Abstracts
More than 140 years after the first description of Friedreich ataxia, autosomal recessive ataxias have become one of the more complex fields in Neurogenetics. Currently this group of diseases contains more than 20 clinical entities and an even larger number of associated genes. Some disorders are very rare, restricted to isolated populations, and others are found worldwide. An expressive number of recessive ataxias are treatable, and responsibility for an accurate diagnosis is high. The purpose of this review is to update the practitioner on clinical and pathophysiological aspects of these disorders and to present an algorithm to guide the diagnosis.
autosomal recessive ataxias; cerebellar ataxia; cerebellum; Friedreich ataxia
Mais de 140 anos após a primeira descrição da ataxia de Friedreich, as ataxias autossômicas recessivas se transformaram em um dos mais complexos campos da Neurogenética. Atualmente, este grupo de doenças é composto por mais de 20 entidades clínicas e possui um número ainda maior de genes associados. Algumas doenças são muito raras, tendo sido observadas apenas em populações isoladas, enquanto que outras são encontradas no mundo todo. Um número expressivo de ataxias é tratável, e a responsabilidade em se fazer um diagnóstico correto é alta. A finalidade desta revisão é a de atualizar o neurologista a respeito dos principais aspectos clínicos e fisiopatológicos destas doenças e de apresentar um algoritmo para auxiliar a sua investigação e o seu diagnóstico.
ataxias autossômicas recessivas; cerebelo; ataxia cerebelar; ataxia de Friedreich
VIEW AND REVIEW
Autosomal recessive ataxias: 20 types, and counting
Ataxias autossômicas recessivas: 20 tipos e muito mais
Emília Katiane EmbiruçuI; Marcília Lima MartynI; David SchlesingerII; Fernando KokIII
IPediatric Neurology Service and Outpatient Neurogenetics Clinic, Hospital das Clínicas, University of São Paulo School of Medicine, São Paulo SP, Brazil: Neurologista Infantil, Doutoranda em Neurologia pela Faculdade de Medicina da Universidade de São Paulo
IIPediatric Neurology Service and Outpatient Neurogenetics Clinic, Hospital das Clínicas, University of São Paulo School of Medicine, São Paulo SP, Brazil: Neurologista Clínico, Médico Preceptor do Serviço de Neurologia do Hospital das Clínicas da Universidade de São Paulo e Doutorando em Genética do Instituto de Biociências da Universidade de São Paulo
IIIPediatric Neurology Service and Outpatient Neurogenetics Clinic, Hospital das Clínicas, University of São Paulo School of Medicine, São Paulo SP, Brazil: Neurologista Infantil, Professor Livre-Docente Médico Assistente do Serviço de Neurologia Infantil do Hospital das Clínicas da Universidade de São Paulo
ABSTRACT
More than 140 years after the first description of Friedreich ataxia, autosomal recessive ataxias have become one of the more complex fields in Neurogenetics. Currently this group of diseases contains more than 20 clinical entities and an even larger number of associated genes. Some disorders are very rare, restricted to isolated populations, and others are found worldwide. An expressive number of recessive ataxias are treatable, and responsibility for an accurate diagnosis is high. The purpose of this review is to update the practitioner on clinical and pathophysiological aspects of these disorders and to present an algorithm to guide the diagnosis.
Key words: autosomal recessive ataxias, cerebellar ataxia, cerebellum, Friedreich ataxia.
RESUMO
Mais de 140 anos após a primeira descrição da ataxia de Friedreich, as ataxias autossômicas recessivas se transformaram em um dos mais complexos campos da Neurogenética. Atualmente, este grupo de doenças é composto por mais de 20 entidades clínicas e possui um número ainda maior de genes associados. Algumas doenças são muito raras, tendo sido observadas apenas em populações isoladas, enquanto que outras são encontradas no mundo todo. Um número expressivo de ataxias é tratável, e a responsabilidade em se fazer um diagnóstico correto é alta. A finalidade desta revisão é a de atualizar o neurologista a respeito dos principais aspectos clínicos e fisiopatológicos destas doenças e de apresentar um algoritmo para auxiliar a sua investigação e o seu diagnóstico.
Palavras-chave: ataxias autossômicas recessivas, cerebelo, ataxia cerebelar, ataxia de Friedreich.
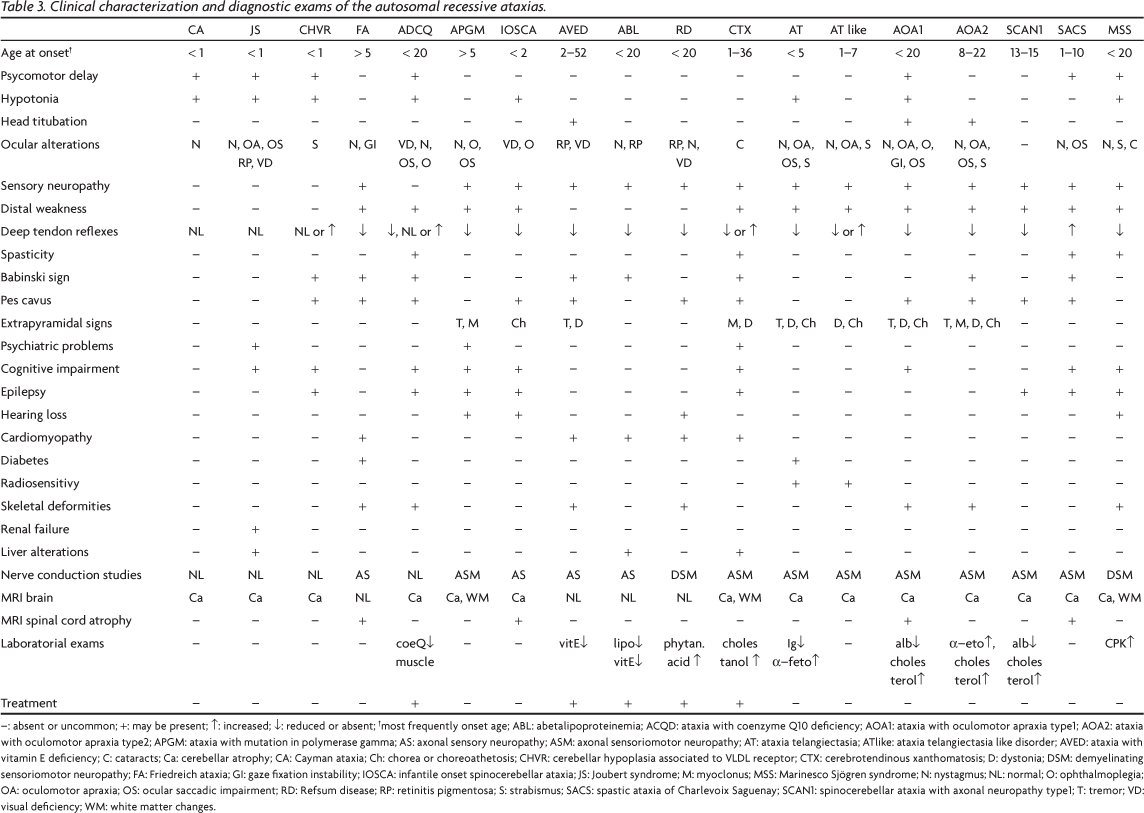
More than 20 different clinical types of autosomal recessive ataxias (ARA) are currently recognized. They are clinically characterized by balance abnormalities, incoordination, kinetic and postural tremor, and dysarthria1. Typically, symptoms start before 25 years of age, and cerebellum, brainstem, and spinocerebellar tracts are involved2. Peripheral neuropathy, ophthalmological abnormalities, and non-neurological signs and symptoms might also be present1. Friedreich ataxia, the most common ARA, was first described in 1863 and is seen worldwide1,2. In the last few years, several other conditions have also been recognized and their loci and genes identified.
