


Activation of the transcription factor nuclear factor-κB (NF-κB) has been suggested to participate in chronic disorders, such as diabetes and its complications. In contrast to the short and transient activation of NF-κB in vitro, we observed a long-lasting sustained activation of NF-κB in the absence of decreased IκBα in mononuclear cells from patients with type 1 diabetes. This was associated with increased transcription of NF-κBp65. A comparable increase in NF-κBp65 antigen and mRNA was also observed in vascular endothelial cells of diabetic rats. As a mechanism, we propose that binding of ligands such as advanced glycosylation end products (AGEs), members of the S100 family, or amyloid-β peptide (Aβ) to the transmembrane receptor for AGE (RAGE) results in protein synthesis–dependent sustained activation of NF-κB both in vitro and in vivo. Infusion of AGE-albumin into mice bearing a β-globin reporter transgene under control of NF-κB also resulted in prolonged expression of the reporter transgene. In vitro studies showed that RAGE-expressing cells induced sustained translocation of NF-κB (p50/p65) from the cytoplasm into the nucleus for >1 week. Sustained NF-κB activation by ligands of RAGE was mediated by initial degradation of IκB proteins followed by new synthesis of NF-κBp65 mRNA and protein in the presence of newly synthesized IκBα and IκBβ. These data demonstrate that ligands of RAGE can induce sustained activation of NF-κB as a result of increased levels of de novo synthesized NF-κBp65 overriding endogenous negative feedback mechanisms and thus might contribute to the persistent NF-κB activation observed in hyperglycemia and possibly other chronic diseases.
Tissue culture models of cellular activation provide easily accessible systems for detailed analysis of mechanisms potentially underlying the pathogenesis of human disease. However, the time course of such in vitro models is usually significantly abbreviated, limited to hours to days, compared with the pace of disorders under study in vivo. This indicates the importance of seeking out mechanisms in cell culture that might bridge the gap that accounts for the chronicity of cellular perturbation observed in the intact organism.
The transcription factor nuclear factor-κB (NF-κB) has been proposed as a critical bridge between oxidant stress and gene expression (1,2,3,4,5,6,7,8). Exposure of cells to inflammatory, infectious, or other stressful stimuli results in rapid phosphorylation and degradation of IκBα and the subsequent release and translocation of NF-κB into the nucleus (1,2,3,4,5,6,7,8,9,10,11). This mechanism ensures quick and finely tuned cellular responses in the absence of de novo protein synthesis. Because transcription of IκBα is positively autoregulated by NF-κB (9,10,11), activation of NF-κB is usually self-terminated within minutes to hours (1,2,3,4,5,6,7,8,9,10,11). Such a scenario lends itself to analysis by short-term in vitro studies in which stimulus-induced responses are transient and the system returns to the baseline state over hours. Consequently, induction of NF-κB and enhanced transcription of its target genes in vitro have been studied mainly in the setting of acute cellular responses.
Reactive oxygen intermediates are generated by processes that occur over seconds. However, increasing evidence suggests a role for oxidative stress in chronic degenerative diseases such as atherosclerosis (1,6,12,13), diabetes (14,15,16), and Alzheimer’s disease (17,18,19). This indicates the relevance of signal transduction systems such as NF-κB, which are capable of transforming the appearance and disappearance of short-lived oxygen free radicals into more sustained signals for cellular activation (8,16). Consistent with this concept, a recent study demonstrated that hyperglycemia-dependent overproduction of mitochondrial superoxide results in NF-κB activation (16). NF-κB, however, also acts as a protective factor against programmed cell death by inducing antiapoptotic genes such as A1, A20, XIAP, Bcl-2, and BcL-XL and thus ensures cell survival in situations of perpetuated cellular activation (20,21,22,23). Therefore, more sustained protein synthesis–dependent pathways of NF-κB activation might be present in addition to immediate, self-controlled protein synthesis–independent pathways of NF-κB activation to rescue cells from cell death in the presence of a persistent stimulus.
Pathophysiologically relevant cellular perturbants associated with oxidant stress and chronic disorders are advanced glycosylation end products (AGEs) (14,16,24,25,26), S100 proteins (27), and amyloid-β peptides (Aβ) (28,29). AGEs are the result of nonenzymatic glycation and glycoxidation that accumulate during normal aging (14,24,25,26). Their deposition is enhanced with concomitant hyperglycemia, oxidant stress, carbonyl stress, and disorders associated with delayed macromolecular turnover, such as renal failure and amyloidosis (16,18,24,25,26,28,29,30,31,32,33,34,35,36,37,38,39,40). Carboxymethyllysine (CML), a rapidly formed glycoxidation product that increases in mononuclear blood cells of healthy volunteers undergoing hyperglycemic clamps already within 2 h (S.S., P.P.N., unpublished observation), is one of the earliest markers of AGE formation (36,37,38,39,40). Aβ is the primary component of the extracellular senile plaques in Alzheimer’s disease (29) and has been strongly linked to the pathogenesis of this devastating neurodegenerative disease (18,29,30). AGEs, S100 proteins, and Aβ are ligands of the receptor for AGEs (RAGE) (24,26,27,28,29,30,31,32,41,42,43,44,45,46,47,48,49). Due to their long-life nature, their ubiquitous presence in the plasma, vessel wall, and tissues, and their recognition by RAGE, whose expression levels seem to increase in the course of chronic disorders (24,29,31,32,41,42,43,44,45,46,47,48,49), we considered the hypothesis that ligand-receptor interaction may alter cellular homeostasis in a self-perpetuating manner.
RAGE is a member of the immunoglobulin superfamily of cell surface molecules (24,27,28,29,31,32,41,50) and is expressed by endothelial cells, monocytes/macrophages, neurons, and a range of other cells (50) whose dysfunction has been linked to chronic disorders such as diabetes, Alzheimer’s disease, inflammation, and atherosclerosis. Binding of ligands to RAGE triggers signal transduction mechanisms, including formation of lipid peroxides, activation of NF-κB, and subsequent expression of genes regulated by NF-κB (27,28,29,31,32,41,42,43,44,45,51,52,53).
There is increasing evidence that NF-κB activation in vivo occurs in a more sustained manner. Constitutive NF-κB activation has been observed in different malignant cells (54,55,56,57,58) as a result of a defect or dysregulation of IκBα and IκBβ or aberrant activation of IκB-kinase (58). A persistent NF-κB activation has also been suggested in atherosclerosis (13,59), Crohn’s disease (60,61,62), and Listeria monocytogenes infection (63). In vitro data have suggested that hypophosphorylated IκBβ (64,65,66,67) or de novo synthesis of c-rel can mediate longer-lasting NF-κB activation (68,69).
However, experiments performed in nonimmune and nonmalignant cells were not directed toward understanding mechanisms underlying chronic NF-κB activation in vivo, and the mechanism by which the normally short-acting response of NF-κB activation is converted to a persistent cell perturbation is not understood. RAGE was recently shown to be involved in chronic disease, such as atherosclerosis, amyloidosis, diabetes, inflammation, tumor proliferation, and metastasis (27,28,29,31,43,44,45,46,47,48,49,53). Therefore, we studied whether ligand-stimulated RAGE activation can act like a master switch to turn short-lasting activation into a prolonged activation of NF-κB.
RESEARCH DESIGN AND METHODS
Reagents.
Reagents were obtained as follows: Dulbecco’s modified Eagle’s medium (DMEM; 4,500 mg/l glucose), RPMI 1648, HEPES buffer solution, l-glutamine, penicillin-streptomycin mixture, and phosphate-buffered saline (PBS; pH 7.4) were from Biowhittaker (Walkerville, MD). Fetal calf serum (FCS) was from Gibco/BRL (Dreieich, Germany). [γ-32P]ATP (3,000 Ci/mmol at 10 mCi/ml), l-[35S]methionine (>1,000 Ci/mmol at 10 mCi/ml), Hybond-N-nylonfilter, enhanced chemiluminescence (ECL)-nitrocellulose membranes, ECL detection reagents, and Hyperfilm X-ray films were obtained from Amersham (Freiburg, Germany). Aβ peptides (1–40 and 25–35), S100B, phenylmethylsulfonyl fluoride (PMSF), actinomycin D, and cycloheximide were purchased from Sigma (Deisenhofen, Germany). Poly dI/dC and oligo(dt) was from Pharmacia (Freiburg, Germany). Anti-p65 (#sc-109), anti-IκBα (#sc-847), anti-IκBβ (#sc-945), anti–c-Jun KM-1 polyclonal antibodies, the respective second antibodies, and protein A/G PLUS-agarose were obtained from Santa Cruz (Heidelberg, Germany). Monoclonal anti-p65 antibodies, specific for active NF-κB, were obtained from Boehringer Mannheim (Mannheim, Germany). Soluble RAGE (sRAGE) preparations used throughout this study have previously been described in detail (29,31,32,45,49,70). Polyclonal anti-RAGE antibodies, generated in goat with recombinant RAGE prepared in Escherichia coli as antigen, were a gift of Dr. M.A. Shearman (Merck, Sharpe & Dome, Essex, U.K.). Anti–heme oxygenase-1 (HO-1) antibodies were from Dr. M.A. Smith (Case Western Reserve University, Cleveland, OH). Vectastain ABC kits were purchased from Vector Laboratories (Burlingame, CA). The vector NF-κB-Luc and primers for human HPRT were from Clontech (Heidelberg, Germany), and Effectene transfection reagent was obtained from Qiagen (Hilden, Germany).
Preparation of AGE-albumin, CML-albumin and Aβ peptides.
AGE-albumin was prepared by incubating bovine or mouse serum albumin with 200 mmol/l glucose-6-phosphate at 37°C for 4 to 8 weeks in 100 mmol/l phosphate (pH 7.4) and 0.5 mmol/l sodium azide. At the end of incubation, AGE-albumin preparations were dialyzed against 5 l of 100 mmol/l phosphate and 10 mmol/l EDTA for 24 h and 0.9% NaCl for 12 h. Control albumin was also dialyzed against these buffers. Alternatively, glycated bovine albumin was purchased from Sigma. Nonglycated bovine albumin, heat-inactivated AGE-bovine albumin, and bovine albumin incubated with the synthetic substrate sorbitol served as negative controls. AGE-albumin preparations were characterized by chromatographic means (see below).
Quantification of furosine, pentosidine, and other amino acids after acid hydrolysis.
AGE-albumin samples were hydrolyzed in the presence of 6 N hydrochloric acid for 23 h at 110°C; the hydrolysate was dried in vacuo on a Speedvac concentrator (Savent, Farmingdale, NY) and dissolved in 0.2N sodium citrate buffer (pH 2.2). Analysis of amino acids, including CML, was performed on an Alpha Plus amino acid analyzer (LKB Biochrom, Cambridge, U.K.), using a stainless steel column (150 × 4 mm) filled with ion-exchange resin, DC4A-spec sodium form (Benson, Reno, NV). Composition of elution buffers, ninhydrin reagent, and running conditions are described elsewhere (71,72). Injection volume was 10–80 μl. After separation on the ion-exchange column, amino acids were initially detected using either a photodiode array detector PDA 996 (Waters, Eschborn, Germany; for furosine quantification, a detection wavelength of 280 nm was used) or a fluorescence detector SFM 25 (Kontron, Eching, Germany; for pentosidine quantification, measuring was achieved at excitation/emission wavelengths of 335/385 nm), connected between the outlet of the column and the ninhydrin reaction coil. After subsequent ninhydrin derivatization, amino acids were detected at 570 and 440 nm. Protein content of the plasma samples was calculated as the sum of the amino acids. From values of furosine, fructoselysine was calculated by multiplying the molar furosine values by 3.1, the known conversion factor for the degradation of fructoselysine during acid hydrolysis (73,74). For quantification of pentosidine, reference material was synthesized according to Henle et al. (75). Furosine was obtained from Neosystem (Strasbourg, France).
Pyrraline analysis after enzymatic hydrolysis.
