 Claire E. Martin
Claire E. Martin Nina Jones
Nina Jones- Department of Molecular and Cellular Biology, University of Guelph, Guelph, ON, Canada
Podocytes are a major component of the glomerular blood filtration barrier, and alterations to the morphology of their unique actin-based foot processes (FP) are a common feature of kidney disease. Adjacent FP are connected by a specialized intercellular junction known as the slit diaphragm (SD), which serves as the ultimate barrier to regulate passage of macromolecules from the blood. While the link between SD dysfunction and reduced filtration selectivity has been recognized for nearly 50 years, our understanding of the underlying molecular circuitry began only 20 years ago, sparked by the identification of NPHS1, encoding the transmembrane protein nephrin. Nephrin not only functions as the core component of the extracellular SD filtration network but also as a signaling scaffold via interactions at its short intracellular region. Phospho-regulation of several conserved tyrosine residues in this region influences signal transduction pathways which control podocyte cell adhesion, shape, and survival, and emerging studies highlight roles for nephrin phospho-dynamics in mechanotransduction and endocytosis. The following review aims to summarize the last 5 years of advancement in our knowledge of how signaling centered at nephrin directs SD barrier formation and function. We further provide insight on promising frontiers in podocyte biology, which have implications for SD signaling in the healthy and diseased kidney.
An Overview of the Glomerular Filtration Barrier (GFB)
The kidneys are responsible for maintenance of blood volume, electrolyte balance, and blood pressure as well as filtering wastes from the blood, which are then excreted from the body as urine. When the kidneys fail, dangerous levels of fluid, electrolytes, and wastes accumulate, wreaking havoc on the body (1). Patients suffering from chronic kidney disease (CKD) display progressive renal dysfunction and irreversible kidney damage. Current CKD treatments focus on limiting disease progression as well as supportive therapies to treat consequences of suboptimal kidney function rather than the underlying cause of disease. Unfortunately, it is difficult to predict which patients will respond well to treatment, or those who will ultimately progress to kidney failure. With the rapidly increasing worldwide burden of CKD, it is of paramount importance to better understand the underlying mechanisms leading to kidney damage, to improve patient stratification and develop novel means to treat all stages of disease.
The nephron is the blood filtering subunit of the kidney and each is made up of a glomerulus, the site of primary filtration, as well as a network of tubules where this filtrate is concentrated and further refined before it passes to the bladder to await excretion (2). The GFB plays a critical role in not only filtering out solutes and excess water but also in retaining essential components, including cells, macromolecules, and proteins within the blood. Loss of GFB selectivity is a hallmark of kidney dysfunction and ultimately results in loss of these components into the urine (proteinuria).
Glomerular dysfunction is a fundamental feature of kidney disease, and it is for this reason that the glomerulus remains the focus of sustained investigation into the pathogenesis and treatment of kidney disease (3). The glomerulus is a ball of tiny capillaries, each lined with a fenestrated endothelium and wrapped by highly specialized epithelial cells called podocytes (Figure 1A). The glomerular basement membrane (GBM), a compilation of proteins produced by podocytes and endothelial cells (4), lies at the interface of the two cell types. These three layers collectively constitute the GFB (5).
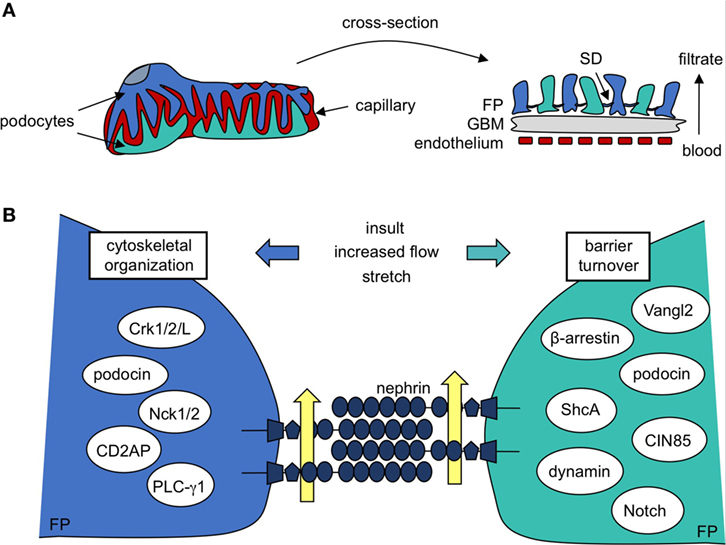
Figure 1. Podocyte protein nephrin is a central component in formation and maintenance of the slit diaphragm (SD). (A) Kidney podocytes wrap finger-like projections around the capillaries of the glomerulus, and these culminate in a network of interacting foot processes (FP). FPs contribute to the glomerular filtration barrier, which is also made up of fenestrated endothelium and the intermediating glomerular basement membrane (GBM). The SD, a specialized podocyte–podocyte junction found between interdigitating FPs, largely determines the size-selectivity of the barrier. (B) A closer look reveals the SD is a molecular sieve made up of nephrin molecules. The repetitive nature and precise patterning of extracellular nephrin–nephrin regions form pores that allow for discerning filtration of the blood (yellow arrows). Nephrin’s cytoplasmic region recruits a variety of signaling molecules that regulate cytoskeletal organization and FP shape as well as barrier turnover, each of which contributes to SD maintenance post-injury.
Central to filtration selectivity is the unique three-dimensional architecture of the podocyte. These highly arborized cells extend a series of processes from their cell body, which branch into an elaborate network of tertiary projections known as “foot processes” (6). Foot processes (FP) from adjacent podocytes interdigitate with each other to surround the glomerular capillaries and these ultimately act as the main barrier to loss of proteins and macromolecules into the urine. The extensive intercellular connections found between adjacent podocytes, which are referred to as filtration slits or slit diaphragms (SD), account for much of the size limitation of the GFB (Figure 1A) (7). The podocyte’s anionic glycocalyx, composed mainly of podocalyxin (8), provides additional charge-dependent resistance against the passage of negatively charged proteins. Together, their contribution toward both the size and charge basis for selective filtration positions podocytes as the key component of the GFB, and they are a primary target of injury in kidney disease (1, 2).
The Unique Ultrastructure of the Podocyte
Recent advances in microscopy have allowed a detailed view of actin’s organization in the podocyte in both health and disease. Super-resolution imaging reveals a network of myosin IIA-containing contractile actin cables within podocyte cell bodies and major processes, which are also rich in intermediate filaments and microtubules. Conversely, myosin IIA-negative, non-contractile actin fibers populate podocyte FP (9), which are largely devoid of other cytoskeletal components.
The injured podocyte is characterized by retraction of FP into broader, more simplified structures in a process known as effacement, and loss of SD structures. Interestingly, super-resolution imaging has demonstrated that, after injury, the podocyte’s contractile acto-myosin network relocates from the major processes to the basolateral surface of the cell, manifesting as sarcomere-like structures juxtaposed to the GBM (9). Serial block face-scanning electron microscopy further demonstrates the formation of podocyte protrusions in the diseased glomerulus, which invade into disordered regions of the GBM (10). Importantly, these substructural changes are consistently observed in animal models with diverse genetic origins of disease, indicating that these alterations may represent common pathomechanisms. These findings suggest new details about the reorganization of the podocyte actin cytoskeleton during disease and the existence of yet-to-be investigated mechanisms that regulate this switch in podocyte architecture. One such mechanism may be the differential proteolytic cleavage of actin and SD proteins during disease, as was suggested by recent work (11).
SD Assembly—Formation of a Molecular Sieve
The SD is a size-selective barrier that protects against loss of essential blood proteins, macromolecules, and cells into the urine during filtration. It is composed of molecules widely expressed in adherens (12) and tight junctions (13), such as P-cadherin, ZO-1, and Fat, as well as those predominantly expressed within podocytes alone, including nephrin (14, 15), nephrin-like (neph) 1 (16, 17) and podocin (18). The unique composition of the SD is central to its function and transition away from its distinct organization ultimately leads to barrier demise. Traditional SDs are often replaced by tight junctions during CKD progression (19, 20). This is potentially an attempt to bolster barrier function in the face of widespread effacement or to prevent the spread of immunogenic chemokines, which likewise lead to further damage. However, transition toward tight junctions creates a barrier that is virtually impermeable to passage of even fluid, prompting the reverse of the desired effect and ultimately leading to increased protein leakage and perpetuation of renal injury. This theory of tight junction-induced barrier dysfunction was recently confirmed in an experimental mouse model using podocyte-specific overexpression of the tight junction protein claudin-1, which caused SD destabilization and disorganization (21). These findings demonstrate not only the importance of the SD’s unique make-up for its function but also the importance of barrier flexibility in blood filtration.
The extracellular portion of the SD acts as a physical barrier and is comprised of two major components—nephrin and the related neph1. Each are members of the Immunoglobulin (Ig) protein superfamily. Nephrin’s large extracellular domain is made up of eight IgG-like motifs and a single fibronectin type 3 repeat (22). Neph1 similarly contains five IgG-like motifs in its extracellular portion (16). IgG domains can aggregate in cis and trans microclusters, and it is these IgG–IgG interfaces that are responsible for formation of the zipper-like SD meshwork that surrounds the glomerular capillaries, creating its sieve-like structure. Nephrin and neph1 also contain a single transmembrane domain and a short cytoplasmic tail. Contained within the intracellular region are several conserved residues that recruit cytosolic signaling partners that are likewise required for formation of the mature SD, which will be discussed in greater detail below.
