


Abstract
B-cell maturation antigen (BCMA) is expressed on normal and malignant plasma cells and represents a potential target for therapeutic intervention. BCMA binds to two ligands that promote tumor cell survival, a proliferation inducing ligand (APRIL) and B-cell activating factor. To selectively target BCMA for plasma cell malignancies, we developed antibodies with ligand blocking activity that could promote cytotoxicity of multiple myeloma (MM) cell lines as naked antibodies or as antibody-drug conjugates. We show that SG1, an inhibitory BCMA antibody, blocks APRIL–dependent activation of nuclear factor-κB in a dose-dependent manner in vitro. Cytotoxicity of SG1 was assessed as a naked antibody after chimerization with and without Fc mutations that enhance FcγRIIIA binding. The Fc mutations increased the antibody-dependent cell-mediated cytotoxicity potency of BCMA antibodies against MM lines by ∼100-fold with a ≥2-fold increase in maximal lysis. As an alternative therapeutic strategy, anti-BCMA antibodies were endowed with direct cytotoxic activity by conjugation to the cytotoxic drug, monomethyl auristatin F. The most potent BCMA antibody-drug conjugate displayed IC50 values of ≤130 pmol/L for three different MM lines. Hence, BCMA antibodies show cytotoxic activity both as naked IgG and as drug conjugates and warrant further evaluation as therapeutic candidates for plasma cell malignancies. [Mol Cancer Ther 2007;6(11):3009–18]
Introduction
B-cell maturation antigen (BCMA, CD269) is a member of the TNF receptor superfamily (1), TNFRSF17. Expression of BCMA is restricted to the B-cell lineage where it is predominantly expressed in the interfollicular region of germinal centers (2) and on differentiated plasma cells (3) and plasmablasts (4). BCMA binds to two distinct ligands, a proliferation inducing ligand (APRIL; ref. 5) and B-cell activating factor (BAFF; ref. 6), the latter also being known as BlyS (7), TALL-1 (8), and THANK (9). The ligands for BCMA bind two additional TNF receptors, transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) and BAFF receptor (BAFF-R; ref. 10), also called BR3 (11). TACI binds APRIL and BAFF, whereas BAFF-R shows restricted but high-affinity binding to BAFF. Together, BCMA, TACI, BAFF-R, and their corresponding ligands regulate different aspects of humoral immunity, B-cell development, and homeostasis (12).
BAFF-R, TACI, and BCMA have unique function and expression during B-cell ontogeny. BAFF-R is expressed on naïve and CD27+ memory B cells (13, 14) but is down-regulated on antibody secreting plasmablasts (4). TACI expression is limited on naïve B cells but is increased on the memory B subset (13, 14). Vital roles for BAFF and BAFF-R in B-cell homeostasis are suggested by in vivo mouse studies where deficiency of either BAFF or BAFF-R leads to reduced B-cell numbers and impaired B-cell differentiation (10, 15). In contrast, TACI acts as both a positive and negative regulator of immune function. This divergent role is shown by TACI mutations in man, resulting in combined variable immunodeficiency (16), and TACI deficiency in mice that leads to impaired response to T1 antigens, coupled with heightened B-cell responsiveness, B-cell accumulation, and predisposition to autoimmunity (17, 18).
BCMA is virtually absent on naïve and memory B cells (13, 14) but it is selectively induced during plasma cell differentiation where it may support humoral immunity by promoting the survival of normal plasma cells and plasmablasts (3, 4). BCMA-mediated survival of plasmablasts and bone marrow plasma cells can be achieved with either BAFF or APRIL (3, 4). BAFF and APRIL are apparently prosurvival factors for malignant plasma cells (13, 19). Consistent expression of BCMA in primary multiple myeloma (MM) samples suggests that BCMA is a candidate receptor for regulating prosurvival pathways in malignant plasma cells. In addition to MM, BCMA has also been detected on the Reed-Sternberg cells (CD30+) from patients with Hodgkin's disease (2). Knockdown technology (small interfering RNA) showed that BCMA contributed to both proliferation and survival of a Hodgkin's disease cell line (2).
The cell survival and immune regulatory functions of TACI, BAFF-R, BCMA, BAFF, and APRIL have drawn considerable interest in these molecules as potential therapeutics for oncology and immunology (20, 21). Anti-BlyS antibodies (22, 23), as well as TACI-Fc and BR3-Fc immunoadhesins, are being evaluated in clinical trials for autoimmunity and B-cell malignancies (20). In contrast, we are unaware of any clinical trials with anti-BCMA antibody–based therapeutics, despite evidence that MM patients in remission with graft-versus-tumor response have BCMA antibodies that may be tumor-lytic in vivo (24).
The goal of this study was to explore antibody targeting of BCMA in vitro as a potential therapeutic strategy for plasma cell malignancies. Because BCMA is known to bind ligands that support tumor cell survival, we screened antibodies for their ligand blocking activity. The ability of BCMA antibodies to support antibody-dependent cellular cytotoxicity (ADCC) was evaluated against tumor cells, including Fc mutations known to enhance ADCC by increasing the affinity for FcγRIIIA (25). Additionally, antibody-drug conjugates (ADC) were generated by conjugation of the BCMA antibodies to the potent cytotoxic drug, monomethyl auristatin F (26). Drug conjugation endowed BCMA antibodies with an alternative and potent means to kill BCMA-positive tumor cells.
Materials and Methods
Cell Lines
MM lines U-266 and JJN3 came from the DSMZ, and NCI-H929 was acquired from the American Type Culture Collection. NCI-H929 and U-266 cells were grown in RPMI supplemented with 10% fetal bovine serum. JJN3 cells were grown in 40% DMEM, 40% Iscove's modified Dulbecco's medium, and 20% fetal bovine serum.