Pathophysiology is quite variable and the defective gene product might be involved with: (A) Cerebellar and/or brain stem development; (B) Mitochondrial energy generation; (C) Intermediate metabolism; (D) DNA repair; and (E) Cerebellar integrity maintenance. Several classifications have been proposed so far, using clinical, neuroimaging, genetic and pathophysiologic data2. In this review, a pathophysiological classification is used (Table 1).
We should be particularly aware of treatable forms of ARA, which includes Refsum disease, ataxia with vitamin E deficiency, coenzyme Q10 deficiency, cerebrotendinous xanthomatosis, and abetalipoproteinemia.
ATAXIA CAUSED BY CEREBELLAR AND/OR BRAINSTEM MALFORMATION
In this group, neuroimaging studies are able to identify cerebellar and/or brainstem malformation and clinically it is characterized by non progressive cerebellar ataxia. Three conditions are discussed in this section: Cayman ataxia, Joubert syndrome and Cerebellar hypoplasia associated to VLDL receptor.
Cayman ataxia
Cayman ataxia (CA) is a condition characterized by variable developmental delay, early onset hypotonia and non-progressive axial cerebellar ataxia, associated to nystagmus, intention tremor, and dysarthria3. MRI presents with cerebellar hypoplasia3. The CA has only been found in Grand Cayman Island, where heterozygote frequency is supposed to be of 18%3. CA is caused by mutation at ATCAY, which codes for caytaxin, a protein involved with glutamate synthesis and also with synaptogenesis of cerebellar granular neurons and Purkinje cells4. Interestingly, ATCAY contains a CRAL-TRIO domain that binds small lipophillic molecules, similar to the alpha-tocopherol transport protein that causes ataxia with vitamin E deficiency3.
Joubert syndrome
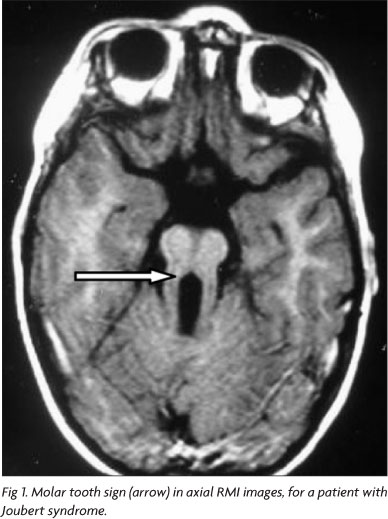
Joubert syndrome (JS) is a rare genetically heterogeneous inherited disorder with an estimated prevalence in the United States of 1 in 100,0005. JS is characterized by congenital ataxia, hypotonia, developmental delay, and at least one of the following features: neonatal respiratory disturbances and abnormal eye movements (nystagmus or oculomotor apraxia). In some cases, Leber congenital amaurosis, pigmentary retinopathy, renal and hepatic abnormalities can also be found. A combination of midline cerebellar vermis hypoplasia, deepened interpeduncular fossa, and thick, elongated superior cerebellar peduncles gives the axial view of the midbrain the appearance known as molar tooth sign (Fig 1), an obligate finding for JS diagnosis5-7.
Recently, Valente, Brancati and Dallapiccola6 proposed a clinical classification of JS in which the molar tooth sign was considered an obligatory criterion. They were able to recognize six subgroups of JS: (1) Pure Joubert syndrome; (2) JS with retinal abnormality; (3) JS with renal disorders; (4) CORS (cerebello-oculo-renal syndrome), or JS with retinal abnormality and kidney involvement; (5) COACH (cerebellar vermis hypoplasia/aplasia, oligophrenia, ataxia, ocular coloboma, and hepatic fibrosis) or JS with mental retardation, ocular coloboma and liver disorder; and (6) Oro-facio-digital syndrome type VI, or JS with orofacial abnormality and polydactylia.
Seven loci and five genes have been identified so far (Table 2)6. It is believed that other loci and genes will be recognized in the future, as mutations in known genes account for only a small fraction of patients. There is no clear correlation between genetic and clinical in JS, nonetheless, AHI1 mutations are usually associated with pure JS and approximately 50% of individuals with cerebello-oculo-renal syndrome have CEP290 mutations6,7. In large series, mutations in AHI1 are found in 10 to 15% of cases, and of CEP290 in 10%5.
Cerebellar hypoplasia associated with VLDL receptor
Cerebellar hypoplasia associated with very low density lipoprotein (VLDL) receptor (CHVR) is clinically characterized by severe developmental delay, hypotonia, global ataxia, flat feet, strabismus, and moderate to severe mental retardation8,9. Epilepsy and short stature might occasionally be seen8. MRI discloses a symmetric cerebellar hypoplasia, mostly of its inferior segment, with variable brainstem and corpus callosum hypoplasia, and plain cortical gyrus8,9. This form of non-progressive cerebellar ataxia was first reported as disequilibrium syndrome among North-Americans Hutterites8. CHVR is caused by mutation in the gene that encodes VLDL receptor (VLDLR)9. This transmembrane protein is part of reelin signaling pathway, which guides neuroblast migration in the developing cerebellum and cerebral cortex9.
ATAXIAS CAUSED BY DEFICIENCY OF MITOCHONDRIAL ENERGY GENERATION
Ataxias caused by deficiency of mitochondrial energy generation includes Friedreich ataxia, Ataxia with CoQ10 deficiency, Mitochondrial recessive ataxic syndrome (MIRAS) and Infantile-onset spinal cerebellar ataxia (IOSCA).
Friedreich ataxia
Friedreich ataxia (FA) is the most common recessive ataxia worldwide, with an estimated prevalence in Caucasian population of 1:30,000 to 1:50,00010 and a carrier frequency of 1:851. Its onset is usually in the second decade of life, but can vary from 2 to 25 years of age. Clinical manifestations are characterized by a combination of sensory and cerebellar symptoms, and gait instability is usually the first recognized abnormality. Relentlessly progressive ataxia is characteristic, and after 10 to 15 years of onset, patients are usually wheelchair bound. Dysarthria is also an early and incapacitating symptom, leading to an almost incomprehensible speech. Vibratory and positional sense is affected, and Romberg sign is usually positive. Deep tendon reflexes are absent, but extensor plantar reflex (Babinski sign) is usually present. Abnormal eye movements and defective fixation are also observed. Cognitive function is preserved, but communication abilities can be affected. Systemic abnormalities as hypertrophic cardiomiopathy, cardiac conduction defects and diabetes can occur. As disease progresses, pes cavus and scoliosis are almost always present. Although there are significant variations in the onset and rate of disease progression, the mean age of death has been reported to be approximately 38 years, with a range as wide as 5 to 70 years. Death usually is secondary to progressive cardiomyopathy10.
