Sarkar M A Kawsar
Tel +88 01762717081; Fax +88 031 2606014
E-mail: akawsarabe@yahoo.com
© 2019 Sift Desk Journals. All Rights Reserved
VOLUME: 4 ISSUE: 4
Page No: 452-462
Sarkar M A Kawsar
Tel +88 01762717081; Fax +88 031 2606014
E-mail: akawsarabe@yahoo.com
Sarkar M. A. Kawsar1*, Mohammed A. Hosen1, Yuki Fujii2, Yasuhiro Ozeki3
1Laboratory of Carbohydrate and Nucleoside Chemistry (LCNC), Department of Chemistry, Faculty of Science, University of Chittagong, Chittagong-4331, Bangladesh.
2Laboratory of Functional Morphology, Graduate School of Pharmaceutical Sciences, Nagasaki International University, 2825-7 Huis Ten Bosch, Sasebo, Nagasaki 859-3298, Japan.
3Laboratory of Glycobiology and Marine Biochemistry, Department of Life and Environmental System Science, Graduate School of Nano Biosciences, Yokohama City University, 22-2 Seto, Kanazawa-ku, Yokohama 236-0027, Japan.
Kawsar SMA, M.A.Hosen, Y.Fujii, Y.Ozeki, Thermochemical, DFT, Molecular Docking and Pharmacokinetic Studies of Methyl β-D-galactopyranoside Esters (2020) Journal of Computational Chemistry & Molecular Modeling 4(4) pp:452-462
Monosaccharide esters have great importance in the field of carbohydrate chemistry because of its effectiveness in the synthesis of biologically active products. As a consequence, the chemistry and biochemistry of carbohydrate derivatives are an essential part of biochemical and medicinal research. Modification of the hydroxyl (-OH) group of monosaccharides by acylation increases their biological activity. In this study, we have optimized a monosaccharide molecule, methyl β-D-galactopyranoside (MGP, 1), and some of its acylated esters employing density functional theory (DFT). All designed esters are optimized at the B3LYP/3-21G level of theory. Electronic energies, enthalpies, Gibbs free energies, heat capacity, entropy, molar refractivity, polarizability dipole moments, HOMO-LUMO gaps, density of states (DOS) and molecular electrostatic potential (MEP) of these modified compounds are also investigated in the subsequent analysis. Then compound (8), which showed a positive test on EAC cell, was subjected to molecular docking analysis with human poly [ADP-ribose] polymerase1 protein 4ZZZ to investigate the molecular interactions widely. Finally, ADMET analysis hints that modified derivatives of MGP (1) are less toxic and have improved pharmacokinetic properties than those of the parent drug.
Keywords: galactopyranoside, esters, EAC, DFT calculation, pharmacokinetic
Carbohydrates (named also sugars or saccharides) are the most abundant biomolecules in nature and they are often conjugated with proteins (glycoproteins) or lipids (glycolipids). These molecules play different roles in the cells, and for this reason, recent studies are focused on carbohydrates as a selective mediator in the molecular recognition processes. They are involved in physiological and pathological events and a variety of intra- and extracellular events. In most of the cases, the initial contacts of saccharides with cells take place at cell-cell and cell-matrix level [1, 2]. However, modifications to the regular cell glycosylation pattern are often observed during pathological conditions, for example in inflammation and cancer development. Therefore, glycans have become a topic of major interest to develop diagnostic tools or therapeutic drugs [3, 4]. Several carbohydrate-based drugs are currently being used to treat several diseases; the most illustrative examples are antibiotics and antivirals [5]. For decades, aminoglycosides, inhibitors of bacterial protein synthesis, have been used as antibiotics especially for infections by aerobic Gram-negative bacteria [6]. Glycans are also related to metabolic drugs e.g., diabetes mellitus type I [7] and type II diabetes [8].
To date, several sugar-based drugs have already been developed and currently employed as therapeutics, diagnostics, vaccines, and many more are expected to come in the next few years. Glycan diversity depends on the monosaccharides (basic unit of saccharide) which are made by, on the linkages that connect monosaccharides (glycosidic linkage), and on other factors such as the anomeric effects, the orientation of all the torsional angles [9, 10], and the intramolecular hydrogen bonding between adjacent OH groups [11]. Therefore, it is of paramount importance to predict and characterize their conformational and structural properties in a systematic manner. Computational techniques have proven to be essential in this aspect [12].
In this study, we have modified the hydroxyl (-OH) group of methyl β-D-galactopyranoside (MGP, 1) (Fig. 1) by some acyl substituents (including aromatic and heteroaromatic group) [13] and these modified esters are optimized to realize their thermal, electrical stability and biochemical behavior based on quantum mechanical methods. The free energy, enthalpy, entropy, heat capacity dipole- moment, HOMO-LUMO gap, DOS plot, polarizability, molar refractivity, and molecular electrostatic potential have been calculated to compare their thermal and chemical characteristic. Compound (8), which showed a positive result on test with EAC cell i.e. anti-cancer activity [13] was subjected to molecular docking to understand the nonbonding interactions, binding affinity, and binding mode with the receptor protein (4ZZZ). Finally, pharmacokinetic enumeration has been performed to compare their absorption, metabolism, and toxicity. The prime intention of our investigation was to understand the thermodynamic, molecular orbital, binding mode, molecular electrostatic potential, physicochemical and, ADMET properties (MGP, 1) and its esters.
