 Mickey J. M. Kuo
Mickey J. M. Kuo Matthew A. Nazari
Matthew A. Nazari Abhishek Jha
Abhishek Jha Karel Pacak
Karel Pacak- 1Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
- 2Section on Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, United States
Although pediatric pheochromocytomas and paragangliomas (PPGLs) are rare, they have important differences compared to those in adults. Unfortunately, without timely diagnosis and management, these tumors have a potentially devastating impact on pediatric patients. Pediatric PPGLs are more often extra-adrenal, multifocal/metastatic, and recurrent, likely due to these tumors being more commonly due to a genetic predisposition than in adults. This genetic risk results in disease manifestations at an earlier age giving these tumors time to advance before detection. In spite of these problematic features, advances in the molecular and biochemical characterization of PPGLs have heralded an age of increasingly personalized medicine. An understanding of the genetic basis for an individual patient’s tumor provides insight into its natural history and can guide clinicians in management of this challenging disease. In pediatric PPGLs, mutations in genes related to pseudohypoxia are most commonly seen, including the von Hippel-Lindau gene (VHL) and succinate dehydrogenase subunit (SDHx) genes, with the highest risk for metastatic disease associated with variants in SDHB and SDHA. Such pathogenic variants are associated with a noradrenergic biochemical phenotype with resultant sustained catecholamine release and therefore persistent symptoms. This is in contrast to paroxysmal symptoms (e.g., episodic hypertension, palpitations, and diaphoresis/flushing) as seen in the adrenergic, or epinephrine-predominant, biochemical phenotype (due to episodic catecholamine release) that is commonly observed in adults. Additionally, PPGLs in children more often present with signs and symptoms of catecholamine excess. Therefore, children, adolescents, and young adults present differently from older adults (e.g., the prototypical presentation of palpitations, perspiration, and pounding headaches in the setting of an isolated adrenal mass). These presentations are a direct result of genetic determinants and highlight the need for pediatricians to recognize these differences in order to expedite appropriate evaluations, including genetic testing. Identification and familiarity with causative genes inform surveillance and treatment strategies to improve outcomes in pediatric patients with PPGL.
Introduction
Pheochromocytomas and paragangliomas (PPGLs) are catecholamine-producing tumors that arise from chromaffin cells of the adrenal gland and extra-adrenal tissue, pheochromocytomas (PCC) and paragangliomas (PGL), respectively. Pediatric PPGLs have important differences compared to those in adults, a consequence of the more frequent genetic predisposition seen in the younger population (80% in pediatric patients compared to 40% in adults, see Table 1) (2, 4). In children with PPGL, disease is more likely to be extra-adrenal (30-60%), bilateral adrenal (12-48% of pediatric vs. 7-14% of adult patients), recurrent (13-38%), and multifocal (17-62%) (2, 4–8). The most common underlying genetic causes of pediatric PPGL are von Hippel-Lindau syndrome (VHL, 27-51%), mutations in succinate dehydrogenase subunit genes (collectively referred to as SDHx, 13-39% in SDHB and 8-10% in SDHD), multiple endocrine neoplasia type 2 (MEN-2, 0.6-10%), and neurofibromatosis type 1 (NF-1, 1-3%) (2, 4, 5, 7–11).

Table 1 Data on pediatric and adult PPGLs from studies directly comparing the two populations.
The underlying pathologic mechanisms that give rise to PPGL are heterogeneous and have been organized into clusters of genes by their effect on different intracellular pathways and gene expression profiling, including pseudohypoxia (cluster 1), kinase signaling (cluster 2), and Wnt signaling (cluster 3) (10, 12–15). The significance of this molecular taxonomy is not only descriptive but also helps to define different aspects of chromaffin cell metabolism that have direct clinical applications, such as the prototypical examples of VHL (cluster 1) and MEN-2 (cluster 2).
PCCs in patients with MEN-2 produce either only epinephrine or both norepinephrine and epinephrine (and their metabolites normetanephrine and metanephrine, respectively; collectively termed metanephrines) and have higher catecholamine levels due to increased expression of tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis (12). PCCs in VHL patients produce norepinephrine almost exclusively, related to decreased expression of the enzyme phenylethanolamine N-methyltransferase (PNMT) which converts norepinephrine to epinephrine (12). Furthermore, it was shown that expression of proteins involved in catecholamine secretion are differentially regulated between MEN-2 and VHL, as VHL-related PCCs show decreased expression of some secretory components, resulting in a less coordinated secretion system and therefore, continuous secretion of catecholamines in sharp contrast to paroxysmal release of catecholamines by MEN-2 PCCs (13). One study reported that, on average, norepinephrine-secreting PPGLs stored 1,760,000 picograms (pg) of norepinephrine/gram (g) tissue with 53% released each day, in contrast to epinephrine-secreting PPGLs that contained 3,801,000 pg of epinephrine/g tissue with only 5% released daily (16). Norepinephrine has a higher affinity for α1-adrenergic receptors, and epinephrine has a higher affinity for β1-adrenergic receptors, which broadly translates into more of an increased risk for hypertension in those with tumors that produce predominantly norepinephrine and more risk for tachycardia and arrhythmias in those tumors that produce predominantly epinephrine (17).
Taken together, these molecular and biochemical features are foundational for understanding the different clinical features in these patients, with paroxysmal hypertension occurring in the context of the adrenergic biochemical phenotype (increased epinephrine and metanephrine) of MEN-2 as compared to sustained hypertension with the noradrenergic biochemical phenotype (increased norepinephrine and normetanephrine) of VHL. Additionally, decreased expression of dopamine β-hydroxylase or tyrosine hydroxylase can result in a dopaminergic or biochemically silent (non-secreting) phenotype, respectively, which may both be seen from mutations in cluster 1 genes (18). Pediatric PPGL is more often due to pathogenic variants in cluster 1 genes (particularly VHL, SDHx, and EPAS1; at least 57-80% of patients with pediatric PPGL when somatic variants are included) and is thus more likely to have a non-adrenergic (noradrenergic, dopaminergic, or non-secreting) biochemical phenotype (93% of pediatric patients without increased plasma metanephrine vs. 57% in adults, see Table 1) (2, 4, 5, 7).
About 10-20% of PPGLs are diagnosed in pediatric patients (19), therefore, it is important for clinicians who care for pediatric patients to be aware of this treatable cause of hypertension, not only to alleviate the symptomatic burden of catecholamine excess, but also to make a timely diagnosis due to the risk of metastatic disease and consequent morbidity and mortality. Among adults under the age of 35 years with PPGL, many similarities to pediatric PPGL are seen (including hereditary, noradrenergic, and multifocal disease), and bilateral tumors were seen even more often than in children or in adults over the age of 35 years, suggesting that these tumors may have evaded clinical detection and were in fact present earlier in childhood (2), thus highlighting the need for clinicians to be aware of the possibility of PPGL in children and adolescents. Retrospective analysis from the Department of Defense Serum Repository found that in adults, biochemical evidence of elevated plasma metanephrines was seen at a median of 6.6 years (elevation above the upper reference limit [URL]) and 4.1 years (3 times above the URL) prior to diagnosis of PCC (20). Therefore, it is reasonable to suspect that pediatric PPGLs may go undetected for many years before diagnosis, putting these patients at risk not only for continued tumor growth and metastatic disease but also catastrophic complications if stored catecholamines in a clinically occult PPGL are suddenly released when provoked by surgery, induction of anesthesia, or as an adverse effect of medication.
While isolated PPGLs are often definitively treated by surgical removal, metastatic disease requires additional treatment modalities, such as chemotherapy, radiotherapy, targeted molecular therapies, or ablative therapies (21). Metastatic disease, defined by the presence of chromaffin tumor cells in tissues without chromaffin cells, is a major aspect of PPGL care and is itself largely genetically determined (1). Metastatic disease can occur in 12% of pediatric patients, in general, but may be up to 70% among SDHB mutation carriers, the second most commonly mutated gene in pediatric PPGL after VHL (19, 22, 23). The sites of involvement are most often bone, lymph nodes, liver, and lungs (though primary PGLs have been described in liver and lungs), which can result in significant impairment in quality of life and prognosis (24, 25). Metastases may be present at the time of initial diagnosis (synchronous) or later (metachronous or non-synchronous); in pediatric PPGL, metastases are less often synchronous as compared to adults, underscoring the importance of long-term surveillance for any child diagnosed with PPGL (2). In addition to those patients who harbor a mutation in SDHB, other established risk factors for metastatic disease include tumor size ≥ 5 cm, extra-adrenal PGL, dopaminergic phenotype (plasma 3-methoxytyramine higher than three times the URL), and a Ki-67 index > 3% (3, 26).
