 after treatment, with greater disruption of the tumor vasculature (Fig. 5A,, right). These data were confirmed by fluorescent immunohistochemistry and confocal laser scanning microscopy of frozen tumor sections stained for p-Stat3 (green), endothelial cells (CD31/PECAM-1, red), and nuclei (Hoechst 33342, blue), which showed that Stat3 was activated in tumor cells and perhaps in tumor endothelial cells (Fig. 5B). Sunitinib treatment reduced Stat3 activity and induced tumor cell death as early as 1 day after treatment (Fig. 5B). Although it is difficult to quantify tumor vasculature, it seemed that CD31+ vessel structure was readily detectable on days 1 and 5 after treatment (Fig. 5B). Taken together, these data indicate that sunitinib inhibited tumor Stat3 activity in vivo and induced tumor cell death before tumor vessel collapse.
Stat3 inhibition by sunitinib in tumor myeloid cells reduces expression of angiogenic genes and number of immunosuppressive cells. A critical role of Stat3 in inhibiting dendritic cell activity in tumors has been shown (17, 19). Inhibition of Stat3 signaling in tumor dendritic cells has also been shown to reduce accumulation of tumor Tregs (17). We therefore used flow cytometry and staining with antibodies against intracellular p-Stat3 to assess whether sunitinib inhibited Stat3 in tumor dendritic cells. Our data suggested that systemic sunitinib treatment reduced tumor dendritic cell Stat3 activity and that the observed reduction was associated with an up-regulation of the costimulatory molecule CD80 (Fig. 6A). These results agreed with previous genetic studies reporting a role for Stat3 in the inhibition of costimulatory molecule expression (17).

Effects of Stat3 inhibition by sunitinib on the tumor microenvironment. A, CD11c+ dendritic cells were enriched from Renca tumors harvested from mice treated with either vehicle or sunitinib (20 or 40 mg/kg orally). Flow cytometric analyses of dendritic cell intracellular levels of p-Stat3 in tumor and surface expression of CD80 from tumor-draining lymph nodes are shown. B, sunitinib inhibits Src and Stat3 activity in macrophages and MDSCs in Renca tumor–bearing mice, leading to reduced expression of several Stat3-regulated angiogenic gene. Shown are Western blots (left) and real-time PCR analyses (right) using samples prepared from CD11b+CD11c− myeloid cells in mice receiving vehicle or sunitinib. Samples were pooled from six mice per group, and assays were done in triplicate. C, reduction of tumor-associated MDSCs in Renca tumor–bearing mice treated with sunitinib. Both splenic and peripheral blood CD11b+Gr1+ myeloid cells were detected by flow cytometry. Columns, mean (n = 12); bars, SE. **, P < 0.01; ***, P < 0.001. D, reduction of p-Stat3 and MDSCs in Renca tumor–bearing mice correlates with decreasing tumor Tregs. Single-cell suspensions prepared from tumors pooled from five to six mice were analyzed by flow cytometry. Columns, mean percentages of FoxP3+ cells in tumor-infiltrating CD4+ T cells (n = 3 independent experiments); bars, SE. P < 0.01.
Effects of Stat3 inhibition by sunitinib on the tumor microenvironment. A, CD11c+ dendritic cells were enriched from Renca tumors harvested from mice treated with either vehicle or sunitinib (20 or 40 mg/kg orally). Flow cytometric analyses of dendritic cell intracellular levels of p-Stat3 in tumor and surface expression of CD80 from tumor-draining lymph nodes are shown. B, sunitinib inhibits Src and Stat3 activity in macrophages and MDSCs in Renca tumor–bearing mice, leading to reduced expression of several Stat3-regulated angiogenic gene. Shown are Western blots (left) and real-time PCR analyses (right) using samples prepared from CD11b+CD11c− myeloid cells in mice receiving vehicle or sunitinib. Samples were pooled from six mice per group, and assays were done in triplicate. C, reduction of tumor-associated MDSCs in Renca tumor–bearing mice treated with sunitinib. Both splenic and peripheral blood CD11b+Gr1+ myeloid cells were detected by flow cytometry. Columns, mean (n = 12); bars, SE. **, P < 0.01; ***, P < 0.001. D, reduction of p-Stat3 and MDSCs in Renca tumor–bearing mice correlates with decreasing tumor Tregs. Single-cell suspensions prepared from tumors pooled from five to six mice were analyzed by flow cytometry. Columns, mean percentages of FoxP3+ cells in tumor-infiltrating CD4+ T cells (n = 3 independent experiments); bars, SE. P < 0.01.