Materials
Peroxidase-conjugated and phycoerythrin-conjugated goat anti-rat IgG was purchased from Jackson ImmunoResearch Laboratories. Peroxidase-conjugated anti-his came from BD Biosciences. Rat anti-hTACI and mouse anti-hBAFF-R were purchased from Axxora, LLC. Rat monoclonal antibody (mAb) isotype controls were obtained from R&D Systems. Phycoerythrin-conjugated anti-6× His was purchased from Martek Biosciences. 3,3′,5,5′-Tetramethylbenzidine was purchased from Pierce. Cell-Titer Glo was from Promega Corporation and the nuclear factor-κB (NF-κB) activation kit was from Active Motif. Basal medium; penicillin; streptomycin; fetal bovine serum; and sodium hypoxanthine, aminopterin, and thymidine were purchased from Invitrogen. Cloning factor and fetal clone I was from Roche Diagnostics and Hyclone, respectively.
Generation of Human BCMA Glutathione S-Transferase Fusion Protein
The human BCMA extracellular domain (ECD; amino acids 5–51; NP_001183) was amplified with forward primer 5-AAGCTTGGATCCATGTTGCAGATGGCTGGGCAGTGCTCC-3 incorporating a BamH1 site (underlined) and reverse primer 5-GAATTCGCGGCCGCTCATCCTTTCACTGAATTGGTCACACTTGCATTAC-3 incorporating a stop codon (italic) and NotI site (underlined) using IMAGE clone 687194 (Invitrogen) as a PCR template. The PCR product was cloned into pGEX4T1 (Stratagene) upstream of glutathione S-transferase (GST), transformed into BL-21 strain (Stratagene), and the induced protein was purified at 4°C on an ÄKTAexplorer (GE Healthcare). The cell pellet was lysed in 1:15 w/v of B-PER buffer (Pierce) containing protease inhibitor and lysozyme. The extract was supplemented with 1 to 2 μg/mL DNase I (Sigma), stirred for an additional 20 min, and adjusted to pH 7.5. The soluble fusion protein was collected after centrifugation at 31,000 × g for 20 min (Beckman) and loaded onto a glutathione Sepharose 4 FF column (GE Healthcare) preequilibrated with B-PER buffer. The column was washed with 4 column volumes (CV) B-PER buffer, 3 CV each of wash buffers 1 and 2 (Pierce), followed by a final column wash with 5 CV 50 mmol/L Tris (pH 8.0), 0.15 mol/L NaCl. The GST-tagged BCMA was eluted with 20 mmol/L reduced glutathione in 50 mmol/L Tris (pH 8.0) and dialyzed against PBS (pH 7.4) using a 3500 MWCO slide-A-lyzer (Pierce). For GST tag removal, BCMA:GST was treated with thrombin in 50 mmol/L Tris (pH 8.0), 0.15 mol/L NaCl, while bound to the glutathione Sepharose. Released thrombin was captured by a benzamidine Sepharose column (GE Healthcare). The GST-cleaved BCMA was eluted from the column with 3 to 5 CV 50 mmol/L Tris (pH 8.0), 0.15 mol/L NaCl, and dialyzed against PBS (pH 7.4). Thrombin removal was confirmed by analyzing fractions for thrombin activity using the chromogenic substrate S-2238 (Chromogenix, DiaPharma). Protein concentration was determined by A280. All purified proteins were analyzed by SDS-PAGE and by TSK-Gel G3000SW HPLC size exclusion chromatography (Tosoh Bioscience).
Generation of Human BCMA 293 Transfectants
Full-length human BCMA was amplified using forward primer 5-GAATTCAAGCTTGCCACCATGTTGCAGATGGCTGGGCAGTGCTCC-3 including a HindIII restriction site (underlined) and Kozak consensus sequence and reverse primer 5-GAATTCTCTAGATTACCTAGCAGAAATTGATTTCTCTATCTCCGTAGC-3 including a 3 stop codon and XbaI restriction site (underlined) using IMAGE clone 687194 (Invitrogen) as a PCR template. The amplification product was cloned into pcDNA3.1, linearized, transfected into 293 cells, and Zeocin selected (Invitrogen). High expressing stable clones were chosen by fluorescence-activated cell sorting analysis.
Generation of Δ-APRIL
Murine APRIL (residues 106–241; NP_076006) was amplified from IMAGE clone 5290965 (Invitrogen) and cloned into bacterial expression vector pET32a fused at the COOH terminus to thioredoxin (Novagen). Mouse APRIL, rather than human APRIL, was synthesized because it has favorable biochemical properties for purification1
I. Grewal, unpublished data.
Antibody Generation
Female Sprague-Dawley rats were immunized s.c. with keyhole limpet hemocyanin–conjugated BCMA ECD (amino acids 5–54; NP_001183) using TiterMax adjuvant (Sigma). Keyhole limpet hemocyanin conjugation was done through a lysine residue using Imject mcKLHV (Pierce). Rats were chosen for antibody production because of the high sequence homology between human and mouse BCMA proteins. B cells were harvested from immunized spleens and fused to P3-X63.Ag8 myeloma cells using a standard polyethylene glycol fusion protocol (27). Hybridomas were cultured in 80% Iscove's modified Dulbecco's medium with 4 mmol/L l-glutamine, 10% fetal clone I, 10% cloning factor supplemented with penicillin, streptomycin and 1× sodium hypoxanthine, aminopterin, and thymidine. Hybridoma culture supernatants were tested for binding to BCMA by ELISA. Positive hybridomas were then screened by flow cytometry for cell-based binding to BCMA transfectants and for ligand blockade activity in a plate-based assay. Top hybridomas went through two rounds of limiting dilution cloning and were expanded for purification.
