

Abstract
Background: Neuroinflammatory processes are increasingly believed to participate in the pathophysiology of a number of major psychiatric diseases, including depression. Immune activation stimulates the conversion of the amino acid tryptophan to kynurenine, leading to the formation of neuroactive metabolites, such as quinolinic acid and kynurenic acid. These compounds affect glutamatergic neurotransmission, which plays a prominent role in depressive pathology. Increased tryptophan degradation along the kynurenine pathway (KP) has been proposed to contribute to disease etiology.
Methods: We used postmortem brain tissue from the ventrolateral prefrontal cortex (VLPFC) to assess tissue levels of tryptophan and KP metabolites, the expression of several KP enzymes and a series of cytokines as well as tissue pathology, including microglial activation. Tissue samples came from nonpsychiatric controls (n = 36) and individuals with depressive disorder not otherwise specified (DD-NOS, n = 45) who died of natural causes, homicide, accident, or suicide.
Results: We found a reduction in the enzymatic conversion of tryptophan to kynurenine, determined using the kynurenine:tryptophan ratio, and reduced messenger RNA expression of the enzymes indoleamine-2,3-dioxygenase 1 and 2 and tryptophan-2,3-dioxygenase in depressed individuals irrespective of the cause of death. These findings correlated with reductions in the expression of several cytokines, including interferon-γ and tumour necrosis factor-α. Notably, quinolinic acid levels were also lower in depressed individuals than controls.
Limitations: Information on the use of antidepressants and other psychotropic medications was insufficient for statistical comparisons.
Conclusion: Contrary to expectations, the present results indicate that depression, in the absence of medical illness or an overt inflammatory process, is associated with compromised, rather than increased, KP metabolism in the VLPFC.
Introduction
Converging lines of evidence indicate that depression associated with medical illness or immunotherapy may develop as a result of the actions of cytokines and inflammatory mediators in the brain.1–4 A key mechanism linking the immune system with mood appears to be the activation of the enzyme indoleamine-2,3-dioxygenase (IDO) and the resultant enhanced conversion of the amino acid tryptophan (TRP) to kynurenine and its neurotoxic downstream metabolites, the free radical generator 3-hydroxykynurenine (3-HK) and the N-methyl-d-aspartate (NMDA) receptor agonist quinolinic acid (QUIN).5–11 Together with kynurenic acid (KYNA), a neuroinhibitory and neuroprotective metabolite that is formed in a side arm of the kynurenine pathway (KP) of TRP degradation and targets a variety of receptors,12 these mainly glia-derived compounds may modulate glutamatergic neurotransmission,13–15 which is involved in mood disorders.16 Supporting evidence linking the KP to depression stems from clinical studies showing that a number of patients receiving cytokine therapy experience changes in mood that correlate with reduced levels of TRP and increased levels of kynurenine and QUIN in the circulation.17,18 Taken together with reports of increased levels of cytokines and other markers of inflammation in the blood of depressed individuals,19,20 these lines of evidence have led to the hypothesis that the pathophysiology of depression, in the absence of an overt inflammatory process, may be related to a subsyndromal activation of the immune system, resulting in chronic stimulation of the KP and altered glutamatergic activity.3,8,9
Currently, the evidence implicating enhanced KP metabolism in depressed individuals in the absence of immunotherapy or medical illness is both limited and inconsistent. Whereas several studies that were performed using specimens from blood or cerebrospinal fluid (CSF) have reported a positive association between various components of the KP and depression or suicide,21–24 others failed to do so25,26 or have revealed qualitatively different changes for various analytes.27 Moreover, evidence for abnormal brain tissue levels of KP metabolites in depressed individuals and individuals who died by suicide is limited or indirect.28–30 The possible role of the KP in the pathophysiology of depression therefore requires further clarification.
In the present study, we conducted a postmortem analysis of KP metabolite levels and the expression of KP enzymes in the ventrolateral prefrontal cortex (VLPFC) of control and depressed individuals whose deaths had resulted from suicide or other causes. In parallel, we examined relevant neuroinflammatory markers, including cytokine gene expression, in the same tissues. The VLPFC was selected because it shows distinct abnormalities in individuals with depressive illness,31–33 in particular increased glucose utilization during TRP depletion.34 Moreover, postmortem studies have consistently shown neurochemical evidence of serotonergic hypofunction in victims of suicide.35–37 We hypothesized that depressed individuals would display an inflammatory profile characterized by increased cytokine expression that would correlate with increased KP activity. Results of the present study did not confirm our assumptions.
