
Cancer stem cells in neuroblastoma therapy resistance


Abstract
Neuroblastoma (NB) is the most common cancer of infancy and accounts for nearly one tenth of pediatric cancer deaths. This mortality rate has been attributed to the > 50% frequency of relapse despite intensive, multimodal clinical therapy in patients with progressive NB. Given the disease’s heterogeneity and developed resistance, attaining a cure after relapse of progressive NB is highly challenging. A rapid decrease in the timeline between successive recurrences is likely due to the ongoing acquisition of genetic rearrangements in undifferentiated NB-cancer stem cells (CSCs). In this review, we present the current understanding of NB-CSCs, their intrinsic role in tumorigenesis, their function in disease progression, and their influence on acquired therapy resistance and tumor evolution. In particular, this review focus on the intrinsic involvement of stem cells and signaling in the genesis of NB, the function of pre-existing CSCs in NB progression and therapy response, the formation and influence of induced CSCs (iCSCs) in drug resistance and tumor evolution, and the development of a CSC-targeted therapeutic approach.
Keywords
Introduction
Neuroblastoma (NB), an extracranial solid tumor, is the most common cancer at infancy (28%)[1,2] and accounts for 6% of all pediatric cancers[3-5]. Clinically, NB disease progression is branded with hematogenous metastasis and frequent relapses, with a rapidly decreasing survival timeline (1st relapse in 18 months, 2nd relapse in 8.7 months, and 3rd relapse in 3.8 months)[6,7]. Given the disease’s heterogeneity and developed resistance, attaining a cure after relapse of progressive NB is highly challenging[3,4,8,9]. The rapid reduction in the timeline between successive recurrences[6,7] is likely due to the ongoing acquisition of genetic rearrangements in undifferentiated cancer stem cells (CSCs)[10-14] with intensive multi-modal clinical therapy (IMCT). The IMCT for high-risk NB comprises induction phase with alternating regimens of high-dose chemotherapeutic drugs [cyclophosphamide, cisplatin or carboplatin, vincristine, doxorubicin (adriamycin), etoposide, topotecan] and load reduction surgery; consolidation phase with more intensive chemotherapy (carboplatin, etoposide, topotecan, busulfan and melphalan, thiotepa) along with radiotherapy (external beam RT, MIBG RT) and stem cell transplant [autologous bone marrow transplantation (ABMT); peripheral blood stem cell reinfusion] and maintenance phase with retinoid drug treatment (13-cis-retinoic acid, isotretinoin), immunotherapy (dinutuximab), and immune-activating cytokine (GM-CSF, IL-2) treatment (www.cancer.org/cancer/neuroblastoma/treating.html; www.childrensoncologygroup.org/index.php/in-treatment-for-neuroblastoma). Appropriately, current studies are focused on understanding the acquired resistance after IMCT, particularly in determining the ongoing acquisitions of genetic/molecular rearrangements in therapy-resistant clones, CSC clonal selection/enrichment, and epithelial-to-mesenchymal transition (EMT) and phenotypic switch endorsing de novo evolution of CSCs[15,16]. Nevertheless, studies have affirmed that the presence of NB-CSCs not only indicates the progressive state of the disease, but also dictates poor response to therapy and poor clinical outcomes[17,18]. In this review, we aim to present the current understanding of NB-CSCs in disease progression, principally in the context of orchestrated resistance to IMCT.
Relevance of stem cells in NB genesis
NB genesis, in general, is considered the accumulation of several mutations in actively dividing cells that form the nervous system during embryogenesis. The heterogeneity of NB indicates the presence of multipotent cells within the tumor, which could be the result of progenitor cell dedifferentiation under anomalous conditions or by accumulation of oncogenic mutations. Such an hypothesis took a definitive turn with the discovery of neural multipotent cells in the adult nervous system[19], recognizing that mutations accumulated in stem cells or in defined progenitors as the trigger points for tumorigenesis. Embryogenesis, in which a single cell evolves into billions, is regarded as the cancer-prone period; indeed, many neural cancers, including NB, are more frequent in infants/children than in adults[20,21]. During embryogenesis, programmed cross-talk between the ectodermal bone morphogenetic protein 4 (BMP4) and notochord noggin and chordin leads to neural tube development and neural crest cell (NCC) migration, which later forms the peripheral nervous system (discussed in detail by de Weille[22]). The downstream fate (peripheral autonomic ganglia, neurons, glia cells, Schwann cells, adrenal medulla, melanocytes, thyroid parafollicular cells, and smooth muscle cells) of NCCs is governed by the orchestrated interplay of sonic hedgehog, BMP, snail family transcriptional repressor 2 (Slug), Snail, fibroblast growth factor (FGF), and wingless-related integration site (Wnt) signaling[23].
A select subset of NCCs in the trunk region shows a sympathoadrenal lineage that contributes to the formation of sympathetic ganglia and medullary region of the adrenal gland. These committed NCCs are designated as sympathoadrenal progenitors (SAPs) and are believed to be the origin of NB [Figure 1]. SAPs constantly undergo a Snail/Slug-dependent EMT augmenting NCCs’ migratory abilities, allowing them to migrate out of the neural tube. The prompted migration is accompanied by DNA repair gene regulation in SAPs, making them vulnerable to genomic alterations[24]. SAPs lose part of their multipotency, and more are designated for neural or melanocyte lineage[25]. Bmp/Notch signals induce differentiation of these cells to the sympathetic ganglion chain. The SAPs express Phox2a/b, which is required for the production of enzymes in catecholamine biosynthesis. Downstream differentiation of SAPs is effected by a complex interplay of FGF, Notch, Wnt, achaete-scute BHLH-transcription factor (ASCL1), paired-like homeobox 2a (Phox2a), and Phox2b[25]. Phox2b mutations cause congenital central hypoventilation syndrome[26], which serves as the predisposition for ganglioneuroma (GN), ganglioneuroblastoma (GNB), and NB. Since the morphologically more differentiated GN and GNB are lower grade with favorable outcomes, the genesis of aggressive NB depends on the stage of the sympathoadrenal progenitor (SAP), with un-/poorly-differentiated SAPs causing more lethal tumors. Accordingly, neuroblastic tumors could be Schwannian stroma-poor (undifferentiated, poorly differentiated, and differentiating), GNB intermixed Schwannian stroma-rich, or GN. Although the presence of catecholamines in an infant’s urine serves as the diagnostic marker for NB, its presence in high levels with much higher frequency than that of NB diagnosis (Dx)[27,28] indicates a considerable spontaneous regression. In this regard, a mass screening study by Sawada assessing the levels of catecholamine metabolites, vanillylmandelic acid (VMA) and homovanillic acid (HVA), the sensitive diagnostic markers for NB in urine indicated that during the development of sympathetic neurons the incidence of in situ NB is higher than the incidence of sporadic cases[28]. Most of these in situ NBs spontaneously regress as the child ages. This is attributed to the VMA-to-HVA ratio where < 1 corresponds to a more aggressive, biologically primitive type of NBL associated with a shorter survival[28]. At the molecular level, NCCs that undergo EMT are characterized by the loss of epithelial morphology, regulation of junctional (E-cadherin, cytokeratin, occludins, claudins) complexes, and induction of migratory ras homolog family member B and matrix [collagenase, matrilysin, urokinase, heparanase, matrix metalloproteinases (MMPs), N-Cadherin] proteins. Consistently, aggressive cells from progressive NB exhibit similar physiognomies[29]. NB of progressive stages have been shown to generate self-renewing and multipotent CSCs that develop into neurons, Schwann-like cells, and melanocytes [Figure 1][17,30].

Figure 1. Schematic representation of cellular and molecular events in embryogenesis and diverted signaling events leading to NB genesis. SAPs undergo a Snail/Slug-dependent EMT that augments NCCs’ migratory abilities, allowing them to migrate out of the neural tube. The prompted migration, accompanied by regulation of DNA repair genes in SAPs, makes them vulnerable to many genomic alterations leading to the genesis of NB. While embryogenesis is a sequential step-down process from pluripotency to differentiation, advancing disease stages of NB progress successively from differentiated to undifferentiated self-renewing multipotent CSCs. NB: neuroblastoma; SAPs: sympathoadrenal progenitors; EMT: epithelial-mesenchymal transition; CSCs: cancer stem cells
The magnificent biogenetic heterogeneity of NB indicates an interplay of more than one gene in tumor initiation and evolution. Researchers have identified that NB could be transmitted as recessive trait at low penetrance (e.g., locus 2p)[31]. Further studies validated the 2p23-24 locus with 104 genes, including v-myc avian myelocytomatosis viral oncogene NB derived homolog (MYCN)[32]. MYCN, the player in neurogenesis that is critical for expansion of progenitor cells, is overexpressed in NB[33]. Transgenic models of animal studies affirmed that overexpression of MYCN causes NB in their progeny[29]. In parallel, researchers also defined the association of the anaplastic lymphoma receptor tyrosine kinase (ALK) gene with the NB predisposition. Thus far, several germline and somatic ALK mutations have been identified (more frequent, R1275, F1174, F1245) in NB. These mutations promote receptor autophosphorylation in the kinase domain, leading to the activation of the ALK pathway. Likewise, genome wide association studies identified the association of BRCA1 associated RING domain 1 β (BARD1β) with high-risk NB[34]. Other studies indicated that polymorphisms at locus of LIM domain only 1 (LMO1) [single nucleotide polymorphism (SNP) rs 2168101][35] and Lin-28 homolog B (LIN28B) are significantly involved in the susceptibility to NB[36]. Loss of chromosome 1p36 region, 3p, 4p, 9p, 11q, and 14q together with gain of 1q, 2p24, 12p, 17q, and mutations of ALK and AXL receptor tyrosine kinase (AXL) and telomerase reverse transcriptase (TERT) re-arrangements may orchestrate the transformation of normal NCCs to NB cells. Recent genome profiling studies in relapsed NB indicated new and acquired recurrent mutations in cadherin 5 (CDH5), dedicator of cytokinesis 8 (DOCK8), protein tyrosine phosphatase non-receptor 14 (PTPN14), HRas proto-oncogene (HRAS), and KRAS proto-oncogene (KRAS), signifying that these acquired mutations contribute to drug resistance and disease evolution[37]. However, it is not clear how and when the mutations occur, or whether these mutations occur in specific clones (e.g., CSCs) or enhance clonal selection and enrichment. In addition to the genetic causes, other factors, including manifestation of other neurocristopathies such as Hirshsprung’s disease[38,39], Klippel-Feil syndrome, Waardenburg’s syndrome[40], Ondine’s curse[39], Beckwith-Weidemann syndrome[41], Cushing’s syndrome[42,43], fetal alcohol syndrome[44-46], fetal hydantoin syndrome caused due to intake of anti-seizure drug phenylhydantoin[47,48], and exposure to products causing high blood pressure, sweating, headache, and abnormal heartbeats in mothers, have been associated with increased susceptibility to NB.
