


Atherosclerosis is a major complication of type 2 diabetes. The pathogenesis of this complication is poorly understood, but it clearly involves production in the vascular wall of macrophage (Mo) lipoprotein lipase (LPL). Mo LPL is increased in human diabetes. Peripheral factors dysregulated in diabetes, including glucose and free fatty acids (FAs), may contribute to this alteration. We previously reported that high glucose stimulates LPL production in both J774 murine and human Mo. In the present study, we evaluated the direct effect of FAs on murine Mo LPL expression and examined the involvement of peroxisome proliferator–activated receptors (PPARs) in this effect. J774 Mo were cultured for 24 h with 0.2 mmol/l unsaturated FAs (arachidonic [AA], eicosapentaenoic [EPA], and linoleic acids [LA]) and monounsaturated (oleic acid [OA]) and saturated FAs (palmitic acid [PA] and stearic acid [SA]) bound to 2% bovine serum albumin. At the end of this incubation period, Mo LPL mRNA expression, immunoreactive mass, activity, and synthetic rate were measured. Incubation of J774 cells with LA, PA, and SA significantly increased Mo LPL mRNA expression. In contrast, exposure of these cells to AA and EPA dramatically decreased this parameter. All FAs, with the exception of EPA and OA, increased extra- and intracellular LPL immunoreactive mass and activity. Intracellular LPL mass and activity paralleled extracellular LPL mass and activity in all FA-treated cells. In Mo exposed to AA, LA, and PA, an increase in Mo LPL synthetic rate was observed. To evaluate the role of PPARs in the modulatory effect of FAs on Mo LPL gene expression, DNA binding assays were performed. Results of these experiments demonstrate an enhanced binding of nuclear proteins extracted from all FA-treated Mo to the peroxisome proliferator–response element (PPRE) consensus sequence of the LPL promoter. PA-, SA-, and OA-stimulated binding activity was effectively diminished by immunoprecipitation of the nuclear proteins with anti–PPAR-α antibodies. In contrast, anti–PPAR-γ antibodies only significantly decreased AA-induced binding activity. Overall, these results provide the first evidence for a direct regulatory effect of FAs on Mo LPL and suggest a potential role of PPARs in the regulation of Mo LPL gene expression by FAs.
Atherosclerosis is the major underlying cause of cardiovascular diseases in adults and the leading cause of morbidity and mortality among diabetic patients (1–6). It has been shown that lipoprotein lipase (LPL), a secretory product of macrophage (Mo) in the arterial wall, contributes to the development and progression of atherosclerosis (7–12). We have previously shown that high glucose enhances LPL production in J774 murine and human Mo (13). Furthermore, we have recently demonstrated that Mo isolated from patients with type 2 diabetes overproduce LPL (14). Besides glucose, other metabolic factors dysregulated in diabetes, such as fatty acids (FAs), may play a key role in the Mo LPL overproduction associated with human diabetes. Indeed, FAs are increased in the plasma of diabetic patients and are produced at high concentrations in the arterial wall through the hydrolysis of triglyceride-rich lipoproteins infiltrating the intima (15).
The role of FAs in atherogenesis has been largely characterized over the last decade. Whereas much evidence supports a proatherogenic effect of saturated FAs (16–20), a protective action with respect to the arterial wall has been attributed to unsaturated FAs (20–25). Lipid metabolism is one major mechanism responsible for the modulatory effect of FAs on the atherogenic process. LPL is a major enzyme involved in the hydrolysis of lipoproteins. Recently, an inhibitory effect of FAs on plasma and adipocyte LPL has been documented. More specifically, it has been shown that rats fed a high-fat diet have decreased plasma and adipocyte LPL activity levels (26) and that high-fat feeding of normal subjects causes an impairment of insulin-stimulated LPL activity in adipose tissue (27). In addition, fasting and catecholamines, which stimulate lipolysis and increase plasma FA levels, have been reported to decrease adipocyte LPL activity (28). Because LPL gene expression in adipose tissue is increased (29) or unaffected by FAs (30,31), posttranslational mechanisms have been implicated in the regulation of adipocyte LPL by these metabolic agents.