Methods
Human postmortem brain samples
The study was reviewed and approved by the Institutional Review Board of the University of Maryland School of Medicine. Samples were obtained from the Section on Neuropathology, Clinical Brain Disorders Branch, National Institute of Mental Health (NIMH). Two board-certified psychiatrists (J.E.K. and T.M.H.) confirmed the diagnosis and classification of depressive disorders based on a review of the medical records and interviews with next of kin. The samples came from a total of 104 individuals, corresponding to 51 nonpsychiatric controls and 53 individuals who had a diagnosis of depressive disorder not otherwise specified (DD-NOS), 26 of whom died by suicide. Individuals with an additional diagnosis of psychotic or bipolar disorders were not included in the sample. For statistical comparisons, we applied the following exclusion criteria: a medical condition of recognized immune involvement, such as cancer, diabetes, infection, asthma, thyrotoxicosis and neurologic disorders (n = 19: 14 controls and 5 depressed); a pH lower than 6.0 (n = 2: 1 control and 1 depressed); and missing key data (n = 2: 1 with manner of death undetermined and 1 missing age at death). This resulted in a sample set from 81 individuals, corresponding to 36 controls and 45 who had DD-NOS. Information on demographic characteristics; tissue characteristics; manner of death; and confounders, including smoking, ethanol at death and death by accidental overdose, are summarized in Table 1. Special consideration was given to cause of death related to cardiovascular diseases (CVDs), such as heart attack, owing to their known association with inflammation. These cases are commonly used as controls in postmortem studies of suicide owing to the sudden death and short agonal state. We conducted exploratory analyses of subgroups defined by the presence or absence of potential confounders to assess, within the limits of the available data, whether they might alter those significant differences in KP or cytokine measures found in adjusted regressions.
Demographic and tissue status characteristics of postmortem brain tissue donors
Tissue processing
All samples consisted of blocks of frozen tissues of at least 2 × 2 × 1 cm corresponding to the VLPFC (approximately Brodmann areas 45 and 47) spanning at least 2 gyri and 2 sulci and containing both grey and white matter. Serial consecutive sections 20 μm thick were used for histochemistry or collected for reverse transcriptase–polymerase chain reaction (RT-PCR) determinations. We dissected adjacent tissue containing grey and white matter for high-performance liquid chromatography (HPLC) and gas chromatography/mass spectrometry (GC/MS) determination of KP metabolites. Prior to analysis, all tissues were stored at –80°C.
Real-Time RT-PCR
Total RNA was isolated from approximately 100 mg of frozen brain tissue using TRIzol (Thermo Fisher Scientific). Quantification and quality of the extraction is detailed in Appendix 1, available at jpn.ca. We reverse-transcribed 500 ng of total RNA per sample into complementary DNA (cDNA) in a 20 μL reaction volume using the iScript cDNA Synthesis Kit (Bio-Rad). The real-time PCR reaction was run on a MyiQ instrument (Bio-Rad) with a 3-step cycling program, as described previously.38 Specific primer sets (Appendix 1, Table S1) were used under the same amplification condition, and relative expression was determined using the 2−ΔΔCt method with multiple control genes for normalization39 (Appendix 1).
KYNA and 3-HK determination
We analyzed KYNA and 3-HK using HPLC, and they were detected fluorometrically and electrochemically,13 respectively (Appendix 1).
TRP, kynurenine and QUIN measurement
We quantified tryptophan, kynurenine and QUIN levels in the brain with GC/MS using a 7890A GC coupled to a 7000 MS/MS (Agilent Technologies), using electron capture negative chemical ionization as described previously40 (Appendix 1).
Histology and immunohistochemistry
Serial consecutive sections were processed for microscopic histological analyses using Nissl and Hematoxylin and Eosin staining for the assessment of vascular integrity and verification of the absence of peripheral cell infiltration. We used acidic toluidine blue to detect mast cells and corpora amylacea, indicative of dead astrocytes and normal aging processes. Sections were also processed for ionized calcium-binding adapter (IBA1) immunohistochemistry using the nickel-DAB method for enhanced detection.41 Microscopic examination and semiquantification was carried out as described in the supplemental methods and in Figures S1 and S2 of Appendix 1.