Finally, metastatic disease has direct implications in the approach to and management of pediatric PPGL, including imaging and treatment. Advances in functional imaging modalities with positron emission tomography/computed tomography (PET/CT) scans using different radionuclides provide insight into how patients from particular genetic backgrounds can receive an increasingly personalized approach to management of these challenging tumors. And while metastatic PPGL is incurable, knowledge of the molecular pathogenesis can direct treatments in some cases, informed by the cellular pathways disrupted by the genetic predisposition. For these reasons, the genetic background may guide management of the pediatric PPGL patient, and genetic testing is essential to providing optimal care for these patients.
Clinical Features
PPGLs are estimated to occur at an incidence of 0.57 per 100,000 person-years, of which, up to 20% of PPGLs are diagnosed in pediatric patients, at an average age of 11 years (19, 22, 23, 27). Most pediatric patients are symptomatic (around 90%), and hypertension is the most common presentation of pediatric PPGL (64-93%), which causes pediatric hypertension in up to 1% of cases (7, 8, 22). Therefore, one should suspect PPGL in pediatric patients with sustained hypertension, with headache (39-95%), diaphoresis (90%), palpitations (53%), and signs/symptoms of mass effect or as an incidental mass (30%) (Table 1) (8, 11, 22).
Pediatric hypertension is defined as an auscultatory blood pressure > 130/80 mmHg for children ages 13 years and above or ≥ 95th percentile for age, sex, and height for children ages 1 to 12 years, on more than 3 occasions, according to the most recent Clinical Practice Guideline from the American Academy of Pediatrics (28). Pediatric hypertension can be further complicated by hypertensive emergency in patients with catecholamine excess, such as retinopathy, resulting in visual disturbances, and hypertensive encephalopathy, resulting in seizures and disorientation (22, 29). Hypertrophic and dilated cardiomyopathy can arise as sequelae of hypertension in pediatric patients with PPGL; among pediatric patients with dilated cardiomyopathy, surgical excision of their tumors led to resolution of hypertension and improved cardiac function (8, 30).
Tachycardia and dysrhythmias, by contrast, are seen more often in adults, which may be related to the relatively higher proportion of cluster 2 mutations in adults resulting in stimulation of cardiac β1-adrenergic receptors by epinephrine (22). Stimulation of β-adrenergic receptors by epinephrine in cluster 2 mutations results in hepatic glycogenolysis and gluconeogenesis, which may account for the higher proportion of adults as opposed to children who present with elevated fasting glucose levels (22, 31).
In addition to the signs and symptoms noted above, other findings related to catecholamine excess may be non-specific and include pallor, orthostatic hypotension and syncope, shortness of breath, abdominal pain, nausea, vomiting, constipation, diarrhea, hyperglycemia, polyuria and polydipsia, anxiety, behavioral symptoms, worsening performance in school, and ADHD (9, 30, 32). Interestingly, among pediatric patients harboring an SDHB mutation, sweating was significantly associated with earlier development of metastatic disease (p = 0.0073), and for those without metastases at initial diagnosis, patients with tumor pain developed metastases at an earlier interval than those without tumor pain (p = 0.0088) (23).
Presenting signs and symptoms may also be related to mass effect of the growing tumor, and patients may present with a palpable abdominal mass on physical examination (30). Abdominal masses may present with abdominal pain and distension or back pain, and bladder masses may result in hematuria or symptoms with voiding (11, 32). Parasympathetic head and neck PGLs (HNPGLs) would not be expected to produce catecholamines, and the clinical presentation may be related to mass effect on cranial nerves and other local structures, resulting in hearing loss, tinnitus, hoarseness, dysphagia, cough, pain, or feeling of fullness in the neck (32). As in adults, PPGLs may also be found as an incidental mass on imaging studies performed for other indications (8, 11). For patients with clinical findings of or genetic predisposition to PPGL, biochemical evaluation is indicated.
Biochemical Foundations
The biosynthesis of catecholamines, metanephrines, and 3-methoxytyramine (3-MT, the O-methylated metabolite of dopamine), as derivatives of tyrosine, is shown in Figure 1. Plasma free metanephrines should be measured, as this highly sensitive test can help to rule out PPGL in children, as in adults (33). Eisenhofer et al. found that the optimal combination of diagnostic sensitivity (97.9%) and specificity (94.2%) was achieved by using age-nonspecific URLs for metanephrine (446 picomoles/liter [pmol/L]) and 3-methoxytyramine (107 pmol/L) with age-specific URLs for normetanephrine, increasing from age 5 years (542 pmol/L) to age 65 years (1092 pmol/L) as modeled by the equation URLNMN = (2.07 × 10-3 × age3) + 545 (pmol/L) (Table 2) (34). Although plasma fractionated metanephrines are the test of choice in children as in adults, urinary free metanephrines may be considered without much loss of sensitivity (97.9% for plasma free metanephrines and 93.4% for urinary free metanephrines), which may be preferable to avoid venipuncture in some children (35).
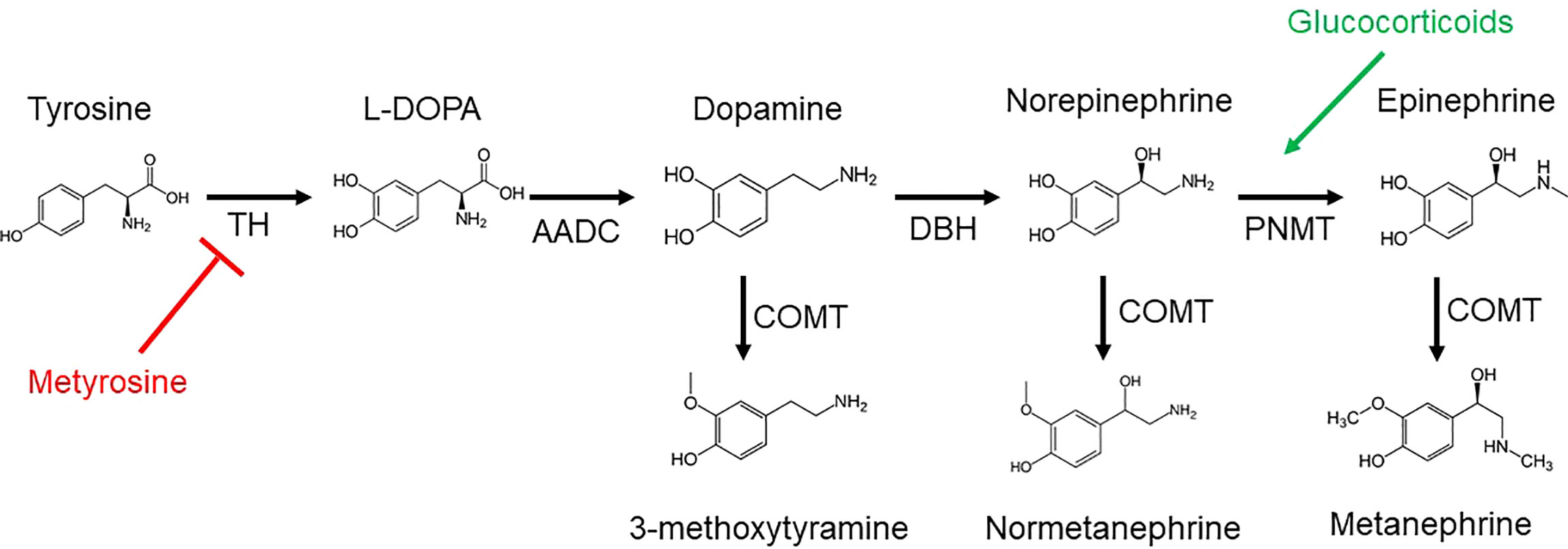
Figure 1 Catecholamines and metanephrines are derived from tyrosine by the enzyme tyrosine hydroxylase (TH), resulting in L-3,4-dihydroxyphenylalanine (L-DOPA). Aromatic L-amino acid decarboxylase (AACD) generates dopamine from L-DOPA. Dopamine β-hydroxylase (DBH) acts on dopamine to produce norepinephrine, followed by the enzyme phenylethanolamine N-methyltransferase (PNMT) to yield epinephrine. Norepinephrine and epinephrine are O-methylated by catechol-O-methyltransferase (COMT) to produce normetanephrine and metanephrine, respectively. The corresponding O-methylated metabolite of dopamine is 3-methyoxytyramine (3-MT). Metyrosine inhibits (red) TH. Glucocorticoids stimulate (green) PNMT.