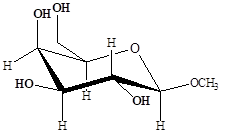
Fig. 1 Chemical structure of the methyl β-D-galactopyranoside (MGP, 1).
2.1. Designing and Optimization of MGP Esters
In computational chemistry, quantum mechanical methods are widely used to calculate thermal, molecular orbital and molecular electrostatic properties [14]. Geometry optimization and further modification of all MGP (1) esters are carried out using the Gaussian 09 program [14]. Density functional theory (DFT) with Beck’s (B) [15] three-parameter hybrid model, Lee, Yang, and Parr’s (LYP) [16] correlation functional under 3-21G basis set has been employed to optimize and predict their thermochemical and molecular orbital properties. Dipole moment, enthalpy, free energy, entropy, heat capacity, molar refractivity, and polarizability were calculated for all the compounds. Besides this, electrostatic potential calculated for MGP (1) and its four esters. Frontier molecular orbital features HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital) were counted at the same level of theory. For each of the MGP (1) esters, HOMO-LUMO energy gap, hardness (η), and softness (S) were calculated from the energies of frontier HOMO and LUMO as reported considering Parr and Pearson interpretation of DFT and Koopmans theorem [17] on the correlation of ionization potential (I) and electron affinities (E) with HOMO and LUMO energy (ε). The following equations are used to calculate hardness (η), softness (S).

2.2. Preparation of Protein and Molecular Docking
The three-dimensional crystal structure of human PARP1 (PDB ID: 4ZZZ) was retrieved in pdb format from the protein data bank [18]. All hetero atoms and water molecules were removed using PyMol (version 1.3) software packages [19]. Swiss-Pdb viewer software (version 4.1.0) was employed for energy minimization of the protein [20]. Then optimized drugs were subjected for molecular docking study against human PARP1 protein (4ZZZ). In computer-aided drug design, molecular docking simulation can predict the binding affinity and mode(s) of ligand with target protein [21]. In fine, molecular docking simulation was rendered by PyRx software (version 0.8) [22] considering the protein as macromolecule and the drug as ligand. AutodockVina was employed for docking analysis, and AutoDock Tools (ADT) of the MGL software package was used to convert pdb into a pdbqt format to input protein and ligands. The size of the grid box in AutoDockVina was kept at 52.1251, 58.4212, and 61.7939 Å for X, Y, Z directions respectively. After the completion docking, both the macromolecule and ligand structures were saved in. pdbqt format needed by Accelrys Discovery Studio (version 4.1) to explore and visualize the docking result and search the nonbonding interactions between ligands and amino acid residues of receptor protein [23].
2.3. Pharmacokinetic Parameters Analysis
To check the pharmacokinetic parameters and toxicity of the modified MGP (1) esters and parent compound, the admetSAR server was utilized [24]. All the latest and most extensive manually curated data predicted by admetSAR with help of structure uniformity search methods for various chemicals linked with known ADMET schemes. For ADMET analysis, the admetSAR program was used in which 96000 matchless compounds with 45 types of ADMET-associated parameters, proteins, and organisms have been cautiously curated from a massive number of diverse literature. Although it is quite difficult to verify all of these compounds and to know whether this program included thymidine-based drugs or not, well known Pt-based cisplatin and carboplatin as well as metal-based drugs approved in the FDA and in clinical trials as test candidates to verify our MGP (1) esters. Moreover, some physicochemical properties (molar refractivity, molecular weight) are also calculated from the swissADME server.
2.4. Strategies and Optimization of Designed MGP (1) Esters
The newly modified esters of MGP (1) used in this study were designated according to the reactions scheme. MGP (1) and its esters (2-10) were designated and optimized in the quantum mechanical method. In the Fig. 2 represented the optimized structure of MGP (1) esters.