Brain MRI in FA is normal; using the multigradient echo sequence it is sometimes possible to detect iron deposits in dentate nuclei of cerebellum. Spinal cord MRI can disclose mild atrophy of its cervical segment, which is explained by the large loss of primary sensory neurons in the dorsal root ganglia, early in the course of the disease. Nerve conduction studies characteristically show axonal sensory neuropathy10. Atypical forms of FA, as late onset or with maintained reflexes, have been proposed, but it is now clear that they are also caused by mutations in the same gene.
FA is caused by mutations in the FRDA gene, which encodes frataxin, a protein involved in mitochondrial iron regulation. Loss of mitochondrial iron-sulfur centers, impairment of mitochondrial respiratory chain, increased mitochondrial iron and increased oxidative damage are observed when frataxin is deficient. Almost all patients are homozygotes for a GAA expansion which occurs in intron 1 of FRDA gene. Normal individuals have up to 40 GAA repeats, and in patients this number can vary from 70 or 90 to over 1,700 repeats. Presence of biallelic expansion confirms diagnosis, independent of clinical phenotype10,11. Close to 2% of patients are compound heterozygotes, with a combination of a GAA expansion in one allele and a point mutation in the other1.
Coenzyme Q10 (CoQ10), and its synthetic analog idebenone, vitamin E and, more recently the iron chelator deferiprone have been used for treating FA, with some promising but still very preliminary results12,13. Deferiprone, an atypical iron chelator, may decrease accumulation of toxic iron in the mitochondria in patients, but the recommended dose and the efficiency of this treatment have not yet been determined12.
Ataxia with coenzyme Q10 deficiency
Primary deficiency of coenzyme Q10 (CoQ10) is a genetically heterogeneous disorder, with a highly variable clinical spectrum, which includes multi-systemic manifestations as well as CNS compromise14-16. Five clinical subtypes have been recognized: (1) Encephalomyophatic, with mitochondrial myopathy, recurrent myoglobinuria and CNS symptoms and signs; (2) Early infantile multi-systemic, with severe visceral and brain manifestations; (3) Leigh syndrome; (4) Pure myopathic; (5) Ataxic14,15,17.
The ataxic subtype is the most common presentation of CoQ10 deficiency14,15. It is characterized by progressive ataxia, cerebellar atrophy and reduced muscle CoQ1014,15. Early symptoms might include developmental delay, hypotonia and frequent falls. Global, progressive ataxia, and dysarthria start before the adolescence17. Epileptic seizures, proximal or distal muscle weakness, dysphagia, ophthalmoparesis, nystagmus, peripheral axonal neuropathy, pyramidal signs and scoliosis might also be present14,15,17. Mental retardation or cognitive decline is also sometimes seen14,17. The adult onset form of ataxia and CoQ10 deficiency is usually associated with hypergonadotrophic hypogonadism14.
CoQ10 (also known as ubiquinone) is a lipophylic compound which is involved in electron transport from complex I and II to complex III of mitochondrial respiratory chain14-17. CoQ10 deficiency impairs proton transfer across the internal mitochondrial membrane and consequently to a reduction in ATP production14,16.
The main source of CoQ10 is endogenous synthesis, which involves a still-uncharacterized complex pathway. Four genes are known to be involved in CoQ10 biosynthesis: PDSS1 e PDSS2 (subunits 1 and 2 of prenyldiphosphate synthase), COQ2 (OH-benzoate polypreniltransferase) and ADCK3, which acts as a chaperone14,16.
Diagnosis is based on reduced amount of CoQ10 in muscle, as plasma CoQ10 levels are usually normal14,15,17. Muscle histopathology is essentially normal and brain MRI discloses global cerebellar atrophy14,17. Treatment with oral CoQ10 should be adjusted according to clinical results, with doses varying from 300 to 3000 mg/day14,17. Treatment outcome is quite variable: in some patients disease stabilized while in others it progressed relentlessly. It is probable that treatment response is dependable of underlying biochemical defect as well as stage of disease14,15,17.
Ataxia with mutation in polymerase gamma
Polymerase gamma (POLG) is a nuclear encoded gene, whose product is responsible for maintaining the integrity of mitochondrial DNA18. Mutations in POLG are associated with a variety of clinical phenotypes, which includes Alpers disease, parkinsonism, and external progressive ophthalmoplegia18. Two similar forms of autosomal recessive ataxias are associated with mutations in POLG: Mithocondrial Recessive Ataxic Syndrome (MIRAS) and Sensory Ataxia, Neuropathy, Dysarthria, and Ophthalmoplegia (SANDO)18,19.
MIRAS is the most frequent recessive ataxia in Finland1,20. Clinical manifestations, which start between 5 to 40 years of age, are characterized by cerebellar ataxia, nystagmus, dysarthria, ophthalmoplegia, tremor, cognitive decline, and myoclonus. Loss of vibratory and position perception is commonly seen19,20. Epilepsy is a frequent manifestation in MIRAS, but not in SANDO, with both partial and generalized seizures, sometimes becoming refractory to antiepileptic drugs and evolving to status epilepticus18-20. Brain MRI discloses cerebellar atrophy and T2 weighed hypersignal on thalamus, and dentate and inferior olivary nuclei19,20. Nerve conduction studies also demonstrate axonal sensory neuropathy19,20. Elevated protein might be detected in CSF19,20. Muscle biopsy is not diagnostic, but Southern blotting analysis might detect multiple deletions in muscle mithocondrial DNA20. Diagnosis is based on sequencing of the POLG gene, with two mutations (p.A467T and p.W748S) beeing responsible for most cases of this disorder in Caucasians19,20. There is no clear genotype-phenotype in this condition.
Infantile-onset spinocerebellar ataxia
Infantile-onset spinocerebellar ataxia (IOSCA) is currently identified only in Finland and is characterized by acute or subacute cerebellar signs triggered by unspecific infection around the age of 1 year21,22. Their clinical features are similar to MIRAS. Hypotonia, athetosis of hands and face, and ataxia with absent reflexes are the early symptoms this disease. Later at pre-school age, ophthalmoplegia and neurosensorial deafness might be seen. Tactile, proprioceptive, and vibratory impairment, without pain or temperature compromise are detected after the first decade. Teenagers are usually wheelchair bound with severe distal muscular atrophy, pes cavus, mild to moderate cognitive impairment and optic atrophy without significant visual impairment. Refractory epilepsy and status epilepticus might contribute to rapid neurological deterioration and death. Further recognized abnormalities are autonomic dysfunction and, in females, primary hypogonadism21,22.