Statistical analyses
We log-transformed KP analytes and mRNA expression data to approximate normal distributions. We compared depressed and control groups using the transformed variables with analysis of covariance (ANCOVA). Covariates selected for inclusion in these models included age, sex and postmortem interval (PMI) as a marker of tissue quality. Tissue pH was considered for inclusion, but excluded owing to very low variability among the samples selected for this study. Age and sex were selected for potential biological relevance to the processes under study. Adjusted means were back-transformed (exponentiated) to yield geometric means with asymmetric 95% confidence intervals (CIs). False discovery rates for statistical multiple comparison tests were controlled using the Benjamini–Hochberg modification of the Bonferroni procedure.42
We used a second approach involving the Wilcoxon rank-sum test of nontransformed, unadjusted data to confirm our results. Exploratory analyses were conducted to examine the distribution of KP metabolites and enzyme activity and of inflammatory cytokines in subgroups defined by variables unevenly distributed between the depressed and control groups or for which information was not completely available (i.e., alcohol at the time of death, death by overdose and death related to a cardiovascular event). All analyses were conducted with either SAS software version 9.3 (SAS Institute Inc.) or STATA software (StataCorp LP).
Results
Kynurenine formation is reduced in the VLPFC of depressed individuals
Age at death, pH and PMI did not differ significantly between the control and depressed groups (Table 1), and none of these parameters correlated with any of the outcome measures analyzed, including KP metabolites and cytokines. All linear regression models comparing geometric means for depressed and control individuals were adjusted for age at death, PMI and race.
We found significant differences between control and depressed individuals for the adjusted means of the kynurenine:TRP ratio (0.045 v. 0.038, t74 = 2.41, p = 0.019; Fig. 1A) and for QUIN levels (1.59 v. 1.11, t74 = 2.46, p = 0.016; Table 2), with the depressed group displaying lower non-adjusted values. No significant differences between adjusted means of control and depressed individuals were found for either TRP or kynurenine. We observed lower values for KYNA and 3-HK levels in the depressed group, but these reductions were not significant (Table 2). Finally, no significant effect of sex was detected when comparing controls with depressed individuals for any of the KP metabolites measured.
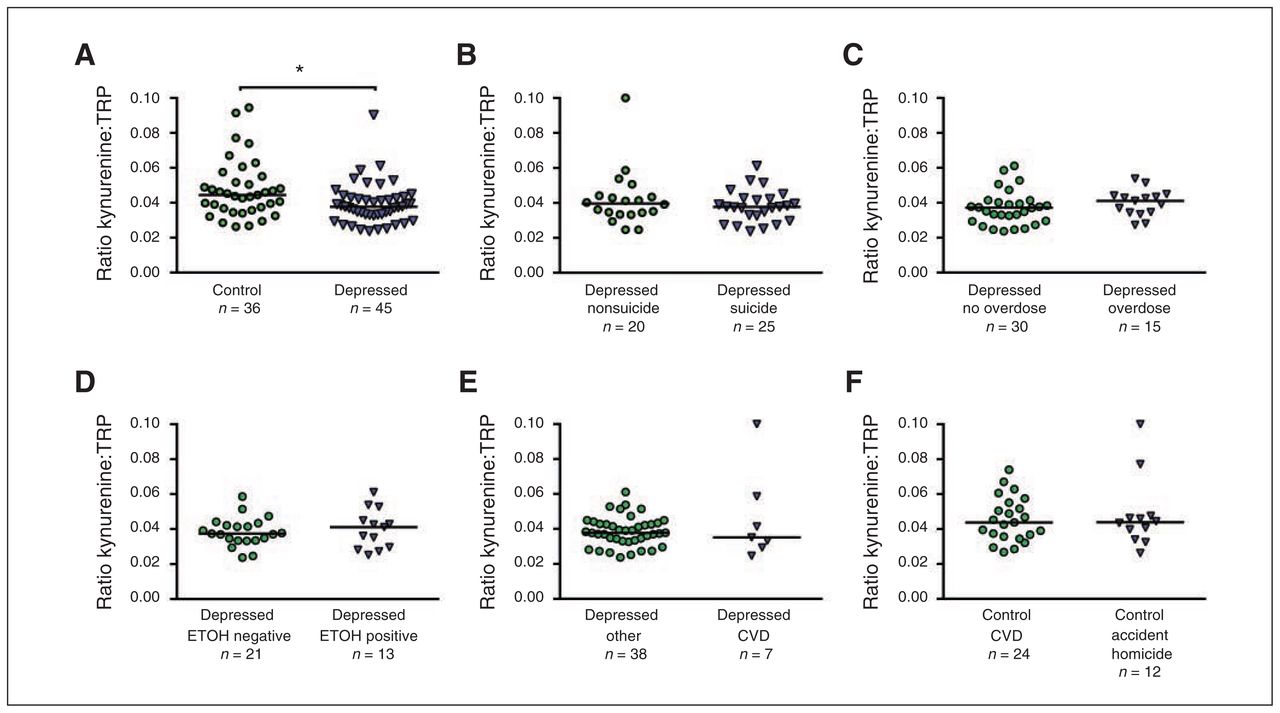
Subgroup comparisons of nontransformed values for the kynurenine:tryptophan (TRP) ratio (kynurenine formation) in the ventrolateral pre-frontal cortex (VLPFC). (A) Scatter plots with median of controls and depressed individuals; (B) depressed individuals who died by suicide versus other causes; (C) depressed individuals who died by overdose versus no overdose; (D) depressed individuals positive for alcohol versus negative for alcohol (ETOH) at the time of death; (E) depressed individuals who died from cardiovascular disease (CVD)–related events versus other causes; (F) and nonpsychiatric controls who died from CVD-related events versus accidents and homicide. *Wilcoxon test, z1 = 2.44, p = 0.015.