NB-CSCs in therapy resistance and disease evolution
Tumor heterogeneity, in general, has been explained by two models: the stochastic model, in which each cancer cell has the ability to contribute to the tumor evolution, and a more accepted CSC model, in which a small subset of cells within the tumor has high tumorigenic potential and can give rise to all other cells of the tumor[49]. Over the last two decades, the functional significance of CSCs in tumor initiation, progression, response to therapy, and poor clinical outcomes has been recognized in different types of tumors, including pancreatic cancer[50], liver cancer[51,52], lung cancer[53], breast cancer[54], head and neck squamous cell carcinoma[55], colon cancer[56,57], brain tumors[58], leukemia[59], and NB[18,60,61]. By definition, CSCs are the indefinitely proliferating subpopulation within the tumor, exhibiting stem-like properties that include self-renewal, multipotency and/or pluripotency maintenance, tumorigenic potential, an unparalleled metastatic state, and anti-apoptotic ability. CSCs are known to play roles in induced resistance to chemotherapy and radiation, resulting in an increased risk of tumor progression, relapse, and recurrence[62-64]. This function is attributed to the possession of multiple mechanisms, including repression of apoptosis, increased DNA damage repair, conserved dormancy, and altered drug response[65]. Traditional treatment modalities, i.e., conventional chemotherapy and radiotherapy, are adopted based on the stochastic model, with a focus on killing all cancer cells[49]. However, later studies indicated that the limitations of such a strategy are mainly attributed to the intrinsic resistance of the CSCs within the tumor[16,66,67]. The current understanding of the role of CSCs in therapy response, tumor evolution, and the development of CSC-targeted therapeutic strategies for various tumors was extensively reviewed [Table 1] and provided a strong basis for similar biology in NB.
Documented reviews on the role of CSCs in therapy response, tumor evolution, and the development of CSC-targeted therapeutic strategies for diverse human tumors, including NB. These compilations clearly define the identification of CSC surface markers, orchestrated signaling events, influence of ECM and TME; portray the functions of CSCs in induced/acquired therapy resistance; and recognize the possibility and benefit of CSC-targeted therapies in cancer treatment
| Title | Ref. |
|---|---|
| Chemoresistance, cancer stem cells, and miRNA influences: The case for neuroblastoma | Buhagiar and Ayers[72] |
| p53, stem cell biology and childhood blastomas | Oh et al.[73] |
| Cancer stem cells and pediatric solid tumors | Friedman and Gillespie[74] |
| Cancer stem cells and their interaction with the tumor microenvironment in neuroblastoma | Garner and Beierle[75] |
| Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma | Alisi et al.[16] |
| CD133: A stem cell biomarker and beyond | Li[76] |
| Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment | Phi et al.[77] |
| Cancer stem cells revisited | Batlle and Clevers[78] |
| Mechanisms of chemoresistance in cancer stem cells | Abdullah and Chow[15] |
| Cancer stem cell metabolism | Pages et al.[62] |
| Cancer stem cells: Implications for cancer therapy | Dawood et al.[79] |
| Cancer stem cell surface markers on normal stem cells | Kim and Ryu[80] |
| Cancer stem cell signaling pathways | Matsui[81] |
| Cancer stem cells: Basic concepts and therapeutic implications | Nassar and Blanpain[82] |
| Cancer stem cells and tumorigenesis | Zhu and Fan[64] |
| The therapeutic promise of the cancer stem cell concept | Frank et al.[83] |
| Drug resistance driven by cancer stem cells and their niche | Vila et al.[63] |
| How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive | Poggi et al.[84] |
| Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications | Ishiguro et al.[85] |
| Therapeutic strategies targeting cancer stem cells | Yoshida and Saya[86] |
| Therapies targeting cancer stem cells: Current trends and future challenges | Dragu et al.[49] |
| Stem cell theory of carcinogenesis | Trosko and Chang[87] |
In the context of progressive NB, several studies have identified that CSCs play a crucial and definite role in disease progression, relapse, and poor prognosis[61]. High-risk NBs consist of small populations of cells with preserved stemness characteristics. These clones exhibit the ability to form highly resistant tumorospheres and have high metastatic potential[60]. NB-CSCs are identified based on morphology, cell efflux property, cell surface markers, and tumorosphere formation in in vitro conditions. On a morphological basis, NB cells can be classified as neuroblastic [neuroblastic type cells (N-type), immature, tumorigenic], Schwann-like (S-type, non-tumorigenic with limited lifespan), and intermediate (I) type cells[30,68]. The malignant NCC-like intermediate type NB cells (I-type) cells are multipotent and can self-renew, as well as differentiate into N and S type morphologies[69]. Beyond the self-renewal and differentiation capabilities, I-type cells are associated with tumor relapse/metastasis[69], possess high tumor-forming capacity in vivo[70], and are characterized by overexpression of stemness-related molecules, including cluster of differentiation 133 (CD133), cKIT, NOTCH 1, GPCR class C group 5 member C (GPRC5C), and tropomyosin receptor kinase B (TRKB)[71]. In addition, the role of NOTCH 1 in maintaining the stem cell phenotype has been recognized. However, I-type cells are only regarded as aggressive NB cells with relatively high plasticity, and hence do not fully meet the criteria for categorization as CSCs.
Drug efflux pumps and chemoresistance
Identification and characterization of NB-CSCs that could be useful for precisely assessing NB Dx/prognosis, therapy response, and clinical outcomes have been continuously evolving over the last two decades. Nestin and ABC subfamily G member 2 (ABCG2) are 2 markers that were regarded as NB-CSC markers very early on. The NB cells presented with drug efflux pumps (that exclude Hoescht dye) that confer multi-drug resistance (MDR) have been consistently identified as a “side-population” (SP). These cells were found to have high expression of ABCG2 and ABC subfamily A member 3 (ABCA3), and are highly [Table 1]. Documented reviews on the role of CSCs in therapy response, tumor evolution, and the development of CSC-targeted therapeutic strategies for diverse human tumors, including NB. These compilations clearly define the identification of CSC surface markers, orchestrated signaling events, influence of extracellular matrix (ECM) and tumor microenvironment (TME); portray the functions of CSCs in induced/acquired therapy resistance; and recognize the possibility and benefit of CSC-targeted therapies in cancer treatment [Table 1].
Concentrated in post-IMCT relapsed tumors (vs. pretreatment controls)[88]. ATP binding cassette (ABC) transporters are the transmembrane proteins that drive efflux of many chemotherapeutic drugs and hence dictate chemoresistance[89,90]. Newton et al.[88] compared the Side populations derived from the same patients at diagnosis and at relapse after IMCT. Their findings showed significantly increased Side populations in post-IMCT relapsed NB (vs. at diagnosis), heightened proliferation and clonal expansion of drug-resistant Side populations, and acquired gain of pluripotency maintaining factors [nanog homeobox (Nanog), POU class 5 homeobox 1 (Oct3/4)] in drug-resistant Side populations. Such critical information clearly indicated the acquired stemness-related genetic alterations in Side populations, their selection and clonal expansion, that subsequently played a crucial role in chemoresistance and tumor relapse[88]. ABCG2, an ABC protein associated with neural stem and progenitor cells, has been shown to influence stemness maintenance. Nestin, on the other hand, is a neuronal stem cell protein that also serves as a putative marker for CSC[91]. In neural stem/progenitor cells, ABCG2 and Nestin exhibits heightened colocalization and in particular the ABCG2+ and Nestin+ cells mimic each other in their ability to form tumorospheres[90]. Consistently, number of studies designated them as putative CSC markers for various tumors including NB[61,91,92]. Similarly, the NB Side population cells expressing high levels of ABCG2 and ABCA3 transporter genes have been shown to possess high capabilities to expel chemotherapy drugs (e.g., mitoxantrone) and promote proliferation[16]. ABCG2 have also been shown to transport a number of common chemotherapy drugs, including anthracyclins, imatinib, and Topoisomerase I and II inhibitors[16].
Cell surface CSC markers and therapy resistance
Identifying the surface expression of select CSC markers clearly provides the basis of the CSC composition in NB as such and for drug response. Recognizing specific surface markers is useful for characterization of CSCs, examining NB biology/evolution, and therapeutic targeting. To date, a number of markers, including CD133, frizzled class receptor 6 (FZD6), leucine rich repeat containing GPCR 5 (LGR5), aldehyde dehydrogenase (ALDH), ALDH1A2, ALDH1A3, cluster of differentiation 114 (CD114), and cluster of differentiation 117 (C-kit), have been used in NB[93].
CD133
NB-CSCs have been shown to pose a genetic profile different from that of non-stem tumor cells, including gains of 16p13.3, 19p13.3, and 19q13.33. Interestingly, the gain in 16p13 is significantly associated with the expression of CD133 in NB[94]. CD133 (Prominin-1), a pentaspan transmembrane protein expressed in neural stem cells, has been indicated as a marker for tumor-initiating cells[76]. CD133 levels were inversely correlated with overall survival (OS) of NB patients[95]. It has been shown that the poor association occurs through Serine/Threonine kinase (AKT) pathway-mediated chemoresistance[96]. Muting CD133 decreases NB cell colony formation and proliferation, increases differentiation in vitro, and decreases tumor burden in vivo[97]. On a therapy response note, studies have shown that CD133+ NB cells efficiently develop tumorospheres, which exhibited high resistance to doxorubicin (DOX) treatment with upmodulation of ABCG2[61]. Conversely, DOX treatment resulted in increased CD133- and ABCG2-expressing Side populations and the ability of these Side populations to generate non-Side population cells[61] exhibiting: (1) drug resistance in CD133-expressing Side populations; (2) clonal selection and enrichment of drug-resistant CSCs; and (3) drug resistance and disease evolution mediated by CSCs. Consistently, studies have indicated the criticality of CD133-expressing Side populations in NB progression and therapy response. For instance, studies demonstrated that (1) non-adherent clumps of tumor cells that express CD133, OCT4, pAKT could grow in a serum-free medium with higher colony and neurosphere formation[98]; (2) CD133 levels were directly associated with NB advanced disease stages and inversely correlated with postoperative survival time[95]; and (3) CD133+ NB cells were more resistant to cisplatin, carboplatin, DOX, and etoposide (vs. CD133- cells) and presented with increased phosphorylation of extracellular signal-regulated kinases (ERK) and P38 mitogen-activated protein kinases (P38)[99]. Consistently, an independent study showed that targeted inhibition of CD133 in NB cells produced increased RET expression and NB differentiation and, this response is mediated through the regulation of the p38 MAPK and phosphoinositide-3-kinase (PI3K)/AKT pathways[97].