There is no biochemical marker for IOSCA. Nerve conduction studies and nerve biopsy demonstrate a severe, mostly sensory, axonal neuropathy. Sensory ganglia are more severely affected than motoneurons21,23. Neuroimaging studies at early stages of disease demonstrate reduced size of cerebellar hemispheres which progress to a more widespread olivopontocerebellar atrophy23. Muscle biopsy is non-diagnostic but mitochondrial DNA depletion might be seen in this tissue. Pathological studies disclose spinal cord atrophy (more intense on the posterior funiculli), cerebellum and brainstem; there is also marked loss of myelinated fibers on peripheral nerve21.
IOSCA is caused by mutation in C10ORF2 gene, which codes for twinkle, a specific mitochondrial DNA helicase, and one of its smaller isoform, twinky. Twinkle is important for maintenance and replication of mitochondrial DNA, and twinky function is currently unknown. The same founder mutation (p.Y508C) was detected in most Finnish patients with classical IOSCA. Mutations in C10ORF2 might also be associated to different phenotypes, such as Alpers disease (early onset encephalopathy with untreatable epilepsy, mtDNA depletion and liver failure) and autosomal dominant progressive external ophthalmoplegia22.
METABOLIC ATAXIAS
Metabolic ataxias are treatable disorders, and it is particularly important to make an early and accurate diagnosis in this group of ARA. Ataxia with vitamin E deficiency, abetalipoproteinemia, hipobetalipoproteinemia, Refsum disease and cerebrotendinous xanthomatosis will be discussed in this section.
Ataxia with vitamin E deficiency
Ataxia with vitamin E deficiency (AVED) is similar to Friedreich ataxia (FA). Age of onset usually varied from 4 to 20 years, with outliers ranging from 2 to 52 years24,25. Clinical manifestations are characterized by progressive trunk and limbs ataxia, dysarthria, disturbance of positional and vibratory lower limbs senses, Babinski sign and abolished deep tendon reflexes. Scoliosis and pes cavus are commonly seen24-26, yet retinopathy is less frequent25,26. Dystonia (13%) and head titubation (28%) are more commonly seen in AVED than in FA24,26. Cardiomyopathy and an acute cardiac event might be associated with premature death among AVED cases25,26.
AVED is caused by mutations in the α-tocopherol transfer protein gene (TTPA), which codes a protein responsible for transferring α-tocopherol from chilomicrons to VLDL26,27. TTPA disfunction causes very low level of circulating α-tocopherol and tissue deficiency of this vitamin27. Several different pathogenic mutations have been reported so far. Two mutations, associated with a severe phenotype, are particularly frequent in Europe, North Africa and North America: c.744delA and c.486delT24,25. On the other hand, the mutation p.H101G was only detected in Japanese families and is associated with later onset and pigmentary retinopathy24. Age of onset, clinical manifestations and progression velocity are quite variable in AVED. It is usually stated that mutations causing profound TTPA protein depletion are responsible for a more severe phenotype and mutations leading to amino acid substitution are associated to a milder form of disease24-26.
Diagnosis in a symptomatic individual is established with the determination of α-tocopherol (vitamin E) serum level, which is always below 2.5 μg/ml (reference values: 5-15 μg/ml)24,25. Brain MRI is usually normal, but mild cerebellar atrophy might also be seen1,26. A pattern of axonal sensitive neuropathy is often observed at nerve conduction studies24.
AVED treatment consists of vitamin E oral administration at a dose of 600 to 2,400 mg/day. Serum levels might be used as guidance for oral dose adjustment24-26. Differential diagnosis of α-tocopherol primary deficiency includes intestinal fat mal-absorption and abetalipoproteinemia26. As a rule, vitamin E serum levels should be checked in all patients with FA clinical phenotype without molecular confirmation for this condition.
Abetalipoproteinemia and hypobetalipoproteinemia
Abetalipoproteinemia (ABL), a multisystem disorder caused by a defect in lipoprotein metabolism, is characterized by acanthocytosis, atypical pigmentary retinopathy and spinocerebellar degeneration28,29. In the first year of life, main manifestations are chronic diarrhea and failure to thrive. Neurological features, present after the first decade of life, include absent deep tendon reflexes, superficial and deep sensory abnormalities, weakness and global ataxia29. As disease progresses, atypical pigmentary retinopathy characterized by small, irregularly distributed white spots, and night and color blindness is detected29. Clinical manifestations of ABL are secondary to deficient absorption of the lipid-soluble vitamins A, D, E, and K1,28.
Apolipoprotein B (ApoB) is the main protein of both VLDL and LDL, and their assembly is dependent on microsomal triglyceride transfer protein (MTP)28. Mutations in the gene coding for the large (88 kD) subunit of MTP is responsible for ABL and determine very low levels of LDL and VLDL cholesterol. Decreased levels of vitamins A, K, and E, anemia, very low sedimentation rate, increased prothrombin time and elevated creatine kinase are also observed. Deficient MTP can also lead to lipid infiltration of small bowel mucosa and hepatic steatosis28,29. Nerve conduction velocity usually discloses sensory axonal peripheral neuropathy1,29.
ABL treatment is done with supplementation of vitamin A (100 to 400 IU/Kg/day), vitamin E (2,400 to 14,400 IU/day), and vitamin K (5 mg/day)29. It is also recommended a low fat diet combined with essential fatty acid supplementation28,29. Coagulation tests are used to monitor vitamin K and serum levels of vitamin A and E to check supplementation adequacy for these vitamins28,29.
Hypobetalipoproteinemia (HBL) is similar to ABL. It is caused by mutations in APOB gene, which encodes apolipoprotein B (ApoB)2. APOB heterozygotes have lower levels of ApoB, VLDL- and LDL-cholesterol, while MTP heterozygotes have normal levels of these substances2,29,30.
Refsum disease
Refsum disease (RD) is a peroxisomal disorder clinically characterized by pigmentary retinopathy, cerebellar ataxia, mixed motor-sensory neuropathy and elevated CSF protein31,32. Its onset usually occurs before 20 years of age, with night blindness secondary to retinopathy, followed by progressive constriction of visual field and optic atrophy, cataracts, vitreous opacities and nystagmus32,33. Other common clinical manifestations are anosmia, cochlear deafness, ichthyosis, bone dysplasia and cardiac abnormalities31-33. Psychiatric disorders are uncommon31,33. If not adequately treated, RD can cause premature cardiac death31,32.
Elevation of serum phytanic acid (>200 μM/L; reference value <30 μM/L) is very suggestive, but not specific of RD31,33. Phytanic acid is a branched long chain fatty acid present on dairy products and red meat33. It is a by-product of chlorophyll catabolism and is not endogenously synthesized31-33. Diagnostic confirmation can be made measuring the activity of phytanoyl-CoA hydroxilase in fibroblasts or by molecular analysis of the responsible gene31.
RD is a genetically heterogeneous disorder. Most cases are caused by mutations in PHYH (encoding phytanoyl-CoA hydroxylase), a peroxisomal matrix enzyme which catalyses ?-oxidation of branched chain fatty acids31-33. Deficiency in PEX7 (encoding peroxin-7), a protein involved in peroxisomal import of some enzymes, including phytanoyl-CoA hydroxylase, also results in this phenotype2,31. PEX7 mutations may also cause a severe peroxisomal biogenesis disorder known as rhizomelic chondrodysplasia punctata32.