Table 2 Age-specific upper reference limits for normetanephrine.
Dopamine-producing PPGLs show elevations of 3-MT; thus, plasma 3-MT should be used to increase the diagnostic sensitivity in PGLs, especially for HNPGLs (22.1% to 50%) using a cut-off of 0.1 nanomoles/liter (nmol/L) (36). Urinary dopamine reflects renal metabolism and is not helpful to identify dopaminergic PPGL (36). Although testing for 3-MT is of more limited clinical availability than that of metanephrines, it is helpful to assess risk of metastatic disease, as plasma elevations above 0.2 nmol/L had a 57% sensitivity and 85% specificity for metastatic disease (37).
Chromogranin A is a biomarker of neuroendocrine tumors, including PPGL, and has been shown to increase the sensitivity of diagnosis when used in conjunction with metanephrines, especially in patients with SDHB-related PCC or sympathetic PGL, though its performance was not as good in patients with HNPGL (38). As its production is independent of the catecholamine metabolism pathway, it is a helpful adjunct to detect non-secreting tumors.
Care must be taken to reduce false-positive biochemical laboratory results. This can be achieved by avoiding circumstances that provoke an adrenergic response at the time of collection. For this reason, the patient should be placed in a supine position with venipuncture performed 20-30 minutes before biochemical labs are obtained – as both non-supine position and venipuncture may lead to catecholamine release (32). Medications that can artifactually increase plasma or urinary fractionated metanephrines include acetaminophen, α-methyldopa, tricyclic antidepressants, monoamine oxidase inhibitors, sympathomimetics, catecholamine reuptake inhibitors, mesalamine/sulfasalazine, phenoxybenzamine, levodopa, anesthetics, neuromuscular blockers, antiemetics, linezolid, peptide hormones, steroids, cocaine, and opioids (17, 39). Glucocorticoids potentiate catecholamine biosynthetic enzymes and should be avoided in patients with PPGL due to risk of causing PCC crisis (40). Therefore, when able, these medications should be held before biochemical labs are obtained.
Cellular consequences of cluster 1 mutations converge on hypoxia-inducible factor 2α (HIF-2α), a transcription factor that dimerizes with HIF-1β and mediates downstream effects such as blocking the glucocorticoid-induced expression of PNMT (41). Gain-of-function mutations in EPAS1 (encoding for HIF-2α) contribute to aberrant neuroendocrine-to-mesenchymal transition which gives rise to PPGLs and contributes to their metastatic behavior (41). Biochemically, this translates to the observation that PNMT and other biosynthetic enzymes are down-regulated in patients with cluster 1 mutations, such that the biochemical phenotype is either noradrenergic, dopaminergic, or non-secreting, but not expected to be adrenergic.
Finally, the metabolic role of succinate dehydrogenase in the tricarboxylic acid (TCA) cycle and electron transport chain is related to diagnostic imaging and may impact treatment. Mutations in TCA cycle genes (cluster 1A) result in decreased function of these metabolic enzymes and increase dependency on glycolysis for energy production (15). Radiologically, this correlates with higher uptake of 18F-fluorodeoxyglucose (18F-FDG) on PET imaging for tumors of a cluster 1 genetic background when compared with cluster 2 (42). In addition to its role in the conversion of succinate to fumarate in the TCA cycle, succinate dehydrogenase also plays a crucial role as complex II in the mitochondrial electron transport chain, using reducing equivalents of FADH2 to ultimately generate ATP (43, 44). Defects in SDH subunit genes (i.e., SDHx) may increase the cell’s dependence on NADH oxidation through complex I, resulting in increased availability of NAD+ to be used by poly (ADP-ribose) polymerase (PARP) in base excision repair of DNA (43). Therefore, SDHx mutations increase chemoresistance to genotoxic agents by stimulating DNA repair mechanisms via PARP. For these reasons, PARP inhibition may deprive these tumors of this protective mechanism and play a role in personalized treatment tailored by genetics (43).
Genetics
Overview
Pediatric PPGLs are often found to have a genetic cause, which is most commonly due to a germline mutation in one of the following susceptibility genes: VHL, SDHx, RET, and NF1. Genes related to pseudohypoxia in cluster 1 are subdivided into cluster 1A with TCA-cycle genes, including the SDHx genes, and cluster 1B related to hypoxia signaling, most significantly VHL and EPAS1. The other two important genetic causes of pediatric PPGL, RET and NF1, are in cluster 2, related to kinase signaling (15, 26).
Genetic syndromes predisposing to PPGL are all inherited in an autosomal dominant manner, but with the caveat that pathogenic variants in SDHD (as well as SDHAF2 and MAX) increase PPGL susceptibility when paternally inherited (18). However, identifying a maternally inherited SDHD variant has implications for screening extended family members and subsequent generations. While a family history of PPGL is often seen in pediatric patients with PPGL, the absence of a family history should not disincline clinicians from referring for genetic counseling and testing, as one must consider the possibility of de novo mutations. All patients with PPGL should be referred for genetic counseling and evaluation (1).
Succinate Dehydrogenase Subunit Defects
Mutations in all four subunits of succinate dehydrogenase (subunits A, B, C, and D) and assembly factor gene SDHAF2 have been described in PPGL. In addition to sequence variants and copy number changes contributing to the genetic pathogenesis of SDHx variants, disease resulting from epigenetic alteration of SDHC by increased promoter methylation (i.e., epimutation) resulting in decreased expression have also been described, especially in PGLs and gastrointestinal stromal tumors (GISTs) (45, 46). Accumulation of succinate results in impaired prolyl hydroxylation of HIF-2α, resulting in the pseudohypoxic expression pattern seen for these cluster 1A genes, as well as other epigenetic effects on gene expression (47). In addition to PPGLs, other tumors, including renal cell carcinoma (RCC), GIST, pituitary adenoma, pulmonary chondroma (as part of Carney triad, which also includes PGL and GIST), and papillary thyroid cancer, as well as thyroid nodules have been reported in patients with SDHx mutations or SDHC epimutation; estimated penetrance of RCC in SDHx carriers is 2-3% (mostly SDHB), but 85% of GISTs diagnosed in childhood are related to SDHx mutations (4, 7, 48). No additional imaging is recommended to screen for these tumors in asymptomatic SDHx carriers (48). While genotype-phenotype correlations have been explored in SDHx genes, particularly for SDHD and SDHB, variable expressivity is seen, such that even among family members with the same variant, the sites and extent of disease and presence of metastases may vary (48).
Mutations in SDHB (on chromosome 1p36.13) are the most well-established genetic risk factor for metastatic PPGL. In a study from the National Institutes of Health that assessed 125 patients with metastatic PPGL, 32 presented before 20 years old, of which 23 (71.9%) were found to have a germline mutation in SDHB (49). Although the penetrance of PPGL in SDHB was initially thought to be higher, a Dutch study using data ascertained from clinical genetics centers (as opposed to PPGL tertiary care centers) found that the penetrance was closer to 21% by age 50 and 42% by age 70 (50). The risk of metastatic disease for SDHB carriers with PPGL is reported to be about 34-71%; in a study of 64 pediatric PPGL patients with SDHB mutations reported by Jochmanova et al., 70% developed metastases at a median age of 16 years (those patients being diagnosed at a median age of 12 years). Among those with metastases, only 19% had synchronous metastases, with the highest risk of metastatic disease within the first 2 years after diagnosis and again between 12-18 years after diagnosis (23), highlighting the critical role of lifelong surveillance and monitoring in these patients. Recurrent disease was seen in 20% of these patients, at a median age of 16 years (median time from diagnosis to recurrence was 2 years); recurrence was more often as sympathetic PGL (sPGL) than PCC (85% compared to 38%, respectively). Tumor size also played an important role in the timing of metastases as patients with tumors ≤ 5 cm developed metastatic disease at a median interval of 7 years compared to 2 years in those with tumors > 5 cm (23). All patients who died (12.5%) had metastatic disease, surviving for a median duration of 7.5 years after diagnosis of metastatic disease; no differences in the time to death were seen by comparison among the 5 most common mutations (one missense, two premature nonsense mutations, and two exonic deletions) (23). A genetic analysis of two European datasets (Germany and Great Britain) comparing different types of mutations in SDHB carriers with PPGL found that truncating mutations in SDHB seemed to confer a higher risk of PPGL than missense variants (62% vs. 38%), which remained statistically significant after removing prominent founder variants from one of the datasets; malignancy was also more common in those with truncating variants (62% vs. 38%), but no significant difference was seen for HNPGL between these different SDHB variants (47% for truncating vs. 53% for missense variants). No difference in age of diagnosis was seen between truncating and missense SDHB variants (46).