CD133+ NB-CSCs exhibit inhibited levels of mitogen-activated protein kinase phosphatase 1 (MKP1) and increased phosphorylation of ERK and P38[99]. The influence of these MAPKs in cell cycle progression, differentiation, and cell death has been extensively documented. It has been realized that chemotherapy drugs such as cisplatin activate JNK, ERK, and other MAPKs through multitude mechanisms that dictate the development of drug resistance[100]. Since MKP1 regulation leads to the phosphorylation of these kinases, the low levels of MKP1 in CD133+ cells directly relate to the induced drug resistance in NB[99]. However, CD133-targeted therapeutic measures for NB warrant in-depth investigation. For instance, although valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, has been shown to induce cell death in NB cells and overcome hypoxia-induced resistance to cisplatin[101], it has also been reported that VPA treatment increases the percentage of CD133+ NB-CSCs and does not induce apoptosis in these cells. Similarly, decitabine, a hypomethylating drug, and trichostatin A, a HDAC inhibitor, have been shown to induce surface expression of CD133 in CD133- cancer cells[66]. Detailed insights on the regulation of CD133 expression and its role in self renewal, tumorigenesis, metastasis, chemo/radio-resistance, metabolism, dedifferentiation, and autophagy have been well documented[76]. Although CD133 is highly regarded as a CSC marker in NB settings, it is pertinent to note that CD133 is only expressed in ~40% of primary tumors, which precludes its use as a stand-alone NB-CSC marker.
FZD6, LGR5 and ALDH
Surface marker FZD6, a WNT receptor, is inversely associated with OS in patients with NB[102]. It has been shown that FZD6+ Side populations were selectively enriched in hypoxic regions, and such Side populations can readily form tumorospheres and develop more aggressive tumors. Likewise, LGR5, a WNT-responsive G-protein-coupled receptors (GPCR) protein, significantly correlates with poor event-free survival (EFS) in high-risk NB subsets[103]. LGR5 is specifically expressed in CSCs, is known to brace WNT/β-catenin signaling as an R-spondins receptor, and drives oncogenesis[104]. Elevated levels of LGR5 in IMCT-resistant cells were associated with aggressive phenotype, and cells presented with high LGR5 levels were highly chemoresistant[104]. Activation of LGR5 with WNT3a ligands promotes WNT-pathway-dependent proliferation. However, muting LRG5 has been shown to mediate apoptosis through mitogen-activated protein kinase (MEK)/ERK signaling, independent of the WNT pathway. Moreover, upmodulation of ALDHs was associated with retinoic acid (RA) tolerance[105]. Consistently, ALDH1A2 and ALDH1A3 showed an inverse relationship with OS in high-risk NB patients. Independent studies have demonstrated that silencing ALDH1A2 or ALDH1A3 reduced clonal expansion of tumor-initiating cells and tumorosphere formation[106,107]. Hartomo et al.[106] assessed the activity and expression of 19 isoforms of ALDH, and identified that ALDH1A2, ALDH1L1, and ALDH3B2 expression consistently induced tumorosphere and colony formation. However, they also recognized that ALDH1A2 is the only candidate to show a significant association with poor prognosis of patients with NB. More importantly, high levels of ALDH1A2 expression correlated with the growth and dedifferentiation of NB xenografts, as well as RA treatment resistance, in NB cells[106]. It is critical to note that there is thus far no definitive in vivo evidence documented for FZD6-, LGR5-, or ALDH1A3-positive Side populations in terms of tumor evolution or drug-response in NB. However, the role of these candidates as drivers, players, or enhancers in acquired disease resistance and disease progression is possible and warrants further investigation. A study by Kuo et al.[108] identified the role of jumonji, AT-rich interactive domain 1 (JARID1B) (KDM5B) a histone lysine demethylase, in modulating stemness-related signaling. High levels of JARID1B were associated with high levels of ALDH activity, and this upmodulation was associated with enriched NB tumorospheres. JARID1B-enriched SP showed the strongest sphere-forming capacity and also presented with high levels of Nestin colocalization. While JARID1B-expressing NB-CSCs have been shown to be less responsive to DOX, etoposide, and cisplatin, silencing JARID1B resulted in decreased invasiveness, CSC phenotype, EMT process, and compromised NOTCH signaling[108].
CD114
Recent studies, identified the surface expression of CD114, a G-CSF receptor, as a putative marker for NB-CSCs. Unlike other surface markers, the CD114-expressing SP is < 1% and exhibit several CSC characteristics with 10 times more tumorigenic capacity[109]. More importantly, incidence of CD114+ Side populations has been reported in all NB cell lines, patient samples, and PDXs tested to date, with a frequency ranging from 0.01%-3%. A drift toward clonal selection and enrichment of CD114+ SP after IMCT in relapsed tumor and in metastases demonstrates the intrinsic chemoresistant capabilities of this SP[109,110]. Growing evidence has documented the characteristics of the CD114+ SP, including the near-identical gene profile [e.g., Sox10, twist family BHLH transcription (TWIST), vimentin (VIM), MMPs] to that of pre-/early migratory NCCs that maintain multipotent capacity[111]. Furthermore, studies have shown that NB pathogenesis and dissemination are dependent on CD114+ SP through downstream activation of signal transducer and activator of transcription 3 (STAT3) target genes[110]. In normal neural tissue, CD114 (G-CSFR) promotes neurogenesis and survival and expansion of the neural stem cells[112,113]. G-CSF activates STAT3 in these receptor-positive CSCs; this signaling promotes stemness maintenance, clonal expansion, tumor formation and dissemination, and chemoresistance[109]. The existence of a granulocyte colony-stimulating factor (GCSF)→STAT3→GCSF positive feedback cycle has also been recognized. Targeted deregulation of this feedback loop with STAT3 inhibitors not only depletes the CSC-SP within tumors, but also prompts tumor regression and profound chemosensitization[110].
Other CSC markers involved in resistance
CFC1, a member of the epidermal growth factor-Cripto/FRL-1/Cryptic (EGF-CFC) family, with designated functions in embryonic development, has been recently recognized as a CSC marker for NB[114]. CFC1, is strongly expressed in sphere-forming CSCs and is associated with unfavorable prognosis in NB patients. In addition, the functional role of CFC1 in NB tumorosphere formation has been realized and further identified that CFC1 directly targets activin-A induced cell differentiation and Smad phosphorylation, resulting in tumor progression. Analysis of activin-A signaling further identified the inhibition of differentiation-inducing [BMP-4, transforming growth factor beta (TGFβ)-1, and TGFβ-3] and tumor suppressor [cyclin dependent kinase 4 (CDK4), cyclin dependent kinase inhibitor 2A (CDKN2A), p14-ARF, and p16-INK4A] molecules in CFC1-overexpressing cells[114]. Naiditch et al.[67] assessed the differential regulation of genes in DOX-resistant NB cells compared with their wild-type counterparts. The high-throughput whole genome approach identified chemotherapy resistance-associated differential regulation of > 1500 candidates. With the network analysis, this study clearly identified the deregulation of EMT pathway, stemness maintenance, and tumor progression signaling. The regulation/deregulation of neuronal, epithelial, mesenchymal, and pluripotency maintenance markers in these DOX-resistant, highly invasive cells illustrates the role of acquired mesenchymal change in induced drug resistance.
B cell-specific Moloney murine leukemia virus integration site 1 (BMI1), a polycomb protein has been shown to have critical roles in stem cell self-renewal, initiation of cancer and chemoresistance in many human malignancies including NB[115-117]. BMI1 has been shown to be highly expressed in NB[117] and is essential for the pathogenesis of the disease[115]. The binding of E2F-1 and MYCN[118] to BMI1 promoter and its activation were documented[117]. It has been shown that BMI1 inflicts NB progression and contributes to therapy resistance/disease evolution through modulating key players in tumorigenesis including Cyclin E1, KIF1Bβ, TSLC1 and others[119,120]. More importantly, the role of BMI1 in the regulation self-renewal capacity and differentiation of I-type NB CSCs has been realized[116]. Consistently, Melone et al.[121] indicated the clinical relevance of BMI1 in NB and defined its association to advanced disease stages. Though the direct role of BMI1+ NB-CSCs in orchestrating therapy resistance is currently unknown, targeting BMI1+ CSCs has been shown to overcome chemoresistance[122-125] and radio-resistance[126-128] in many human cancers.
Induced CSCs and therapy resistance
As discussed above, drug resistance and cancer recurrence are majorly affected by the preexisting CSCs (tumor-initiating cells) that are derived from normal stem cells under certain environments. The overall hypothesis is that these preexisting CSCs cause therapy resistance and/or disease relapse with their unique abilities of clonal selection, self-renewal, clonal expansion, stemness maintenance, and plasticity. In parallel, recent findings indicated that CSCs can be formed from non-stem cancer cells exposed to radiation or chemotherapeutic drugs, creating an SP of induced-CSCs (iCSCs)[129]. The transformation of non-stem cancer cells into iCSCs involves reprogramming factors [OCT4, SOX2, myelocytomatosis viral oncogene homolog (C-MYC), kruppel like factor 4 (KLF4)][130-133] depended dedifferentiation. Such transformation always displays significant CSC properties, sphere formation, drug resistance, and tumorigenicity[134,135]. In addition to radiation and chemotherapy drugs, many other driving factors, including temperature, external cytokines/transformation factors, inhibitor of DNA binding 4 (ID4), and interleukins 6 (IL6) have been causally linked to dedifferentiation of non-stem cancer cells[136-138]. In NB, epigenetic modifiers have been shown to endorse iCSCs formation and maintenance[139]. With regard to the role of CSCs in induced drug resistance, studies across human tumors, including NB, widely agreed that CSCs are inherently resistant to radiotherapy and/or chemotherapy[140-143]. However, the understanding of the acquired genetic alterations in non-stem cancer cells and their transformation into iCSCs after radiochemotherapy is recently on an upsurge (reviewed in detail elsewhere[144]). ICSCs formation was documented with common chemotherapy drugs [fluorouracil (5-FU), DOX] and with various qualities of radiation. Assessment of the mechanism(s) involved in the formation of iCSCs revealed that a complex interplay of multiple signaling pathways, small non-coding RNAs (microRNAs), and the appropriate TME facilitates dedifferentiation. Interestingly, both the transformation of normal stem cells in to CSCs and dedifferentiation of non-stem cancer cells into iCSCs employ near-identical pathways (e.g., NFkB, Notch, Wnt, Hedgehog). During the formation of iCSCs from non-stem cancer cells exposed to radiotherapy, these signaling events have been shown to coincide with other complementary pathways, including pluripotency maintenance reprogramming (SOX-2, OCT4, NANOG) and plasticity, and to configure iCSCs.
