Meiotic Recombination Defects and Premature Ovarian Insufficiency
 Chengzi Huang
Chengzi Huang Ting Guo
Ting Guo Yingying Qin
Yingying Qin- 1Center for Reproductive Medicine, Cheeloo College of Medicine, Shandong University, Jinan, China
- 2National Research Center for Assisted Reproductive Technology and Reproductive Genetics, Shandong University, Jinan, China
- 3Key Laboratory of Reproductive Endocrinology of Ministry of Education, Shandong University, Jinan, China
- 4Shandong Provincial Clinical Medicine Research Center for Reproductive Health, Shandong University, Jinan, China
Premature ovarian insufficiency (POI) is the depletion of ovarian function before 40 years of age due to insufficient oocyte formation or accelerated follicle atresia. Approximately 1–5% of women below 40 years old are affected by POI. The etiology of POI is heterogeneous, including genetic disorders, autoimmune diseases, infection, iatrogenic factors, and environmental toxins. Genetic factors account for 20–25% of patients. However, more than half of the patients were idiopathic. With the widespread application of next-generation sequencing (NGS), the genetic spectrum of POI has been expanded, especially the latest identification in meiosis and DNA repair-related genes. During meiotic prophase I, the key processes include DNA double-strand break (DSB) formation and subsequent homologous recombination (HR), which are essential for chromosome segregation at the first meiotic division and genome diversity of oocytes. Many animal models with defective meiotic recombination present with meiotic arrest, DSB accumulation, and oocyte apoptosis, which are similar to human POI phenotype. In the article, based on different stages of meiotic recombination, including DSB formation, DSB end processing, single-strand invasion, intermediate processing, recombination, and resolution and essential proteins involved in synaptonemal complex (SC), cohesion complex, and fanconi anemia (FA) pathway, we reviewed the individual gene mutations identified in POI patients and the potential candidate genes for POI pathogenesis, which will shed new light on the genetic architecture of POI and facilitate risk prediction, ovarian protection, and early intervention for POI women.
Introduction
Premature ovarian insufficiency (POI) is the depletion or dysfunction of ovarian follicles before the age of 40, which is characterized by menstrual disturbance (amenorrhea or oligomenorrhea) for at least 4 months, with raised gonadotrophins (FSH > 25 IU/I on two occasions > 4 weeks apart) and estrogen deficiency (European Society for Human et al., 2016). Approximately 1–5% of women under 40 years old are affected by POI, demonstrated with isolated or syndromic phenotype (Desai and Rajkovic, 2017). The etiologies of POI are heterogeneous, including genetic factors, autoimmune diseases, infection, iatrogenic factors, and environmental toxins. However, most of the cases are still unexplained, known as idiopathic POI. Genetic defects account for approximately 20–25% of POI patients, including chromosomal abnormalities (10–15%) and monogenic mutations (Qin et al., 2015; Jiao et al., 2017). Until now, more than 75 genes have been found to cause POI, which were involved in various processes, including gonadal development, meiosis, DNA damage repair, follicle development, hormone metabolism, and mitochondrial function (Patino et al., 2017; Franca and Mendonca, 2020). Recently, advances in next-generation sequencing (NGS) allow more identification in DNA damage repair genes. Most of the newly identified genes play predominate roles in meiotic homologous recombination (HR), such as STAG3 (Xiao et al., 2019), while other genes, although participating in DNA damage repair in somatic cells, are found to be essential for meiotic HR as well, such as MCM8 (AlAsiri et al., 2015) and BRCA2 (Weinberg-Shukron et al., 2018). Therefore, the role of meiotic HR genes in POI pathogenesis is indispensable.
Females are born with fixed number of oocytes within the ovaries. The fertile lifespan depends on the size of oocyte pool at birth and the rapidity of follicle depletion. The initial oocyte pool is determined by the number of primordial germs cells migrating to the genital ridge, followed by germ cell proliferation and functional meiosis, established as the number of primordial follicles at puberty. The human germ cells enter into meiosis from week 9 postconception, go through leptotene, zygotene, and pachytene, and then transitorily arrest at diplotene stage from the time of birth until puberty when primordial follicles are activated and meiosis continues secondary to FSH and LH secretion. During meiotic prophase I, the key processes are deliberate generation of DNA double-strand breaks (DSBs) and subsequent HR, which laid the foundations of stability and diversity of oocyte genome (Handel and Schimenti, 2010). Disturbance of meiotic HR leads to meiosis blocking before diplotene and DSB accumulation. Animal models defective at DSB formation and HR resulted in early exhaustion of follicle pool and infertility, which were similar to the phenotypes of human POI. While only a few genes have been identified with mutations in POI patients, such as MSH4 (Carlosama et al., 2017) and MSH5 (Guo et al., 2017), here, we categorized the genes participating in meiotic HR, candidate genes for human POI, and further reviewed the mutations in detail, Which have been identified in POI patients (Figure 1 and Table 1).
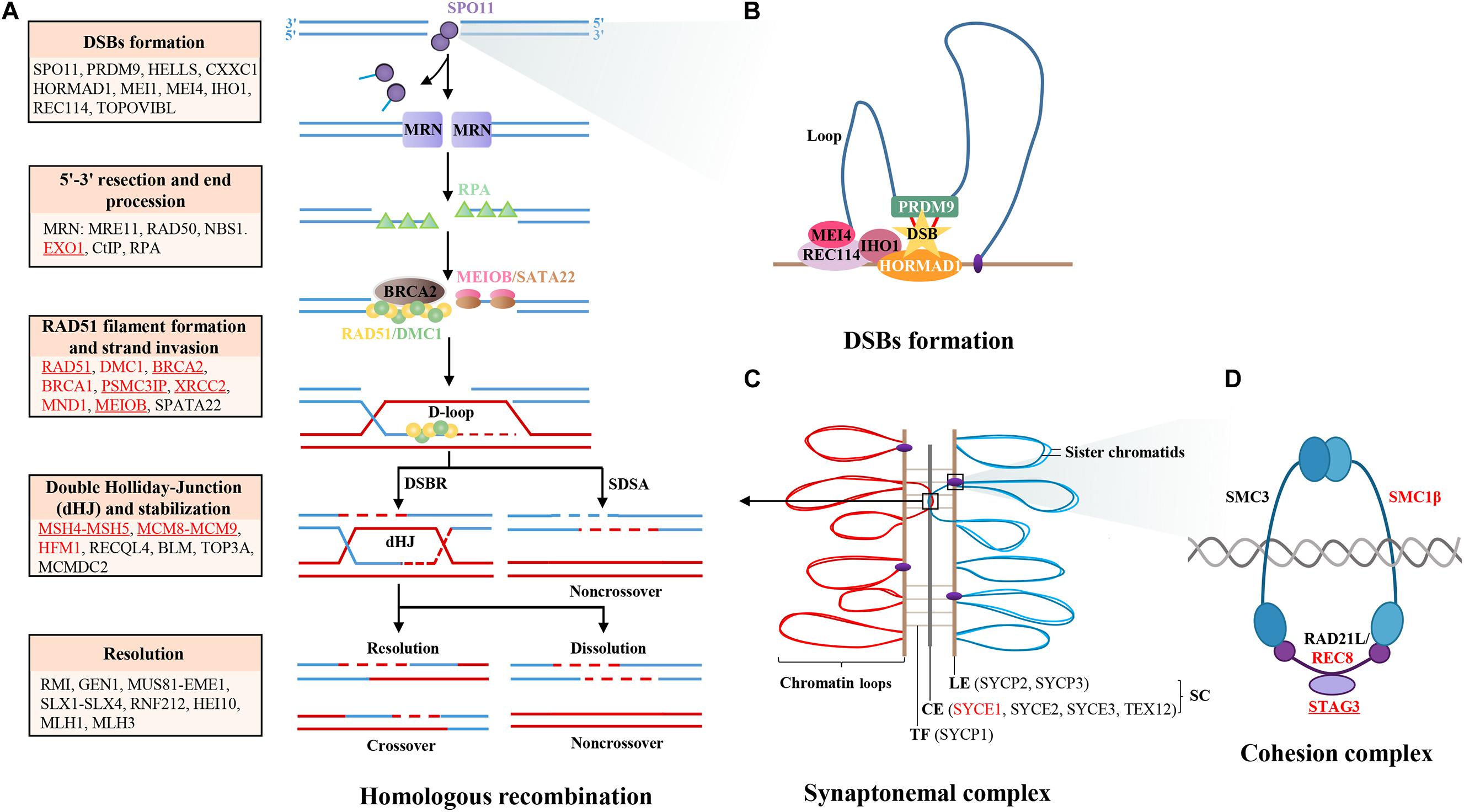
Figure 1. Diagram of meiotic HR genes. (A) Major steps of meiotic prophase I, including DSBs formation, 5′-3′ resection, end procession, RAD51 filament formation, strand invasion, intermediates formation, and resolution. The key genes in different steps are labeled in the box, those genes that have been identified in POI patients are labeled with red color, and those that have been functionally validated are underlined. (B) The formation of DSBs is initiated by PRDM9, which binds to chromatin and catalyzes H3K4 trimethylation to mark hotspots. Then, the complex MEI4/REC114/IHO1 binds to HORMAD1 on the axis and activates SPO11 to cut chromatin to form DSBs. (C) The synapsis complex is installed by CE and LE with TF connections in each pair of homologous chromosomes, which establish the platform of HR. (D) The cohesion complex regulates sister chromatid cohesion and SC formation, which is consisted of meiosis-specific subunits STAG3, RAD21L, and SMC1B and non-specific subunits SMC3 and REC8. Notes: DSBs, double-strand breaks; SC, synaptonemal complex; LE, lateral element; CE, central element; TFs, transverse filaments; HR, homologous recombination.
