Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance Through Integrin αv in Glioblastoma Cells
 Qi Yu
Qi Yu Weikun Xiao1
Weikun Xiao1 - 1Department of Bioengineering, University of California, Los Angeles, Los Angeles, CA, United States
- 2Department of Neurosurgery, Shengjing Hospital of China Medical University, Shenyang, China
- 3Jonsson Comprehensive Cancer Center, University of California, Los Angeles, Los Angeles, CA, United States
- 4Brain Research Institute, University of California, Los Angeles, Los Angeles, CA, United States
- 5Broad Stem Cell Research Center, University of California, Los Angeles, Los Angeles, CA, United States
Chemotherapy resistance to glioblastoma (GBM) remains an obstacle that is difficult to overcome, leading to poor prognosis of GBM patients. Many previous studies have focused on resistance mechanisms intrinsic to cancer cells; the microenvironment surrounding tumor cells has been found more recently to have significant impacts on the response to chemotherapeutic agents. Extracellular matrix (ECM) proteins may confer cell adhesion-mediated drug resistance (CAMDR). Here, expression of the ECM proteins laminin, vitronectin, and fibronectin was assessed in clinical GBM tumors using immunohistochemistry. Then, patient-derived GBM cells grown in monolayers on precoated laminin, vitronectin, or fibronectin substrates were treated with cilengitide, an integrin inhibitor, and/or carmustine, an alkylating chemotherapy. Cell adhesion and viability were quantified. Transcription factor (TF) activities were assessed over time using a bioluminescent assay in which GBM cells were transduced with lentiviruses containing consensus binding sites for specific TFs linked to expression a firefly luciferase reporter. Apoptosis, mediated by p53, was analyzed by Western blotting and immunocytofluorescence. Integrin αv activation of the FAK/paxillin/AKT signaling pathway and effects on expression of the proliferative marker Ki67 were investigated. To assess effects of integrin αv activation of AKT and ERK pathways, which are typically deregulated in GBM, and expression of epidermal growth factor receptor (EGFR), which is amplified and/or mutated in many GBM tumors, shRNA knockdown was used. Laminin, vitronectin, and fibronectin were abundant in clinical GBM tumors and promoted CAMDR in GBM cells cultured on precoated substrates. Cilengitide treatment induced cell detachment, which was most pronounced for cells cultured on vitronectin. Cilengitide treatment increased cytotoxicity of carmustine, reversing CAMDR. ECM adhesion increased activity of NFκB and decreased that of p53, leading to suppression of p53-mediated apoptosis and upregulation of multidrug resistance gene 1 (MDR1; also known as ABCB1 or P-glycoprotein). Expression of Ki67 was correlative with activation of the integrin αv-mediated FAK/paxillin/AKT signaling pathway. EGFR expression increased with integrin αv knockdown GBM cells and may represent a compensatory survival mechanism. These results indicate that ECM proteins confer CAMDR through integrin αv in GBM cells.
Introduction
Glioblastoma (GBM) is the most common primary brain tumor and the most aggressive in nature. For many decades, the standard therapy for GBM remains the same, including maximum feasible surgical resection, followed by radiation (XRT) plus concomitant chemotherapy with temozolomide (TMZ), and then followed by adjuvant TMZ (Ellor et al., 2014; Patel et al., 2014). The combined therapy resulted in an improved median overall survival from 12.1 to 14.6 months and an increase in the 2 years survival rate from 10 to 27% (Miranda et al., 2017). The Gliadel wafer (Arbor Pharmaceuticals, Atlanta, GA) is a carmustine-impregnated wafer that is placed in the surgical cavity after maximal tumor resection. Prospective data regarding use of carmustine wafers included all high-grade gliomas, and some of the survival benefits were largely a result of grade 3 patients with long-term survival (Chowdhary et al., 2015). Some retrospective studies also suggest a modest gain of 2–3 months for patients with GBM (Dixit et al., 2011). Nevertheless, the rising chemotherapeutic agent resistance leading to the treatment failure is still a challenge and has been one of the priorities in neuroscience. Additional concomitant chemotherapeutics for newly diagnosed GBM have not shown an incremental survival benefit. Concomitant bevacizumab with TMZ against newly diagnosed GBM in two large phase III trials showed prolonged progression-free survival but failed to show survival benefit (Chinot et al., 2014; Gilbert et al., 2014). Similarly, cilengitide, as an selective αvβ3 and αvβ5 integrin inhibitor, als although cilengitide exhibited antitumor activity in phase II trials. it failed to show overall survival improvement in patients with methylated MGMT promoter in phase III trials (Stupp et al., 2014; Nabors et al., 2015). Rindopepimut is a peptide vaccine against the most common epidermal growth factor receptor (EGFR) deletion mutation (EGFR variant III) in GBM. Although single-arm phase II studies showed promising results, a randomized phase III trail (ACT IV) failed to show benefit over the control group (Weller et al., 2017).
Advanced knowledge has been established with regard to the mechanism of chemoresistance in GBM. One reason why GBM has a very poor prognosis is related with the lack of successful drug delivery across the physiologic barriers, especially the blood–brain barrier (BBB), which protects the central nervous system (CNS) from the passage of foreign and harmful substances through the bloodstream (Pourgholi et al., 2016). The relevant mechanisms underlying the barrier function include enzymatic barrier, transport barrier (para-cellular and transcellular), immunologic barrier, and efflux transport systems (Hendricks et al., 2015). Mutation of DNA repair systems is another cause responsible for the chemoresistance such as enhanced MGMT activity, impaired DNA mismatch repair system, and enhanced base excision repair system (Thon et al., 2013; Messaoudi et al., 2015; Motegi et al., 2019). In addition, other factors have been shown to interfere with TMZ activity, contributing to the poor prognosis of GBM. Such factors include EGFR, PI3K/AKT/mTOR pathway (the mechanistic target of rapamycin), galectin-1, p53, murine double minute 2 (Mdm2), ATP-binding cassette transports, phosphatase and tensin homolog (PTEN), isocitrate dehydrogenase (IDH-1), and cell cycle checkpoint pathways (Messaoudi et al., 2015; Tivnan et al., 2015; Pourgholi et al., 2016; Yan et al., 2016; Kim and Kim, 2020; Pine et al., 2020). More recently, a series of publications identified cell adhesion to the extracellular matrix (ECM) as a key determinant among the myriad of microenvironmental factors impacting cancer cell resistance (Xiao et al., 2017). It has been reported that adhesion to laminin, vitronectin, or fibronectin confers cell adhesion-mediated drug resistance (CAMDR) in various cancer models (Fei et al., 2014; Nakagawa et al., 2014; Sun et al., 2014; Sanchez et al., 2019). Data have revealed that αvβ3 and αvβ5 integrins mediate the interaction between endothelial cells and components of the ECM (Varner et al., 1995). Integrin αvβ3 binds to Arg-Gly-Asp (RGD) in vitronectin, fibronectin, and fibrinogen, among other substrates. Cilengitide, one of the cyclic RGD peptides, was found to disrupt VE-cadherin localization at cell junctions, increase endothelia monolayer permeability, and restrain angiogenesis in vitro and in vivo (Chowdhary et al., 2015).
The p53 transcription factor (TF) was initially known as the guardian of the genome due to the fact that it prevents the proliferation of cells with damaged nuclear DNA (Olotu and Soliman, 2019; Horikawa, 2020). However, p53 acts as a TF to regulate the expression of a variety of genes that coordinate the DNA damage responses. On the one hand, it can initiate apoptosis through death receptor and mitochondrial pathway. On the other hand, it can arrest growth by holding the cell cycle at the G1/S regulation point through p53-dependent p21WAF1/CIP1, which binds to the G1-S/CDK complex and inhibits their activity.
Carmustine [1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU)] was first introduced for chemotherapy against malignant gliomas in the 1980’s (Walker et al., 1978; Walker et al., 1980). During the past 30 years, the evaluation of the role of chemotherapy has not produced impressive results to date. Beyond TMZ, the US Food and Drug Administration (FDA) has approved two other agents for treatment of newly diagnosed malignant glioma till now: carmustine wafers and bevacizumab (Affronti et al., 2009). Gliadel wafers (Arbor Pharmaceuticals, Atlanta, GA) are commercial products of biodegradable copolymers (prolifeprospan 20) impregnated with carmustine. Wafer efficacy has been well documented (Stewart, 2002). Subsequent trials revealed increased benefit from combining Gliadel wafers with XRT/TMZ. Median overall survival tended to be improved by 3–4 months beyond that observed for Gliadel wafers or TMZ when used alone in the respective III trials (Ashby et al., 2016).
In this study, we found that laminin, vitronectin, and fibronectin, three main components of ECM proteins, could affect CAMDR in GBM cells. Enrichment of these proteins in the microenvironment promotes tumor cell proliferation through integrin αv-mediated FAK/paxillin/AKT signaling pathway and suppresses p53-mediated apoptosis. In addition, we found that the efflux transporter ABCB1 was elevated with ECM adhesion. Compensatory activation of EGFR occurred when integrin αv was knocked down. These findings will provide promising insights to overcome chemotherapeutic resistance for GBM.
Materials and Methods
Cell Lines, Cell Culture, and Treatment Setup
The GBM cell line U87MG was obtained from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Primary GBM cell line HK308 (patient background: a 50-year-old female with recurrent GBM with wild-type IDH 1/2 received XRT, TMZ, and Avastin treatment) was established from patient tumors in accordance with UCLA Institutional Review Board protocol 10-000655 and generously gifted by Dr. Harley Kornblum at UCLA. Authentication was conducted by immunoblot studies (Laks et al., 2016). Primary GBM6 (patient background: a 65 years old male with newly diagnosed GBM with mutant p53 and amplification of EGFR vIII mutant received XRT and TMZ treatment) was obtained from Dr. David Nathanson and authenticated by DNA fingerprinting (Sarkaria et al., 2006; Akhavan et al., 2013). DMEM/F12 with G21 (Gemini Bio, Sacramento, CA, United States, 1:50), 50 ng/ml of EGF (PeproTech, Rocky Hill, NJ, United States), 20 ng/ml of FGF-2 (PeproTech), 25 μg/ml of heparin (Sigma-Aldrich, St. Louis, MO, United States), and 1% penicillin/streptomycin was used for primary GBM cell line culture. All cell lines were used fewer than 30 passages and incubated in a 5% CO2-humidified incubator at 37°C.
ECM proteins including laminin (Thermo Fisher Scientific, Waltham, MA, United States), vitronectin and fibronectin (Sigma-Aldrich, St. Louis, MO, United States) were used, respectively, as coating protein in a 12-well plate at a dilution of 10 μg/ml. The plate was then incubated at 37°C for over 4 h before the cells were seeded. Carmustine was purchased from Sigma-Aldrich and dissolved at 100 mmol/L in 100% ethanol for stocking. It was then added in culture medium at a final concentration of 100 μmol/L. Ethanol alone was used as the negative control. Cilengitide (Sigma-Aldrich, St. Louis, MO, United States) was dissolved in phosphate-buffered saline (PBS) as 10 mmol/L of stock and then added in culture medium at a final concentration of 50 μmol/L.