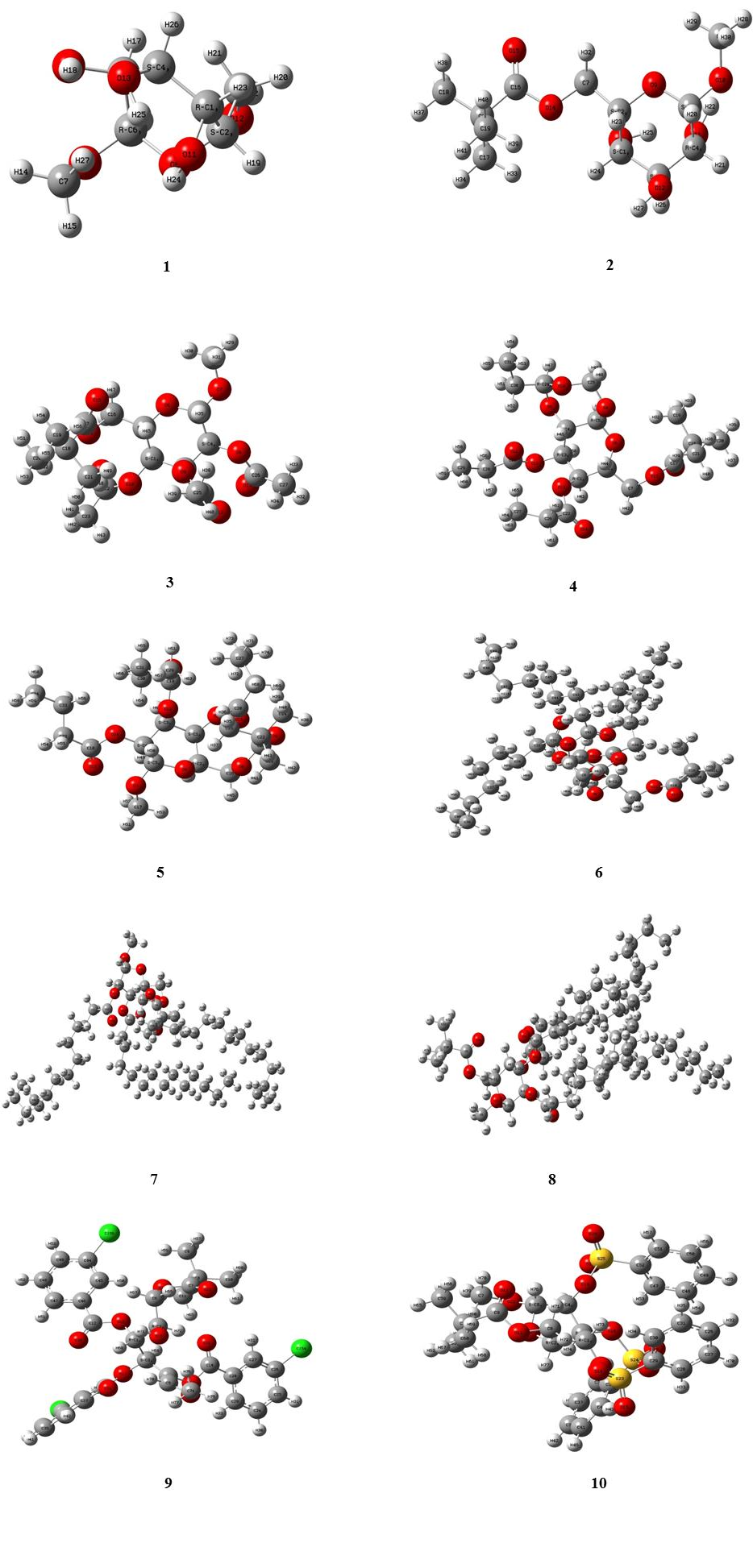
Fig. 2 Optimized structure of MGP (1) and its esters (2-10). Optimized with DFT-B3LYP/3-21g.
3.1. Characterization
We have modified a group of MGP (1) esters (Fig. 3) and attempt to performed computational study to realize the mode of their different properties. Gaussian 09 is a way of molecular modeling program which consents to develop and analyze diverse molecular structures and determine their thermophysical, thermo-chemical, frontier orbital, and molecular electrostatic potential. To perform this work, core i7 computer and Gaussian 09 software were employed and the optimized structures are provided in Fig. 2. To setup reactivity computing based on Gaussian 09, molecular symmetry is a very potent tool. Molecular optimization revealed that all modified derivatives containing more than one symmetry axis. The molecules belong to the class asymmetry, and non-planar and they have two or more elements of symmetry and the plane of the molecule.
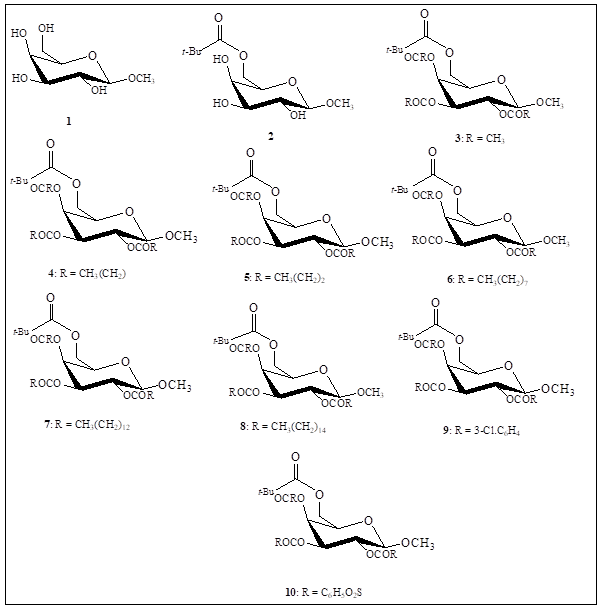
Fig. 3 Chemical structures of the MGP (1) esters 2-10.
3.2. Thermochemical Analysis
Usual alterations of the molecular structure significantly influence the structural properties including thermal and molecular orbital parameters. Spontaneously of a reaction and stability of a product can be elucidated from the free energy and enthalpy values [25]. Highly negative values are more suitable for thermal stability. In computational molecular modeling, the dipole moment influences non-bonded interactions hydrogen bond formation smoothly. Binding property can also be improved by the increasing of dipole moment [26]. Table 1 represents the thermochemical properties of methyl β-D-galactopyranoside and it’s all synthesized esters. The highest free energy is (-3655.5316 Hartree) observed for ester 10 which also showed the highest enthalpy (-3655.3896 Hartree), highest electronic energy (-3655.3906 Hartree) and the highest dipole moment (15.7081Debye).