Antibody Purification
Rat antibodies were purified using protein G-Sepharose chromatography (GE Healthcare). Briefly, the antibody-containing conditioned medium were concentrated ∼10-fold and buffer-exchanged into 125 mmol/L boric acid, 100 mmol/L NaCl (pH 9.0), by tangential flow filtration (Millipore). The samples were then adjusted to 50 mmol/L boric acid and 3 mol/L NaCl (pH 9.0; BN buffer) and loaded onto a protein G column preequilibrated with BN buffer. The column was washed with 5 to 6 CV 50 mmol/L BN buffer. Rat mAbs were eluted with either PBS (pH 7.4; IgG1), 50 mmol/L sodium citrate (pH 3.4; IgG2b), or 0.1 mol/L glycine (pH 2.7; IgG2a), and pools were dialyzed against PBS (pH 7.4). Antibody concentrations were estimated spectrophotometrically [ε280 (1%) = 1.4]. Endotoxin levels in antibody and ADC preparations was determined by quantitative kinetic Limulus amebocyte lysate assay as described (28).
ELISA and Plate-Based Ligand Blockade
Immunosorb 96-well plates were coated with 1.5 μg/mL of GST-BCMA-ECD, washed with PBS + 1% Tween (PBS-T), and blocked with PBS-T plus 1% (w/v) bovine serum albumin. BCMA-coated plates were incubated with hybridoma culture supernatants for 2 h at room temperature, washed 5× with PBS-T, and incubated with peroxidase-conjugated goat–anti-rat IgG. Following incubation with secondary antibody, plates were washed, incubated with 3,3,5,5-tetramethylbenzidine substrate, and stopped with an equal volume of 1 mol/L H2SO4. For plate-based ligand blockade, plates were coated with 1 μg/mL of GST-BCMA-ECD as described above. Coated plates were preincubated with purified antibodies at the specified concentrations, washed with PBS-T, and then incubated with 3 μg/mL of recombinant human MegaAPRIL (Alexis Biochemicals). APRIL binding was detected using peroxidase-conjugated anti-FLAG followed by development with 3,3′,5,5′-tetramethylbenzidine as described above.
Flow Cytometry and Cell-Based Ligand Blockade
Flow cytometry was done by incubating cells with specified concentrations of primary antibody. Cells were washed and antibody binding was detected with a phycoerythrin-conjugated secondary antibody. For saturation binding studies, antibodies were directly conjugated with Alexa Fluor 546 (Invitrogen) and added at the specified concentrations to determine saturation. To assess ligand blockade, cells were preincubated with 10 μg/mL antibodies, washed, and incubated with Δ-APRIL. Δ-APRIL was detected using a phycoerythrin-conjugated anti-6× His. All flow cytometry was done on a FACScan (BD Biosciences) and the data were analyzed with CellQuest software.
Chimerization and Fc Mutagenesis of SG1
SG1 was chimerized using RNA isolated from hybridoma cells as previously described (29). Amplified VH and VL fragments were cloned into pUC19, and DNA from five VH and five VL clones was sequenced to obtain the consensus sequence. The SG1 VL domain was fused to the human κ constant domain within a mammalian expression vector containing a cytomegalovirus promoter. Likewise, the SG1 VH domain was fused to the human IgG1 constant domains. The triple Fc mutation, S293D:A330L:I332E (25), was introduced into the SG1 IgG1 constructs using a Quikchange kit (Stratagene). Briefly, the S293D mutation was introduced into IgG1 using mutagenic sense and antisense primers (sense shown, mutated residue underlined) 5-CTCCTGGGGGGACCGGACGTCTTCCTCTTCCCC-3 followed by introduction of the A330L and I332E mutations using sense and antisense mutagenic primer (sense shown, mutated residues underlined) 5-CCAACAAAGCCCTCCCACTGCCCGAGGAGAAAACCATCTCCAAAGCC-3. The wild-type and mutant forms of recombinant chimeric antibodies were expressed transiently in 293 cells and purified using protein A.
NF-κB Activation Assays
H929 cells were washed and incubated in RPMI plus 0.25% fetal bovine serum for 24 h before treatment. Next, cells untreated or treated for 20 min with 0.1 μg/mL TNFα, 1 μg/mL heat-treated HT-Δ-APRIL, 1 μg/mL Δ-APRIL, 1 to 40 μg/mL rat IgG2a isotope control, or 1 to 40 μg/mL SG1. To assess ligand blockade, cells were treated with 1 μg/mL of Δ-APRIL after a 20-min pretreatment with 1 to 40 μg/mL of SG1 or an isotype control antibody. Cells were harvested, washed, and lysed with 50 mmol/L Tris-HCl (pH 7.5), 1% NP40, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA plus protease, and phosphatase inhibitors. Protein extracts were analyzed for NF-κB activity using a TransAM chemiluminescent assay kit (Active Motif). Luminescent signal was read using a Fusion HT plate reader (Packard Instruments).
ADCC Assay
ADCC activity was measured using a 51Cr release assay as previously described (30). 51Cr-labeled target tumor cells were preincubated with antibodies 15 to 30 min before the addition of effector cells. CD16+ effector cells were isolated from a normal FcγRIIIA-158V donor as described (30) and added at a ratio of 10:1 relative to the target cell. After 4-h incubation, the 51Cr released from lysed cells was measured and the percent specific lysis calculated as (test sample cpm − spontaneous cpm) / (total cpm − spontaneous cpm) × 100. Spontaneous release of isotope was determined from the supernatant of target cells incubated in medium alone. Total counts were determined from target cells lysed with 1% Triton X-100.