Treatment is based on phytanic acid dietary restriction, combined, if necessary, with plasmapheresis. With reduction of phytanic acid serum levels, RD symptoms stabilize and there may be some improvement of ataxia and ichthyosis, albeit effects on pigmentary retinopathy are uncertain32,33.
Cerebrotendinous xanthomatosis
Cerebrotendinous xanthomatosis (CTX) is a rare bile acid synthesis disorder34. Its main clinical manifestations are juvenile cataracts, chronic diarrhea, and tendinous xanthomas34-36. In the neonatal period, a potentially lethal cholestatic syndrome has been reported34. After the second decade of life, progressive neurological deterioration may occur, characterized by cognitive decline, psychiatric manifestations, cerebellar ataxia, progressive spastic paraplegia, dysphagia, and less frequently, seizures and peripheral neuropathy34,36,37. Exceptionally, neurological manifestations are restricted to the spinal cord35. There is a wide intra- and inter-familial clinical variability34,37. Coronary heart disease without elevated cholesterol is an important cause of morbidity and mortality in adults34,35.
CTX is caused by mutations in CYP27A1 gene, which codes for sterol 27- hydroxylase, a protein expressed mostly in liver34,36. This enzyme is essential for bile acids synthesis, including chenodeoxycholic acid36. In the absence of sterol 27- hydroxylase, the substrate of this enzyme is converted to cholestanol by the action of 7 α-hydroxylase34,36. Elevated serum cholestanol is the biochemical hallmark of CTX35,37. Increased urinary excretion of bile alcohol glucuronides might also be present37. This disorder can be treated by oral administration of chenodeoxycholic acid, which inhibits 7 α-hydroxylase and consequentely cholestanol synthesis34-37. Liver transplant is another therapeutic alternative34. Cholestanol determination in asymptomatic siblings of CTX patients is recommended to improve clinical outcome34,37.
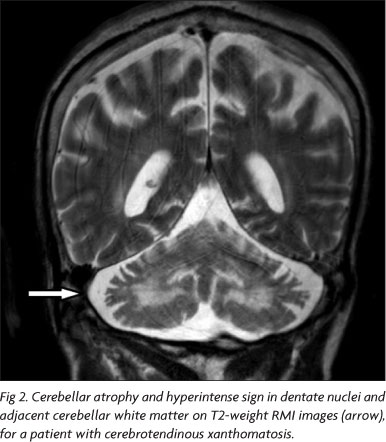
Brain MRI most distinctive abnormalities are detected at T2-weighed and FLAIR sequences, which demonstrate bilateral, heterogeneous and hyperintense sign in dentate nuclei and adjacent cerebellar white matter (Fig 2)35,36. Other less characteristic abnormalities which might be detected are brain stem, cerebellar and cerebral atrophy and diffuse hyperintense cerebral white matter lesions35,36. MR spectroscopy (MRS) of CTX patients discloses reduction of N-acetylaspartate and increase in lactate36.
ATAXIAS WITH DNA REPAIR DEFECTS
This group has as a common pathogeny a defect in double or single strand DNA repair; besides ataxia, extrinsic ocular movements are frequently affected. Ataxia-telangiectasia, ataxia telangiectasia-like, apraxia and oculomotor apraxia types 1, 2 and 3, and spinocerebellar ataxia with axonal neuropathy type 1 belong to this group.
Ataxia-telangiectasia
Ataxia-telangiectasia (AT) has an estimated frequency in the USA of 1/40,000 individuals38 and it is predicted that 0.5% of UK population carries one mutation in ATM gene, which is responsible for AT39. Progressive ataxia with onset before 3 years of age is the main clinical characteristic38,40. Telangiectasias (Fig 3), another hallmark of disease, are seen in at least 90% of affected individuals and their age onset ranges from 2 to 8 years; they are more easily seen in conjunctivas, ears, face and neck38,40,41. A large range of ophthalmological abnormities might also be detected, including: optokinetic nystamus (present in 81% of cases), gaze induced nystamus (seen in 29%), hypometric or delayed saccades (76%), delayed eye tracking (63%), strabismus (38%), and oculomotor apraxia (30%)41. Dysarthria, dysphagia, facial hypomimia, generalized hypotonia, peripheral neuropathy and movement disorder, as tremor or choreoathetosis, are seen after five years of age38,40,41. Cognitive level is usually normal, even though severe dysarthria and incoordination might leave an impression of mental retardation. Independent gait is lost by the end of first decade38,40. Immunodeficiency (mainly humoral) leading to chronic sinopulmonary infection and increased susceptibility to cancer are other important features in AT. Risk of lymphoproliferative disorders are dramatically increased in AT38,40. Treating cancer in AT patients is particularly challenging, because of their increased radiosensitivity and adverse side effects to chemotherapy40. Female carriers of one mutated copy of the gene have 3-4 fold increased risk of breast cancer when compared to general population39.
ATM gene encodes for a ATM serine/threonine kinase, a large protein with 3,056 amino acids which is part of the phosphatidyl-inositol-3-kinase (PI3-K) complex, responsible for DNA repair during the cell cycle, avoiding incorporation of deleterious mutations38,39. ATM gene is very large, containing 66 exons, and its sequencing with current technology is still cumbersome, but feasible38,39. Most patients are compound heterozygotes for ATM mutations and a large variety of sequence variants have been recognized, thus making interpretation of results difficult. Pathogenic mutations are usually nonsense mutations (85%), and missense mutations are responsible for 10% of detected pathogenic changes38,39,40.
Some laboratory tests might help AT investigation: serum alpha-fetoprotein is elevated in more than 95% of cases and low levels of IgA, IgE and reduced T lymphocyte count, with normal or elevated B lymphocytes are usually detected. Karyotype might show translocation between chromosomes 7 and 1438-41 and radiosensivity test might be used to demonstrate chromosomal breakage predisposition.
Brain MRI discloses, except in early stages of disease, cerebellar atrophy, which is initially more evident on the hemispheres and superior vermis and later becomes diffuse38. CT-scan and plain radiography should be avoided because of increased X-ray sensitivity.
Ataxia-telangiectasia like
Ataxia-telangiectasia like (ATL) is a very rare clinical condition which is characterized by slowly progressive ataxia with onset between 1 and 7 years of age, associated to oculomotor apraxia and dysarthria42. No ocular or facial telangiectasias are detected and cognition is preserved39,42,43. Reflexes might be initially brisk and became reduced42. At advanced stages of disease, tongue and facial dyskinesia, choreoathetosis, and dystonia, suggesting basal ganglia compromise, might be seen42,43. ATL is progressive up to the adolescence, when it stabilizes43. There is no increased risk for infections or neoplasias, as is seen in AT, but occasionally microcephaly is present39,42.
Brain MRI detects cerebellar atrophy, and laboratory tests are non-informative43. Radiosensitivity test is usually present but in a lesser degree than in AT35,42-44.
ATL is caused by mutation in MRE11 gene, located in chromosome 11q21, near the ATM gene. Its product is part of the MRN complex, which recognizes DNA double strand breakage. Both missense and null mutations have been reported. Severity varies according to the type of molecular defect. Most of reported cases were original from Saudi Arabia 39,42-44.