Mutations in SDHD (on chromosome 11q23.1) have a higher penetrance than those in SDHB (> 80%), and the autosomal dominant inheritance pattern is modified by maternal imprinting such that the disease is usually inherited from the paternal allele (46). Cases involving confirmed maternal inheritance are rare and involve additional genetic events, such as post-zygotic loss of chromosome 11 material as demonstrated in one study of 20 such patients with only one who was confirmed to have PPGL (35 years woman with PCC) (51). SDHD mutations are commonly seen in HNPGLs, but thoracic PGLs and PCCs are also seen (4, 18). Metastatic risk in SDHD is reported to be around 15-29% (26). In a study of 32 pediatric PPGL patients who developed metastatic disease, the three patients with a primary HNPGL were all identified to have a SDHD mutation (49), however in three other studies of pediatric PPGL patients, 17 of 177 patients, 5 of 88 patients, and 2 of 25 patients were found to have SDHD mutations, and only 2 of 17 later developed metastatic disease (4, 5, 7).
Pathogenic variants in SDHC (on chromosome 1q23.3) and SDHA (on chromosome 5p15.33) contributing to PPGL are much less common, with estimates of lifetime penetrance for PPGL at 8.3% and 1.7%, respectively (52). While metastatic disease risk is estimated to be low for SDHC mutations, metastatic risk in SDHA is 30-66% (26). Bausch et al. reported that among 177 pediatric PPGL patients, there was one patient who carried pathogenic variants in each of these genes: a 12 years male with head and neck PGL (SDHC) who did not develop metastases and a 15 years male with PCC (SDHA) who died within 13 years of follow-up (4). Interestingly, SDHC and SDHD share structural (anchoring the SDH complex to the inner mitochondrial membrane) and bioenergetic roles (the SDHC-SDHD dimer transfers electrons from iron-sulfur clusters of SDHB to ubiquinone in the electron transport chain) as well as both commonly presenting as head and neck PGLs (18, 44).
International consensus guidelines were published for PPGL screening in asymptomatic SDHx carriers (SDHA, SDHB, SDHC, and paternally-inherited SDHD variants), which should start in childhood at age 6-10 years for SDHB carriers and otherwise at 10-15 years and include clinical evaluation with symptom assessment, physical examination, and measurement of blood pressure; biochemical testing with plasma or urine metanephrines; and magnetic resonance imaging (MRI) studies of the head and neck as well as of the thorax, abdomen, and pelvis (48). If the initial evaluation is negative, follow-up should include annual clinical evaluation, biochemical testing every 2 years, and MRI every 2-3 years. Functional imaging by PET/CT was included for initial screening in adults, but no recommendation was made for screening by functional imaging in children (48).
Von Hippel-Lindau Syndrome
Due to pathogenic variants in VHL (on chromosome 3p25.3), VHL syndrome is due to loss of function of this tumor suppressor gene that encodes for an E3 ubiquitin ligase that targets HIF-2α for degradation (53). VHL is a cluster 1B gene and is associated with a noradrenergic biochemical phenotype. PPGL develops in about 10-25% of VHL patients, typically presenting as PCC, though sympathetic and parasympathetic PGLs can also be seen, and the risk of metastatic disease is 5-8% (26, 54). Other tumors seen include angiomas and hemangioblastomas of the retina and central nervous system, renal and pancreatic cysts, RCC, endolymphatic sac tumor, pancreatic neuroendocrine tumor, and papillary cystadenoma of the epididymis or broad ligament (54). Genotype-phenotype correlations have been reported in VHL: type 2 VHL, associated with higher risk of PCC, is usually due to missense variants, whereas type 1 VHL, with a lower risk for PCC, includes truncating variants and exon deletions as well as missense mutations (53, 54). In about 20% of cases, patients will have a de novo mutation; interestingly, a study of Spanish PPGL patients found a much higher de novo rate of 60% overall and 50% for the pediatric group, though the authors attributed this to sample size (29, 54). VHL is the most common genetic cause of pediatric PPGL and tends to present the earliest at an average age of 11-12 years (4, 5, 7, 8, 22), in contrast to those syndromes arising from pathogenic variants in cluster 2 – specifically, MEN-2 (14-20 years) and NF1 (16-17 years); PCC may be the first manifestation of VHL disease (4, 5, 7, 8, 22). While screening recommendations for VHL may vary, PCC has been identified in a girl as young as 2 years, so measurement of blood pressure at each medical appointment and annual plasma free metanephrines or 24-hour urine fractionated metanephrines starting as early as 2 years but no later than 5 years has been proposed (54). Other aspects of screening for VHL-associated tumors, including retinal examination, audiology evaluation, and MRI studies are not discussed here but should be included as part of the comprehensive care for these patients.
EPAS1 Gain-of-Function Syndrome
One notable exception to the general rule of germline susceptibility is Pacak-Zhuang syndrome (PZS), characterized by polycythemia, PPGL, and duodenal somatostatinoma (55, 56). PZS is usually due to post-zygotic somatic mutations (rarely associated with germline inheritance) in the EPAS1 gene (on chromosome 2p21), encoding for the transcription factor HIF-2α (55, 56). This syndrome was first described in two female patients with congenital polycythemia and onset of PGL during adolescence (55, 56). In PZS, gain-of-function mutation results in impaired prolyl hydroxylation of HIF-2α, increasing its stability by interfering with ubiquitination by the VHL protein (pVHL). Transcriptional effects include expression of erythropoietin, resulting in polycythemia, and vascular endothelial growth factor (VEGF), contributing to a pro-angiogenic phenotype, and resulting in decreased expression of PNMT, characteristic of cluster 1 mutations (41, 55). This increased stability also affects differentiation of neural crest progenitor cells that may promote metastatic behavior such as facilitating the neuroendocrine-to-mesenchymal transition (41). Pamporaki et al. reported that 4 of 92 pediatric patients (4.3%) compared to 5 of 519 adult patients (1.0%) with PPGL were found to have a somatic mutation in EPAS1 from tumor tissue, emphasizing how frequently cluster 1 mutations, even post-zygotic mutations, are seen in the pediatric population (2). In another cohort, Redlich et al. identified 1 of 88 (1.6%) pediatric PPGL patients with an EPAS1 somatic mutation and clinical features of PZS (including abdominal PGL) but without evidence of metastatic disease (7). A study of 7 patients with PZS showed that PGLs were multiple and recurrent in all patients but only 2 of 7 (29%) had metastatic disease (57). These patients highlight the benefit of somatic – in addition to germline – genetic testing when patient tumor tissue is available, as it may provide additional clues to the underlying etiology and raise awareness of associated clinical features and risk of multiplicity, recurrence, and malignancy.