Despite the potential key role of Mo LPL and FAs in atherogenesis associated with diabetes, the regulatory effect of FAs on Mo LPL expression has not been investigated. Based on our previous results showing a similar metabolic regulation of LPL expression in murine and human Mo (13), in the present study, we investigated the direct regulatory effect of FAs on LPL in J774 murine Mo. Our data demonstrate that FAs directly modulate Mo LPL expression.
RESEARCH DESIGN AND METHODS
Reagents.
Fetal calf serum (FCS) was purchased from Wisent (St-Bruno, Quebec, Canada). Dulbecco’s minimal essential medium (DMEM), penicillin-streptomycin, and heparin were obtained from Gibco BRL (Grand Island, NY). Sodium salt FAs, FA-free bovine serum albumin (BSA) fraction V, and dextran sulfate were obtained from Sigma (St. Louis, MO). Cesium chloride, glycin, sodium dodecyl sulfate (SDS), and formaldehyde solution were obtained from Fischer (Fair Lawn, NJ). [32P]dCTP (specific activity 3,000 Ci/mmol) and [35S]methionine (specific activity 1,000 Ci/mmol) were provided by ICN Biochemicals (Costa Mesa, CA).
Murine Mo.
The J774 murine Mo cell line was obtained from American Type Cell Collection (Rockville, MD). Murine Mo 7 × 106 were cultured in DMEM containing 10% FCS and 1% penicillin-streptomycin. Mo were incubated for 24 h in serum-free DMEM in the presence of 0.2 mmol/l of various FAs, including polyunsaturated FAs (arachidonic [AA], eicosapentaenoic [EPA], and linoleic acids [LA]) and monounsaturated (oleic acid [OA]) and saturated FAs (palmitic [PA] and stearic acids [SA]) bound to 2% BSA. Serum-free DMEM supplemented with 2% BSA was used as control. Under these experimental conditions, the FA-to-albumin molar ratio was ∼1, a value that is within physiological levels.
Analysis of LPL mRNA expression
Northern blot analysis.
Following treatment with FAs, Mo were lysed with guanidine isothiocyanate. Total RNA was purified by centrifugation through a cesium chloride gradient (32), and 20 μg total RNA was separated on a 1.2% agarose gel containing 2.2 mol/l formaldehyde (33). The blots were prehybridized for 8 h in prehybridization buffer. The mRNA expression was analyzed by hybridization with [32P]dCTP-labeled murine LPL and S28 cDNA probes. Hybridization was detected by autoradiography with Kodak Biomax Film (Rochester, NY). mRNA expression was quantified by high-resolution optical densitometry (Alpha Imager 2000; Packard Instruments, Meriden, CT). LPL mRNA levels were normalized to the levels of S28 mRNA and expressed as percentages of control values.
Electrophoretic mobility shift assay.
The isolation of the nuclei was performed as previously described (34). Briefly, 5 × 107 J774 cells were collected, washed with cold phosphate-buffered salt solution (PBS), and lysed in 1 ml ice-cold buffer A (15 mmol/l KCl, 2 mmol/l MgCl2, 10 mmol/l HEPES, 0.1% phenylmethylsulfonyl fluoride [PMSF], and 0.5% Nonidet P-40). After a 10-min incubation on ice, lysed cells were centrifuged, and the nuclei were washed with buffer A without Nonidet P-40. The nuclei were then lysed in a buffer containing 2 mol/l KCl, 25 mmol/l HEPES, 0.1 mmol/l EDTA, and 1 mmol/l dithiothreitol (DTT). After a 15-min incubation period, a dialysis buffer (25 mmol/l HEPES, 1 mmol/l DTT, 0.1% PMSF, 2 μg/ml aprotinin, 0.1 mmol/l EDTA, and 11% glycerol) was added to the nuclei preparation. Nuclei were collected by centrifugation for 20 min at 13,000 rpm. Aliquots (50 μl) of the supernatants were frozen at −70°C, and protein concentration was determined. DNA retardation (mobility shift) electrophoresis assays were performed as previously described by Fried and Crothers (35). Briefly, 5 μg nuclear extracts were incubated for 15 min in the presence of 5× binding buffer (125 mmol/l HEPES, pH 7.5, 50% glycerol, 250 mmol/l NaCl, 0.25% Nonidet P-40, and 5 mmol/l DTT) in the presence or absence of 200 ng anti–PPAR-α, -β, (Santa Cruz Biotechnology, Santa Cruz, CA), or -γ (Calbiochem, La Jolla, CA) antibodies. End-labeled double-stranded consensus sequences of the LPL promoter PPAR-enhancing element (20,000 cpm per sample) were then added to the samples for 30 min. Samples were analyzed on a 4% nondenaturating PAGE containing 0.01% Nonidet P-40. The specificity of the nuclear protein binding was assessed by incubating the nuclear proteins isolated from murine Mo with a labeled DNA probe in the presence of a 1,000-fold molar excess of unlabeled DNA probe.