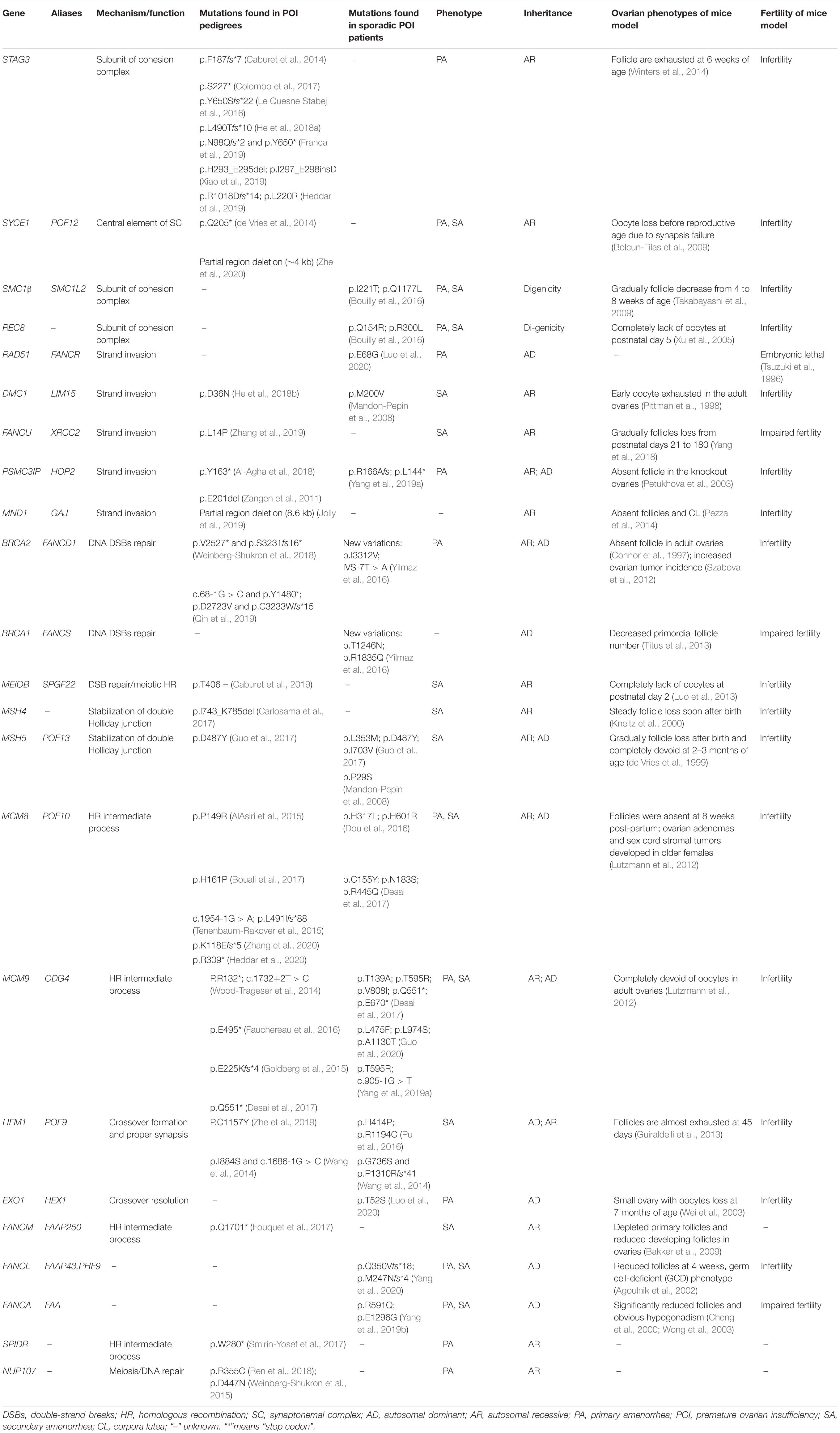
Table 1. Variants of meiosis HR genes identified in POI patients.
Subsections Relevant for the Subject
Programmed Double-Strand Break Formation
At the beginning of meiotic prophase I, accurate DSB localization and formation are the basis of homologous chromosome recognition and synapsis, and indispensable for crossover, which is crucial for chromosome segregation and formation of euploid gametes. The predominant protein determining potential DSB sites is PRDM9, which recognizes the DSB hotspots on the chromosome loops, catalyzes H3K4 trimethylation (Sun et al., 2015; Chen et al., 2020), and binds to the chromosome axis through interaction with protein CXXC1, HORMAD1 (Daniel et al., 2011), MEI4 (Kumar et al., 2010), REC114 (Kumar et al., 2018), and IHO1 (Stanzione et al., 2016; Kumar et al., 2018). Then, HELLS and PRDM9 form a pioneer complex to open chromatin at hotspots, permitting correct placement and repair of DSBs (Spruce et al., 2020). Then, the endonuclease SPO11 is recruited at PRDM9-binding sites before or after the loop axis interaction and catalyzes DSB formation at the hotspots (Romanienko and Camerini-Otero, 2000). Moreover, other proteins required along with SPO11 to generate DSBs including MEI1 (Reinholdt and Schimenti, 2005) and TOPOVIBL (Robert et al., 2016). The knockout mouse models of the above genes demonstrate female infertility and premature depletion of oocytes due to defective DSB formation and homologous synapsis, except for CXXC1 and HORMAD1. The conditional knockout mice of Cxxc1 are fertile (Tian et al., 2018). Hormad1 deficiency does not affect folliculogenesis but disrupts homologous chromosome segregation, resulting in infertility due to gemmate aneuploidy (Shin et al., 2010). Although most of the genes involving DSB formation might be candidate genes for human POI, no causative mutation has been identified in POI patients. Interestingly, there are findings that bi-allelic deleterious mutations of TOROVIBL, MEI1, and REC114 could result in recurrent androgenetic complete hydatidiform moles due to extrusion of all maternal chromosomes with the spindles into the first polar body during meiosis metaphase I. These findings indicated that DSB-formed genes were essential for stabilization of gemmate genome. Defects in these genes might be pleiotropic and responsible for heterogeneous reproductive phenotypes (Nguyen et al., 2018).
DSB End Processing
After DSB formation, DNA ends are engaged in a process of maturation, involving the release of SPO11-oligonucleotide covalent complexes and exonucleolytic degradation on the same strand, which leads to extended overhanging of 3’ single strands on both sides of the DSBs. This process is facilitated by the MRN complex, EXO1, CtIP, and RPA.
The multiprotein complex MRN is consisted of MRE11, RAD50, and NBS1, which are evolutionarily conserved in 5′-end resection of DSBs (Anand et al., 2019). Disruption of the N-terminal exons of Nbs1 in mice resulted in female infertility due to oogenesis failure (Kang et al., 2002). The female mice with a mutation in Mre11 exhibited premature oocyte elimination attributing to defects in homologous chromosome pairing and DSB repair during meiotic prophase I (Inagaki et al., 2016). Rad50 heterozygous mutant mice demonstrated ovarian atrophy as well (Roset et al., 2014). These animal models indicated the essential role of the MRN complex in maintenance of the primordial follicle pool. In humans, mutations in MRN subunits caused Nijmegen Breakage Syndrome in recessive pattern, in which POI was one of the degenerative changes (Chrzanowska et al., 2012). Although they are potential causative genes for POI, no mutation has been identified in isolated POI yet. Besides that, CtIP is an important cofactor of MRN in catalyzing the 5′-end resection (Sartori et al., 2007). CtIP mutations cause Seckel and Jawad syndromes in a recessive manner, while no ovarian abnormality was noticed (Qvist et al., 2011). Therefore, although CtIP performs an important role in DSB end processing, it might not be a potential causative gene of POI.
EXO1 has 5′ to 3′ exonuclease activity, which is recruited to DSBs by MRN and promotes the formation of 3′-tailed single-strand DNA (ssDNA) (Garcia et al., 2011). Exo1 knockout female mice were infertile due to dynamic loss of chiasmata during meiosis prophase I (Wei et al., 2003). A meta-analysis of 53 GWASs with nearly 70,000 women found EXO1 polymorphism associated with the age of natural menopause (Day et al., 2015). Recently, through whole-exome sequencing (WES) in 50 patients with POI, one heterozygous mutation in EXO1 was identified, which impaired meiosis by disrupting recruitment of RPA and RAD51 onto DSB sites (Luo et al., 2020). These findings confirmed the role of EXO1 in POI, and discussed the dosage-dependent effect of oocyte-non-specific HR gene on ovarian function.
When 3′-tailed ssDNAs are established, RPA is recruited to prevent ssDNA degradation or formation of secondary structure (Soustelle et al., 2002). Recent study found that the loss of RPA completely abrogated the loading of recombinases RAD51 and DMC1 on DSBs sites, blocked strand invasion, and chromosome synapsis (Shi et al., 2019). However, because RPA is ubiquitously expressed, the Rpa1 null mice showed embryonic lethality. Although the heterozygotes displayed defects in DSB repair, ovarian phenotype had not been observed (Wang et al., 2005). Therefore, the evidence of RPA participating in POI pathogenesis was insufficient yet.
Strand Invasion
As the proceeding of HR, RPA is replaced by the recombination proteins RAD51 and DMC1, which catalyze homology search and strand invasion, establishing the basis of synapsis (Brown and Bishop, 2014). Besides RAD51 and DMC1, the dynamic process is also regulated by other recombination factors, including BRCA2, PSMC3IP, MND1, MEIOB, and SPATA22.
RAD51 and its meiotic paralog DMC1 execute the critical step of strand invasion (Bishop et al., 1992; Park et al., 2008). In the Dmc1-deficient mice, gametogenesis arrested in meiotic prophase I, resulting in germ cell depletion in the adult ovaries and infertility. Recently, a homozygous mutation p.D36N in DMC1 was identified in one consanguineous pedigree having one patient with POI and one patient with non-obstructive azoospermia (NOA) (He et al., 2018b). Histological study found that spermatogenesis was blocked at zygotene stage in the patient with NOA, indicating that POI in women might be caused by dysfunctional meiosis prophase I of oocytes (Pittman et al., 1998). Besides that, another homozygous mutation DMC1 p.M200V was identified by Sanger sequencing in sporadic POI (Hikiba et al., 2008), while the point mutant mice showed normal ovarian morphology, highlighting the importance of functional studies in verifying the pathogenicity of variations (Hikiba et al., 2008; Tran and Schimenti, 2018). Absolutely, loss of RAD51 resulted in embryo lethality in mice (Tsuzuki et al., 1996). However, a WES study in sporadic POI patients found one heterozygous missense mutation of RAD51, which resisted the protein localization in the nucleus. In vitro experiments found that heterozygous mutation affected HR efficiency by haploinsufficiency, indicating that the pathogenic effect of RAD51 on POI might be dosage dependent (Luo et al., 2020).