Neurofibromatosis Type 1
Mutations in the NF1 gene (encoding for neurofibromin), located on chromosome 17q11.2, are responsible for neurofibromatosis type 1. Though both are in cluster 2, in contrast to the RET gene, NF1 is a tumor suppressor that negatively regulates the RAS-MAPK signaling pathway. Partly due to historical challenges in sequencing related to the size of this gene, clinical diagnostic criteria were developed at the National Institutes of Health and include the presence of café-au-lait macules, axillary or inguinal freckling, neurofibromas, optic glioma, Lisch nodules, distinctive bone lesions, and an affected first-degree relative (58). While an often-cited retrospective analysis showed that PCCs were seen in 0.1 to 5.7% of patients with NF-1, 3.3 to 13.0% of NF-1 patients were found to have PCC on autopsy (59). A prospective study of adults with NF-1 identified PCC in 7.7% of patients, suggesting that these tumors are often found incidentally in asymptomatic patients (60). Unilateral adrenal disease is most common (78-84%), with bilateral disease seen in only 9.6 to 16.6% of patients with PCC, and metastatic disease is seen in up to about 11.5% of patients, though these estimates are derived mostly from adult NF-1 patients. Extra-adrenal locations include the organ of Zuckerkandl, abdominal sympathetic chain, and bladder (59–61). Adrenal findings in NF-1 include benign adrenal nodules, adrenal hyperplasia, and PCCs, which may be found histologically as composite tumors (with ganglioneuroma, ganglioneuroblastoma, neuroblastoma, and mixed neuroendocrine-neural tumor); GIST, renal oncocytoma, and carcinoid tumors have also been reported in NF-1 (59, 60). By contrast, pediatric PPGL in NF-1 is far less commonly seen, with reports indicating that these patients comprise only 1-3% of pediatric PPGL cohorts and present as teenagers (15-17 years), though 3 of 8 patients developed metastatic disease (one of which had microscopic residual tumor on histopathological analysis) (2, 4, 7). An important consideration is that up to 50% of the cases of NF–1 arise de novo, another reason why the absence of a family history per se should not preclude NF–1 from the differential diagnosis in the presence of suggestive clinical findings (59).
Multiple Endocrine Neoplasia Type 2
MEN-2 is due to pathogenic variants in the RET gene (on chromosome 10q11.21), a cluster 2 proto-oncogene encoding for a receptor tyrosine kinase that plays an important role in the development of neural crest cells (62). MEN-2A is associated with the combination of medullary thyroid cancer (MTC, > 90%), hyperparathyroidism (15-30%), and PCC (57%) and accounts for up to 95% of MEN-2 (including familial MTC [FMTC], a variant of MEN-2A), whereas MEN-2B constitutes about 5% of MEN-2 and is characterized by early and aggressive MTC, PCC (50%), mucosal neuromas, and a marfanoid habitus (63, 64). As a proto-oncogene, disease arises due to gain-of-function mutations in RET, often due to missense mutations at cysteine residues, such as at codons 609, 618, and 620 in exon 10 or 634 in exon 11, which result in MEN-2A (63, 64). In contrast, about 95% of individuals with MEN-2B have the p.M918T mutation in exon 16, and about 75% of patients have disease resulting from a de novo mutation (63, 64). Genotype-phenotype correlations are particularly important in MEN-2, and the 2015 Revision of the Guidelines for Management of Medullary Thyroid Cancer by the American Thyroid Association has classified RET variants into three risk categories on the basis of risk for MTC, which for the “high” and “highest” risk categories also correspond to those mutations for the greatest risk of PCC: "highest" (only the p.M918T mutation), "high" (p.A883F and p.C634 mutations), and "moderate" (other mutations) (63). With mutations of the cysteine residue at codon 634, the risk of PCC, hyperparathyroidism, and cutaneous lichen amyloidosis is higher, and specifically for the p.C634R mutation, there is a higher risk of metastatic MTC at diagnosis (64). For RET mutations at codons 918, 883, and 634, screening for PCC should begin at age 11 years, but for those moderate risk mutations, screening may begin at age 16 years (63). PCCs in MEN-2 are often multicentric and bilateral, developing in the setting of diffuse nodular adrenal medullary hyperplasia (63). Metastatic risk of PCC is < 5% (26). Among 6 studies of pediatric PPGL patients with MEN-2 due to a RET mutation, ten patients were identified (representing 0.6-3.3% of each cohort except for 13.0% in one study), all with PCC Age at PCC diagnosis ranged from 14-20 years; eight patients had bilateral or recurrent disease, and no patients were reported to have metastatic disease (2, 4, 5, 7, 8, 29). Though not discussed here, other aspects of MEN-2, including hyperparathyroidism and MTC, require clinical consideration and management (63).
Imaging
Anatomic imaging is essential to the diagnosis for any patient being evaluated for PPGL and is typically performed by CT or MRI. In pediatric patients, high signal-intensity T2-weighted MRI is preferable over CT to reduce radiation exposure and may be better suited to identify extra-adrenal tumors, invasion into the spinal canal, and involvement of major vessels (2, 9, 11, 65). Children may not tolerate MRI as well as adults and may require sedation, so clinicians should be mindful regarding the choice of sedative so as not to cause precipitous catecholamine release; medications such as opioids (except for fentanyl) can cause histamine release, which may stimulate catecholamine secretion (66).
Anatomical imaging is used for tumor localization and detection of metastases after positive biochemical testing but also for screening, as non-functional tumors may be negative on biochemical evaluation, such as in patients with SDHx mutations or those with parasympathetic PGLs arising in the head and neck (48). For follow-up imaging in the setting of metastases, CT and MRI can be complementary, as CT is better for visualizing lung lesions, and MRI is better for liver lesions (26).
Functional imaging provides insight into the molecular features of PPGLs by use of different radiopharmaceuticals, which is especially informative in metastatic disease. “Theranostics” refers to this unification of therapeutic and diagnostic utility with the same radiopharmaceutical platform, for example using 123I-labeled metaiodobenzylguanidine (MIBG) scintigraphy to identify PPGLs which can then be treated by 131I-MIBG (67). MIBG is an analog of norepinephrine and enters tumor cells via the norepinephrine transporter (42, 68). Medications such as over-the-counter cough and cold treatments, calcium channel blockers, labetalol, and tricyclic antidepressants can interfere with MIBG uptake and should be avoided before treatment. Special preparation is also required to reduce accumulation of radioiodine in the thyroid (68). While highly specific, MIBG may not be as sensitive as other functional imaging modalities, particularly in hereditary PPGL, and especially in those with SDHx mutations. Additionally, sensitivity is higher for PCC than PGL (88% vs 67%, respectively), and in metastatic PPGL, per-lesion sensitivity is < 60% (42). Despite these limitations in the pediatric population with metastatic PPGL, MIBG may still have a useful theranostic role in patients where 123I-MIBG avidity is observed in all lesions.
The other relevant application of the theranostic paradigm is for tumors expressing somatostatin receptors – especially type 2 (SSTR2) – that bind to somatostatin analogs (SSAs) linked to a chelator (DOTA) for the radionuclide. Typically, 68Ga- or 64Cu-DOTA-SSA is used for imaging, and 90Y- and 177Lu-DOTA-SSAs deliver the radiation dose to the sites of tumors, with the latter referred to as peptide receptor radionuclide therapy (PRRT) (42, 69). 68Ga-DOTATATE is commonly used since DOTATATE binds preferentially to SSTR2 over other somatostatin receptors, as opposed to DOTATOC or DOTANOC (42). 68Ga-DOTATATE is particularly useful for cluster 1A mutations (especially SDHx), HNPGLs, metastatic PPGL, and pediatric PPGL (42). Among nine pediatric patients with SDHx mutations, both 18F-FDG and 68Ga-DOTATATE detected lesions in all of the patients but 68Ga-DOTATATE had superior sensitivity on a per-lesion basis (94% compared to 79% on 18F-FDG) and was also more sensitive than anatomic imaging by CT or MRI with contrast enhancement (74%). 68Ga-DOTATATE was especially superior to CT/MRI for mediastinal lesion detection and superior to 18F-FDG for detection of adrenal and liver lesions, though 18F-FDG and CT/MRI outperformed 68Ga-DOTATATE for detection of other abdominal lesions (Figure 2) (70). These data offer a compelling case for performing 68Ga-DOTATATE PET/CT or 68Ga-DOTATATE PET/MRI with contrast enhancement to detect such abdominal lesions. Currently both functional and anatomic imaging are required for staging and for assessing treatment response in pediatric metastatic PPGL patients and, therefore, simultaneous PET/MRI may be considered in this cohort due to decreased radiation exposure, fewer instances requiring sedation or general anesthesia, fewer appointments, and simultaneous imaging with two advanced diagnostic imaging techniques (whole-body PET and MRI) (71). Recommended imaging modalities for different types of PPGL based on genetic and clinical features are summarized in Table 3. A 2015 meta-analysis by Han et al. pooled patients of unknown genetic background, finding a significantly superior detection rate of 68Ga-DOTA-conjugated somatostatin receptor-targeting peptide (68Ga-DOTA-SST) PET (93%) as compared to other functional imaging modalities (72). Therefore, in cases where a genetic etiology is not known, 68Ga-DOTATATE PET may be performed.