The mature SD junction is not formed until the later stages of glomerular development (23). Podocytes originate in the S-shaped body as columnar epithelial cells containing, instead, apically localized tight junctions. During the early capillary loop stage, the apical membrane area of the podocyte expands and these junctions begin migrating baso-laterally. It is at this point that nephrin begins to be expressed (24). As the capillary loop stage proceeds, the apical domain of the podocyte continues to increase in size, and both nephrin and neph1 migrate toward the basal domain coincident with the start of podocyte process formation. Tight junctions containing nephrin form between these nascent processes (24). As the glomerulus continues to mature, podocytes extend additional processes as they wrap around the newly forming capillary loops and only then do the mature SD structures containing tyrosine phosphorylated nephrin and neph1 appear (25).
Nephrin/neph-like complex formation is an evolutionarily conserved adhesion module in which heterodimeric trans neph1-nephrin interactions occur between two distinct cell types, with one cell expressing nephrin and the other expressing neph1 (26, 27). In the glomerulus, however, interactions occur between adjacent podocyte cells which each express both nephrin and neph1 proteins, thereby allowing for nephrin–nephrin, neph–neph, and nephrin–neph pairings. The likely arrangement of nephrin and neph1 components in the SD was recently illuminated using high-resolution ultrastructural imaging (17). Unlike in other modules, nephrin and neph1 appear to minimally interact in podocytes. Instead, distinct segments of trans nephrin–nephrin and neph1–neph1 multimers largely comprise the SD in an approximate 2:5 ratio, with neph1 molecules spanning the lower part of the junction, closer to the GBM with a width of 23 nm, while nephrin molecules contribute to the apical region of the SD, with a width of 45 nm. These nephrin and neph1 complexes are spaced 7-nm apart, creating a two-tiered configuration, setting the SD apart from other nephrin–neph-like cell–cell adhesion modules.
Nephrin localization to the SD is dependent on its cytosolic interaction with podocin (28). Podocin (encoded by NPHS2) interacts with nephrin’s R1160 residue and directs its localization to lipid rafts at the cell surface (14, 15, 18, 29), enabling it to form the SD and perform its signaling functions. The podocin R138Q mutation is the most common NPHS2 disease-causing variant (28, 30) and, similar to other NPHS2 mutations, it disrupts podocin folding and glycosylation, resulting in severe congenital nephropathy associated with podocin’s retention in the endoplasmic reticulum (ER) (29, 31). Interestingly, expression of R138Q with nephrin in culture also results in nephrin’s retention in the ER (29), demonstrating a clear requirement for appropriate podocin trafficking in nephrin’s localization to the membrane.
Neph1 likewise depends on cytoplasmic interactions for its localization to the plasma membrane. Although neph1 has also been described to bind podocin (16), whether podocin mutations negatively influence neph1 localization remains to be investigated. Studies have alternatively focused on the role of the motor protein myosin 1c (Myo1c) in supporting neph1’s recruitment to and turnover at the plasma membrane (32, 33). Interestingly, Myo1c inhibits nephrin’s localization at the plasma membrane (32) in culture, a finding of unknown relevance in vivo.
Mouse knockout models have clearly demonstrated that loss of either nephrin (34, 35), podocin (36), or neph1 (37) expression is sufficient to disrupt SD formation and induce severe disease within days of birth and even in utero. Furthermore, over 250 distinct genetic mutations in NPHS1 (38) and over 100 in NPHS2 (39) have been identified to date. The overall importance of nephrin–podocin communication in development is further highlighted in instances of congenital nephrotic syndrome (CNS) in which single mutations in either NPHS1 or NPHS2 are benign in respective parents, but their digenic heterozygosity leads to congenital disease (40), a relatively uncommon phenomenon (41). Maintenance of nephrin and podocin expression throughout adulthood is also required (42, 43), as is their communication throughout life, which was recently demonstrated using a mouse model with inducible expression of a CNS-related NPHS2 mutation (44). Adult animals induced to express the human-corresponding R138Q mutation, which causes retention of nephrin in the ER, rapidly develop nephrotic syndrome. This demonstrates an ongoing requirement for nephrin–podocin interaction throughout life and, more specifically, nephrin’s dependence on podocin for trafficking to the SD.
Nephrin Phosphorylation Establishes a Signaling Platform
Beyond its role as a physical barrier, nephrin acts a central signaling platform within the podocyte, facilitated by an extensive network of cytoplasmic binding partners which have been identified over the past two decades (Figure 1B) (18, 29, 45–76). These proteins represent key components of diverse signaling cascades that affect podocyte polarity, cell survival, calcium mechano-signaling, membrane trafficking, and actin organization. Nephrin complexes with many of its binding partners via phosphorylation of various tyrosine and threonine residues found in specific binding motifs within its cytoplasmic region. The complex interplay between SD signaling and foot process morphology is accomplished in large part through cell signaling events centered at these conserved residues.
The signaling pathways induced by nephrin tyrosine phosphorylation can be loosely defined based on their dependence on two different sets of residues. Group A tyrosines include Y1114 and Y1138/9, while group B tyrosines encompass Y1176, Y1193, and Y1217 (human nephrin numbering system) (Figure 2). These tyrosines often coordinate signal propagation independently of each other, but at times, can also work in concert. Both group A and B tyrosines can be phosphorylated by Src family kinases, including Src (77), Fyn (46, 47, 77), Lyn (77), and Yes (46, 77), although each residue may not be phosphorylated to the same extent (47, 48, 53, 62). Fyn kinase also directly binds group B phospho-tyrosine residues (46), which may provide better access to target tyrosine residues (78) as well as protection of these sites from de-phosphorylation (79).
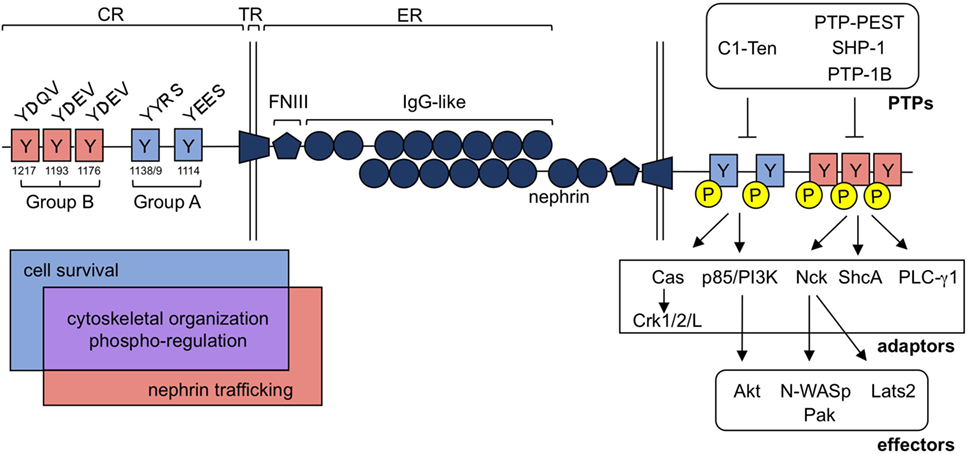
Figure 2. Nephrin tyrosine phosphorylation regulates a diverse group of signaling processes within the podocyte. Nephrin is a single pass transmembrane protein and contains a singular extracellular (ER), transmembrane (TR), and cytoplasmic (CR) region. Nephrin’s ER contains 1 Fibronectin-like III (FNIII) motif and eight Immunoglobulin (IgG)-like regions, which allow for homophilic interactions of nephrin molecules in trans. Tyrosine residues embedded in nephrin’s CR can be loosely classified into two categories, denoted as group A or B tyrosines, based largely on their flanking sequences. These consensus sequences are conserved and influence the adaptor molecules and downstream signaling effectors that can be recruited to nephrin upon their tyrosine phosphorylation. Interactions with each group contributes to cytoskeletal organization (through Cas-Crk/1/2/L, Nck-N-WASp/Pak and/or PLC-γ1) and nephrin’s phospho-regulation [through protein tyrosine phosphatases (PTPs)], denoted in purple. Alternatively, cell survival signaling appears to be largely restricted to signaling at group A residues (through p85/PI3K-AKT), denoted in blue, while nephrin trafficking is influenced by group B signaling (through ShcA), denoted in red.
Phosphorylation of group A tyrosines 1114 and 1138/9 induces binding of p85/PI3K (57, 80, 81), ultimately leading to activation of Akt and Rac1 and the recruitment of cofilin (57, 81). These residues also recruit Cas, which, once phosphorylated via group B-mediated signaling, allows for the engagement of Crk1/2/L (71, 73), which affects actin and focal adhesion dynamics within the podocyte.
The impact of phosphorylation of group B tyrosines 1176, 1193, and 1217 is the most widely characterized, potentially due to the commercial availability of phospho-specific antibodies for these sites (82). Phosphorylation at these sites results in the recruitment of Nck, PLC-γ1, and ShcA. Nck subsequently recruits both Pak (83, 84) and N-WASp (85), allowing for actin polymerization at nephrin (Figure 2).
Dysregulation of the actin cytoskeleton by mutations in proteins that link the SD to actin [α-actinin-4, FAT Atypical Cadherin (FAT)1, CD2AP, PLC-ε1, nephrin, and podocin] or that regulate actin polymerization [inverted formin 2, myosin 1e (Myo1e), Rho GTPase activating protein (ARHGAP) 24, Rho GDP Dissociation Inhibitor Alpha (ARHGDIA), and TRPC6] or podocyte contractility [myosin heavy chain 9, Myo1e, laminin subunit β2, integrin β4, integrin α3, Lamin A/C, tetraspanin (CD151) and several type IV collagens] give rise to spontaneous kidney disease in humans characterized by loss of filtration selectivity [reviewed recently Ref. (86)]. Connections between nephrin and actin, mediated both by or independently of group A and B tyrosines, allow for steady communication between the extracellular barrier and the core of the podocyte FP. Nephrin’s tyrosine phosphorylation appears to modulate two forms of actin in culture: lamellipodia formation, which is often associated with pathogenic FP effacement, and the growth of actin polymers (“tails” or “comets”) at nephrin, which are believed to stabilize foot process ultrastructure and the SD laterally. Lamellipodia formation appears to be modulated by p85/PI3K/Akt/Cas/Crk through phosphorylation of group A tyrosines (57, 71, 73, 81), while the production of actin tails at nephrin is dependent on phosphorylation of group B residues and recruitment of Nck (53, 85).