Enzymatic hydrolysis was performed using four enzymes (pepsin, pronase, aminopeptidase, and prolidase) as described previously (75). For pyrraline detection, amino acid analysis was performed as described above with the combination of the PDA (set at a wavelength of 297 nm) and ninhydrin derivatization (75). Alternatively, pyrraline analysis was performed via isocratic ion-pair reversed-phase high-performance liquid chromatography (HPLC) with UV detection (76). Pyrraline reference material was synthesized according to Henle and Bachmann (77).
Preparation of CML-albumin and Aβ peptides.
In vitro synthesis of CML-albumin was performed as described by Schleicher et al. (36). Assays for endotoxin showed AGE-albumin and CML preparations to contain virtually undetectable levels of lipopolysaccharide (LPS; <10 pg/ml at a protein concentration of 5 mg/ml according to the Limulus assay [Sigma]). Synthetic Aβ peptides (1–40) were purchased from Sigma and made up freshly every time. To allow fibril formation, we kept preparations at room temperature (rt) for at least 4 h before use.
Isolation of CML-modified proteins from erythrocytes.
Erythrocytes were lysed in 0.9% NaCl, 1.5 mmol/l PMSF, 0.1 mmol/l leupeptin, 20 mg/ml soybean inhibitor, and 2 mmol/l benzamidine by three freeze-thaw cycles followed by pulsed ultrasonication for 3 min and a final freeze-thaw cycle. Insoluble material was removed by a 5-min centrifugation at 6,000g at 4°C, and the supernatant was used for extraction of CML-modified proteins as previously described (70). Ten micrograms of total erythrocyte lysate was loaded onto a 1-ml anti–CML-Sepharose column (36,70) and incubated for 2 h at rt (70). After extensive washing, CML-modified proteins were eluted with 1 mol/l glycine (pH 3.0), neutralized, and dialyzed in DMEM. Protein concentration was determined using the BCA assay system (Pierce, Rockford, IL).
Plasmids.
The SV-40 driven luciferase control plasmid pGL2-control, the promoterless plasmid pGL2-basic, and the β-galactosidase (β-Gal) control plasmid pSV-β-Gal were obtained from Promega (Heidelberg, Germany). The plasmid NF-κB-Luc, which contains four tandem copies of the NF-κB consensus sequence fused to a TATA-like promoter region from the herpes simplex virus thymidine kinase promoter, was from Clontech (Heidelberg, Germany).
Transgenic mouse model.
Mice transgenic for an NF-κB–driven β-globin reporter gene (tg14) were provided by Dr. Thomas Wirth (Würzburg, Germany) and have been previously characterized in detail (78). Eight-week-old female mice were left untreated or treated once with 1,000 μg of mouse AGE-albumin (500 μg i.p. and 500 μg i.v.) at time point 0 and kept for an additional 6 days. Where indicated, mice received sRAGE (25 μg/mouse) or anti-RAGE antibodies (40 μg/mouse) as described (29,31,32). At the end of the experiments, mice were killed and organs were removed, immediately snap-frozen, and analyzed by reverse transcription–polymerase chain reaction (RT-PCR) and electrophoretic mobility shift assays (EMSAs).
Diabetic rat model.
The kidneys of diabetic rats and of nondiabetic Sprague-Dawley rats were provided by Dr. I. Köting (Karlsburg, Germany). Diabetic BB/O(ttawa)K(arlsburg) rats were bred as previously described in detail (79), developed diabetes at the age of 104 days (±16 days), and were kept for 57 days (±9 days) before harvest. During this time, rats were treated with sustained-release insulin implants (Linplant; Mollegaard, Skensved, Denmark) with an insulin rate of 2 units/24 h. With this treatment, the mean plasma glucose amounted to 136 ± 24 mg/dl.
Patient characteristics.
Blood from six patients with newly manifested diabetes type 1 (two men, four women; age 35.1 ± 11.7 years; HbA1c 12.4 ± 2.3%) was collected on the day the diagnosis was made. Four healthy volunteers (three men, one woman; age 31.5 ± 5.5 years) served as control subjects. From three of these patients, repetitive blood sampling was performed over 3 consecutive days. HbA1c levels were routinely determined by HPLC (Diamat; Biorad, Munich, Germany; normal range 4.5–6.1%) in the central laboratory unit of the hospital. The study was approved by the ethical committee of the Department of Medicine, University of Tübingen, and informed consent had been obtained from all patients studied.
Preparation of peripheral blood mononuclear cells and immunocytological detection of activated NF-κBp65.
Immediately after venipuncture, peripheral blood mononuclear cells (PBMCs) were separated as previously described in detail (80,81,82). Isolated PBMC were washed three times in cold PBS (pH 7.4, 4°C), seeded on coverslips, and fixed in 4% paraformaldehyde according to standard methods. Staining for NF-κBp65 was performed as described below using a monoclonal antibody (Boehringer Mannheim), which exclusively recognizes the activated form of NF-κB (13).
Immunocytochemistry.
Immunocytochemistry was performed by indirect immunoperoxidase techniques using PBMC fixed onto poly-l-lysine–coated glass slides with 4% paraformaldehyde; slides were washed twice in PBS (pH 7.4) for 5 min. After the last wash, slides were treated with 0.3% hydrogen peroxide, dissolved in methanol, for 30 min at rt. Blocking was performed with 0.6% normal goat or horse serum for 30 min (rt) before the affinity-purified polyclonal rabbit antibody against IκBα (concentration: 0.01 μg/μl; 60 min; rt) or the monoclonal mouse antibody for activated NF-κBp65 (concentration: 0.01 μg/μl; 60 min; rt) were used as primary antibodies. Slides were washed 3 times for 10 min in PBS (pH 7.4), and detection of signals was performed using Vectastain ABC kits (Vector Laboratories) according to the manufacturer’s instructions, as previously described in detail (61,80,81). Peroxidase activity was visualized with 0.05% 3,3-diaminobenzidine-tetrahydrochloride (Vector Laboratories) before the slides were counterstained with Mayer’s hematoxylin (Vector Laboratories). Controls for immunospecificity were included in all experiments by omission of the primary antibody and its replacement by PBS and matching concentrations of normal rabbit serum (data not shown).
RT-PCR.
RT and PCR for β-globin, NF-κBp65, hypoxanthine guanine phosphoribosyl transferase (HPRT), and β-actin were performed as described by Lernbecher et al. (78). cDNA was reverse-transcribed from 2 μg of total RNA with oligo-dT primers (Pharmacia), AMV-reverse transcriptase, and Taq-polymerase (Promega). When RT-PCR was performed from PBMC, Taq-polymerase was replaced by PFU-polymerase (Stratagene, Heidelberg, Germany). Amplification was performed using the following primer pairs: β-globin transgene forward 5′-TTTCTGATAGGCAGCCTGCACTGGT-3′, β-globin transgene reverse 5′-CATAGTTGTGTTCAGATCGATCTGG-3′; p65-forward-1 5′-GCTACAAGTGCGAGGGGC-3′, p65-reverse-1 5′-GGGGTCTGCGTAGGGAGGG-3′; p65-reverse-2 5′-GGCCTGCCTGATGGGTCCC-3; actin-forward 5′-AGAGGTATCCTGACCCTGAAGTACC-3′, actin-reverse 5′-CCACCAGACAACACTGTGTTGGCAT-3′; HPRT-forward-1 5′-GGCGTCGTGATTAGTGATGATGAACC-3′, HPRT-reverse 5′-CTTGCGACCTTGACCATCTTTGGA-3′ The following conditions were used: 1× [95°C, 360 s]; 40× [94°C, 40 s; 65°C, 60 s.; 72°C, 120 s]; 1× [72°C, 600 s] for β-globin and actin, 1× [95°C, 360 s]; 35× [94°C, 40 s; 68°C, 60 s; 72°C, 120 s]; 1× [72°C, 600 s] for NF-κBp65, 1× [95°C, 360 s]; 1× [94°C, 60 s; 52°C, 120 s; 72°C, 120 s]; 1× [94°C, 60 s; 58°C, 60 s; 72°C, 120 s]; 32× [94°C, 40 s; 62°C, 60 s; 72°C, 60 s]; 1× [72°C, 600 s] for HPRT and separated onto 2% agarose gels. Verification of PCR products was performed by reamplification with different primers recognizing sequences internal to the amplification products and by Southern blot analysis. Reactions that lacked template RNA or AMV-reverse transcriptase served as internal controls.
EMSA.
Nuclear proteins were harvested as described elsewhere (51,52,61,80,81,82,83,84) and assayed for transcription factor binding activity using the NF-κB consensus sequence 5′-AGTTGAGGGGACTTTCCCAGGC-3′. Specificity of binding was ascertained by competition with a 160-fold molar excess of unlabeled consensus oligonucleotides and by supershift experiments (data not shown).
Immunoblot (Western blot) analysis.
Cytoplasmic and nuclear fractions were prepared as previously described in detail (51,52,61,84). Twenty micrograms of cytoplasmic extracts or 10 μg of nuclear extracts was separated onto 10–12% SDS-PAGE, followed by electroblotting to ECL-nitrocellulose membranes. Membranes were incubated with primary antibodies for NF-κBp65, IκBα, and IκBβ (1:100) for 60 min at rt. After washing (2 × 7 min in Tris-buffered saline [TBS], 0.05% Tween), the secondary antibody (horseradish peroxidase–coupled rabbit IgG, 1:2000) was added, and incubation was continued for 30 min at rt. Membranes were washed 3× 5 min as above followed by a last 5-min wash in TBS. Immunoreactive proteins were detected with the ECL-Western blot system and subsequent autoradiography for 2 min.
In vitro phosphorylation of IκBα.
For in vitro phosphorylation, 5 × 106 bovine aortic endothelial cells (BAECs) were incubated in the presence of 500 nmol/l AGE-albumin for the times indicated in the figure legends. At the end of incubation, cells were washed 3 times in 0.9% NaCl, harvested by scraping, collected by a 5-min centrifugation at 500g, 4°C, and resuspended in 1 ml of 0.9% NaCl. Preparations were counted in a Neugebauer chamber and adjusted for the same number of cells, collected by centrifugation as above, and resuspended in 500 μl of 0.9% NaCl. Whole-cell lysate was obtained by mechanical disruption with an Ultrathurrax (Wheaton, Milleville, NJ), cleared by centrifugation at 10,000g at 4°C for 5 min, and immediately supplemented with 5 μl of 100 mmol/l PMSF (80). In vitro phosphorylation of a 70-kDa IκBα-GST fusion protein (Santa Cruz) was performed for 30 min at 37°C in 35 mmol/l Tris (pH 7.5), 15 mmol/l MgCl2, 1 mmol/l MnCl2, 2 μl of 100 mmol/l rATP, and 1 μl of γ-32P-ATP (10 μCi) with 15 μl of the above whole-cell extract (85). The reaction was completed by the addition of 180 μl of IP buffer (20 mmol/l Tris [pH 7.5], 150 mmol/l NaCl, 5 mmol/l EDTA, 0.2% SDS, 1% Triton-X100, 0.1 mmol/l PMSF, 1 unit/ml aprotinin) supplemented with 1 mmol/l sodium orthovanadate (85). After addition of 2 μl of IκBα (FL) antibody and 10 μl of protein A/G Sepharose, incubation was continued for 16 h at 4°C. At the end of incubation, precipitates were collected by centrifugation at 10,000g for 10 min at 4°C; washed twice in IP buffer followed by a final wash in 150 mmol/l NaCl, 50 mmol/l Tris-HCl (pH 7.5), and 5 mmol/l EDTA; and then resuspended in 40 μl of denaturing buffer (125 mmol/l Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 10% 2-mercaptoethanol) (86). The immunoprecipitate was analyzed by SDS-PAGE and autoradiography (85,86).
Intrinsic labeling and immunoprecipitation of NF-κBp65.
