The innate immune response is mediated via critical stimulation of the inflammatory response through the release of cytokines and consequent recruitment of macrophages to the site of infection/injury. A key event in this response is the formation of the inflammasome, a multi-protein complex that serves as the scaffold for the activation of the central protease in this response, caspase-1. Caspase-1 subsequently leads to the maturation of the proinflammatory cytokines and to pyroptosis, a form of cell death that is induced by bacterial pathogens. Members of the Nod-like receptor family are critical components of the inflammasome and link microbial and endogenous danger signals to caspase-1 activation. By utilizing certain upstream and downstream events in the assembly and activation process, researchers are able to assess the engagement of the innate immune response. The article reviews the research methods for evaluating inflammasome activation including priming and induction methods, in vivo study models and inflammasome inhibitors.
In response to microbial infection the inflammasome is activated and contributes to host protection by inducing immune responses that limit microbial invasion. Thus, the inflammasome assembly into a multi-protein complex and certain events that are associated with this process have been utilized to evaluate the status of the immune response. A key event in this process is activation of caspase-1, the central protease required for the stimulation of pro-cytokines and pro-gasdermin-D into their mature forms. Cytokines attract phagocytes to facilitate the removal of cellular debris and clearance of microbes, stimulating wound healing and homeostasis restoration. Mature gasdermin-D molecules assemble into N-terminal fragments that form pores in the plasma membrane and promote pyroptosis - an inflammatory form of cell death that also leads to the release of mature cytokines [4]. More recently, Z Zhou et al showed that granzyme A from cytotoxic T lymphocytes and natural killer cleaved and activated gasdermin B to induce target cell pyroptosis [5] Although the inflammasome complex formation is a desired inflammatory response it may be damaging and contribute to an acceleration of pathology and to worsening outcome. For example, Zewinger S et al identified apolipoprotein C3 as an inducer of sterile inflammation and organ damage through an alternative inflammasome activation pathway [6]. Increasing interest in assessing the inflammasome complex activity has therefore been generated in autoinflammatory syndromes and autoimmune diseases and in study of host defense response system against microbes. The unique inflammasome components, assembly process, activated enzymes, consequent changes in cytokine levels and associated changes in cellular processes serve as important background features and the basis for the development of research methods.
Inflammasomes are molecular complexes that are comprised of three basic protein units:
- • A sensor molecule, which may include one of these; NLRP1, NLRP2, NLRP3, NLRC4, AIM2, or pyrin.
- • The adaptor ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain or CARD). The ASC adaptor is associated with all inflammasomes and its role is to recruit pro-caspase-1 into the inflammasome. The ASC is pivotal for activation and perpetuation of inflammation.
- • Pro-caspase 1. Caspase-1 is a cysteine protease that initiates or executes cellular programs, leading to inflammation or cell death. Caspases are synthesized as inactive zymogens, and their potent cellular activities are tightly controlled by proteolytic activation. Caspase-1 is the most fully characterized. Its catalytic activity is tightly regulated by signal-dependent autoactivation of inflammasome complex that mediate caspase-1- dependent processing of cytokines such as IL-1β [7].
Among the inflammasome components (see Table 1), the sensor molecule is unique. Each sensor molecule responds to different stimuli and creates a unique inflammasome which is named by the sensor molecule. For example, the best known is the NLRP3 inflammasome which utilizes the NLRP3 sensor molecule. Immune sensing is mediated in the innate immune system through engagement of an array of germline-encoded pattern-recognition receptors (PRRs) that detect invariant microbial motifs. NOD-like receptors (NLRs) such as NLRP3 recognize pathogen-associated molecular patterns (PAMPs), as well as host-derived danger signals (danger-associated molecular patterns, DAMPs). The NLR family is characterized by the presence of a central nucleotide-binding and oligomerization (NACHT) domain, the only domain common to all NLR family members. NACHT is commonly flanked by C-terminal leucine-rich repeats (LRRs) and N-terminal caspase recruitment (CARD) or pyrin (PYD) domains. The LRRs function in ligand sensing and autoregulation, and CARD and PYD domains mediate homotypic protein-protein interactions for downstream signaling. The NACHT domain enables activation of the signaling complex via ATP-dependent oligomerization. Unlike the NLRs, the sensor molecule AIM2 belongs to intracellular nucleic-acid sensing PRRs that provide cytosolic surveillance by sensing DNA [8], and AIM2 inflammasome is essential for neurodevelopment [9]. A more detailed coverage of the signals that induce the sensor molecules of the inflammasome is provided discussed later.
| Sym | Protein | Top three suppliers |
|---|---|---|
| AIM2 | absent in melanoma 2 | Cell Signaling Technology 12948 (7), Abcam ab204995 (1), BioLegend 652804 (1) |
| CARD8 | caspase recruitment domain family member 8 | Santa Cruz Biotechnology sc-81213 (1) |
| CASP1 | caspase 1 | Adipogen AG-20B-0042 (34), Santa Cruz Biotechnology sc-56036 (24), Cell Signaling Technology 3866 (15) |
| CASP4 | caspase 4 | Novus Biologicals NB120-10454 (19), Santa Cruz Biotechnology sc-56056 (8), MBL International M029-3 (4) |
| CASP5 | caspase 5 | Sino Biological 10292-R018 (2), Cell Signaling Technology 46680 (2), Santa Cruz Biotechnology sc-393346 (1) |
| CASP12 | caspase 12 | Santa Cruz Biotechnology sc-21747 (1) |
| DDX3X | DEAD-box helicase 3 X-linked | LifeSpan Biosciences LS-C64576 (3), Santa Cruz Biotechnology sc-81247 (2), Cell Signaling Technology 8192 (2) |
| GSDMD | gasdermin D | Abcam ab209845 (18), Santa Cruz Biotechnology sc-81868 (5) |
| NLRC4 | NLR family CARD domain containing 4 | Abcam ab201792 (3), Cell Signaling Technology 12421 (2), BioLegend 659702 (1) |
| NLRP1 | NLR family pyrin domain containing 1 | MBL International M032-3 (2) |
| NLRP3 | NLR family pyrin domain containing 3 | Adipogen AG-20B-0014-C100 (43), Cell Signaling Technology 15101 (30), Abcam ab263899 (11) |
| PYCARD | PYD and CARD domain containing | Santa Cruz Biotechnology sc-514414 (13), MBL International D086-3 (5), LifeSpan Biosciences LS-C175123 (4) |
In response to pathogens and other damage-associated signals that induce the inflammasome, inactive NLRP3 senses a stress signal which trigger structural changes in NLRP3 and activates its oligomerization domain (NACHT)-rendering it active. Active NLRP3 can then oligomerize and form a wheel-shaped signaling hub with the LRR domains on the outside of the wheel. The PYD domains form a disk at the center, and generate a platform for the recruitment of the inflammasome adaptor ASC. ASC binds to the PYD ring via its PYD domain. Recruited ASC molecules polymerize to form long helical filaments [10]. The caspase recruited domain (CARD) of ASC now recruits pro-caspase-1, and the inflammasome is fully formed.