In addition to its several conserved tyrosine motifs, nephrin contains a S/TX4S/T region between residues 1120 and 1125, a consensus motif for serine/threonine phosphorylation. Protein kinase C alpha (PKCα), which also interacts with nephrin in a phosphorylation-independent manner, can phosphorylate nephrin at these residues (87, 88). This in turn enhances nephrin’s interaction with β-arrestin and ultimately induces its endocytosis in a clathrin (68, 89) and dynamin-dependent (90) fashion. Activation of this pathway has been characterized in diabetic (87), hypertensive (88), and acute (91) renal injury models, indicating that it may represent a central pathomechanism in disease. Exactly how phosphorylation of this residue is regulated is largely unknown, although there is some evidence that β-arrestin competes for nephrin with phospho-tyrosine dependent binding partners including Nck (88) as well as with podocin (52), a mechanism warranting further investigation.
Phosphatase-Mediated Regulation of Nephrin Tyrosine Phosphorylation
Nephrin’s tyrosine phosphorylation is modulated by several protein tyrosine phosphatases that influence Fyn activation. De-phosphorylation of Fyn by the SHP-2 phosphatase (92), which also binds group B nephrin phospho-tyrosines (75), releases the intramolecular inhibition of Fyn, ultimately enhancing its kinase activity on nephrin (75). Conversely, the active site of Fyn (Y418) was found to be a substrate for the phosphatase PTP-PEST, and de-phosphorylation of this site leads to reduced Fyn activity, indirectly diminishing nephrin’s phosphorylation (67). Interestingly, Nck and ShcA, adaptor proteins known to bind group B phospho-tyrosines, work in a feed-forward mechanism by promoting activation of Fyn (76, 93). In the case of Nck, this is dependent on its ability to complex with nephrin and Fyn (93).
Tyrosine phosphatases can also act directly on nephrin (Figure 2). PTP-1B, which is upregulated in the puromycin aminonucleoside (PAN) model of membranous nephropathy (MN), can directly dephosphorylate rat tyrosine residues that correlate to the human sites 1193 and 1217 (67). Likewise, the SHP-1 phosphatase, which binds group A tyrosine residues, dephosphorylates tyrosines 1176, 1193, and 1217 (74) and its upregulation has been observed in instances of hyperglycemia and diabetes by several groups (74, 94–98). In podocytes, hyperglycemia induces a persistent increase in SHP-1 expression, due to epigenetic modification in the SHP-1 promoter (97), leading to insulin signaling resistance, podocyte dysfunction, and cell death. Interestingly, PTP-1B and SHP-1 both are unable to dephosphorylate nephrin’s Y1138 site (67, 74), indicating that these phosphatases likely do not exert their influence by modulating PI3K-Akt signaling, but rather through tyrosine residues that bind Nck, ShcA, and PLC-γ1.
C1-Ten is the most recently identified nephrin tyrosine phosphatase (99) and it uniquely targets the Y1114 and Y1138 motifs, disrupting nephrin-PI3K complex formation. Interestingly, C1-Ten is upregulated in diabetic nephropathy (DN) models and is associated with induction of podocyte hypertrophy (99). Increased levels of SHP-1 phosphatase have likewise been identified in models of DN (74, 94–98) resulting in reduced phosphorylation of Y1176, Y1193, and Y1217, and disruption of nephrin and Nck binding. These recent findings position global reductions in nephrin tyrosine phosphorylation as a mechanism of damage in DN, the most common form of CKD.
Disruption of Nephrin Tyrosine Phosphorylation in Podocyte-Based Kidney Diseases
The role of nephrin signaling during disease has remained an intense area of interest since the discovery of reduced phosphorylation of group B tyrosine residues in instances of human disease and in various disease models. Alterations in phosphorylation of group B tyrosines has been described in minimal change disease (MCD) (100) and MN (101) as well as in the PAN (76, 82), protamine sulfate (54, 102), nephrotoxic serum (NTS) (75, 88, 103), and lipopolysaccharide (102) rodent injury models and type I diabetic Akita mice (74, 98). Similarly, altered levels of several phosphatases known to influence nephrin’s phospho-status have also been identified in instances of disease (104). Unfortunately, antibodies targeting group A tyrosines or nephrin’s 1120/1125 serine/threonine regions remain unavailable, making it difficult to fully understand the role of signaling at these sites.
Historically, there has been a lack of consensus on whether tyrosine phosphorylation of group A residues is up- or downregulated during disease, and whether these changes are associated with promoting podocyte damage or protection from injury. These discrepancies may be attributed to the use of multiple unique sets of phospho-specific antibodies by different groups, which variably recognize the three similar tyrosine-based motifs. However, the recent development of the nephrin-Y3F mouse model appears to have solidified an overall requirement for nephrin’s tyrosine phosphorylation in SD maintenance (102, 105). In this knockin model, all three group B tyrosine residues were converted to phenylalanine (denoted Y3F), a change which retains protein structure while preventing phosphorylation. Although nephrin-Y3F animals are born with no obvious impairments, they quickly develop progressive disease characterized by foot process effacement, GBM thickening and proteinuria. Furthermore nephrin-Y3F animals display a reduced ability to recover after damage induced by several acute injury models. This collective evidence supports the essential role of signaling through group B tyrosines in both SD barrier maintenance and podocyte repair.
Nck’s ability to recruit actin to nephrin may be central to the requirement of group B tyrosines in barrier stability. Inducible deletion of Nck in adult mice results in reduced nephrin tyrosine phosphorylation (93) and loss of actin recruitment to nephrin, leading to podocyte effacement and loss of SD structures (82), thereby supporting its role in barrier maintenance. Complex formation between nephrin and Nck is likewise interrupted in several acute injury models coincident with damage (88, 102) implicating a central importance for their interaction in SD function. Elegant biophysical studies have provided context for the overlapping nature of group B tyrosines on nephrin, all of which can bind Nck, and the iterative SH3 domains of Nck, all of which can bind N-WASp and activate Arp2/3 (106–108). Threshold levels of nephrin-Nck signaling appear to be necessary to effectively induce formation of biomolecular signaling nodes on synthetic membranes which promote actin polymerization (106, 107). Future studies in which these concepts are applied to cell systems may provide insight into the physiological relevance of these mechanisms and the unique requirement for multivalent interactions between nephrin, Nck, and actin in the podocyte.
Endocytosis: An Emerging Focus in Podocyte Biology
As a function of their barrier role, SD components require regular replacement to maintain SD integrity. Endocytosis is the process by which cells internalize membrane-bound components including embedded surface receptors and their ligands. In the podocyte, two endocytic pathways have been identified: clathrin-mediated endocytosis (CME) and clathrin-independent endocytosis (CIE) (109). Generally, CME mediates the regular turnover of receptors (110) and appears to predominate within podocytes (111). Clathrin is recruited in the initiation stage of pit invagination, supported by various proteins including α-adaptin, synaptojanin and endophilin-1, and actin. Subsequently, the GTPase dynamin is recruited to the vesicle where it wraps and constricts the neck until the small membrane-encapsulated vesicle is cut free, completing its internalization. The majority of cargo endocytosed in this manner, about 95%, is believed to be recycled back to the plasma membrane and this pathway thereby defines a constitutive turnover mechanism in cells (112). The remaining 5% that is targeted for degradation may be damaged, requiring replacement by synthesizing mechanisms, or lost as a means to finesse receptor activity.
Reports characterizing the effects of podocyte-specific deletion of dynamin1/2 (111), synaptojanin (111), and endophilin (111) have clearly established the functional importance of endocytic machinery within podocytes. Evidence further indicates that dynamin can complex with nephrin indirectly during its own endocytosis (111, 113). Investigation of nephrin trafficking mechanisms has become a keen area of interest in recent years owing to the recognition of nephrin mislocalization in a broad range of human diseases including MN (101), CNS (114), steroid-resistant nephropathy (28), MCD (115), DN (63), and hypertensive nephropathy (88). However, relatively little is known about the specific mechanisms that dictate SD protein trafficking (112).
β-arrestin2 was the first nephrin binding partner identified to influence nephrin trafficking (52), over 10 years ago. It binds phosphorylated T1120/T1125 on nephrin’s cytoplasmic tail, which facilitates nephrin endocytosis in a CME fashion (88). β-arrestin mediated nephrin endocytosis is relatively well-characterized within podocytes and several pathways seem to converge on this mechanism. PKCα mediates nephrin phosphorylation of T1120/1125, a phenomenon that is enhanced by PLC-γ1 (87, 88), leading to β-arrestin recruitment and nephrin internalization. This pathway has been shown to be relevant in several diseases including in diabetes (116) and in response to the vascular protein angiotensin II (ANGII) (87, 88), which is upregulated with hypertension.
Activation of the planar cell polarity (PCP) pathway during glomerular development also appears to stimulate nephrin endocytosis via a clathrin/β-arrestin-dependent mechanism (68, 89). Disruption of Vangl2 activity, which is involved in the PCP pathway, results in increased surface expression of nephrin in podocytes, and this leads to disruption of glomerular maturation in mice (117). Notch activation likewise has been shown to induce nephrin internalization via a β-arrestin/dynamin-dependent, raft-independent route (90) and animals overexpressing activated Notch display proteinuria and damage that is associated with enhanced nephrin endocytosis and loss of SDs.