DNA probes.
The cDNA probes for detection of murine LPL were prepared by the polymerase chain reaction technique. cDNA was obtained from total RNA using a reverse transcription reaction. Two synthetic primers spanning bases 255–287 and 1117–1149 of the LPL cDNA were used to enzymatically amplify a 894-bp region of the LPL probe. The cDNA probe for S28 was obtained from American Type Cell Collection. A 20-mer double-stranded oligonucleotide containing the peroxisome proliferator–responsive element (PPRE) consensus sequence of the human LPL gene promoter (36) (sense: 5′-CGTCTGCCCTTTCCCCCTCT-3′; antisense: 5′-GAGAAGAGGGGGAAAGGGCA-3′) was synthesized with the aid of an automated DNA synthesizer. After annealing, the double-stranded oligonucleotide was labeled with [γ-32P]ATP using the Boehringer-Mannheim 5′ end–labeling kit (Indianapolis, IN).
Determination of murine LPL immunoreactive mass and activity.
J774 cells (2 × 106) were incubated for 24 h in DMEM containing 0.2 mmol/l FA. Then, 1 h before the end of the incubation period, 100 U/ml heparin was added to the medium. Extra- and intracellular LPL immunoreactive mass and activity were determined in the supernatants and in 0.5N NaOH cell lysates using the Markit-F-LPL kit (Dainippon Pharmaceutical, Osaka, Japan) (37) and the confluolip kit (Progen, Heidelberg, Germany) (38), respectively. LPL mass and activity were normalized to the levels of total cell proteins.
Immunoprecipitation assay.
Following treatment with the appropriate FA, 1 × 106 cells were washed twice with PBS and incubated for 1 h with methionine-free DMEM. Cells were then metabolically labeled with 100 μCi [35S]methionine for 4 h and chased for 1 h with complete DMEM. Radiolabeling was ended by the addition of lysis buffer (50 mmol/l Tris-HCl [pH 7.5], 150 mmol/l NaCl, 100 μg/ml PMSF, and 1% Triton X-100). Immunoprecipitation of LPL in cell lysates was performed after centrifugation of homogenates at 17,000g for 20 min at 4°C; the supernatant was collected and used for immunoprecipitation. The samples (200 μg total cytoplasmic proteins) were incubated at 4° for 4 h with 10 μg of the monoclonal anti-LPL antibody 5D2 (received from J. D. Brunzell, University of Washington, Seattle, WA) followed by incubation with 10 μl goat anti-mouse IgG antiserum (Bio Rad, Hercules, CA). The immunocomplexes were collected on protein A/G PLUS-agarose beads (Santa Cruz Biotechnology), washed twice with Tris-buffered saline (TBS) containing 0.2% NP-40, 2 mmol/l EDTA, and 500 mmol/l NaCl, then with TBS only. The pellet was resuspended in 1× SDS-loading buffer (50 mmol/l Tris-HCl [pH 6.8], 100 mmol/l DTT, 2% SDS, 0.1% bromophenol blue, and 10% glycerol) and boiled for 10 min, and the samples were subjected to 8% SDS-PAGE (39). Immunoprecipitation of cytoplasmic proteins isolated from heparin-stimulated Mo was used as positive control (40). Immunoprecipitation with the irrelevant anti-human interleukin-6 antibody (10 μg) (Santa Cruz Biotechnology) was used as negative control. After autoradiography, the intensity of the bands was quantified by densitometry using an image analysis scanning system (Alpha Imager 2000; Packard Instruments).