Table 1. Molecular formula, Molecular weight (g/mol), Electronic energy, enthalpy, Gibbs free energy (Hartree) & dipole moment (Debye) of MGP (1) esters
|
Compounds no. |
Molecular formula |
Molecular weight |
Electronic energy |
Enthalpy |
Gibbs free energy |
Dipole moment |
|
1 |
C7H14O6 |
194.18 |
-722.2093 |
-722.2084 |
-722.2608 |
4.7712 |
|
2 |
C12H22O7 |
278.30 |
-991.2505 |
-991.2495 |
-991.3208 |
3.8025 |
|
3 |
C18H28O10 |
404.41 |
-1446.5981 |
-1446.5972 |
-1446.6928 |
4.0591 |
|
4 |
C21H34O10 |
446.49 |
-1563.7944 |
-1563.7934 |
-1563.8927 |
4.1739 |
|
5 |
C24H40O10 |
488.57 |
-1681.0208 |
-1681.0198 |
-1681.1321 |
3.1126 |
|
6 |
C39H70O10 |
698.97 |
-2267.0782 |
-2267.0773 |
-2267.2356 |
2.5455 |
|
7 |
C54H100O10 |
909.37 |
-2853.1492 |
-2853.1482 |
-2853.3563 |
3.3707 |
|
8 |
C60H112O10 |
993.53 |
-3249.3087 |
-3249.3078 |
-3249.4455 |
5.3101 |
|
9 |
C33H31Cl3O10 |
693.95 |
-3390.6553 |
-3390.6543 |
-3390.7839 |
5.0127 |
|
10 |
C30H34O13S3 |
698.78 |
-3655.3906 |
-3655.3896 |
-3655.5316 |
15.7081 |
Table 2. Molar refractivity (Å3), Polarizability (a.u.), Heat capacity (cal/mol-kelvin), Entropy (cal/mol-kelvin) and Total energy (Hartree)
|
Compounds |
Molar Refractivity |
Polarizability |
Heat capacity |
Entropy |
Total energy |
|
1 |
40.47 |
85.3296 |
49.303 |
110.240 |
-722.4470 |
|
2 |
64.36 |
138.3413 |
77.785 |
149.967 |
-991.6197 |
|
3 |
93.58 |
201.4366 |
113.232 |
201.255 |
-1447.0918 |
|
4 |
108.00 |
232.8350 |
123.704 |
208.931 |
-1564.3807 |
|
5 |
122.42 |
263.6293 |
140.956 |
236.339 |
-1681.6970 |
|
6 |
194.52 |
415.4296 |
212.121 |
333.346 |
-2268.2077 |
|
7 |
266.63 |
570.1433 |
283.525 |
437.868 |
-2854.7312 |
|
8 |
295.47 |
631.8790 |
314.425 |
469.859 |
-3250.0814 |
|
9 |
168.31 |
383.7566 |
166.283 |
272.731 |
-3391.2924 |
|
10 |
161.98 |
424.9966 |
191.798 |
298.880 |
-3656.0891 |
In Table 2 represents some other thermophysical properties where the compound (8) showed the highest value for molar refractivity, polarizability, heat capacity, and entropy. Presence of bulky acylating groups also suggesting the possible improvement of polarizability. In Table 1 and Table 2, the values of all the properties increase with the increase of molecular weight from compound (2) to (8) and these esters contain long-chain acyl substituents. This analysis may reveal that with the increased molecular weight of MGP (1) esters, their stability may increase. But in the case of compound (9) and (10), which heteroaromatic ring showed the highest value for energies than the long-chain acyl substituents. In a word, all the esters showed better characteristics than the parent molecule (1).
3.3. Molecular Orbitals Analysis
The frontier molecular orbitals are the most important orbitals in a molecule and they are considered to characterize the chemical reactivity and kinetic stability (Table 3). These frontier molecular orbitals are known as the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) (Fig. 4).
Table 3. Energy (eV) of HOMO, LUMO, energy gap, hardness and softness of esters
|
Compounds |
εHOMO |
εLUMO |
Gap |
Hardness (η) |
Softness (S) |
|
1 |
-6.1918 |
1.3761 |
7.5679 |
3.7839 |
0.2643 |
|
2 |
-6.5189 |
-0.0474 |
6.4715 |
3.2357 |
0.3091 |
|
3 |
-7.0076 |
-0.1346 |
6.8730 |
3.4365 |
0.2909 |
|
4 |
-6.7812 |
-0.1806 |
6.6006 |
3.3003 |
0.3031 |
|
5 |
-6.8892 |
-0.1690 |
6.7202 |
3.3601 |
0.2976 |
|
6 |
-6.9978 |
0.1268 |
6.8710 |
3.4355 |
0.2911 |
|
7 |
6.8974 |
-0.4283 |
6.4691 |
3.2345 |
0.3092 |
|
8 |
-6.4016 |
-1.8344 |
4.5672 |
2.2836 |
0.4379 |
|
9 |
-6.2307 |
-1.8755 |
4.3552 |
2.1776 |
0.4593 |
|
10 |
6.7488 |
-1.7865 |
4.9623 |
2.4812 |
0.4031 |
The electronic absorption relates to the transition from the ground to the first excited state and mainly described by one electron excitation from HOMO to LUMO [27]. Kinetic stability increases with the increase of the HOMO-LUMO gap. As a result, the removal of electrons from ground state HOMO to excited state LUMO requires more energy. Table 3 represents the values of orbital energies, along with the two global chemical descriptors, hardness, and softness, which are also calculated for all compounds. The highest softness was observed for the compound (9). Compound (9) also showed the lowest HOMO-LUMO gap (4.3552 eV) and hardness (2.1776eV), indicating the molecule is more reactive than other compounds, according to Hoque et al [28, 29]. On the other hand, compound (3) showed the highest HOMO-LUMO gap (6.8730 eV) which less than our parent compound MGP (1) (7.5679 eV) which indicates that ester (3) closely stable as parent compound (1). Fig. 5 represents the density of states (DOS) plot for the highest and lowest energy gap of the modified derivatives.