BRCA2 regulates the localization of RAD51 onto ssDNA to form an RAD51-ssDNA filament, promoting HR repair for DSBs both in somatic cells and in germ cells (Davies et al., 2001; Xia et al., 2001). Somatic BRCA2 mutations impaired chromosome integrity, manifesting with an increased risk of tumor (Daum et al., 2018), whereas recent studies found its crucial role in ovarian development mediated by functional meiotic recombination (Miao et al., 2019). Until now, four pairs of compound heterozygous mutations and one homozygous mutation in BRCA2 have been identified in POI pedigrees through WES analysis (Weinberg-Shukron et al., 2018; Qin et al., 2019; Caburet et al., 2020). Among them, four mutation carriers demonstrated with microcephaly, leukemia, thyroid cancer, or breast carcinoma, while other three carries presented with isolated POI. The widely varying severity of clinical profiles of bi-allelic BRCA2 mutation carriers confirmed the complicated function of BRCA2, also highlighted the necessity of long-term follow-up for them. Recent studies in Caenorhabditis elegans found BRCA1 influenced RAD51 dynamics and combined with SYCP3 and MSH5 to promote synapsis and crossover resolution (Janisiw et al., 2018). Although its function in mammalian meiosis was unclear, Brca1 mutant mice had impaired reproductive capacity and decreased primordial follicle counts (Titus et al., 2013). Women with BRCA1 variations also presented with accelerated ovarian reserve decline (Lin et al., 2017; Porcu et al., 2020). Therefore, BRCA1 was a potential causative gene of POI, which required comprehensive evaluation of somatic characteristics like BRCA2.
PSMC3IP (also known as HOP2) and MND1 are meiosis-specific factors in all organisms expressing DMC1. PSMC3IP-MND1 complex facilitates strand invasion and D-loop formation by promoting DMC1/RAD51 capturing of double-strand DNA (dsDNA) (Chi et al., 2007; Pezza et al., 2007). Absence of them resulted in non-homologous synapses and DSB accumulation (Sansam and Pezza, 2015). Psmc3ip and Mnd1 knockout mice showed severely reduced ovarian size and defective gametogenesis (Petukhova et al., 2003; Pezza et al., 2014). In previous studies, two-point mutations of PSMC3IP and one microdeletion of MND1 inherited in recessive patterns have been identified in consanguineous pedigrees with POI or XX female gonadal dysgenesis (Zangen et al., 2011; Zhao and Sung, 2015; Al-Agha et al., 2018; Jolly et al., 2019), confirming their crucial roles in gametogenesis and POI pathogenesis.
MEIOB and SPATA22 are ssDNA-binding proteins predominately expressed in meiosis prophase I, which form a complex that interacts with RPA to recruit RAD51 and DMC1 to the ssDNA (La Salle et al., 2012; Luo et al., 2013; Souquet et al., 2013; Ishishita et al., 2014). Both Meiob-null mice and Spata22-null mice exhibited small ovaries devoid of oocytes in any developmental stage due to uncompleted meiotic HR (Luo et al., 2013; Hays et al., 2017). Recent WES study with a POI pedigree identified one homozygous splicing mutation in MEIOB, which resulted in a truncated MEIOB protein, thus interrupting the interaction with SPATA22 (Caburet et al., 2019). POI might be caused by defective MEIOB-SPATA22 complex-induced insufficient DNA single-strand invasion during meiotic HR. Although no mutation has been found in SPATA22, it still is a potential candidate gene of POI.
Intermediate Processing and Homologous Recombination
During strand invasion, the presynaptic filaments recognize the template strands, invade into the duplex DNA, displace the original strand, and bind to their complementary sequence, forming the intermediate of HR repair. The intermediate processing is performed by two pathways: synthesis-dependent strand annealing and double Holliday junction (dHJ). SDSA is a pathway for non-crossover repair, in which a D-loop intermediate is formed and the broken DNA is synthesized using the homologous chromosome as a template (Ranjha et al., 2018). In the dHJ pathway, two DSB ends participate in the invasion that forms a classic double junction intermediate, which facilitates crossover formation and resolution. During the process, MSH4–MSH5 heterodimer, MCM8–MCM9 helicase complex, HFM1, RECQL4, BLM, and MCMDC2 are involved.
The meiotic specially expressed proteins MSH4 and MSH5 form a heterodimeric complex (Acharya et al., 1996; Snowden et al., 2004), which clamps on homologous chromosomes to stabilize the Holliday junctions (Nishant et al., 2010). In the Msh5/Msh4 deficient mice, chromosome pairing was failed and crossover was absent, resulting in atrophic ovaries, which were similar to the phenotype of human POI (de Vries et al., 1999; Kneitz et al., 2000). Through WES in two POI pedigrees, homozygous mutations of MSH4 and MSH5 were identified, pathogenicity of which was confirmed by in vitro studies and knock-in mice models (Carlosama et al., 2017; Guo et al., 2017). These results implied the recessive mode of inheritance for MSH4 and MSH5 in POI. Interestingly, four heterozygous mutations of MSH5 have been reported in sporadic cases, indicating that their effects on meiosis and oogenesis might be dominated or dosage dependent as well (Mandon-Pepin et al., 2008; Guo et al., 2017).
MCM8 and MCM9 form a helicase complex regulating DNA repair and genome integrity both in somatic cells and in germ cells (Nishimura et al., 2012). They not only promote MRN-mediated ssDNA maturation, but also participate in intermediate processing of HR. Mcm8 or Mcm9 knockout mice suffered meiosis blocking at prophase I (Lutzmann et al., 2012). Bi-allelic mutations of MCM8 and MCM9 have been identified in POI patients with or without familial history (Wood-Trageser et al., 2014; AlAsiri et al., 2015; Goldberg et al., 2015; Tenenbaum-Rakover et al., 2015; Fauchereau et al., 2016; Bouali et al., 2017; Desai et al., 2017). The prevalence of bi-allelic mutations of MCM9 in sporadic cases was variable among different studies, ranging from 1.6 to 6.1% (Yang et al., 2019a; Guo et al., 2020). However, heterozygous variations were also found in 1.0–4.6% of sporadic POI, making the inheritance pattern of MCM8/MCM9 in recessive or dominate to be ambiguous (Dou et al., 2016; Desai et al., 2017; Guo et al., 2020). Interestingly, some patients were found to carry digenic heterozygous variants in both MCM8 and MCM9 or in MCM8/MCM9 and other DNA repair genes (Desai et al., 2017). Moreover, researchers observed MCM8 had a dosage-dependent effect on the severity of POI phenotypes. These findings indicated that heterozygous variations of HR genes might establish a genetic background susceptive to DNA damage, which would affect meiosis when additional variations in the related genes or environmental toxin existed (Heddar et al., 2020; Wang et al., 2020). Besides the essential role in meiosis, MCM8 and MCM9 were involved in DNA replication, DNA damage response and cell cycle regulation in somatic cells. Some of the mutation carriers presented with short stature, and an MCM8 carrier was reported to have pilomatricomas (Heddar et al., 2020). Mitomycin-induced DNA breaks and aberrant metaphases in the patient’s lymphoblastoid cells suggested that the patients carrying MCM8 or MCM9 mutations were susceptive to tumor or growth retardation due to impaired DNA repair and genome instability in somatic cells. Therefore, long-term follow-up of cancers for those mutation carriers is needed.
HFM1 is a DNA helicase preferentially expressed in germline cells. Absence of HFM1 resulted in aberrant intermediate processing and reduced crossover formation (Guiraldelli et al., 2013). Two-compound heterozygous mutations of HFM1 were identified in two familial POI and one sporadic case (Wang et al., 2014). Moreover, heterozygous pathogenic mutations were found in a POI pedigree and 1.5% of the sporadic case, indicating that HFM1 mutants might cause POI through both recessive and dominate modes (Pu et al., 2016; Zhe et al., 2019).
RECQL4 and BLM are pleiotropic helicases expressed non-specifically, which unwind dsDNA into ssDNA during HR repair for DSBs (Singh et al., 2012). They are essential for maintenance of genome stability in both somatic and germline cells. Therefore, their defects mostly cause syndromic POI, such as Rothmund–Thomson syndrome (Siitonen et al., 2009) and Bloom syndrome (Arora et al., 2014), in which POI is one of the complicated symptoms.
Besides the helicases above, MCMDC2 is an atypical yet conserved MCM protein, which also plays an important role in ssDNA invasion that promotes homolog alignment and inter-homolog crossover formation. Mcmdc2 knockout mice were infertile, demonstrated to have atrophic ovaries completely devoid of oocyte at 6 weeks post-natal (Finsterbusch et al., 2016). Although no mutation of MCMDC2 has been reported in POI patient yet, it still is a potential causative gene for POI.
Synaptonemal Complex and Cohesion Complex
Throughout meiosis prophase I, the chromosomes are reorganized as linear arrays of chromatin loops anchored to a central axis. The chromosome axis forms a platform for the assembly of synaptonemal complex (SC), which plays a central role in homologous pairing, recombination, and chromosome segregation. The SC is installed by five central elements linked to two lateral elements by a transverse filament in each pair of homologous chromosomes.
The central elements of SC include SYCE1-3, C14ORF39, and TEX12. Female knockout mice of those genes were affected by infertility and oocyte loss before reproductive age due to different degrees of synapsis failure (Bolcun-Filas et al., 2007, 2009; Hamer et al., 2008; Schramm et al., 2011; Davies et al., 2012; Lu et al., 2014). In POI patients, except for one microdeletion and two homozygous mutations of SYCE1 which have been identified (McGuire et al., 2011; Zhen et al., 2013; de Vries et al., 2014; Zhe et al., 2020), no causative mutation has been found in other genes, indicating that the mutations in central elements of SC might not be a common genetic causation for POI.
SYCP1 is the transverse filament of SC that connects central elements SYCE1–2 to the lateral elements localized in each homologous chromosome. Absence of Sycp1 disturbed chromosomal synapsis, resulting in oocytes arrested at pachytene stage and apoptosis (de Vries et al., 2005). SYCP2 and SYCP3 are lateral elements of SC, which interact with each other (Winkel et al., 2009) and stabilize the linear array of chromatin loops during SC assembly (Yang et al., 2006; Syrjanen et al., 2014, 2017). The Sycp2 and Sycp3 mutant mice were subfertile (Yuan et al., 2000; Yang et al., 2006), which might be explained by insufficient SC formation, contrasting to the absolute loss of SC in Sycp1 null mice (Yang et al., 2006). That dosage-dependent meiosis dysfunction could also be a potential explanation for heterogeneous clinical phenotypes of human POI. Furthermore, Sycp3 mutant female mice exhibited increased aneuploidy in oocytes and embryos. In human beings, heterozygous variations of SYCP3 were associated with miscarriage and increased predisposition to infertility (Bolor et al., 2009; Nishiyama et al., 2011). Therefore, although no SYCP mutation has been identified in POI patient yet, their roles in oogenesis and embryo development should be further explored.