A recent study also showed that Stat3 was constitutively activated in tumor macrophages and MDSCs, promoting tumor angiogenesis (15). Other studies identified Stat3 as an important molecule in MDSC accumulation in tumor-bearing mice (20). Because sunitinib inhibits Stat3 in tumor cells, we tested whether it also inhibited Stat3 in tumor-associated myeloid cells. Western blot analysis of p-Stat3 of MDSCs isolated from tumor-free and Renca tumor–bearing mice indicated that Stat3 activity was elevated in tumor-associated CD11b+/CD11c− myeloid cells (Fig. 6B), which include MDSCs and macrophages. Treating mice systemically with sunitinib inhibited Stat3 activity in MDSCs and macrophages (Fig. 6B). However, sunitinib did not have notable inhibitory effects on p-AKT and p-ERK in the tumor-associated MDSCs (Fig. 6B). Consistent with the antiangiogenic effects of Stat3 ablation in tumor macrophages and MDSCs (15), sunitinib-induced Stat3 inhibition correlated with down-regulation of several Stat3-regulated angiogenic genes, as determined by real-time PCR (Fig. 6B,, right). Supporting a critical role of Stat3 in promoting tumor MDSC accumulation, flow cytometric analyses to detect CD11b+/Gr1+ MDSCs indicated that inhibition of Stat3 by sunitinib was accompanied by a significant reduction in MDSCs (Fig. 6C) as well as tumor-infiltrating Tregs (Fig. 6D).
Discussion
Our results showed that sunitinib inhibited Stat3 and induced direct RCC tumor cell apoptosis, independent of tumor vasculature destruction. Furthermore, sunitinib inhibition of Stat3 in tumor-associated myeloid cells was accompanied by a reduction of MDSCs and tumor-infiltrating Tregs. These findings warrant evaluation of Stat3 as a biomarker for sunitinib response and resistance in RCC clinical trials and establish the evolving paradigm of using these targeted agents to assist in the combined targeted/immunologic and potential curative approaches to this disease.
Our data indicated that RCC tumor cell apoptosis correlated with Stat3 inhibition and that persistent Stat3 activation rescued tumor cells from sunitinib-induced death. Although activated AKT, MAPK, and JAK2 were not significantly affected by sunitinib in RCC tumor cells, it is possible that these pathways might be affected by sunitinib in other RCC tumor cells. Because RCC tumor cells, like all cancer cells, have different genetic backgrounds with various mutations, the cause of Stat3 activation is variable. Although our results showed that inhibition of p-Src by sunitinib was important for Stat3 down-regulation, other tyrosine kinases upstream of Stat3, including VEGFR and PDGFR, in RCC tumor cells (12) can also be targets of sunitinib, leading to inhibition of Stat3. At the same time, whether sunitinib can inhibit Stat3 in tumor cells with activated interleukin-6 (IL-6) receptor and other cytokine receptor signaling remains to be determined. Further studies in this area should allow for the rational design of clinical trials to effectively block Stat3 in RCC tumor cells. For example, if sunitinib does not effectively block IL-6 receptor-induced Stat3 activation, it might be desirable to combine sunitinib with IL-6–directed therapy (e.g., antibody therapy).