Ataxia with oculomotor apraxia type 1
Ataxia with oculomotor apraxia type 1 (AOA1) is a condition characterized by involuntary movements (chorea and dystonia) and/or progressive global ataxia, with dysarthria associated with hands and head tremor. Onset can vary between 1 to 20 years of age and developmental delay might be seen before clinical symptoms became apparent. As disease progresses, movement disorder are attenuated and peripheral neuropathy signs, as distal atrophy, pes cavus, superficial and deep sensory impairment, hypo/arreflexia, become apparent. The most distinctive clinical signs in AOA1 are related to external eye movements: gaze-evoked nystagmus (found in all patients), oculomotor apraxia (seen in 86%), saccadic pursuit, hypometric saccades, fixation instability, and excessive blinking45-47. In advanced stages, oculomotor apraxia might be masked by progressive external ophthalmoparesis, which starts with upward gaze paralysis45. Optic atrophy and retinal exsudative lesions have been occasionally reported (Barbot, 2001; Le Ber, 2003). Variable cognitive impairment might be seen and mental retardation is not uncommon46,47.
Laboratory findings include hypoalbuminemia and hypercholesterolemia46,47. Elevated creatine kinase is occasionally detected47. Nerve conduction velocity studies disclose sensory-motor axonal neuropathy. MRI reveals marked cerebellar atrophy, mild brainstem atrophy and, in advanced cases, cortical atrophy45-47. Loss of myelinated fibers with maintenance of amyelinic ones is seen at sural nerve45,47.
AOA1 is caused by mutation in APTX gene, which encodes aprataxin, a nuclear protein involved in single-strand DNA repair, acting in the same pathway of the ATM protein39,46. Several mutations have been reported so far, most of them in exons 5, 6, and 7 of APTX gene47. This condition was originally reported in Japan (where it is the most common cause of ARA) but is found worldwide. In Portugal, AOA1 is the second most common cause of ARA46.
Ataxia with oculomotor apraxia type 2
Ataxia with oculomotor apraxia type 2 (AOA2) is characterized by global progressive ataxia with onset usually between 8 and 25 years of age48,49, dysarthria, axonal motor sensory neuropathy, and oculomotor apraxia, which is seen in less than 50% of cases48-50. Saccadic pursuit is seen in all patients, gaze evoked nystagmus in 89%, and bilateral limited abduction of the eyes with strabismus in 61% of the patients48. Dystonia, head and postural tremor, chorea, dysphagia, pes cavus, and scoliosis are occasionally seen. Cognitive function is usually preserved, but executive dysfunction is sometimes observed48-50. Premature ovarian failure was also reported in some patients48. Progression is slow, and most patients are wheelchair bound 10 years after its onset48,49.
Serum alpha-fetoprotein is mildly to moderately elevated in all patients with AOA239,48-50. Increased serum creatine kinase, cholesterol, and immunoglobulin IgG and IgA, and reduced serum albumin are inconstantly seen48,50. Brain MRI discloses diffuse cerebellar atrophy, more intense in the vermis, occasionally associated with pontine atrophy50. Nerve conduction studies detect sensory-motor axonal neuropathy and nerve biopsy demonstrates that large myelinated fibers are more severely affected than thin ones48,50.
AOA2 is caused by mutation in SETX gene (encoding senataxin), a protein with DNA and RNA helicase activity and which is involved in RNA processing and DNA repair39,49,50. Amyotrophic lateral sclerosis type 4 (ALS4), is caused by dominant mutations in senataxin50.
Ataxia with oculomotor apraxia type 3
Ataxia with oculomotor apraxia type 3 (AOA3) is a recently described ARA with a phenotype similar to ataxia-telangiectasia, but with onset after 8 years of age. Reported clinical features are ataxic gait, dysarthria, oculomotor apraxia and cerebral atrophy. No telangiectasia, biochemical abnormalities, or nerve conduction impairment was detected. Other forms of ataxia with oculomotor apraxia were excluded. Studies performed in fibroblasts demonstrated a defect in repairing DNA, making these cells sensitive to agents that cause single strand breaks in DNA. Nevertheless, locus for AOA3 remains elusive51.
Spinocerebellar ataxia with axonal neuropathy type 1
Spinocerebellar ataxia with axonal neuropathy type 1 (SCAN1) is a rare disorder recognized in 2002 in a large consanguineous family of Saudi Arabia52. Age of onset is around 14 years, characterized by moderate ataxia, dysarthria, muscular weakness, distal atrophy, pes cavus and reduction of vibratory and postural sense. Epilepsy may occur, but there is no cognitive decline or oculomotor abnormality. Nerve conduction studies disclose motor-sensory axonal neuropathy and biochemical tests are non diagnostic, but low serum albumin and elevated cholesterol are occasionally seen52. Mild cerebellar and cerebral atrophy might be present on MRI studies. SCAN1 is caused by mutation in TDP1 gene, which codes for tyrosil DNA phosphodiesterase 1 (TDP1), a protein involved in single strand DNA repair39,52,53. SCAN1 is an additional example of nervous system vulnerability to impaired DNA repair, as occurs in AOA1, AOA2 and AT52,53.
DEGENERATIVE ATAXIAS
Degenerative ataxias have as a common feature the compromise of a protein that acts as a chaperone, helping protein folding. Two conditions belong to this group: autossomal recessive ataxia of Charlevoix-Saguenay and Marinesco-Sjögren syndrome.
Autosomal recessive spastic ataxia of Charlevoix Saguenay
Spastic ataxia of Charlevoix-Saguenay (SACS) was first reported in Charlevoix and Saguenay region of northeast Quebec province, Canada1,54. In this area, its incidence was estimated in 1/1,932 newborns, and 1 in every 22 of its inhabitants are supposed to be carrier of the mutation responsible for SACS2,55. This condition has now been reported worldwide, but the largest series remains from Canada1,54-56.
Clinically, SACS is characterized by delay in acquiring independent walk, frequent falls and gait instability55. Disease progression is slow and ataxic gait; dysarthria and spastic paraplegia are the major manifestations in the first two decades. Later, lower limb peripheral neuropathy can also be detected. As disease evolves, pyramidal signs can be masked by progression of peripheral neuropathy, with the exception of the Babinski sign, which is usually present even in later stages of disease. Distal atrophy, pes cavus, and hammer toes are commonly seen as disease advances54-56. In some patients, fundoscopy discloses hypermyelination of fibers radiating from optic disk and embedding the retinal vessels, a very peculiar finding. Horizontal nystagmus, saccadic alteration of smooth ocular pursuuit and miccional urgency might be present54,55. Mild mental retardation and cognitive decline were occasionally reported54. Patients usually become wheelchair bound after the 3rd or 4th decades of life and life expectancy is reduced as they become bedridden. During pregnancy, disease progression is apparently accelerated in affected women55.