Tissue Microarray
Tissue microarray (TMA) containing 34 GBM samples and 19 other low-grade CNS tumors were employed to analyze laminin, vitronectin, and fibronectin expressions. All the patients had given informed consent, and collection of these tissue samples had been studied for other researches before (Choe et al., 2003; Guo et al., 2009; Lu et al., 2009). TMA was a high-throughput screening platform that enables a bunch of patient tumor samples to be analyzed on the same slide. It was constructed using a 0.6 mm needle to exact 54 representative tumor tissue cores from the paraffin-embedded tissue blocks (Akhavan et al., 2013). Theses cores were placed in a grid pattern into two recipient paraffin blocks, from which tissue sections were cut for immunohistochemistry (IHC) analysis, as previously described (Zhou et al., 2015). Briefly, the slides were first deparaffinized, followed by blocking with 30% normal donkey serum for 10 min. Then the sections were incubated with the primary laminin, vitronectin, and fibronectin antibodies (Thermo Fisher, PA1-16730/PA5-27909/PA1-26205, respectively; Waltham, MA, United States, dilution 1:500) overnight at 4°C. Appropriate secondary antibodies were applied for 30 min. Negative controls were carried out by replacement of the primary antibody with substituting PBS. Images were scored and calculated by Fisher’s exact test.
Cell Adhesion Assay
A 96-well plate was coated with PBS (as negative control) and 10 μg/ml of laminin, vitronectin, and fibronectin, over 4 h at 37°C. To block any remaining protein binding sites on the plate, coating solutions were removed, and then 1% bovine serum albumin (BSA) was added for another hour. Appropriate density of GBM cell suspension (10,000 cells/well for GBM6; 5,000 cells/well for U87MG and HK308) was seeded, followed by another 2 h incubation. Non-adherent GBM cells were removed by careful washing two times with PBS. Then, 100% ethanol was used to fix the adherent cells for 15 min followed by 0.1% crystal violet (Thermo Fisher Scientific, Pittsburgh, PA, United States, dissolved in 100% ethanol) staining for another 30 min. After excessive stain was removed and 0.3% Triton-X was applied to lyse cells, the absorbance was measured at 570 nm using a microplate reader (BioTek, Winooski, VT, United States). The percentage of adhesion was determined by dividing the corrected (background subtracted) optical density of adherent cells by the total corrected optical of cells added to each microplate well and multiplying by 100%. Experiments were repeated three times with five replications per experiment.
Cell Viability Assay
Cell viability and drug–response curves were assessed using a CellTiter 96 AQueous One Solution (MTS) kit (Promega, Madison, WI, United States) as described before (Kong et al., 2015). Briefly, GBM cells (5,000 cells/well for GBM6; 3,000 cells/well for U87MG and HK308) were cultured on a 96-well plate precoated with PBS, laminin, vitronectin, or fibronectin for 24 h. Cells were then treated with 100 μM of carmustine or ethanol (as control). At the end of the treatment, 20 μl/well of MTS solution was added and incubated at 37°C for 2 h. Absorbance was measured at 490 nm. All data points were set up with five replicates for each experiment. The IC50 was determined by GraphPad Prism Software Version 7 (San Diego, CA, United States).
Immunofluorescent Staining
GBM cells were fixed with 4% paraformaldehyde for 20 min at room temperature and permeabilized with a blocking solution containing 5% BSA and 0.01% Triton X-100 diluted in PBS. Then, cells were incubated overnight at 4°C with anti-Ki67 primary antibody (1:200, Thermo Fisher Scientific, Waltham, MA, United States). Goat anti-rabbit secondary antibody was then added at a dilution 1:500 for 2 h. Secondary antibody alone without primary antibody was used as negative control. All the GBM cells were later counterstained with Hoechst 33242 (Thermo Fisher Scientific, Waltham, MA, United States). Glass coverslips were mounted with fluorescence Mounting Medium (SouthernBiotech, Birmingham, AL, United States). Images were captured with an AXIO-Observer inverted microscope equipped for wide-field fluorescence and phase contrast (Zeiss, Oberkochen, Germany).
Lentiviral Construction and Transfection
Plasmids pTA-/p53-/NFκB-/c-myc-FLuc were kindly obtained from Dr. Lonnie D. Shea at Northwestern University. The plasmids were designed so that each contained a consensus binding sequence for a particular TF in the enhancer region upstream of a minimal TATA-box promoter driving expression of the reporter gene firefly luciferase (Supplementary Table 1; Penalver Bernabe et al., 2016). TP53bp1 was a gift from Nicola Burgess-Brown (Addgene plasmid #73252). ITGAV shRNA was obtained from Dharmacon (V2LHS_133468, Lafayette, CO, United States). Lentivirus was produced by co-transfecting HEK-293T cells with a third-generation packaging system (Dull et al., 1998). As quantified by lentix-rtPCR kit (Takara), transduction was performed with a virus concentration of 2,000 physical particles/cell. Fresh media were replaced 24 h after transfection. TA-/p53-/NFκB-/c-myc-FLuc, integrin αv knockdown, and TP53bp1+ stable cell lines were created and continuously cultured for 3 days before use in the subsequent assay. All experiments with regard to the virus were performed on BSL2 laboratory under relevant management regulations.
Bioluminescence Assay
TF activity was assessed by bioluminescence imaging of firefly luciferase using an IVIS imaging system (Caliper Life Sciences, Hopkinton, MA, United States) as described previously (Bellis et al., 2011). D-Luciferin (Sigma-Aldrich, St. Louis, MO, United States) was added as the substrate for Fluc at 1 μM/well, followed by incubation for 1 h. Exposure time was 5 min, and images were taken every 24 h for 3 days. After each time point of any dynamic imaging experiment, the medium was refreshed. Normalized TF activity was determined by dividing the normalized light emission for target TFs by the average normalized light emission for TA. Each condition was performed in triplicate.
Western Blotting Antibody Information Table
Proteins were extracted by lysing GBM cell lines in radioimmunoprecipitation assay (RIPA) buffer with a protease/phosphatase inhibitor cocktail. Then the sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transfer were performed, followed by blocking in 5% BSA as described previously (Xiao et al., 2018; Yu et al., 2018). Relevant primary antibodies were used for detecting target bands overnight at 4°C (integrin αv/β1/β3/β5, t-/p-FAK, p-paxillin, t-/p-AKT, t-/p-EGFR, p53, cleaved PARP, ABCB1, p-ERK1/2, and cyclin D1, obtained from Cell Signaling Technology, Danvers, MA, United States, dilution 1:1,000). Horseradish peroxidase (HRP) goat anti-mouse IgG or anti-rabbit IgG were used as secondary antibodies (dilution 1:2,000). Immunoreactive bands were visualized using Clarity ECL substrate (Bio-Rad, Hercules, CA, United States) and imaged (MyECL imager) without overexposing the target bands. Equal loading was assessed after probing the same membrane with anti-GAPDH antibody (Thermo Fisher Scientific, Waltham, MA, United States). Images of blots were analyzed using ImageJ (NIH).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism Software Version 7 (San Diego, CA, United States). Two-way ANOVA test was applied to examine the statistical significance followed by Dunnett’s test as a post hoc test for within- or between-group comparisons. Probability values less than 0.05 were regarded as statistically significant.
Results
Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance in Glioblastoma Cell Lines
To investigate the impact of ECM on GBM tumor cells, IHC of patient TMAs was used to analyze expression of laminin, vitronectin, and fibronectin in GBM and low-grade CNS tumors (Figure 1A). Compared with tumors in the low-grade CNS group, GBM tumors showed higher expression of laminin (p = 0.0178) and vitronectin (p < 0.0001). Fibronectin was widely expressed in both GBM and low-grade CNS tumor groups. An in vitro assay showed that fibronectin induced the highest degree of attachment for the U87 GBM cell line and two patient-derived GBM cell lines (GBM6 and HK308), followed by vitronectin and laminin, respectively (Figure 1B). All ECM proteins investigated significantly increased attachment over non-coated substrates (laminin, p < 0.05 for all; vitronectin, p < 0.05 for all; fibronectin, p < 0.05 for GBM6; p < 0.01 for U87MG and HK308).
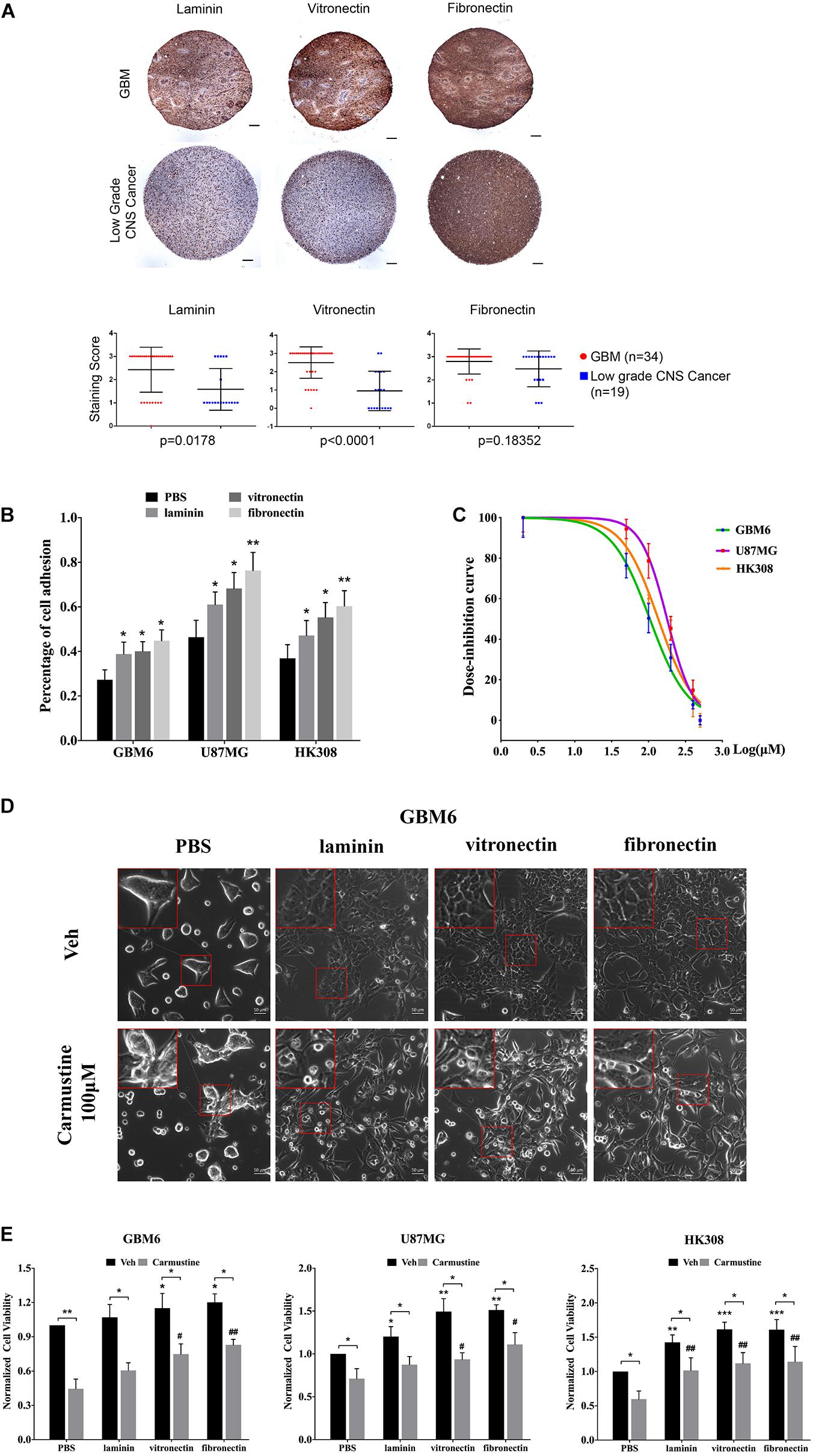
Figure 1. Extracellular matrix (ECM) proteins confer cell adhesion-mediated drug resistance (CAMDR) in glioblastoma (GBM) cell lines. (A) Tissue microarray (TMA) of expressions of laminin, vitronectin, and fibronectin in patient-derived GBM and low-grade central nervous system (CNS) tumors. Scale bars, 100 μm. GBM group, n = 34; low-grade CNS cancer, n = 19. (B) Attachment assay of GBM cell lines GBM6, U87MG, and HK308 on fibronectin, laminin, and vitronectin. Error bars, SD (n = 3). (C) Dose inhibition curve of carmustine on GBM6, U87MG, and HK308. Error bars, SD (n = 3). (D) Representative light images of GBM6 with or without carmustine [100 μM, phosphate-buffered saline (PBS) as control] treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm. (E) Cell viability assay of carmustine on GBM6, U87MG, and HK308. Error bars, SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0.01.