Expressional deregulation of many other proteins are also causally linked to the dedifferentiation of non-stem cancer cells and formation of iCSCs[118,145-147]. For instance, Lamin A/C, the type V intermediate filaments of nuclear lamina, is often reduced or absent in proliferative cells of various tumors[148,149]. Lack of lamin A/C predisposes cells towards an immature phenotype and influences the presence of tumor-initiating cells in NB[150]. Selective silencing of lamin A/C triggers the formation of NB-iCSCs with self-renewing ability. Loss of lamin A/C is also MYCN expression-dependent[150]. SOX2, a member of the SRY-related high mobility group box, is a transcription factor that is mostly expressed during embryonic development. It has been shown to affect cell fate and differentiation[151], self-renewal, and proliferation[152-156]. In NB, SOX2 levels were heightened and its expression correlated with advanced disease stage[157]. Studies with SOX2 stably over-expressed NB cells not only recognized amplified tumorigenicity, but also showed clonal selection and expansion of stem-like cells (with loss of N and S type). Selective silencing of SOX2 in highly malignant I-type CSCs greatly reduced its tumorigenicity and enriched N and S type cells[158]. Targeting SOX2 resulted in cell-cycle arrest at G0/G1 phase and hence drove decreased cell proliferation. Similarly, tailless-like receptors (TLX), which is a nuclear receptor and a transcription factor, plays a critical role in self-renewing, undifferentiated, and proliferative states of neural stem cells[159]. During NB genesis, PHOX2B dictates dedifferentiation of SAPs by upmodulating T cell leukemia homeobox 3 (TLX3) and neurotrophin receptor P75 (P75). TLX has been causally linked to NB dedifferentiation, resulting in tumorigenesis by activating MMP2 and MMP9. Consistently, TLX has been identified to be overexpressed in NB, involved in progression of tumorigenesis and correlated with shorter survival rates[160].
Role of TME in influencing CSC status and therapy resistance
The TME also serves as a critical regulator of stem cell differentiation, dedifferentiation of non-stem cancer cells, and tumorigenesis[161,162]. The TME contributes hypoxia, inflammation, acidic stress (pH), and remodeling of ECM, coordinates CSC self-renewal, and inhibits differentiation. For instance, NB exposure to hypoxia led to the upmodulation of NCC markers (C-Kit, Notch1) and hypoxia-induced response genes [hypoxia inducible factor (HIF)-1α, HIF-2α][163,164]. Similarly, studies have showed a greater concentration of tumor associated macrophages (TAMs) in metastatic NB than in locoregional disease[165]. The role and functions of these microenvironmental factors are reviewed in detail elsewhere[75]. With regard to their function in orchestrated drug resistance and NB evolution, mesenchymal stromal cell-derived cancer-associated fibroblasts (CAF) contributed significantly to clonal expansion and chemoresistance. Once induced, CAFs maintain their activated state; increased CAFs were linked with induced microvascular proliferation and Schwannian stroma-poor histology. This study showed that the pro-tumorigenic activity of mesenchymal stem cells (MSC)-CAF occurs through the co-activation of the janus kinase 2 (JAK2)/STAT3 and MEK/ERK1/2 pathways. Further, treatment with Ruxolitinib (JAK2/STAT3 inhibitor) or Trametinib (MEK/ERK1/2 inhibitor) significantly enhanced the response of NB tumors to etoposide[166]. The ECM small leucine-rich proteoglycans (SLRPs), including decorin (DCN) and lumican (LUM), exhibited acquired upmodulation during NB-CSC enrichment[146]. Further, these small leucine rich proteoglycans (SLRP)-positive NB-CSCs were highly resistant to temozolomide (TMZ), demonstrating that (1) CSCs promote huge quantities of DCN and LUM; and (2) increased SLRPs promote acquired TMZ resistance, cellular heterogeneity, and a quiescent phenotype. The outcomes of this study clearly identified the pivotal role of SLRPs in drug resistance, the cell plasticity of NB-CSCs that dictates cell survival, and ECM/ TME modulation potential[146].
CSC-targeted therapy
Considering the significance of targeting CSCs or the formation of iCSCs in countering acquired therapy resistance and in the treatment of high-risk aggressive NB [Figure 2], recent studies are appropriately focused on developing improved therapeutic strategies. These proposed strategies include targeting specific surface markers, modulation of signaling pathways, adjustment of the microenvironment signals, inhibition of drug-efflux pumps, manipulation of miRNA expression, and induction of CSCs apoptosis and differentiation. Immunotherapy is often used in combination with chemotherapy and radiotherapy. It involves the use of antibodies that target specific cancer stem cell markers[49]. However, the limitations must be considered when selecting the appropriate target surface markers, as most of the documented markers are (1) not ubiquitously expressed in all (100%) NB-CSCs; and (2) many CSC markers are also expressed in normal stem cells[60,74,167]. Researchers have focused in identifying and characterizing agents [Rapamycin, dequalinium analogue, C-14 linker (DECA-14)] that could selectively target CSCs while sparing normal stem cells. Differentiating agents, such as RA alone or in combination with proteasome inhibitor (MG132), have been shown to decrease stem cell markers (Nestin, Sox2, Oct3/4) and suppress spheroid formation[168,169]. Recently, Bahmad et al.[170] showed that targeting AKT/MTOR signaling in NB-CSCs could be beneficial. They reported that triciribine and rapamycin, which inhibit at 2 different points of the AKT/MTOR pathway, decrease cell survival and tumorosphere formation. Further, virotherapy can be an effective approach to kill CSCs. A study by Mahller et al.[61] showed that engineered oncolytic virus Nestin-targeted oHSV significantly killed DOX-resistant CD133+ NB-SP. Interestingly in this study authors demonstrated the existence of CD34 and CD133 double positive cells within all patient derived cell-lines. The tumorospheres derived from these cells exhibited CD133 and ABCG2 enrichment, multi-lineage potential and are resistant to Doxorubicin. In an attempt to kill both the differentiated and tumor initiating NB cells, this study demonstrated that a nestin targeted engineered clinically safe oncolytic virus (rQNestin34.5) promoted a profound cell death of both bulk and tumor initiating tumorsphere-derived NB cells. More importantly, this study showed new virus production within in tumorsphere, and prompted a significant delay in tumor formation in vivo[61]. Induced telomerase activity (TA) also directly relates to clinical management of NB. Interestingly, studies found that the TA is selectively confined to the CSC-SP (CD15+) and is not detectable in most of the non-stem cancer cells and normal tissue stem cells[171]. Such a finding designates the TA as highly specific, and it could serve as a suitable candidate for anti-CSC therapy. It has also been shown that telomerase inhibition with Imetelstat will result in CSC exhaustion through its ability to irreversibly alter self-renewal capacity and cell growth[171].
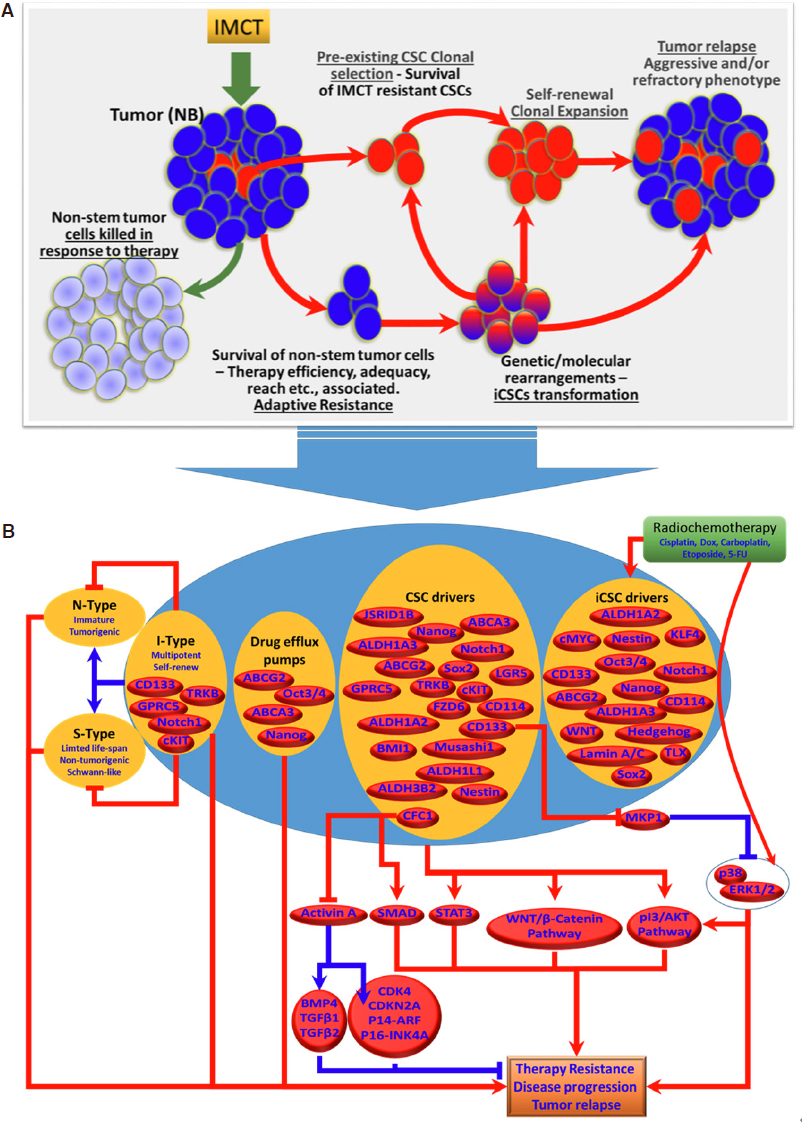
Figure 2. A: Cartoon showing the models of NB-CSCs-associated tumor resistance and tumor relapse. Pre-existing NB-CSCs survive IMCT and undergo self-renewal, clonal expansion and development of non-stem tumor cells, resulting in tumor relapse. In parallel, the non-stem tumor cells that survive IMCT under unique circumstances undergo extensive genetic and molecular rearrangements that lead to their transformation into induced CSCs (iCSCs). Generation of iCSCs with unique stem-cell characteristics of self-renewal and expansion results in tumor maintenance and relapse. B: Schematic representation of the molecular characteristics of NB-CSCs (pre-existing NB-CSCs and induced iCSCs) and their signaling flow-through that dictates therapy resistance and NB disease evolution. IMCT: intensive multi-modal clinical therapy; NB: neuroblastoma; CSCs: cancer stem cells; iCSCs: induced cancer stem cells; N-Type: neuroblastic type cells; I-Type: intermediate type neuroblastoma cells; S-Type: Schwann-type cells
The FDA approved drug vorinostat (histone deacetylase inhibitor) increases chemosensitization, inhibits NB-CSCs’ tumorosphere formation capacity, reduced tumor cell invasion and deplete SP[172]. Further in doxorubicin-resistant NB-CSCs, vorinostat treatment resulted in the regulation of stemness maintenance and drug-resistance candidates including ABCB1, ABCC4, LMO2, SOX2, ERCC5, S100A10, IGFBP3, TCF3, and VIM[172]. Like-wise, the inhibition of L1-Cam (CD171), the driver for tumorigenic invasion and motility[173] in CD133+ cells showed reduced cell survival, tumorosphere formation, self-renewal capacity, increased apoptosis and, suppressed tumor growth in vivo[174]. It was indicated that the effect is through Olig2 regulation and parallel activation of the tumor suppressor, p21 (WAF1/CIP1). In NB, studies have documented that CE7 epitope of L1-CAM is an useful tool in CAR-T targeted therapy[175,176]. The safety of targeting the CE7 epitope CD171 with CE7-CAR T cells has also been realized and, more importantly the potential to generate bioactive CAR-T cells from patients with recurrent/refractory disease after current IMCT[176]. Small molecule kinase inhibitor screening by Grinshtein et al.[177] identified Polo-like kinase 1 (PLK-1) as the potential target for NB tumor initiating cells. To that end, a study by Pajtler et al.[178] demonstrated that imidazotriazine (GSK461364), a competitive inhibitor for ATP binding to PLK1 which is in clinical development promoted cell death, inhibited clonal expansion and exerted anti-tumoral activity in vivo.