Pro-capase-1 clustering within the inflammasome triggers dimerization, and pro-caspase-1 cleaves itself to generate the fully active protease. Capsase-1 then cleaves substrates such as pro-IL-1β and pro-IL-18 to generate the mature cytokines. Caspase-1 also cleaves pro-gasdermin-D to generate N-terminal fragments. These fragments begin to assemble together and form gasdermin-D pores in the plasma membrane which enables cytokine secretion, water influx, and osmotic swelling, leading to cell rupture and to the release of the cytoplasmic content including alarmins. Alarmins and inflammasome-dependent cytokines attract phagocytes to the site to mediate inflammatory responses, removing cellular debris, clearing microbes, and promoting wound healing to restore homeostasis. An excellent movie that tracks the events that lead to a fully active inflammasome complex and the ensuing pyroptosis may be visualized.
A recent study has described complexes of disc-shaped active NLRP3 oligomers with adenosine 5'-O-(3-thio)triphosphate, the centrosomal NIMA-related kinase 7 (NEK7) and the adaptor protein ASC [11]. The NACHT-associated domain conserved in NLRP3 was shown to mediate most interactions in the inflammasome disc. Mutations on these interactions suppress NLRP3-mediated caspase-1 activation. Although NEK7 is not directly involved in NLRP3 oligomerization, NEK7 was found to act as a molecular switch in NLRP3 activation by disrupting the cage to allow the formation of the active NLRP3 disc. The results of the study indicated that the role of NEK7 is to transform NLRP3 into the active NLRP3 inflammasome disc.
The analysis of inflammasome formation is generally conducted by monitoring downstream processes and outcomes. These methods are not direct assessments of the formation of an inflammasome structure, and in some cases can be outcomes of non-inflammasome processes. Hence, determination of inflammasome activity is best done by a combination of techniques, and by usage of cells with gene knockouts or specific inhibitors in order to confirm that responses require inflammasome function. These methods include the following assembly-process associated features:
- • Expression of the main inflammasome components; NLRP3 (or other sensory molecules), ASC, and caspase-1.
- • Detection of ASC specks - unique localized 3D structures that form upon inflammasome activation
- • The presence of cleaved/active caspase-1 enzyme - the gold-standard method for detecting inflammasome activation.
- • Caspase-1 activity on substrates such as pro-IL-1β, pro-IL-18 and pro-gasdermin-D
- • Cell lysis and the following increase in the level of mature IL-1β, IL-18 and gasdermin-D
Figure 1 provides a reference for these 5 assembly-associated inflammasome features.

Immunoblotting with antibodies that are raised against the main inflammasome components is an efficient detection method. These antibodies are available from antibody producing companies. Conveniently, kits for inflammasome detections are also available, and most of these include several components that also assess the caspase-1 activity status.
| Priming agent | Receptor | Details |
|---|---|---|
| Lipopolysaccharide (LPS) | TRL4 | LPS is the principal component of Gram-negative bacteria that activates the innate immune response through its recognition by TLR4, leading to signaling that activate the NF-κB signaling [12] |
| Synthetic triacylated lipopeptide | TRL2/TRL1 | Bacterial lipoproteins are proinflammatory cell wall components found in Gram positive and negative bacteria. The acylated amino terminus Pam3CSK4 mimics the acylated amino terminus of bacterial lipoproteins. It is a potent activator of NF-κB signaling which is mediated by cytoplasmic domain of the heterodimer TRL2/TRL1 [13, 14] |
| Polyinosine-polycytidylic acid (poly(I:C)) | TRL3 | poly(I:C) is a synthetic analog of double stranded RNA (dsRNA), a molecular pattern associated with viral infection which induces type I interferons (IFN) and other cytokines production through subsequent activation of the transcription factors NF-κB and AP-1 [15-17]. |
Important criteria for detecting some of the inflammasome components is the priming of the cells prior to detection. Inflammasome priming specifically affects NLRP3 at the transcriptional level and also serves to trigger posttranslational modifications of inflammasome components that allow for oligomerization [18]. Priming may include stimulation of receptor signaling that results in the activation of the transcription factor NF-κB, such as ligands for IL-1R1, TLRs, NLRs, and the cytokine receptors TNFR1 and TNFR2 [19, 20]. Table 2 describes common NLRP3 priming agents. NF-κB signaling activation is also critical for upregulating the transcription of pro-IL-1β and NLRP3. In contrast, ASC, pro-caspase-1, and the caspase-1 substrate - pro-IL-18, are often found in adequate concentrations in the steady state [19-21].
The assessment of ASC specks in cells has the obvious advantage of being a direct measure of inflammasome formation, and detects an early step in inflammasome formation at a single cell level. Activated danger or pathogen sensors trigger assembly of the inflammasome adaptor ASC into specks - large signaling platforms which are considered the hallmarks of inflammasome activation. All inflammasomes require ASC for the activation of caspase-1. ASC speck can be observed using the light microscope as it reaches a size of around 1 μm. In most cells only one speck forms upon inflammasome activation. Recently, more advanced flow cytometry and in vivo methods have been developed for ASC speck formation, expanding our ability to assess the inflammasome formation in animal models.
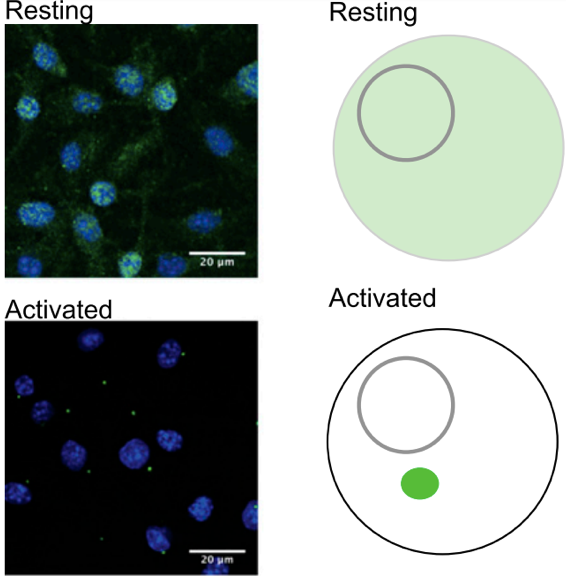
Visualization by light microscopy requires the expression of ASC fused to a fluorescent protein [22-24]. As indicated in Figure 2, the distinct switch from a diffused fluorescent signal that is present throughout the cell into a single bright point upon immune activation is considered a readout and a proxy for inflammasome activation [1, 24-26]. However, because expression of the ASC constructs also increases the cellular concentration of ASC, which has high tendency to aggregate when overexpressed [27], the risk that speck formation may occur without an inflammatory stimulus also increases [25].