Phospho-regulation clearly allows for dynamic control of various signaling processes in the podocyte, downstream of nephrin, and emerging evidence indicates that this regulatory ability extends to modulation of nephrin endocytosis (109). However, there are conflicting reports regarding the potential role for site-specific nephrin phosphorylation events. Reduced phosphorylation of Y1193 has been shown to induce binding of β-arrestin and promote rapid removal of nephrin from the cell surface by CME (52), while phosphorylation of this same tyrosine promotes podocin binding to nephrin, which is proposed to localize nephrin to lipid raft microdomains where it is turned over at a slower rate by CIE (29, 109). Others have reported that mutation of Y1217 or compound mutation of Y1193/Y1176 decreases nephrin internalization (109, 113), and that enhanced dynamin-mediated phosphorylation of nephrin promotes its endocytosis (113). Interestingly, ShcA is the only nephrin phospho-tyrosine binding partner identified to date that actually binds tyrosine phosphorylated nephrin and contributes to nephrin internalization (76). ShcA, however, is expressed at low levels in the mature podocyte and its association with nephrin endocytosis appears to be a pathogenic one, as was recently uncovered in focal segmental glomerulosclerosis (FSGS), MCD, and immunoglobulin A nephropathies (76). It is likely then that other yet-to-be-identified phospho-nephrin binding partners are responsible for nephrin endocytosis in the healthy state during barrier maintenance.
Disrupted Nephrin Trafficking—An Overlooked Mechanism of Hereditary Disease?
Despite the multitude of disease-causing NPHS1 mutations identified, relatively few have been characterized for precisely how they affect nephrin function from a cell biology or cell signaling perspective. From the studies available, it appears that most of the NPHS1 mutations lead to abnormal retention of nephrin in the ER, and thus failed trafficking to the cell surface (118, 119). This suggests that the majority of NPHS1 mutations result in a loss-of-function of nephrin due to the inability for it to localize to the SD, which leads to the early-onset and severe disease often associated with NPHS1 mutations (118). Assumedly, this also affects the ability of nephrin to be phosphorylated and there is some evidence that disruption of nephrin localization to the membrane results in its sub-maximal phosphorylation (29). Although less commonly reported, some mutations, such as V822M, do not appear to affect nephrin trafficking to the cell membrane, but rather they affect normal nephrin trafficking from the plasma membrane (120). This likewise causes CNS (121), indicating that bidirectional trafficking is important for normal nephrin function. Interestingly, although nephrin-V822M localizes normally to the cell membrane, it displays altered nephrin phosphorylation at group B residues and is unable to reorganize actin filaments (120). We question whether abnormal nephrin endocytosis may be an overlooked contributing factor in other instances of hereditary kidney disease.
Emerging Evidence of SD-GBM Communication in Response to Mechanical Strain
In addition to the SD, foot process morphology and barrier integrity are also critically dependent on basally localized adhesive complexes, which secure the podocyte to the underlying basement membrane. Just as mutations in the lateral SD proteins nephrin and podocin are well-established to contribute to disease, mutations in α-actinin-4, integrin α3, type IV collagens, and laminins, all of which contribute to signaling within podocyte adhesomes, similarly result in congenital kidney disease (86).
The podocyte’s SD and focal adhesions are both stabilized through extensive connections with the underlying actin cytoskeleton. However, little is known about the mechanisms by which inter-compartment communication occurs. The Crk family of proteins, whose recruitment and activity are collectively modulated by both group A and group B tyrosine residues, influences lamellipodia formation and focal adhesion reorganization in podocytes in culture (71, 73). Arf6, a GTPase, was also recently shown to be activated downstream of nephrin tyrosine phosphorylation (122), leading to similar effects on lamellipodia and focal adhesion reorganization. A recent report showed that activation of β1 integrin can likewise induce nephrin tyrosine phosphorylation (123). These studies collectively link signaling between the basal cell compartment and the SD. Supporting the importance of compartmental crosstalk in barrier function, digenic predisposition to disease has been recently identified in individuals with heterozygous mutations in collagen IV and podocin (22, 124) or collagen IV and myosin1e. Further investigation characterizing SD-GBM communication is required to more fully elucidate the mechanisms by which FP maintain their unique structure and thereby the filtration barrier.
The podocyte is continually exposed to hemodynamic strain and shear stress, and dynamic signaling between the SD and focal adhesions is proposed to allow the cell to remain adhered to the GBM while maintaining its barrier function (125–127). It was recently revealed that nephrin signaling directly modulates Hippo mechano-signaling by regulating the stability of its pro-survival component YAP. Phosphorylated nephrin can recruit and sequester Lats2, its negative regulator, at group B tyrosines through an indirect mechanism involving Nck and WTIP (103), thereby limiting YAP’s degradation. During the initiation of NTS disease in mice, a reduction in nephrin tyrosine phosphorylation is observed at group B residues, reducing Lats2 inhibition, which ultimately leads to YAP’s degradation (103). Such activation of Hippo signaling has also been shown to induce podocyte cell death (128). The importance of YAP was further highlighted by the development of a podocyte-specific YAP knockout mouse, which developed kidney disease soon after birth, and reduced levels of YAP are associated with FSGS (129). Interestingly, expression of podocyte adhesive proteins localized to both its SD (nephrin, podocin, and α-actinin-4) and focal adhesions (integrins α3 αv, β1, β5), as well as components that are central players in Hippo mechanosensing (WWTR1, TEAD1, Yap1, and Lats2) were recently identified to be collectively regulated by the transcription factor WT-1. The presence of mutations in WT-1 in hereditary CNS has become more apparent in recent years (130–132) and this points at a potential central role for WT-1 in regulating the podocyte’s ability to respond to mechanical strain.
The significance of mechanical force sensation in the podocyte is further made evident by the discovery of human disease-causing mutations in TRPC5 and TRPC6 (50, 133), which encode the main calcium channels of the podocyte. Aberrant calcium signaling is linked to the pathogenic remodeling of the actin cytoskeleton that results in foot process effacement, although much debate remains about the ultimate impact of the activity of these channels on podocyte function (50, 134–139). Nephrin also appears to crosstalk with TRPC6, as previous studies have demonstrated that phosphorylation of nephrin Y1193 promotes its interaction with PLC-γ1, stimulating its activation and triggering calcium mobilization at TRPC6 channels (62, 140). Interestingly, nephrin may also impede calcium signaling by directly binding and inhibiting TRPC6 activity in a non-tyrosine-dependent fashion (140). In support of this, several disease-causing TRPC6 mutations leave the calcium channel unresponsive to nephrin inhibition in cell culture experiments (140), identifying an important role for nephrin-mediated regulation of TRPC6 in podocytes.
Frontiers in Podocyte Biology—Toward Novel Treatments Targeting the Podocyte
Personalized medicine offers the promise of tailored therapeutics, reduced side effects and, ultimately, superior outcomes for patients. However, the movement toward more individualized treatment can only proceed once the molecular signatures underlying disease have been identified and characterized. With the diverse nature of renal pathologies, and the sparse treatment options available, the nephrology field is relying on basic research to uncover a more detailed understanding of renal disease etiologies in order to establish effective new therapies (141).
Given the importance of the SD in kidney function, the capacity for treatments to stabilize the SD barrier remains a keen area of interest for drug development. Expression of nephrin within the SD is essential for barrier maintenance throughout life (42, 44) and its aberrant endocytosis increasingly appears to be a central mechanism of disease (42, 44, 76, 88, 142). ANGII inhibition, which is commonly used in the treatment of hypertension, reportedly inhibits nephrin endocytosis, stabilizing the barrier and protecting mice from disease (88). Small molecule-facilitated preservation of the interaction between neph-1 and ZO-1, which is often lost at the SD during injury, has also been shown to promote barrier function in mice (143).
Since neph1 and nephrin are directly exposed to the circulating blood, and their roles predominantly have been characterized to be kidney-specific, they remain tempting targets for drug development (144). Nonetheless, both neph1 (145) and nephrin (34, 146–148) are expressed in a variety of other tissues and it is unclear whether pathologic non-renal effects of targeted treatments will arise. Recent studies have uncovered a role for nephrin in the trafficking and release of insulin from pancreatic beta cells (113, 149–151), and new evidence identifies previously unrecognized deficiencies in blood glucose regulation in CNS patients with known NPHS1 mutations (150). This may have implications for the use of ANGII inhibition or other treatments that inhibit nephrin trafficking during renal disease, as it may cause negative consequences in the pancreas where a similar signaling pathway appears to be required for insulin release.
Targeting pathogenic actin polymerization (152, 153), abnormal TRPC5 and TRPC6 channel activation (136, 154, 155), aberrant integrin activation (156, 157), and depletion of laminin-521 in the GBM (158) remain other emerging prospective treatments, all of which have shown significant promise in animal models. GDC-0879, a B-RAFV600E inhibitor, was also recently identified in a high-throughput screen of approved drugs to promote podocyte cell survival (159). Although the utility of this compound as a treatment for renal disease in vivo has not yet been explored, GDC-0879 has successfully been used in mouse models to inhibit tumor growth (160), making it an exciting candidate for future investigations in the kidney.
Summary
Precise regulation of signaling at the SD is crucial for formation and maintenance of the GFB. Nephrin acts as the core of the SD and also as a signaling hub in the podocyte, modulating cell polarity, survival, adhesion, cytoskeletal organization, mechanosensing, and SD turnover. These roles have been revealed using a variety of cell and animal models that continue to improve in their sophistication as well as inferences from diverse human kidney diseases. The next frontier is to mobilize the insight gained from over two decades of “nephr(in)ology” research into novel therapies that target this unique indispensable protein and the molecules it communicates with.