Cytotoxicity Assay
Maleimidocaproyl-valine-citrulline-p-aminobenzoyl-monomethyl auristatin F (vcMMAF) was synthesized and conjugated to cysteine residues on purified rat mAb after DTT reduction as previously described (26). Cells were plated at 5,000 per well in 96-well plates in the presence or absence of antibody or ADCs. Cell viability was assessed 96 h after exposure to antibody or ADCs using a luminescent cell viability assay (CellTiter-Glo, Promega). Luminescent signal was read using a Fusion HT plate reader (Packard Instruments). The IC50 values were determined as the drug concentration that results in 50% of cell growth or viability of the untreated control wells.
Results
Generation of BCMA-Selective mAbs for Tumor Cell Targeting
We generated a panel of BCMA-specific antibodies to explore mechanisms of targeting BCMA for oncology. Hybridomas were screened based on (a) BCMA-specific reactivity, (b) ligand blockade activity, and (c) cell-based binding. Three of 14 hybridomas that displayed robust binding to BCMA were advanced for further characterization, namely, SG1, SG2, and SG3. These antibodies showed similar binding to BCMA by ELISA (Fig. 1A) but differ in their ability to inhibit ligand binding (Fig. 1B). SG1 and SG3 significantly inhibited APRIL binding to BCMA, whereas SG2 did not (Fig. 1B). SG1 was chosen for subsequent ligand blockade studies because it showed greater potency at lower antibody concentrations when compared with SG3. Binding of SG-1 to CD138+ B cells was confirmed by immunohistochemistry using frozen sections of normal human tonsil (data not shown).
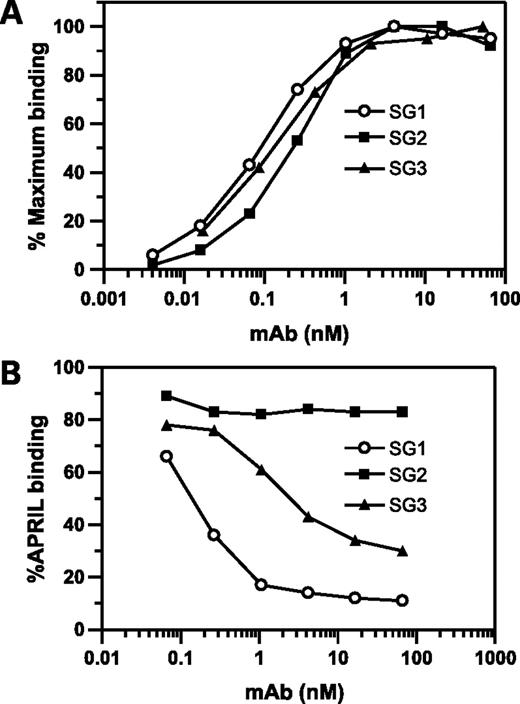
mAb binding to BCMA and their blockade of APRIL binding. A, direct binding of mAb SG1, SG2, and SG3 to immobilized GST-hBCMA-ECD as detected by ELISA. B, APRIL binding after mAb pretreatment of immobilized BCMA. SG1 and SG3 resulted in a dose-dependent inhibition of APRIL binding, whereas no significant inhibition was detected with SG2. APRIL binding to GST-hBCMA-ECD was detected with a horseradish peroxidase–conjugated anti-FLAG antibody. APRIL did not show any binding to GST alone (data not shown).
mAb binding to BCMA and their blockade of APRIL binding. A, direct binding of mAb SG1, SG2, and SG3 to immobilized GST-hBCMA-ECD as detected by ELISA. B, APRIL binding after mAb pretreatment of immobilized BCMA. SG1 and SG3 resulted in a dose-dependent inhibition of APRIL binding, whereas no significant inhibition was detected with SG2. APRIL binding to GST-hBCMA-ECD was detected with a horseradish peroxidase–conjugated anti-FLAG antibody. APRIL did not show any binding to GST alone (data not shown).
BCMA-Specific mAbs Block APRIL Binding and Inhibit NF-κB Signaling
We sought to develop an in vitro system to selectively examine APRIL interactions with the receptor, BCMA. To eliminate confounding signals derived from the receptor, TACI, we screened malignant B-cell lines to identify cell lines that express BCMA but not TACI. H929 cells fit the desired profile showing high levels of BCMA expression while lacking TACI expression (Fig. 2A). Moderate expression of BAFF-R was observed but this was not a concern because full-length and truncated forms of APRIL do not bind significantly to BAFF-R (31, 32). Full-length APRIL binds heparin sulfate proteoglycans in addition to BCMA and TACI (31, 32). For this reason, we generated a truncated form of APRIL that lacks the heparin sulfate proteoglycan binding site (Δ-APRIL) so that binding of APRIL to H929 cells will be BCMA dependent and heparin sulfate proteoglycan independent (31, 32). Size exclusion chromatography showed that purified Δ-APRIL is a trimeric protein,2
K. Kim, unpublished observations.
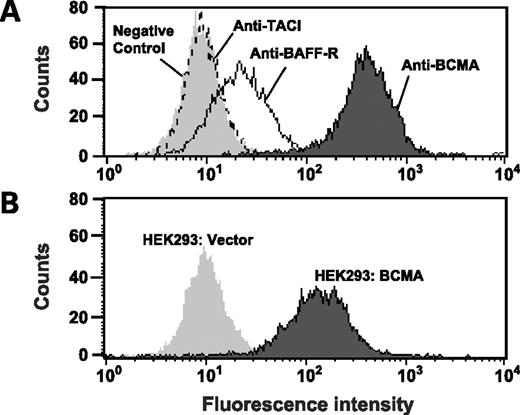
BCMA is highly expressed on H929 cells and HEK 293 stable transfectants. A, H929 cells were incubated with control IgG, anti-TACI, anti-BAFF-R, and anti-BCMA mAb SG1 followed by detection with a phycoerythrin-conjugated secondary antibody. H929 cells expressed high levels of BCMA, moderate levels of BAFF-R, but no TACI. B, HEK 293 cells were stably transfected with either a full-length BCMA construct (HEK 293:BCMA) or vector control (HEK 293:vector). Anti-BCMA mAb SG1 showed strong binding to HEK 293:BCMA but not HEK 293:vector.