Geometric means with 95% confidence intervals for kynurenine pathway metabolites and gene expression of associated enzymes in the VLPFC of controls and depressed individuals
Significant differences were also found for mRNA levels of the enzymes IDO1 (0.96 v. 0.40, t68 = 4.39, p < 0.001), IDO2 (0.84 v. 0.40, t59 = 2.95, p < 0.001) and tryptophan-2,3-dioxygenase (TDO2; 0.92 v. 0.37, t68 = 4.39, p < 0.001; Table 2). The reduced kynurenine:TRP ratio in the depressed group indicates a decreased conversion of TRP into kynurenine and is in agreement with the lower expression of the IDO1, IDO2 and TDO2 genes.
Reduced cytokine mRNA expression in the VLPFC of depressed individuals
We found significant differences in mRNA expression between control and depressed individuals, with higher values in controls, for the cytokines tumour necrosis factor (TNF)-α (1.04 v. 0.64, t62 = 2.86, p = 0.006), interferon (IFN)-γ (0.96 v. 0.50, t62 = 2.85, p = 0.006), interleukin (IL) 13 (0.92 v. 0.64, t62 = 2.23, p = 0.029), IL33 (1.00 v. 0.52, t49 = 3.57, p < 0.001), IL2 (0.66 v. 0.27, t61 = 2.15, p = 0.036) and the chemokine CCL2 (1.29 v. 0.76, t62 = 2.86, p = 0.031; Table 3). No differences between groups were observed for the expression of the cytokines IL1-β, IL6, IL4 and IL5, and for the enzyme cyclooxygenase-2 (COX2). These results indicate that, overall, depressed individuals showed reduced mRNA expression levels in some of the cytokines analyzed in the VLPFC when compared with nonpsychiatric controls.
Geometric means with 95% confidence intervals for cytokine gene expression in the VLPFC of control and depressed individuals
Correlation analyses
Pearson’s correlation coefficients of log-transformed values showed that mRNA levels for the cytokines IFN-γ, TNF-α IL13 and IL33 had the strongest correlations with a majority of the KP measures analyzed in the combined data from controls and depressed individuals (Table 4). Both IFN-γ and TNF-α correlated with the kynurenine:TRP ratio (r = 0.32 and r = 0.38, respectively) and with mRNA expression of the enzymes IDO1, IDO2 and TDO2 (Table 4). In the overall sample, we found positive correlations for the kynurenine:TRP ratio and the expression of IDO1 (r = 0.36, p = 0.001), IDO2 (r = 0.34, p = 0.029) and TDO2 (r = 0.32, p = 0.028; Appendix 1, Table S2). Significant and positive correlations also existed between kynurenine and all of its measured metabolites (3-HK: r = 0.54; QUIN: r = 0.61; KYNA: r = 0.50) as well as between the levels of KYNA, 3-HK and QUIN. These positive correlations between the kynurenine:TRP ratio and the expression of the converting enzymes support the accuracy and consistency of the biochemical and RT-PCR determinations and indicate that the main effect is driven by a reduced enzymatic conversion of TRP to kynurenine, which is likely a consequence of decreased IDO1, IDO2 and TDO2 expression in depressed individuals.