BAEC in 35-mm2 dishes that had been confluent for 3 days were stimulated with AGE-albumin (500 nmol/l) for the times given in the figure legends. Four hours before harvest, the medium was changed to methionine-free medium for 1 h. Thereafter, the medium was changed again to DMEM supplemented with 7 μCi/ml l-[35S]methionine. Where indicated, BAECs were cultivated in the presence of 0.1 μmol/l antisense-p65 oligonucleotides, added 24 h before harvest. At the end of incubation, BAECs were lysed in 400 μl of IP buffer (20 mmol/l Tris [pH 7.5], 150 mmol/l NaCl, 5 mmol/l EDTA, 0.2% SDS, 1% Triton-X100, 0.1 mmol/l PMSF, and 1 unit/ml aprotinin) for 20 min at 4°C, before lysates were cleared by centrifugation (10,000g, 10 min, 4°C) (86). The protein concentration in the supernatant was determined according to Bradford. Total protein lysate (200 μg) was precleared by adding 1 μg of an irrelevant monoclonal mouse antibody (c-jun KM; Santa Cruz) and 20 μl of protein A/G PLUS-agarose. The reaction was incubated at 4°C for 30 min and centrifuged for 5 min at 10,000g, 4°C. The supernatant was incubated with 2 μg of a polyclonal anti-p65 antibody (Santa Cruz) for 1 h at 4°C before 20 μl agarose conjugate was added and incubation was continued for 16 h at 4°C. At the end of incubation, precipitates were collected by centrifugation at 10,000g for 10 min at 4°C; washed three times in IP buffer, followed by a last wash in 150 mmol/l NaCl, 50 mmol/l Tris-HCl (pH 7.5), and 5 mmol/l EDTA, and then resuspended in 40 μl of denaturing buffer (125 mmol/l Tris-HCl [pH 6.8], 4% SDS, 20% glycerol, 10% β-mercaptoethanol) (86). Four microliters were counted in a β-counter to determine incorporation of l-[35S]methionine into each sample. Determination was repeated twice. The remaining 32 μl was subjected to SDS-gel electrophoresis and subsequent autoradiography to confirm the incorporation data.
Tissue culture and transient transfection experiments.
BAEC were cultured as previously described (83,87). Bovine arterial smooth muscle cells (SMCs) prepared from bovine aorta were cultured in DMEM containing 10% FCS (88). hT neurons (Stratagene), derived from Ntera/D1(NT2) precursor cells (Stratagene), were cultured according to the manufacturer’s instructions. THP-1 monocytic cells were cultured in RPMI 1648 medium supplemented with 10% FCS. BAECs and SMCs were cultured to confluence and kept so for 3 days in the respective medium containing 10% FCS before the experiments were started. Experiments with hT-neurons were started 1 day after the cells had reached confluence. THP-1 cells were seeded 24 h before the test protein was added. All experiments were performed without serum deprivation before stimulation.
For transfection experiments, BAECs growing in the logarithmic phase were transfected with an NF-κB–driven luciferase reporter gene (Clontech) using the Effectene Transfection Reagent (Qiagen) according to the manufacturer’s instructions by adding 0.4 μg of NF-κB DNA for each well at a concentration of 0.2 μg/ml of culture medium. After applying the DNA to the cells, cells were cultivated for 1 h before stimulation by AGE-albumin (500 nmol/l) or Aβ (1 μmol/l) was performed for 48 h. To correct for efficiency in transfection, we included 0.05 μg of pSV-β-Gal plasmid/ml medium (83). The ratio of luciferase activity to β-gal activity served to normalize luciferase activity (83). Each experiment was performed in triplicate.
Antisense-phosphorothioate oligodeoxynucleotides.
Antisense-RAGE-phosphorothioate oligodeoxynucleotides ([PS]ODNs) (5′-G-[PTO]-ACCACTGCCCCTGCTGC-3′); sense-RAGE-[PS]ODNs-(5′-G-[PTO]-CAGCAGGGGCAGTGGTC-3′), antisense-NF-κBp65-[PS]ODNs (5′-G-[PTO]-GGGAACAG-[PTO]-TTCGTCCATGG-[PTO]-C-3), scrambled-antisense-NF-κBp65-[PS]ODNs (5′-A-[PTO]-GTCTGGAG-[PTO]-CCATCGAGGTG-[PTO]-C-3), and sense-NF-κBp65-[PS]ODNs (5′-G-[PTO]-CCATGGACGAA-[PTO]-CTGTTCCC-[PTO]-C-3′) derived from the published DNA sequences for bovine RAGE and human NF-κBp65 were synthesized and purified by HPLC (MWG-Biotech, Ebersberg, Germany). [PS]ODNs were chemically modified by introducing phosphorothioate linkages in which a nonbridging phosphate oxygen atom was substituted with a sulfur atom to protect oligonucleotides from serum- and nuclease-mediated degradation, as previously described in detail (52). Uptake of ODNs by the cells occurred passively without any further pretreatment of the cells (52).
Statistical analysis.
All values are given as mean, with the bars showing standard deviations. The means of groups were compared by analysis of variance using the Student’s t test to correct for multiple comparisons. P < 0.05 was considered to be statistically significant.
RESULTS
Sustained NF-κB activation in vivo.
Hyperglycemia promotes leukocyte adhesion to the endothelium at least in part through NF-κB–dependent upregulation of adhesion molecules (1,6,89,90). This implicates a hyperglycemia-associated continuous activation of NF-κB in both leukocytes and endothelial cells. Consistently, the presence of activated NF-κBp65 in PBMCs isolated from diabetic patients has been reported to depend on the state of glycemic control (80). When PBMCs were isolated from newly manifested diabetic patients with poor glycemic control (HbA1c >13), almost all PBMCs were recognized by a monoclonal antibody that exclusively binds to activated NF-κBp65 (Fig. 1A, left) (13). Activated NF-κBp65 was localized within the cells and more pronounced in the nuclear region (Fig. 1A, left, insert), thereby indicating that most of the activated NF-κBp65 was present in the nucleus. In contrast, minor staining in much fewer cells, indicating only a little activated NF-κBp65, was observed in healthy control subjects (Fig. 1A, right). In these cells, staining for activated NF-κBp65 was located outside the nucleus in the cytoplasm, close to the outer cell membrane (Fig. 1A, right, insert). Repetitive sampling of PBMC from three patients with poor metabolic control also showed activated NF-κBp65 in almost all cells over 3 days (Fig. 1C). In contrast, no difference was found when PBMCs of the above diabetic patients and healthy control subjects were analyzed for full-length IκBα (Figs. 1B and C). An average of 86% PBMCs derived from diabetic patients were positive for IκBα, similar to the 87% found in healthy control subjects (Fig. 1C). Consequently, the sustained NF-κBp65 activation observed in PBMCs of diabetic patients (Figs. 1A and C) cannot be explained by a loss of IκBα. Therefore, pathways of sustained NF-κB activation must be present in vivo, overriding the IκBα-dependent autoregulatory loop, and allow activation of NF-κBp65 in the presence of substantial amounts of IκBα. This hypothesis is consistent with an upregulation of NF-κBp65 mRNA in PBMCs of diabetic patients: When total RNA was isolated from blood samples at the same time PBMCs were prepared and subjected to RT-PCR for NF-κBp65, NF-κBp65 mRNA was markedly increased in PBMC of patients with diabetes compared with PBMCs of healthy control subjects (Fig. 1D). RT-PCR for HPRT served as an internal control and confirmed comparable RNA input in each reaction (Fig. 1D).
Endothelial cells cannot be obtained from patients with diabetes. To define whether a comparable NF-κB activation associated with severe diabetes can also be observed in endothelial cells, we studied NF-κB activation in kidneys of diabetic BB/OK rats (79), compared to kidneys of age-matched euglycemic Sprague Dawley-rats. The BB/OK rats manifested diabetes at the age of 104 ± 16 days, and the duration of diabetes was 57 ± 9 days, sufficient to allow significant AGE formation (91,92). A total of nine diabetic kidneys and five control kidneys were subjected to immunocytochemistry, with identical results (Fig. 2). Using a polyclonal rabbit-derived antibody recognizing NF-κBp65, antigen-pronounced cytoplasmic and nuclear immunoreactive NF-κBp65 was observed in the renal vascular endothelium of diabetic BB/OK rats (Fig. 2A, left, arrowhead), whereas vessels in nondiabetic control kidneys demonstrated only minor staining for NF-κBp65 (Fig. 2A, right, arrowhead). When immunocytochemistry was repeated on consecutive sections of diabetic kidneys using the monoclonal antibody for activated NF-κBp65, pronounced staining was found in large blood vessel endothelia and close to small blood vessels (Fig. 2B). In almost all of the endothelial cells, NF-κBp65 staining was visible in the cytoplasm and within the nuclear region (Fig. 2B, arrowheads). If a self-regulated, transient, protein synthesis–independent NF-κB activation were the only pathway present in vivo, however, one would not expect all endothelial cells (Fig. 2B) or all PBMCs (Figs. 1A and D) to react positively. This implicates that protein synthesis–dependent pathways of sustained NF-κB activation may exist.
RAGE-mediated sustained activation of NF-κB.
Incubation of BAECs with tumor necrosis factor-α (TNF-α; 1 nmol/l) induces only a transient activation of NF-κB, lasting ∼1 h (83 and data not shown), whereas exposure of BAECs to AGE-albumin (Fig. 3A) (or in vitro synthesized carboxymethyllysine-modified albumin [see below]) or Aβ peptide (Fig. 3B) induced an NF-κB activation that lasted for >6 days. Similar prolonged activation of NF-κB was observed with several different AGE-albumin preparations, in which 36% of lysine residues were modified as described in Table 1. Incubation with similar molar concentrations of control albumin (Fig. 3C), heat-inactivated AGE-albumin (data not shown), or a scrambled version of Aβ (Fig. 3D), which does not bind to RAGE (29), did not result in activation of NF-κB, excluding a general protein overload (93) or a nonspecific membrane effect as the cause for the observed persistent NF-κB activation. Furthermore, incubation of BAECs with AGE-albumin (or Aβ) for 1 day followed by washing and removal of the ligands resulted in NF-κB activation still present 3 days later (data not shown). Therefore, a transient ligand-receptor interaction rather than restimulation or carryover of an excess of ligands triggers sustained NF-κB activation. Prolonged NF-κB activation was not restricted to endothelial cells but was also observed in neuronal cells (Fig. 3E), SMCs (Fig. 3F), and THP-1 monocytic cells (Fig. 3G). In all experiments, NF-κB binding activity was specifically competed by a 160-fold molar excess of the unlabeled NF-κB oligonucleotides (see Fig. 3, last lane in each gel) but not by nonspecific or mutated NF-κB oligonucleotides (data not shown).
AGE-albumin–induced NF-κB binding activity (Fig. 4A, lane 2) was suppressed by the addition of an excess of sRAGE (Fig. 4A, lane 3), a truncated form of the receptor (27,29,31,32,45,53,70), and thereby implicated that engagement of RAGE is central for activation of NF-κB. However, sRAGE might also act by inhibition of RAGE dimerization or by competing for ligand binding to not-yet-identified non-RAGE receptors. To prove directly the involvement of RAGE in AGE-albumin–induced sustained NF-κB activation, BAECs were preincubated with antisense oligonucleotides directed against RAGE, previously shown to suppress RAGE expression (52). Sustained NF-κB activation was reduced (but not entirely blocked) in the presence of antisense RAGE oligonucleotides (Fig. 4A, lane 4), whereas oligonucleotides in the sense orientation had no effect (data not shown) and thus confirmed the involvement of RAGE. Because not all cells uniformly internalized the oligonucleotides, the effect of antisense RAGE was only partial, whereas sRAGE and neutralizing anti-RAGE antibodies (data not shown) blocked all cell surface interactions with RAGE.