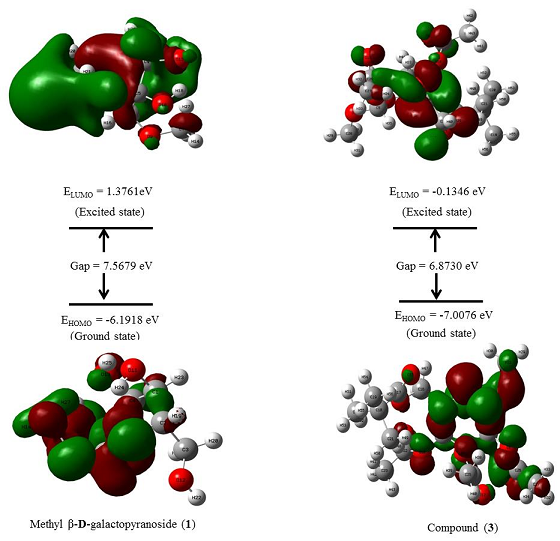
Fig. 4 Molecular orbital distribution diagram of HOMO & LUMO of (1) and ester (3).
In Fig. 4, the LUMO diagram of the compound (3) showed that the electron was localized at the modified acylating group regions only, while the HOMO diagram showed that the electron was localized on the upper part of both the monosaccharide ring and acyl substituents.
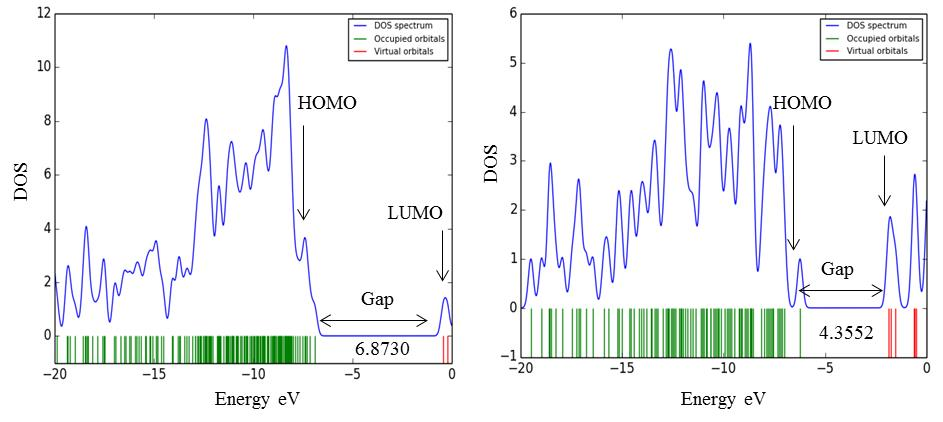
Fig. 5 DOS plot and HOMO-LUMO energy gap of the compounds (3) and (9).
3.4. Molecular Electrostatic Potential (MEP) Analysis
Molecular electrostatic potential (MEP) is globally preferred as a map of reactivity that exhibits the most probable region for organic molecules to performing electrophilic and nucleophilic reactions of charged points like reagents [30]. It helps to interpret the biological recognition process and hydrogen bonding interaction [31]. MEP counter map provides a simple way to predict how different geometry could interact. The MEP of the title compound is obtained based on the B3LYP with basis set 3-21G optimized result and shown in Figure 5. The prime significant of MEP is that it simultaneously displays positive, negative, and neutral electrostatic potential regions as well as molecular size, shape by color grading, and very helpful in the study of molecular structure with physicochemical features relationship [32]. Molecular electrostatic potential (MEP) was calculated to forecast the reactive sites for electrophilic and nucleophilic attack of the optimized structure of MGP (1) esters 3, 6, 9, and 10 (Fig. 6). The different colors of electrostatic potential indicate different values. Potentiality of attacking zone decreases in the sequence of blue ˃ green ˃ yellow ˃ orange ˃ red. The maximum negative area is displayed by red color where electrophiles can easily attack and the maximum positive area indicated by blue color which is suitable for nucleophilic attack. Moreover, the green color showed the zero potential zones.
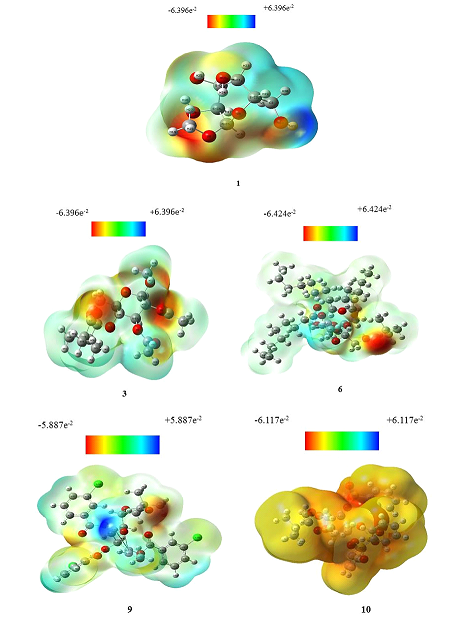
Fig. 6 Molecular electrostatic potential map of MGP (1) esters of 3, 6, 9, and 10.