Pearson correlations of log-transformed data on kynurenine pathway metabolites and enzymes with cytokines
Subgroup analyses
We conducted subgroup comparisons of variables showing significant differences in the main analyses. These included the kynurenine:TRP ratio, QUIN, IDO1, IDO2, TDO2 and the cytokines IFN-γ, TNF-α, IL13, IL33 and CCL2. Subgroup analyses were performed for manner of death (suicide v. nonsuicide) in the depressed group and for groups defined by potential confounders (e.g., accidental overdose v. other causes of death, death from CVDs v. accident/homicide) in the control and depressed groups; additional subgroup comparisons are described in Appendix 1. To explore additional potential confounders, we also performed unadjusted group and subgroup comparisons using Wilcoxon rank-sum tests for nontransformed data. Similar to the results of our linear regression models, we found statistically significant differences between depressed individuals and controls for the kynurenine:TRP ratio (z1 = 2.44, p = 0.015; Fig. 1A) and for QUIN levels (z1 = 2.56, p = 0.009; Appendix 1, Fig. S3). Subgroup comparisons for the kynurenine:TRP ratio showed no significant differences between depressed individuals who died by suicide versus other causes (Fig. 1B), between depressed individuals who died by overdose versus nonover-dose (Fig. 1C), or in ethanol at the time of death (Fig. 1D), or between depressed individuals who died from CVD events versus other causes (Fig. 1E) and controls who died from CVD versus accident/homicide (Fig. 1F). Similarly, no subgroup differences were found for QUIN and the enzymes IDO1, IDO2 and TDO. Additional subgroup comparisons for cytokines showed no significant differences. Since information on antidepressant medications was incomplete in the depressed sample, we assessed the effects of fluoxetine on KP metabolites in the cerebral cortex of rats under chronic administration. We found no effect on any KP metabolite after 21 days of fluoxetine treatment (Appendix 1, Table S3).
Histopathology and immunohistochemistry
Microscopic analyses confirmed the absence of infiltrating peripheral leukocytes, the integrity of vascular elements and the presence of a low number of the acidic reactive elements defined as corpora amylacea. Scattered and very low numbers of IBA1-positive amoeboid microglial cells were found in both controls and depressed individuals. Semiquantitative analyses of these cells revealed significant group differences when comparing the adjusted means of controls versus depressed individuals (geometric mean 38.7/cm2 [95% CI 31.6–47.2] v. 73.7/cm2 [95% CI 60.7–89.3]), with the latter group showing a significant increase (p < 0.001). Nevertheless, the proportion of these amoeboid microglia was very low, corresponding to less than 5% of the total microglial population for controls and less than 9% for depressed individuals. We identified no specific anatomic distribution when comparing grey versus white matter, and no differences between depressed individuals who died by suicide versus other causes were observed. Intensely stained hypertrophic microglia, characterized by long, thick hyper-ramified processes, were primarily restricted to the boundaries between the nervous tissue and vascular and meningeal regions (Appendix 1, Fig. S2D). These conspicuous microglia were observed in 24 of 36 controls (67%) and in 29 of 47 depressed individuals (62%), with no differences between groups or correlations with any of the markers assessed in the present study. In line with the KP and cytokine data obtained in these samples, our results confirmed the absence of an overt M1 type of proinflammatory process in adjacent tissue.
Discussion
Contrary to our expectations, the kynurenine:TRP ratio as well as QUIN levels and the expression of major KP enzymes were significantly decreased in the VLPFC of depressed individuals compared with nonpsychiatric controls. The expression of TNF-α, IFN-γ and other proinflammatory cytokines was also unexpectedly reduced in this brain region. Parallel microscopic analyses revealed that none of the depressed individuals had evidence of overt, ongoing inflammatory processes. A small increase in amoeboid, probably phagocytic, microglial cells in the VLPFC of depressed individuals was not deemed pathological, as the density of these cells was very low. Notably, all these abnormalities were independent of the cause of death. Statistical analyses showed that several components of the KP correlated both with each other and with markers of inflammation in both controls and depressed individuals. These findings confirm the existence of biochemical links between immune factors and KP metabolism in the human brain, but also highlight the heterogeneity of depressive illness and indicate that, in the VLPFC, immune activation by a classical proinflammatory M1 type of response does not appear to be part of disease pathology.