Determination of cell viability.
Cell viability after treatment with FAs was assessed by trypan blue exclusion. Viability was consistently found to be >90%.
Determination of protein concentrations.
Protein concentration was measured according to the Bradford method (41) using a colorimetric assay (Bio-Rad, Mississauga, Ontario, Canada) and BSA as standard.
Statistical analysis.
Statistical analysis of the results were done by one-way analysis of variance followed by the Student-Newman-Keuls test for multiple comparisons. All values were expressed as means ± SE. P < 0.05 was considered significant.
RESULTS
Effect of FAs on Mo LPL mRNA expression.
The kinetic effect of FAs on LPL mRNA expression was determined by incubating J774 cells in the presence of FAs for 3–48 h. The earliest changes in LPL mRNA levels in response to saturated and polyunsaturated FAs were observed after a 6 h- and a 12 h-incubation period, respectively (data not shown). Maximal effect of all tested FAs on Mo LPL gene expression was observed at 24 h (Fig. 1A). Incubation of J774 cells in presence of LA, PA, and SA for 24 h significantly increased Mo LPL mRNA levels. Maximal effect was observed with saturated FAs (LPL mRNA levels [% over basal values]: PA 150 ± 20% and SA 140 ± 15%, P < 0.001). LPL mRNA levels were also positively modulated by LA and OA (LPL mRNA levels [% over basal values]: LA 133 ± 6%, P < 0.001, and OA 116 ± 22%, P > 0.05). In contrast, exposure of Mo to AA and EPA dramatically decreased LPL mRNA levels (LPL mRNA levels [% of basal values]: AA 48 ± 6%, P < 0.001, and EPA 60 ± 5%, P < 0.001). Under these experimental conditions, no modulation of the rRNA expression of S28 was observed (Fig. 1B). LPL mRNA levels normalized to the levels of S28 rRNA are presented in Fig. 1C.
Effect of FAs on Mo LPL immunoreactive mass and activity.
The levels of extra- and intracellular LPL immunoreactive mass and activity, normalized to the levels of total cell proteins in FA-treated Mo, are illustrated in Fig. 2. Treatment of J774 cells for 24 h with AA, LA, PA, and SA increased extra- and intracellular LPL immunoreactive mass (Figs. 2A and B) and activity (Figs. 2C and D). In contrast, exposure of Mo to OA and EPA did not significantly alter these parameters (Figs. 2A–D). Relative content of LPL mass and activity within versus heparin released from cells remained constant following treatment of the cells by FA (data not shown).
Effect of FAs on Mo LPL synthetic rate.
To determine the effect of FAs on Mo LPL synthesis, Mo were pulse-labeled in the presence of FAs, and cell extracts were immunoprecipitated to yield the radiolabeled LPL protein. Heparin-stimulated cells and immunoprecipitation with an irrelevant antibody were used as positive and negative controls, respectively. As illustrated in Fig. 3, treatment of J774 cells with AA, LA, and PA for 24 h led to a significant increase in LPL synthetic rate (LPL synthetic rate [% over basal values]: AA 153 ± 18%, LA 168 ± 8%, and PA 142 ± 5%, P < 0.05). In contrast, EPA, OA, and SA did not affect this parameter (Fig. 3). LPL synthesis in FA-treated Mo, expressed as percentages of control values, is illustrated in Fig. 3B.
Effect of FAs on the binding of nuclear proteins to the regulatory PPRE sequence of the LPL gene promoter.