Table 4. Binding affinity (kcal/mol) and nonbonding interactions of MGP (1) and ester (8)
|
Compounds |
Protein |
Binding affinity |
Residues in contact |
Interaction types |
Distance (Å) |
|
1 |
4ZZZ |
-4.6 |
GLY888 |
Conventional H-Bond |
2.11116 |
|
TYR907 |
Conventional H-Bond |
2.94769 |
|||
|
MET890 |
Conventional H-Bond |
2.3132 |
|||
|
MET890 |
Conventional H-Bond |
2.14398 |
|||
|
8 |
4ZZZ |
-6.9 |
GL N759 |
Conventional H-Bond |
2.37494 |
|
GLN759 |
C-H Bond |
3.38979 |
|||
|
MET890 |
Alkyl |
4.52133 |
|||
|
LYS903 |
Alkyl |
4.88986 |
|||
|
LEU985 |
Alkyl |
4.55185 |
|||
|
TYR907 |
Pi-Alkyl |
4.92231 |
3.5. Molecular Docking Analysis
As compound (8) showed anti-cancer activity on EAC cell [13], that’s why it subjected to molecular docking against receptor protein 4ZZZ to check its binding mode. Molecular docking analysis reveal that, compound (8) showed higher binding affinity (-6.9 kcal/mol) than the parent compound MGP (1). It has been seen from the structural contrast compound (8) has four additional acyl substituents in the monosaccharide ring, providing a high density of electron in the molecule leading to the highest binding affinity (Fig. 7). To predict the shape and behavior of molecules nonbonding interactions are oftentimes used (Fig. 8). Among all sorts of nonbonding interactions such as CH/O, CH/N, and CH/π, the CH/O is the most noted interaction found in the drug-protein interactions. Parent MGP (1) molecule showed interactions with glycine moiety of the protein including one intensive interaction within shorter distance 2.11116Å. Besides, methionine and tyrosine interactions are observed where methionine exhibited a closed distance (2.14398 Å) interaction due to the interaction of branched alkyl chain with monosaccharide ring. In contrast with parent molecule (1), compound (8) exhibited mostly hydrophobic interactions such as alkyl and Pi-Alkyl. Again, compound (8) showed most interactions in the case of glutamine with a closed distance 2.37494 Å. Moreover, hydrogen and hydrophobic both types of interaction observed in the case of derivative (8). From molecular docking analysis, the major and common residue of 4ZZZ active site is Met890 and Tyr907 from diverse nonbonding interactions with the ligand in Table 4.
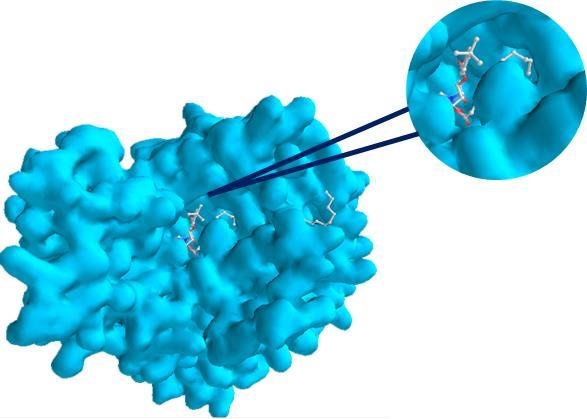
Fig. 7 Docked conformation of ester 8 at inhibition bounding site of 4ZZZ.
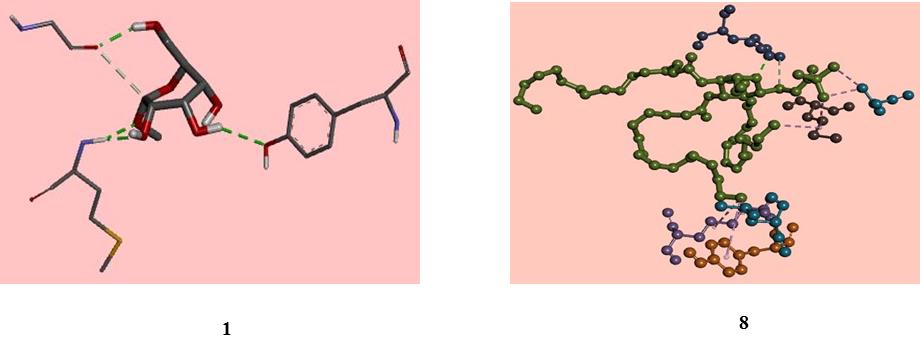
Fig. 8 Non-bonding interactions of MGP (1) and ester (8) with the amino acid residues of 4ZZZ generated by Discovery Studio.