BCMA is highly expressed on H929 cells and HEK 293 stable transfectants. A, H929 cells were incubated with control IgG, anti-TACI, anti-BAFF-R, and anti-BCMA mAb SG1 followed by detection with a phycoerythrin-conjugated secondary antibody. H929 cells expressed high levels of BCMA, moderate levels of BAFF-R, but no TACI. B, HEK 293 cells were stably transfected with either a full-length BCMA construct (HEK 293:BCMA) or vector control (HEK 293:vector). Anti-BCMA mAb SG1 showed strong binding to HEK 293:BCMA but not HEK 293:vector.

mAb SG1 blocks binding of APRIL to HEK 293:BCMA transfectants and H929 cells. A, HEK 293:vector and HEK 293:BCMA transfectants were incubated with and without Δ-APRIL either alone or after pretreatment with SG1 or an isotype-matched IgG1 control. APRIL binding was detected with a phycoerythrin-conjugated anti-His antibody. Ligand binding of Δ-APRIL was detected in HEK 293:BCMA transfectants but not HEK 293:vector control. Pretreatment with SG1 but not the isotype control blocks Δ-APRIL binding to HEK 293:BCMA transfectants. B, H929 cells were incubated with or without Δ-APRIL after preincubation with an isotype control or SG1. Detection of Δ-APRIL binding with a phycoerythrin-conjugated anti-His antibody showed that SG1, but not the control antibody, blocked ligand binding.
mAb SG1 blocks binding of APRIL to HEK 293:BCMA transfectants and H929 cells. A, HEK 293:vector and HEK 293:BCMA transfectants were incubated with and without Δ-APRIL either alone or after pretreatment with SG1 or an isotype-matched IgG1 control. APRIL binding was detected with a phycoerythrin-conjugated anti-His antibody. Ligand binding of Δ-APRIL was detected in HEK 293:BCMA transfectants but not HEK 293:vector control. Pretreatment with SG1 but not the isotype control blocks Δ-APRIL binding to HEK 293:BCMA transfectants. B, H929 cells were incubated with or without Δ-APRIL after preincubation with an isotype control or SG1. Detection of Δ-APRIL binding with a phycoerythrin-conjugated anti-His antibody showed that SG1, but not the control antibody, blocked ligand binding.
Next, we determined the functional consequences of SG1 on signaling in H929 cells. Because several TNF superfamily ligands promote cell survival and gene expression through NF-κB activation, we examined whether Δ-APRIL could activate NF-κB signaling in H929 cells. Both TNFα and Δ-APRIL stimulated NF-κB activation when compared with the no treatment control (Fig. 4). The activation of NF-κB by Δ-APRIL could be blocked by heat inactivation (Fig. 4) or by adding BCMA-Fc (data not shown). SG1 does not possess agonist activity as judged by its inability to activate NF-κB in the absence of ligand. Moreover, SG1 in the presence of a cross-linking antibody also failed to induce NF-κB activation (data not shown). In contrast, when SG1 was added as an antagonist antibody to block APRIL binding, NF-κB activity was inhibited in a dose-dependent manner whereas addition of a mAb isotype control had no effect. Thus, APRIL promotes NF-κB signaling in tumor cells through BCMA, and this activity can be specifically blocked with functionally antagonistic antibodies generated against BCMA. Maximal NF-κB inhibition was observed at an antibody concentration of 67 nmol/L, with significant inhibition achieved even at 6.7 nmol/L. The apparent binding affinity (KD) of SG1 to H929 cells was estimated to be 51 nmol/L by saturation binding. These data suggest that maximal inhibition of NF-κB may not require complete blockade of BCMA binding.

SG1 blocks APRIL-dependent NF-κB activation in H929 cells. Nuclear extracts were harvested from quiescent H929 cells that were treated with serum-free medium (vehicle), TNFα, Δ-APRIL heat-treated HT-Δ-APRIL, SG1, or an isotype control. Alternatively, nuclear extracts were harvested from quiescent H929 cells that were treated with Δ-APRIL after preincubation with increasing amounts (6.7, 33, 67, or 268 nmol/L) of SG1 or an isotype control. NF-κB activity was assayed using a functional ELISA that detects chemiluminescent signal from p65 bound to the NF-κB consensus sequence.
SG1 blocks APRIL-dependent NF-κB activation in H929 cells. Nuclear extracts were harvested from quiescent H929 cells that were treated with serum-free medium (vehicle), TNFα, Δ-APRIL heat-treated HT-Δ-APRIL, SG1, or an isotype control. Alternatively, nuclear extracts were harvested from quiescent H929 cells that were treated with Δ-APRIL after preincubation with increasing amounts (6.7, 33, 67, or 268 nmol/L) of SG1 or an isotype control. NF-κB activity was assayed using a functional ELISA that detects chemiluminescent signal from p65 bound to the NF-κB consensus sequence.