To prove that the effects observed with AGEs synthesized in vitro can be compared with natural RAGE ligands that occur in vivo, we isolated CML-modified proteins (36,37,38,39,40,44,70) from erythrocytes from the above patients with diabetes, as previously described, and assayed for NF-κB activating activity (70). Consistent with CML being currently recognized as a major AGE rapidly formed within a short time in the presence of oxidative and carbonyl stress (36,37,38,39,40), CML-modified proteins were already present at diabetes onset and could be isolated. A comparable induction of NF-κB binding activity was detected when BAECs were stimulated for 5 days either with different ligands, such as Aβ (1 μmol/l; Fig. 3B) and S100B (400 nmol/l; Fig. 4B), or with 400 nmol/l CML-modified proteins (Fig. 4C; lanes 2, 4, and 6) as in vitro synthesized, highly modified CML-albumin (36) (Fig. 4C, lane 2), 400 nmol/l patient-derived CML-modified proteins (Fig. 4C, lane 4), or 400 nmol/l in vitro synthesized minimally modified CML-albumin (36) (Fig. 4C, lane 6). In contrast, CML-modified hippuric acid (Fig. 4C, lane 8) did not result in significant induction of NF-κB binding activity. Consistent with a recent report (44), CML-mediated NF-κB activation was dependent on RAGE, because blockade of RAGE with a threefold molar excess of sRAGE resulted in reduced NF-κB binding activity (Fig. 4C, lanes 3, 5, 7, and 9).
The observations in cell culture led us to predict that AGE/RAGE-dependent NF-κB activation would also occur in vivo. Mice transgenic for an NF-κB–controlled β-globin transgene (78) were used. Under physiological conditions, constitutive expression of the transgene is restricted to lymphoid tissues, in which its expression is driven by constitutively present NF-κB(p50/c-Rel) heterodimers (78). Activation of the p50/p65 heterodimer, however, can confer inducible transgene activation in all cell types (78). These mice received a single administration of 1,000 μg of AGE-albumin (500 μg i.v., 500 μg i.p.) or 1,000 μg of control albumin (500 μg i.v., 500 μg i.p.). Tissue was harvested 6 days later. The AGE concentration used was in the range of AGE serum levels determined in diabetic patients with end-stage renal disease (94,95,96,97,98). As a result of degradation and urinary AGE clearance (25,96,97,98), AGE concentrations present in these mice after 6 days probably gives rise to mouse serum levels even below AGE serum levels found in diabetic patients with good glycemic control (40,94,95,96). Total RNA was isolated from kidneys and subjected to RT-PCR for the β-globin reporter gene (Fig. 5A). RNA isolated from the spleen served as control and demonstrated constitutive β-globin transcription in all animals (data not shown). Despite the presence of endogenous AGE-degrading systems (25,97,98), an AGE-dependent induction of the NF-κB–driven β-globin transcription was still observed after 6 days (Fig. 5A, lane 2) and paralleled by an increase in NF-κBp65 mRNA (data not shown). No signal could be seen in mice that were infused with control albumin (Fig. 5A, lane 1) or in untreated control animals (data not shown). The strong β-globin transgene signal in mice that were treated with AGE-albumin (Fig. 5A, lane 2) was reduced when mice were treated with anti-RAGE IgGs (40 μg/mouse i.v; Fig. 5A, lane 3) or an excess of sRAGE (25 μg/mouse i.v.; Fig. 5A, lane 4) at days 0 and 3 after stimulation. RT-PCR for actin served as an internal control and confirmed comparable RNA input in each reaction (Fig. 5A, bottom). In-parallel–performed EMSA with whole-cell extracts from these kidneys demonstrated prominent NF-κB binding activity only in mice that had received AGE-albumin (Fig. 5B, lane 2). NF-κB binding activity was significantly reduced in the kidney nuclear extracts of mice that had received anti-RAGE IgGs (40 μg/mouse i.v.; Fig. 5B, lane 3) or sRAGE (25 μg/mouse i.v.) (Fig. 5B, lane 4). Inhibition of β-globin expression (Fig. 5A) and NF-κB binding activity (Fig. 5B) by RAGE antibodies, however, was lower than the inhibition observed in the presence of sRAGE. Because sRAGE competes for AGE binding with all cellular AGE receptors, whereas blocking RAGE antibodies specifically inhibit ligation to RAGE, this implicates that other AGE receptors (25,43,99) or not-yet-identified RAGE-like receptors might also contribute to AGE-mediated sustained NF-κB, though to a much lesser extent than RAGE. RAGE antibodies inhibit >80% of the AGE-dependent NF-κB activation, thereby indicating that RAGE is the predominant receptor in mediating sustained NF-κB activation.
AGE- and Aβ-induced sustained NF-κB activation mediating sustained gene expression.
The functional significance of RAGE-dependent continuous NF-κB activation was demonstrated in transient transfection studies in which BAECs were transfected with a luciferase expression plasmid controlled by four tandem copies of the NF-κB consensus sequence. Luciferase expression in BAECs stimulated with unmodified control albumin served as control and was regarded as baseline expression (Fig. 6A, column 1). Whereas longer incubation with nonglycated control albumin (500 nmol/l) did not change gene expression (Fig. 6A, column 2), stimulation with AGE-albumin (500 nmol/l) induced NF-κB–dependent gene expression over time, increasing to the highest levels 48 h after stimulation (Fig. 6A, columns 3 and 4). A similar increase in luciferase expression was observed in cells stimulated with Aβ (1 μmol/l) but not in BAECs incubated with scrambled Aβ (1 μmol/l) (Fig. 6A, columns 5–8). Because of the experimental limitations of transient transfection studies, expression of the luciferase reporter gene could not be followed for >48 h. However, immunoblot analysis for the NF-κB–controlled HO-1 (100) demonstrated that HO-1 expression remained elevated over 5 days (Fig. 6B). A similar increase was observed with other NF-κB–regulated genes, such as the procoagulant tissue factor (101 and data not shown) or the NF-κB–autoregulated inhibitor IκBα (see below).
AGE and Aβ induce increased nuclear translocation of NF-κBp65.
To define the NF-κB proteins induced by AGE-albumin and Aβ in cultured endothelial cells, we performed supershift experiments using antibodies specific for NF-κBp65 and NF-κBp50, and the supershifted bands were quantified by densitometry. In unstimulated cells, 90% of the densitometrically measured binding activity was due to NF-κBp50, whereas NF-κBp65 contributed only 10% (Fig. 7A). These amounts changed after 24 h as a result of a relative increase of NF-κBp65 compared to NF-κBp50 (Figs. 7A and B). The increased contribution of NF-κBp65 to the DNA binding complex continued over time: 3 days after AGE-albumin (Figs. 7A and B) or Aβ induction (data not shown), NF-κB anti-p50 and anti-p65 both yielded prominent supershifts. At later time points, however, the portion of NF-κBp50 decreased, whereas only the contribution of NF-κBp65 to the observed shift further increased, reaching a maximum 5 days after the initial stimulus and being still evident at day 6 (Fig. 7B). These data implicate an increased availability or stability of nuclear NF-κBp65 at later time points.
Immunoblot analysis confirmed an increase in nuclear NF-κBp65 after prolonged induction by AGE-albumin (Fig. 8A). Despite persistent high nuclear NF-κBp65 levels, we did not observe a loss of NF-κBp65 immunoreactivity in the cytoplasm (Fig. 8B). The increase of NF-κBp65 antigen observed after 3 days in the cytoplasm (Fig. 8B) correlated with an increase in IκBα (Fig. 8C) and IκBβ (Fig. 8D) and also with the observation in diabetic patients (Fig. 1). A decrease of cytoplasmic IκBα and IκBβ was evident only within the first 48 h of AGE-albumin induction (Figs. 8C and D). The reappearance of IκBα and IκBβ and the simultaneous increase in nuclear and cytoplasmic NF-κBp65 suggest that mechanisms such as de novo protein synthesis of NF-κB might be operative in the maintenance of sustained NF-κB activity. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown).
Because degradation of IκBα also depends on its phosphorylation state, recombinant IκBα-GST fusion protein was coincubated with cell extracts from control or AGE-albumin–treated BAECs in the presence of [γ-32P]ATP. Increased phosphorylation of IκBα was evident between 24 and 48 h (Fig. 9), demonstrating that AGE-albumin stimulates a kinase activity able to phosphorylate IκBα within the first 48 h of induction, which corresponds to the loss of IκBα during this time (Fig. 8C). Although this implicates that phosphorylation might tag IκBα for subsequent degradation, it cannot be excluded that phosphorylation might also have other effects.
AGE- and Aβ-induced sustained NF-κB activation is dependent on newly synthesized NF-κBp65.
Twelve hours after AGE-albumin stimulation of BAECs, RT-PCR analysis demonstrated an increase in NF-κBp65 mRNA (Fig. 10A, left), occurring parallel to the increase in NF-κBp65 protein observed in immunoblot analysis (Figs. 8A and B). This time span corresponds to the increased NF-κBp65 mRNA observed in diabetic patients (Fig. 1D). mRNA levels reached a maximum between 72 h and 5 days and slightly decreased after 6 days (Fig. 10A, left). Control albumin did not induce NF-κBp65 mRNA over time (Fig. 10A, right). Intrinsic labeling of BAECs with l-[35S]methionine was performed to prove whether de novo protein synthesis of NF-κBp65 also occurred in response to RAGE-mediated cell stimulation (Fig. 10B). Subsequent immunoprecipitation of the labeled proteins with antibodies to NF-κBp65 demonstrated an AGE-albumin–induced increase in anti–NF-κBp65–precipitated material, reaching a maximum 72 h after stimulation (Fig. 10B). Consistent with the results in EMSA (Fig. 3), immunoblot (Figs. 8A and B), and RT-PCR (Fig. 10A), the amount of NF-κBp65–precipitated material decreased at day 6 but was still threefold higher than in unstimulated control cells (Fig. 10B, left). No significant induction of NF-κBp65–precipitated material was observed (Fig. 10b, right) when BAECs were preincubated in the presence of antisense NF-κBp65 oligonucleotides before induction with AGE-albumin. Therefore, de novo NF-κBp65 transcription and expression seem to be an important step in maintaining sustained NF-κB activation.
AGE-albumin–dependent NF-κB activation was reduced at time points later than 72 h but not at the earlier time points (EMSA) (Fig. 11) as a result of blocking mRNA synthesis by pulsing BAECs for 3 h with actinomycin D (15 μg/ml) before harvest. Pulsing for 3 h with cycloheximide (1 μg/μl) reduced NF-κB activation only after ≥3 days (data not shown), further confirming that sustained NF-κB activation is dependent, at least in part, on de novo mRNA synthesis. However, these reagents uniformly suppress NF-κBp65 and IκBβ synthesis, whereas the latter has been demonstrated to contribute to sustained NF-κB activation (64,65,66,67).
Therefore, we applied oligonucleotides directed against NF-κBp65 to prove directly the contribution of newly synthesized NF-κBp65 to the observed NF-κB binding activity. Applied 24 h before harvest, antisense NF-κBp65 oligonucleotides reduced AGE-albumin–dependent NF-κB activation at time points later than 24 h of AGE-albumin stimulation (EMSA) (Fig. 12A, top, lanes 4–8) but not earlier (Fig. 12A, top, lanes 2 and 3). Sense (EMSA) (Fig. 12A, bottom) or scrambled oligonucleotides (data not shown) had no effect. Specificity of oligonucleotides was confirmed in immunoblot experiments, which demonstrated reduction of AGE-albumin–induced NF-κBp65 expression, but not of NF-κBc-Rel expression in the presence of antisense p65 oligonucleotides (data not shown).
Consistently, AGE-albumin–induced NF-κB binding activity and NF-κBp65 mRNA were not affected by sense p65 oligonucleotides (Fig. 12B, left, lane 2), whereas upregulation of NF-κB binding activity and NF-κBp65 mRNA was significantly reduced in the presence of antisense p65 oligonucleotides (Fig. 12B, left, lane 3). In contrast, NF-κB binding activity induced by 3 h of stimulation with 1 nmol/l TNFα was not reduced in the presence of antisense p65 oligonucleotides (Fig. 12B, right, lane 6), and RT-PCR from BAECs stimulated with TNFα (1 nmol/l, 3 h) in the presence of sense (Fig. 12B, right, lane 5) or antisense p65 oligonucleotides (Fig. 12B, right, lane 6) failed to detect NF-κBp65 mRNA (Fig. 12B, right, lane 5).