Adhered cells were treated with carmustine, an alkylating agent that forms interstrand cross-links in DNA to prevent its replication and transcription. GBM cells were inhibited by carmustine in a dose-dependent manner, with a half maximal inhibitory concentration (IC50) of 106 ± 13 μM for GBM6, 179 ± 29 μM for U87MG, and 137 ± 27 μM for HK308 (Figure 1C). To assess whether ECM proteins conferred CAMDR, GBM cells were cultured on substrates precoated with PBS (as a negative control), laminin, vitronectin, or fibronectin for 24 h prior to treatment with carmustine (or vehicle control) for 48 h. In the absence of precoated ECM, carmustine treatment significantly reduced numbers of adhered cells (Figure 1D and Supplementary Figure S1). While some rounded cells were observed on ECM-coated substrates, more rounded cells were apparent on non-coated substrates after treatment. Quantitative assays showed a significant increase in cell viability when cultured on either vitronectin- or fibronectin-coated (compared with uncoated) substrates (vitronectin, p < 0.05 for GBM6, p < 0.01 for U87MG, p < 0.001 for HK308; fibronectin, p < 0.05 for GBM6, p < 0.01 for U87MG, p < 0.001 for HK308), while adhesion to laminin significantly increased viability of U87MG and HK308 (p < 0.05 for U87MG; p < 0.01 for HK308), but not GBM6, cells (Figure 1E). Treatment with carmustine for 48 h significantly decreased cell viability in all cases; however, ECM adhesion provided significant protection over non-coated substrates (Figure 1E, PBS, p < 0.05 for all cell lines; laminin, p < 0.05 for GBM6 and U87MG, p < 0.01 for HK308; vitronectin, p < 0.05 for U87MG, p < 0.01 for GBM6 and HK308; fibronectin, p < 0.01 for all).
Cilengitide Reverses Cell Adhesion-Mediated Drug Resistance Through Integrin αv in a Matrix-Specific Manner
Cilengitide, a selective inhibitor of the integrin αv receptor, decreases adhesion of GBM6, U87MG, and HK308 cells on precoated laminin and vitronectin, but not fibronectin (Figure 2A).
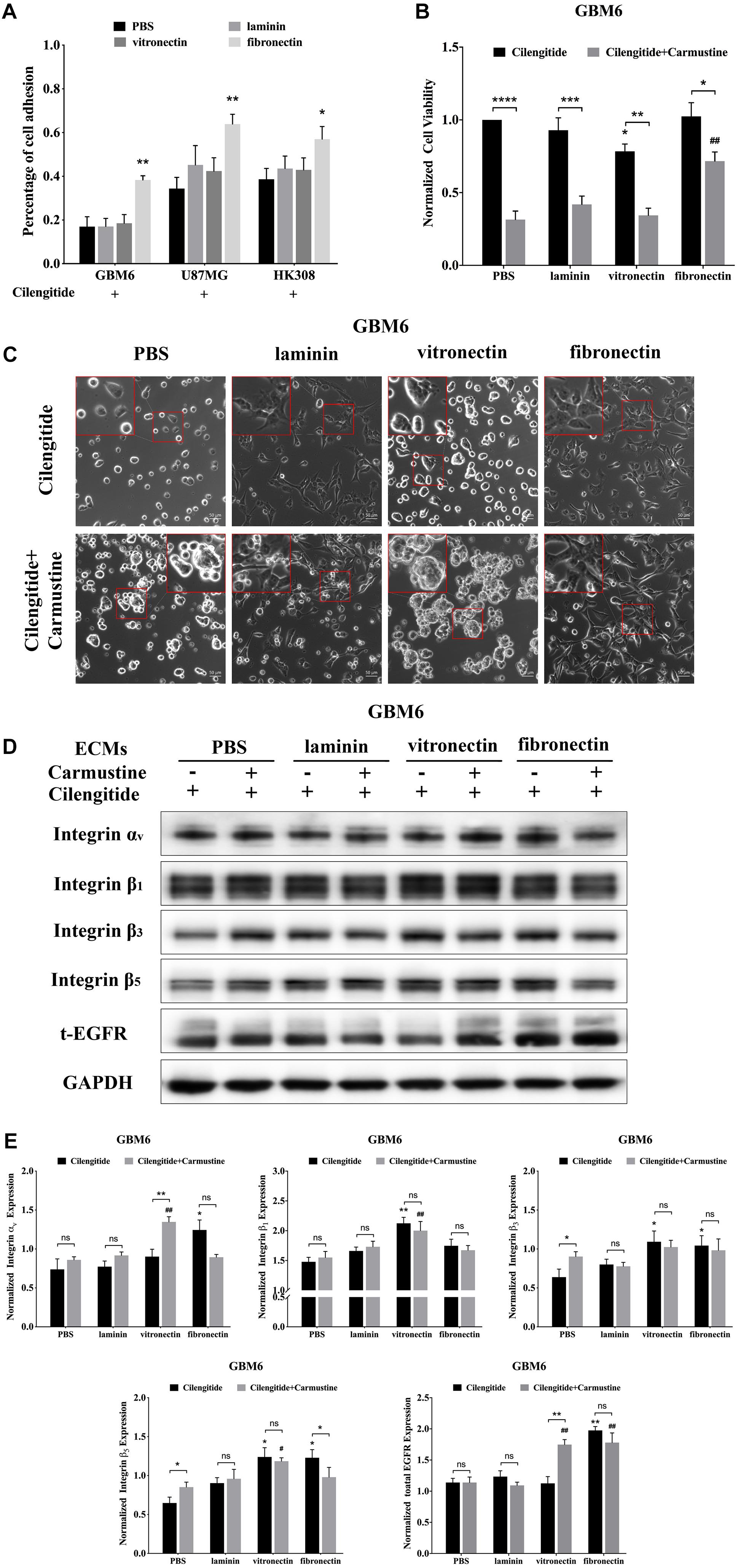
Figure 2. Cilengitide reverses cell adhesion-mediated drug resistance (CAMDR) through of integrin αv in a matrix-specific manner. (A) Attachment assay of cilengitide treated glioblastoma (GBM) cell lines GBM6, U87MG, and HK308 on fibronectin, laminin, and vitronectin. Error bars, SD (n = 3). (B) Cell viability assay of carmustine and cilengitide on GBM6 cells. Error bars, SD (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 0.05, ##p < 0.01). (C) Representative light images of GBM6 cells with or without carmustine and cilengitide [100 μM, phosphate-buffered saline (PBS) as control] treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm. (D,E) Representative western blot of GBM6 with or without carmustine and cilengitide (100 μM, PBS as control) treatment on integrin subunits (αv, β1, β3, and β5) and epidermal growth factor receptor (EGFR) expression (n = 3).
Compared with cells grown on substrates without precoated ECM protein, only coating with fibronectin results in higher levels of GBM6 cell viability (Figure 2B, p < 0.01). However, treatment with cilengitide only reduced viability of cells when cultured on vitronectin-coated substrates (Figure 2B, p < 0.05). Similarly, coating with fibronectin, but not laminin or vitronectin, leads to restoration of U87MG cell viability when treated with cilengitide plus carmustine (Supplementary Figure S2A, p < 0.05). U87MG shows less viability, with cilengitide on precoated laminin and vitronectin substrates (Supplementary Figure S2A, p < 0.05). On the contrary, cilengitide does not reverse cell viability of HK308 on any precoated substrates (Supplementary Figure S2A). In general, a loss of cell adhesion and spreading after cilengitide treatment (Figure 2C and Supplementary Figure S2B) was associated with increased efficacy carmustine treatment (Figure 2B and Supplementary Figure S2A). Cell responses after detachment by cilengitide are also different between cell types. For GBM6, detachment was only observed on precoated vitronectin, which resulted in higher chemotherapeutic toxicity. For U87MG, detachment occurred in all substrates except fibronectin, which leads to lower cell viability on relative substrates. Cilengitide did not affect cell adhesion and carmustine efficacy in HK308. Next, we investigated candidate integrins, which were required as the receptor for GBM6 cells responding to cilengitide administration on varying ECMs (Figures 2D,E). All integrin subunits evaluated were upregulated by cells cultured on fibronectin, when compared with those cultured on laminin or vitronectin (Figures 2D,E). Moreover, culture fibronectin-coated substrates also induced upregulation of EGFR expression.
Compared with GBM6, cells grown on uncoated substrates, cilengitide treatment induced upregulation of integrin subunits in an ECM-dependent way. Specifically, integrin αv expression was higher in cells grown on fibronectin, integrin β1 in cells grown on vitronectin, and integrins β3 and β5 with cells grown on fibronectin or vitronectin (Figure 2E, p < 0.05). When treated with a combination of cilengitide and carmustine, shifts in integrin expression were again dependent on the type of ECM coating. Cells grown on uncoated substrates increased expression of integrins β3 and β5, while those on fibronectin downregulated integrins αv and β5. When cultured on vitronectin, dual treatment with cilengitide and carmustine increased integrin αv and total EGFR expression.
Extracellular Matrix Induces Activation of NFκB and Deactivation of p53
Binding events to consensus sequences for the TFs NFκB, p53, and c-myc in response to treatments were assessed using GBM6 cells transduced with bioluminescent reporters of TF activity when cultured on ECM-coated substrates (Figure 3A) as previously reported (Penalver Bernabe et al., 2016). In all cases, there was a trend towards increased NFκB reporter during the 48 h after seeding (Figure 3B). This trend was more pronounced with carmustine treatment. Activity of the p53 reporter was only affected in cells seeded on uncoated substrates and treated with both carmustine and cilengitide. There were no obvious effects of any condition on c-myc activity.