Author Contributions
CM and NJ contributed to the concept, design, and literature research for this review. CM prepared the figures and drafted the manuscript, in consultation with NJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by operating grants from the Canadian Institutes of Health Research and the Kidney Foundation of Canada (to NJ) and studentships from the Natural Sciences and Engineering Research Council (to CM). NJ holds a Tier 2 Canada Research Chair in Eukaryotic Cellular Signaling.
References
1. Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol (2012) 74:299–323. doi:10.1146/annurev-physiol-020911-153238
2. Scott RP, Quaggin SE. Review series: the cell biology of renal filtration. J Cell Biol (2015) 209:199–210. doi:10.1083/jcb.201410017
3. New LA, Martin CE, Jones N. Advances in slit diaphragm signaling. Curr Opin Nephrol Hypertens (2014) 23:420–30. doi:10.1097/01.mnh.0000447018.28852.b6
4. Striker GE, Smuckler EA. An ultrastructural study of glomerular basement membrane synthesis. Am J Pathol (1970) 58:531–55.
5. Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun (2010) 396:164–9. doi:10.1016/j.bbrc.2010.04.069
6. Schell C, Huber TB. The evolving complexity of the podocyte cytoskeleton. J Am Soc Nephrol (2017) 28:3166–74. doi:10.1681/ASN.2017020143
7. Menon MC, Chuang PY, He CJ. The glomerular filtration barrier: components and crosstalk. Int J Nephrol (2012) 2012:749010. doi:10.1155/2012/749010
8. Kerjaschki D, Sharkey DJ, Farquhar MG. Identification and characterization of podocalyxin – the major sialoprotein of the renal glomerular epithelial cell. J Cell Biol (1984) 98:1591–6. doi:10.1083/jcb.98.4.1591
9. Suleiman HY, Roth R, Jain S, Heuser JE, Shaw AS, Miner JH. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. JCI Insight (2017) 2. doi:10.1172/jci.insight.94137
10. Randles MJ, Collinson S, Starborg T, Mironov A, Krendel M, Königshausen E, et al. Three-dimensional electron microscopy reveals the evolution of glomerular barrier injury. Sci Rep (2016) 6:35068. doi:10.1038/srep35068
11. Rinschen MM, Hoppe A-K, Grahammer F, Kann M, Völker LA, Schurek E-M, et al. N-degradomic analysis reveals a proteolytic network processing the podocyte cytoskeleton. J Am Soc Nephrol (2017) 28:2867–78. doi:10.1681/ASN.2016101119
12. Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol (2000) 11:1–8.
13. Schnabel E, Anderson JM, Farquhar MG. The tight junction protein ZO-1 is concentrated along slit diaphragms of the glomerular epithelium. J Cell Biol (1990) 111:1255–63. doi:10.1083/jcb.111.3.1255
14. Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestilä M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A (1999) 96:7962–7. doi:10.1073/pnas.96.14.7962
15. Wartiovaara J, Ofverstedt L-G, Khoshnoodi J, Zhang J, Mäkelä E, Sandin S, et al. Nephrin strands contribute to a porous slit diaphragm scaffold as revealed by electron tomography. J Clin Invest (2004) 114:1475–83. doi:10.1172/JCI22562
16. Sellin L, Huber TB, Gerke P, Quack I, Pavenstädt H, Walz G. NEPH1 defines a novel family of podocin interacting proteins. FASEB J (2003) 17:115–7. doi:10.1096/fj.02-0242fje
17. Grahammer F, Wigge C, Schell C, Kretz O, Patrakka J, Schneider S, et al. A flexible, multilayered protein scaffold maintains the slit in between glomerular podocytes. JCI Insight (2016) 1:e86177. doi:10.1172/jci.insight.86177
18. Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest (2001) 108:1621–9. doi:10.1172/JCI200112849
19. Kurihara H, Anderson JM, Kerjaschki D, Farquhar MG. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am J Pathol (1992) 141:805–16.
20. Succar L, Boadle RA, Harris DC, Rangan GK. Formation of tight junctions between neighboring podocytes is an early ultrastructural feature in experimental crescentic glomerulonephritis. Int J Nephrol Renovasc Dis (2016) 9:297–312. doi:10.2147/IJNRD.S113071
21. Gong Y, Sunq A, Roth RA, Hou J. Inducible expression of claudin-1 in glomerular podocytes generates aberrant tight junctions and proteinuria through slit diaphragm destabilization. J Am Soc Nephrol (2017) 28:106–17. doi:10.1681/ASN.2015121324
22. Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82. doi:10.1016/S1097-2765(00)80057-X
23. Schell C, Wanner N, Huber TB. Glomerular development – shaping the multi-cellular filtration unit. Semin Cell Dev Biol (2014) 36:39–49. doi:10.1016/j.semcdb.2014.07.016
24. Holzman LB, St John PL, Kovari IA, Verma R, Holthofer H, Abrahamson DR. Nephrin localizes to the slit pore of the glomerular epithelial cell. Kidney Int (1999) 56:1481–91. doi:10.1046/j.1523-1755.1999.00719.x
25. Ruotsalainen V, Patrakka J, Tissari P, Reponen P, Hess M, Kestilä M, et al. Role of nephrin in cell junction formation in human nephrogenesis. Am J Pathol (2000) 157:1905–16. doi:10.1016/S0002-9440(10)64829-8
26. Helmstädter M, Höhne M, Huber TB. A brief overview on IRM function across evolution. J Neurogenet (2014) 28:264–9. doi:10.3109/01677063.2014.918976
27. Linneweber GA, Winking M, Fischbach K-F. The cell adhesion molecules roughest, Hibris, kin of irre and sticks and stones are required for long range spacing of the Drosophila wing disc sensory sensilla. PLoS One (2015) 10:e0128490. doi:10.1371/journal.pone.0128490
28. Nishibori Y, Liu L, Hosoyamada M, Endou H, Kudo A, Takenaka H, et al. Disease-causing missense mutations in NPHS2 gene alter normal nephrin trafficking to the plasma membrane. Kidney Int (2004) 66:1755–65. doi:10.1111/j.1523-1755.2004.00898.x
29. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet (2003) 12:3397–405. doi:10.1093/hmg/ddg360
30. Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol (2008) 19:365–71. doi:10.1681/ASN.2007040452
31. Philippe A, Weber S, Esquivel EL, Houbron C, Hamard G, Ratelade J, et al. A missense mutation in podocin leads to early and severe renal disease in mice. Kidney Int (2008) 73:1038–47. doi:10.1038/ki.2008.27
32. Arif E, Wagner MC, Johnstone DB, Wong HN, George B, Pruthi PA, et al. Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol Cell Biol (2011) 31:2134–50. doi:10.1128/MCB.05051-11
33. Arif E, Sharma P, Solanki A, Mallik L, Rathore YS, Twal WO, et al. Structural analysis of the Myo1c and Neph1 complex provides insight into the intracellular movement of Neph1. Mol Cell Biol (2016) 36:1639–54. doi:10.1128/MCB.00020-16
34. Putaala H, Soininen R, Kilpeläinen P, Wartiovaara J, Tryggvason K. The murine nephrin gene is specifically expressed in kidney, brain and pancreas: inactivation of the gene leads to massive proteinuria and neonatal death. Hum Mol Genet (2001) 10:1–8. doi:10.1093/hmg/10.1.1
35. Rantanen M, Palmén T, Pätäri A, Ahola H, Lehtonen S, Aström E, et al. Nephrin TRAP mice lack slit diaphragms and show fibrotic glomeruli and cystic tubular lesions. J Am Soc Nephrol (2002) 13:1586–94. doi:10.1097/01.ASN.0000016142.29721.22
36. Roselli S, Heidet L, Sich M, Henger A, Kretzler M, Gubler M-C, et al. Early glomerular filtration defect and severe renal disease in podocin-deficient mice. Mol Cell Biol (2004) 24:550–60. doi:10.1128/MCB.24.2.550-560.2004
37. Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol (2001) 21:4829–36. doi:10.1128/MCB.21.14.4829-4836.2001
38. Guaragna MS, Cleto TL, Souza ML, Lutaif ACGB, de Castro LCG, Penido MGMG, et al. NPHS1 gene mutations confirm congenital nephrotic syndrome in four Brazilian cases: a novel mutation is described. Nephrology (Carlton) (2016) 21:753–7. doi:10.1111/nep.12667
39. Bouchireb K, Boyer O, Gribouval O, Nevo F, Huynh-Cong E, Morinière V, et al. NPHS2 mutations in steroid-resistant nephrotic syndrome: a mutation update and the associated phenotypic spectrum. Hum Mutat (2014) 35:178–86. doi:10.1002/humu.22485
40. Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet (2002) 11:379–88. doi:10.1093/hmg/11.4.379
41. Schäffer AA. Digenic inheritance in medical genetics. J Med Genet (2013) 50:641–52. doi:10.1136/jmedgenet-2013-101713
42. Li X, Chuang PY, D’Agati VD, Dai Y, Yacoub R, Fu J, et al. Nephrin preserves podocyte viability and glomerular structure and function in adult kidneys. J Am Soc Nephrol (2015) 26:2361–77. doi:10.1681/ASN.2014040405