DISCUSSION
The NF-κB system triggers a self-terminating acute-phase response in situations in which a rapid protein synthesis–independent activation of defense mechanisms is critical for survival (1,2,3,4,5,6,7,8,9,10,11). Recent studies, however, describe the presence of activated NF-κB in injured arteries (102), atherosclerotic plaques (6,13), Crohn’s disease bowel tissue (61,62,63,103), and circulating mononuclear cells of patients with septicemia (82) and of diabetic patients with a new onset of diabetes or a long history of diabetes (Fig. 1) (80,81). Under these conditions, NF-κB seems to be activated in almost all cells, indicating the possibility that NF-κB activation also occurs over longer time periods in vivo. Here we show that sustained NF-κB activation in the absence of significantly decreased IκBα expression is associated with an increase in NF-κBp65 mRNA in PBMCs of patients with type 1 diabetes (Fig. 1) and in the renal endothelium of diabetic rats (Fig. 2). In vitro studies show that ligand-RAGE interaction results in sustained NF-κB activation dependent on de novo synthesis of NF-κBp65 in all cell types tested.
To our knowledge, RAGE-mediated sustained NF-κB activation provides, for the first time, evidence that prolonged NF-κB activation, which is dependent on newly synthesized NF-κBp65 mRNA and results in perpetuated NF-κB–dependent gene expression, occurs as a cellular response to a nonviral stimulus. Recently, upregulation of NF-κBp65 mRNA was demonstrated in hypoxia-exposed oligodendrocytes (104); however, no detectable expression of NF-κB target genes was observed in this model (104). Consistent with the in vitro data, the in vivo experiments presented show that NF-κB–dependent β-globin reporter gene expression is elevated for 6 days in mice treated with a single dose of AGE-albumin (Fig. 5). Furthermore, the animal model supports the hypothesis that RAGE is central for sustained NF-κB activation in vivo (Fig. 5). This animal model does not, however, allow us to define whether the observed increase in NF-κBp65– and NF-κB–driven gene expression results from cells in which NF-κB activation is sustained or from cells that have been recently activated by primary or secondary activation through AGE-albumin–induced cytokines or immune modulators. Reactive oxygen species (ROS) produced by the mitochondrial respiratory chain have been described as one major mediator of hyperglycemia-dependent NF-κB activation (16). Although monocytes do not contain mitochondria, ligation to RAGE results in antioxidant-inhibitable NF-κB activation (data not shown). Therefore, additional routes of ROS production also seem to be involved. A recent study demonstrated that upon AGE engagement of RAGE, NADPH-oxidase is one major source of ROS in endothelial cells (105) and thus implicates that multiple sources of oxidative stress contribute to RAGE-mediated NF-κB activation (43,105). Thioredoxin, which is rapidly activated by oxidative stress, has been demonstrated to activate NF-κB by c-Jun NH2-terminal kinase–mediated redox-dependent degradation of IκBα in human pulmonary artery endothelial cells (106). Because oxidative stress also induces nuclear translocation of thioredoxin, thioredoxin might further cooperate with redox-factor 1 to facilitate the DNA binding of NF-κB, which requires reduced cysteines in the NF-κBp65 subunit (106,107). In addition, ligands of RAGE might induce NF-κB coactivators, such as CBP/p300 or HMGY (108), that support and enhance sustained NF-κB activation.
One pathway of RAGE-dependent cellular perturbation includes oxidative stress–mediated activation of p21ras and subsequent activation of mitogen-activated protein kinases, encoded by the extracellular signal-regulated kinases (ERK) 1 and 2 (43,109). A recent study in SMCs demonstrated that ERK-1 and ERK-2 promote NF-κB activity independent of IκBα degradation, most probably by modulating NF-κB binding to the DNA (110). Additional studies are required to elucidate whether ligands of RAGE also modulate NF-κB DNA recognition. Besides ERK signal transduction pathways, phosphorylation of IκBα at tyrosine residue 42 has been demonstrated to mediate NF-κB translocation in the absence of IκBα degradation (111). Although preliminary data in our laboratory indicate that AGE-albumin stimulation increases phosphotyrosine modifications on IκBα and IκBβ in vitro (A.B., P.P.N., unpublished observations), we do not know whether these modifications also contribute to RAGE-mediated sustained NF-κB activation. The time course of the AGE-induced phosphorylating activity (Fig. 9) implicates that phosphorylation might tag IκBα for subsequent degradation. However, we did not characterize the locus of AGE-dependent IκBα phosphorylation and thus do not know whether phosphorylation occurs at serine residues, thereby initiating subsequent IκBα degradation (4,7,9,10,11), or at tyrosine residues, thereby promoting NF-κB translocation in the absence of IκBα degradation (111). Future studies will address this issue. New synthesis of IκBβ is evident at time points later than 3 days (Fig. 8D). Newly synthesized IκBβ has been shown to complex with NF-κB without inhibiting its nuclear translocation or transactivation capacity (64,65,66,67). Therefore, it is likely that the newly generated IκBβ prevents NF-κB from being trapped by newly synthesized IκBα and thus might also contribute to RAGE-dependent sustained NF-κB activation. However, additional studies are required to evaluate the role of IκBβ in mediating sustained NF-κB activation induced by ligands of RAGE and to define the extent of sustained NF-κB activation in the absence of newly synthesized IκBβ.
From the data presented here, we conclude that RAGE-dependent sustained NF-κB activation occurs in two phases. In the first phase, lasting up to 48 h, free NF-κBp65 protein translocates to the nucleus as a result of stimulus-induced degradation of IκBα and IκBβ. This phase is independent of mRNA and protein synthesis, because neither actinomycin D nor cycloheximide or antisense NF-κBp65 oligonucleotides blocked NF-κB activation (Figs. 11 and 12). Because IκBα expression itself is controlled by NF-κB (9,11), the increase in nuclear NF-κBp65 results in elevated IκBα gene transcription and expression, which replenishes the pool of cytoplasmic IκBα at time points later than 48 h (Fig. 8C). The second phase of NF-κB activation occurs at time points later than 48 h and is associated with elevated NF-κBp65 mRNA levels, as observed in AGE-albumin–stimulated cells (Figs. 10 and 12), AGE-albumin–treated mice (data not shown), and diabetic patients (Fig. 1D). Inhibition of mRNA and/or protein synthesis by actinomycin D, cycloheximide, or antisense NF-κBp65 oligonucleotides results in loss of sustained NF-κB activation (Figs. 11 and 12), thereby confirming that new synthesis rather than stabilization of NF-κBp65 mRNA is critical for perpetuating and maintaining NF-κB activation. New synthesis of NF-κBp65 results in a constantly growing pool of free NF-κBp65 in vitro and in vivo, which provides newly generated transcriptionally active NF-κBp65 that can override the NF-κB–dependent IκBα-autoregulatory loop. Thus, delineation of mechanisms underlying increased NF-κBp65 transcription and expression upon engagement of RAGE might be crucial to understanding sustained NF-κB activation. The hypothesis that newly synthesized NF-κBp65 plays an essential role in sustained NF-κB activation is further supported by studies of human cytomegalovirus infections (112), in which persistent NF-κB activation, critical to the viral life cycle, is also mediated through induction of NF-κBp65 mRNA (112).
The RAGE promoter itself contains two functional NF-κB sites (113), and RAGE expression has been demonstrated to be induced in the presence of AGE-albumin (52,113). Therefore, it seems likely that the sustained NF-κB activation described here results in elevated RAGE expression and, in turn, upregulation of the receptor ensures that sustained NF-κB activation is not only maintained but also amplified. Therefore, the data suggest a link between acute activation of NF-κB observed in response to many stimuli, such as LPS and cytokines and sustained activation of NF-κB observed in vivo in chronic disorders. The nature of such mechanisms may be critical in settings where endogenous negative feedback pathways, those responsible for returning cellular behavior to homeostasis, are replaced by an upwardly spiraling cycle of cellular perturbation due, in part, to sustained NF-κBp65 transcription and inappropriate NF-κB activation. However, complete and persistent NF-κB inhibition has been linked directly to apoptosis (20,21,22,23), and sustained NF-κB activation might represent a mechanism necessary to ensure survival in the presence of an ongoing proinflammatory challenge accompanying chronic disorders (16,24,114,115). Additional studies are required to define whether the sustained NF-κB activation described here has to be understood as supportive for complications or as a protective mechanism in the course of diabetes.

Immunocytochemistry of NF-κBp65 and IκBα in circulating cells of patients with diabetes and in healthy control subjects. A: Immunocytochemistry for activated NF-κBp65 in PBMCs of a patient with newly manifested type 1 diabetes (left) and in a healthy control subject (right) (magnification: ×40). Positivity for NF-κBp65 is indicated by the dark brown color. Higher magnification (×200) of single cells demonstrated nuclear localization of NF-κBp65 in PBMCs derived from diabetic patients (left insert, arrow), whereas staining for NF-κBp65 in control PBMCs is restricted to cytoplasmic areas close to the cell wall (right insert, arrow). B: Immunocytochemistry for full-length IκBα in PBMCs of a patient with newly manifested type 1 diabetes (left) and a healthy control subject (right). Positivity for IκBα is indicated by the dark brown color. C: Compared with healthy control subjects (n = 6), staining for activated NF-κBp65 was significantly increased in PBMCs of patients with diabetes (n = 6; P = 0.0006 at day 1, diabetes versus control). In contrast, there was no significant difference in IκBα expression (P = 0.846 at day 1, diabetes versus control). NF-κBp65 and IκBα levels did not change within 3 days. D: PBMCs were isolated from four healthy volunteers and four patients with diabetes; total RNA was prepared, and 1 μg of each preparation was analyzed by RT-PCR with primers specific for NF-κBp65 (top) and HPRT (bottom), respectively. Representative RT-PCR results from two diabetic patients (lanes 1 and 2) and two control subjects (lanes 3 and 4) are shown at the left. The signal intensity of the NF-κBp65 representing band and the HPRT band was quantified by densitometry, and the ratio of NF-κBp65 to HPRT was calculated for each patient (n = 4) and each control subject (n = 4). The results are summarized at the right side.
Immunocytochemistry of NF-κBp65 and IκBα in circulating cells of patients with diabetes and in healthy control subjects. A: Immunocytochemistry for activated NF-κBp65 in PBMCs of a patient with newly manifested type 1 diabetes (left) and in a healthy control subject (right) (magnification: ×40). Positivity for NF-κBp65 is indicated by the dark brown color. Higher magnification (×200) of single cells demonstrated nuclear localization of NF-κBp65 in PBMCs derived from diabetic patients (left insert, arrow), whereas staining for NF-κBp65 in control PBMCs is restricted to cytoplasmic areas close to the cell wall (right insert, arrow). B: Immunocytochemistry for full-length IκBα in PBMCs of a patient with newly manifested type 1 diabetes (left) and a healthy control subject (right). Positivity for IκBα is indicated by the dark brown color. C: Compared with healthy control subjects (n = 6), staining for activated NF-κBp65 was significantly increased in PBMCs of patients with diabetes (n = 6; P = 0.0006 at day 1, diabetes versus control). In contrast, there was no significant difference in IκBα expression (P = 0.846 at day 1, diabetes versus control). NF-κBp65 and IκBα levels did not change within 3 days. D: PBMCs were isolated from four healthy volunteers and four patients with diabetes; total RNA was prepared, and 1 μg of each preparation was analyzed by RT-PCR with primers specific for NF-κBp65 (top) and HPRT (bottom), respectively. Representative RT-PCR results from two diabetic patients (lanes 1 and 2) and two control subjects (lanes 3 and 4) are shown at the left. The signal intensity of the NF-κBp65 representing band and the HPRT band was quantified by densitometry, and the ratio of NF-κBp65 to HPRT was calculated for each patient (n = 4) and each control subject (n = 4). The results are summarized at the right side.

Immunocytochemistry of NF-κBp65 in resting cells. A: Nine diabetic kidneys and five control kidneys were examined by immunocytochemistry with a rabbit-derived polyclonal antibody for NF-κBp65, which recognizes both the activated and the inactive protein. Selected representative stainings are shown. Blood vessel endothelia of diabetic kidneys demonstrated pronounced immunoreactivity for NF-κBp65 (left), whereas blood vessel endothelia of control kidneys showed no NF-κBp65 staining (right). Positivity for NF-κBp65 is indicated by the brown color. B: Immunocytochemistry of the above diabetic kidneys using the monoclonal antibody for activated NF-κBp65, demonstrating nuclear translocation of activated NF-κBp65 in the majority of endothelial cells (arrowheads). Positivity for NF-κBp65 is indicated by the dark brown color.