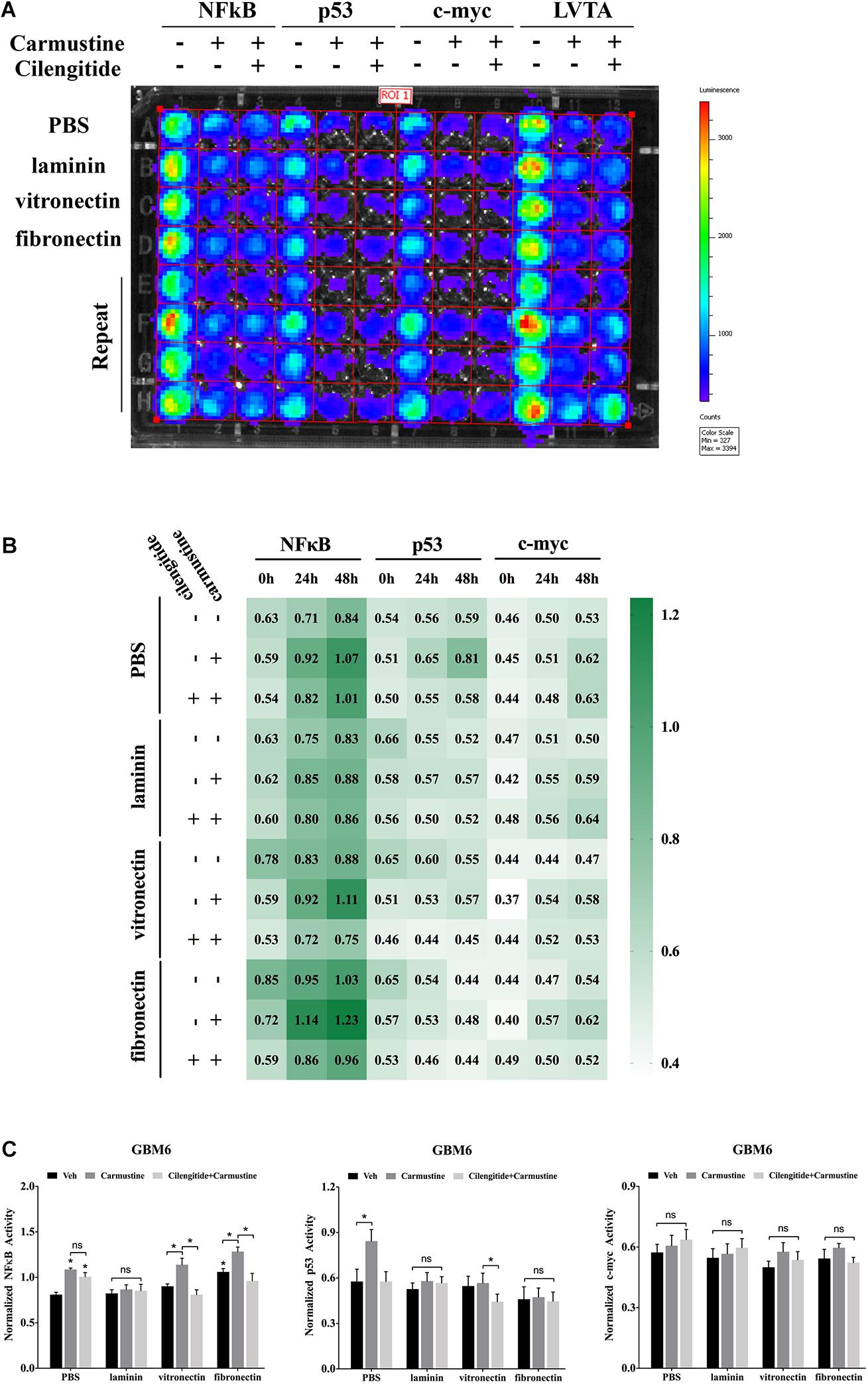
Figure 3. Extracellular matrix induces activation of NFκB and deactivation of p53. (A) Bioluminescence assay of GBM6 cells with or without carmustine and cilengitide on precoated extracellular matrix (ECM) proteins (n = 3). (B) Heatmap of sequential expression of transcription factors (TFs) in GBM6 cells over time with or without carmustine and cilengitide (n = 3). (C) Normalized TFs activity in GBM6 cells after 48 h treatment with or without carmustine and cilengitide on precoated ECM proteins (n = 3). *p < 0.05.
After 48 h of treatment with carmustine or carmustine and cilengitide, GBM6 cells on uncoated substrates exhibited significantly higher NFκB reporter activity (Figure 3C, p < 0.05). Activity of the p53 reporter increased in cells on uncoated substrates with carmustine treatment, but this effect was lost with combined treatment with carmustine and cilengitide. When seeded on vitronectin or fibronectin, NFκB activity increased significantly with carmustine treatment alone, but this effect was lost when treated with both carmustine and cilengitide (Figure 3C, p < 0.05). Activity of the p53 reporter significantly decreased with affected by dual treatment with carmustine and cilengitide in cells seeded on vitronectin (Figure 3C, p < 0.05). No other differences in p53 reporter activity were observed. Likewise, no significant changes in activities of NFκB, p53, and c-myc reporters were observed with laminin coating. No significant differences in c-myc reporter activities were observed in any conditions.
Extracellular Matrix Suppresses p53-Mediated Apoptosis and Increases ABCB1 Expression
Translocation of p53 to the nucleus, where it acts as a TF to regulate gene expression, is associated with apoptosis in response to DNA damage (Castrogiovanni et al., 2018). Western blots were performed to characterize levels of p53 in the nucleus. In untreated GBM6 cells, total p53 expression was elevated for cells when grown on vitronectin or fibronectin (Figure 4A). However, nuclear expression of p53 was unaffected by the ECM substrate in cells grown on any ECM protein. Expression of nuclear p53 was significantly higher with carmustine treatment in cells all conditions (p < 0.05). However, carmustine treatment induced higher expression of total p53 only in cells cultured on laminin and vitronectin. Dual treatment with carmustine and cilengitide significantly reduced total p53 levels, compared with carmustine alone, in GBM6 cells cultured on precoated laminin or vitronectin, but not fibronectin. Nuclear expression of p53 increased in cells on vitronectin substrates with dual treatment, compared with carmustine alone. Dual treatment had no effects on nuclear p53 levels on laminin- or fibronectin-coated substrates. Consistent with TF reporter and Western blot results, quantification of numbers of cells with nuclear TP53bp1 from immunofluorescence images showed elevated nuclear p53 with carmustine treatment and decreased by precoated laminin, vitronectin, and fibronectin, consistent with the nuclear p53 results (Figures 4B,C).
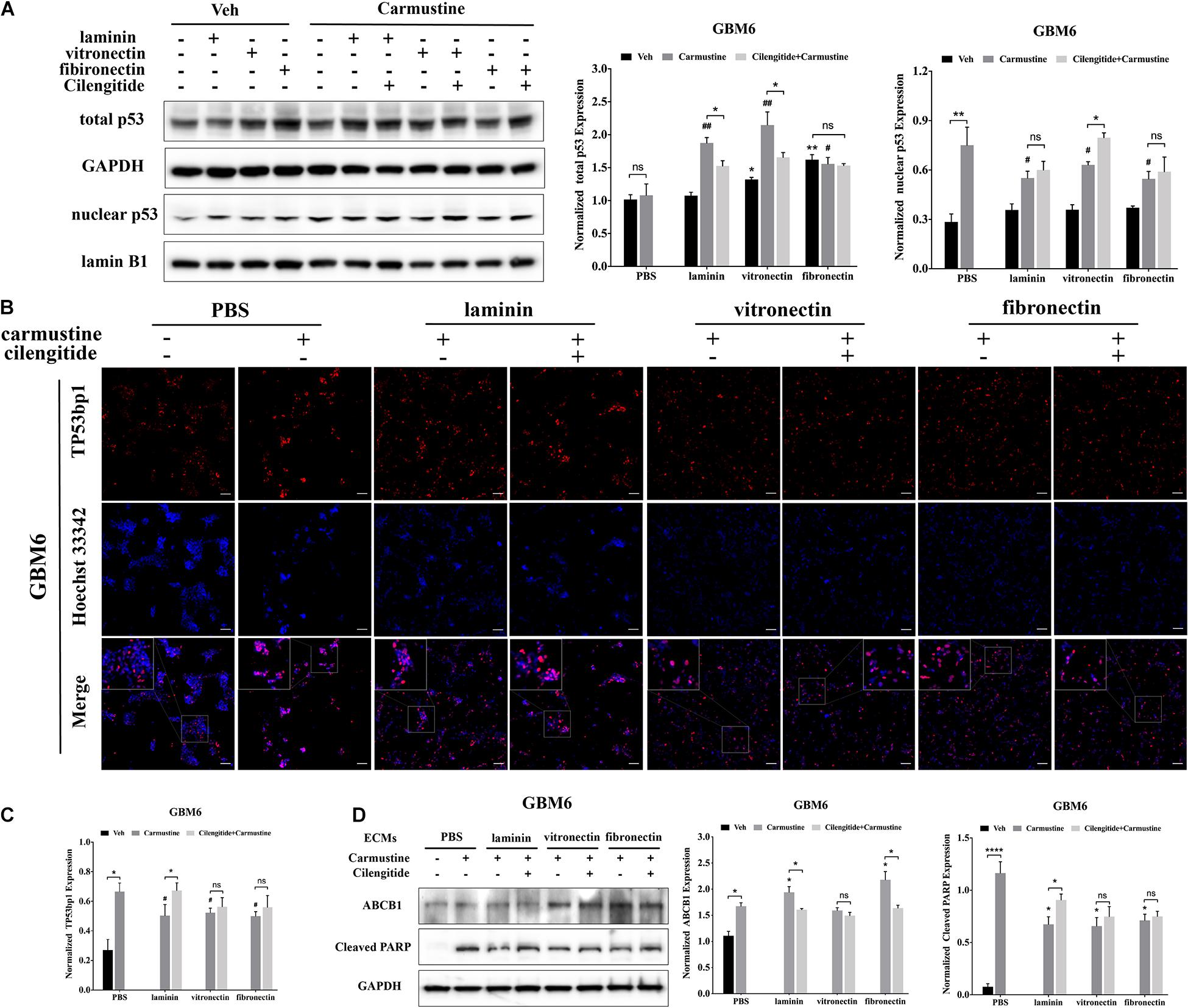
Figure 4. Extracellular matrix (ECM) suppresses p53-mediated apoptosis and increases ABCB1 expression. (A) Representative western blot of GBM6 with or without carmustine and cilengitide [100 μM, phosphate-buffered saline (PBS) as control] treatment on total p53 and nuclear p53 expression (n = 3). (B,C) Immunofluorescent staining assay of Tp53bp1 expression in GBM6 cells on precoated ECMs. Scale bars, 100 μm. (D) Western blot assay of GBM6 with or without carmustine and cilengitide (100 μM, PBS as control) treatment on ABCB1 and cleaved PARP expression (n = 3, *p < 0.05, ****p < 0.0001, #p <0.05, ##p < 0.01).
Western blots for c-PARP expression, indicative of apoptotic cells, showed an increased in c-PARP with carmustine treatment in all conditions for GBM6 (Figure 4D), and U87 and HK308 cells (Supplementary Figure S3). However, culture on ECM-coated substrates significantly reduced c-PARP, which was significantly compared with cells on uncoated substrates. In GBM6 cells cultured on laminin, dual treatment with carmustine and cilengitide significantly increased c-PARP levels.
Expression of efflux transporter ABCB1, a product of the MDR1 gene, increased with carmustine treatment in all cases (Figure 4D and Supplementary Figure S3). For carmustine-treated GBM6 cells cultured on precoated laminin or fibronectin, but not vitronectin, substrates, ABCB1 expression was significantly higher than cells on uncoated substrates. Addition of cilengitide to carmustine reduced this increase in ABCB1 expression. Similar patterns of expression for cleaved PARP and ABCB1 were detected in U87MG and HK308 (Supplementary Figure S3).
Integrin αv-Mediated FAK/Paxillin/AKT Signaling Pathway Is Essential for Glioblastoma Cell Proliferation
The FAK/paxillin signaling pathway, downstream of integrin αv, is involved in tumor progression, angiogenesis, and metastasis (Eke and Cordes, 2015; Noh et al., 2020). In the current study, Western blots showed decreased expression of integrin αv by GBM6 cells cultured on all substrates after 48 h of treatment with carmustine, when compared with non-treated cells (Figures 5A,B). Integrin αv expression was significantly elevated by carmustine-treated GBM6 cells cultured on vitronectin or fibronectin, compared with uncoated or laminin-coated substrates. On all substrates, combined treatment with carmustine and cilengitide significantly reduced expression of integrin αv compared with carmustine treatment alone (Figure 5 and Supplementary Figure S6). While integrin αv expression was significantly elevated by carmustine treatment in HK308 cells cultured on uncoated or laminin-coated substrates, expression decreased on fibronectin and was unchanged on vitronectin (Supplementary Figure S5). For HK308 cells that adhered to laminin, but not fibronectin or vitronectin, dual treatment with carmustine and cilengitide significantly reduced expression of integrin αv compared with carmustine treatment alone. Similarly, expression of integrin αv decreased with carmustine treatment in U87 cells on uncoated substrates; however, no significant effects were observed in cells on ECM-coated substrates (Supplementary Figure S4). In contrast to patient-derived GBM6 and HK308 cells, combined treatment with carmustine and cilengitide significantly upregulated expression of integrin αv in immortalized U87 cells cultured on vitronectin or fibronectin, when compared with carmustine treatment alone.