Table 5. Pharmacokinetic properties of MGP (1) esters
|
Compound No. |
BBB |
Human intestinal absorption |
P-glycoprotein inhibitor |
hERG |
Carcinogen |
Acute oral toxicity |
|
1 |
+(0.8193) |
+(0.9782) |
NI(0.9612) |
WI(0.5371) |
NC(0.9857) |
III |
|
2 |
+(0.8193) |
+(0.9782) |
NI(0.9612) |
WI(0.5371) |
NC(0.9857) |
III |
|
3 |
+(0.9479) |
+(0.6389) |
NI(0.5963) |
WI(0.5051) |
NC(0.9000) |
III |
|
4 |
+(0.9532) |
+ (0.4042) |
NI(0.7355) |
WI(0.4495) |
NC(0.8714) |
III |
|
5 |
+(0.9634) |
+ (0.4042) |
NI(0.7641) |
WI(0.4627) |
NC(0.8714) |
III |
|
6 |
+(0.9667) |
+ (0.4042) |
NI(0.7949) |
WI(0.4091) |
NC(0.8714) |
III |
|
7 |
+(0.9693) |
+(0.8102) |
NI(0.7553) |
WI(0.3916) |
NC(0.9143) |
III |
|
8 |
+(0.9667) |
+ (0.4042) |
NI(0.7641) |
WI(0.6682) |
NC(0.8714) |
III |
|
9 |
+(0.9606) |
+(0.9208) |
NI(0.8798) |
WI(0.9007) |
NC(0.8857) |
III |
|
10 |
+(0.9732) |
+(0.8513) |
NI(0.8510) |
WI(0.8055) |
NC(0.8429) |
III |
+ = Positive, I = Inhibitor, NI = Non-Inhibitor, WI = Weak Inhibitor, NC = Non-Carcinogenic, III = Category III includes compounds with LD50 greater than 500 mg/kg but less than 5000 mg/kg.
3.6. ADMET Analysis
Pharmacokinetic properties have been enumerated to compare the absorption, metabolism, and toxicity of all modified MGP (1) esters. AdmetSAR calculation (Table 5) predicts these monosaccharide derivatives are non-carcinogenic and possess category III oral toxicity, so MGP (1) esters can be suggested to be relatively harmless for oral administration. All drugs are P-glycoprotein non-inhibitor where P-glycoprotein inhibitor can obstruct the absorption, permeability, and retention of the drugs. All drugs exhibit favorable positivity allowing for the blood-brain barrier. However, all the MGP (1) esters have shown weak inhibitory characteristics for human ether –a go-go-Related Gene (hERG). Inhibitory characteristic of hERG can lead to long QT syndrome [33], that’s why further investigation is required on this aspect.
In this study, the intrinsic stability, thermochemical features and biochemical interactions of monosaccharide MGP (1) and its modified esters are studied. The most important properties for biological chemistry, thermophysical properties, chemical reactivity, and molecular orbital study like HOMO, LUMO, HOMO-LUMO gap, and molecular electrostatic potential in molecule were optimized which indicates that it may a potential drug molecules. Most of the modified monosaccharide derivatives have the HOMO-LUMO gap closer to MGP (1) and have exalted pharmacokinetic properties than the parent molecule (1). That’s why it may be said that some modified compounds were may more reactive than thymidine, as the showed lower HOMO-LUMO gaps and some may stable like MGP (1). The electrostatic potential map showed probable electrophilic and nucleophilic attacking zone of some derivatives. Besides that, the docked complex of ester (8) and 4ZZZ shows a better binding affinity with significant nonbonding interactions than the parent structure. ADMET analysis suggested that the modified esters were less toxic and have improved pharmacokinetic scheme than the parent drug. Finally, this study may be useful to realize the thermochemical, physicochemical, biological, and pharmacological properties of MGP (1) esters.
The authors are very much thankful to the Ministry of Science and Technology (MOST), Government of Bangladesh (Ref.: 39.00.0000.09.06.79.2017/Phy’s-437, 2017-2018) for providing financial support to carry out this research project. Also, this study was supported by JSPS KAKENHI under grant No. JP19K06239 (Y.O. and Y.F.), and also by the Research Promotion Fund of Yokohama City University, and Nagasaki International University.
Abbreviations
MGP: Methyl β-D-galactopyranoside (1); EAC: Ehrlich Ascites Carcinoma; PARP1: poly [ADP-ribose] polymerase1; DFT: Density Functional Theory; HOMO: Highest Occupied Molecular Orbital; LUMO: Lowest Unoccupied Molecular Orbital; QM: Quantum Mechanical; LYP: Lee, Yang and Parr’s; DOS: Density of states; MEP: Molecular electrostatic potential; hERG: Human Ether-A-Go-Go-Related Gene; BBB: Blood-Brain Barrier.
Gabius HJ, Siebert HC, André S, Jiménez-Barbero J, Rudiger H (2004) Chemical biology of the sugar code. Chem Bio Chem 5:740-764. PMid:15174156
View Article PubMed/NCBIFraser-Reid BO, Tatsuta K, Thiem J (Eds.) Glycoscience: Chemistry and Chemical Biology, Springer-Verlag Berlin Heidelberg, 2008, 2nd Edition. PMid:18989931
View Article PubMed/NCBIVarki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Essential of Glycobiology. Cold Spring Harbor Laboratory Press, 2009, 2nd Edition.