Immunocytochemistry of NF-κBp65 in resting cells. A: Nine diabetic kidneys and five control kidneys were examined by immunocytochemistry with a rabbit-derived polyclonal antibody for NF-κBp65, which recognizes both the activated and the inactive protein. Selected representative stainings are shown. Blood vessel endothelia of diabetic kidneys demonstrated pronounced immunoreactivity for NF-κBp65 (left), whereas blood vessel endothelia of control kidneys showed no NF-κBp65 staining (right). Positivity for NF-κBp65 is indicated by the brown color. B: Immunocytochemistry of the above diabetic kidneys using the monoclonal antibody for activated NF-κBp65, demonstrating nuclear translocation of activated NF-κBp65 in the majority of endothelial cells (arrowheads). Positivity for NF-κBp65 is indicated by the dark brown color.

Ligands of RAGE induce sustained NF-κB DNA binding activity. A–D: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l; A) or Aβ peptide (1 μmol/l; 1–40; B), unmodified control-albumin (500 nmol/l; C), or scrambled Aβ peptide (1 μmol/l; 40–1; D) for 12 h to 6 days. Nuclear extracts were prepared as described (see research design and methods) and assayed for NF-κB binding activity, monitored in EMSA. Radioactive labeled oligonucleotides, spanning the consensus NF-κB recognition motif, were incubated with equal protein amounts of nuclear extracts, and complexes were separated onto nondenaturing 5% PAA gels. For confirming NF-κB binding, binding observed after 72 h was competed with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (cons.). The position of NF-κB is indicated by an arrow. E–G: Nuclear extract from hNT-neurons (E), SMC (F), or THP-1 cells (G), harvested before (0 h) or 6 days after AGE-albumin stimulation (500 nmol/l) (6 days) were subjected to NF-κB–specific EMSA. NF-κB binding was confirmed by competing the shift observed after 6 days with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (cons.). The position of NF-κB is indicated by an arrow.
Ligands of RAGE induce sustained NF-κB DNA binding activity. A–D: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l; A) or Aβ peptide (1 μmol/l; 1–40; B), unmodified control-albumin (500 nmol/l; C), or scrambled Aβ peptide (1 μmol/l; 40–1; D) for 12 h to 6 days. Nuclear extracts were prepared as described (see research design and methods) and assayed for NF-κB binding activity, monitored in EMSA. Radioactive labeled oligonucleotides, spanning the consensus NF-κB recognition motif, were incubated with equal protein amounts of nuclear extracts, and complexes were separated onto nondenaturing 5% PAA gels. For confirming NF-κB binding, binding observed after 72 h was competed with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (cons.). The position of NF-κB is indicated by an arrow. E–G: Nuclear extract from hNT-neurons (E), SMC (F), or THP-1 cells (G), harvested before (0 h) or 6 days after AGE-albumin stimulation (500 nmol/l) (6 days) were subjected to NF-κB–specific EMSA. NF-κB binding was confirmed by competing the shift observed after 6 days with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (cons.). The position of NF-κB is indicated by an arrow.

AGE-mediated induction of NF-κB DNA binding activity in vitro is dependent on RAGE. A: BAECs were left untreated (lane 1) or were incubated for 6 days with AGE-albumin (500 nmol/l) (lane 2) or with AGE-albumin (500 nmol/l) in the presence of either a 10-fold molar excess of sRAGE (lane 3) or 0.1 μmol/l antisense RAGE oligonucleotides (lane 4) before NF-κB binding activity was monitored in EMSA. For confirming NF-κB binding, binding observed at day 6 was competed with a 160-fold molar excess of unlabeled NF-κB consensus oligonucleotides (lane 5). The result shown is representative of three independent experiments. B: BAECs were incubated for 5 days with S100B (400 nmol/l) in the absence (lane 1) or presence (lane 2) of a threefold molar excess of sRAGE, and NF-κB binding activity was monitored in EMSA as above. C: BAECs were left untreated (lane 1) or incubated for 5 days with 400 nmol/l highly modified CML-albumin (lane 2), CML-modified proteins isolated from erythrocytes of a patient with diabetes and poor glycemic control (HbA1c 14.3%; lane 4), minimally modified CML-albumin (lane 6), or CML-modified hippuric acid (lane 8) in the absence (lanes 1, 2, 4, 6, 8) or presence (lanes 3, 5, 7, 9) of a threefold molar excess of sRAGE. NF-κB binding activity was monitored in EMSA. The NF-κB signal intensity was quantified by densitometry and is given below in the autoradiogram.
AGE-mediated induction of NF-κB DNA binding activity in vitro is dependent on RAGE. A: BAECs were left untreated (lane 1) or were incubated for 6 days with AGE-albumin (500 nmol/l) (lane 2) or with AGE-albumin (500 nmol/l) in the presence of either a 10-fold molar excess of sRAGE (lane 3) or 0.1 μmol/l antisense RAGE oligonucleotides (lane 4) before NF-κB binding activity was monitored in EMSA. For confirming NF-κB binding, binding observed at day 6 was competed with a 160-fold molar excess of unlabeled NF-κB consensus oligonucleotides (lane 5). The result shown is representative of three independent experiments. B: BAECs were incubated for 5 days with S100B (400 nmol/l) in the absence (lane 1) or presence (lane 2) of a threefold molar excess of sRAGE, and NF-κB binding activity was monitored in EMSA as above. C: BAECs were left untreated (lane 1) or incubated for 5 days with 400 nmol/l highly modified CML-albumin (lane 2), CML-modified proteins isolated from erythrocytes of a patient with diabetes and poor glycemic control (HbA1c 14.3%; lane 4), minimally modified CML-albumin (lane 6), or CML-modified hippuric acid (lane 8) in the absence (lanes 1, 2, 4, 6, 8) or presence (lanes 3, 5, 7, 9) of a threefold molar excess of sRAGE. NF-κB binding activity was monitored in EMSA. The NF-κB signal intensity was quantified by densitometry and is given below in the autoradiogram.
![FIG. 5. AGE-mediated induction of NF-κB DNA binding activity in vivo is dependent on RAGE. A: Mice transgenic for an NF-κB–controlled β-globin reporter gene (78) were infused once with 1,000 μg of AGE-albumin (500 μg i.v.; 500 μg i.p.) or control albumin (500 μg i.v.; 500 μg i.p.). At time points 0 and 3 days, mice were treated with RAGE IgGs (40 μg/mouse i.v.) or sRAGE (25 μg/mouse i.v.). After 6 days, mice were killed and total RNA from the kidney was prepared and 2 μg were analyzed by RT-PCR for β-globin transgene and β-actin expression (β-globin transgene [top, 440 bp] and β-actin [bottom, 720 bp]). B: Nuclear extracts were prepared from the other kidney of the above β-globin transgenic mice. Ten micrograms of each nuclear extract were analyzed for NF-κB (p50/p65) binding activity in EMSA using a 32P-radiolabeled oligonucleotide with the high-affinity NF-κB(p50/p65) consensus sequence. The inducible NF-κB complex is indicated by an arrow. For confirming NF-κB–binding, kidney nuclear extracts from AGE-treated transgenic mice was competed with a 160-fold molar excess of unlabeled NF-κB consensus oligonucleotides (lane 5).](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/50/12/10.2337_diabetes.50.12.2792/2/m_db1210800005.jpeg?Expires=1716400796&Signature=bhixC0tU-BqNKzugF-FdYDwtl1PgmYoTuyqJ4fVJoDQQXHxtCVPlQS7kv3soCp1vDB2dl1rCX2rrnEW6zt9neSV7emLCJIU8GmdQau5Gjq06feWYK1hRJm2hoGvBG9S6g9HmrDzlEyt2O5ap7C~l3gEumDbXUOCSJTzSEKe4HWP1lECvdUcwKoqqdcivne61481RS-dii-6nT-XBTrevTfTm~OJFz9cOUmqqH5s6W5BizhLpH53EAV3fTNIolTQfrLHDHOLXcX69452v7HRKP50hdKHD52tpFz4JW-BAvNEgSQfXr9TqGNiX5y0rSGZ4ZAKHkGGM5Zvjky-1mfECFw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AGE-mediated induction of NF-κB DNA binding activity in vivo is dependent on RAGE. A: Mice transgenic for an NF-κB–controlled β-globin reporter gene (78) were infused once with 1,000 μg of AGE-albumin (500 μg i.v.; 500 μg i.p.) or control albumin (500 μg i.v.; 500 μg i.p.). At time points 0 and 3 days, mice were treated with RAGE IgGs (40 μg/mouse i.v.) or sRAGE (25 μg/mouse i.v.). After 6 days, mice were killed and total RNA from the kidney was prepared and 2 μg were analyzed by RT-PCR for β-globin transgene and β-actin expression (β-globin transgene [top, 440 bp] and β-actin [bottom, 720 bp]). B: Nuclear extracts were prepared from the other kidney of the above β-globin transgenic mice. Ten micrograms of each nuclear extract were analyzed for NF-κB (p50/p65) binding activity in EMSA using a 32P-radiolabeled oligonucleotide with the high-affinity NF-κB(p50/p65) consensus sequence. The inducible NF-κB complex is indicated by an arrow. For confirming NF-κB–binding, kidney nuclear extracts from AGE-treated transgenic mice was competed with a 160-fold molar excess of unlabeled NF-κB consensus oligonucleotides (lane 5).
AGE-mediated induction of NF-κB DNA binding activity in vivo is dependent on RAGE. A: Mice transgenic for an NF-κB–controlled β-globin reporter gene (78) were infused once with 1,000 μg of AGE-albumin (500 μg i.v.; 500 μg i.p.) or control albumin (500 μg i.v.; 500 μg i.p.). At time points 0 and 3 days, mice were treated with RAGE IgGs (40 μg/mouse i.v.) or sRAGE (25 μg/mouse i.v.). After 6 days, mice were killed and total RNA from the kidney was prepared and 2 μg were analyzed by RT-PCR for β-globin transgene and β-actin expression (β-globin transgene [top, 440 bp] and β-actin [bottom, 720 bp]). B: Nuclear extracts were prepared from the other kidney of the above β-globin transgenic mice. Ten micrograms of each nuclear extract were analyzed for NF-κB (p50/p65) binding activity in EMSA using a 32P-radiolabeled oligonucleotide with the high-affinity NF-κB(p50/p65) consensus sequence. The inducible NF-κB complex is indicated by an arrow. For confirming NF-κB–binding, kidney nuclear extracts from AGE-treated transgenic mice was competed with a 160-fold molar excess of unlabeled NF-κB consensus oligonucleotides (lane 5).

Sustained activation of NF-κB by AGE-albumin and Aβ induces NF-κB–dependent gene expression. A: BAECs were transiently transfected with the plasmid NF-κB-Luc, which contains four tandem copies of the NF-κB consensus sequence fused to a TATA-like promoter region from the herpes simplex virus thymidine kinase promoter using the Effectene transfection reagent. One hour after application of DNA, cells were stimulated with control albumin (500 nmol/l), AGE-albumin (500 nmol/l), scrambled Aβ (1 μmol/l), or Aβ (1 μmol/l) for either 6 or 48 h. After harvest, luciferase activity was determined in the cell lysates and normalized for transfection efficiency by the amount of β-gal activity expressed by the co-transfected control plasmid pSV-β-Gal (83). Corrected values were expressed as relative luciferase (LUC) units and are given as percentage of luciferase expression determined in BAECs, stimulated with control albumin for 6 h. Three independent experiments were performed six times with identical results. The results are given ± SD. Statistical analysis was performed using a two-tailed Student’s t test. B: Immunoblot analysis of HO-1 in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated time-dependent induction of HO-1. HO-1 (molecular weight 39 kDa) is indicated by an arrow. The result shown is representative of three independent experiments. The HO-1 signal intensity was quantified by densitometry and is given below in the autoradiogram. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown).