The cohesion complex regulates sister chromatids cohesion and SC formation, which is composed of meiosis-specific subunits STAG3, RAD21L, and SMC1B and non-specific subunits SMC3 and REC8 (Ishiguro, 2019). Female mice deficient in Stag3 were sterility and follicle exhausted at a young age (Caburet et al., 2014). To date, seven bi-allelic mutations of STAG3 have been found in POI pedigrees. All the affected patients manifested with primary amenorrhea and streak ovaries (Caburet et al., 2014; Le Quesne Stabej et al., 2016; Colombo et al., 2017; He et al., 2018a; Franca et al., 2019; Heddar et al., 2019; Xiao et al., 2019), indicating that recessive mutations in STAG3 were relatively common genetic causation for primary POI. Furthermore, mice deficient in other cohesion genes demonstrated with similar ovarian morphology of Stag3 null mice (Xu et al., 2005; Takabayashi et al., 2009; Herran et al., 2011). Through target gene screening of sporadic POI patients, heterozygous mutations in STAG3, SMC1B, and REC8 have been found (Bouilly et al., 2016), indicating that the recessive and dominate causative modes of cohesion genes in POI might coexist. Moreover, age-dependent decrease of cohesion protein is associated with increased rate of aneuploidy oocytes, while mutations of SMC3 were reported in Cornelia de Lange syndrome without ovarian abnormalities (Deardorff et al., 2007). Therefore, besides the indispensable contribution to POI, the pleiotropic effects of cohesion genes in reproductive and somatic diseases should be considered as well.
Resolution of Recombination Intermediates
In germ cells, the essential step for accurate separation of homologous chromosomes at the first meiotic division is resolution of recombination intermediates, including non-crossover pathway and crossover pathway. In the non-crossover pathway, the final products are generated by annealing the invaded strand to the complementary break end of single Holliday junction or dissolution of the dHJs (Bizard and Hickson, 2014; Daley et al., 2014). That process is the major route for dissipation of HR intermediate, which limits chromosomal rearrangements and heterozygosity of oocytes. This reaction requires the RecQ helicase BLM (Wu et al., 2000), topoisomerase TOP3A (Martin et al., 2018), RMI complex (Raynard et al., 2008), structure-selective endonucleases GEN1 (Shah Punatar et al., 2017), MUS81-EME1, and SLX1–SLX4 (Matos and West, 2014; Wyatt and West, 2014). Absence of the above genes resulted in syndromic disease or embryonic lethality. Therefore, their pleiotropic effect on meiosis and ovarian function was illusive and needs more detailed exploration.
Crossover pathway is the meiosis-specific resolution of dHJs that contributes to the genetic diversity of species. Although the process resolves less recombination than non-crossover pathway, the occurrence of at least one crossover in every pair of homologous chromosomes is essential for precise separation of chromosomes in the first meiotic division. Crossover pathway involves RNF212 (Qiao et al., 2014), HEI10 (Ward et al., 2007), MLH1 (Baker et al., 1996), and MLH3 (Lipkin et al., 2002). The knockout mice of the above genes had normal ovarian morphology; oocytes show proficient synapsis but deficient crossover, presenting with abnormal chromosome alignment at metaphase I and disturbed extrusion of polar bodies. Those female mice were infertile due to a decreased number of mature MII oocytes and increased number of aneuploidy embryos. Therefore, these gene defects are responsible for disorders of oocyte maturation or early embryo development rather than POI.
Fanconi Anemia Pathway Genes in Meiotic HR
Fanconi anemia (FA) is usually a recessive genetic disease associated with bone marrow failure, increased cancer susceptibility, and severe germline defects. There are 22 identified FA genes, which are involved in DNA interstrand crosslink repair, including the FA core complex which catalyzes the mono-ubiquitination of FANCD2 and FANCI, and DSB repair genes—BRCA1 (FANCS), BRCA2 (FANCD1), BRIP1 (FANCJ), PALB2 (FANCN), RAD51C (FANCO), SLX4 (FANCP), RAD51 (FANCR), and XRCC2 (FANCU) (Tsui and Crismani, 2019). Although all mice models of FA genes reported to date have different degrees of reduction in fertility, the links between their roles in DNA repair and fertility have not been extensively explained. Recent studies found that BRCA2 promoted the localization of RAD51 and DMC1 to meiotic DSBs. As a member of RAD51 paralogs, FANCU (XRCC2) might be involved in the RAD51-mediated strand invasion during meiotic HR (Yang et al., 2018). Besides that, FA core factors FANCA, FANCB, and FANCC were reported to facilitate the recruitment of FANCD2 on sex chromosomes and regulate the histone modification during meiotic HR (Alavattam et al., 2016). FANCM has also been shown to limit crossover frequencies, which promoted the conservatism of gametes (Crismani et al., 2012). The increasing research of FA genes highlighted their roles in the resolution of meiotic DSBs, giving more indications of oogenesis as well. Up to date, several FA genes had identifications in POI, such as homozygous mutations in FANCM (Fouquet et al., 2017) and FANCU (Zhang et al., 2019) and heterozygous mutations in FANCA (Yang et al., 2019b) and FANCL (Yang et al., 2020). Interestingly, heterozygous FANCA knockout mice showed a declined follicle number and reduced fertility; in vitro studies found that single-allelic defects of FANCA and FANCL compromised DNA repair ability by haploinsufficiency, indicating that the adverse effects of FA gene variations on meiosis and ovarian function might be dosage dependent.
Discussion
Identifying causative genes of POI and elucidating their molecular mechanisms are important for the genetic diagnosis of POI. As an increasing number of women prefer to conceive after their mid-30s, the genetic counseling of POI predisposition will be instructive for their childbearing plans. To date, more than 75 genes have been found to be responsible for POI, among which 24 genes were involved in meiotic HR process. With the widespread use of NGS and whole-genome sequencing, the identification of novel genes will be increased in the near future. Furthermore, with the development of data analysis strategies, more non-synonymous mutations with high risk of pathogenicity, microdeletion or interruption, and rearrangement of gene sequences will be identified, expanding the mutation spectrum and genetic architecture of POI.
Along with the increasing genes and variations identified, more challenges are emerging to determine the causative patterns of meiotic HR genes in POI. Most of the meiotic HR genes were found in familial POI by recessive modes, while heterozygous mutations were more common in sporadic cases, and the mutation frequencies varied significantly among different cohorts. These observations indicated that the genetic architecture of sporadic POI would be more complicated than that in familial cases. With more and more di-genetic or multigenetic variations reported and dosage-dependent effect confirmed by functional studies, sporadic POI seemed to be a complex disease, which occurred as a result of multiple genomic variants paired with environmental influences. Furthermore, many HR genes had pleiotropic effects in proliferation and apoptosis of somatic cells. The relationships between pleiotropic genes and heterogeneous phenotypes of isolated or syndromic POI should be further explored as well.
Meiotic HR genes not only participate in oogenesis but also facilitate oocyte maturation, fertilization, and early embryo development. Dysfunction of several genes might be responsible for unexplained infertility or early pregnancy loss, such as members of SC and cohesion complex influenced chromosome separation and aneuploidy of oocytes. Therefore, besides the benefits of early diagnosis, intervention, and treatment of POI, further studies on the meiotic HR genes will give advice to other diseases of infertility and adverse pregnancy outcomes. Moreover, considering the increased cancer susceptibility of HR gene defects, long-term follow-up for cancer risks and healthcare should be suggested.
Author Contributions
YQ and TG contributed to the conceptual idea. CH performed the data collection. TG and CH wrote the manuscript. YQ reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Developmental Program of China (2018YFC1003702); the Science Foundation for Distinguished Young Scholars of Shandong (JQ201720); the National Natural Science Foundation of China (32070847, 81771541, and 81671413); Basic Science Center Program of NFSC (31988101); and Shandong University Education Foundation Public Welfare Project (23460047102008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Acharya, S., Wilson, T., Gradia, S., Kane, M. F., Guerrette, S., Marsischky, G. T., et al. (1996). hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. U.S.A. 93, 13629–13634. doi: 10.1073/pnas.93.24.13629
Agoulnik, A. I., Lu, B., Zhu, Q., Truong, C., Ty, M. T., Arango, N., et al. (2002). A novel gene, Pog, is necessary for primordial germ cell proliferation in the mouse and underlies the germ cell deficient mutation, gcd. Hum. Mol. Genet. 11, 3047–3053. doi: 10.1093/hmg/11.24.3047
Al-Agha, A. E., Ahmed, I. A., Nuebel, E., Moriwaki, M., Moore, B., Peacock, K. A., et al. (2018). Primary ovarian insufficiency and azoospermia in carriers of a homozygous PSMC3IP stop gain mutation. J. Clin. Endocrinol. Metab. 103, 555–563. doi: 10.1210/jc.2017-01966
AlAsiri, S., Basit, S., Wood-Trageser, M. A., Yatsenko, S. A., Jeffries, E. P., Surti, U., et al. (2015). Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. J. Clin. Invest. 125, 258–262. doi: 10.1172/JCI78473
Alavattam, K. G., Kato, Y., Sin, H. S., Maezawa, S., Kowalski, I. J., Zhang, F., et al. (2016). Elucidation of the fanconi anemia protein network in meiosis and its function in the regulation of histone modifications. Cell Rep. 17, 1141–1157. doi: 10.1016/j.celrep.2016.09.073
Anand, R., Jasrotia, A., Bundschuh, D., Howard, S. M., Ranjha, L., Stucki, M., et al. (2019). NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 38:e101005. doi: 10.15252/embj.2018101005
Arora, H., Chacon, A. H., Choudhary, S., McLeod, M. P., Meshkov, L., Nouri, K., et al. (2014). Bloom syndrome. Int. J. Dermatol. 53, 798–802. doi: 10.1111/ijd.12408
Baker, S. M., Plug, A. W., Prolla, T. A., Bronner, C. E., Harris, A. C., Yao, X., et al. (1996). Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 13, 336–342. doi: 10.1038/ng0796-336
Bakker, S. T., van de Vrugt, H. J., Rooimans, M. A., Oostra, A. B., Steltenpool, J., Delzenne-Goette, E., et al. (2009). Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum. Mol. Genet. 18, 3484–3495. doi: 10.1093/hmg/ddp297
Bishop, D. K., Park, D., Xu, L., and Kleckner, N. (1992). DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456. doi: 10.1016/0092-8674(92)90446-j
Bizard, A. H., and Hickson, I. D. (2014). The dissolution of double Holliday junctions. Cold Spring Harb. Perspect. Biol. 6:a016477. doi: 10.1101/cshperspect.a016477
Bolcun-Filas, E., Costa, Y., Speed, R., Taggart, M., Benavente, R., De Rooij, D. G., et al. (2007). SYCE2 is required for synaptonemal complex assembly, double strand break repair, and homologous recombination. J. Cell Biol. 176, 741–747. doi: 10.1083/jcb.200610027
Bolcun-Filas, E., Hall, E., Speed, R., Taggart, M., Grey, C., de Massy, B., et al. (2009). Mutation of the mouse Syce1 gene disrupts synapsis and suggests a link between synaptonemal complex structural components and DNA repair. PLoS Genet. 5:e1000393. doi: 10.1371/journal.pgen.1000393
Bolor, H., Mori, T., Nishiyama, S., Ito, Y., Hosoba, E., Inagaki, H., et al. (2009). Mutations of the SYCP3 gene in women with recurrent pregnancy loss. Am. J. Hum. Genet. 84, 14–20. doi: 10.1016/j.ajhg.2008.12.002
Bouali, N., Francou, B., Bouligand, J., Imanci, D., Dimassi, S., Tosca, L., et al. (2017). New MCM8 mutation associated with premature ovarian insufficiency and chromosomal instability in a highly consanguineous Tunisian family. Fertil. Steril. 108, 694–702. doi: 10.1016/j.fertnstert.2017.07.015
Bouilly, J., Beau, I., Barraud, S., Bernard, V., Azibi, K., Fagart, J., et al. (2016). Identification of multiple gene mutations accounts for a new genetic architecture of primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 101, 4541–4550. doi: 10.1210/jc.2016-2152
Brown, M. S., and Bishop, D. K. (2014). DNA strand exchange and RecA homologs in meiosis. Cold Spring Harb. Perspect. Biol. 7:a016659. doi: 10.1101/cshperspect.a016659
Caburet, S., Arboleda, V. A., Llano, E., Overbeek, P. A., Barbero, J. L., Oka, K., et al. (2014). Mutant cohesin in premature ovarian failure. N. Engl. J. Med. 370, 943–949. doi: 10.1056/NEJMoa1309635
Caburet, S., Heddar, A., Dardillac, E., Creux, H., Lambert, M., Messiaen, S., et al. (2020). Homozygous hypomorphic BRCA2 variant in primary ovarian insufficiency without cancer or Fanconi anaemia trait. J. Med. Genet. doi: 10.1136/jmedgenet-2019-106672 [Epub ahead of print].