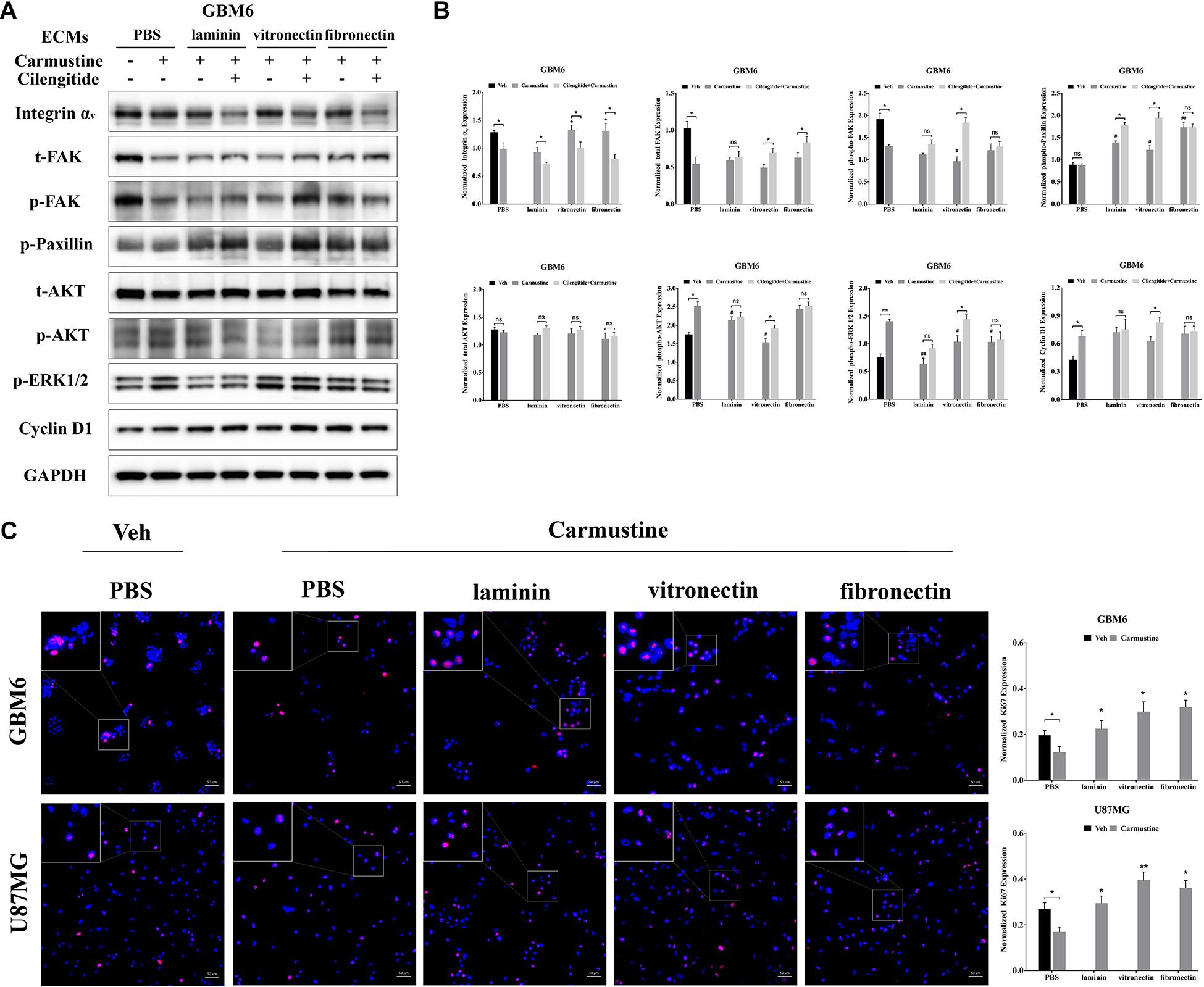
Figure 5. Integrin αv-mediated FAK/paxillin/AKT signaling pathway is essential for glioblastoma (GBM) cell proliferation. (A,B) Western blot assay of GBM6 cells with or without carmustine and cilengitide [100 μM, phosphate-buffered saline (PBS) as control] treatment on integrin αv, total/p-FAK, paxillin, total/p-AKT, p-ERK 1/2, and cyclin D1 (n = 3, *p < 0.05, **p < 0.01, #p < 0.05, ##p < 0.01). (C) Immunofluorescent staining assay of Ki67 expression in GBM6 and U87MG cells on precoated extracellular matrices (ECMs) Ki67 positive cells were normalized to total cell numbers. Scale bars, 50 μm (n = 3, *p < 0.05, **p < 0.01).
While integrin αv levels in GBM6 cells decreased when cilengitide was added to carmustine treatment, total levels of FAK increased in cultures on vitronectin and fibronectin (Figure 5). Levels of phosphorylated FAK increased significantly in GBM6 cells cultured on vitronectin, but not laminin, fibronectin, or PBS control (Supplementary Figure S6). Similarly, t-/p-FAK was decreased after carmustine treatment in cells growing on precoated PBS and was not found restored in cells growing on precoated laminin, vitronectin, or fibronectin. Phosphorylation of paxillin, a focal adhesion protein recruited to the intracellular domain of ECM-engaged integrins, was upregulated significantly when GBM6 cells on all ECM-coated, but not uncoated, substrates were treated with carmustine (Figure 5). With dual carmustine and cilengitide treatment, p-paxillin was further increased in cells on laminin and vitronectin.
In addition, p-AKT, p-ERK 1/2, and cyclin D1 were increased in a large degree by carmustine without ECM protein presence. Cilengitide induced a significant elevation of p-AKT, p-ERK 1/2, and cyclin D1 in cells growing on vitronectin. U87MG and HK308 showed variant changes but the same activation trend in FAK/paxillin/AKT signaling pathway (Supplementary Figures S4, S5). In GBM6 and U87MG cells, numbers of proliferating Ki67 positive cells were significantly decreased with carmustine treatment on precoated PBS, but this effect was lost when cells were cultured on precoated laminin, vitronectin, and fibronectin (Figure 5C).
Increased Activity of Epidermal Growth Factor Receptor-Mediated Pathways Correlates With Increased Cell Survival in Integrin αv Knockdowns
To investigate the relationship between integrin αv and EGFR-mediated oncogenic pathways, integrin αv was knocked down in GBM6 cells using a lentiviral vector encoding shRNA against integrin αv. Green fluorescent protein (GFP) was used to identify transfected cells expressing the shRNA (Figure 6A). Compared with uncoated or laminin-coated substrates, culture on vitronectin- or fibronectin-coated substrates significantly reduced efficacy of the shRNA knockdown of integrin αv, indicating its importance for cell survival (Figures 6B,C). When treated with carmustine, levels of integrin αv in knockdown cultures on uncoated and laminin substrates were elevated to those found in wild-type cultures on uncoated substrates. In contrast, knockdown cultures treated with carmustine on fibronectin further decreased integrin αv expression. Strikingly, when integrin αv expression was knocked down in GBM6 cells on all substrates, carmustine treatment induced robust apoptosis, with levels of nuclear c-PARP significantly increased over treated, wild-type cells on uncoated substrates.
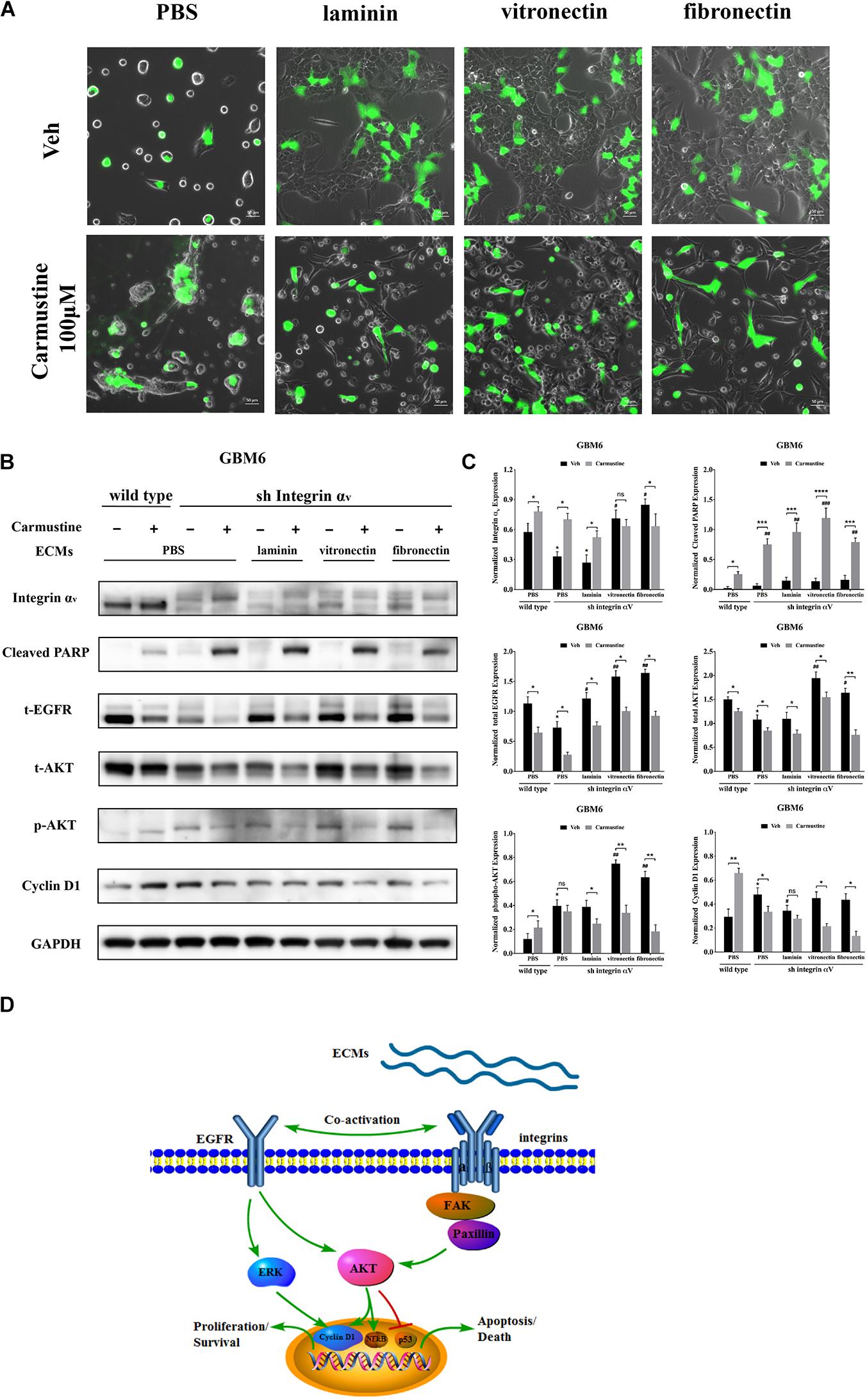
Figure 6. Increased activity of epidermal growth factor receptor (EGFR)-mediated pathways correlates with increased cell survival in integrin αv knockdowns. (A) Representative light images of GBM6 cells transfected with integrin αv shRNA [identified by green carmustine [100 μM, phosphate-buffered saline (PBS) as control] treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm. (B,C) Western blot assay of wild-type and sh integrin αv transfected GBM6 cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on integrin αv, c-PARP, total-EGFR, total/p-AKT, and cyclin D1 (n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 0.05, ##p < 0.01, ###p < 0.001). (D) Hypothetic schematic diagram illustrated the mechanism of extracellular matrices (ECMs) induced cell adhesion-mediated drug resistance (CAMDR) by activation of integrin αv-mediated FAK/paxillin/AKT signaling pathway and co-activation of EGFR-mediated pathways when integrin αv was knocked down.