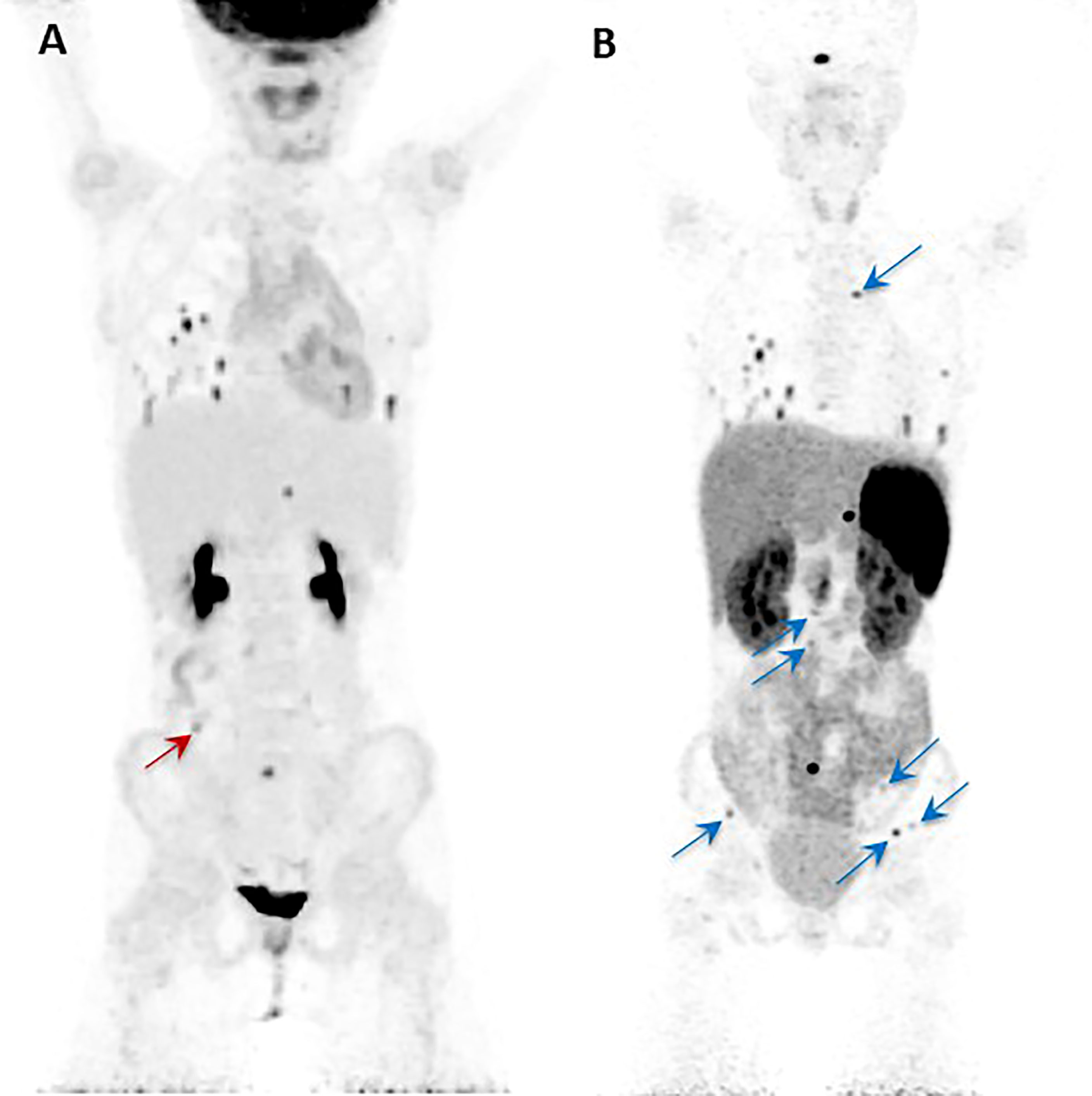
Figure 2 Anterior maximal intensity projection (MIP) images of the 18F-FDG PET/CT (A) and 68Ga-DOTATATE PET/CT (B) studies of a 10-year-old SDHB positive girl. She was diagnosed initially with metastatic disease at the age of 8 years. Her right paraaortic, retroperitoneal primary paraganglioma was surgically resected. On presentation to our institution, the progression of her disease was demonstrated by metastatic lesions in bone, lungs, and abdomen as shown in the images (A, B). The single red arrow on image (A) indicates the one lesion (abutting bowel) localized by 18F-FDG PET/CT that is not visualized by the 68Ga-DOTATATE PET/CT. Similarly, all the additional lesions (transverse process of T4 spine, L2-L5 vertebral bodies, left ilium, and left and right iliac wings) localized by the 68Ga-DOTATATE PET/CT (blue arrows) are not visualized by 18F-FDG PET/CT (B). This figure was adapted from the figure that was initially published as Figure 2 by Jha et al. (70).
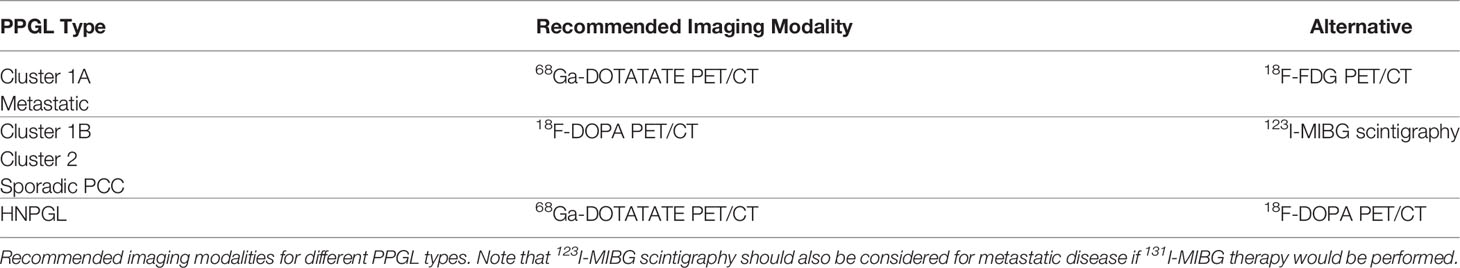
Table 3 Functional imaging recommendations for pediatric PPGL.
18F-fluorodihydroxyphenylalanine (18F-FDOPA) enters cells via the large neutral amino acid transporter (LAT-1) and enters into the catecholamine synthesis pathway (42). Preparation for 18F-FDOPA PET/CT involves fasting and administration of carbidopa to block decarboxylation of DOPA to dopamine, improving uptake in target tissue. There are no known interfering medications (42). 18F-FDOPA PET/CT is particularly helpful for imaging PPGLs in clusters 1B (VHL and PZS) and 2 (MEN-2 and NF-1) and has an advantage over MIBG in that normal adrenal tissue has lower uptake, increasing the sensitivity to detect nonmetastatic PCC (94% in patients of known genetic background and up to 100% in patients with apparently sporadic nonmetastatic PCC); indeed, the detection of metastatic PPGL may also vary on the basis of the genetic background with greater sensitivity in non-SDHx PPGL (42, 73).
18F-FDG is a radiolabeled form of glucose and is taken up by glucose transporters (particularly GLUT-1) and enters into the glycolytic pathway (42). Preparation for 18F-FDG involves fasting, controlling hyperglycemia, and avoidance of ambient cold temperature to prevent artifacts from glucose metabolism in thermogenic brown fat (42, 74). The sensitivity of 18F-FDG in detection of metastatic PPGL is superior to MIBG (usually > 80%), particularly in SDHx patients, with sensitivity in metastatic disease for SDHx patients ranging from 83-92% compared to 62% for non-SDHx (42). Furthermore, while there are no studies of direct comparisons, GIST appears to be better appreciated on 18F-FDG compared to 68Ga-DOTATATE, and therefore, some SDHx patients would benefit from 18F-FDG in addition to 68Ga-DOTATATE PET/CT scan when GIST is suspected (75, 76).
Management
After biochemical studies and tumor localization by imaging, definitive treatment of PPGL should be addressed. In addition to surgery of the primary tumor, when appropriate, patients with metastatic disease require other treatment modalities to address disease at sites of metastasis, such as chemotherapy, radiotherapy, ablative therapy, or other targeted therapies, such as somatostatin analogs or small molecule inhibitors (21). Clinical trials for pediatric patients with advanced/metastatic PPGL that are recruiting, active, or approved are listed in Table 4.