Caburet, S., Todeschini, A. L., Petrillo, C., Martini, E., Farran, N. D., Legois, B., et al. (2019). A truncating MEIOB mutation responsible for familial primary ovarian insufficiency abolishes its interaction with its partner SPATA22 and their recruitment to DNA double-strand breaks. EBioMedicine 42, 524–531. doi: 10.1016/j.ebiom.2019.03.075
Carlosama, C., Elzaiat, M., Patino, L. C., Mateus, H. E., Veitia, R. A., and Laissue, P. (2017). A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Hum. Mol. Genet. 26, 3161–3166.
Chen, Y., Lyu, R., Rong, B., Zheng, Y., Lin, Z., Dai, R., et al. (2020). Refined spatial temporal epigenomic profiling reveals intrinsic connection between PRDM9-mediated H3K4me3 and the fate of double-stranded breaks. Cell Res. 30, 256–268. doi: 10.1038/s41422-020-0281-1
Cheng, N. C., van de Vrugt, H. J., van der Valk, M. A., Oostra, A. B., Krimpenfort, P., de Vries, Y., et al. (2000). Mice with a targeted disruption of the Fanconi anemia homolog Fanca. Hum. Mol. Genet. 9, 1805–1811. doi: 10.1093/hmg/9.12.1805
Chi, P., San Filippo, J., Sehorn, M. G., Petukhova, G. V., and Sung, P. (2007). Bipartite stimulatory action of the Hop2-Mnd1 complex on the Rad51 recombinase. Genes. Dev. 21, 1747–1757. doi: 10.1101/gad.1563007
Chrzanowska, K. H., Gregorek, H., Dembowska-Baginska, B., Kalina, M. A., and Digweed, M. (2012). Nijmegen breakage syndrome (NBS). Orphanet. J. Rare. Dis. 7:13. doi: 10.1186/1750-1172-7-13
Colombo, R., Pontoglio, A., and Bini, M. (2017). A STAG3 missense mutation in two sisters with primary ovarian insufficiency. Eur. J. Obstet. Gynecol. Reprod. Biol. 216, 269–271. doi: 10.1016/j.ejogrb.2017.08.005
Connor, F., Bertwistle, D., Mee, P. J., Ross, G. M., Swift, S., Grigorieva, E., et al. (1997). Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation. Nat. Genet. 17, 423–430. doi: 10.1038/ng1297-423
Crismani, W., Girard, C., Froger, N., Pradillo, M., Santos, J. L., Chelysheva, L., et al. (2012). FANCM limits meiotic crossovers. Science 336, 1588–1590. doi: 10.1126/science.1220381
Daley, J. M., Gaines, W. A., Kwon, Y., and Sung, P. (2014). Regulation of DNA pairing in homologous recombination. Cold Spring Harb. Perspect. Biol. 6:a017954. doi: 10.1101/cshperspect.a017954
Daniel, K., Lange, J., Hached, K., Fu, J., Anastassiadis, K., Roig, I., et al. (2011). Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat. Cell Biol. 13, 599–610. doi: 10.1038/ncb2213
Daum, H., Peretz, T., and Laufer, N. (2018). BRCA mutations and reproduction. Fertil. Steril. 109, 33–38. doi: 10.1016/j.fertnstert.2017.12.004
Davies, A. A., Masson, J. Y., McIlwraith, M. J., Stasiak, A. Z., Stasiak, A., Venkitaraman, A. R., et al. (2001). Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 7, 273–282. doi: 10.1016/s1097-2765(01)00175-7
Davies, O. R., Maman, J. D., and Pellegrini, L. (2012). Structural analysis of the human SYCE2-TEX12 complex provides molecular insights into synaptonemal complex assembly. Open Biol. 2:120099. doi: 10.1098/rsob.120099
Day, F. R., Ruth, K. S., Thompson, D. J., Lunetta, K. L., Pervjakova, N., Chasman, D. I., et al. (2015). Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 47, 1294–1303. doi: 10.1038/ng.3412
de Vries, F. A., de Boer, E., van den Bosch, M., Baarends, W. M., Ooms, M., Yuan, L., et al. (2005). Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev. 19, 1376–1389. doi: 10.1101/gad.329705
de Vries, L., Behar, D. M., Smirin-Yosef, P., Lagovsky, I., Tzur, S., and Basel-Vanagaite, L. (2014). Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 99, E2129–E2132. doi: 10.1210/jc.2014-1268
de Vries, S. S., Baart, E. B., Dekker, M., Siezen, A., de Rooij, D. G., de Boer, P., et al. (1999). Mouse MutS-like protein Msh5 is required for proper chromosome synapsis in male and female meiosis. Genes Dev. 13, 523–531. doi: 10.1101/gad.13.5.523
Deardorff, M. A., Kaur, M., Yaeger, D., Rampuria, A., Korolev, S., Pie, J., et al. (2007). Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 80, 485–494. doi: 10.1086/511888
Desai, S., and Rajkovic, A. (2017). Genetics of reproductive aging from gonadal dysgenesis through menopause. Semin. Reprod. Med. 35, 147–159. doi: 10.1055/s-0037-1599086
Desai, S., Wood-Trageser, M., Matic, J., Chipkin, J., Jiang, H., Bachelot, A., et al. (2017). MCM8 and MCM9 nucleotide variants in women with primary ovarian insufficiency. J. Clin. Endocrinol. Metab. 102, 576–582. doi: 10.1210/jc.2016-2565
Dou, X., Guo, T., Li, G., Zhou, L., Qin, Y., and Chen, Z. J. (2016). Minichromosome maintenance complex component 8 mutations cause primary ovarian insufficiency. Fertil. Steril. 106, 1485–1489e2. doi: 10.1016/j.fertnstert.2016.08.018
European Society for Human, R., Embryology Guideline Group on, P. O. I., Webber, L., Davies, M., Anderson, R., Bartlett, J., et al. (2016). ESHRE Guideline: management of women with premature ovarian insufficiency. Hum. Reprod. 31, 926–937. doi: 10.1093/humrep/dew027
Fauchereau, F., Shalev, S., Chervinsky, E., Beck-Fruchter, R., Legois, B., Fellous, M., et al. (2016). A non-sense MCM9 mutation in a familial case of primary ovarian insufficiency. Clin. Genet. 89, 603–607. doi: 10.1111/cge.12736
Finsterbusch, F., Ravindranathan, R., Dereli, I., Stanzione, M., Trankner, D., and Toth, A. (2016). Alignment of homologous chromosomes and effective repair of programmed DNA double-strand breaks during mouse meiosis require the minichromosome maintenance domain containing 2 (MCMDC2) protein. PLoS Genet. 12:e1006393. doi: 10.1371/journal.pgen.1006393
Fouquet, B., Pawlikowska, P., Caburet, S., Guigon, C., Makinen, M., Tanner, L., et al. (2017). A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. Elife 6:e30490. doi: 10.7554/eLife.30490
Franca, M. M., and Mendonca, B. B. (2020). Genetics of primary ovarian insufficiency in the next-generation sequencing era. J. Endocr. Soc. 4:bvz037. doi: 10.1210/jendso/bvz037
Franca, M. M., Nishi, M. Y., Funari, M. F. A., Lerario, A. M., Baracat, E. C., Hayashida, S. A. Y., et al. (2019). Two rare loss-of-function variants in the STAG3 gene leading to primary ovarian insufficiency. Eur. J. Med. Genet. 62, 186–189. doi: 10.1016/j.ejmg.2018.07.008
Garcia, V., Phelps, S. E., Gray, S., and Neale, M. J. (2011). Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244. doi: 10.1038/nature10515
Goldberg, Y., Halpern, N., Hubert, A., Adler, S. N., Cohen, S., Plesser-Duvdevani, M., et al. (2015). Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 208, 621–624. doi: 10.1016/j.cancergen.2015.10.001
Guiraldelli, M. F., Eyster, C., Wilkerson, J. L., Dresser, M. E., and Pezza, R. J. (2013). Mouse HFM1/Mer3 is required for crossover formation and complete synapsis of homologous chromosomes during meiosis. PLoS Genet. 9:e1003383. doi: 10.1371/journal.pgen.1003383
Guo, T., Zhao, S., Zhao, S., Chen, M., Li, G., Jiao, X., et al. (2017). Mutations in MSH5 in primary ovarian insufficiency. Hum. Mol. Genet. 26, 1452–1457. doi: 10.1093/hmg/ddx044
Guo, T., Zheng, Y., Li, G., Zhao, S., Ma, J., and Qin, Y. (2020). Novel pathogenic mutations in minichromosome maintenance complex component 9 (MCM9) responsible for premature ovarian insufficiency. Fertil. Steril. 113, 845–852. doi: 10.1016/j.fertnstert.2019.11.015
Hamer, G., Wang, H., Bolcun-Filas, E., Cooke, H. J., Benavente, R., and Hoog, C. (2008). Progression of meiotic recombination requires structural maturation of the central element of the synaptonemal complex. J. Cell Sci. 121(Pt 15), 2445–2451. doi: 10.1242/jcs.033233