Along the line of targeting select CSC markers for NB cure, number of potential drug candidates are in the developmental stages and/or undergoing functional (molecular) characterization. For instance, Naveen et al.[179] recognized the potential of Berberine, a plant alkaloid in EMT reversal, inhibiting cancer stemness, and prompting the neuronal differentiation. In NB, Berberine exerts programmed (DAXX, p53, etc., dependent) cell death and inhibits tumor cell growth[180-182]. Berberine has been shown to revert EMT by inhibiting vimentin and fibronectin (mesenchymal markers) and restore E-cadherin, laminin and Smad[179]. Further this study identified the Berberine associated EMT reversal is through the downregulation of PI3/Akt and Ras-Raf-ERK signalling and subsequent upregulation of p38-MAPK. More importantly Berberine promotes neuronal differentiation as evidenced with upmodulation of MAP2, β-III tubulin and NCAM. In another study, Tian et al.[183] recognized the clonal enrichment of NB-CSCs (CD133+) with chemodrug (etoposide) identified the potential of XAV939 a small molecule tankyrase (TNKS) inhibitor in alleviating the stemness physiognomy and migration of NB-CSCs. Cyclopamine (11-deoxojervine), a steroidal alkaloid (teratogen) derived from corn lily exerted a dramatic decrease in the CD133 and CD15 double positive populations that preserve more stemness characteristics[184]. Further, it has been shown that cyclopamine targets autocrine activation of hedgehog signaling and hence affect survival, clonal expansion and tumorosphere formation[184]. Like-wise, CD114+ NB-CSCs that exhibit high tumorigenicity, self-renewal capacity, and evasive phenotype has always remained as an attractive therapeutic candidate for stem-cell targeted therapy. In this regard, it has been shown that CD114 anti-body therapy and/or targeting its downstream driver STAT3 depletes CSC subpopulation within NB and corresponded with reduced metastatic state, increased chemosensitization and decreased tumor growth[110]. Targeting STAT3 has been considered for NB cure by many investigators and numerous promising candidates were investigated. For instance, a study from Goel and Aggarwal[185] indicated that curcumin, a natural STAT3 inhibitor from turmeric promoted chemosensitization in multifarious tumors including NB. In addition, Cucurbitacin I, a triterpenoid that acts as a potent inhibitor of the STAT3/JAK has been shown to exert anti-tumor effect in NB[186]. Similarly, Honokiol, a biphenolic compound derived from the magnolia bark has been shown to target STAT3 signaling pathway and inhibit NB growth[187]. Owing to the criticality of developing effective drug deliverables in targeting tumor initiating CSCs, their clonal enrichment, resistance to current IMCT, the acquired formation of iCSCs with IMCT, investigations around the globe are appropriately focused in identifying/characterizing promising drug candidates. See Table 2 for the summation of a list of such candidates in the pipeline and are characterized. Identifying novel adjuvants that selectively deplete CSCs or target the formation of iCSCs could lead to the development of improved therapeutic strategies for the cure NB.
Short list of drug candidates that were investigated for their efficacy in killing and/or differentiation of NB-CSCs
| Drug | Targets | Ref. |
|---|---|---|
| Retinoic Acid + proteasome inhibitor MG132 | Nestin, Sox2, Oct4 | Hämmerle et al.[168] |
| Dequalinium analogue, C-14 | Metabolic Pathways | Smith et al.[169] |
| Rapamycin | p70S6K, S6RP | Smith et al.[169] |
| Rapamycin | AKT | Bahmad et al.[170] |
| Triciribine | mTOR | Bahmad et al.[170] |
| Oncolytic virus | Nestin | Mahller et al.[61] |
| Imetelstat, RNA TR oligonucleotide antagonist | hTERT | Castelo-Branco et al.[171] |
| Vorinostat, histone deacetylase inhibitor | ABCB1, ABCC4, LMO2, SOX2, ERCC5, S100A10, IGFBP3, TCF3, VIM | Zheng et al.[172] |
| imidazotriazine (GSK461364) | PLK-1 | Pajtler et al.[178] |
| Berberine | Vimentin, fibronectin, E-cadherin, laminin, Smad, PI3/Akt, Ras-Raf-ERK | Naveen et al.[179] |
| XAV939 a small molecule tankyrase (TNKS) inhibitor | TNKS, CD133 | Tian et al.[183] |
| Cyclopamine (11-deoxojervine) | Hedgehog signaling | Schiapparell et al.[184] |
| Curcumin | Stat3 | Goel and Aggarwal[185] |
| Cucurbitacin I (triterpenoid) | STAT3/JAK | Gheeya et al.[186] |
| Honokiol (biphenolic compound) | STAT3 signaling | Prasad et al.[187] |
| Aqueous ethanolic extract of T. cordifolia | NFkB, NCAM, MMPs | Mishra and Kaur[189] |
| Aspirin (acetylsalicylic acid) | p21Waf1, hypo-pRb1 | Pozzoli et al.[190] |
| Rexinoid + IIF + EGCG | MMP-2, MMP-9 and COX-2 | Farabegoli et al.[191] |
On the current clinical perspective for NB CSC-targeted therapy, autologous stem-cell rescue after myeloablative chemotherapy with/without radiotherapy has been shown to relatively improve patient survival[192,193]. Further, studies have indicated the effectiveness of high-dose therapy with tandem or triple autologous stem-cell rescue in treating high-risk NB, with encouraging long-term survival, slashed CNS relapse and secondary malignancies[194,195]. The benefit of such a major therapeutic advancement is limited by the risk of reinfusing NB cancer cells that could lead to and/or attributable to the post-transplant relapse[196,197]. However, significant number of strategies are currently under consideration or revisited to betterment the purging efficacy that could benefit in long-term clinical outcome for NB. The strategy, effect, clinical practice, modifications and pitfalls are adequately documented/reviewed elsewhere[198-202] and hence is not discussed in this review.
Conclusions and perspectives
The existence, clonal selection, and enrichment of CSCs contributes to NB disease progression, resistance to therapeutic measures, and poor prognosis [Figure 2A]. It has been widely recognized that the treatment modalities that spare these CSC clones allow them to self-renew and recapitulate the non-stem tumor cell mass, subsequently leading to tumor relapse. In addition, the outcomes of recent investigations have recognized the formation of iCSCs and their influence in treatment resistance and disease evolution. It is apparent that some, if not all, current treatment components (radiation, chemotherapy drugs) inflict acquired genetic and molecular rearrangements in non-stem tumor cells and prompt the transformation into iCSCs that are extremely tumorigenic and equipped with self-renewing capacity, stemness maintenance, and drug resistance [Figure 2A]. It is critical to develop therapeutic measures that target both the pre-existing CSCs and the iCSCs, if we are to counter therapy resistance and successfully treat NB. Although a number of promising CSC surface markers for NB have been characterized and their cellular and molecular functions in stemness, therapy response, and disease progression have been realized (schematically summarized in Figure 2B), two crucial factors, (1) the lack of ubiquitous availability of specific candidate in all NB-CSCs within the tumor and (2) the presence of candidate marker(s) in normal non-tumorigenic stem cells highly limit their use in developing a CSC-targeted approach. Preliminary studies of the CSC-targeted approach to counter therapy resistance and as an adjuvant with current clinical therapy are encouraging. We believe that this review will provide an up-to-date understanding of NB-CSCs in disease evolution and drug resistance. Overall, the documented evidence supports the enormous clinical potential of targeting CSCs to counter therapy resistance and disease evolution, and warrants further rigorous investigation.
Declarations
AcknowledgmentsThe authors acknowledge the OUHSC Staff Editor (Ms. Kathy Kyler) for the help in critically reviewing this manuscript.
Authors’ contributionsMade substantial contributions to conception and design of the study and performed literature analysis and interpretation, wrote the manuscript: Aravindan N
Performed literature collection, literature analysis and helped in drafting the manuscript: Jain D
Performed literature analysis, technical support and critically reviewed the manuscript: Somasundaram DB
Contribute to the conception and design of the study and critically reviewed the manuscript: Herman TS
Contribute to the conception and design of the study, performed literature interpretation and critically reviewed the manuscript: Aravindan S
Availability of data and materialsNot applicable.
Financial support and sponsorshipThe authors are supported by the research funding from the National Institutes of Health (NIH 1P20GM103639-01) from the COBRE Program of NIH; OUHSC Department of Radiation Oncology Research Development Funds.
Conflicts of interestAll authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2019.
REFERENCES
1. Marc TG, Gurney JG, Smith MA, Olshan AF. Sympathetic nervous system tumors. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, editors. Cancer incidence and survival among children and adolescents: United States SEER program 1975-1995. USA NCI Pub; 1999. pp. 65-72.
2. Gurney JG, Smith MA, Ross JA. Cancer among infants. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, editors. Cancer incidence and survival among children and adolescents: United States SEER program 1975-1995. USA NCI Pub; 1999. pp. 149-56.
3. Morgenstern DA, Baruchel S, Irwin MS. Current and future strategies for relapsed neuroblastoma: challenges on the road to precision therapy. J Pediatr Hematol Oncol 2013;35:337-47.
4. Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, et al. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 2010;28:2625-34.
5. American Cancer Society. Cancer facts & figures 2019. Atlanta: American Cancer Society. 2019;1:1-76.
6. Santana VM, Furman WL, McGregor LM, Billups CA. Disease control intervals in high-risk neuroblastoma. Cancer 2008;112:2796-801.