Exposure of Mo for 24 h to all FAs resulted in a dramatic increase in the binding of nuclear proteins to the PPRE consensus sequence of the LPL promoter (Figs. 4A and B). This binding was specifically competed in the presence of a 1,000-fold molar excess of the unlabeled PPRE oligonucleotide (data not shown). Whereas immunoneutralization of nuclear proteins with anti–PPAR-γ antibodies only effectively reduced AA-stimulated binding activity (Fig. 4A), incubation of nuclear extracts in presence of anti–PPAR-α antibodies decreased PA-, SA-, and OA-stimulated binding activity to the PPRE sequence (Fig. 4B). In contrast, no effect of anti–PPAR-β antibodies on FA-stimulated binding activity to the PPRE sequence was observed (data not shown).
DISCUSSION
The present study demonstrates that FAs exert a direct regulatory effect on Mo LPL expression. Key elements in the lipid signal transduction pathway include transporting of exogenous FAs into cells, metabolism of FAs to second messengers, trafficking of the messengers to the nucleus, and regulation of gene transcription via association with nucleus coactivators or repressors. A wide number of adipocyte lipid–related genes, including the adipocyte lipid binding protein (42,43), have been shown to be regulated by exogenous FAs (43,44). Our results, demonstrating that FAs exert a direct modulatory effect on Mo LPL mRNA levels, indicate that LPL is also a target gene for FAs in Mo. Regulation of Mo LPL gene expression by FAs may involve transcriptional factors such as PPARs (45) or FA-activated receptors (FAARs) (42), which both recognize the putative PPRE sequence present in the LPL gene. Alternatively, based on the well-documented stimulatory effect of FAs on protein kinase C (PKC) (46) and on the key role of PKC and c-fos in the regulation of the LPL gene (46–48), a role of c-fos in the regulation of Mo LPL gene expression by FAs may be postulated.
These results suggest that FAs, including PA, SA, and AA, might exert their regulatory effect on Mo LPL gene expression, at least partly, through a PPAR-dependent mechanism. Indeed, we found that these well-characterized ligands and activators of PPARs (49) directly regulate Mo LPL gene expression and enhance the binding activity of nuclear proteins to the PPRE element present in the LPL gene in a PPAR-specific manner. The hypothesis that PPARs may play a role in the control of Mo LPL by FAs awaits future studies aimed at evaluating the role of FAs in LPL transcription and reporter construct activity. Apart PPARs, FAARs, which also recognize the putative PPRE sequence present in the LPL gene promoter, are likely to be involved in the regulation of Mo LPL gene expression by exogenous FA. This possibility may apply to LA and EPA, which enhance the binding activity to the PPRE sequence in a PPAR-independent manner. Alternatively, a role of c-fos in the regulation of Mo LPL by FAs may be proposed. This hypothesis merits particular consideration in the case of LA and EPA, which regulate the PKC/c-fos signaling pathway (50–52) and induce parallel changes of Mo c-fos and LPL mRNA expression (S.E.M., G.R., unpublished observations).
Saturated FAs bind to PPAR-α with high affinity, and unsaturated FAs bind to all three PPAR subtypes with varying affinities. Of the unsaturated FAs tested in the present study, LA and OA show higher affinity for PPAR-α, whereas AA and EPA interact more efficiently with PPAR-γ (49). Our findings that FAs that function as PPAR-α ligands enhance Mo LPL gene expression and our observations that anti–PPAR-α decreases the enhanced binding of nuclear proteins isolated from SA-, PA-, and OA-treated Mo to the PPRE regulatory domain of the LPL gene suggest a positive relationship between PPAR-α activation and Mo LPL gene expression. Whether a PPAR signaling pathway is involved in the suppressive effect of AA and EPA on Mo LPL gene regulation is currently unclear. Indeed, despite the previous evidence for a role of PPAR-γ as a negative regulator of Mo gene expression (53,54), the net effect of PPAR-γ ligands on Mo LPL gene expression has not been studied. Although our results demonstrate that PPAR-γ binding to the PPRE sequence occurs in AA-treated Mo, this does not provide evidence for a role of this transcription factor in the EPA-induced DNA binding activity to the PPRE sequence. Apart from PPAR, various transcription factors, including FAAR (42), hepatocyte nuclear factor-4 (55), and chicken ovalbumin upstream promoter transcription factor (56), may bind to the PPRE DNA target site. Whether these transcription factors may contribute to the suppressive effect of AA and EPA on Mo LPL gene expression remains to be investigated.