Handel, M. A., and Schimenti, J. C. (2010). Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat. Rev. Genet. 11, 124–136. doi: 10.1038/nrg2723
Hays, E., Majchrzak, N., Daniel, V., Ferguson, Z., Brown, S., Hathorne, K., et al. (2017). Spermatogenesis associated 22 is required for DNA repair and synapsis of homologous chromosomes in mouse germ cells. Andrology 5, 299–312. doi: 10.1111/andr.12315
He, W. B., Banerjee, S., Meng, L. L., Du, J., Gong, F., Huang, H., et al. (2018a). Whole-exome sequencing identifies a homozygous donor splice-site mutation in STAG3 that causes primary ovarian insufficiency. Clin. Genet. 93, 340–344. doi: 10.1111/cge.13034
He, W. B., Tu, C. F., Liu, Q., Meng, L. L., Yuan, S. M., Luo, A. X., et al. (2018b). DMC1 mutation that causes human non-obstructive azoospermia and premature ovarian insufficiency identified by whole-exome sequencing. J. Med. Genet. 55, 198–204. doi: 10.1136/jmedgenet-2017-104992
Heddar, A., Beckers, D., Fouquet, B., Roland, D., and Misrahi, M. (2020). A novel phenotype combining primary ovarian insufficiency growth retardation and pilomatricomas with MCM8 mutation. J. Clin. Endocrinol. Metab. 105:dgaa155. doi: 10.1210/clinem/dgaa155
Heddar, A., Dessen, P., Flatters, D., and Misrahi, M. (2019). Novel STAG3 mutations in a Caucasian family with primary ovarian insufficiency. Mol. Genet. Genomics 294, 1527–1534. doi: 10.1007/s00438-019-01594-4
Herran, Y., Gutierrez-Caballero, C., Sanchez-Martin, M., Hernandez, T., Viera, A., Barbero, J. L., et al. (2011). The cohesin subunit RAD21L functions in meiotic synapsis and exhibits sexual dimorphism in fertility. EMBO J. 30, 3091–3105. doi: 10.1038/emboj.2011.222
Hikiba, J., Hirota, K., Kagawa, W., Ikawa, S., Kinebuchi, T., Sakane, I., et al. (2008). Structural and functional analyses of the DMC1-M200V polymorphism found in the human population. Nucleic Acids Res. 36, 4181–4190. doi: 10.1093/nar/gkn362
Inagaki, A., Roset, R., and Petrini, J. H. (2016). Functions of the MRE11 complex in the development and maintenance of oocytes. Chromosoma 125, 151–162. doi: 10.1007/s00412-015-0535-8
Ishiguro, K. I. (2019). The cohesin complex in mammalian meiosis. Genes Cells 24, 6–30. doi: 10.1111/gtc.12652
Ishishita, S., Matsuda, Y., and Kitada, K. (2014). Genetic evidence suggests that Spata22 is required for the maintenance of Rad51 foci in mammalian meiosis. Sci. Rep. 4:6148. doi: 10.1038/srep06148
Janisiw, E., Dello Stritto, M. R., Jantsch, V., and Silva, N. (2018). BRCA1-BARD1 associate with the synaptonemal complex and pro-crossover factors and influence RAD-51 dynamics during Caenorhabditis elegans meiosis. PLoS Genet. 14:e1007653. doi: 10.1371/journal.pgen.1007653
Jiao, X., Zhang, H., Ke, H., Zhang, J., Cheng, L., Liu, Y., et al. (2017). Premature ovarian insufficiency: phenotypic characterization within different etiologies. J. Clin. Endocrinol. Metab. 102, 2281–2290. doi: 10.1210/jc.2016-3960
Jolly, A., Bayram, Y., Turan, S., Aycan, Z., Tos, T., Abali, Z. Y., et al. (2019). Exome sequencing of a primary ovarian insufficiency cohort reveals common molecular etiologies for a spectrum of disease. J. Clin. Endocrinol. Metab. 104, 3049–3067. doi: 10.1210/jc.2019-00248
Kang, J., Bronson, R. T., and Xu, Y. (2002). Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J. 21, 1447–1455. doi: 10.1093/emboj/21.6.1447
Kneitz, B., Cohen, P. E., Avdievich, E., Zhu, L., Kane, M. F., Hou, H. Jr., et al. (2000). MutS homolog 4 localization to meiotic chromosomes is required for chromosome pairing during meiosis in male and female mice. Genes Dev. 14, 1085–1097.
Kumar, R., Bourbon, H. M., and de Massy, B. (2010). Functional conservation of Mei4 for meiotic DNA double-strand break formation from yeasts to mice. Genes Dev. 24, 1266–1280. doi: 10.1101/gad.571710
Kumar, R., Oliver, C., Brun, C., Juarez-Martinez, A. B., Tarabay, Y., Kadlec, J., et al. (2018). Mouse REC114 is essential for meiotic DNA double-strand break formation and forms a complex with MEI4. Life Sci. Alliance 1:e201800259. doi: 10.26508/lsa.201800259
La Salle, S., Palmer, K., O’Brien, M., Schimenti, J. C., Eppig, J., and Handel, M. A. (2012). Spata22, a novel vertebrate-specific gene, is required for meiotic progress in mouse germ cells. Biol. Reprod. 86:45. doi: 10.1095/biolreprod.111.095752
Le Quesne Stabej, P., Williams, H. J., James, C., Tekman, M., Stanescu, H. C., Kleta, R., et al. (2016). STAG3 truncating variant as the cause of primary ovarian insufficiency. Eur. J. Hum. Genet. 24, 135–138. doi: 10.1038/ejhg.2015.107
Lin, W., Titus, S., Moy, F., Ginsburg, E. S., and Oktay, K. (2017). Ovarian aging in women with BRCA germline mutations. J. Clin. Endocrinol. Metab. 102, 3839–3847. doi: 10.1210/jc.2017-00765
Lipkin, S. M., Moens, P. B., Wang, V., Lenzi, M., Shanmugarajah, D., Gilgeous, A., et al. (2002). Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat. Genet. 31, 385–390. doi: 10.1038/ng931
Lu, J., Gu, Y., Feng, J., Zhou, W., Yang, X., and Shen, Y. (2014). Structural insight into the central element assembly of the synaptonemal complex. Sci. Rep. 4:7059. doi: 10.1038/srep07059
Luo, M., Yang, F., Leu, N. A., Landaiche, J., Handel, M. A., Benavente, R., et al. (2013). MEIOB exhibits single-stranded DNA-binding and exonuclease activities and is essential for meiotic recombination. Nat. Commun. 4:2788. doi: 10.1038/ncomms3788
Luo, W., Guo, T., Li, G., Liu, R., Zhao, S., Song, M., et al. (2020). Variants in homologous recombination genes EXO1 and RAD51 related with premature ovarian insufficiency. J. Clin. Endocrinol. Metab. 105:dgaa505. doi: 10.1210/clinem/dgaa505
Lutzmann, M., Grey, C., Traver, S., Ganier, O., Maya-Mendoza, A., Ranisavljevic, N., et al. (2012). MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Mol. Cell 47, 523–534. doi: 10.1016/j.molcel.2012.05.048
Mandon-Pepin, B., Touraine, P., Kuttenn, F., Derbois, C., Rouxel, A., Matsuda, F., et al. (2008). Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur. J. Endocrinol. 158, 107–115. doi: 10.1530/EJE-07-0400
Martin, C. A., Sarlos, K., Logan, C. V., Thakur, R. S., Parry, D. A., Bizard, A. H., et al. (2018). Mutations in TOP3A cause a bloom syndrome-like disorder. Am. J. Hum. Genet. 103, 221–231. doi: 10.1016/j.ajhg.2018.07.001
Matos, J., and West, S. C. (2014). Holliday junction resolution: regulation in space and time. DNA Repair (Amst) 19, 176–181. doi: 10.1016/j.dnarep.2014.03.013
McGuire, M. M., Bowden, W., Engel, N. J., Ahn, H. W., Kovanci, E., and Rajkovic, A. (2011). Genomic analysis using high-resolution single-nucleotide polymorphism arrays reveals novel microdeletions associated with premature ovarian failure. Fertil. Steril. 95, 1595–1600. doi: 10.1016/j.fertnstert.2010.12.052
Miao, Y., Wang, P., Xie, B., Yang, M., Li, S., Cui, Z., et al. (2019). BRCA2 deficiency is a potential driver for human primary ovarian insufficiency. Cell Death Dis. 10:474. doi: 10.1038/s41419-019-1720-0
Nguyen, N. M. P., Ge, Z. J., Reddy, R., Fahiminiya, S., Sauthier, P., Bagga, R., et al. (2018). Causative mutations and mechanism of androgenetic hydatidiform moles. Am. J. Hum. Genet. 103, 740–751. doi: 10.1016/j.ajhg.2018.10.007