To provide a rapid and sensitive technique for investigating molecular interactions in inflammasome formation, David P. Sester et al developed a flow cytometric assay for inflammasome formation - time of flight inflammasome evaluation - which detects the change in ASC distribution within the cell. The transit of ASC into the speck is detected in this technique by a decreased width or increased height of the pulse of emitted fluorescence [1]. Upon induction treatment with inflammasome stimuli such as the NLRP3 agonist nigericin, a potassium ionophore, ASC rapidly relocates to form the inflammasome speck in an all or nothing response. The flow cytometry approach allows analysis of the data in a window below the threshold of ASC expression at which specks tend to form spontaneously, and it also exclude poorly transfected cells in which ASC levels are not sufficient to permit speck formation, thus, minimizing the effects of variable transfection efficiency and the effects of any subsequent cell treatments that may alter the expression of the ASC plasmid, as well as reducing background speck numbers [1, 23]. In addition, this approach provides a rapid, sensitive, and quantitative technique for assessing the molecular interactions that govern inflammasome initiation.
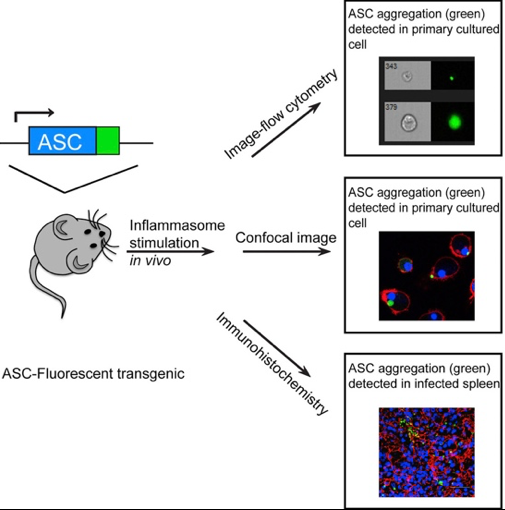
Tzeng et al developed a transgenic mouse model expressing ASC fluorescent protein (ASC-citrine) that upon treatment with inflammasome activators showed increased ASC aggregation, and formation of specks in primary cells and tissues. The ASC-citrine fusion protein is expressed in the Rosa26 locus, a knock-in ASC-citrine chimera with a proximal floxed stop site that allows for conditional expression of ASC-citrine in a lineage specific manner. Bone marrow-derived macrophages or bone marrow-derived dendritic cells from this reporter mouse showed ASC speck formation after inflammasome stimulation. Speck formation was detectable in the tissues of infected mice, both intracellularly and extracellularly. The authors showed that both hematopoietic cells and nonhematopoietic cells are capable of expressing specks and stimulating the downstream signal pathway when activated [2]. Figure 3 summarizes this in vivo detection system of activated inflammasome complex. Recently, speck formation within a tissue was also visualized by intravital imaging of macrophages derived from retrovirally transduced, ASC–GFP hematopoietic stem cells in bone marrow chimeric mice [28].
Endogenous ASC dynamics was also conducted in zebrafish in a CRISPR/Cas9 ASC reporter model that tagged the endogenous ASC gene. The authors used a toxic stimulus to trigger speck formation and observed rapid pyroptosis in keratinocytes in vivo. Using this model, the authors concluded that the formation of a single, compact speck and rapid cell-death induction in vivo requires the full-length ASC protein [29].
The presence of active caspase-1 enzyme is the gold-standard method for detecting inflammasome activation since caspase-1 activation is a highly regulated process. Several methods are utilized to assay for the active caspase-1 enzyme, however sensitivity and specificity challenges indicate that there is further need to develop novel methods for this area of research. Caspase-1 cleavage measured by Western blot provides good evidence for inflammasome activation, but it is generally only semiquantitative. Assessment by ELISA for released caspase-1 does not directly monitor active caspase-1. Another method to monitor active caspases, FLICA (Fluorescently Labeled Inhibitors of CAspase), requires a wash step, an inherent problem for capturing active caspase-1 since it is typically released from cells following inflammasome activation. In addition, analysis of caspase activity by means of fluorogenic inhibitor-based substrates may be disadvantaged by the limited specificity of the substrates for caspase-1 and the possibility of measuring other non-inflammasome-related caspase or protease activities. Genetic constructs that are used as biosensors for caspase-1 activity have also been described, and these require to be transfected/transduced into cells and may impact the endogenous pathway [3, 30].

Martha O’Brien et al developed a bioluminescent, plate-based caspase-1 activity assay that provides a one-step, high-throughput method to directly monitor inflammasome activity using a bioluminescent substrate in a coupled-enzyme system. This assay utilizes the substrate, Z-WEHD-aminoluciferin, which is cleaved by caspase-1, releasing a substrate for luciferase (aminoluciferin) resulting in the luciferase reaction and light production. In the assay, the two reactions occur simultaneously resulting in a stable signal (Figure 4). The assay specificity is conferred by inclusion of a proteasome inhibitor in the lytic reagent and caspase-1 inhibitor to confirm the activity, an approach that enables a specific and rapid determination of caspase-1 activation. Caspase-1 activation has been determined with this assay in THP-1 cells, a human monocytic cell line following treatment with inflammasome inducers such as α-hemolysin, LPS, nigericin, gramicidin, MSU, R848, Pam3CSK4 and flagellin. In addition, caspase-1 activation has also been demonstrated in treated J774A.1 mouse macrophage, bone marrow-derived macrophages, and in human primary monocytes [3]. An additional advantage of this assay is that it can be multiplexed with other assays to monitor additional parameters from the same cells, such as IL-1β release or cell death [3]. This method is available from Promega (Caspase-Glo(R) 1 Inflammasome Assay TM456). The selective cleaved substrate, Z-WEHD, releases Luciferin, which reacts chemically with Luciferase and results in light production that is corralated with caspase 1 activity.