LPL-producing cell types have the capacity to control this enzyme at a variety of levels (28–30). Whereas adipocyte LPL regulation in response to FAs has been proposed to take place mainly at the posttranscriptional level (30), our results suggest that Mo LPL regulation may occur at both the transcriptional and posttranscriptional level in response to FAs. Because changes in LPL mRNA levels on exposure of Mo to LA and PA are of the same size as changes in the LPL synthetic rate and secretion, it may be hypothesized that Mo LPL regulation in response to these agents occurs primarily at the transcriptional level. Although we did not perform run-on assays, data showing that nuclear proteins isolated from Mo exposed to these FAs bind to the LPL promoter PPRE sequence seem to support this possibility. In contrast, our results, demonstrating that changes in LPL gene expression following treatment of Mo with SA, AA, and EPA do not correlate with changes in LPL synthetic rate and/or secretion, indicate that these FAs regulate Mo LPL at the posttranscriptional level. Our finding that LPL synthetic rate paralleled protein activity in AA- and EPA-treated Mo indicates that translational events are involved in the effect of these FAs on Mo LPL. In contrast, our observation that LPL protein synthesis is unaffected by SA, whereas LPL secretion is increased in SA-treated Mo, reflects posttranslational control of LPL by this FA. Although the mechanisms involved in the translational control of Mo LPL by AA and EPA are unknown, it is possible that these FAsmay increase LPL translation processing by activating cytoplasmic trans-stimulating proteins that bind to the mRNA 3′ untranslated region. This possibility is supported by the observation of Kern et al. (57) that LPL translational regulation by thyroid hormone is exerted via a cytoplasmic protein interacting with the 3′ untranslated region of the LPL mRNA.
Multiple evidence indicates that dietary saturated FAs represent a risk factor for atherosclerosis (16–20). Based on the proatherogenic effects of LPL secreted by Mo in the subendothelial space, our results, demonstrating that saturated FAs increase Mo LPL production, suggest a new mechanism by which these metabolic factors may promote atherogenesis. In contrast to saturated FAs, intake of polyunsaturated n-3 FA has been associated with a lower incidence of cardiovascular events (24,58,59). n-3 FA may favorably affect the atherogenic process by reducing the secretion of proatherogenic cytokines by Mo. In accordance with our previous observation showing a reduction in LPL mRNA levels in Mo isolated from mice fed a fish oil–enriched diet (60) we found that incubation of Mo with n-3 FA for 24 h decreases Mo LPL gene expression and that prolonged incubation (48 h) of Mo with this FA reduces Mo LPL secretion (data not shown). Overall, these data suggest that a relative reduction in Mo LPL production may contribute to the cardioprotective effect of n-3 FA. Macrophages are able to use glucose, glutamine, and FAs as energy sources (61). Because complete oxidation of glucose and glutamine is limited in Mo, FA oxidation may provide, under various circumstances, a significant proportion of the energy required by these cells. In particular, these metabolic factors may constitute, in the energy-limited environment of the atherosclerotic plaque, the crucial fuel for activated Mo energy expenditure. Infiltration of triglyceride-rich lipoproteins into the arterial wall and hydrolysis of these particles by LPL are likely to release high concentrations of FAs in the subendothelial space of the vessels. Although the levels of FAs to which Mo are exposed in vivo are unknown, increased serum levels of unbound free FAs have been documented in patients undergoing coronary angioplasty (62). Whether the concentrations of unbound free FAs used in our experimental conditions reflect the levels of FAs to which macrophages are exposed in the arterial wall is currently uncertain.
Data generated by the current study suggest that production of FAs in the arterial wall may regulate the production of Mo LPL at vascular sites. Because lipoproteins isolated from diabetic patients show increased levels of saturated FAs and LA (63) and low levels of n-3 FA, infiltration of these particles in the arterial wall may favor the induction of Mo LPL that we previously reported in human type 2 diabetes (13). Enhanced production of Mo LPL in the arterial wall may contribute to the accelerated atherosclerosis associated with diabetes.