Sustained activation of NF-κB by AGE-albumin and Aβ induces NF-κB–dependent gene expression. A: BAECs were transiently transfected with the plasmid NF-κB-Luc, which contains four tandem copies of the NF-κB consensus sequence fused to a TATA-like promoter region from the herpes simplex virus thymidine kinase promoter using the Effectene transfection reagent. One hour after application of DNA, cells were stimulated with control albumin (500 nmol/l), AGE-albumin (500 nmol/l), scrambled Aβ (1 μmol/l), or Aβ (1 μmol/l) for either 6 or 48 h. After harvest, luciferase activity was determined in the cell lysates and normalized for transfection efficiency by the amount of β-gal activity expressed by the co-transfected control plasmid pSV-β-Gal (83). Corrected values were expressed as relative luciferase (LUC) units and are given as percentage of luciferase expression determined in BAECs, stimulated with control albumin for 6 h. Three independent experiments were performed six times with identical results. The results are given ± SD. Statistical analysis was performed using a two-tailed Student’s t test. B: Immunoblot analysis of HO-1 in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated time-dependent induction of HO-1. HO-1 (molecular weight 39 kDa) is indicated by an arrow. The result shown is representative of three independent experiments. The HO-1 signal intensity was quantified by densitometry and is given below in the autoradiogram. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown).

Relative contribution of NF-κBp50 and NF-κBp65 to the binding complexes formed during AGE-albumin–mediated sustained NF-κB binding activity monitored by supershifting experiments. Characterization of the NF-κB subunits, contributing to the observed shift formed at the NF-κB consensus sequence, was performed by adding 2.5 μg of an irrelevant IgG or of anti–NF-κBp50 and anti–NF-κBp65 antibodies into the binding reactions with nuclear extracts of unstimulated BAECs or of BAECs stimulated with AGE-albumin for 24 h to 6 days. Signals obtained were quantified by densitometry. A: Signal intensity determined for the NF-κB binding complex in the presence of a nonspecific IgG was taken as 100% at each time point investigated and compared with signal intensity for NF-κB binding determined in the presence of anti-p50 or anti-p65 antibodies. Because of supershifted complexes and blockade of NF-κB binding, the NF-κB binding complex was significantly reduced in the presence of anti-p65 and anti-p50 antibodies. The extent of reduction served as a measure to calculate the relative contribution of NF-κBp65 and NF-κBp50 to each NF-κB binding complex. Two different experiments were evaluated with identical results, and the data of one representative experiment are shown. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). B: Ratio formed by the amount of NF-κBp65 divided by the amount of NF-κBp50 contributing to the NF-κB binding activity observed over time.
Relative contribution of NF-κBp50 and NF-κBp65 to the binding complexes formed during AGE-albumin–mediated sustained NF-κB binding activity monitored by supershifting experiments. Characterization of the NF-κB subunits, contributing to the observed shift formed at the NF-κB consensus sequence, was performed by adding 2.5 μg of an irrelevant IgG or of anti–NF-κBp50 and anti–NF-κBp65 antibodies into the binding reactions with nuclear extracts of unstimulated BAECs or of BAECs stimulated with AGE-albumin for 24 h to 6 days. Signals obtained were quantified by densitometry. A: Signal intensity determined for the NF-κB binding complex in the presence of a nonspecific IgG was taken as 100% at each time point investigated and compared with signal intensity for NF-κB binding determined in the presence of anti-p50 or anti-p65 antibodies. Because of supershifted complexes and blockade of NF-κB binding, the NF-κB binding complex was significantly reduced in the presence of anti-p65 and anti-p50 antibodies. The extent of reduction served as a measure to calculate the relative contribution of NF-κBp65 and NF-κBp50 to each NF-κB binding complex. Two different experiments were evaluated with identical results, and the data of one representative experiment are shown. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). B: Ratio formed by the amount of NF-κBp65 divided by the amount of NF-κBp50 contributing to the NF-κB binding activity observed over time.
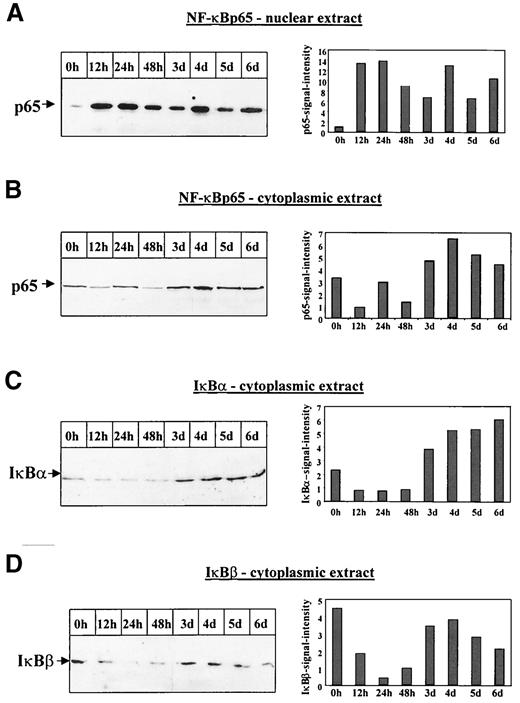
AGE-albumin induces sustained translocation of NF-κBp65 at time points at which IκB proteins already reappeared in the cytoplasm. A and B: Immunoblot analysis of nuclear (A) and cytoplasmic (B) extracts of BAECs, stimulated with AGE-albumin for the times indicated, demonstrated AGE-albumin–dependent prolonged translocation of NF-κBp65 antigen into the nucleus (A). The NF-κBp65–specific complex is indicated by an arrow. The NF-κB signal intensity was quantified by densitometry and is given on the right side of each autoradiogram. The result shown is representative of six independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). C: Immunoblot analysis of IκBα in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated a decrease in cytoplasmic IκBα antigen between 12 and 48 h. The IκBα-specific complex is indicated by an arrow. The IκBα signal intensity was quantified by densitometry and is given on the right side of the autoradiogram. The result shown is representative of four independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). D: Immunoblot analysis of IκBβ in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated a decrease of cytoplasmic IκBβ antigen between 12 and 48 h. The IκBβ-specific complex is indicated by an arrow. The IκBβ signal intensity was quantified by densitometry and is given on the right side of the autoradiogram. The result shown is representative of four independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown).
AGE-albumin induces sustained translocation of NF-κBp65 at time points at which IκB proteins already reappeared in the cytoplasm. A and B: Immunoblot analysis of nuclear (A) and cytoplasmic (B) extracts of BAECs, stimulated with AGE-albumin for the times indicated, demonstrated AGE-albumin–dependent prolonged translocation of NF-κBp65 antigen into the nucleus (A). The NF-κBp65–specific complex is indicated by an arrow. The NF-κB signal intensity was quantified by densitometry and is given on the right side of each autoradiogram. The result shown is representative of six independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). C: Immunoblot analysis of IκBα in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated a decrease in cytoplasmic IκBα antigen between 12 and 48 h. The IκBα-specific complex is indicated by an arrow. The IκBα signal intensity was quantified by densitometry and is given on the right side of the autoradiogram. The result shown is representative of four independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown). D: Immunoblot analysis of IκBβ in cytoplasmic extracts from BAECs that were untreated (0 h) or treated with AGE-albumin (500 nmol/l) for the times indicated demonstrated a decrease of cytoplasmic IκBβ antigen between 12 and 48 h. The IκBβ-specific complex is indicated by an arrow. The IκBβ signal intensity was quantified by densitometry and is given on the right side of the autoradiogram. The result shown is representative of four independent experiments. Similar data were obtained after stimulation with 1 μmol/l Aβ (data not shown).
![FIG. 9. AGE-albumin induces an IκBα phosphorylating kinase activity. BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 12 h to 6 days. Total cell extracts were prepared and incubated with recombinant IκBα for 30 min at 37°C in the presence of 10 μCi [γ-32P]ATP. Subsequently, the reaction was immunoprecipitated with IκBα-specific antibodies and analyzed by SDS-PAGE and autoradiography.](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/50/12/10.2337_diabetes.50.12.2792/2/m_db1210800009.jpeg?Expires=1716400796&Signature=tLQEPr5ipnmvPvBaLvdl7jY8gA16LwiJ5SxLtc1dExJKXEmqTh5~-iwifCYzC2Y~6rFO9SPjtVkxBtdW2d652qvUYWVPqqe40jgdD3NjO1Mo9jRxHk4pWZdIpmafIWhJQUGfJzr4-7s6sppysa2Fu7wE2ebtZ4BPOHJB0batldFTbKJME1ulsSKybdODru84nASvJLpvGtP7Md2u5qTnNqxSwOLsJ7wRbEB9eYWPxznaIZBpxcgDJmJItteAaZTf0vZlEMsiC2SoXKjhtCNEEmI2T7Xk68iAy7v8Dfo73dlgSAvoLZcoJLXco-Cg-BIswStcj-qSXhlocwOetG1DzQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AGE-albumin induces an IκBα phosphorylating kinase activity. BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 12 h to 6 days. Total cell extracts were prepared and incubated with recombinant IκBα for 30 min at 37°C in the presence of 10 μCi [γ-32P]ATP. Subsequently, the reaction was immunoprecipitated with IκBα-specific antibodies and analyzed by SDS-PAGE and autoradiography.
AGE-albumin induces an IκBα phosphorylating kinase activity. BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 12 h to 6 days. Total cell extracts were prepared and incubated with recombinant IκBα for 30 min at 37°C in the presence of 10 μCi [γ-32P]ATP. Subsequently, the reaction was immunoprecipitated with IκBα-specific antibodies and analyzed by SDS-PAGE and autoradiography.
![FIG. 10. AGE-albumin induces accumulation of NF-κBp65 mRNA and de novo synthesis of NF-κBp65 antigen in endothelial cells. A: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l, left) or control albumin (500 nmol/l, right) for 12 h to 6 days. Total RNA was prepared, and 2 μg of each preparation was analyzed by RT-PCR with primers specific for NF-κBp65 (top, 680 bp) and β-actin (bottom, 720 bp), respectively (for details, see research design and methods). The left lane (SM) shows the 564- to 947-bp fragment of a λ-DNA EcoRI-HindIII digest. The signals obtained in AGE-albumin–stimulated BAECs were analyzed by densitometry and are expressed as the ratio of NF-κBp65 to β-actin. B: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 24 h, 72 h, or 6 days in the absence (left) or presence (right) of 0.1 μmol/l antisense p65 oligonucleotides. Four hours before harvest, the medium was changed to methionine-free medium for 1 h. Thereafter, medium was changed again to DMEM supplemented with 7 μCi/ml l-[35S]methionine. At the end of incubation, intrinsic labeled cells were harvested, lysed, and subjected to NF-κBp65–specific immunoprecipitation (for details, see research design and methods). Incorporation of l-[35S]methionine into NF-κBp65–precipitated material was determined in a β-counter. The experiment was repeated twice with similar results.](https://ada.silverchair-cdn.com/ada/content_public/journal/diabetes/50/12/10.2337_diabetes.50.12.2792/2/m_db1210800010.jpeg?Expires=1716400796&Signature=fEIreisfHFzDfgVo0O9HqxQrD1pKMkLDQm-ww1KKue5Sv-RI9Utxpfs5gNY1mdMU-gU6yweaGZ~sC~tU~fdEKAcb017e6bCsex4XQDX1JwaaHfiEMQ8NOi1DKhSEQTczEiyh16jtP2aDUpqMvoNY2oilr-mJM8KpJgPreIEkOI~Y0fm~7EZvbyyIIace8mY5jl-aSMUJmB-3CnrfCpd4HqswnWMnQvEWzF6wQvZjCjM2bHnNw8Z73QyznAdgbI6qO-FOsSef99g2GLy8uBAwCrImCIL25sa9bwDO1KTBRAJYYsYcj~5~tUcu6m8FL9yUhhdv-uah8N9c6M8lRo41kQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
AGE-albumin induces accumulation of NF-κBp65 mRNA and de novo synthesis of NF-κBp65 antigen in endothelial cells. A: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l, left) or control albumin (500 nmol/l, right) for 12 h to 6 days. Total RNA was prepared, and 2 μg of each preparation was analyzed by RT-PCR with primers specific for NF-κBp65 (top, 680 bp) and β-actin (bottom, 720 bp), respectively (for details, see research design and methods). The left lane (SM) shows the 564- to 947-bp fragment of a λ-DNA EcoRI-HindIII digest. The signals obtained in AGE-albumin–stimulated BAECs were analyzed by densitometry and are expressed as the ratio of NF-κBp65 to β-actin. B: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 24 h, 72 h, or 6 days in the absence (left) or presence (right) of 0.1 μmol/l antisense p65 oligonucleotides. Four hours before harvest, the medium was changed to methionine-free medium for 1 h. Thereafter, medium was changed again to DMEM supplemented with 7 μCi/ml l-[35S]methionine. At the end of incubation, intrinsic labeled cells were harvested, lysed, and subjected to NF-κBp65–specific immunoprecipitation (for details, see research design and methods). Incorporation of l-[35S]methionine into NF-κBp65–precipitated material was determined in a β-counter. The experiment was repeated twice with similar results.