View ArticleGabius HJ, André S, Kaltner H, Siebert HC (2002) The sugar code: functional lectinomics. Biochim Biophys Acta 1572:165-177. 00306-9
View ArticleFernandez-Tejada A, Canada FJ, Jiménez-Barbero J (2015) Glycans in medicinal chemistry: An underexploited resource. Chem Med Chem 10:1291-1295. PMid:25974358
View Article PubMed/NCBIMingeot-Leclercq MP, Glupczynski Y, Tulkens PM (1999) Aminoglycosides: activity and resistance. Antimicrob Agents Chemother 43:727-737. PMid:10103173
View Article PubMed/NCBICampo VL, Aragao-Leoneti V, Carvalho I (2013) Glycosidases and diabetes: metabolic changes, mode of action and therapeutic perspectives. Carbohydr Chem 39:181-203.
View ArticleDabhi AS, Bhatt NR, Shah MJ (2013) Voglibose: an alpha glucosidase inhibitor. J Clin Diagn Res 7:3023-3027.
Cummings RD (2009) The repertoire of glycan determinants in the human glycome. Mol Biosyst 5:1087-1104. PMid:19756298
View Article PubMed/NCBIGabius HJ, André S, Jiménez-Barbero J, Romero A, Solís D (2011) From lectin structure to functional glycomics: principles of the sugar code. Trends Biochem Sci 36: 298-313. PMid:21458998
View Article PubMed/NCBIMallajosyula SS, MacKerell AD (2011) Influence of solvent and intramolecular hydrogen bonding on the conformational properties of O-linked glycopeptides. J Phys Chem B 115:11215-11229. PMid:21823626
View Article PubMed/NCBIFadda E, Woods RJ (2010) Molecular simulations of carbohydrates and proteincarbohydrate interactions: motivation, issues and prospects. Drug Discov Today 15:596-609. PMid:20594934
View Article PubMed/NCBIMisbah MH, Ferdous J, Bulbul MZH, Chowdury TS, Kawsar SMA (2020) An efficient, easy route for the synthesis of methyl β-D-galactopyranoside derivatives. J Bang Chem Soc 32: In press.
Gaussian09 RA, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA et al, Gaussian, Inc, Wallingford CT., 2009.
View ArticleBecke AD (1988) Density-functional exchange-energy approximation with correct asymptotic behaviour. Physical Review A 38:3098-3100. PMid:9900728
View Article PubMed/NCBILee C, Yang W, Parr RG (1988) Development of the colle-Salvetti correlation-energy formula into a functional of the electron density. Physical Review B 37:785-789. PMid:9944570
View Article PubMed/NCBIPearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci 83:8440-8441. PMid:16578791
View Article PubMed/NCBIBerman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H (2000) The Protein Data Bank. Nucleic Acids Res 28:235-242. PMid:10592235
View Article PubMed/NCBIDelano wl, The PyMOL Molecular Graphics System. De-Lano Scientific, San Carlos, CA, USA, 2002.
View ArticleGuex N, Peitsch MC (1997) Swiss-model and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 18:2714-2723. PMid:9504803
View Article PubMed/NCBISeeliger D, De Groot BL (2010) Conformational transitions upon ligand binding: holo-structure prediction from apo conformations. PLoS Comput Biol 6:e1000634. PMid:20066034
View Article PubMed/NCBIDallakyan A, Olson J Small-Molecule Library Screening by Docking with Py Rx. In: Hempel JE, Williams CH, Hong CC (Eds.). Chemical Biology: Methods and Protocols. Springer New York, USA, (2015) 243-250. PMid:25618350
View Article PubMed/NCBIVersion ADS 4.0, Accelrys, San Diego, USA, 2017.
Cheng F, Li W, Zhou Y, Shen J, Wu Z, Liu G, et al (2012) admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099-3105. PMid:23092397
View Article PubMed/NCBICohen N, Benson SW (1993) Estimation of heats of formation of organic compounds by additivity methods. Chem Rev 93:2419-2438.
View ArticleLien EJ, Guo ZR, Li RL, Su CT (1982) Use of dipole moment as a parameter in drug-receptor interaction and quantitative structure-activity relationship studies. J Pharm Sci 71:641-655. PMid:7097526
View Article PubMed/NCBISaravanan S, Balachandran V (2014) Quantum chemical studies, natural bond orbital analysis and thermodynamic function of 2, 5-dichlorophenylisocyanate. Spectrochim Acta Part A Mol Biomol Spectrosc 120:351-364. PMid:24200649
View Article PubMed/NCBIHoque MM, Halim MA, Sarwar MG, Khan M (2015) Palladium‐catalyzed cyclization of 2-alkynyl‐N‐ethanoyl anilines to indoles: synthesis, structural, spectroscopic, and mechanistic study. J Phys Org Chem 28:732-742.
View ArticleParr RG, Zhou Z (1993) Absolute hardness: unifying concept for identifying shells and subshells in nuclei, atoms, molecules, and metallic clusters. Acc Chem Res 26:256-258.
View ArticleAmin ML (2013) P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013:27-34. PMid:24023511
View Article PubMed/NCBIPolitzer P, Murray JS (1991) Molecular electrostatic potentials and chemical reactivity. Rev Comput Chem 2:273-312.
View ArticlePolitzer P, Truhlar DG (Eds.), Chemical applications of atomic and molecular electrostatic potentials, Plenum Press, NY, 1981.
View ArticleSanguinetti MC, Firouz MT (2006) hERG potassium channels and cardiac arrhythmia. Nature 440:463-469. PMid:16554806
View Article PubMed/NCBI