Our in vivo studies involving murine RCC Renca tumors indicated that sunitinib inhibited Stat3 activity in tumor cells, perhaps also in tumor endothelial cells. Tumor cell apoptosis correlated with inhibition of Stat3 in growing tumors but not with p-AKT or p-ERK. In addition, tumor cell apoptosis occurred before tumor vasculature collapse, suggesting tumor cell apoptosis was an independent factor affecting RCC response to sunitinib. Recent studies have also indicated the importance of Stat3 for tumor angiogenesis and metastasis (12, 28). Stat3 is constitutively activated not only in tumor cells but also in tumor endothelial and myeloid cells, including tumor-associated macrophages and MDSCs (15, 17), promoting the expression of a large number of angiogenic and metastatic factors (12, 28). Inhibition of Stat3 activity in tumor, endothelial, or myeloid cells reduces endothelial migration and potential to form capillary tubes in vitro and in vivo (15). Because Stat3 is also downstream of VEGFR, basic fibroblast growth factor receptor, and PDGFR (12), it is likely that Stat3 inhibition contributes to the antiangiogenic effects of sunitinib. However, additional studies are required to further test this hypothesis. On the other hand, our results showed that sunitinib inhibits Src/Stat3 activity in tumor-associated MDSCs, reducing expression of several Stat3-regulated angiogenic genes in MDSCs derived from sunitinib-treated mice. These findings support our previous results showing a role of Stat3 in tumor macrophages and MDSCs in promoting tumor angiogenesis (15).
A recent published study showed that sunitinib induces Th-1 immune response (IFN-γ expression) while reducing Tregs in RCC cancer patients, although the underlying mechanism is unknown (8). It seems that in patients, sunitinib has an indirect inhibitory effect on Tregs (8). In a related study, it was suggested that sunitinib treatment in RCC patients reduced the number of MDSCs (11). It is possible that the reduction of Tregs by sunitinib treatment in RCC patients is mediated by a down-regulation of MDSCs and/or increase in IFN-γ production. Constitutive activation of Stat3 in tumor-infiltrating dendritic cells and MDSCs has been shown in several murine tumor models and human tumor specimens (15, 17). Inhibiting Stat3 in tumor-infiltrating dendritic cells has also been correlated with their activation, reduction of tumor-infiltrating Tregs, and activation of CD8+ effector cells (17). Furthermore, recent studies showed that tumor-infiltrating MDSC/macrophages displayed highly activated Stat3 (15) and that Stat3 was critical for up-regulation of myeloid-related protein S100A9, which inhibited dendritic cell differentiation and MDSC accumulation in cancer (20). Our current study further showed that sunitinib administration in Renca tumor–bearing mice led to the inhibition of Stat3 in tumor-associated myeloid cells, including dendritic cells and MDSCs, which was accompanied by a reduction of tumor Tregs. These results suggest that the immunomodulating effects of sunitinib in RCC patients might be mediated by Stat3 inhibition and that the direct antiapoptotic effects of sunitinib against RCC tumor cells were, at least in part, associated with Stat3 inhibition. These findings warrant further clinical investigation to evaluate Stat3 as biomarker for sunitinib response and resistance in RCC patients and to explore the use of combinatory approaches, including molecular targeting and immunotherapies, to enhance the antitumor efficacy of sunitinib against immunologically responsive tumor such as RCC.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
H. Xin, C. Zhang, and A. Herrmann contributed equally to this work.
Acknowledgments
Grant support: National Cancer Institute (H. Yu), Nancy & Richard Bloch, Vicki Lynn Piha Ashberg, Paul and Marcia Mazzocchi, and The Kure It! Kidney Cancer Research Funds at City of Hope.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank the City of Hope Light Microscopy Digital Imaging and Flow Cytometry cores, the Animal Facility at the Beckman Research Institute at City of Hope for their superb technical assistance, and Drs. Silvia da Costa and Katherine Henderson for reading and editing.