7. Simon T, Berthold F, Borkhardt A, Kremens B, De Carolis B, et al. Treatment and outcomes of patients with relapsed, high-risk neuroblastoma: results of German trials. Pediatr Blood Cancer 2011;56:578-83.
8. Lau L, Tai D, Weitzman S, Grant R, Baruchel S, et al. Factors influencing survival in children with recurrent neuroblastoma. J Pediatr Hematol Oncol 2004;26:227-32.
9. Berthold F, Hero B, Breu H, Christiansen H, Erttmann R, et al. The recurrence patterns of stages I, II and III neuroblastoma: experience with 77 relapsing patients. Ann Oncol 1996;7:183-7.
10. Molenaar JJ, Ebus ME, Koster J, van Sluis P, van Noesel CJ, et al. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res 2008;68:2599-609.
11. Moll UM, LaQuaglia M, Bénard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A 1995;92:4407-11.
12. Barbieri E, De Preter K, Capasso M, Chen Z, Hsu DM, et al. Histone chaperone CHAF1A inhibits differentiation and promotes aggressive neuroblastoma. Cancer Res 2014;74:765-74.
13. Bhaskara VK, Mohanam I, Rao JS, Mohanam S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS One 2012;7:e30905.
14. Schleiermacher G, Janoueix-Lerosey I, Delattre O. Recent insights into the biology of neuroblastoma. Int J Cancer 2014;135:2249-61.
15. Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med 2013;2:3.
16. Alisi A, Cho WC, Locatelli F, Fruci D. Multidrug resistance and cancer stem cells in neuroblastoma and hepatoblastoma. Int J Mol Sci 2013;14:24706-25.
18. Ross RA, Biedler JL, Spengler BA. A role for distinct cell types in determining malignancy in human neuroblastoma cell lines and tumors. Cancer Lett 2003;197:35-9.
19. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992;255:1707-10.
20. Linabery AM, Ross JA. Trends in childhood cancer incidence in the US (1992-2004). Cancer 2008;112:416-32.
21. Kohler BA, Ward E, McCarthy BJ, Schymura MJ, Ries LA, et al. Annual report to the nation on the status of cancer, 1975-2007, featuring tumors of the brain and other nervous system. J Natl Cancer Inst 2011;103:714-36.
22. de Weille J. On the genesis of neuroblastoma and glioma. Int J Brain Sci 2014;2014:14.
23. Kerosuo L, Bronner-Fraser M. What is bad in cancer is good in the embryo: importance of EMT in neural crest development. Semin Cell Dev Biol 2012;23:320-32.
24. Albino D, Brizzolara A, Moretti S, Falugi C, Mirisola V, et al. Gene expression profiling identifies eleven DNA repair genes down-regulated during mouse neural crest cell migration. Int J Dev Biol 2011;55:65-72.
25. Crane JF, Trainor PA. Neural crest stem and progenitor cells. Annu Rev Cell Dev Biol 2006;22:267-86.
26. Mohlin SA, Wigerup C, Påhlman S. Neuroblastoma aggressiveness in relation to sympathetic neuronal differentiation stage. Seminars Cancer Biol 2011;21:276-82.
27. Craft AW, Parker L. Poor prognosis neuroblastoma: is screening the answer? Br J Cancer Suppl 1992;18:S96-101.
28. Sawada T. Past and future of neuroblastoma screening in Japan. Am J Pediatr Hematol Oncol 1992;14:320-6.
29. Jiang M, Stanke J, Lahti JM. The connections between neural crest development and neuroblastoma. Curr Top Dev Biol 2011;94:77-127.
30. Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, et al. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004;6:838-45.
31. Longo L, Panza E, Schena F, Seri M, Devoto M, et al. Genetic predisposition to familial neuroblastoma: identification of two novel genomic regions at 2p and 12p. Hum Hered 2007;63:205-11.
32. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:930-5.
33. Melotte V, Qu X, Ongenaert M, Van Criekinge W, de Bruïne AP, et al. The N-myc downstream regulated gene (NDRG) family: diverse functions, multiple applications. FASEB J 2010;24:4153-66.
34. Bosse KR, Diskin SJ, Cole KA, Wood AC, Schnepp RW, et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res 2012;72:2068-78.
35. Oldridge DA, Wood AC, Weichert-Leahey N, Crimmins I, Sussman R, et al. Genetic predisposition to neuroblastoma mediated by a LMO1 super-enhancer polymorphism. Nature 2015;528:418-21.
36. Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, et al. A LIN28B-RAN-AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer cell 2015;28:599-609.
37. Schramm A, Koster J, Assenov Y, Althoff K, Peifer M, et al. Mutational dynamics between primary and relapse neuroblastomas. Na Genet 2015;47:872-7.
38. Gaisie G, Oh KS, Young LW. Coexistent neuroblastoma and Hirschsprung’s disease - another manifestation of the neurocristopathy? Pediatr Radiol 1979;8:161-3.
39. Roshkow JE, Haller JO, Berdon WE, Sane SM. Hirschsprung’s disease, Ondine’s curse, and neuroblastoma-manifestations of neurocristopathy. Pediatr Radiol 1988;19:45-9.
40. Quackenbush KE, Luna-Fineman S, Magee JF, Gundogan M, Golobi M, et al. Neuroblastoma involvement of the falx cerebri. Pediatr Blood Cancer 2009;53:1337-9.
41. Emery LG, Shields M, Shah NR, Garbes A. Neuroblastoma associated with beckwith-wiedemann syndrome. Cancer 1983;52:176-9.
42. Kenny FM, Stavrides A, Voorhess ML, Klein R. Cushing’s syndrome associated with an adrenal neuroblastoma: a case in an infant with clinical, pathological, and adrenal cortical and medullary hormone excretion studies. Am J Dis Child 1967;113:611-5.
43. Arnesen MA, Scheithauer BW, Freeman S. Cushing’s syndrome secondary to olfactory neuroblastoma. Ultrastruct Pathol 1994;18:61-8.
44. Kinney H, Faix R, Brazy J. The fetal alcohol syndrome and neuroblastoma. Pediatrics 1980;66:130-2.
45. Yang Q, Olshan AF, Bondy ML, Shah NR, Pollock BH, et al. Parental smoking and alcohol consumption and risk of neuroblastoma. Cancer Epidemiol Biomarkers Prev 2000;9:967-72.
46. Schüz J, Kaletsch U, Meinert R, Kaatsch P, Spix C, et al. Risk factors for neuroblastoma at different stages of disease. Results from a population-based case-control study in Germany. J Clin Epidemiol 2001;54:702-9.
47. Al-Shammri S, Guberman A, Hsu E. Neuroblastoma and fetal exposure to phenytoin in a child without dysmorphic features. Can J Neurol Sci 1992;19:243-5.
49. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: Current trends and future challenges. World J Stem Cells 2015;7:1185-201.
50. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007;130:194-5.
51. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest 2013;123:1911-8.
52. Wang Y, He L, Du Y, Zhu P, Huang G, et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015;16:413-25.
53. Alamgeer M, Peacock CD, Matsui W, Ganju V, Watkins DN. Cancer stem cells in lung cancer: evidence and controversies. Respirology 2013;18:757-64.
54. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983-8.
55. Han J, Fujisawa T, Husain SR, Puri RK. Identification and characterization of cancer stem cells in human head and neck squamous cell carcinoma. BMC cancer 2014;14:173.
56. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A 2007;104:10158-63.
57. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007;445:111-5.
58. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396-401.
59. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730-7.
60. Hansford LM, McKee AE, Zhang L, George RE, Gerstle JT, et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res 2007;67:11234-43.
61. Mahller YY, Williams JP, Baird WH, Mitton B, Grossheim J, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PLoS One 2009;4:e4235.
62. Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res 2016;18:55.
63. Prieto-Vila M, Takahashi RU, Usuba W, Kohama I, Ochiya T. Drug resistance driven by cancer stem cells and their niche. Int J Mol Sci 2017;18:E2574.
65. Dean M. Cancer stem cells: Implications for cancer causation and therapy resistance. Discov Med 2005;5:278-82.
66. Khalil MA, Hrabeta J, Cipro S, Stiborova M, Vicha A, et al. Neuroblastoma stem cells - mechanisms of chemoresistance and histone deacetylase inhibitors. Neoplasma 2012;59:737-46.
67. Naiditch JA, Jie C, Lautz TB, Yu S, Clark S, et al. Mesenchymal change and drug resistance in neuroblastoma. J Surg Res 2015;193:279-88.
68. Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res 1973;33:2643-52.
69. Ross RA, Spengler BA, Domenech C, Porubcin M, Rettig WJ, et al. Human neuroblastoma I-type cells are malignant neural crest stem cells. Cell Growth Differ 1995;6:449-56.
70. Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, et al. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004;6:838-45.
71. Ross RA, Walton JD, Han D, Guo HF, Cheung NK. A distinct gene expression signature characterizes human neuroblastoma cancer stem cells. Stem Cell Res 2015;15:419-26.
72. Buhagiar A, Ayers D. Chemoresistance, cancer stem cells, and miRNA influences: the case for neuroblastoma. Anal Cell Pathol (Amst) 2015;2015:150634.
73. Oh L, Hafsi H, Hainaut P, Ariffin H. P53, stem cell biology and childhood blastomas. Curr Opin Oncol 2019;31:84-91.
74. Friedman GK, Gillespie GY. Cancer stem cells and pediatric solid tumors. Cancers (Basel) 2011;3:298-318.
75. Garner EF, Beierle EA. Cancer stem cells and their interaction with the tumor microenvironment in neuroblastoma. Cancers (Basel) 2015;8:e5.
77. Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, et al. Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int 2018;2018:5416923.
79. Dawood S, Austin L, Cristofanilli M. Cancer stem cells: implications for cancer therapy. Oncology 2014;28:1101.
82. Nassar D, Blanpain C. Cancer stem cells: basic concepts and therapeutic implications. Annu Rev Pathol 2016;11:47-76.
83. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest 2010;120:41-50.
84. Poggi A, Varesano S, Zocchi MR. How to hit mesenchymal stromal cells and make the tumor microenvironment immunostimulant rather than immunosuppressive. Front Immunol 2018;9:262.
85. Ishiguro T, Ohata H, Sato A, Yamawaki K, Enomoto T, et al. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci 2017;108:283-9.
86. Yoshida GJ, Saya H. Therapeutic strategies targeting cancer stem cells. Cancer Sci 2016;107:5-11.
88. Newton TC, Wolcott K, Roberts SS. Comparison of the side populations in pretreatment and postrelapse neuroblastoma cell lines. Transl Oncol 2010;3:246-51.
89. Hirschmann-Jax C, Foster AE, Wulf GG, Nuchtern JG, Jax TW, et al. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc Natl Acad Sci U S A 2004;101:14228-33.