When integrin αv was knocked down, expression of total EGFR increased in GBM6 cells that adhered to laminin, vitronectin, or fibronectin, compared with cells on uncoated substrates (Figures 6B,C). Although total AKT was decreased in integrin αv knockdown cells, vitronectin and fibronectin promoted total AKT expression. There was also a significant elevation of phospho-AKT in integrin αv knockdown cells growing on precoated vitronectin and fibronectin (Figures 6B,C). Cyclin D1 was decreased remarkably in integrin αv knockdown cells treated with carmustine (Figures 6B,C). As shown in Figure 6D, schematic diagram depicted the involvement of integrin αv and EGFR for the survival pathway in the CAMDR. Although knockdown of integrin αv might prevent the survival of GBM cells, compensatory activation of EGFR and driving its downstream signal molecule AKT and ERK acted as a survival pathway, preventing GBM cells from being killed by carmustine.
Discussion
Cancer cells may exhibit intrinsic resistance, where they are resistant at the time of initial treatment, and acquired resistance, where resistance develops during the treatment, leading to therapeutic failure. Although a series of publications have revealed the possible mechanisms by which cancer cells become resistant to chemotherapeutic agents, such as decreased loss of receptor or transporter, specific metabolism, enhanced efflux pumps, and limited drug uptake (Zaal and Berkers, 2018), it remains unclear how the tumor microenvironment confers drug resistance to cancer cells. In the current study, our data show three major components of ECM, laminin, vitronectin, and fibronectin, induce GBM cells to become drug resistant by activation of integrin αv and EGFR. First, upregulated integrin αv activates FAK/paxillin/AKT pathway, leading to a suppression of p53-mediated apoptosis and promoting proliferation by altered cell cycle checkpoints. At the same time, increased expression of ABCB1 indicates that more drug was pumped out by the cells. Second, a compensatory survival pathway was required through activation of EGFR, especially when the integrin αv was missing or inhibited by the integrin receptor antagonist cilengitide.
The composition of the ECM in tumors is vastly different from that found in the normal part tissue, but little was known on their exact role in the tumor progression. As the main soluble particles in the ECM, laminin, vitronectin, and fibronectin were found present in the GBM biopsy samples (Schiffer et al., 1984; Shinoda et al., 1989; Gladson and Cheresh, 1991). Similar findings were observed in the current study; by using the TMA, laminin, vitronectin, and fibronectin were expressed in the GBM at a high level. On the contrary, only fibronectin was abundant in the low-grade CNS tumors, indicating that ECM evolved with tumor progression. It was clear from our data that cell attachment was increased when growing on laminin, vitronectin, and fibronectin, enabling GBM cells to acquire the chemotherapeutic resistance, which was also called CAMDR. Our data revealed a variant cell adhesion between cell lines on the same ECM protein, probably as a result of the distribution of certain integrin receptors on the cell surface. Furthermore, integrin receptors have also been shown to bind ECM proteins on sites other than RGD sequences (Albelda and Buck, 1990). Integrins are not the only cell surface receptors for ECM molecules (Venstrom and Reichardt, 1993). CD44 can also mediate glioma cell adhesion to hyaluroonic acid in the ECM and invasion in vitro (Xiao et al., 2018). As shown in Figure 1E, fibronectin increased the cell viability in all three cell lines, revealing its strong CAMDR effect, which was also in accordance with the TMA results. It has been reported that fibronectin induced CAMDR in multiple myeloma and bladder cancer (Gao et al., 2017; Wu et al., 2019). Laminins are high-molecular-weight proteins of ECM and are thought as an important and biologically active part of basal lamina, influencing cell differentiation, migration, and adhesion (Yap et al., 2019; Barros et al., 2020). It is also documented that laminin was involved in the acquired chemotherapeutic resistance in gastric cancer (Sun et al., 2014). Only HK308 attached to laminin developed drug resistance in our study.
Cilengitide is based on the cyclin peptide cyclo (-RGDfV-), which is a selective for integrin αv and is reported beneficial for GBM patients only in phase II trials (Reardon et al., 2008; Khasraw et al., 2016). Here, cilengitide had different antagonistic effects on GBM6, U87MG, and HK308. As shown in Supplementary Figure S2 and Figure 2, cilengitide detached U87MG completely when cells were not grown on ECM, and more cells detached after vitronectin treatment. A similar result was found in GBM6 in that cells grown on vitronectin more easily detached after carmustine treatment. On the contrary, only attachment to fibronectin was not influenced by cilengitide. Hence, we assume that predominance of integrin receptors of vitronectin is more enriched over other types of receptors on these GBM cells. Expression of vitronectin and its integrin receptor αvβ3 have been described in human GBM in vivo (Franovic et al., 2015). Other integrin receptors such as integrins β1 and β8 are also reported in literature (Tchaicha et al., 2011; Cosset et al., 2012). Although integrins β1, β3 and β5 were upregulated in cells grown on vitronectin, there were no changes after the cells were detached by cilengitide. Meanwhile, integrin αv was boosted, likely due to the negative feedback response when the cells lost attachment. Since the trend of integrins α and β is not exactly the same, we cannot accurately determine which β subunit plays a major role. Previous literature and research show that fibronectin laminin and vitronectin have different specific integrin receptors. Integrin αvβ3, is the main binding receptor of vitronectin (Hermann et al., 1999). Therefore, cilengitide has the most obvious antagonistic effect on cell adhesion to vitronectin compared to on laminin or fibronectin, which is consistent with our current results. In addition to αvβ3, αvβ1 is also found as a receptor of vitronectin (Ding et al., 2002), whereas the main binding receptors of fibronectin are αvβ1 (Ye et al., 2020), αvβ6 (Mould et al., 2014), and α5β1 (Mould et al., 2014) and the main binding receptor of laminin is α6β1 (Corsini and Martin-Villalba, 2010). Previous research also confirmed that M21 human melanoma cells not only lost the ability to attach to vitronectin but showed a dramatic reduction in tumorigenicity when lacking integrin αv gene expression (Lacaria et al., 2020). In addition, accompanied with the elevation of integrin αv, in our study EGFR was also found upregulated in carmustine- and cilengitide-treated cells growing on vitronectin. Other researchers have proposed that integrin αv associates with the EGFR on the cell membrane in a macromolecular complex including the adaptor protein p130Cas and the c-Src kinase that could lead to phosphorylation of specific EGFR tyrosine residues (Kosibaty et al., 2020).
Many studies have addressed the association between NFκB and cell adhesion events. NFκB activity is required for the expression of several cell adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), and endothelial leukocyte adhesion molecule-1 (ELAM-1) (Ampofo et al., 2019). Previous publication also reported that over-expression of NFκB subunits in GBM cells elevated the levels of fibronectin gene expression, indicating a positive loop in the regulatory role for NFκB in ECM protein–cell communication. Activation of NFκB also led to an increase in the levels of mRNA for the αv and β3 integrin subunits, which was in accordance with our present results (Ritchie et al., 2000). Bioluminescent analysis of genetic reporters has been previously used in live cells to assess dynamic changes in TF activity (Pannier et al., 2007; Penalver Bernabe et al., 2016). Our data indicated that activity of NFκB was promoted by either drug treatment as a feedback response or increased attachment to vitronectin and fibronectin, but detachment induced by cilengitide dramatically suppressed activity of NFκB. The result was in accordance with the prior studies showing that NFκB was involved in the proliferation progress in many tumors (Zeligs et al., 2016). Another interesting thing was that the activity of p53 was only elevated by carmustine in cells growing on precoated PBS but decreased in cells growing on laminin, vitronectin, and fibronectin. Along with the downregulation, p53-mediated apoptosis was reduced accordingly. It has been reported that high expression of fibronectin is associated with the cell proliferation and malignancy via NFκB/p53-apoptosis signaling pathway in colorectal cancer (Yi et al., 2016).
A number of studies have demonstrated that chemotherapeutic resistance against alkylating agents is often associated with the overexpression of ABCB1 (Munoz et al., 2014; Stavrovskaya et al., 2016). Generally, ABCB1 serves as a drug efflux pump actively reducing intracellular drug concentrations in resistant tumor cells, but its biological regulation remains unclear. We found that ABCB1 was elevated by the treatment of carmustine without precoated ECM proteins, which is probably due to negative feedback to the alkylating agent. ABCB1 went to an even higher-level expression with precoated fibronectin, which was correlated with the upregulation of EGFR, as was also reported in the prior work (Munoz et al., 2014). Among microenvironment–cell interaction-mediated regulation of ABCB1, a family of ECM proteins called CCN (CYR61/CTGF/NOV) was demonstrated to regulate ABCB1 and to confer vinblastine resistance in renal cell carcinoma cells targeting αv β3 (Long et al., 2013). HA–CD44 interactions have been shown to be involved in multidrug resistance in breast tumor cells and are linked to a positive feedback circuit involving HA, phosphoinositide 3-kinase (PI3K), and ErbB2 (Ma et al., 2018).
As shown in the present study, activation of integrin αv-mediated FAK/paxillin/AKT signaling pathway was required for the proliferation, which was consistent with studies in other tumor cells that integrin αv facilitates a proliferative role (Hayashido et al., 2014; Hung et al., 2014). Some research also indicated the integrin αv was involved in the GBM neurosphere formation. Integrin αv-mediated cell–cell adhesion limited cell dispersion from spheroids in a fibronectin-poor microenvironment. However, in a fibronectin-rich microenvironment, αv promoted cell dispersion (Blandin et al., 2016). EGFR has been well identified as a predictor for the chemoresistance of GBM in the last decade (Taylor et al., 2012). Here, we show that with integrin αv knock down, compensatory activation of EGFR may enable the cells to escape apoptotic effects of alkylating agents. Overall, this study emphasizes that importance of interactions of the ECM and a corresponding profile of cell surface receptors to regulation of cancer cell survival.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The patient-derived GBM cell lines were in accordance with UCLA Institutional Review Board protocol 10-000655.
Author Contributions
SS and QY conceived the studies and wrote the manuscript. QY and WX designed the methodology. QY, WX, SS, AS, and JL conducted the experiments and acquired and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This work was supported with funding from the National Institutes of Health (NIH) (R21NS093199) and the UCLA ARC 3R’s Award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Harley Kornblum for the provision of the HK308 cell lines; Dr. David Nathanson for the provision of the GBM6 cell line; and Dr. William Yong, Director of the Brain Tumor Tissue Resource at UCLA, for the provision of the patient tumor specimens for TMA. We also thank UCLA Crump Institute for Molecular Imaging using the IVIS imaging system. TP53bp1 was a gift from Nicola Burgess-Brown (Addgene plasmid #73252). ITGAV shRNA was obtained from Dharmacon (V2LHS_133468, Lafayette, CO, United States).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2021.616580/full#supplementary-material
Supplementary Figure 1 | (A) Representative light images of U87MG cells with or without carmustine (100 μM, PBS as control) treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm. (B) Representative light images of HK308 cells with or without carmustine (100 μM, PBS as control) treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm.
Supplementary Figure 2 | (A) Cell viability assay of carmustine and cilengitide on U87MG and HK308 cells. Error bars, SD (n = 3, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.05, ##p < 0.01). (B) Representative light images of U87MG cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50 μm. (C) Representative light images of HK308 cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on precoated fibronectin, laminin, and vitronectin. Scale bars, 50μm.