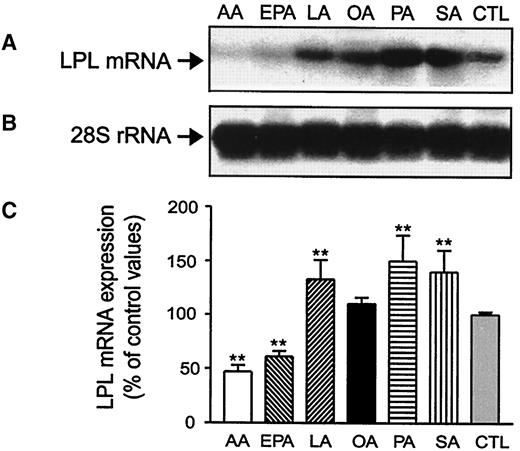
Effect of FAs on murine Mo LPL mRNA expression. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of the incubation period, cells were lysed and total RNAs were extracted and analyzed by Northern blot analysis for LPL mRNA (A) and S28 rRNA (B). C: LPL mRNA levels normalized to the levels of S28 RNA. Data are means ± SE of five experiments. **P < 0.01 vs. controls.
Effect of FAs on murine Mo LPL mRNA expression. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of the incubation period, cells were lysed and total RNAs were extracted and analyzed by Northern blot analysis for LPL mRNA (A) and S28 rRNA (B). C: LPL mRNA levels normalized to the levels of S28 RNA. Data are means ± SE of five experiments. **P < 0.01 vs. controls.
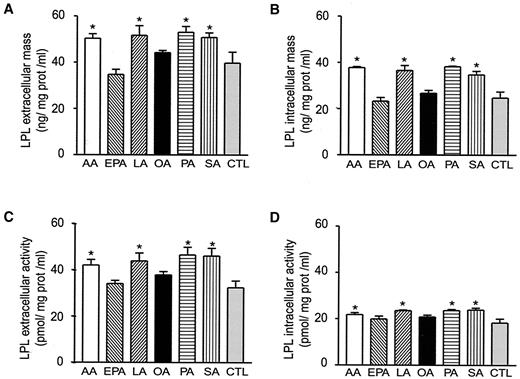
Effect of FAs on murine Mo LPL immunoreactive mass and activity. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of the incubation period, extracellular LPL immunoreactive mass (A), intracellular LPL immunoreactive mass (B), extracellular LPL activity (C), intracellular LPL activity (D) were measured in the culture medium and in the cell lysates. All values represent LPL mass and activity normalized to the levels of total cell proteins. Data are means ± SE of five experiments. *P < 0.05 vs. controls.
Effect of FAs on murine Mo LPL immunoreactive mass and activity. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of the incubation period, extracellular LPL immunoreactive mass (A), intracellular LPL immunoreactive mass (B), extracellular LPL activity (C), intracellular LPL activity (D) were measured in the culture medium and in the cell lysates. All values represent LPL mass and activity normalized to the levels of total cell proteins. Data are means ± SE of five experiments. *P < 0.05 vs. controls.