AGE-albumin induces accumulation of NF-κBp65 mRNA and de novo synthesis of NF-κBp65 antigen in endothelial cells. A: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l, left) or control albumin (500 nmol/l, right) for 12 h to 6 days. Total RNA was prepared, and 2 μg of each preparation was analyzed by RT-PCR with primers specific for NF-κBp65 (top, 680 bp) and β-actin (bottom, 720 bp), respectively (for details, see research design and methods). The left lane (SM) shows the 564- to 947-bp fragment of a λ-DNA EcoRI-HindIII digest. The signals obtained in AGE-albumin–stimulated BAECs were analyzed by densitometry and are expressed as the ratio of NF-κBp65 to β-actin. B: BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for 24 h, 72 h, or 6 days in the absence (left) or presence (right) of 0.1 μmol/l antisense p65 oligonucleotides. Four hours before harvest, the medium was changed to methionine-free medium for 1 h. Thereafter, medium was changed again to DMEM supplemented with 7 μCi/ml l-[35S]methionine. At the end of incubation, intrinsic labeled cells were harvested, lysed, and subjected to NF-κBp65–specific immunoprecipitation (for details, see research design and methods). Incorporation of l-[35S]methionine into NF-κBp65–precipitated material was determined in a β-counter. The experiment was repeated twice with similar results.

AGE-albumin induction of NF-κB activation at time points later than 2 days is dependent on mRNA. BAECs were stimulated with AGE-albumin (500 nmol/l) for the times indicated. Three hours before harvest, cells received a pulse of actinomycin D (15 μg/ml). Nuclear extracts were prepared as described and assayed for NF-κB binding activity in EMSA using radioactive labeled oligonucleotides, spanning the consensus NF-κB recognition motif. For confirming NF-κB binding, the observed shift was competed with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (last lane). To exclude the possibility that EtOH influenced NF-κB binding activity, BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for the times indicated in the presence of 0.4% EtOH (final concentrations used in actinomycin D experiments; right). The position of NF-κB is indicated by an arrow.
AGE-albumin induction of NF-κB activation at time points later than 2 days is dependent on mRNA. BAECs were stimulated with AGE-albumin (500 nmol/l) for the times indicated. Three hours before harvest, cells received a pulse of actinomycin D (15 μg/ml). Nuclear extracts were prepared as described and assayed for NF-κB binding activity in EMSA using radioactive labeled oligonucleotides, spanning the consensus NF-κB recognition motif. For confirming NF-κB binding, the observed shift was competed with a 160-fold molar excess of cold consensus NF-κB oligonucleotides (last lane). To exclude the possibility that EtOH influenced NF-κB binding activity, BAECs were left untreated (0 h) or were stimulated with AGE-albumin (500 nmol/l) for the times indicated in the presence of 0.4% EtOH (final concentrations used in actinomycin D experiments; right). The position of NF-κB is indicated by an arrow.
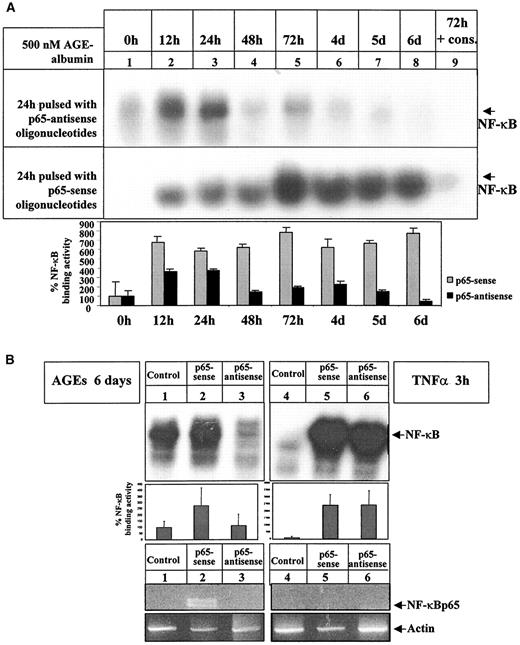
AGE-albumin induction of NF-κB activation at time points later than 2 days is dependent on NF-κBp65 mRNA de novo synthesis. A: BAECs were stimulated with AGE-albumin (500 nmol/l) for the times indicated. Twenty-four hours before harvest, a pulse of 0.1 μmol/l antisense (top) or sense p65 oligonucleotides (bottom) was added to the medium. Uptake was in a passive manner, as previously described in detail (52). NF-κB binding activity was assayed in EMSA as above; the position of NF-κB is indicated by an arrow. The experiment was repeated twice, with similar results, and the densitometric analysis of both experiments is summarized below in the autoradiogram. The position of NF-κB is indicated by an arrow. B: BAECs were stimulated with AGE-albumin (500 nmol/l) for 6 days (left) or TNF-α (1 nmol/l) for 3 h (right). Twenty-four hours before harvest, a pulse of 0.1 μmol/l antisense or sense p65 oligonucleotides was added to the medium. From one-half of the cells, nuclear extracts were prepared and NF-κB binding activity was assayed in EMSA as above (top). Intensity of NF-κB binding activity was quantified by densitometry (middle). The second half of cells were used for RNA isolation, and 2 μg of each preparation was subsequently subjected to cDNA synthesis and RT-PCR for NF-κBp65 and β-actin (bottom).
AGE-albumin induction of NF-κB activation at time points later than 2 days is dependent on NF-κBp65 mRNA de novo synthesis. A: BAECs were stimulated with AGE-albumin (500 nmol/l) for the times indicated. Twenty-four hours before harvest, a pulse of 0.1 μmol/l antisense (top) or sense p65 oligonucleotides (bottom) was added to the medium. Uptake was in a passive manner, as previously described in detail (52). NF-κB binding activity was assayed in EMSA as above; the position of NF-κB is indicated by an arrow. The experiment was repeated twice, with similar results, and the densitometric analysis of both experiments is summarized below in the autoradiogram. The position of NF-κB is indicated by an arrow. B: BAECs were stimulated with AGE-albumin (500 nmol/l) for 6 days (left) or TNF-α (1 nmol/l) for 3 h (right). Twenty-four hours before harvest, a pulse of 0.1 μmol/l antisense or sense p65 oligonucleotides was added to the medium. From one-half of the cells, nuclear extracts were prepared and NF-κB binding activity was assayed in EMSA as above (top). Intensity of NF-κB binding activity was quantified by densitometry (middle). The second half of cells were used for RNA isolation, and 2 μg of each preparation was subsequently subjected to cDNA synthesis and RT-PCR for NF-κBp65 and β-actin (bottom).
Characterization of AGE-albumin used throughout the experiments
| Amino acid | Quantity (pmol/mg) |
|---|---|
| Unmodified lysine | 688.360 |
| Fructoselysine | 181.470 |
| Carboxymethyllysine | 204.320 |
| Pyrraline | 190 |
| Pentosidine | 6 |
| Total | 1,074.346 |
| Lysine derivatization: | |
\[\frac{{[}sum\ of\ lysine\ derivates{]}}{{[}sum\ of\ lysine\ derivates{]}\ {+}\ {[}unmocified\ lysine{]}}\ {\times}\ 100{\%}\ {=}\ 35.9{\%}\] | |
| Amino acid | Quantity (pmol/mg) |
|---|---|
| Unmodified lysine | 688.360 |
| Fructoselysine | 181.470 |
| Carboxymethyllysine | 204.320 |
| Pyrraline | 190 |
| Pentosidine | 6 |
| Total | 1,074.346 |
| Lysine derivatization: | |
\[\frac{{[}sum\ of\ lysine\ derivates{]}}{{[}sum\ of\ lysine\ derivates{]}\ {+}\ {[}unmocified\ lysine{]}}\ {\times}\ 100{\%}\ {=}\ 35.9{\%}\] | |
Article Information
This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (P.P.N., M.S., M.H.); the Bundesministerium für Forschung und Technik (B.W., M.K.); the Fortune program (A.B., P.P.N.) and the IZKF program of the University of Tübingen (P.P.N.); Stiftung Verum (P.P.N.); the University of Heidelberg (A.B.); and ASTA-Medica (A.B., P.P.N.). P.P.N. performed this work during the tenure of a Schilling professorship. A.M.S. and D.S. were supported by grants from the U.S. Public Health Service, the American Heart Association (New York affiliate), and the Juvenile Diabetes Foundation.
Part of this work was presented at the 17th meeting of the International Society for Thrombosis and Hemostasis (ISTH), Florence, Italy, 10–16 June 1997; at the 42nd meeting of the German Society for Endocrinology, Freiburg, Germany, 4–7 March 1998; and at the 33rd meeting of the German Diabetes Society, Leipzig, Germany, 21–23 May 1998.
We thank Dr. T. Wirth (Würzburg, Germany) for providing the β-globin transgenic mice and Dr. I. Besenthal (Tübingen, Germany) for help in identifying patients with newly manifested diabetes. TNF-α was a gift from Knoll (Ludwigshafen, Germany). Anti-RAGE antibodies were kindly provided by Dr. M.A. Shearman (Merck, Sharpe & Dome, Essex, U.K.). The authors also thank M. Kanitz (Heidelberg, Germany), S. Langer (Dresden, Germany), and A. Oehmichen (Dresden, Germany) for excellent technical assistance and B. Andrassy for editorial help.
REFERENCES
Address correspondence and reprint requests to Peter P. Nawroth, MD, Department of Medicine I, University of Heidelberg, Bergheimer Strasse 58, 69115 Heidelberg, Germany. E-mail: peter_nawroth@med.uni-heidelberg.de.
Received for publication 19 October 2000 and accepted in revised form 12 September 2001.
M.S. and G. P. serve as consultants/collaborators for Panacea Pharmaceuticals, Prion Development Laboratories, and Voyager Laboratories. A.-M.S. and D.M.S. are consultants for and have received a research grant from TransTech Pharma. P.P.N. has received a research grant from ASTA-Medica.
Aβ, amyloid-β peptide; AGE, advanced glycosylation end product; BAEC, bovine aortic endothelial cell; β-Gal, β-galactosidase; CML, carboxymethyllysine; DMEM, Dulbecco’s modified Eagle’s medium; ECL, enhanced chemiluminescence; EMSA, electrophoretic mobility shift assay; ERK, extracellular signal-regulated kinases; FCS, fetal calf serum; HO-1, heme oxygenase-1; HPLC, high-performance liquid chromatography; HPRT, hypoxanthine guanine phosphoribosyl transferase; LPS, lipopolysaccharide; NF-κB, nuclear factor-κB; ODN, oligodeoxynucleotide; PBMC, peripheral blood mononuclear cell; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; PMSF, phenylmethylsulfonyl fluoride; [PS]ODN, phosphorothioate oligodeoxynucleotide; RAGE, receptor for AGE; ROS, reactive oxygen species; RT, reverse transcription; rt, room temperature; SMC, smooth muscle cell; sRAGE, soluble RAGE; TBS, Tris-buffered saline; TNF-α, tumor necrosis factor-α.