Supplementary Figure 3 | (A) Western blot assay of U87MG cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on ABCB1 and Cleaved PARP expression (n = 3, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.05, ###p < 0.001). (B) Western blot assay of HK308 cells with or without carmustine and cilengitide (100μM, PBS as control) treatment on ABCB1 and Cleaved PARP expression (n = 3, ∗p < 0.05, ∗∗∗p < 0.001, #p < 0.05, ###p < 0.001).
Supplementary Figure 4 | (A,B) Western blot assay of U87MG cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on integrin αv, total/p-FAK, paxillin, total/p-AKT, p-ERK 1/2, and cyclin D1 (n = 3, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.05).
Supplementary Figure 5 | (A,B) Western blot assay of HK308 cells with or without carmustine and cilengitide (100 μM, PBS as control) treatment on integrin αv, total/p-FAK, paxillin, total/p-AKT, p-ERK 1/2, and cyclin D1 (n = 3, ∗p < 0.05, ∗∗p < 0.01, #p < 0.05).
Supplementary Figure 6 | (A,B) Western blot assay of GBM6 cells with dual treatment of carmustine and cilengitide on uncoated ECMs (100 μM, PBS as control) (n = 3, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, #p < 0.05, ##<0.01, ###<0.001).
References
Affronti, M. L., Heery, C. R., Herndon, J. E. II, Rich, J. N., Reardon, D. A., Desjardins, A., et al. (2009). Overall survival of newly diagnosed glioblastoma patients receiving carmustine wafers followed by radiation and concurrent temozolomide plus rotational multiagent chemotherapy. Cancer 115, 3501–3511. doi: 10.1002/cncr.24398
Akhavan, D., Pourzia, A. L., Nourian, A. A., Williams, K. J., Nathanson, D., Babic, I., et al. (2013). De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 3, 534–547. doi: 10.1158/2159-8290.cd-12-0502
Albelda, S. M., and Buck, C. A. (1990). Integrins and other cell adhesion molecules. FASEB J. 4, 2868–2880. doi: 10.1096/fasebj.4.11.2199285
Ampofo, E., Berg, J. J., Menger, M. D., and Laschke, M. W. (2019). Maslinic acid alleviates ischemia/reperfusion-induced inflammation by downregulation of NFkappaB-mediated adhesion molecule expression. Sci. Rep. 9:6119.
Ashby, L. S., Smith, K. A., and Stea, B. (2016). Gliadel wafer implantation combined with standard radiotherapy and concurrent followed by adjuvant temozolomide for treatment of newly diagnosed high-grade glioma: a systematic literature review. World J. Surg. Oncol. 14:225.
Barros, D., Amaral, I. F., and Pego, A. P. (2020). Laminin-inspired cell-instructive microenvironments for neural stem cells. Biomacromolecules 21, 276–293. doi: 10.1021/acs.biomac.9b01319
Bellis, A. D., Penalver-Bernabe, B., Weiss, M. S., Yarrington, M. E., Barbolina, M. V., Pannier, A. K., et al. (2011). Cellular arrays for large-scale analysis of transcription factor activity. Biotechnol. Bioeng. 108, 395–403. doi: 10.1002/bit.22916
Blandin, A. F., Noulet, F., Renner, G., Mercier, M. C., Choulier, L., Vauchelles, R., et al. (2016). Glioma cell dispersion is driven by alpha5 integrin-mediated cell-matrix and cell-cell interactions. Cancer Lett. 376, 328–338. doi: 10.1016/j.canlet.2016.04.007
Castrogiovanni, C., Waterschoot, B., De Backer, O., and Dumont, P. (2018). Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 25, 190–203. doi: 10.1038/cdd.2017.143
Chinot, O. L., Wick, W., and Cloughesy, T. (2014). Bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370:2049.
Choe, G., Horvath, S., Cloughesy, T. F., Crosby, K., Seligson, D., Palotie, A., et al. (2003). Analysis of the phosphatidylinositol 3’-kinase signaling pathway in glioblastoma patients in vivo. Cancer Res. 63, 2742–2746.
Chowdhary, S. A., Ryken, T., and Newton, H. B. (2015). Survival outcomes and safety of carmustine wafers in the treatment of high-grade gliomas: a meta-analysis. J. Neurooncol. 122, 367–382. doi: 10.1007/s11060-015-1724-2
Corsini, N. S., and Martin-Villalba, A. (2010). Integrin alpha 6: anchors away for glioma stem cells. Cell Stem Cell 6, 403–404. doi: 10.1016/j.stem.2010.04.003
Cosset, E. C., Godet, J., Entz-Werle, N., Guerin, E., Guenot, D., Froelich, S., et al. (2012). Involvement of the TGFbeta pathway in the regulation of alpha5 beta1 integrins by caveolin-1 in human glioblastoma. Int. J. Cancer 131, 601–611. doi: 10.1002/ijc.26415
Ding, Q., Stewart, J. Jr., Prince, C. W., Chang, P. L., Trikha, M., Han, X., et al. (2002). Promotion of malignant astrocytoma cell migration by osteopontin expressed in the normal brain: differences in integrin signaling during cell adhesion to osteopontin versus vitronectin. Cancer Res. 62, 5336–5343.
Dixit, S., Hingorani, M., Achawal, S., and Scott, I. (2011). The sequential use of carmustine wafers (Gliadel(R)) and post-operative radiotherapy with concomitant temozolomide followed by adjuvant temozolomide: a clinical review. Br. J. Neurosurg. 25, 459–469. doi: 10.3109/02688697.2010.550342
Dull, T., Zufferey, R., Kelly, M., Mandel, R. J., Nguyen, M., Trono, D., et al. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998
Eke, I., and Cordes, N. (2015). Focal adhesion signaling and therapy resistance in cancer. Semin. Cancer Biol. 31, 65–75. doi: 10.1016/j.semcancer.2014.07.009
Ellor, S. V., Pagano-Young, T. A., and Avgeropoulos, N. G. (2014). Glioblastoma: background, standard treatment paradigms, and supportive care considerations. J. Law Med. Ethics 42, 171–182. doi: 10.1111/jlme.12133
Fei, M., Hang, Q., Hou, S., He, S., and Ruan, C. (2014). Adhesion to fibronectin induces p27(Kip1) nuclear accumulation through down-regulation of Jab1 and contributes to cell adhesion-mediated drug resistance (CAM-DR) in RPMI 8,226 cells. Mol. Cell Biochem. 386, 177–187. doi: 10.1007/s11010-013-1856-7
Franovic, A., Elliott, K. C., Seguin, L., Camargo, M. F., Weis, S. M., and Cheresh, D. A. (2015). Glioblastomas require integrin alphavbeta3/PAK4 signaling to escape senescence. Cancer Res. 75, 4466–4473. doi: 10.1158/0008-5472.can-15-0988
Gao, F., Xu, T., Wang, X., Zhong, S., Chen, S., Zhang, M., et al. (2017). CIP2A mediates fibronectin-induced bladder cancer cell proliferation by stabilizing beta-catenin. J. Exp. Clin. Cancer Res. 36:70.
Gilbert, M. R., Dignam, J. J., Armstrong, T. S., Wefel, J. S., Blumenthal, D. T., Vogelbaum, M. A., et al. (2014). A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 370, 699–708.
Gladson, C. L., and Cheresh, D. A. (1991). Glioblastoma expression of vitronectin and the alpha v beta 3 integrin. Adhesion mechanism for transformed glial cells. J. Clin. Invest. 88, 1924–1932. doi: 10.1172/jci115516
Guo, D., Prins, R. M., Dang, J., Kuga, D., Iwanami, A., Soto, H., et al. (2009). EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2:ra82. doi: 10.1126/scisignal.2000446
Hayashido, Y., Kitano, H., Sakaue, T., Fujii, T., Suematsu, M., Sakurai, S., et al. (2014). Overexpression of integrin alphav facilitates proliferation and invasion of oral squamous cell carcinoma cells via MEK/ERK signaling pathway that is activated by interaction of integrin alphavbeta8 with type collagen. Int. J. Oncol. 45, 1875–1882. doi: 10.3892/ijo.2014.2642
Hendricks, B. K., Cohen-Gadol, A. A., and Miller, J. C. (2015). Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg. Focus 38:E10.
Hermann, P., Armant, M., Brown, E., Rubio, M., Ishihara, H., Ulrich, D., et al. (1999). The vitronectin receptor and its associated CD47 molecule mediates proinflammatory cytokine synthesis in human monocytes by interaction with soluble CD23. J. Cell Biol. 144, 767–775. doi: 10.1083/jcb.144.4.767
Horikawa, I. (2020). Balancing and differentiating p53 activities toward longevity and no cancer? Cancer Res. 80, 5164–5165. doi: 10.1158/0008-5472.can-20-3080
Hung, C. J., Hsu, H. I., Lin, C. C., Huang, T. H., Wu, B. C., Kao, C. T., et al. (2014). The role of integrin alphav in proliferation and differentiation of human dental pulp cell response to calcium silicate cement. J. Endod. 40, 1802–1809. doi: 10.1016/j.joen.2014.07.016
Khasraw, M., Lee, A., Mccowatt, S., Kerestes, Z., Buyse, M. E., Back, M., et al. (2016). Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J. Neurooncol. 128, 163–171. doi: 10.1007/s11060-016-2094-0
Kim, H. J., and Kim, D. Y. (2020). Present and future of anti-glioblastoma therapies: a deep look into molecular dependencies/features. Molecules 25:4641. doi: 10.3390/molecules25204641
Kong, L. R., Chua, K. N., Sim, W. J., Ng, H. C., Bi, C., Ho, J., et al. (2015). MEK inhibition overcomes cisplatin resistance conferred by SOS/MAPK pathway activation in squamous cell carcinoma. Mol. Cancer Ther. 14, 1750–1760. doi: 10.1158/1535-7163.mct-15-0062
Kosibaty, Z., Murata, Y., Minami, Y., Noguchi, M., and Sakamoto, N. (2020). ECT2 promotes lung adenocarcinoma progression through extracellular matrix dynamics and focal adhesion signaling. Cancer Sci. 00, 1–12.