![FIG. 3. Effect of FAs on Mo LPL synthesis. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA and then incubated for 1 h in methionine-free medium. Cells were next labeled with [35S]methionine for 4 h and chased for 1 h with complete DMEM. LPL was immunoprecipitated by incubating total cytoplasmic proteins with 10 μg of the monoclonal anti-LPL antibody 5D2. A: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs or heparin (CTL+) and immunoprecipitated with the anti-LPL antibody. CTL–: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs and immunoprecipitated with the irrelevant anti-human interleukin-6 antibody. B: Graph bar showing LPL synthesis in FA-treated Mo. Data are means ± SE of three independent experiments. **P < 0.01 vs. controls.](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/50/3/10.2337_diabetes.50.3.660/2/m_db0310494003.jpeg?Expires=1716310127&Signature=EF0GdrNTTrXQk4Ibtq4DUyruk7rCkfKfhHy79IEJLK~Y-r4d-jkLgKaE0Ii4BgeVHsB8T5hiDkjnQh3sIi~Wkg1cuNKvuTUHc73gL6TrO7zY3Cr4wpMs9I4IR7QcKei5sBKjleT-afUj5XlAxZnT-6Qy8fkTsrPHLiuHWI4ANkLAyeweiDEBhaHaSRJk~QqVWmrTlE9r5YaZIOeZWY-Q3oGNAdxsnRqDhtWTStecCbrqKoCc3IiHK10i7B0wN3h74zdRC3vvvmtMUuiJ0122qO2MTpxrDSLqH22UFmzQRbhxmu-R8FdvUwW2DVljG01U1P7ucPg8OG5KAgBeQV0ykw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effect of FAs on Mo LPL synthesis. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA and then incubated for 1 h in methionine-free medium. Cells were next labeled with [35S]methionine for 4 h and chased for 1 h with complete DMEM. LPL was immunoprecipitated by incubating total cytoplasmic proteins with 10 μg of the monoclonal anti-LPL antibody 5D2. A: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs or heparin (CTL+) and immunoprecipitated with the anti-LPL antibody. CTL–: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs and immunoprecipitated with the irrelevant anti-human interleukin-6 antibody. B: Graph bar showing LPL synthesis in FA-treated Mo. Data are means ± SE of three independent experiments. **P < 0.01 vs. controls.
Effect of FAs on Mo LPL synthesis. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA and then incubated for 1 h in methionine-free medium. Cells were next labeled with [35S]methionine for 4 h and chased for 1 h with complete DMEM. LPL was immunoprecipitated by incubating total cytoplasmic proteins with 10 μg of the monoclonal anti-LPL antibody 5D2. A: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs or heparin (CTL+) and immunoprecipitated with the anti-LPL antibody. CTL–: SDS-PAGE of cytoplasmic proteins isolated from Mo treated with FAs and immunoprecipitated with the irrelevant anti-human interleukin-6 antibody. B: Graph bar showing LPL synthesis in FA-treated Mo. Data are means ± SE of three independent experiments. **P < 0.01 vs. controls.

Effect of FAs on the binding of nuclear proteins to the regulatory PPRE sequence of the LPL gene promoter. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of this incubation period, nuclear proteins were isolated from the cells and incubated with end-labeled double-stranded consensus sequences of the LPL promoter PPAR-enhancing element. In some experiments, nuclear proteins were incubated in presence of anti–PPAR-α and -γ antibodies. Retardation was assessed by gel electrophoresis in 4% PAGE. Data are the results of one representative experiment of four. A: Binding activity of nuclear proteins extracted from AA-, EPA-, and LA-treated Mo. B: Binding activity of nuclear proteins extracted from OA-, PA-, and SA-treated Mo.
Effect of FAs on the binding of nuclear proteins to the regulatory PPRE sequence of the LPL gene promoter. J774 cells were cultured for 24 h in presence of 0.2 mmol/l AA, EPA, LA, OA, PA, and SA. At the end of this incubation period, nuclear proteins were isolated from the cells and incubated with end-labeled double-stranded consensus sequences of the LPL promoter PPAR-enhancing element. In some experiments, nuclear proteins were incubated in presence of anti–PPAR-α and -γ antibodies. Retardation was assessed by gel electrophoresis in 4% PAGE. Data are the results of one representative experiment of four. A: Binding activity of nuclear proteins extracted from AA-, EPA-, and LA-treated Mo. B: Binding activity of nuclear proteins extracted from OA-, PA-, and SA-treated Mo.
Article Information
This work was supported by grants from the Medical Research Council of Canada and the Association Diabète Québec.
REFERENCES
Address correspondence and reprint requests to Dr. Geneviève Renier, Notre-Dame Hospital, Research Center, 3rd floor, y-3622, 1560 Sherbrooke St. E., Montreal, Quebec, Canada H2L 4M1. E-mail: renierg@ere.umontreal.ca.
Received for publication 27 June 2000 and accepted in revised form 7 December 2000.