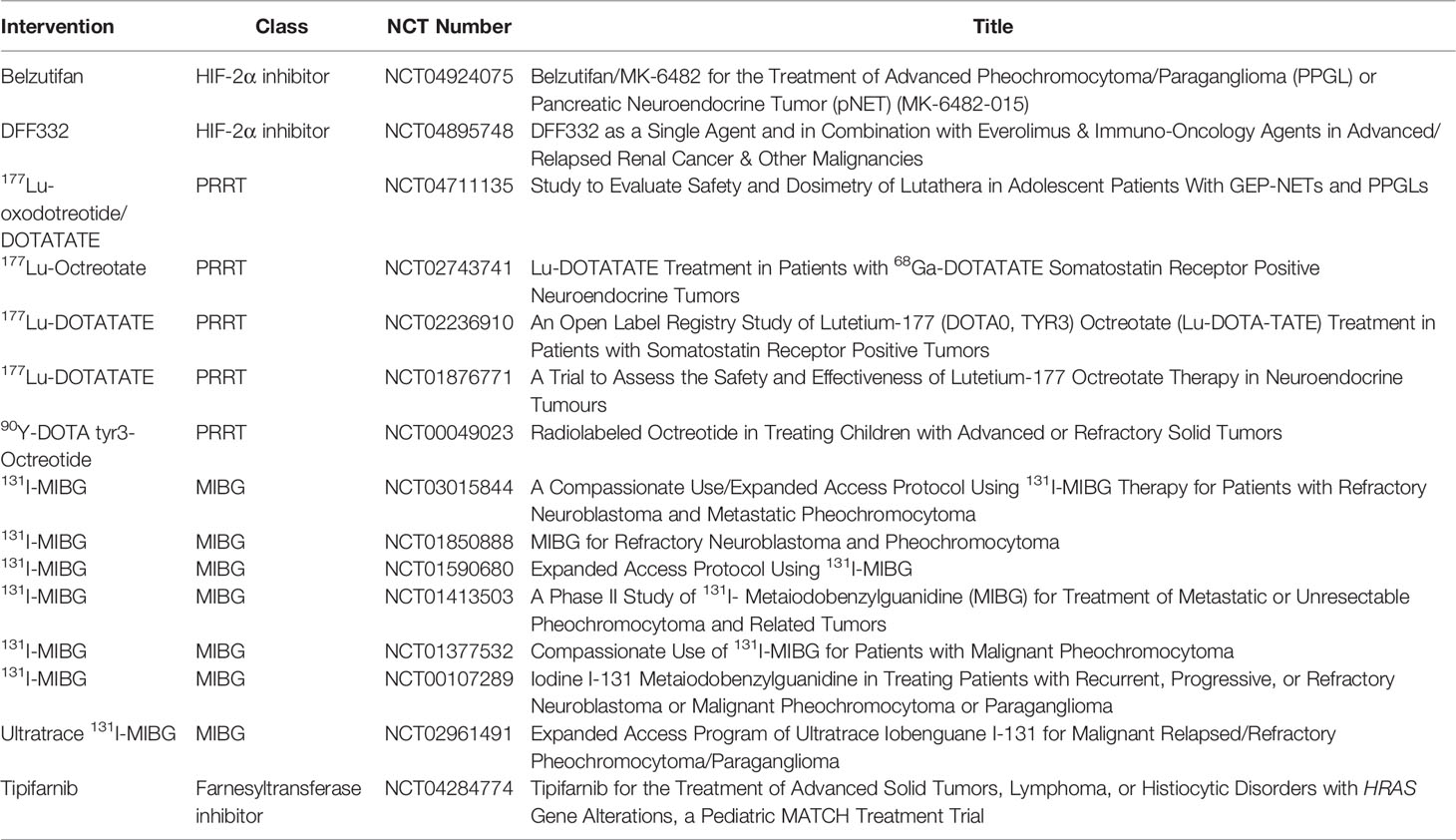
Table 4 Clinical trials for metastatic PPGL therapies in pediatric patients that are recruiting, active, or approved.
The first medication clinicians should consider in pediatric PPGL is an α1-adrenergic antagonist, such as the non-competitive long-acting phenoxybenzamine or competitive short-acting drugs, typically doxazosin. Alpha-adrenergic antagonists help to reduce the symptoms associated with catecholamine excess, improve hypertension, and prepare patients for therapeutic interventions, therefore α-adrenergic antagonists should be started in all PPGL patients (1, 26). Blood pressure and heart rate should be monitored, and β-adrenergic blockade should be considered in patients who have persistent tachycardia but only after starting α-adrenergic blockade for at least 2 to 3 days to avoid the effect of unopposed α-adrenergic stimulation (16). To prevent complications related to catecholamine release during treatment, patients should start α-adrenergic blockade 7 to 14 days before surgery or before non-surgical treatments, such as chemotherapy or radiotherapy, as these may all result in catastrophic surges of stored catecholamines (26). Competitive inhibition of tyrosine hydroxylase by α-methyl-L-tyrosine (metyrosine, or Demser®) results in decreased synthesis of all downstream catecholamines and can be helpful for patients with highly elevated levels of catecholamines to improve hemodynamic stability before and after surgery but must be used cautiously as it is contraindicated in patients with severe depression and suicidal ideation (17, 26). Calcium channel blockers, especially amlodipine, can also be helpful in controlling hypertension in pediatric patients with PPGL (77).
Surgical interventions should be considered even for patients with metastatic disease, to reduce the tumor burden contributing to catecholamine excess and to improve radiopharmaceutical uptake in remaining metastatic lesions (26). Genetic testing preoperatively can be helpful to inform the surgical approach, as cortical-sparing adrenalectomy is preferred in younger patients to reduce the risk of adrenal insufficiency (and requirement for exogenous steroids) and does not seem to increase the risk of recurrence for patients with mutations in VHL, RET, or NF1, but in patients with higher risk for metastatic disease, such as those with SDHB mutations, more extensive resection may be needed (21).
The approach to the patient with metastatic disease depends on the extent of disease and the rate of progression. For patients with very extensive disease or rapid progression, chemotherapy should be tried first, with first-line therapy consisting of cyclophosphamide, vincristine, and dacarbazine (CVD); temozolomide as monotherapy may also be considered as an alternative or a second-line agent in these cases (78). The impact of SDH deficiency on chemoresistance, as previously mentioned, suggests that PARP inhibitors such as olaparib may be useful in combination with temozolomide, and a clinical trial is currently recruiting to evaluate this combination in adults (NCT04394858). Hypoxia-inducible factors increase expression of growth factors such as VEGF, platelet-derived growth factor (PDGF), and transforming growth factor α (TGFα), which bind to their respective receptor tyrosine kinases (53). Therefore, additional therapies to consider include tyrosine kinase inhibitors with anti-VEGF activity, such as sunitinib (53). As a last resort, additional approaches to consider include immunotherapy with checkpoint inhibitors (e.g., pembrolizumab or combination nivolumab-ipilimumab) in patients with cluster 1 mutations (consequent to impaired immune recognition related to pseudohypoxia) or mTORC1 inhibitors (e.g., everolimus) in patients with cluster 2 mutations (mTORC1 regulates cellular activity downstream of kinase signaling pathways), but additional studies are needed in children with metastatic PPGL (78).
For patients with less rapid progression or smaller tumor burden, systemic radiotherapy can be particularly useful as part of a personalized approach when lesions have avidity on imaging scans for the corresponding diagnostic radionuclides. For patients with DOTA-SSA-avid lesions, as is often seen with cluster 1 mutations, PRRT with 177Lu-DOTA-SSA (“hot SSA”) or SSAs that do not carry radiopharmaceuticals (referred to as “cold SSAs”) may also be helpful, especially for SDHx patients with DOTATATE-avid lesions, and additional clinical trials are evaluating these in children (Table 4). Conventional or high-specific activity (HSA) 131I-MIBG (referred to as Ultratrace™, or Azedra®) may be particularly useful for patients with cluster 2 mutations who are more likely to have adrenal disease but requires positive findings on diagnostic 123I-MIBG scan before use (69). When considering systemic radiotherapy, clinicians should ideally perform both 123I-MIBG and 68Ga-DOTATATE scans to determine which radiopharmaceutical best shows all the metastatic lesions in order to guide which radiotherapy should be used to treat the patient (79).