Nerve conduction velocity studies usually disclose an axonal neuropathy with mild demyelination, sensory fibers are more severely affected that motor fibers. The most consistent neuroimaging finding is cerebellar vermis atrophy, mostly from its superior portion54-56. Cervical and thoracic spinal cord thinning are occasionally reported55.
At an early stage, SACS can be misdiagnosed as cerebral palsy55. Diagnosis is based on clinical manifestations and confirmed by mutation analysis of SACS gene located on 13q1154,56. Putative role of its product, sacsin, is to help protein folding, acting as a chaperone56. How sacsin deficiency causes neurodegeneration it is not known, but it has been reported to interact with Ataxin-1, the cause of Autosomal Dominant Spinocerebellar Ataxia type 156.
Marinesco-Sjögren syndrome
Marinesco-Sjögren syndrome (MSS) is a rare, multisystem disorder, characterized by congenital or early-onset cataracts, developmental delay, cerebellar ataxia and mild to severe mental retardation. Microcephaly, nystagmus, short stature, scoliosis, hypergonadotrophic hypogonadism and myopathy are common additional features57,58. Peripheral neuropathy, deafness, optic atrophy, strabismus, spasticity and seizures might be present57. Disease progression is slow and long survival can be expected2.
Brain MRI usually discloses cerebellar atrophy or hypoplasia. Additional uncommon findings are cortical atrophy and leucoencephalopathy. Serum creatine kinase is usually elevated and muscle biopsy show chronic myopathy with rimmed subsarcolemmal vacuoles57,58.
MSS is caused by mutations in SIL1 gene (encoding a nucleotide exchange factor for heat-shock protein 70 family member HSPA5). Heat-shock protein 70 family members are the highly conserved molecular chaperones that assist in stabilization and folding of newly synthesized polypeptides. Decrease of SIL1 gene product leads to a reduction of protein synthesis in endoplasmic reticulum58,59.
CONCLUSION
Differential diagnosis of ARA is a difficult task, as there is a clear overlap of clinical manifestations among several previously discussed conditions. Table 3 presents the main characteristics of each these disorders and Fig 4 is a proposed algorithm to help investigation of this group of diseases. Nevertheless, we should be aware that ataxia might be a symptom in many other progressive disorders, affecting primarily white matter (e.g., metachromatic leukodystrophy, leukoencephalopathy with vanishing white matter, Paelizeus-Merzbacher disease, X-linked adrenoleukodystrophy), neurons (e.g. neuronal lipofuscinosis ceroid, juvenile Tay-Sachs disease), or leading to a more widespread brain malformation or systemic manifestations, as it happens in pontocerebellar hypoplasia and congenital disorders of glycosilation (CDG). Non progressive cerebellar symptoms are also prominent in ataxic cerebral palsy, an important differential diagnosis for early-onset ARA. It is also important to remind that all spinocerebellar ataxias (SCAs) inherited as a dominant trait are out of the scope of this review.
Currently, neuroimaging studies, especially brain MRI, are particularly important in ARA work-up and to help detection of cerebellar malformations and atrophy. Normal MRI is expected in some disorders, as FA and AVED. Nerve conduction velocity studies and, in some cases, electromyography are useful for evaluation of some patients, even in the absence of clinical signs of peripheral neuropathy or myopathy. Elevation of serum alpha-fetoprotein is characteristic of AT and AOA2. More specific biochemical tests, as determination of vitamin E and phytanic acid should not be neglected, once they can help the diagnosis of some treatable forms of ARA. Determination of serum cholestanol and muscle CoQ10 are performed in a few specialized centers around the world but both CTX and ataxia with CoQ deficiency are potentially treatable.
Molecular analysis access is limited, but it is feasible for diseases as FA and, in lesser degree, AOA1 and AOA2. Many patients with putative ARA remain undiagnosed, and is expected that new forms of ARA will be recognized in the near future. The number of recessive ataxias is already high, and we will probably keep counting new arrivals for the forthcoming years.
Received 11 May 2009, received in final form 3 August 2009. Accepted 22 September 2009.
Dr. Fernando Kok - Serviço de Neurologia Infantil / Hospital das Clínicas da Faculdade de Medicina da USP - Av. Dr. Enéas de Carvalho Aguiar 255 / 5011 - 05403-000 São Paulo SP - Brasil. E-mail: fernando.kok@fleury.com.br
- 1. Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 2007;6:245-257.
- 2. Palau F, Espinós C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Diseases 2006;1:47.
- 3. Bomar JM, Benke PJ, Slattery EL, et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nature Genet 2003;35:264-269.
- 4. Hayakawa Y, Itoh M, Yamada A, Mitsuda T, Nakagawa T. Expression and localization of Cayman ataxia-related protein, Caytaxin, is regulated in a developmental- and spatial-dependent manner. Brain Res 2007;1129:100-109.
- 5. Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM213300). Eur J Hum Genet 2007;15:511-521.
- 6. Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet 2008;51:1-23.
- 7. Zaki MS, Abdel-Aleem A, Abdel-Salam G, et al. The molar tooth sign. A new Joubert syndrome and related cerebellar disorders classification system tested in Egyptian families. Neurology 2008;70:556-565.
- 8. Glass HC, Boycott KM, Adams C, et al. Autosomal recessive cerebellar hypoplasia in the Hutterite population. Dev Med Child Neurol 2005; 47:691-695.
- 9. Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet 2008;16:1070-1074.
- 10. Pandolfo M. Friedreich ataxia. Arch Neurol 2008;65:1296-1303.
- 11. Hebert MD, Whittom AA. Gene-based approaches toward Friedreich ataxia therapeutics. Cell Mol Life Sci 2007;64:3034-3043.
- 12. Gonçalves S, Paupe V, Dassa EP, Rustin P. Deferiprone targets aconitase: Implication for Friedreich's ataxia. BMC Neurology 2008;8:1-4.
- 13. Cooper JM, Korlipara LVP, Korlipara PE, Hart PE, Bradley JL, Schapira AHV. Coenzyme Q10 and vitamin E in Friedreich's ataxia: predictor of efficacy of vitamin E and coenzyme Q10 therapy. Eur J Neurol 2008; 15:1371-1379.
- 14. Montero R, Pineda M, Aracil A, et al. Clinical, biochemical and molecular aspects of cerebellar ataxia and coenzyme Q10 deficiency. Cerebellum 2007;6:118-122.
- 15. Rötig A, Mollet J, Rio M, Munnich A. Infantile and pediatric quinine deficiency diseases. Mitochondrion 2007;7(Suppl):S112-S121.
- 16. Quinzii CM, López LC, Naini A, DiMauro S, Hirano M. Human CoQ10 deficiencies. BioFactors 2008;32:113-118.
- 17. Musumeci O, Naini A, Slonim AE, et al. Familial cerebellar ataxia with muscle coenzyme Q10 deficiency. Neurology 2001;56:849-855.
- 18. Milone M, Brunetti-Pierri N, Tang L-Y, et al. Sensory ataxic neuropathy with ophthalmoparesis caused by POLG mutations. Neuromuscul Disord 2008;18:626-632.
- 19. Van Goethem G, Luoma P, Rantamäki M, et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology 2004;63:1251-1257.