Nishant, K. T., Chen, C., Shinohara, M., Shinohara, A., and Alani, E. (2010). Genetic analysis of baker’s yeast Msh4-Msh5 reveals a threshold crossover level for meiotic viability. PLoS Genet. 6:e1001083. doi: 10.1371/journal.pgen.1001083
Nishimura, K., Ishiai, M., Horikawa, K., Fukagawa, T., Takata, M., Takisawa, H., et al. (2012). Mcm8 and Mcm9 form a complex that functions in homologous recombination repair induced by DNA interstrand crosslinks. Mol. Cell 47, 511–522. doi: 10.1016/j.molcel.2012.05.047
Nishiyama, S., Kishi, T., Kato, T., Suzuki, M., Bolor, H., Nishizawa, H., et al. (2011). A rare synaptonemal complex protein 3 gene variant in unexplained female infertility. Mol. Hum. Reprod. 17, 266–271. doi: 10.1093/molehr/gaq098
Park, J. Y., Yoo, H. W., Kim, B. R., Park, R., Choi, S. Y., and Kim, Y. (2008). Identification of a novel human Rad51 variant that promotes DNA strand exchange. Nucleic Acids Res. 36, 3226–3234. doi: 10.1093/nar/gkn171
Patino, L. C., Beau, I., Carlosama, C., Buitrago, J. C., Gonzalez, R., Suarez, C. F., et al. (2017). New mutations in non-syndromic primary ovarian insufficiency patients identified via whole-exome sequencing. Hum. Reprod. 32, 1512–1520. doi: 10.1093/humrep/dex089
Petukhova, G. V., Romanienko, P. J., and Camerini-Otero, R. D. (2003). The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev. Cell 5, 927–936. doi: 10.1016/s1534-5807(03)00369-1
Pezza, R. J., Voloshin, O. N., Vanevski, F., and Camerini-Otero, R. D. (2007). Hop2/Mnd1 acts on two critical steps in Dmc1-promoted homologous pairing. Genes Dev. 21, 1758–1766. doi: 10.1101/gad.1562907
Pezza, R. J., Voloshin, O. N., Volodin, A. A., Boateng, K. A., Bellani, M. A., Mazin, A. V., et al. (2014). The dual role of HOP2 in mammalian meiotic homologous recombination. Nucleic Acids Res. 42, 2346–2357. doi: 10.1093/nar/gkt1234
Pittman, D. L., Cobb, J., Schimenti, K. J., Wilson, L. A., Cooper, D. M., Brignull, E., et al. (1998). Meiotic prophase arrest with failure of chromosome synapsis in mice deficient for Dmc1, a germline-specific RecA homolog. Mol. Cell 1, 697–705. doi: 10.1016/s1097-2765(00)80069-6
Porcu, E., Cillo, G. M., Cipriani, L., Sacilotto, F., Notarangelo, L., Damiano, G., et al. (2020). Impact of BRCA1 and BRCA2 mutations on ovarian reserve and fertility preservation outcomes in young women with breast cancer. J. Assist. Reprod. Genet. 37, 709–715. doi: 10.1007/s10815-019-01658-9
Pu, D., Wang, C., Cao, J., Shen, Y., Jiang, H., Liu, J., et al. (2016). Association analysis between HFM1 variation and primary ovarian insufficiency in Chinese women. Clin. Genet. 89, 597–602. doi: 10.1111/cge.12718
Qiao, H., Prasada Rao, H. B., Yang, Y., Fong, J. H., Cloutier, J. M., Deacon, D. C., et al. (2014). Antagonistic roles of ubiquitin ligase HEI10 and SUMO ligase RNF212 regulate meiotic recombination. Nat. Genet. 46, 194–199. doi: 10.1038/ng.2858
Qin, Y., Jiao, X., Simpson, J. L., and Chen, Z. J. (2015). Genetics of primary ovarian insufficiency: new developments and opportunities. Hum. Reprod. Update 21, 787–808. doi: 10.1093/humupd/dmv036
Qin, Y., Zhang, F., and Chen, Z. J. (2019). BRCA2 in ovarian development and function. N. Engl. J. Med. 380:1086. doi: 10.1056/NEJMc1813800
Qvist, P., Huertas, P., Jimeno, S., Nyegaard, M., Hassan, M. J., Jackson, S. P., et al. (2011). CtIP mutations cause seckel and jawad syndromes. PLoS Genet. 7:e1002310. doi: 10.1371/journal.pgen.1002310
Ranjha, L., Howard, S. M., and Cejka, P. (2018). Main steps in DNA double-strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214. doi: 10.1007/s00412-017-0658-1
Raynard, S., Zhao, W., Bussen, W., Lu, L., Ding, Y. Y., Busygina, V., et al. (2008). Functional role of BLAP75 in BLM-topoisomerase IIIalpha-dependent Holliday junction processing. J. Biol. Chem. 283, 15701–15708. doi: 10.1074/jbc.M802127200
Reinholdt, L. G., and Schimenti, J. C. (2005). Mei1 is epistatic to Dmc1 during mouse meiosis. Chromosoma 114, 127–134. doi: 10.1007/s00412-005-0346-4
Ren, Y., Diao, F., Katari, S., Yatsenko, S., Jiang, H., Wood-Trageser, M. A., et al. (2018). Functional study of a novel missense single-nucleotide variant of NUP107 in two daughters of Mexican origin with premature ovarian insufficiency. Mol. Genet. Genomic. Med. 6, 276–281. doi: 10.1002/mgg3.345
Robert, T., Nore, A., Brun, C., Maffre, C., Crimi, B., Bourbon, H. M., et al. (2016). The TopoVIB-Like protein family is required for meiotic DNA double-strand break formation. Science 351, 943–949. doi: 10.1126/science.aad5309
Romanienko, P. J., and Camerini-Otero, R. D. (2000). The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol. Cell 6, 975–987. doi: 10.1016/s1097-2765(00)00097-6
Roset, R., Inagaki, A., Hohl, M., Brenet, F., Lafrance-Vanasse, J., Lange, J., et al. (2014). The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes Dev. 28, 451–462. doi: 10.1101/gad.236745.113
Sansam, C. L., and Pezza, R. J. (2015). Connecting by breaking and repairing: mechanisms of DNA strand exchange in meiotic recombination. FEBS J. 282, 2444–2457. doi: 10.1111/febs.13317
Sartori, A. A., Lukas, C., Coates, J., Mistrik, M., Fu, S., Bartek, J., et al. (2007). Human CtIP promotes DNA end resection. Nature 450, 509–514. doi: 10.1038/nature06337
Schramm, S., Fraune, J., Naumann, R., Hernandez-Hernandez, A., Hoog, C., Cooke, H. J., et al. (2011). A novel mouse synaptonemal complex protein is essential for loading of central element proteins, recombination, and fertility. PLoS Genet. 7:e1002088. doi: 10.1371/journal.pgen.1002088
Shah Punatar, R., Martin, M. J., Wyatt, H. D., Chan, Y. W., and West, S. C. (2017). Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc. Natl. Acad. Sci. U.S.A. 114, 443–450. doi: 10.1073/pnas.1619790114
Shi, B., Xue, J., Yin, H., Guo, R., Luo, M., Ye, L., et al. (2019). Dual functions for the ssDNA-binding protein RPA in meiotic recombination. PLoS Genet. 15:e1007952. doi: 10.1371/journal.pgen.1007952
Shin, Y. H., Choi, Y., Erdin, S. U., Yatsenko, S. A., Kloc, M., Yang, F., et al. (2010). Hormad1 mutation disrupts synaptonemal complex formation, recombination, and chromosome segregation in mammalian meiosis. PLoS Genet. 6:e1001190. doi: 10.1371/journal.pgen.1001190
Siitonen, H. A., Sotkasiira, J., Biervliet, M., Benmansour, A., Capri, Y., Cormier-Daire, V., et al. (2009). The mutation spectrum in RECQL4 diseases. Eur. J. Hum. Genet. 17, 151–158. doi: 10.1038/ejhg.2008.154
Singh, D. K., Popuri, V., Kulikowicz, T., Shevelev, I., Ghosh, A. K., Ramamoorthy, M., et al. (2012). The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 40, 6632–6648. doi: 10.1093/nar/gks349
Smirin-Yosef, P., Zuckerman-Levin, N., Tzur, S., Granot, Y., Cohen, L., Sachsenweger, J., et al. (2017). A biallelic mutation in the homologous recombination repair gene SPIDR is associated with human gonadal dysgenesis. J. Clin. Endocrinol. Metab. 102, 681–688. doi: 10.1210/jc.2016-2714
Snowden, T., Acharya, S., Butz, C., Berardini, M., and Fishel, R. (2004). hMSH4-hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol. Cell 15, 437–451. doi: 10.1016/j.molcel.2004.06.040
Souquet, B., Abby, E., Herve, R., Finsterbusch, F., Tourpin, S., Le Bouffant, R., et al. (2013). MEIOB targets single-strand DNA and is necessary for meiotic recombination. PLoS Genet. 9:e1003784. doi: 10.1371/journal.pgen.1003784
Soustelle, C., Vedel, M., Kolodner, R., and Nicolas, A. (2002). Replication protein A is required for meiotic recombination in Saccharomyces cerevisiae. Genetics 161, 535–547.