BCMA Antibodies Support ADCC
Having established that a BCMA antibody can specifically block APRIL signaling in tumor cells, we then tested the potency of SG1 in ADCC assays to determine if a BCMA antibody can also support immune-mediated killing of tumor cells. To this end, SG1 was converted into a rat-human chimeric IgG by fusing the rat VH and VL domains to wild-type human IgG1 heavy chain and κ light chain constant domains, respectively. The chimerized antibody, designated cSG1 wild-type, showed similar antigen binding properties when compared with the parental antibody SG1 (Fig. 5A). Next, we installed Fc mutations, S239D:A330L:I332E, known to enhance ADCC (25), to generate cSG1 mutant. Similar to cSG1 wild-type, generation of the Fc triple mutant did not alter the antigen-binding properties of cSG1 mutant (data not shown).
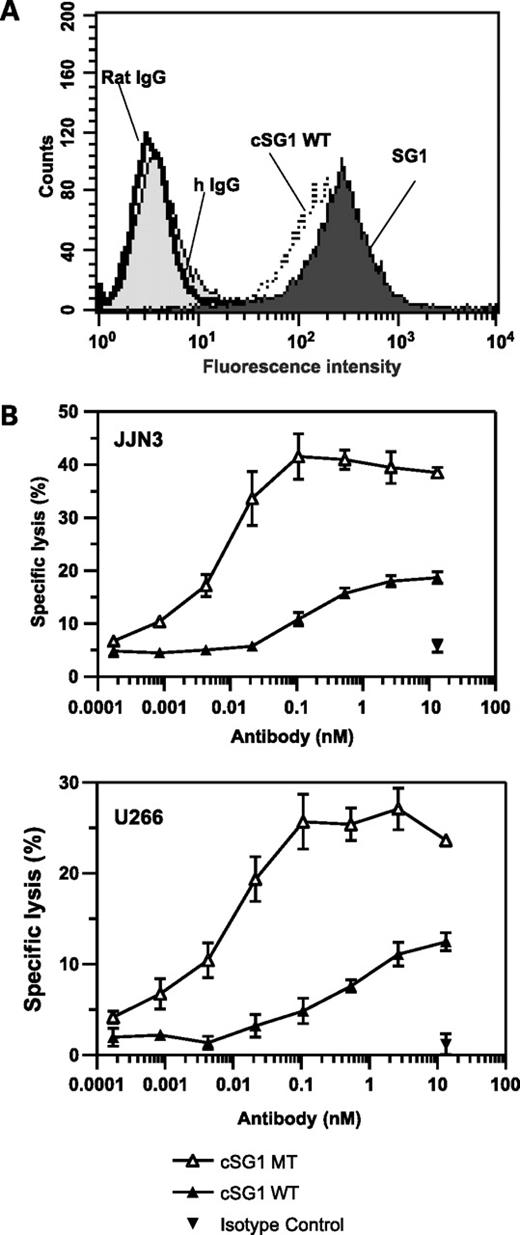
Antigen binding and ADCC activity of cSG1. A, flow cytometry shows that the antigen-binding activity of cSG1 wild-type is similar to that of the parental antibody SG1 on HEK 293:BCMA transfectants. Isotype controls (rat IgG and hIgG) show no binding. B, ADCC assays showing specific lysis of U266 cells or JJN3 cells using cSG1 wild-type (WT) and cSG1 mutant (MT). The nonbinding control IgG1 did not show any significant lysis. Target cells were mixed with natural killer cell–enriched peripheral blood mononuclear cells at an effector-to-target ratio of 10:1. Specific lysis was determined by 51Cr release assay.
Antigen binding and ADCC activity of cSG1. A, flow cytometry shows that the antigen-binding activity of cSG1 wild-type is similar to that of the parental antibody SG1 on HEK 293:BCMA transfectants. Isotype controls (rat IgG and hIgG) show no binding. B, ADCC assays showing specific lysis of U266 cells or JJN3 cells using cSG1 wild-type (WT) and cSG1 mutant (MT). The nonbinding control IgG1 did not show any significant lysis. Target cells were mixed with natural killer cell–enriched peripheral blood mononuclear cells at an effector-to-target ratio of 10:1. Specific lysis was determined by 51Cr release assay.
Evaluation of cSG1 wild-type and cSG1 mutant in an ADCC assay with purified natural killer cells resulted in dose-dependent lysis of JJN3 and U266 cells whereas no significant lysis was observed with a nonbinding human IgG control. The cSG1 wild-type antibody displayed limited ADCC activity on JJN3 cells, which was increased ∼100-fold in potency and >2-fold in efficacy (maximal lysis) by cSG1 mutant (Fig. 5B). Similarly, for U266 cells, the ADCC activity of cSG1 mutant was enhanced ∼100-fold in potency and 2-fold in efficacy compared with the parent chimeric antibody. The concentration of cSG1 mutant required for maximal lysis of both JJN3 and U266 cells was ∼100 pmol/L. In contrast, the dissociation constant (KD) of cSG1 on JJN3 and U266 cells was estimated as 15 and 10 nmol/L, respectively. Thus, maximal lysis by cSG1 mutant was achieved at concentrations well below those required to reach saturation binding. This enhanced ADCC activity is in agreement with published data showing that Fc triple mutant S239D:A330L:I332E increases the affinity of human IgG1 Fc domain for FcγRIIIA and corresponding potency in ADCC assays (25). Thus, BCMA antibodies have the potential to kill tumor cells where ligand-blockade effects may be overridden by redundant APRIL and BAFF receptors or by constitutive activation of NF-κB (33, 34).