Western blots of processed IL-1β and IL-18 or ELISA of released cytokines are frequently used as indicators for inflammasome activation and presume caspase-1 activation, for example, [31]. However, when assessing inflammasome activation indirectly with IL-1β ELISA assay, cross-reactivity with the pro-IL-1β species and the possibility that IL-1β may be released from cells when there is cytotoxicity without inflammasome activation [32], may provide inaccurate readouts [33, 34]. In addition, most ELISA assays require washing steps which interfere with the ability to detect secreted cytokines. To circumvent the washing step, some ELISA assays offer an unwashed protocol option, however the use of such protocol may result in loss of sensitivity which may vary for each cytokine. A joint assessment with immunoblot analysis to confirm caspase-1 cleavage to its bioactive form is therefore required. While the role of caspase-1 in processing of IL-1β and IL-18 is well defined [35, 36], how these cytokines are released is not completely understood [37]. Evidence suggests that pyroptosis is the mechanism for IL-1β and IL-18 release [38], however there is also evidence indicating that IL-1β and IL-18 are processed and released in the absence of pyroptosis [39, 40], particularly in neutrophils [41].
IL-1β/IL-18 Sensor Cells present an alternative to assay these caspase-1 processed cytokines. Reporter cells derived from HEK293 cells were developed by InvivoGen. HEK-Blue IL-1 β and HEK-Blue IL-18 cells are useful in monitoring bioactive IL-1β and IL-18 that are released by test cells upon inflammasome activation. These cells express a secreted alkaline phosphatase (SEAP) reporter gene which is induced by NF-κB/AP-1 upon activation of the cytokine signaling cascade. Levels of SEAP in the supernatant can then be easily monitored using QUANTI-Blue - a SEAP detection medium.
Inflammasome activation leads to pyroptotic and apoptotic cell death, but only pyroptosis is unique to the inflammasome. Pyroptotic death can be used to measure inflammasome responses, but its differentiation from other cell-death modalities can be problematic. One way of detecting pyroptotic cell death is by measuring the release of cytosolic lactate dehydrogenase (LDH) into the medium. A JOVE protocol using mouse bone marrow-derived macrophages in a 96-well plate format describes rapid detection of cell lysis using a LDH release assay [42]. The method measures the release of LDH into the cell supernatant, which occurs during pyroptotic macrophage lysis following caspase-1 activation. This approach allows for rapid analysis of samples to determine if there is caspase-1-mediated cell death.
A potential disadvantage of the LDH release assay is that it measures lysis at a cell population level and at a single time point per assay. It is important to note that LDH release indicates loss of plasma membrane integrity and cell lysis, which occurs during many other forms of cell death, and is not specific to pyroptosis. However, the use of pyroptosis inhibition that are based on caspase 1, 11, 4 or 5 ablations in this assay could help determine if cell death still occurs. This can be done by either chemical inhibitors or by using transgenic models. In the JOVE protocol, pyroptotic cell death is distinguished through the use of caspase-1/11 deficient cells, C1/C11 KO [42]. Chemical inhibitors such as Ac-YVAD-cmk, a cell-permeable, selective and irreversible inhibitor of the cysteine protease caspase-1, can also be used to block inflammasome activation, and hence to display anti-inflammatory, anti-apoptotic and anti-pyroptotic effects [43, 44].
An alternative approach is to quantify the uptake of cell impermeant dyes by microscopy, which identifies loss of membrane integrity on a single cell basis. Combined with live cell imaging platforms, this approach can also provide a temporal assessment of pyroptosis [45]. Specifically, small DNA binding fluorescent dyes such as propidium iodide or ethidium bromide can pass through the gasdermin-D pore, bind cellular nucleic acids and fluoresce [45-47]. Pore formation and uptake of these small dyes temporally precedes terminal cell lysis and can serve as pyroptosis indicators.
The generally accepted model of producing a functional inflammasome is a two-step process that includes both priming and activating signals. The priming signal leads to the activation of the transcription factor NF-κB and subsequent upregulation of NLRP3 and pro-IL-1β, also stimulating NLRP3 post-translational modifications that license its activation. The activation signal fuels multiple molecular or cellular events, including ionic flux, mitochondrial dysfunction and reactive oxygen species (ROS) generation, and lysosomal damage, leading to activation of the NLRP3 inflammasome, caspase-1 activation and IL-1β maturation and secretion [48].
The initial upstream priming step affects the sensor molecules of the inflammasome such as NLRP3, and initiates changes in the transcription level and in posttranslational modifications of inflammasome components. This step is paramount since inflammasome-activation signals in the absence of priming results in either a response of a nominal magnitude or in a complete failure to generate an inflammasome [18]. In general, priming stimuli are mediated through receptors that stimulate signaling of the transcription factor NF-κB which is critical for upregulating the transcription of both pro-IL-1β and NLRP3. Some of these stimuli include ligands for the pattern recognition receptors such as TLR4 and NOD2, or cytokine receptors, such as TNFR and IL-1R [18-20]. Although NLRP3 inflammasome activation is possible if priming and activation signals are provided simultaneously, the kinetics and extent of inflammasome activation are greatly enhanced with increased availability of NLRP3 and pro-IL-1β [49, 50]. Table 2 outlines three commonly used inflammasome priming agents.
Assembly signal follows the initial priming phase and was found to include acetylation of NLRP3 at lysine 24, which is vital for the oligomerization and the assembly of NLRP3 [51]. NLRP3-K24R knock-in mice showed diminished NLRP3 inflammasome activation. Also, KAT5 acetyltransferase acetylated NLRP3, and pharmacological suppression of KAT5 led to decreased NLRP3 activation.
Once primed, the inflammasome can be activated through a diverse group of agonists that trigger conformation change in the stimulated sensor molecules, leading to the assembly of the inflammasome complex and culminating in caspase-1 activation. The inflammasome inducers described in Table 3 may be classified as canonical (via caspase-1) and non-canonical inducers (via caspase 4/5 in humans and caspase-11 in mice). Both the canonical and noncanonical inflammasome activation modes eventually lead to cell lysis and the release of proinflammatory cytokines. In humans, caspase-4 is constitutively expressed in many non-monocytic cells and in monocytes; therefore, cytosolic LPS can activate the noncanonical inflammasome without the priming step [52]. The noncanonical inflammasome can sense a number of Gram-negative bacteria, but not Gram-positive bacteria, which indicates that the outer membrane LPS of Gram-negative bacteria may be the critical activator of the noncanonical inflammasome. In addition, NLRP3 inflammasome can be activated by both exogenous (including tissue damage, infection, and metabolic dysregulation) and endogenous inducers such as crystalline molecules, extracellular ATP- through its cell surface receptor, P2X7R, A fibrils, LPS, hyaluronan, and uric acid crystals [53].