Spruce, C., Dlamini, S., Ananda, G., Bronkema, N., Tian, H., Paigen, K., et al. (2020). HELLS and PRDM9 form a pioneer complex to open chromatin at meiotic recombination hot spots. Genes Dev. 34, 398–412. doi: 10.1101/gad.333542.119
Stanzione, M., Baumann, M., Papanikos, F., Dereli, I., Lange, J., Ramlal, A., et al. (2016). Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat. Cell Biol. 18, 1208–1220. doi: 10.1038/ncb3417
Sun, F., Fujiwara, Y., Reinholdt, L. G., Hu, J., Saxl, R. L., Baker, C. L., et al. (2015). Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma 124, 397–415. doi: 10.1007/s00412-015-0511-3
Syrjanen, J. L., Heller, I., Candelli, A., Davies, O. R., Peterman, E. J., Wuite, G. J., et al. (2017). Single-molecule observation of DNA compaction by meiotic protein SYCP3. Elife 6:e22582. doi: 10.7554/eLife.22582
Syrjanen, J. L., Pellegrini, L., and Davies, O. R. (2014). A molecular model for the role of SYCP3 in meiotic chromosome organisation. Elife 3:e02963. doi: 10.7554/eLife.02963
Szabova, L., Yin, C., Bupp, S., Guerin, T. M., Schlomer, J. J., Householder, D. B., et al. (2012). Perturbation of Rb, p53, and Brca1 or Brca2 cooperate in inducing metastatic serous epithelial ovarian cancer. Cancer Res. 72, 4141–4153. doi: 10.1158/0008-5472.CAN-11-3834
Takabayashi, S., Yamauchi, Y., Tsume, M., Noguchi, M., and Katoh, H. (2009). A spontaneous smc1b mutation causes cohesin protein dysfunction and sterility in mice. Exp. Biol. Med. (Maywood) 234, 994–1001. doi: 10.3181/0808-RM-244
Tenenbaum-Rakover, Y., Weinberg-Shukron, A., Renbaum, P., Lobel, O., Eideh, H., Gulsuner, S., et al. (2015). Minichromosome maintenance complex component 8 (MCM8) gene mutations result in primary gonadal failure. J. Med. Genet. 52, 391–399. doi: 10.1136/jmedgenet-2014-102921
Tian, H., Billings, T., and Petkov, P. M. (2018). CXXC1 is not essential for normal DNA double-strand break formation and meiotic recombination in mouse. PLoS Genet. 14:e1007657. doi: 10.1371/journal.pgen.1007657
Titus, S., Li, F., Stobezki, R., Akula, K., Unsal, E., Jeong, K., et al. (2013). Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci. Transl. Med. 5:172ra21. doi: 10.1126/scitranslmed.3004925
Tran, T. N., and Schimenti, J. C. (2018). A putative human infertility allele of the meiotic recombinase DMC1 does not affect fertility in mice. Hum. Mol. Genet. 27, 3911–3918. doi: 10.1093/hmg/ddy286
Tsui, V., and Crismani, W. (2019). The fanconi anemia pathway and fertility. Trends Genet. 35, 199–214. doi: 10.1016/j.tig.2018.12.007
Tsuzuki, T., Fujii, Y., Sakumi, K., Tominaga, Y., Nakao, K., Sekiguchi, M., et al. (1996). Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl. Acad. Sci. U.S.A. 93, 6236–6240. doi: 10.1073/pnas.93.13.6236
Wang, F., Guo, S., and Li, P. (2020). Two novel mutations in the MCM8 gene shared by two Chinese siblings with primary ovarian insufficiency and short stature. Mol. Genet. Genomic Med. 8:e1396. doi: 10.1002/mgg3.1396
Wang, J., Zhang, W., Jiang, H., Wu, B. L., and Primary Ovarian Insufficiency, C. (2014). Mutations in HFM1 in recessive primary ovarian insufficiency. N. Engl. J. Med. 370, 972–974. doi: 10.1056/NEJMc1310150
Wang, Y., Putnam, C. D., Kane, M. F., Zhang, W., Edelmann, L., Russell, R., et al. (2005). Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 37, 750–755. doi: 10.1038/ng1587
Ward, J. O., Reinholdt, L. G., Motley, W. W., Niswander, L. M., Deacon, D. C., Griffin, L. B., et al. (2007). Mutation in mouse hei10, an e3 ubiquitin ligase, disrupts meiotic crossing over. PLoS Genet. 3:e139. doi: 10.1371/journal.pgen.0030139
Wei, K., Clark, A. B., Wong, E., Kane, M. F., Mazur, D. J., Parris, T., et al. (2003). Inactivation of Exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 17, 603–614. doi: 10.1101/gad.1060603
Weinberg-Shukron, A., Rachmiel, M., Renbaum, P., Gulsuner, S., Walsh, T., Lobel, O., et al. (2018). Essential role of BRCA2 in ovarian development and function. N. Engl. J. Med. 379, 1042–1049. doi: 10.1056/NEJMoa1800024
Weinberg-Shukron, A., Renbaum, P., Kalifa, R., Zeligson, S., Ben-Neriah, Z., Dreifuss, A., et al. (2015). A mutation in the nucleoporin-107 gene causes XX gonadal dysgenesis. J. Clin. Invest. 125, 4295–4304. doi: 10.1172/JCI83553
Winkel, K., Alsheimer, M., Ollinger, R., and Benavente, R. (2009). Protein SYCP2 provides a link between transverse filaments and lateral elements of mammalian synaptonemal complexes. Chromosoma 118, 259–267. doi: 10.1007/s00412-008-0194-0
Winters, T., McNicoll, F., and Jessberger, R. (2014). Meiotic cohesin STAG3 is required for chromosome axis formation and sister chromatid cohesion. EMBO J. 33, 1256–1270. doi: 10.1002/embj.201387330
Wong, J. C., Alon, N., McKerlie, C., Huang, J. R., Meyn, M. S., and Buchwald, M. (2003). Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum. Mol. Genet. 12, 2063–2076. doi: 10.1093/hmg/ddg219
Wood-Trageser, M. A., Gurbuz, F., Yatsenko, S. A., Jeffries, E. P., Kotan, L. D., Surti, U., et al. (2014). MCM9 mutations are associated with ovarian failure, short stature, and chromosomal instability. Am. J. Hum. Genet. 95, 754–762. doi: 10.1016/j.ajhg.2014.11.002
Wu, L., Davies, S. L., North, P. S., Goulaouic, H., Riou, J. F., Turley, H., et al. (2000). The Bloom’s syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 275, 9636–9644. doi: 10.1074/jbc.275.13.9636
Wyatt, H. D., and West, S. C. (2014). Holliday junction resolvases. Cold Spring Harb. Perspect. Biol. 6:a023192. doi: 10.1101/cshperspect.a023192
Xia, F., Taghian, D. G., DeFrank, J. S., Zeng, Z. C., Willers, H., Iliakis, G., et al. (2001). Deficiency of human BRCA2 leads to impaired homologous recombination but maintains normal nonhomologous end joining. Proc. Natl. Acad. Sci. U.S.A. 98, 8644–8649. doi: 10.1073/pnas.151253498
Xiao, W. J., He, W. B., Zhang, Y. X., Meng, L. L., Lu, G. X., Lin, G., et al. (2019). In-frame variants in STAG3 gene cause premature ovarian insufficiency. Front. Genet. 10:1016. doi: 10.3389/fgene.2019.01016
Xu, H., Beasley, M. D., Warren, W. D., van der Horst, G. T., and McKay, M. J. (2005). Absence of mouse REC8 cohesin promotes synapsis of sister chromatids in meiosis. Dev. Cell 8, 949–961. doi: 10.1016/j.devcel.2005.03.018
Yang, F., De La Fuente, R., Leu, N. A., Baumann, C., McLaughlin, K. J., and Wang, P. J. (2006). Mouse SYCP2 is required for synaptonemal complex assembly and chromosomal synapsis during male meiosis. J. Cell Biol. 173, 497–507. doi: 10.1083/jcb.200603063
Yang, X., Touraine, P., Desai, S., Humphreys, G., Jiang, H., Yatsenko, A., et al. (2019a). Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J. Assist. Reprod. Genet. 36, 39–45. doi: 10.1007/s10815-018-1349-4
Yang, X., Zhang, X., Jiao, J., Zhang, F., Pan, Y., Wang, Q., et al. (2019b). Rare variants in FANCA induce premature ovarian insufficiency. Hum. Genet. 138, 1227–1236. doi: 10.1007/s00439-019-02059-9
Yang, Y., Guo, J., Dai, L., Zhu, Y., Hu, H., Tan, L., et al. (2018). XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J. Med. Genet. 55, 628–636. doi: 10.1136/jmedgenet-2017-105145
Yang, Y., Guo, T., Liu, R., Ke, H., Xu, W., Zhao, S., et al. (2020). FANCL gene mutations in premature ovarian insufficiency. Hum. Mutat. 41, 1033–1041. doi: 10.1002/humu.23997
Yilmaz, N. K., Karagin, P. H., Terzi, Y. K., Kahyaoglu, I., Yilmaz, S., Erkaya, S., et al. (2016). BRCA1 and BRCA2 sequence variations detected with next-generation sequencing in patients with premature ovarian insufficiency. J. Turk. Ger. Gynecol. Assoc. 17, 77–82. doi: 10.5152/jtgga.2016.16035
Yuan, L., Liu, J. G., Zhao, J., Brundell, E., Daneholt, B., and Hoog, C. (2000). The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol. Cell 5, 73–83. doi: 10.1016/s1097-2765(00)80404-9
Zangen, D., Kaufman, Y., Zeligson, S., Perlberg, S., Fridman, H., Kanaan, M., et al. (2011). XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am. J. Hum. Genet. 89, 572–579. doi: 10.1016/j.ajhg.2011.09.006
Zhang, Y. X., He, W. B., Xiao, W. J., Meng, L. L., Tan, C., Du, J., et al. (2020). Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol. Genet. Genomic Med. 8:e1165. doi: 10.1002/mgg3.1165
Zhang, Y. X., Li, H. Y., He, W. B., Tu, C., Du, J., Li, W., et al. (2019). XRCC2 mutation causes premature ovarian insufficiency as well as non-obstructive azoospermia in humans. Clin. Genet. 95, 442–443. doi: 10.1111/cge.13475
Zhao, W., and Sung, P. (2015). Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res. 43, 4055–4066. doi: 10.1093/nar/gkv259
Zhe, J., Chen, S., Chen, X., Liu, Y., Li, Y., Zhou, X., et al. (2019). A novel heterozygous splice-altering mutation in HFM1 may be a cause of premature ovarian insufficiency. J. Ovarian. Res. 12:61. doi: 10.1186/s13048-019-0537-x
Zhe, J., Ye, D., Chen, X., Liu, Y., Zhou, X., Li, Y., et al. (2020). Consanguineous Chinese familial study reveals that a gross deletion that includes the SYCE1 gene region is associated with premature ovarian insufficiency. Reprod. Sci. 27, 461–467. doi: 10.1007/s43032-019-00037-0
Keywords: premature ovarian insufficiency, meiosis, homologous recombination, mutations, next-generation sequencing
Citation: Huang C, Guo T and Qin Y (2021) Meiotic Recombination Defects and Premature Ovarian Insufficiency. Front. Cell Dev. Biol. 9:652407. doi: 10.3389/fcell.2021.652407
Received: 12 January 2021; Accepted: 05 February 2021;
Published: 08 March 2021.
Edited by:
Wei Li, State Key Laboratory of Stem Cell and Reproductive Biology, Institute of Zoology, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Caixia Guo, Key Laboratory of Genome and Precision Medicine, Beijing Institute of Genomics (CAS), ChinaZhenbo Wang, State Key Laboratory of Stem Cell and Reproductive Biology, Institute of Zoology, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Huang, Guo and Qin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingying Qin, qinyingying1006@163.com; Ting Guo, gtlyp2008@126.com