In vitro Potency and Selectivity of Auristatin-Conjugated BCMA mAbs
An alternative strategy for enhancing the antitumor activity of antibodies is by conjugation to potent cytotoxic drugs (35), such as a monomethyl auristatin F, a potent inhibitor of tubulin polymerization (26). Robust antitumor activity through this strategy requires internalization of the antibody followed by drug release. We assessed the ability of our panel of BCMA antibodies to induce cytotoxicity as ADCs using vcMMAF (26) with a stoichiometry of eight drugs per antibody. SG1-vcMMAF8, SG2-vcMMAF8, and SG3-vcMMAF8 were potently cytotoxic against H929 cells (Fig. 6A). As expected, no decline in cell viability was observed using a nonbinding control ADC (Fig. 6A) or unconjugated antibodies (data not shown). We also examined the potency of BCMA ADCs across other MM cell lines, including JJN3 and U266 cell lines. SG1-vcMMAF8 showed consistent and high potency (IC50 values ≤130 pmol/L) across all three MM cell lines, whereas SG2-vcMMAF8 and SG3-vcMMAF8 showed more variability and less overall potency across the three cell lines with IC50 values that ranged from 210 to 430 pmol/L and 120 to 1,000 pmol/L, respectively (Fig. 6B).

Anti-BCMA drug conjugates have cytotoxic activity against BCMA-positive myeloma cell lines. H929 (A and B), JJN3 (B), and U266 (B) cell lines were incubated with 0.2 pmol/L to 13 nmol/L of SG1-vcMMAF8, SG2-vcMMAF8, or SG3-vcMMAF8. Cell viability was assessed by luminescent detection of ATP levels. B, IC50 values (mean ± SE) are shown from three independent experiments done in quadruplicate with the exception of JJN3 cells. The mean IC50 value for JJN3 cells is based on two independent experiments done in quadruplicate. No activity was observed using IgG-vcMMAF7 as a nonbinding control ADC (A).
Anti-BCMA drug conjugates have cytotoxic activity against BCMA-positive myeloma cell lines. H929 (A and B), JJN3 (B), and U266 (B) cell lines were incubated with 0.2 pmol/L to 13 nmol/L of SG1-vcMMAF8, SG2-vcMMAF8, or SG3-vcMMAF8. Cell viability was assessed by luminescent detection of ATP levels. B, IC50 values (mean ± SE) are shown from three independent experiments done in quadruplicate with the exception of JJN3 cells. The mean IC50 value for JJN3 cells is based on two independent experiments done in quadruplicate. No activity was observed using IgG-vcMMAF7 as a nonbinding control ADC (A).
Discussion
Here, we investigated BCMA as a potential therapeutic target for oncology by creating ligand-blocking BCMA antibodies with in vitro antitumor activity against MM cell lines. We focused on MM because current treatments for this disease are rarely curative and patients eventually relapse (36).
Much evidence provides a strong rationale for targeting BCMA in plasma cell malignancies. First, BCMA has limited expression on normal cells but is well established as a signature molecule expressed on malignant plasma cells (13, 19, 37). Differential analyses of genes expressed on myeloma cells identified BCMA as up-regulated when compared with nonmyeloma cell lines (37). Moreover, most primary myeloma isolates express either BCMA or TACI with more limited expression of BAFF-R (13, 19). The prevalence of BCMA and TACI on MM samples suggests that either receptor could be targeted. Targeting either receptor has the potential to cause immunodeficiency by affecting normal cellular counterparts, including BCMA+ plasma cells. However, BCMA, unlike TACI, is absent from memory B cells (13, 14). Hence, any immunosuppressive effects are expected to be short-lived and reversible.
A second major rationale for targeting BCMA is that this is a target antigen in graft versus tumor response in MM patients after donor lymphocyte infusion (24). Postinfusion serum from responders induced ADCC and complement-dependent cytotoxicity in BCMA-transfected cells and BCMA-positive primary myeloma cells (24). In contrast to postdonor lymphocyte serum, which presumably contains polyclonal antibodies to BCMA, we evaluated the ADCC activity of SG1, an antagonist mAb to BCMA. The chimeric antibody, cSG1 mutant, has potent ADCC activity against MM lines, supporting BCMA as a target for tumor antigen-specific immunotherapy. cSG1 mutant was engineered with the triple mutations S239D:A330L:I332E, which were previously shown by others to enhance the binding affinity of IgG1 for FcγRIIIA and increase the in vitro potency of mAbs in ADCC assays (25). Consistently, cSG1 mutant showed an ∼100-fold increase in ADCC potency and a ≥2-fold increase in maximal lysis of tumor cells when compared with the wild-type counterpart, cSG1 wild-type. Our BCMA antibodies have yet to be evaluated in complement-dependent cytotoxicity assays. For rituximab, the triple mutant S239D:A330L:I332E enhances ADCC but abolishes complement-dependent cytotoxicity, whereas a related double mutant, S239D: I332E, enhances ADCC while preserving complement-dependent cytotoxicity (25).
Engineering of antibody Fc protein sequence or tailoring of Fc glycosylation have become a focus of intense investigation for enhancing the effector functions of antibody therapeutics (38). Clinical studies with rituximab have underscored the importance of Fc-dependent interactions in patients with polymorphisms in the FCGR3A gene (39). FCGR3A polymorphisms result in either valine (V) or phenylalanine (F) being incorporated at position 158 of the corresponding FcγRIIIA protein. Natural killer cells homozygous for V158 have increased affinity for human IgG1 and display more potent ADCC activity in vitro (40). Consistently, follicular NHL patients that were homozygous for the V158 polymorphism in the FCGR3A gene showed improved responsiveness to rituximab (39).