The VLPFC is part of the orbitofrontal cortex region, which is involved in higher emotional function33 and is considered a critical neural substrate in the pathology of depression.31,32 Postmortem studies in this region have shown reductions in serotonin transporter function associated with a loss of serotonergic terminals, concurrent with a presumably compensatory increase in serotonin receptor binding in individuals who died by suicide, suggesting reduced serotonergic activity.35,36 This serotonergic hypofunction was associated with increased impulsivity and impaired decision-making preceding suicidal behaviour.35 Remarkably, the metabolic activity of the lateral portions of the orbitofrontal area, including the VLPFC, was previously found to be negatively correlated with depression severity31,33 and to be specifically affected during a TRP depletion (TD) test in depressed individuals.34 Interestingly, while the protocol of inducing TD likely results in reduced levels of serotonin in the brain, there is a consensus that TD involves other mechanisms as well.43 It is possible that reductions in KP metabolites during TD44 also contribute to VLPFC dysfunction and the manifestation of depressive symptomatology.
Increased formation of kynurenine and downstream KP metabolites has been repeatedly described in the blood and CSF of individuals suffering from depressive disorders and suicide attempters.21–23,45 Nevertheless, this has not been consistently reported in the literature,25,26,46,47 and changes in KP metabolites and their corresponding enzymes in human serum and CSF do not necessarily parallel events in the brain.14 Furthermore, to our knowledge only 2 groups had examined the KP in postmortem brains of individuals with depressive illnesses prior to the present study. In the anterior cingulate cortex (ACC), 1 group reported a higher density of TDO- positive glial cells in depressed individuals compared with controls29 but revealed no change in the tissue levels of several KP metabolites,28 whereas the other investigators described subregion-specific changes in the density of QUIN-positive microglial cells in depressed individuals.30,48 The present study, which revealed a reduction rather than an increase in brain KP metabolism in depressed individuals, indicates that an etiological link between enhanced KP metabolism and depression does not appear to exist in the absence of overt signs of inflammatory processes in the VLPFC. Taken together, these studies and ours strongly suggest that regulation of the KP in the human brain may be region-specific and that the consequence of compensatory mechanisms between different brain regions is affected in individuals with depression.
Although the expression of pro- and anti-inflammatory cytokines and immune markers in the brain has often been reported as increased in individuals with depression and suicidality, the results from different brain repositories and laboratories have been quite inconsistent in this respect.38,49–53 A previous study from our group reported increased expression of IL4 and IL13, and no differences in IL6, TNF-α and IL1-β, in the orbitofrontal cortex of individuals who died by suicide compared with controls.38 This variability in the expression of certain cytokines in the brain suggests that caution is indicated regarding their use as biological markers of mood disorders. Future clarification of the association between brain inflammatory markers and depressive illness should carefully consider diagnostic subgroups based on variables affecting inflammation, such as body mass index, nutritional status and sleep.
The unexpected results in the present study may be attributable to a number of different factors. The most obvious explanation is that the specific brain region studied and/or the diagnosis classification of the depressed sample influenced the overall results. Whereas our study focused on the VLPFC, most postmortem studies that have reported increased cytokine expression in depressed individuals and individuals who died by suicide were conducted in other areas of the brain, including different regions of the prefrontal cortex (PFC).49,51,53 Additionally, the diagnosis of DD-NOS, while prevalent among the general population,54 constitutes a heterogeneous category of depressive disorders. Alternatively, considering that depression is not uniform, the results of this study may be dependent on the distribution of proinflammatory and nonproinflammatory depressive phenotypes in our sample set. Finally, a reassessment of the association between the KP, inflammation and depression may be warranted based on inconsistencies across studies. For example, a recent study reported that depressive episodes were not associated with increased KP metabolites in medication-free patients with major depressive disorder (MDD).46 At the same time, a functional imaging study reported increased microglial activation in the PFC and ACC in patients with MDD after a depressive episode.55 An interpretation of these apparently contradictory results is that the absence of KP activation may indicate an alternative microglial activation process, which likely reflects dysfunction and dystrophy in depression, as shown in studies of rats and mice that developed depressive-like behaviour.56 Two recent postmortem studies showed that microglial abnormalities in individuals with MDD were restricted to the neurovascular interface,57,58 with no evidence of M1 activation in the brain parenchyma. Our histopathological results also indicated the absence of an M1 inflammatory process in the parenchyma of the brain. Nevertheless, our approach using Ni-DAB for signal enhancement, although effective in detecting amoeboid microglia in the parenchyma, did not allow an adequate quantification of abnormalities at the perivascular and meningeal spaces. Delineating whether microglial activation in depression is part of a detrimental proinflammatory process or a compensatory mechanism leading to dystrophy may help identify specific depression phenotypes associated with high versus low inflammatory biomarkers.