| Canonical Inducers | Description |
|---|---|
| Alum- Aluminum salts such as aluminum hydroxide, aluminum phosphate or potassium aluminum sulfate | Aluminum salts are commonly used vaccine adjuvants. Alum induces an NLRP3 inflammasome-dependent induction of IL-1β and IL-18 [54]. |
| ATP- Adenosine triphosphate | ATP is a potassium efflux agent that triggers NLRP3 inflammasome activation in response to PAMPs such as LPS and peptidoglycan. ATP stimulates the caspase-1-dependent cleavage and secretion of IL-1β and triggers the opening of the non- selective cation channel of the purinergic P2X7 receptor, followed by the gradual opening of a larger pore [55]. |
| Calcium Pyrophosphate Dihydrate crystals (CPPD Crystals) | CPPD crystals can act as endogenous danger signals that stimulate the innate immune system to produce inflammatory cytokines such as IL-1β [56]. Recently, CPPD crystals were shown to activate caspase-1, which is required for IL-1β maturation, through the NRLP3 inflammasome [57]. |
| Synthetic heme crystal (Hemozoin) | Hemozoin is produced by the intraerythrocytic parasite Plasmodium, the causative agent of malaria and it possess adjuvant properties [58]. Hemozoin is taken up by macrophages, initiating signals that lead to activation of the NLRP3 inflammasome and IL-1β production [59]. |
| Monosodium Urate Crystals (MSU Crystals) | MSU crystals induce inflammation by triggering the production of IL-1β by activating NLRP3 inflammasome supported by the finding that macrophages from mice deficient in various components of the inflammasome are defective in crystal-induced IL-1β induction [57]. |
| Nanoparticles of silica dioxide (Nano-SiO2) | An inorganic metal oxide, with a diameter of less than 100 nm. Nano-SiO2 triggers caspase-1 cleavage and IL-1β secretion in human macrophages and keratinocytes [60]. |
| Nigericin, sodium salt | Derived from Streptomyces hygroscopicus, this microbial toxin acts as a potassium ionophore and leads to the release of IL-1β in a NLRP3-dependent mechanism [61]. Similar to ATP, nigericin induces a net decrease in intracellular levels of potassium which is crucial for the activation of caspase-1 [55]. Sample uses: [6, 62] |
| Synthetic analog of the cord factor (TBD) | TDB is the most studied immune stimulatory component of Mycobacterium tuberculosis [63]. TDB binds the C-Type lectin, Mincle (macrophage-inducible C-type lectin) and interacts with the Fc receptor common γ-chain which triggers NF-κB signaling activation [64]. |
| Non-canonical inducers | Description |
| E. Coli Outer Membrane Vesicles (OMVs) | OMVs are small, spherical bodies secreted by Gram-negative bacteria, that potently induce caspase 11-4/5 inflammasome. They contain various PAMPs such as lipoproteins and endotoxins that initiate a multitude of immune responses through cell-surface receptors such as Toll-like receptors (TLRs) [65] and intracellular receptors leading to inflammasome activation [66]. |
| Beta-1,3-glucan from A. faecalis (Curdlan), Dectin-1 ligand | Curdlan is a high molecular weight linear polymer consisting of β-(1 -> 3)-linked glucose residues. Curdlan is produced as a water-insoluble polysaccharide by the soil bacterium, Alcaligenes faecalis. Curdlan is recognized by the membrane bound Dectin-1 receptor leading to NF-κB activation [67]. Curdlan is also recognized by the cytosolic NLRP3 inflammasome complex which cooperates with Dectin-1 resulting in a robust activation of IL-1β–mediated inflammatory response [68]. |
| Heat-killed C. albicans (HKCA) | HKCA is a heat-killed preparation of C. albicans that activates β-glucan, a specific dectin-1 receptor, which is expressed on phagocytes [69]. β-Glucans are glucose polymers found in the cell walls of fungi, such as zymosan (a cell wall preparation of Saccharomyces cerevisiae) and C. albicans. Stimulated Dectin-1 mediates the production of reactive oxygen species and activation of NF-κB, leading to secretion of proinflammatory cytokines. |
| Beta-D-glucan from lichen Lasallia pustulata (Pustulan) | Pustulan is a linear (1-6) linked β-D-glucan from lichen Lasallia pustulata which is recognized by the membrane bound Dectin-1, leading to activation of NF-κB [67, 70]. Pustulan can stimulate the innate immune response, induce heat shock protein expression, elicit phagocytosis and production of proinflammatory cytokines [71, 72]. |
| Zymosan Depleted (Hot alkali treated zymosan) | Zymosan Depleted is obtained by treating zymosan (an insoluble preparation of Saccharomyces cerevisiae cell wall) with hot alkali to remove its Toll-like receptor -stimulating properties to selectively activate the C-type lectin receptor Dectin-1 but not TLR2 [73]. |
Most reports that characterize the inflammasomes have focused on cells of the myeloid lineage, such as macrophages or dendritic cells. One cell line that appears in many inflammasome studies is THP-1. THP-1 is a human monocytic cell line that is derived from an acute monocytic leukemia patient. This cell line is readily differentiated into human macrophages with phorbol 12-myristate 13-acetate (PMA) and thus provides a model for evaluating macrophages in pre- and post-differentiated states [74]. In addition, THP-1 cells stably express ASC-cerulean and can be used to evaluate ASC speck formation and IL-1β release as endpoints. THP-1 cells may be primed with bacterial endotoxins (LPS), and induced to form inflammasomes with nigericin stimulation. Prior to induction, THP-1 cells may also be treated with inhibitors and help determine the molecular mechanism of inflammasome activation [74] and assessment of inhibitors' efficacy [75].
In myeloid cells, suppression of serum/glucocorticoid-inducible kinase 1 activates NLRP3 inflammasome in macrophage-like cells from patients with Ménière's disease [76]. Inhibition of serum/glucocorticoid-inducible kinase 1 significantly activated IL-1β production damaging inner ear hair cells. Sgk-/- mice, which lack the expression of an NLRP3 inflammasome suppressor, showed amplified auditory symptoms and increased inflammasome activation.
The role of Polyribonucleotide nucleotidyltransferase 1 (Pnpt1) in inflammasome activation, was studied using myeloid-specific Pnpt1-knockout mice [77]. The results of the study have shown that Pnpt1 deficiency stimulated interleukin-1 beta (IL-1β) and interleukin-18 (IL-18) production. Pnpt1 was observed to control NLRP3 inflammasome-dependent IL-1β secretion in response to lipopolysaccharide (LPS), and Pnpt1 deficient macrophages elevated glycolysis following exposure to LPS.