90. Islam MO, Kanemura Y, Tajria J, Mori H, Kobayashi S, et al. Functional expression of ABCG2 transporter in human neural stem/progenitor cells. Neurosci Res 2005;52:75-82.
92. Xing LL, Sha YL, Wu YM, Hu JM, Zhang M, et al. Preliminary analysis of stem cell-like cells in human neuroblastoma. World J Pediatr 2015;11:54-60.
93. Shohet JM, Ghosh R, Coarfa C, Ludwig A, Benham AL, et al. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer Res 2011;71:3841-51.
94. Cournoyer S, Nyalendo C, Addioui A, Belounis A, Beaunoyer M, et al. Genotype analysis of tumor-initiating cells expressing CD133 in neuroblastoma. Genes Chromosomes Cancer 2012;51:792-804.
95. Tong QS, Zheng LD, Tang ST, Ruan QL, Liu Y, et al. Expression and clinical significance of stem cell marker CD133 in human neuroblastoma. World J Pediatr 2008;4:58-62.
96. Sartelet H, Imbriglio T, Nyalendo C, Haddad E, Annabi B, et al. CD133 expression is associated with poor outcome in neuroblastoma via chemoresistance mediated by the AKT pathway. Histopathology 2012;60:1144-55.
97. Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, et al. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 2011;30:97-105.
98. Khalil MA, Hrabeta J, Groh T, Prochazka P, Doktorova H, et al. Valproic acid increases CD133 positive cells that show low sensitivity to cytostatics in neuroblastoma. PLoS One 2016;11:e0162916.
99. Vangipuram SD, Wang ZJ, Lyman WD. Resistance of stem-like cells from neuroblastoma cell lines to commonly used chemotherapeutic agents. Pediatr Blood Cancer 2010;54:361-8.
100. Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci 2005;26:113-6.
101. Cipro S, Hrebackova J, Hrabeta J, Poljakova J, Eckschlager T. Valproic acid overcomes hypoxia-induced resistance to apoptosis. Oncol Rep 2012;27:1219-26.
102. Cantilena S, Pastorino F, Pezzolo A, Chayka O, Pistoia V, et al. Frizzled receptor 6 marks rare, highly tumourigenic stem-like cells in mouse and human neuroblastomas. Oncotarget 2011;2:976-83.
103. Vieira GC, Chockalingam S, Melegh Z, Greenhough A, Malik S, et al. LGR5 regulates pro-survival MEK/ERK and proliferative Wnt/beta-catenin signalling in neuroblastoma. Oncotarget 2015;6:40053-67.
104. Forgham H, Johnson D, Carter N, Veuger S, Carr-Wilkinson J. Stem cell markers in neuroblastoma - an emerging role for LGR5. Front Cell Dev Biol 2015;3:77.
105. Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle 2011;10:1378-84.
106. Hartomo TB, Van Huyen Pham T, Yamamoto N, Hirase S, Hasegawa D, et al. Involvement of aldehyde dehydrogenase 1A2 in the regulation of cancer stem cell properties in neuroblastoma. Int J Oncol 2015;46:1089-98.
107. Flahaut M, Jauquier N, Chevalier N, Nardou K, Balmas Bourloud K, et al. Aldehyde dehydrogenase activity plays a key role in the aggressive phenotype of neuroblastoma. BMC Cancer 2016;16:781.
108. Kuo YT, Liu YL, Adebayo BO, Shih PH, Lee WH, et al. JARID1B expression plays a critical role in chemoresistance and stem cell-like phenotype of neuroblastoma cells. PLoS One 2015;10:e0125343.
109. Hsu DM, Agarwal S, Benham A, Coarfa C, Trahan DN, et al. G-CSF receptor positive neuroblastoma subpopulations are enriched in chemotherapy-resistant or relapsed tumors and are highly tumorigenic. Cancer Res 2013;73:4134-46.
110. Agarwal S, Lakoma A, Chen Z, Hicks J, Metelitsa LS, et al. G-CSF promotes neuroblastoma tumorigenicity and metastasis via STAT3-dependent cancer stem cell activation. Cancer Res 2015;75:2566-79.
111. Baggiolini A, Varum S, Mateos JM, Bettosini D, John N, et al. Premigratory and migratory neural crest cells are multipotent in vivo. Cell Stem Cell 2015;16:314-22.
112. Kirsch F, Kruger C, Schneider A. The receptor for granulocyte-colony stimulating factor (G-CSF) is expressed in radial glia during development of the nervous system. BMC Dev Biol 2008;8:32.
113. Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest 2005;115:2083-98.
114. Chikaraishi K, Takenobu H, Sugino RP, Mukae K, Akter J, et al. CFC1 is a cancer stemness-regulating factor in neuroblastoma. Oncotarget 2017;8:45046-59.
115. Cui H, Hu B, Li T, Ma J, Alam G, et al. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol 2007;170:1370-8.
116. Cui H, Ma J, Ding J, Li T, Alam G, et al. Bmi-1 regulates the differentiation and clonogenic self-renewal of I-type neuroblastoma cells in a concentration-dependent manner. J Biol Chem 2006;281:34696-704.
117. Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, et al. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res 2006;34:1745-54.
119. Ochiai H, Takenobu H, Nakagawa A, Yamaguchi Y, Kimura M, et al. Bmi1 is a MYCN target gene that regulates tumorigenesis through repression of KIF1Bbeta and TSLC1 in neuroblastoma. Oncogene 2010;29:2681-90.
120. Mao L, Ding J, Perdue A, Yang L, Zha Y, et al. Cyclin E1 is a common target of BMI1 and MYCN and a prognostic marker for neuroblastoma progression. Oncogene 2012;31:3785-95.
121. Melone MA, Giuliano M, Squillaro T, Alessio N, Casale F, et al. Genes involved in regulation of stem cell properties: studies on their expression in a small cohort of neuroblastoma patients. Cancer Biol Ther 2009;8:1300-6.
122. Chen D, Wu M, Li Y, Chang I, Yuan Q, et al. Targeting BMI1+ cancer stem cells overcomes chemoresistance and inhibits metastases in squamous cell carcinoma. Cell Stem Cell 2017;20:621-34.
123. Ren H, Du P, Ge Z, Jin Y, Ding D, et al. TWIST1 and BMI1 in cancer metastasis and chemoresistance. J Cancer 2016;7:1074-80.
124. Yin T, Zhang Z, Cao B, Duan Q, Shi P, et al. Bmi1 inhibition enhances the sensitivity of pancreatic cancer cells to gemcitabine. Oncotarget 2016;7:37192-204.
125. Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells 2012;30:372-8.
126. Yang XX, Ma M, Sang MX, Zhang XY, Liu ZK, et al. BMI-1 suppression increases the radiosensitivity of oesophageal carcinoma via the PI3K/Akt signaling pathway. Oncol Rep 2018;39:667-78.
127. Yan Y, Wang Y, Zhao P, Ma W, Hu Z, et al. BMI-1 promotes self-renewal of radio- and temozolomide (TMZ)-resistant breast cancer cells. Reprod Sci 2017;24:1620-9.
128. Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci 2010;30:10096-111.
129. Jandial R, Waters DJ, Chen MY. Cancer stem cells can arise from differentiated neoplastic cells. Neurosurgery 2011;69:N22.
130. Friedmann-Morvinski D, Bushong EA, Ke E, Soda Y, Marumoto T, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science 2012;338:1080-4.
131. Nishi M, Sakai Y, Akutsu H, Nagashima Y, Quinn G, et al. Induction of cells with cancer stem cell properties from nontumorigenic human mammary epithelial cells by defined reprogramming factors. Oncogene 2014;33:643-52.
132. Scaffidi P, Misteli T. In vitro generation of human cells with cancer stem cell properties. Nat Cell Biol 2011;13:1051-61.
133. Vares G, Cui X, Wang B, Nakajima T, Nenoi M. Generation of breast cancer stem cells by steroid hormones in irradiated human mammary cell lines. PLoS One 2013;8:e77124.
134. Kang SK, Park JB, Cha SH. Multipotent, dedifferentiated cancer stem-like cells from brain gliomas. Stem Cells Dev 2006;15:423-35.
135. Oshima N, Yamada Y, Nagayama S, Kawada K, Hasegawa S, et al. Induction of cancer stem cell properties in colon cancer cells by defined factors. PLoS One 2014;9:e101735.
136. Shi L, Fei X, Sun G, Wang Z, Wan Y, et al. Hypothermia stimulates glioma stem spheres to spontaneously dedifferentiate adjacent non-stem glioma cells. Cell Mol Neurobiol 2015;35:217-30.
137. Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev 2008;22:2028-33.
138. Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A 2011;108:1397-402.
139. Ikegaki N, Shimada H, Fox AM, Regan PL, Jacobs JR, et al. Transient treatment with epigenetic modifiers yields stable neuroblastoma stem cells resembling aggressive large-cell neuroblastomas. Proc Natl Acad Sci U S A 2013;110:6097-102.
140. Colak S, Medema JP. Cancer stem cells - important players in tumor therapy resistance. FEBS J 2014;281:4779-91.
141. Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, et al. Identification of cells initiating human melanomas. Nature 2008;451:345-9.
142. Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 2008;10:R25.
143. Hashimoto N, Tsunedomi R, Yoshimura K, Watanabe Y, Hazama S, et al. Cancer stem-like sphere cells induced from de-differentiated hepatocellular carcinoma-derived cell lines possess the resistance to anti-cancer drugs. BMC Cancer 2014;14:722.
144. Chen X, Liao R, Li D, Sun J. Induced cancer stem cells generated by radiochemotherapy and their therapeutic implications. Oncotarget 2017;8:17301-12.
145. Seo Y, Kim YS, Lee KE, Park TH, Kim Y. Anti-cancer stemness and anti-invasive activity of bitter taste receptors, TAS2R8 and TAS2R10, in human neuroblastoma cells. PLoS One 2017;12:e0176851.
146. Farace C, Oliver JA, Melguizo C, Alvarez P, Bandiera P, et al. Microenvironmental modulation of decorin and lumican in temozolomide-resistant glioblastoma and neuroblastoma cancer stem-like cells. PLoS One 2015;10:e0134111.
147. Diaz-Carballo D, Acikelli AH, Bardenheuer W, Gustmann S, Malak S, et al. Identification of compounds that selectively target highly chemotherapy refractory neuroblastoma cancer stem cells. Int J Clin Pharmacol Ther 2014;52:787-801.
148. Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar DZ, et al. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 2009;13:1059-85.
149. Wu Z, Wu L, Weng D, Xu D, Geng J, et al. Reduced expression of lamin A/C correlates with poor histological differentiation and prognosis in primary gastric carcinoma. J Exp Clin Cancer Res 2009;28:8.