Antibodies in oncology are commonly used in combination with cytotoxic chemotherapy to increase their antitumor activity (41). An alternative strategy to increase the potency of antitumor antibodies is by conjugation to a cytotoxic drug. These so-called ADCs are an emerging class of therapeutics that aim to combine the potency of cytotoxic drugs with the targeting selectivity of antibodies (35). We generated potent ADCs by conjugating BCMA antibodies to the cytotoxic drug, monomethyl auristatin F (26). The most potent ADC identified, SG1-vcMMAF8, resulted in IC50 values of ≤130 pmol/L across all three BCMA-positive MM lines evaluated. Moreover, exquisite selectivity was shown with a control ADC lacking cytotoxic activity, IC50 >2,000 nmol/L.
Auristatin-containing ADCs directed at >20 different tumor antigens have shown robust antitumor activity in xenograft models (35). Moreover, two auristatin-containing ADCs have advanced into phase I clinical trials: SGN-35 for CD30+ hematologic malignancies (42, 43) and CR011-vcMMAE targeting glycoprotein NMB in metastatic melanoma (44).
Recently, an alternative approach was developed to target malignant B-cell lines (45) or chronic lymphocytic leukemia (46) by fusing the ligand BlyS (also known as BAFF) with a xenogeneic recombinant protein toxin, gelonin. This ligand-toxin fusion protein, rGel/BlyS, was tested for potency and selectivity on several different B-cell lines that included mantle cell lymphoma, diffuse large B-cell lymphoma, and MM (45). The greatest potency of rGel/BlyS was observed on mantle cell lymphoma and diffuse large B-cell lymphoma cell lines, with IC50 values of 1 to 5 pmol/L. In contrast, the rGel/BlyS fusion protein showed less potency on MM lines with IC50 values of 200 to 280 nmol/L. Furthermore, unlike the exquisite selectivity of the ADCs described here, the selectivity for rGel/BlyS was limited as judged by IC50 values that were only 3.5-fold lower than those observed for rGel alone. The much greater selectivity of the BCMA ADCs over the ligand-toxin fusion protein seems to be a significant advantage for clinical applications.
Here, we generated a panel of BCMA-specific antibodies that were categorized as either blocking or nonblocking based on a differential ability to inhibit APRIL binding. The lead ligand-blocking antibody, SG1, was assayed in vitro to evaluate BCMA signaling in H929 cells. A truncated form of APRIL, designated here as Δ-APRIL, shows no detectable binding to cell-surface heparin sulfate proteoglycans (31), and enabled evaluation of signaling through TNF superfamily receptors. Because BCMA is the only TNF receptor superfamily member on H929 cells capable of binding APRIL, we could evaluate signaling through endogenous BCMA. This approach is distinct from previous studies that examined BCMA-dependent signaling by introducing constructs of BCMA into a nonexpressing cell line such as HEK 293T cells (47–49) or by triggering BCMA signaling with an agonistic antibody (2). A drawback to the use of HEK 293 transfectants is that overexpression of full-length BCMA results in ligand-independent NF-κB activation (47, 48). Likewise, the use of agonistic antibodies does not discriminate between signaling through APRIL or BAFF. Our studies are, to our knowledge, the first to show APRIL-dependent NF-κB activation through endogenous BCMA and show that a function blocking antibody can directly inhibit BCMA-specific signaling. Hence, our ligand blockade studies not only confirm biological signaling of APRIL through endogenous BCMA but they also provide supportive data for therapeutic strategies aimed at targeting of BCMA alone or in combination with other therapies. The importance of NF-κB signaling in MM is highlighted by the clinical success of bortezomib, a proteasome inhibitor that targets this pathway (50). However, the prevalence of mutations that constitutively activate NF-κB in MM (33, 34) suggest that antagonistic BCMA antibodies should have significant ADCC activity or be conjugated with cytotoxic drugs to achieve optimal potency for oncology indications.
Beyond plasma cell malignancies, a BCMA antagonist antibody warrants evaluation for autoimmune applications as suggested by recent publications. Ligation of BCMA expressed on transfected A20 cells or ex vivo culturing of BCMA-positive spleen cells resulted in up-regulation of proteins required for antigen presentation and an improved antigen presentation of ovalbumin (49). BCMA, but not TACI or BAFF-R, was capable of inducing antigen presentation. Although all three receptors triggered NF-κB activation, only BCMA was capable of activating c-Jun N-terminal kinase (JNK) in this system. JNK then transduced downstream signals that conferred a BCMA-specific signaling pathway for antigen presentation (49). Our results show that SG1 can block NF-κB signaling in H929 cells without inducing agonistic activity—critical for a BCMA-based antibody therapeutic. Together, these data suggest that a pure antagonist antibody such as SG1 could be used to modulate BCMA signaling for oncology as well as immunologic applications.
In summary, we explored BCMA as a target for tumor-antigen–specific immunotherapy by developing and engineering BCMA antibodies with in vitro antitumor activity. We confirmed that BCMA antibodies can act on MM cell lines through multiple mechanisms that include inhibition of APRIL-dependent NF-κB activation, promotion of tumor cell lysis by natural killer cell–mediated ADCC activity, and induction of cytotoxicity by ADCs. Together, these findings support further evaluation of BCMA as a target for plasma cell malignancies using naked antibodies and ADCs.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Current address for D. Peckham: Amgen, Inc., Seattle, Washington. Current address for C.F. McDonagh: Merrimack Pharmaceuticals, Inc., Cambridge, Massachusetts. Current address for R.F. Zabinski: deCODE Biostructures, Inc., Bainbridge Island, Washington.
Acknowledgments
We thank Kim Kissler, Dana Chace, and Ivan Stone for help with animal studies; Jeff Johnson for expert technical assistance; Julie McEarchern for guidance on ADCC assays; Tim Lewis for advice on NF-κB activation assays; and Che-Leung Law for critical reading of the manuscript.