43. Mollet G, Ratelade J, Boyer O, Muda AO, Morisset L, Lavin TA, et al. Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J Am Soc Nephrol (2009) 20:2181–9. doi:10.1681/ASN.2009040379
44. Tabatabaeifar M, Wlodkowski T, Simic I, Denc H, Mollet G, Weber S, et al. An inducible mouse model of podocin-mutation-related nephrotic syndrome. PLoS One (2017) 12:e0186574. doi:10.1371/journal.pone.0186574
45. Shih NY, Li J, Cotran R, Mundel P, Miner JH, Shaw AS. CD2AP localizes to the slit diaphragm and binds to nephrin via a novel C-terminal domain. Am J Pathol (2001) 159:2303–8. doi:10.1016/S0002-9440(10)63080-5
46. Verma R, Wharram B, Kovari I, Kunkel R, Nihalani D, Wary KK, et al. Fyn binds to and phosphorylates the kidney slit diaphragm component nephrin. J Biol Chem (2003) 278:20716–23. doi:10.1074/jbc.M301689200
47. Li H, Lemay S, Aoudjit L, Kawachi H, Takano T. SRC-family kinase Fyn phosphorylates the cytoplasmic domain of nephrin and modulates its interaction with podocin. J Am Soc Nephrol (2004) 15:3006–15. doi:10.1097/01.ASN.0000146689.88078.80
48. Liu XL, Kilpeläinen P, Hellman U, Sun Y, Wartiovaara J, Morgunova E, et al. Characterization of the interactions of the nephrin intracellular domain. FEBS J (2005) 272:228–43. doi:10.1111/j.1432-1033.2004.04408.x
49. Lehtonen S, Ryan JJ, Kudlicka K, Iino N, Zhou H, Farquhar MG. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc Natl Acad Sci U S A (2005) 102:9814–9. doi:10.1073/pnas.0504166102
50. Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat Genet (2005) 37:739–44. doi:10.1038/ng1592
51. Hirabayashi S, Mori H, Kansaku A, Kurihara H, Sakai T, Shimizu F, et al. MAGI-1 is a component of the glomerular slit diaphragm that is tightly associated with nephrin. Lab Invest (2005) 85:1528–43. doi:10.1038/labinvest.3700347
52. Quack I, Rump LC, Gerke P, Walther I, Vinke T, Vonend O, et al. beta-Arrestin2 mediates nephrin endocytosis and impairs slit diaphragm integrity. Proc Natl Acad Sci U S A (2006) 103:14110–5. doi:10.1073/pnas.0602587103
53. Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature (2006) 440:818–23. doi:10.1038/nature04662
54. Verma R, Kovari I, Soofi A, Nihalani D, Patrie K, Holzman LB. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J Clin Invest (2006) 116:1346–59. doi:10.1172/JCI27414
55. Coward RJM, Welsh GI, Koziell A, Hussain S, Lennon R, Ni L, et al. Nephrin is critical for the action of insulin on human glomerular podocytes. Diabetes (2007) 56:1127–35. doi:10.2337/db06-0693
56. Hartleben B, Schweizer H, Lübben P, Bartram MP, Möller CC, Herr R, et al. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem (2008) 283:23033–8. doi:10.1074/jbc.M803143200
57. Zhu J, Sun N, Aoudjit L, Li H, Kawachi H, Lemay S, et al. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int (2008) 73:556–66. doi:10.1038/sj.ki.5002691
58. Kim EY, Choi K-J, Dryer SE. Nephrin binds to the COOH terminus of a large-conductance Ca2+-activated K+ channel isoform and regulates its expression on the cell surface. Am J Physiol Renal Physiol (2008) 295:F235–46. doi:10.1152/ajprenal.00140.2008
59. Hirose T, Satoh D, Kurihara H, Kusaka C, Hirose H, Akimoto K, et al. An essential role of the universal polarity protein, aPKClambda, on the maintenance of podocyte slit diaphragms. PLoS One (2009) 4:e4194. doi:10.1371/journal.pone.0004194
60. Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol (2009) 20:798–806. doi:10.1681/ASN.2008080871
61. Hussain S, Romio L, Saleem M, Mathieson P, Serrano M, Moscat J, et al. Nephrin deficiency activates NF-kappaB and promotes glomerular injury. J Am Soc Nephrol (2009) 20:1733–43. doi:10.1681/ASN.2008111219
62. Harita Y, Kurihara H, Kosako H, Tezuka T, Sekine T, Igarashi T, et al. Phosphorylation of nephrin triggers Ca2+ signaling by recruitment and activation of phospholipase C-{gamma}1. J Biol Chem (2009) 284:8951–62. doi:10.1074/jbc.M806851200
63. Tossidou I, Teng B, Drobot L, Meyer-Schwesinger C, Worthmann K, Haller H, et al. CIN85/RukL is a novel binding partner of nephrin and podocin and mediates slit diaphragm turnover in podocytes. J Biol Chem (2010) 285:25285–95. doi:10.1074/jbc.M109.087239
64. Wu F, Saleem MA, Kampik NB, Satchwell TJ, Williamson RC, Blattner SM, et al. Anion exchanger 1 interacts with nephrin in podocytes. J Am Soc Nephrol (2010) 21:1456–67. doi:10.1681/ASN.2009090921
65. Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int (2010) 77:989–99. doi:10.1038/ki.2010.64
66. Bertuccio C, Veron D, Aggarwal PK, Holzman L, Tufro A. Vascular endothelial growth factor receptor 2 direct interaction with nephrin links VEGF-A signals to actin in kidney podocytes. J Biol Chem (2011) 286:39933–44. doi:10.1074/jbc.M111.241620
67. Aoudjit L, Jiang R, Lee TH, New LA, Jones N, Takano T. Podocyte protein, nephrin, is a substrate of protein tyrosine phosphatase 1B. J Signal Transduct (2011) 2011:376543. doi:10.1155/2011/376543
68. Babayeva S, Zilber Y, Torban E. Planar cell polarity pathway regulates actin rearrangement, cell shape, motility, and nephrin distribution in podocytes. Am J Physiol Renal Physiol (2011) 300:F549–60. doi:10.1152/ajprenal.00566.2009
69. Wasik AA, Polianskyte-Prause Z, Dong M-Q, Shaw AS, Yates JR, Farquhar MG, et al. Septin 7 forms a complex with CD2AP and nephrin and regulates glucose transporter trafficking. Mol Biol Cell (2012) 23:3370–9. doi:10.1091/mbc.E11-12-1010
70. Kajiho Y, Harita Y, Kurihara H, Horita S, Matsunaga A, Tsurumi H, et al. SIRPα interacts with nephrin at the podocyte slit diaphragm. FEBS J (2012) 279:3010–21. doi:10.1111/j.1742-4658.2012.08682.x
71. George B, Verma R, Soofi AA, Garg P, Zhang J, Park T-J, et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J Clin Invest (2012) 122:674–92. doi:10.1172/JCI60070
72. Reidy KJ, Aggarwal PK, Jimenez JJ, Thomas DB, Veron D, Tufro A. Excess podocyte semaphorin-3A leads to glomerular disease involving plexinA1-nephrin interaction. Am J Pathol (2013) 183:1156–68. doi:10.1016/j.ajpath.2013.06.022
73. George B, Fan Q, Dlugos CP, Soofi AA, Zhang J, Verma R, et al. Crk1/2 and CrkL form a hetero-oligomer and functionally complement each other during podocyte morphogenesis. Kidney Int (2014) 85:1382–94. doi:10.1038/ki.2013.556
74. Denhez B, Lizotte F, Guimond M-O, Jones N, Takano T, Geraldes P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J Biol Chem (2015) 290:350–8. doi:10.1074/jbc.M114.612721
75. Verma R, Venkatareddy M, Kalinowski A, Patel SR, Salant DJ, Garg P. Shp2 associates with and enhances nephrin tyrosine phosphorylation and is necessary for foot process spreading in mouse models of podocyte injury. Mol Cell Biol (2015) 36:596–614. doi:10.1128/MCB.00956-15
76. Martin CE, Petersen KA, Aoudjit L, Tilak M, Eremina V, Hardy WR, et al. ShcA adaptor protein promotes nephrin endocytosis and is upregulated in proteinuric nephropathies. J Am Soc Nephrol (2018) 29:92–103. doi:10.1681/ASN.2017030285
77. Lahdenperä J, Kilpeläinen P, Liu XL, Pikkarainen T, Reponen P, Ruotsalainen V, et al. Clustering-induced tyrosine phosphorylation of nephrin by Src family kinases. Kidney Int (2003) 64:404–13. doi:10.1046/j.1523-1755.2003.00097.x
78. Mayer BJ. Perspective: dynamics of receptor tyrosine kinase signaling complexes. FEBS Lett (2012) 586:2575–9. doi:10.1016/j.febslet.2012.05.002
79. Jadwin JA, Curran TG, Lafontaine AT, White FM, Mayer BJ. Src homology 2 domains enhance tyrosine phosphorylation in vivo by protecting binding sites in their target proteins from dephosphorylation. J Biol Chem (2018) 293:623–37. doi:10.1074/jbc.M117.794412
80. Huber TB, Hartleben B, Kim J, Schmidts M, Schermer B, Keil A, et al. Nephrin and CD2AP associate with phosphoinositide 3-OH kinase and stimulate AKT-dependent signaling. Mol Cell Biol (2003) 23:4917–28. doi:10.1128/MCB.23.14.4917-4928.2003
81. Garg P, Verma R, Cook L, Soofi A, Venkatareddy M, George B, et al. Actin-depolymerizing factor cofilin-1 is necessary in maintaining mature podocyte architecture. J Biol Chem (2010) 285:22676–88. doi:10.1074/jbc.M110.122929
82. Jones N, New LA, Fortino MA, Eremina V, Ruston J, Blasutig IM, et al. Nck proteins maintain the adult glomerular filtration barrier. J Am Soc Nephrol (2009) 20:1533–43. doi:10.1681/ASN.2009010056