With regard to macrophage functions in allergy, a recombinant fusion protein combining the adjuvant and TLR5-ligand flagellin with the birch pollen allergen Bet v 1 (rFlaA:Betv1) activated macrophages and enhance IL-1β secretion [78]. rFlaA:Betv1-mediated stimulation of THP-1 macrophages depended on NLRP3- and NLRC4-inflammasomes. NFκB and SAP/JNK MAP kinases affected rFlaA:Betv1-induced inflammasome activation by modulating pro-Caspase-1- and pro-IL-1β-expression in macrophages.
In studies of neuroinflammation relevant to neurodegenerative diseases, brain resident macrophages - microglia, are also utilized as an efficient model to trigger an appropriate immune response involving secretion of cytokines and recruitment of peripheral immune cells. These cells form functional NLRP3 inflammasomes and secrete IL-1β in the microglial compartment of the mouse brain [79]. Preparation of adult microglia is commonly derived from adult C57BL/6 mice and these are even compared to neonatal glia that are cultures from young pups of the same strain. Detailed protocols for the preparation of both neonatal glia cultures and primary adult microglia from the animal brains are described by Elena Redondo-Castro et al [80].
A recent study on chronic itch revealed that neurons expressing gastrin-releasing peptide receptor (GRPR) are activated by microglia [81]. The study also observed activation of NLRP3 inflammasome and increased release of IL-1β in spinal microglia. The authors further found that by inhibiting microglial stimulation and the NLRP3/caspase-1/IL-1β axis, chronic itch could be reduced. IL-1β+ microglia were observed in close proximity to GRPR+ neurons expressing type 1 IL-1 receptor (IL-1R1). These findings indicate that the microglial NLRP3/caspase-1/IL-1β axis contributes to the activation of GRPR+ neurons.
Although the inflammasome function has been mainly studied in cells of the innate immune system such as in macrophages, cells outside the myeloid compartment were also shown to activate inflammasomes. One example is epithelial cells. Many pathogens and toxic agents first enter the body through epithelia which forms the interface between the body and the environment. Evidently, this exposure requires the innate immune-surveillance mechanisms [82]. Inflammasome activation in keratinocytes has been implicated in response to several stimuli [83-85], and strong expression of ASC in the skin and in other epithelia-like gills and intestines suggest that the activation of inflammasome is of particular importance in those tissues. Moreover, studies indicate that keratinocytes respond to inflammatory conditions by forming ASC specks and triggering pyroptosis, thus highlighting the relevance of inflammasome signaling in epithelia in vivo [82].
While the use of cell lines has significantly contributed to the dissection of the molecular interactions and mechanisms involved in the inflammasome, deeper understanding of how inflammasome activation occurs in cells within their native environment could be enhanced in studies that utilize whole organisms [86, 87]. One whole organism study model in keratinocytes was developed in zebrafish . The zebrafish (Danio rerio) is a genetically and optically accessible model organism for in vivo innate immune responses studies [86, 88]. The zebrafish genome contains over 10 times more NLR genes than in the mouse or human genome [89-91]. However, it has only one gene encoding ASC with a PYD–CARD domain structure. A zebrafish homologue of caspase-1 has also been identified, although it has an N-terminal PYD domain, instead of a CARD domain, but when co-transfected with zebrafish ASC in mammalian cells, the two proteins colocalize into a single speck [92]. Particularly, the transparency of the zebrafish makes this model especially well-suited to study ASC-mediated inflammasome formation using speck formation as the readout [29].
In addition to myeloid and epithelial cell subsets, the expression of Gasdermin E (GSDME) by human helper type 17 T (TH17) cells is regulated by a T cell-intrinsic NLRP3 inflammasome [93]. The expression of GSDME is associated with prolonged viability and is involved in the production of the alarmin interleukin (IL)-1α. The caspase-8–caspase-3–GSDME axis was reported to act in TH17 cells on TCR activation. Thus, NLRP3 inflammasome regulates the release of IL-1α in TCR-activated TH17 cells.
Mammalian inflammasome study models were also developed recently in two studies that analyzed inflammasome activation within mouse living tissues. ASC speck formation were visualized by intravital imaging in macrophages that were derived from retrovirally transduced, ASC–GFP hematopoietic stem cells in bone marrow chimeric mice [28]. The second study generated a transgenic mouse carrying ASC–citrine (Figure 3), which can be expressed in a lineage-specific manner [2]. Together, these in vivo tools helped advance our understanding of inflammasome stimulation and activity in whole organisms.
Various molecular compounds have been shown to affect the activation of NLRP3 inflammasome. N,N-dimethylformamide (DMF) exposure led to focal necrosis of hepatocytes and NLRP3 inflammasome activation before the onset of obvious liver damage and macrophage infiltration of the liver tissue [94]. Paeoniflorin improved abnormal liver function and steatosis and suppressed the production of inflammatory cytokines in diabetic db/db mice. The observed effects were mediated via the suppression of the TXNIP/NLRP3 signaling pathway by PF [95]. Melatonin has been shown to decrease high-fat diet-induced non-alcoholic steatohepatitis by preventing the high-fat diet-induced stimulation of the inflammasome and acting via TLR4/NF-κB signaling pathway [96].
Excessive and/or chronic activation of the immune system can lead to immune-related inflammatory disorders. Indeed, there are over 80 inflammatory disorders such as type 1 diabetes, rheumatoid arthritis, multiple sclerosis, Crohn’s disease and others that have been linked to elevated IL-1β, and some are known as cryopyrin/NLRP3-associated periodic syndromes (CAPS), that are directly associated with NLRP3 mutations. Better understanding of the mechanisms of NLRP3 activation will enable the development of specific inhibitors that can be used to treat NLRP3-related diseases. In addition, these inhibitors may be used to advance our knowledge of the molecular mechanisms that are associated with NLRP3 activity. Treatment of NLRP3-related diseases often target IL-1β with IL-1β antibodies, or recombinant IL-1β receptor antagonist such as canakinumab and anakinra respectively. However, IL-1β can be produced by inflammasome-independent pathways or other inflammasomes, thus inhibitors targeting IL1β may lead to unintended immunosuppressive effects. Pharmacological inhibitors that are specific to NLRP3 inflammasome and that are being used in clinical trials with high safety profiles are described below [75].