- 20. Winterhun S, Ferrari G, He L, Taylor RW, et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase γ mutations. Neurology 2005;64:1204-1208.
- 21. Lönnqvist T, Paetau A, Nikali K, von Boguslawski K, Pihko H. Infantile onset spinocerebellar ataxia with sensory neuropathy (IOSCA): neuropathological features. J Neurol Sci 1998;161:57-65.
- 22. Nikali K, Suomalainen A, Saharien J, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet 2005;14:2981-2990.
- 23. Koskinen T, Valanne L, Ketonen LM, Pihko H. Infantile-onset spinocerebellar ataxia: MR and CT findings. Am J Neuroradiol 1995;16:1427-1433.
- 24. Cavalier L, Ouahchi K, Kayden HJ, et al. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet 1998;62:301-310.
- 25. Marzouki N, Benomar A, Yahyaoui M, et al. Vitamin E deficiency ataxia with (744del A) mutation on α-TTP gene: genetic and clinical peculiarities in Moroccan patients. Eur J Med Genet 2005;48:21-28.
- 26. Mariotti C, Gellera C, Rimoldi M, et al. Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci 2004;25:130-137.
- 27. Arita M, Sato Y, Miyata A, et al. Human α-tocoferol transfer protein: cDNA cloning, expression and chromosomal localization. Biochem J 1995;306:437-443.
- 28. Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma ME, Wetterau JR. The role of the microsomal triglyceride transfer protein in abetalipoproteinemia. Annu Rev Nutr 2000;20:663-697.
- 29. Di Leo E, Magnolo L, Bertolotti M, et al. Variable phenotype expression of homozygous familial hypobetalipoproteinemia due to novel APOB gene mutations. Clin Genet 2008;74:267-273.
- 30. Zamel R, Khan R, Pollex RL, Hegele RA. Abetalipoproteinemia: two case reports and literature review. Orphanet J Rare Dis 2008;3:19.
- 31. Wanders RJA, Jansen GA, Skjeldal OH. Refsum disease, Peroxisomes and phytanic acid oxidation: a review. J Neuropathol Exp Neurol 2001; 60:1021-1031.
- 32. Jansen GA, Waterham HR, Wanders RJA. Molecular basis of refsum disease: sequence variations in phytanoyl-CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Hum Mutat 2004;23:209-218.
- 33. Wierzbicki AS, Lloyd MD, Schofield CJ, Feher MD, Gibberd FB. Refsum's disease: a peroxisomal disorder affecting phytanic acid α-oxidation. J Neurochem 2002;80:727-735.
- 34. Federico A, Dotti MT. Cerebrotendinous xanthomatosis: clinical manifestations, diagnostic criteria, pathogesnesis, and therapy. J Child Neurol 2003;18:633-638.
- 35. Barkhof F, Verrips A, Wesseling P, et al. Cerebrotendinous xanthomatosis: the spectrum of imaging findings and the correlation with neuropathogic findings. Neuroradiology 2000;217:869-876.
- 36. Gallus GN, Dotti MT, Federico A. Clinical and molecular diagnosis of cerebrotendinous xanthomatosis with a review of the mutations in the CYP27A1 gene. Neurol Sci 2006;27:143-149.
- 37. Berginer VM, Gross B, Morad K, et al. Chronic diarrhea and juvenile cataracts: think cerebrotendinous xanthomatosis and treat. Pediatrics 2009;123:143-147.
- 38. Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair 2004;3:1187-1196.
- 39. Taylor AMR, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol 2005;58:1009-1015.
- 40. Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: diagnosis and treatment. Semin Pediatr Neurol 2003;10:173-182.
- 41. Farr AK, Shalev B, Crawford TO, Lederman HM, Winkelstein JA, Repka MX. Ocular manifestations of ataxia-telangiectasia. Am J Ophthalmol 2002;134:891-896.
- 42. Fernet M, Gribaa M, Salih MAM, Seidahmed MZ, Hall J, Koenig M. Identification and functional consequences of a novel MRE11 mutation affecting 10 Saudi Arabian patients with the ataxia telangiectasia-like disorder. Hum Mol Genet 2005;14:307-318.
- 43. Delia D, Piane M, Buscemi G, et al. MRE11 mutations and impaired ATM-dependent responses in na Italian family with ataxia-telangiectasia-like disorder. Hum Mol Genet 2004;13:2155 -2163.
- 44. Taylor AMR, Groom A, Byrd PJ. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair 2004; 3:1219-1225.
- 45. Barbot C, Coutinho P, Chorão R, et al. Recessive ataxia with ocular apraxia: review of 22 portuguese patients. Arch Neurl 2001;58:201-205.
- 46. Le Ber I, Moreira M-C, Rivaud-Péchoux S, et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain 2003;126: 2761-2772.
- 47. Ferrarini M, Squintani G, Cavallaro T, Ferrari S, Rizzuto N, Fabrizi GM. A novel mutation of aprataxin associated with ataxia ocular apraxia type 1: Phenotypical ang genotypical characterization. J Neurol Sci 2007;260:219-224.
- 48. Le Ber I, Bouslam N, Rivaud-Péchoux S, et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic stydy in 18 patients. Brain 2004;127:759-767.
- 49. Tazir M, Ali-Pacha L, M'Zahem A, et al. Ataxia with ocolumotor apraxia type 2: a clinical and genetic study of 19 patients. J Neurol Sci 2009; 278:77-81.
- 50. Duquette A, Roddier K, McNabb-Baltar J, et al. Muattions in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol 2005;57:408-414.
- 51. Gueven N, Chen P, Nakamura J, et al. A subgroup of spinocerebellar ataxias defective in DNA damage responses. Neuroscience 2007;145: 1418-1425.
- 52. Takashima H, Boerkoel CF, John J, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 2002;32:267-272.
- 53. El-Khamisy SF, Caldecott KW. TDP1-dependent DNA single-strand break repair and neurodegeneration. Mutagenesis 2006;21:219-224.
- 54. Gücüyener K, Özgul K, Paternotte C, et al. Autosomal recessive spastic of Charlevoix-Saguenay in two unrelated Turkish families. Neuropediatrics 2001;32:142-146.
- 55. Bouchard J-P, Richter A, Mathieu J, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Neuromuscul Disord 1998;8:474-479.
- 56. Ouyang Y, Segers K, Bouquiaux O, et al. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J Neurol Sci 20008;264:73-76.
- 57. Lagier-Tourenne C, Tranebjaerg L, Chaigne D, et al. Homozygosity mapping of Marinesco-Sjögren syndrome to 5q31. Eur J Hum Genet 2003;11:770-778.
- 58. Senderek J, Krieger M, Stendel C, et al. Mutations in SIL1 cause Marinesco-Sjögren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet 2005;37 (12):1312-1314.
- 59. Anttonen A-K, Mahjneh I, Hämäläinen RH, et al. The gene disrupted in Marinesco-Sjögren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet 2005;37:1309-1311.
Publication Dates
-
Publication in this collection
17 Dec 2009 -
Date of issue
Dec 2009
History
-
Reviewed
03 Aug 2009 -
Received
11 May 2009 -
Accepted
22 Sept 2009