83. Zhu J, Attias O, Aoudjit L, Jiang R, Kawachi H, Takano T. p21-activated kinases regulate actin remodeling in glomerular podocytes. Am J Physiol Renal Physiol (2010) 298:F951–61. doi:10.1152/ajprenal.00536.2009
84. Venkatareddy M, Cook L, Abuarquob K, Verma R, Garg P. Nephrin regulates lamellipodia formation by assembling a protein complex that includes Ship2, filamin and lamellipodin. PLoS One (2011) 6:e28710. doi:10.1371/journal.pone.0028710
85. Blasutig IM, New LA, Thanabalasuriar A, Dayarathna TK, Goudreault M, Quaggin SE, et al. Phosphorylated YDXV motifs and Nck SH2/SH3 adaptors act cooperatively to induce actin reorganization. Mol Cell Biol (2008) 28:2035–46. doi:10.1128/MCB.01770-07
86. Ha T-S. Genetics of hereditary nephrotic syndrome: a clinical review. Korean J Pediatr (2017) 60:55–63. doi:10.3345/kjp.2017.60.3.55
87. Quack I, Woznowski M, Potthoff SA, Palmer R, Königshausen E, Sivritas S, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem (2011) 286:12959–70. doi:10.1074/jbc.M110.204024
88. Königshausen E, Zierhut UM, Ruetze M, Potthoff SA, Stegbauer J, Woznowski M, et al. Angiotensin II increases glomerular permeability by β-arrestin mediated nephrin endocytosis. Sci Rep (2016) 6:39513. doi:10.1038/srep39513
89. Babayeva S, Rocque B, Aoudjit L, Zilber Y, Li J, Baldwin C, et al. Planar cell polarity pathway regulates nephrin endocytosis in developing podocytes. J Biol Chem (2013) 288:24035–48. doi:10.1074/jbc.M113.452904
90. Waters AM, Wu MYJ, Huang Y-W, Liu GY, Holmyard D, Onay T, et al. Notch promotes dynamin-dependent endocytosis of nephrin. J Am Soc Nephrol (2012) 23:27–35. doi:10.1681/ASN.2011010027
91. Haase R, Potthoff SA, Meyer-Schwesinger C, Frosch C, Wiech T, Panzer U, et al. A novel in vivo method to quantify slit diaphragm protein abundance in murine proteinuric kidney disease. PLoS One (2017) 12:e0179217. doi:10.1371/journal.pone.0179217
92. Fang X, Lang Y, Wang Y, Mo W, Wei H, Xie J, et al. Shp2 activates Fyn and Ras to regulate RBL-2H3 mast cell activation following FcεRI aggregation. PLoS One (2012) 7:e40566. doi:10.1371/journal.pone.0040566
93. New LA, Keyvani Chahi A, Jones N. Direct regulation of nephrin tyrosine phosphorylation by Nck adaptor proteins. J Biol Chem (2013) 288:1500–10. doi:10.1074/jbc.M112.439463
94. Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, et al. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med (2009) 15:1298–306. doi:10.1038/nm.2052
95. Mima A, Kitada M, Geraldes P, Li Q, Matsumoto M, Mizutani K, et al. Glomerular VEGF resistance induced by PKCδ/SHP-1 activation and contribution to diabetic nephropathy. FASEB J (2012) 26:2963–74. doi:10.1096/fj.11-202994
96. Drapeau N, Lizotte F, Denhez B, Guay A, Kennedy CR, Geraldes P. Expression of SHP-1 induced by hyperglycemia prevents insulin actions in podocytes. Am J Physiol Endocrinol Metab (2013) 304:E1188–98. doi:10.1152/ajpendo.00560.2012
97. Lizotte F, Denhez B, Guay A, Gévry N, Côté AM, Geraldes P. Persistent insulin resistance in podocytes caused by epigenetic changes of SHP-1 in diabetes. Diabetes (2016) 65:3705–17. doi:10.2337/db16-0254
98. Denhez B, Geraldes P. Regulation of nephrin phosphorylation in diabetes and chronic kidney injury. Adv Exp Med Biol (2017) 966:149–61. doi:10.1007/5584_2017_62
99. Lee J, Koh A, Jeong H, Kim E, Ha T-S, Saleem MA, et al. C1-Ten is a PTPase of nephrin, regulating podocyte hypertrophy through mTORC1 activation. Sci Rep (2017) 7:12346. doi:10.1038/s41598-017-12382-8
100. Uchida K, Suzuki K, Iwamoto M, Kawachi H, Ohno M, Horita S, et al. Decreased tyrosine phosphorylation of nephrin in rat and human nephrosis. Kidney Int (2008) 73:926–32. doi:10.1038/ki.2008.19
101. Ohashi T, Uchida K, Asamiya Y, Tsuruta Y, Ohno M, Horita S, et al. Phosphorylation status of nephrin in human membranous nephropathy. Clin Exp Nephrol (2010) 14:51–5. doi:10.1007/s10157-009-0241-z
102. New LA, Martin CE, Scott RP, Platt MJ, Keyvani Chahi A, Stringer CD, et al. Nephrin tyrosine phosphorylation is required to stabilize and restore podocyte foot process architecture. J Am Soc Nephrol (2016) 27:2422–35. doi:10.1681/ASN.2015091048
103. Keyvani Chahi A, Martin CE, Jones N. Nephrin suppresses hippo signaling through the adaptor proteins Nck and WTIP. J Biol Chem (2016) 291:12799–808. doi:10.1074/jbc.M116.724245
104. Geraldes P. Protein phosphatases and podocyte function. Curr Opin Nephrol Hypertens (2018) 27:49–55. doi:10.1097/MNH.0000000000000376
105. Carney EF. Podocyte biology: phosphorylation preserves podocytes. Nat Rev Nephrol (2016) 12:197. doi:10.1038/nrneph.2016.22
106. Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, et al. Phase transitions in the assembly of multivalent signalling proteins. Nature (2012) 483:336–40. doi:10.1038/nature10879
107. Banjade S, Rosen MK. Phase transitions of multivalent proteins can promote clustering of membrane receptors. Elife (2014) 3:e04123. doi:10.7554/eLife.04123
108. Banjade S, Wu Q, Mittal A, Peeples WB, Pappu RV, Rosen MK. Conserved interdomain linker promotes phase separation of the multivalent adaptor protein Nck. Proc Natl Acad Sci U S A (2015) 112:E6426–35. doi:10.1073/pnas.1508778112
109. Qin X-S, Tsukaguchi H, Shono A, Yamamoto A, Kurihara H, Doi T. Phosphorylation of nephrin triggers its internalization by raft-mediated endocytosis. J Am Soc Nephrol (2009) 20:2534–45. doi:10.1681/ASN.2009010011
110. Kirchhausen T, Owen D, Harrison SC. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol (2014) 6:a016725. doi:10.1101/cshperspect.a016725
111. Soda K, Balkin DM, Ferguson SM, Paradise S, Milosevic I, Giovedi S, et al. Role of dynamin, synaptojanin, and endophilin in podocyte foot processes. J Clin Invest (2012) 122:4401–11. doi:10.1172/JCI65289
112. Swiatecka-Urban A. Endocytic trafficking at the mature podocyte slit diaphragm. Front Pediatr (2017) 5:32. doi:10.3389/fped.2017.00032
113. Jeon J, Leibiger I, Moede T, Walter B, Faul C, Maiguel D, et al. Dynamin-mediated nephrin phosphorylation regulates glucose-stimulated insulin release in pancreatic beta cells. J Biol Chem (2012) 287:28932–42. doi:10.1074/jbc.M112.389452
114. Shimizu J, Tanaka H, Aya K, Ito S, Sado Y, Seino Y. A missense mutation in the nephrin gene impairs membrane targeting. Am J Kidney Dis (2002) 40:697–703. doi:10.1053/ajkd.2002.35676
115. Wernerson A, Dunér F, Pettersson E, Widholm SM, Berg U, Ruotsalainen V, et al. Altered ultrastructural distribution of nephrin in minimal change nephrotic syndrome. Nephrol Dial Transplant (2003) 18:70–6. doi:10.1093/ndt/18.1.70
116. Tossidou I, Teng B, Menne J, Shushakova N, Park J-K, Becker JU, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One (2010) 5:e10185. doi:10.1371/journal.pone.0010185
117. Rocque BL, Babayeva S, Li J, Leung V, Nezvitsky L, Cybulsky AV, et al. Deficiency of the planar cell polarity protein Vangl2 in podocytes affects glomerular morphogenesis and increases susceptibility to injury. J Am Soc Nephrol (2015) 26:576–86. doi:10.1681/ASN.2014040340
118. Liu L, Doné SC, Khoshnoodi J, Bertorello A, Wartiovaara J, Berggren PO, et al. Defective nephrin trafficking caused by missense mutations in the NPHS1 gene: insight into the mechanisms of congenital nephrotic syndrome. Hum Mol Genet (2001) 10:2637–44. doi:10.1093/hmg/10.23.2637
119. Liu XL, Doné SC, Yan K, Kilpeläinen P, Pikkarainen T, Tryggvason K. Defective trafficking of nephrin missense mutants rescued by a chemical chaperone. J Am Soc Nephrol (2004) 15:1731–8. doi:10.1097/01.ASN.0000129826.28932.FD
120. Shono A, Tsukaguchi H, Kitamura A, Hiramoto R, Qin X-S, Doi T, et al. Predisposition to relapsing nephrotic syndrome by a nephrin mutation that interferes with assembly of functioning microdomains. Hum Mol Genet (2009) 18:2943–56. doi:10.1093/hmg/ddp232
121. Kitamura A, Tsukaguchi H, Hiramoto R, Shono A, Doi T, Kagami S, et al. A familial childhood-onset relapsing nephrotic syndrome. Kidney Int (2007) 71:946–51. doi:10.1038/sj.ki.5002110
122. Lin JS, Jeon JS, Fan Q, Wong HN, Palmer MB, Holzman LB. ARF6 mediates nephrin tyrosine phosphorylation-induced podocyte cellular dynamics. PLoS One (2017) 12:e0184575. doi:10.1371/journal.pone.0184575
123. Verma R, Venkatareddy M, Kalinowski A, Patel SR, Garg P. Integrin ligation results in nephrin tyrosine phosphorylation in vitro. PLoS One (2016) 11:e0148906. doi:10.1371/journal.pone.0148906