MCC950 is a small-molecule inhibitor of the NLRP3 inflammasome, first described by Coll et al in 2015 [97]. MCC950 specifically inhibits both the canonical and noncanonical NLRP3 inflammasome activation and IL-1β secretion by preventing NLRP3-induced ASC oligomerization. In vivo studies demonstrated that MCC950 reduces IL-1β and IL-18 secretion, and alleviate the severity of experimental autoimmune encephalomyelitis in a mouse model of human multiple sclerosis [97]. Interestingly, MCC950 does not inhibit TLR signaling or NLRP3 priming, thus, it is hypothesized that MCC950 may act by regulating a key step in NLRP3 activation such as posttranslational modifications [98]. MCC950 and VX765 significantly decreased liver injury and local inflammation and suppressed the NLRP3 production, and Caspase-1 in rats exposed to ketamine [99]. Additional studies are needed to further define the MCC950 mechanism of action, however current knowledge suggest that MCC950 may be used for treating conditions that involve either canonical and/or noncanonical NLRP3 inflammasome [62].
These inhibitors have been characterized as inhibitors of NLRP3 ATPase. Jiang et al found that the cystic fibrosis transmembrane conductance regulator (CFTR) channel inhibitor-C172, CY-09, has significant anti-inflammatory effects on NLRP3 inflammasome activation [100]. CY-09 specifically inhibits NLRP3 inflammasome activation, and the post-translational modifications step (NLRP3 ubiquitination). Jiang and colleagues demonstrated that CY-09 directly binds to the Walker A motif of NLRP3, and abrogates the ATP binding of NLRP3, thus inhibiting NLRP3 ATPase activity [100]. The results of this study were consistent with a previous study which showed that ATPase activity of NLRP3 is critical for the oligomerization of NLRP3 and its activation [101]. Importantly, CY-09 displayed a good pharmacokinetic profile in terms of safety, stability, and oral bioavailability, and it can be used for blocking NLRP3 inflammasome activation as found in a mouse model of NLRP3-related diseases (C57BL6J) [100].
OLT1177, an active β-sulfonyl nitrile, was initially identified as a candidate for the topical treatment of degenerative arthritis [102]. Marchetti et al showed that OLT1177 suppressed proinflammatory IL-1β and IL-6 secretion in the mouse model of zymosan- and monosodium urate-induced arthritis [103]. Like CY-09, OLT1177 exerts its anti-inflammatory effect on NLRP3 inflammasome activation independently of signal 1 (NLRP3 and pro-IL-1β expression) or K+ efflux, but directly binds to NLRP3 and inhibits ATPase activity [104]. Future work is needed to investigate the effect of CY-09 and OLT1177 on other inflammasomes activation.
Tranilast, N-(3′,4′-dimethoxycinnamoyl)-anthranilic acid, is an analog of a tryptophan metabolite that was initially recognized as an anti-allergic agent used to treat a variety of inflammatory diseases [105]. Tranilast was shown to specifically prevent IgE-induced histamine secretion from mast cells [106]. Molecularly, Tranilast does not interfere with the upstream signaling pathways of NLRP3 inflammasome, but it directly binds to the NACHT domain of NLRP3 and inhibits NLRP3–NLRP3 interaction and subsequent ASC oligomerization [107]. In vivo experiments indicate that Tranilast has significant therapeutic and preventive effects on the mouse models of NLRP3-related diseases and may provide a pharmacological avenue for treating NLRP3 inflammasome-related diseases.
Oridonin is the major bioactive constituent of Rabdosia Rubescens, a widely used over-the-counter herbal medicine for the treatment of inflammatory diseases. Studies demonstrated that Oridonin may suppress NF-κB activation and inhibit inflammasome independent proinflammatory cytokines release, such as TNF-α and IL-6 [108, 109]. He et al clarified the underlying mechanisms of Oridonin in anti-inflammatory activity and showed that Oridonin can specifically inhibit NLRP3 inflammasome activation by directly binding to cysteine 279 of NLRP3 NACHT domain forming a covalent bond that prevents NEK7–NLRP3 interaction and the subsequent NLRP3 inflammasome activation [110]. Importantly, oridonin exerts therapeutic effects on peritonitis, gouty arthritis, and type 2 diabetes in a NLRP3 inflammasome-dependent manner [110].
In vivo studies have shown that TGF-β-activated kinase-1 (TAK1) inhibitor LL-Z1640-2 (LLZ) could block the priming and activation of NLRP3 inflammasome in macrophages. LLZ also inhibited LPS-induced synthesis of TNF-α in bone marrow macrophages and suppressed IL-1β-induced production of MMP-3, IL-6 and RANKL in synovial fibroblasts. Furthermore, the TAK1 inhibitor suppressed synovial hypertrophy and pannus formation and therefore reduces pain and inflammation in mice with collagen-induced arthritis [111].
Nucleotide-binding domain leucine-rich repeat and pyrin domain containing receptor 3 (NLRP3) inflammasome activation was suppressed in mice with collagen-induced arthritis and upregulated expression of β-Arrestin-2 [112]. The mice showed the improvement in the symptoms of arthritis and decreased levels of pro-inflammatory cytokines in serum.
LicoB has protective effects in several mouse models of NLRP3 inflammasome-mediated diseases. In particular, LicoB has been reported to inhibit the activation of NLRP3 inflammasome in macrophages by direct binding to NEK7 and blocking the interaction between NLRP3 and NEK7 [113].
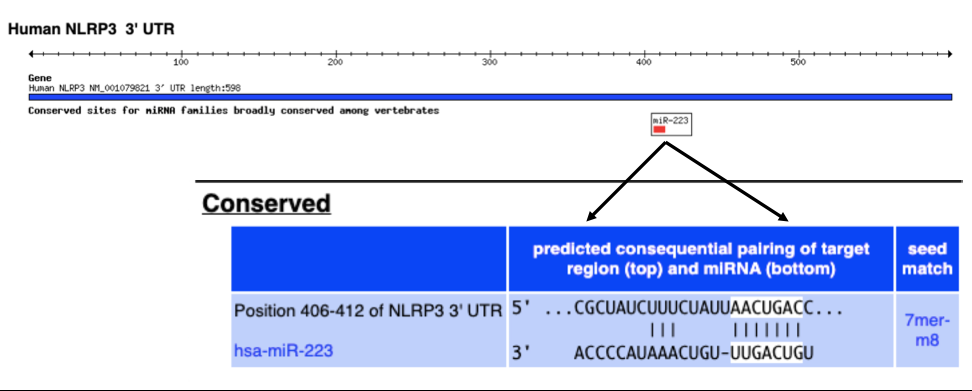
Aside from pharmacological inhibitors, there is accumulating evidence that NLRP3 may also be negatively regulated by microRNAs (miRNAs). miRNAs play important roles in controlling gene expressing through complementary base pairing to the 3’ untranslated region (3’ UTRs) of protein coding mRNAs [114]. Figure 5 shows this type of base pairing relationship between the 3’UTR sequence of NLRP3 and miR-223 as predicted by the TargetScan web prediction tool. This specific prediction was validated in a few studies that showed that miR-223 deregulates the NLRP3 inflammasome by targeting the NLRP3 3’-UTR and preventing the accumulation of the NLRP3 protein [115]. This was shown in a variety of immune-related inflammatory disorders such as sarcoidosis [116], diabetic cardiomyopathy [117], airway inflammation during neutrophilic asthma [118]. Together these studies helped establish miR-223 as a negative regulator of NLRP3; miR-223 decreases NLRP3 mRNA, suppresses the NLRP3 inflammasome activation, and deregulates the immunoinflammatory responses, suggesting that miR-223 antagomirs may serve as potent inhibitors in attenuating NLRP3 levels and inflammation. Interestingly, evaluation of the 3’UTR of NLRP3 in TargetScan, indicated that the only conserved miRNA for this transcript is miR-223. However, additional miRNAs with lower conservation scores were implicated in NLRP3 regulation including miR-193a in traumatic brain injury that is associated with microglia-regulated neuroinflammation [119] and miR-21 in neutrophilic asthma [120]. MiR-223 in the exosomes secreted by bone marrow mesenchymal stem cells, inhibited NLRP3 expression and progression of arthritis [121].