Lacaria, L., Lange, J. R., Goldmann, W. H., Rico, F., and Alonso, J. L. (2020). alphavbeta3 integrin expression increases elasticity in human melanoma cells. Biochem. Biophys. Res. Commun. 525, 836–840. doi: 10.1016/j.bbrc.2020.02.156
Laks, D. R., Crisman, T. J., Shih, M. Y., Mottahedeh, J., Gao, F., Sperry, J., et al. (2016). Large-scale assessment of the gliomasphere model system. Neuro Oncol. 18, 1367–1378. doi: 10.1093/neuonc/now045
Long, Q. Z., Zhou, M., Liu, X. G., Du, Y. F., Fan, J. H., Li, X., et al. (2013). Interaction of CCN1 with alphavbeta3 integrin induces P-glycoprotein and confers vinblastine resistance in renal cell carcinoma cells. Anticancer Drugs 24, 810–817. doi: 10.1097/cad.0b013e328363046d
Lu, K. V., Zhu, S., Cvrljevic, A., Huang, T. T., Sarkaria, S., Ahkavan, D., et al. (2009). Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 69, 6889–6898. doi: 10.1158/0008-5472.can-09-0347
Ma, J. W., Wang, X., Chang, L., Zhong, X. Y., Jing, H., Zhu, X., et al. (2018). CD44 collaborates with ERBB2 mediate radiation resistance via p38 phosphorylation and DNA homologous recombination pathway in prostate cancer. Exp. Cell Res. 370, 58–67. doi: 10.1016/j.yexcr.2018.06.006
Messaoudi, K., Clavreul, A., and Lagarce, F. (2015). Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discov. Today 20, 899–905. doi: 10.1016/j.drudis.2015.02.011
Miranda, A., Blanco-Prieto, M., Sousa, J., Pais, A., and Vitorino, C. (2017). Breaching barriers in glioblastoma. Part I: molecular pathways and novel treatment approaches. Int. J. Pharm. 531, 372–388. doi: 10.1016/j.ijpharm.2017.07.056
Motegi, A., Masutani, M., Yoshioka, K. I., and Bessho, T. (2019). Aberrations in DNA repair pathways in cancer and therapeutic significances. Semin. Cancer Biol. 58, 29–46. doi: 10.1016/j.semcancer.2019.02.005
Mould, A. P., Craig, S. E., Byron, S. K., Humphries, M. J., and Jowitt, T. A. (2014). Disruption of integrin-fibronectin complexes by allosteric but not ligand-mimetic inhibitors. Biochem. J. 464, 301–313. doi: 10.1042/bj20141047
Munoz, J. L., Rodriguez-Cruz, V., Greco, S. J., Nagula, V., Scotto, K. W., and Rameshwar, P. (2014). Temozolomide induces the production of epidermal growth factor to regulate MDR1 expression in glioblastoma cells. Mol. Cancer Ther. 13, 2399–2411. doi: 10.1158/1535-7163.mct-14-0011
Nabors, L. B., Fink, K. L., Mikkelsen, T., Grujicic, D., Tarnawski, R., Nam, D. H., et al. (2015). Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 17, 708–717. doi: 10.1093/neuonc/nou356
Nakagawa, Y., Nakayama, H., Nagata, M., Yoshida, R., Kawahara, K., Hirosue, A., et al. (2014). Overexpression of fibronectin confers cell adhesion-mediated drug resistance (CAM-DR) against 5-FU in oral squamous cell carcinoma cells. Int. J. Oncol. 44, 1376–1384. doi: 10.3892/ijo.2014.2265
Noh, K., Bach, D. H., Choi, H. J., Kim, M. S., Wu, S. Y., Pradeep, S., et al. (2020). The hidden role of paxillin: localization to nucleus promotes tumor angiogenesis. Oncogene 40, 384–395. doi: 10.1038/s41388-020-01517-3
Olotu, F. A., and Soliman, M. E. S. (2019). Dynamic perspectives into the mechanisms of mutation-induced p53-DNA binding loss and inactivation using active perturbation theory: structural and molecular insights toward the design of potent reactivators in cancer therapy. J. Cell Biochem. 120, 951–966. doi: 10.1002/jcb.27458
Pannier, A. K., Ariazi, E. A., Bellis, A. D., Bengali, Z., Jordan, V. C., and Shea, L. D. (2007). Bioluminescence imaging for assessment and normalization in transfected cell arrays. Biotechnol. Bioeng. 98, 486–497. doi: 10.1002/bit.21477
Patel, M. A., Kim, J. E., Ruzevick, J., Li, G., and Lim, M. (2014). The future of glioblastoma therapy: synergism of standard of care and immunotherapy. Cancers (Basel) 6, 1953–1985. doi: 10.3390/cancers6041953
Penalver Bernabe, B., Shin, S., Rios, P. D., Broadbelt, L. J., Shea, L. D., and Seidlits, S. K. (2016). Dynamic transcription factor activity networks in response to independently altered mechanical and adhesive microenvironmental cues. Integr. Biol. (Camb) 8, 844–860. doi: 10.1039/c6ib00093b
Pine, A. R., Cirigliano, S. M., Nicholson, J. G., Hu, Y., Linkous, A., Miyaguchi, K., et al. (2020). Tumor microenvironment is critical for the maintenance of cellular states found in primary glioblastomas. Cancer Discov. 10, 964–979. doi: 10.1158/2159-8290.cd-20-0057
Pourgholi, F., Hajivalili, M., Farhad, J. N., Kafil, H. S., and Yousefi, M. (2016). Nanoparticles: novel vehicles in treatment of Glioblastoma. Biomed. Pharmacother. 77, 98–107. doi: 10.1016/j.biopha.2015.12.014
Reardon, D. A., Fink, K. L., Mikkelsen, T., Cloughesy, T. F., O’neill, A., Plotkin, S., et al. (2008). Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J. Clin. Oncol. 26, 5610–5617. doi: 10.1200/jco.2008.16.7510
Ritchie, C. K., Giordano, A., and Khalili, K. (2000). Integrin involvement in glioblastoma multiforme: possible regulation by NF-kappaB. J. Cell Physiol. 184, 214–221. doi: 10.1002/1097-4652(200008)184:2<214::aid-jcp9>3.0.co;2-z
Sanchez, V. E., Nichols, C., Kim, H. N., Gang, E. J., and Kim, Y. M. (2019). Targeting PI3K signaling in acute lymphoblastic leukemia. Int. J. Mol. Sci. 20:412. doi: 10.3390/ijms20020412
Sarkaria, J. N., Carlson, B. L., Schroeder, M. A., Grogan, P., Brown, P. D., Giannini, C., et al. (2006). Use of an orthotopic xenograft model for assessing the effect of epidermal growth factor receptor amplification on glioblastoma radiation response. Clin. Cancer Res. 12, 2264–2271. doi: 10.1158/1078-0432.ccr-05-2510
Schiffer, D., Giordana, M. T., Mauro, A., and Migheli, A. (1984). GFAP, F VIII/RAg, laminin, and fibronectin in gliosarcomas: an immunohistochemical study. Acta Neuropathol. 63, 108–116. doi: 10.1007/bf00697192
Shinoda, J., Hirayama, H., Araki, Y., Andoh, T., Sakai, N., and Yamada, H. (1989). [Morphological changes in basement membrane associated with endothelial proliferation in astrocytic tumors–an immunohistochemical study of laminin]. No To Shinkei 41, 263–271.
Stavrovskaya, A. A., Shushanov, S. S., and Rybalkina, E. Y. (2016). Problems of glioblastoma multiforme drug resistance. Biochemistry (Mosc) 81, 91–100. doi: 10.1134/s0006297916020036
Stewart, L. A. (2002). Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet 359, 1011–1018. doi: 10.1016/s0140-6736(02)08091-1
Stupp, R., Hegi, M. E., Gorlia, T., Erridge, S. C., Perry, J., Hong, Y. K., et al. (2014). Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 15, 1100–1108. doi: 10.1016/s1470-2045(14)70379-1
Sun, L., Liu, L., Liu, X., Wang, Y., Li, M., Yao, L., et al. (2014). Gastric cancer cell adhesion to laminin enhances acquired chemotherapeutic drug resistance mediated by MGr1-Ag/37LRP. Oncol. Rep. 32, 105–114. doi: 10.3892/or.2014.3184
Taylor, T. E., Furnari, F. B., and Cavenee, W. K. (2012). Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr. Cancer Drug Targets 12, 197–209. doi: 10.2174/156800912799277557
Tchaicha, J. H., Reyes, S. B., Shin, J., Hossain, M. G., Lang, F. F., and Mccarty, J. H. (2011). Glioblastoma angiogenesis and tumor cell invasiveness are differentially regulated by beta8 integrin. Cancer Res. 71, 6371–6381. doi: 10.1158/0008-5472.can-11-0991
Thon, N., Kreth, S., and Kreth, F. W. (2013). Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 6, 1363–1372. doi: 10.2147/ott.s50208
Tivnan, A., Zakaria, Z., O’leary, C., Kogel, D., Pokorny, J. L., Sarkaria, J. N., et al. (2015). Inhibition of multidrug resistance protein 1 (MRP1) improves chemotherapy drug response in primary and recurrent glioblastoma multiforme. Front. Neurosci. 9:218. doi: 10.3389/fnins.2015.00218
Varner, J. A., Brooks, P. C., and Cheresh, D. A. (1995). REVIEW: the integrin alpha V beta 3: angiogenesis and apoptosis. Cell Adhes. Commun. 3, 367–374.
Venstrom, K. A., and Reichardt, L. F. (1993). Extracellular matrix. 2: role of extracellular matrix molecules and their receptors in the nervous system. FASEB J. 7, 996–1003. doi: 10.1096/fasebj.7.11.8370483
Walker, M. D., Alexander, E. Jr., Hunt, W. E., Maccarty, C. S., Mahaley, M. S. Jr., and Mealey, J. Jr. et al. (1978). Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J. Neurosurg. 49, 333–343. doi: 10.3171/jns.1978.49.3.0333
Walker, M. D., Green, S. B., Byar, D. P., Alexander, E. Jr., Batzdorf, U., Brooks, W. H., et al. (1980). Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N. Engl. J. Med. 303, 1323–1329. doi: 10.1056/nejm198012043032303
Weller, M., Butowski, N., Tran, D. D., Recht, L. D., Lim, M., Hirte, H., et al. (2017). Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 18, 1373–1385.
Wu, Y., Zhu, X., Shen, R., Huang, J., Xu, X., and He, S. (2019). miR-182 contributes to cell adhesion-mediated drug resistance in multiple myeloma via targeting PDCD4. Pathol. Res. Pract. 215:152603.
Xiao, W., Sohrabi, A., and Seidlits, S. K. (2017). Integrating the glioblastoma microenvironment into engineered experimental models. Future Sci. OA 3:FSO189.
Xiao, W., Zhang, R., Sohrabi, A., Ehsanipour, A., Sun, S., Liang, J., et al. (2018). Brain-mimetic 3D culture platforms allow investigation of cooperative effects of extracellular matrix features on therapeutic resistance in glioblastoma. Cancer Res. 78, 1358–1370.
Yan, Y., Xu, Z., Dai, S., Qian, L., Sun, L., and Gong, Z. (2016). Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 35:23.
Yap, L., Tay, H. G., Nguyen, M. T. X., Tjin, M. S., and Tryggvason, K. (2019). Laminins in cellular differentiation. Trends Cell Biol. 29, 987–1000.
Ye, Y., Zhang, R., and Feng, H. (2020). Fibronectin promotes tumor cells growth and drugs resistance through a CDC42-YAP-dependent signaling pathway in colorectal cancer. Cell Biol. Int. 44, 1840–1849.
Yi, W., Xiao, E., Ding, R., Luo, P., and Yang, Y. (2016). High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the NF-kappaB/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 36, 3145–3153.
Yu, Q., Xue, Y., Liu, J., Xi, Z., Li, Z., and Liu, Y. (2018). Fibronectin promotes the malignancy of glioma stem-like cells via modulation of cell adhesion, differentiation, proliferation and chemoresistance. Front. Mol. Sci. 11:130. doi: 10.3389/fnmol.2018.00130
Zaal, E. A., and Berkers, C. R. (2018). The influence of metabolism on drug response in cancer. Front. Oncol. 8:500.
Zeligs, K. P., Neuman, M. K., and Annunziata, C. M. (2016). Molecular pathways: the balance between cancer and the immune system challenges the therapeutic specificity of targeting nuclear factor-kappaB signaling for cancer treatment. Clin. Cancer Res. 22, 4302–4308.
Keywords: cell adhesion-mediated drug resistance, integrin αv, glioblastoma, extracellular matrix, laminin, vitronectin, fibronectin
Citation: Yu Q, Xiao W, Sun S, Sohrabi A, Liang J and Seidlits SK (2021) Extracellular Matrix Proteins Confer Cell Adhesion-Mediated Drug Resistance Through Integrin αv in Glioblastoma Cells. Front. Cell Dev. Biol. 9:616580. doi: 10.3389/fcell.2021.616580
Received: 12 October 2020; Accepted: 26 January 2021;
Published: 23 March 2021.
Edited by:
Liwu Fu, Sun Yat-sen University, ChinaReviewed by:
Vanessa Morais Freitas, University of São Paulo, BrazilJian-ye Zhang, Guangzhou Medical University, China
Copyright © 2021 Yu, Xiao, Sun, Sohrabi, Liang and Seidlits. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie K. Seidlits, seidlits@ucla.edu