150. Nardella M, Guglielmi L, Musa C, Iannetti I, Maresca G, et al. Down-regulation of the Lamin A/C in neuroblastoma triggers the expansion of tumor initiating cells. Oncotarget 2015;6:32821-40.
151. Kamachi Y, Uchikawa M, Kondoh H. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet 2000;16:182-7.
152. Fang X, Yu W, Li L, Shao J, Zhao N, et al. ChIP-seq and functional analysis of the SOX2 gene in colorectal cancers. OMICS 2010;14:369-84.
153. Gen Y, Yasui K, Zen Y, Zen K, Dohi O, et al. SOX2 identified as a target gene for the amplification at 3q26 that is frequently detected in esophageal squamous cell carcinoma. Cancer Genet Cytogenet 2010;202:82-93.
154. Ji J, Zheng PS. Expression of Sox2 in human cervical carcinogenesis. Hum Pathol 2010;41:1438-47.
155. Nakatsugawa M, Takahashi A, Hirohashi Y, Torigoe T, Inoda S, et al. SOX2 is overexpressed in stem-like cells of human lung adenocarcinoma and augments the tumorigenicity. Lab Invest 2011;91:1796-804.
156. Xiang R, Liao D, Cheng T, Zhou H, Shi Q, et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br J Cancer 2011;104:1410-7.
157. Yang S, Zheng J, Ma Y, Zhu H, Xu T, et al. Oct4 and Sox2 are overexpressed in human neuroblastoma and inhibited by chemotherapy. Oncol Rep 2012;28:186-92.
158. Yang S, Zheng J, Xiao X, Xu T, Tang W, et al. SOX2 promotes tumorigenicity and inhibits the differentiation of I-type neuroblastoma cells. Int J Oncol 2015;46:317-23.
159. Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, et al. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature 2004;427:78-83.
160. Chavali PL, Saini RK, Zhai Q, Vizlin-Hodzic D, Venkatabalasubramanian S, et al. TLX activates MMP-2, promotes self-renewal of tumor spheres in neuroblastoma and correlates with poor patient survival. Cell Death Dis 2014;5:e1502.
161. Fuchs E, Tumbar T, Guasch G. Socializing with the neighbors: stem cells and their niche. Cell 2004;116:769-78.
162. Ye J, Wu D, Wu P, Chen Z, Huang J. The cancer stem cell niche: cross talk between cancer stem cells and their microenvironment. Tumour Biol 2014;35:3945-51.
163. Jögi A, Øra I, Nilsson H, Lindeheim A, Makino Y, et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A 2002;99:7021-6.
164. Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006;10:413-23.
165. Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol 2012;30:3525-32.
166. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res 2017;77:5142-57.
167. Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997;90:5002-12.
168. Hämmerle B, Yañez Y, Palanca S, Cañete A, Burks DJ, et al. Targeting neuroblastoma stem cells with retinoic acid and proteasome inhibitor. PLoS One 2013;8:e76761.
169. Smith KM, Datti A, Fujitani M, Grinshtein N, Zhang L, et al. Selective targeting of neuroblastoma tumour-initiating cells by compounds identified in stem cell-based small molecule screens. EMBO Mol Med 2010;2:371-84.
170. Bahmad HF, Mouhieddine TH, Chalhoub RM, Assi S, Araji T, et al. The Akt/mTOR pathway in cancer stem/progenitor cells is a potential therapeutic target for glioblastoma and neuroblastoma. Oncotarget 2018;9:33549-61.
171. Castelo-Branco P, Zhang C, Lipman T, Fujitani M, Hansford L, et al. Neural tumor-initiating cells have distinct telomere maintenance and can be safely targeted for telomerase inhibition. Clin Cancer Res 2011;17:111-21.
172. Zheng X, Naiditch J, Czurylo M, Jie C, Lautz T, et al. Differential effect of long-term drug selection with doxorubicin and vorinostat on neuroblastoma cells with cancer stem cell characteristics. Cell Death Dis 2013;4:e740.
173. Zaatiti H, Abdallah J, Nasr Z, Khazen G, Sandler A, et al. Tumorigenic proteins upregulated in the MYCN-amplified IMR-32 human neuroblastoma cells promote proliferation and migration. Int J Oncol 2018;52:787-803.
174. Bao S, Wu Q, Li Z, Sathornsumetee S, Wang H, et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer Res 2008;68:6043-8.
175. Hong H, Stastny M, Brown C, Chang WC, Ostberg JR, et al. Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J Immunother 2014;37:93-104.
176. Kunkele A, Taraseviciute A, Finn LS, Johnson AJ, Berger C, et al. Preclinical assessment of CD171-directed CAR T-cell adoptive therapy for childhood neuroblastoma: CE7 epitope target safety and product manufacturing feasibility. Clin Cancer Res 2017;23:466-77.
177. Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, et al. Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res 2011;71:1385-95.
178. Pajtler KW, Sadowski N, Ackermann S, Althoff K, Schönbeck K, et al. The GSK461364 PLK1 inhibitor exhibits strong antitumoral activity in preclinical neuroblastoma models. Oncotarget 2017;8:6730-41.
179. Naveen CR, Gaikwad S, Agrawal-Rajput R. Berberine induces neuronal differentiation through inhibition of cancer stemness and epithelial-mesenchymal transition in neuroblastoma cells. Phytomedicine 2016;23:736-44.
180. Kim DW, Ahan SH, Kim TY. Enhancement of arsenic trioxide [As(2)O(3)]-mediated apoptosis using berberine in human neuroblastoma SH-SY5Y cells. J Korean Neurosurg Soc 2007;42:392-9.
181. Choi MS, Yuk DY, Oh JH, Jung HY, Han SB, et al. Berberine inhibits human neuroblastoma cell growth through induction of p53-dependent apoptosis. Anticancer Res 2008;28:3777-84.
182. Li J, Gu L, Zhang H, Liu T, Tian D, et al. Berberine represses DAXX gene transcription and induces cancer cell apoptosis. Lab Invest 2013;93:354-64.
183. Tian X, Hou W, Bai S, Fan J, Tong H, et al. XAV939 inhibits the stemness and migration of neuroblastoma cancer stem cells via repression of tankyrase 1. Int J Oncol 2014;45:121-8.
184. Schiapparelli P, Shahi MH, Enguita-Germán M, Johnsen JI, Kogner P, et al. Inhibition of the sonic hedgehog pathway by cyplopamine reduces the CD133+/CD15+ cell compartment and the in vitro tumorigenic capability of neuroblastoma cells. Cancer Lett 2011;310:222-31.
185. Goel A, Aggarwal BB. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer 2010;62:919-30.
186. Gheeya JS, Chen QR, Benjamin CD, Cheuk AT, Tsang P, et al. Screening a panel of drugs with diverse mechanisms of action yields potential therapeutic agents against neuroblastoma. Cancer Biol Ther 2009;8:2386-95.
187. Prasad R, Katiyar SK. Honokiol, an active compound of magnolia plant, inhibits growth, and progression of cancers of different organs. Adv Exp Med Biol 2016;928:245-65.
188. Marcucci F, Rumio C, Lefoulon F. Anti-cancer stem-like cell compounds in clinical development - an overview and critical appraisal. Front Oncol 2016;6:115.
189. Mishra R, Kaur G. Tinospora cordifolia induces differentiation and senescence pathways in neuroblastoma cells. Mol Neurobiol 2015;52:719-33.
190. Pozzoli G, Petrucci G, Navarra P, Marei HE, Cenciarelli C. Aspirin inhibits proliferation and promotes differentiation of neuroblastoma cells via p21(Waf1) protein up-regulation and Rb1 pathway modulation. J Cell Mol Med 2019;23:7078-87.
191. Farabegoli F, Govoni M, Spisni E, Papi A. Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene limit BE(2)-C neuroblastoma cell growth and neurosphere formation in vitro. Nutrients 2018;10:1141.
192. Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med 1999;341:1165-73.
193. Frappaz D, Michon J, Coze C, Berger C, Plouvier E, et al. LMCE3 treatment strategy: results in 99 consecutively diagnosed stage 4 neuroblastomas in children older than 1 year at diagnosis. J Clin Oncol 2000;18:468-76.
194. George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, et al. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. J Clin Oncol 2006;24:2891-6.
195. Kletzel M, Katzenstein HM, Haut PR, Yu AL, Morgan E, et al. Treatment of high-risk neuroblastoma with triple-tandem high-dose therapy and stem-cell rescue: results of the Chicago Pilot II Study. J Clin Oncol 2002;20:2284-92.
196. Moss TJ, Cairo M, Santana VM, Weinthal J, Hurvitz C, et al. Clonogenicity of circulating neuroblastoma cells: implications regarding peripheral blood stem cell transplantation. Blood 1994;83:3085-9.
197. Rill DR, Santana VM, Roberts WM, Nilson T, Bowman LC, et al. Direct demonstration that autologous bone marrow transplantation for solid tumors can return a multiplicity of tumorigenic cells. Blood 1994;84:380-3.
198. Yalcin B, Kremer LC, van Dalen EC. High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst Rev 2013:CD006301.
199. Kaffenberger J, Basak SA, Ioffreda M. Autologous graft-versus-host disease in a child with stage IV neuroblastoma. Pediatr Dermatol 2015;32:276-9.
200. Grupp SA, Asgharzadeh S, Yanik GA. Neuroblastoma: issues in transplantation. Biol Blood Marrow Transplant 2012;18:S92-100.
201. Fish JD, Grupp SA. Stem cell transplantation for neuroblastoma. Bone Marrow Transplant 2008;41:159-65.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Aravindan N, Jain D, Somasundaram DB, Herman TS, Aravindan S. Cancer stem cells in neuroblastoma therapy resistance. Cancer Drug Resist 2019;2:948-67. http://dx.doi.org/10.20517/cdr.2019.72
AMA Style
Aravindan N, Jain D, Somasundaram DB, Herman TS, Aravindan S. Cancer stem cells in neuroblastoma therapy resistance. Cancer Drug Resistance. 2019; 2(4): 948-67. http://dx.doi.org/10.20517/cdr.2019.72
Chicago/Turabian Style
Aravindan, Natarajan, Drishti Jain, Dinesh Babu Somasundaram, Terence S. Herman, Sheeja Aravindan. 2019. "Cancer stem cells in neuroblastoma therapy resistance" Cancer Drug Resistance. 2, no.4: 948-67. http://dx.doi.org/10.20517/cdr.2019.72
ACS Style
Aravindan, N.; Jain D.; Somasundaram DB.; Herman TS.; Aravindan S. Cancer stem cells in neuroblastoma therapy resistance. Cancer Drug Resist. 2019, 2, 948-67. http://dx.doi.org/10.20517/cdr.2019.72
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 10 clicks
Cite This Article 10 clicks

 Like This Article 17
likes
Like This Article 17
likes








Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.