Our expanded knowledge of the inflammasome assembly and activation process, and specific details of associated upstream and downstream events, helped increase our ability to develop sensitive assessment methods for this important complex. Research methods that are designed to assess the inflammasome components and evaluate inflammasome priming and activation methods have significantly improved our understanding of the molecular mechanisms that are involved. In vivo study models, and potent inflammasome specific inhibitors also improved the evaluation of potent therapies for autoinflammatory syndromes and autoimmune diseases that result from excess and damaging inflammatory response. Because the inflammasome is activated in a multi-step process and some of the key features may occur independent of the inflammasome activity, it is essential to probe for it with more than one method and in more than one step within the process. Such approach would provide a more accurate assessment of the inflammasome role and further enhance our understanding of the engagement of the innate immune response.
- Aliprantis A, Yang R, Mark M, Suggett S, Devaux B, Radolf J, et al. Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science. 1999;285:736-9 pubmed
- Ozinsky A, Underhill D, Fontenot J, Hajjar A, Smith K, Wilson C, et al. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc Natl Acad Sci U S A. 2000;97:13766-71 pubmed
- Alexopoulou L, Holt A, Medzhitov R, Flavell R. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732-8 pubmed
- Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochem Biophys Res Commun. 2002;293:1364-9 pubmed
- Fernandes Alnemri T, Wu J, Yu J, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007;14:1590-604 pubmed
- Prysi A, Sargent L, Franklin J. Rigid facial skeletal fixation: advances in treatment. South Med J. 1989;82:727-32, 735 pubmed
- Thornberry N, Bull H, Calaycay J, Chapman K, Howard A, Kostura M, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768-74 pubmed
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417-26 pubmed
- Pelegrin P, Barroso Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. 2008;180:7147-57 pubmed
- Fink S, Cookson B. Pillars Article: Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006. 8: 1812-1825. J Immunol. 2019;202:1913-1926 pubmed
- Li H, Willingham S, Ting J, Re F. Cutting edge: inflammasome activation by alum and alum's adjuvant effect are mediated by NLRP3. J Immunol. 2008;181:17-21 pubmed
- Locovei S, Scemes E, Qiu F, Spray D, Dahl G. Pannexin1 is part of the pore forming unit of the P2X(7) receptor death complex. FEBS Lett. 2007;581:483-8 pubmed
- Shi Y, Evans J, Rock K. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516-21 pubmed
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237-41 pubmed
- Mariathasan S, Weiss D, Newton K, McBride J, O Rourke K, Roose Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228-32 pubmed
- Brown G, Herre J, Williams D, Willment J, Marshall A, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197:1119-24 pubmed
- Willment J, Gordon S, Brown G. Characterization of the human beta -glucan receptor and its alternatively spliced isoforms. J Biol Chem. 2001;276:43818-23 pubmed
- Rubin Bejerano I, Abeijon C, Magnelli P, Grisafi P, Fink G. Phagocytosis by human neutrophils is stimulated by a unique fungal cell wall component. Cell Host Microbe. 2007;2:55-67 pubmed
- Ikeda Y, Adachi Y, Ishii T, Miura N, Tamura H, Ohno N. Dissociation of Toll-like receptor 2-mediated innate immune response to Zymosan by organic solvent-treatment without loss of Dectin-1 reactivity. Biol Pharm Bull. 2008;31:13-8 pubmed
- Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer H. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr Biol. 2007;17:1140-5 pubmed
- Stein C, Caccamo M, Laird G, Leptin M. Conservation and divergence of gene families encoding components of innate immune response systems in zebrafish. Genome Biol. 2007;8:R251 pubmed
- Masumoto J, Zhou W, Chen F, Su F, Kuwada J, Hidaka E, et al. Caspy, a zebrafish caspase, activated by ASC oligomerization is required for pharyngeal arch development. J Biol Chem. 2003;278:4268-76 pubmed
- Duncan J, Bergstralh D, Wang Y, Willingham S, Ye Z, Zimmermann A, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A. 2007;104:8041-6 pubmed
- Platten M, Ho P, Youssef S, Fontoura P, Garren H, Hur E, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005;310:850-5 pubmed
- Tenshin H, Teramachi J, Ashtar M, Hiasa M, Inoue Y, Oda A, et al. TGF-β-activated kinase-1 inhibitor LL-Z1640-2 reduces joint inflammation and bone destruction in mouse models of rheumatoid arthritis by inhibiting NLRP3 inflammasome, TACE, TNF-α and RANKL expression. Clin Transl Immunology. 2022;11:e1371 pubmed publisher
- Materials and Methods [ISSN : 2329-5139] is a unique online journal with regularly updated review articles on laboratory materials and methods. If you are interested in contributing a manuscript or suggesting a topic, please leave us feedback.
- genemethod
- Activators and Inhibitors in Cell Biology Research
- Apoptosis Assays
- Bats as Experimental Animal Models for the Study of Human Diseases
- CRISPR and Genomic Engineering
- Cell Proliferation Assays and Cell Viability Assays
- Extracellular Traps – NETosis and METosis
- Macrophage Markers
- MicroRNA Experimental Protocols
- MicroRNA Research Reagents
- MicroRNA Research Web Resources
- Microglia Markers
- Protease Inhibitors
- Protein/Peptide Tags