124. Stefanou C, Pieri M, Savva I, Georgiou G, Pierides A, Voskarides K, et al. Co-inheritance of functional podocin variants with heterozygous collagen IV mutations predisposes to renal failure. Nephron (2015) 130:200–12. doi:10.1159/000432406
125. Kriz W, Lemley KV. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J Am Soc Nephrol (2015) 26:258–69. doi:10.1681/ASN.2014030278
126. Kriz W, Lemley KV. Mechanical challenges to the glomerular filtration barrier: adaptations and pathway to sclerosis. Pediatr Nephrol (2017) 32:405–17. doi:10.1007/s00467-016-3358-9
127. Kriz W, Lemley KV. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int (2017) 91:1283–6. doi:10.1016/j.kint.2017.02.032
128. Wennmann DO, Vollenbröker B, Eckart AK, Bonse J, Erdmann F, Wolters DA, et al. The Hippo Pathway is controlled by angiotensin II signaling and its reactivation induces apoptosis in podocytes. Cell Death Dis (2014) 5:e1519. doi:10.1038/cddis.2014.476
129. Schwartzman M, Reginensi A, Wong JS, Basgen JM, Meliambro K, Nicholas SB, et al. Podocyte-specific deletion of yes-associated protein causes FSGS and progressive renal failure. J Am Soc Nephrol (2016) 27:216–26. doi:10.1681/ASN.2014090916
130. Kann M, Ettou S, Jung YL, Lenz MO, Taglienti ME, Park PJ, et al. Genome-wide analysis of Wilms’ tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J Am Soc Nephrol (2015) 26:2097–104. doi:10.1681/ASN.2014090940
131. Ahn YH, Park EJ, Kang HG, Kim SH, Cho HY, Shin JI, et al. Genotype-phenotype analysis of pediatric patients with WT1 glomerulopathy. Pediatr Nephrol (2017) 32:81–9. doi:10.1007/s00467-016-3395-4
132. Tan W, Lovric S, Ashraf S, Rao J, Schapiro D, Airik M, et al. Analysis of 24 genes reveals a monogenic cause in 11.1% of cases with steroid-resistant nephrotic syndrome at a single center. Pediatr Nephrol (2018) 33:305–14. doi:10.1007/s00467-017-3801-6
133. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi:10.1126/science.1106215
134. Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol (2015) 309:F393–7. doi:10.1152/ajprenal.00186.2015
135. Wieder N, Greka A. Calcium, TRPC channels, and regulation of the actin cytoskeleton in podocytes: towards a future of targeted therapies. Pediatr Nephrol (2016) 31:1047–54. doi:10.1007/s00467-015-3224-1
136. Zhou Y, Castonguay P, Sidhom E-H, Clark AR, Dvela-Levitt M, Kim S, et al. A small-molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science (2017) 358:1332–6. doi:10.1126/science.aal4178
137. van der Wijst J, Bindels RJM. Renal physiology: TRPC5 inhibition to treat progressive kidney disease. Nat Rev Nephrol (2018) 14:145–6. doi:10.1038/nrneph.2018.4
138. Riehle M, Büscher AK, Gohlke B-O, Kaßmann M, Kolatsi-Joannou M, Bräsen JH, et al. TRPC6 G757D loss-of-function mutation associates with FSGS. J Am Soc Nephrol (2016) 27:2771–83. doi:10.1681/ASN.2015030318
139. Greka A, Mundel P. Balancing calcium signals through TRPC5 and TRPC6 in podocytes. J Am Soc Nephrol (2011) 22:1969–80. doi:10.1681/ASN.2011040370
140. Kanda S, Harita Y, Shibagaki Y, Sekine T, Igarashi T, Inoue T, et al. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-γ1 and nephrin: effect of mutations associated with focal segmental glomerulosclerosis. Mol Biol Cell (2011) 22:1824–35. doi:10.1091/mbc.e10-12-0929
141. Greka A, Gibson D, Mundel P, Demetri G, Hildebrandt F, Pollak M, et al. Personalized comments on challenges and opportunities in kidney disease therapeutics: the glom-NExT symposium. Semin Nephrol (2016) 36:448. doi:10.1016/j.semnephrol.2016.09.005
142. Yang Q, Ma Y, Liu Y, Liang W, Chen X, Ren Z, et al. Angiotensin II down-regulates nephrin-Akt signaling and induces podocyte injury: role of c-Abl. Mol Biol Cell (2016) 27:197–208. doi:10.1091/mbc.E15-04-0223
143. Sagar A, Arif E, Solanki AK, Srivastava P, Janech MG, Kim S-H, et al. Targeting Neph1 and ZO-1 protein-protein interaction in podocytes prevents podocyte injury and preserves glomerular filtration function. Sci Rep (2017) 7:12047. doi:10.1038/s41598-017-12134-8
144. Li X, He JC. An update: the role of nephrin inside and outside the kidney. Sci China Life Sci (2015) 58:649–57. doi:10.1007/s11427-015-4844-1
145. Völker LA, Petry M, Abdelsabour-Khalaf M, Schweizer H, Yusuf F, Busch T, et al. Comparative analysis of Neph gene expression in mouse and chicken development. Histochem Cell Biol (2012) 137:355–66. doi:10.1007/s00418-011-0903-2
146. Moeller MJ, Kovari IA, Holzman LB. Evaluation of a new tool for exploring podocyte biology: mouse Nphs1 5’ flanking region drives LacZ expression in podocytes. J Am Soc Nephrol (2000) 11:2306–14.
147. Wagner N, Morrison H, Pagnotta S, Michiels J-F, Schwab Y, Tryggvason K, et al. The podocyte protein nephrin is required for cardiac vessel formation. Hum Mol Genet (2011) 20:2182–94. doi:10.1093/hmg/ddr106
148. Liu L, Aya K, Tanaka H, Shimizu J, Ito S, Seino Y. Nephrin is an important component of the barrier system in the testis. Acta Med Okayama (2001) 55:161–5. doi:10.18926/AMO/32022
149. Fornoni A, Jeon J, Varona Santos J, Cobianchi L, Jauregui A, Inverardi L, et al. Nephrin is expressed on the surface of insulin vesicles and facilitates glucose-stimulated insulin release. Diabetes (2010) 59:190–9. doi:10.2337/db09-0655
150. Villarreal R, Mitrofanova A, Maiguel D, Morales X, Jeon J, Grahammer F, et al. Nephrin contributes to insulin secretion and affects mammalian target of rapamycin signaling independently of insulin receptor. J Am Soc Nephrol (2016) 27:1029–41. doi:10.1681/ASN.2015020210
151. Lehtonen S, Jalanko H. Nephrin trafficking beyond the kidney – role in glucose-stimulated insulin secretion in β cells. J Am Soc Nephrol (2016) 27:965–8. doi:10.1681/ASN.2015080960
152. Gu C, Chang J, Shchedrina VA, Pham VA, Hartwig JH, Suphamungmee W, et al. Regulation of dynamin oligomerization in cells: the role of dynamin-actin interactions and its GTPase activity. Traffic (2014) 15:819–38. doi:10.1111/tra.12178
153. Schiffer M, Teng B, Gu C, Shchedrina VA, Kasaikina M, Pham VA, et al. Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat Med (2015) 21:601–9. doi:10.1038/nm.3843
154. Kim J-H, Xie J, Hwang K-H, Wu Y-L, Oliver N, Eom M, et al. Klotho may ameliorate Proteinuria by targeting TRPC6 channels in podocytes. J Am Soc Nephrol (2017) 28:140–51. doi:10.1681/ASN.2015080888
155. Sonneveld R, Hoenderop JG, Isidori AM, Henique C, Dijkman HB, Berden JH, et al. Sildenafil prevents podocyte injury via PPAR-γ-mediated TRPC6 inhibition. J Am Soc Nephrol (2017) 28:1491–505. doi:10.1681/ASN.2015080885
156. Greka A, Weins A, Mundel P. Abatacept in B7-1-positive proteinuric kidney disease. N Engl J Med (2014) 370:1263–6. doi:10.1056/NEJMc1400502
157. Zhou X, Zhang J, Haimbach R, Zhu W, Mayer-Ezell R, Garcia-Calvo M, et al. An integrin antagonist (MK-0429) decreases proteinuria and renal fibrosis in the ZSF1 rat diabetic nephropathy model. Pharmacol Res Perspect (2017) 5. doi:10.1002/prp2.354
158. Lin M-H, Miller JB, Kikkawa Y, Suleiman HY, Tryggvason K, Hodges BL, et al. Laminin-521 protein therapy for glomerular basement membrane and podocyte abnormalities in a model of pierson syndrome. J Am Soc Nephrol (2018) 29(5):1426–36. doi:10.1681/ASN.2017060690
159. Sieber J, Wieder N, Clark A, Reitberger M, Matan S, Schoenfelder J, et al. GDC-0879, a BRAFV600EInhibitor, protects kidney podocytes from death. Cell Chem Biol (2018) 25:175–184.e4. doi:10.1016/j.chembiol.2017.11.006
160. Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res (2009) 69:3042–51. doi:10.1158/0008-5472.CAN-08-3563
Keywords: podocyte, nephrin, phosphorylation, signal transduction, chronic kidney disease
Citation: Martin CE and Jones N (2018) Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. 9:302. doi: 10.3389/fendo.2018.00302
Received: 12 April 2018; Accepted: 22 May 2018;
Published: 05 June 2018
Edited by:
Sanna Helena Lehtonen, University of Helsinki, FinlandReviewed by:
Rachel Lennon, University of Manchester, United KingdomGavin Iain Welsh, University of Bristol, United Kingdom
Copyright: © 2018 Martin and Jones. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Jones, jonesmcb@uoguelph.ca