Limitations
Although the brain donors were well-matched demographically and tissue characteristics were uniform in relevant categories, the present results must be considered within the limitations of studies using postmortem human brain tissue, in which gene expression and metabolites may represent end points of pathological processes. Moreover, conclusions derived from the analysis of a single brain region do not necessarily extend to other brain areas, such as the ACC or the amygdala, which are also involved in the pathophysiology of depression. This study involved conducting a large number of hypothesis tests, raising the possibility of false-positive findings of group differences due to chance. The group differences we found were in the direction of higher KP activity and cytokine expression in the control group rather than the depressed group, the reverse of the directional differences we expected to find, possibly limiting concern about false-positive results in our data. We attempted to address some potential confounders in exploratory subgroup analyses, which, while lacking power, showed very similar patterns in differences (or lack thereof) between depressed individuals and controls across subgroups. Information on antidepressants and other psychotropic medications was insufficient for use in the present study. While we attempted to overcome this limitation by evaluating the effects of the common antidepressant fluoxetine on KP metabolites in rats, different classes of antidepressant medications may have affected the present results.59–61 Other variables known to affect inflammatory markers, such as body mass index, were not available for statistical corrections. While our histopathological analysis revealed no major signs of activated M1 amoeboid microglia, other microglial abnormalities, particularly at the neurovascular interface, could not be adequately quantified in the present study.
Conclusion
In the absence of an overt inflammatory process, medical illness and/or immunotherapy, KP metabolism in the VLPFC of depressed individuals compared with non-psychiatric controls is hypoactive. This effect was paralleled by reduced expression of several cytokines that strongly correlated with several components of the KP in both controls and depressed individuals. The present study reinforces the notion that depression is a highly heterogeneous condition and highlights the importance of further studies, which may lead to a more personalized pharmacotherapy for the treatment of this disorder.
Acknowledgements
This study was supported in part by grants from the National Institute of Mental Health (MH097676 to L.H.T. and MH083729 to R.S.). Ryan Mulford and Partam Manalai are acknowledged for their technical assistance with tissue processing and mRNA purification.
Footnotes
↵* These authors contributed equally to this work.
Competing interests: R. Schwarcz reports equity from Vistagen outside the submitted work. He also holds a patent “Small Inhibitors of Kynurenine 3-Monooxygenase” and has a patent “A new class of 3-hydroxyanthranilate-3,4-dioxygenase (3-HAO) inhibitors as potential neuroprotective drugs” pending. L. Tonelli is supported by a grant from the National Institute of Health. No other competing interests declared.
Contributors: T. Postolache, R. Schwarcz, J. Stiller and L. Tonelli designed the study. S. Clark, A. Pocivavsek, J. Nicholson, F. Notarangelo, P. Langenberg, J. Kleinman, T. Hyde, R. Schwarcz and L. Tonelli acquired the data, which S. Clark, A. Pocivavsek, J. Nicholson, F. Notarangelo, P. Langenberg, R. McMahon, T. Postolache, R. Schwarcz and L. Tonelli analyzed. S. Clark, A. Pocivavsek, R. McMahon, T. Postolache, R. Schwarcz and L. Tonelli wrote the article, which all authors reviewed and approved for publication.
- Received June 19, 2015.
- Revision received December 16, 2015.
- Accepted December 18, 2015.
References
In this issue

{kind=link}
Article tools