Locoregional approaches may be helpful for certain aspects of metastatic disease. External radiation therapy can be useful for head and neck primary disease due to a more favorable risk profile as compared to surgical intervention and for rapidly-growing metastatic tumors, such as in bone, to provide symptomatic relief (1, 26, 78). Treatment of a primary HNPGL by external beam radiation therapy, therefore, may be helpful for those with SDHD mutations. Bone metastases are also treated with bisphosphonates or denosumab (3, 26). Interventional procedures such as radiofrequency ablation, cryoablation, or ethanol injection can be used in cases for treatment of a single metastatic lesion or oligo-metastases (3, 26). Furthermore, patients with inoperable primary tumors can be considered for potential treatment with a cold-SSA if lesions are 68Ga-DOTATATE-avid, as reported in an adult SDHB patient with pterygopalatine fossa PGL, who was successfully controlled for 36 months with octreotide (80).
Belzutifan is one of the newer targeted therapies that specifically inhibits HIF-2α and has been approved to treat VHL-associated tumors (81). Given the central role that HIF-2α plays in hypoxia signaling, this therapy could be incredibly impactful for patients with cluster 1 mutations. A recent case report demonstrated the efficacy of belzutifan in treating multiple PGLs in an adolescent patient with PZS, with improvement in biochemical (normetanephrine and chromogranin A) and hematologic (erythropoietin and hemoglobin) markers and reduced tumor size on imaging. Belzutifan was well-tolerated in this patient with minimal side effects, most notably, anemia that did not require transfusion (81).
While many studies examining prognosis, disease progression, and outcomes in PPGL are focused on adults or include both adults and children, it seems that the most important prognostic factor for disease-free survival (DFS) is extent of surgical resection of the primary tumor (for pediatric PPGL: 45.6% for complete resection vs. 24.1% for incomplete resection, p < 0.001) (7, 21, 30). Redlich et al. described other significant differences in 10-year DFS in pediatric patients with PPGL, including the presence of metastatic disease (29.6% vs. 43.5% in those without metastatic disease, p = 0.014), and PGL (36.6% vs. 47.8% in PCC, p = 0.039), whereas the presence of an SDHB mutation was associated with lower DFS but did not reach statistical significance (45.1% in non- SDHB vs. 24.4% in SDHB, p = 0.063) (7). Outcomes in pediatric patients with SDHB mutations have been well-described by Jochmanova et al., and specific outcomes are also discussed above (23).
Perspectives and Future Directions
This review highlights important features to consider in pediatric patients with PPGL, with an emphasis on metastatic disease. Pediatric PPGL is overwhelmingly due to an underlying genetic predisposition, especially from cluster 1 genes associated with pseudohypoxia. The central role of hypoxia signaling by HIF-2α in these patients results in a pro-metastatic phenotype and disruption of normal development of chromaffin cell precursors that can result in increased susceptibility to PPGL in multiple tissues. Indeed, the biochemical and secretory features could be described as more primitive or underdeveloped in pediatric patients with cluster 1 mutations predisposing them to PPGL. All pediatric PPGL patients require lifelong surveillance and monitoring, even those with cluster 2 mutations who may not present with PPGL until their teenage years. To the extent possible, these rare and challenging patients should be managed at centers of expertise with multidisciplinary teams that can address the different aspects of care.
Rather than viewing the pediatric patient with a genetic mutation as having an immutable risk factor for PPGL, the knowledge of this risk can guide clinicians to develop an appropriate screening and management plan with their patients and families, especially since the penetrance of PPGL is incomplete for all of the susceptibility genes (26). Genetic counseling can play an important supportive role in addition to contributing valuable information about risk of disease. Reducing anxiety by dispelling misconceptions about genetic risks as well as emphasizing preventive strategies and forming a screening plan to help reduce anxiety and empower patients and families is a key part of the approach to genetic counseling as shown in a study of patients with SDHx mutations. Out of 164 patients, only 2 (1.2%) had increased anxiety in response to genetic testing results, and only 1 (0.6%) did not proceed with any preventive measures nor any screening for her positive children (82).
Though not as prevalent a cause of metastatic disease as cluster 1 mutations, additional studies of targeted therapies for patients with cluster 2 mutations are needed in the pediatric population as our increased understanding of cellular pathways has generated specific therapeutic targets. Mweempwa et al. demonstrated efficacy of a selective RET inhibitor (selpercatinib) in metastatic PCC due to an activating gene fusion of RET-SEPTIN9 in an adult with recurrent PCC with biopsy-confirmed metastatic disease to the lungs and liver with negative urinary metanephrines but highly elevated chromogranin A (33,710 µg/L). After 12 weeks of therapy with selpercatinib, chromogranin A had decreased to 598 µg/L, and pulmonary and hepatic tumors demonstrated reduction in size on CT (46% in the sum of diameters of lesions), raising the possibility that this targeted therapy may also be useful for pediatric patients with activating RET mutations; however additional studies are needed (83). Similarly, targeted therapy directed by the genetic background may also inform the approach to patients with NF-1. Selumetinib selectively inhibits MEK in the RAS-MAPK pathway, and in children with inoperable plexiform neurofibromas, treatment with selumetinib resulted in a reduction in tumor volume (median reduction of 27.9%) and progression-free survival of 84% (compared to 15% in natural history controls) at 3 years from treatment initiation. MEK inhibitors have also shown responses in other NF1-related tumors (e.g., optic pathway glioma), suggesting that PPGLs driven by NF1 mutations might also be considered for this targeted therapy (84).
PPGLs have been described as the tumors that are the most highly genetically determined of any human tumor, and in children, around 80% have a germline predisposition. When accounting for those with additional somatic changes (e.g., EPAS1 gain-of-function mutations), an even higher proportion is realized. Germline genetic testing to determine an underlying cause of PPGL is mandatory, especially in children with metastatic disease, but somatic tumor genetic testing has clear benefits as well and may identify new genes and targetable mechanisms that can personalize the care of these vulnerable patients.
Author Contributions
MK drafted and revised the initial manuscript, figures, and tables. MN aided in conceptualizing and contributing to the manuscript. He also revised the manuscript. AJ aided in conceptualizing, contributing, and revising the manuscript. KP conceived, conceptualized, critically reviewed the manuscript for intellectual content, and aided in revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported, by the Intramural Research Program of the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, Diagnosis, Management and Future Directions of Research of Phaeochromocytoma and Paraganglioma: A Position Statement and Consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens (2020) 38(8):1443–56. doi: 10.1097/HJH.0000000000002438
2. Pamporaki C, Hamplova B, Peitzsch M, Prejbisz A, Beuschlein F, Henri JLM, et al. Characteristics of Pediatric vs Adult Pheochromocytomas and Paragangliomas. J Clin Endocrinol Metab (2017) 102(4):1122–32. doi: 10.1210/jc.2016-3829
3. Angelousi A, Kassi E, Zografos G, Kaltsas G. Metastatic Pheochromocytoma and Paraganglioma. Eur J Clin Invest (2015) 45(9):986–97. doi: 10.1111/eci.12495
4. Bausch B, Wellner U, Bausch D, Schiavi F, Barontini M, Sanso G, et al. Long-Term Prognosis of Patients With Pediatric Pheochromocytoma. Endocr Relat Cancer (2014) 21(1):17–25. doi: 10.1530/ERC-13-0415
5. Petenuci J, Guimaraes AG, Fagundes GFC, Benedetti AFF, Afonso ACF, Pereira MAA, et al. Genetic and Clinical Aspects of Paediatric Pheochromocytomas and Paragangliomas. Clin Endocrinol (Oxf) (2021) 95(1):117–24. doi: 10.1111/cen.14467
6. Kittah NE, Gruber LM, Bancos I, Hamidi O, Tamhane S, Iñiguez-Ariza N, et al. Bilateral Pheochromocytoma: Clinical Characteristics, Treatment and Longitudinal Follow-Up. Clin Endocrinol (Oxf) (2020) 93(3):288–95. doi: 10.1111/cen.14222
7. Redlich A, Pamporaki C, Lessel L, Frühwald MC, Vorwerk P, Kuhlen M, et al. Pseudohypoxic Pheochromocytomas and Paragangliomas Dominate in Children. Pediatr Blood Cancer (2021) 68(7):e28981. doi: 10.1002/pbc.28981
8. Park H, Kim MS, Lee J, Kim JH, Jeong BC, Lee S, et al. Clinical Presentation and Treatment Outcomes of Children and Adolescents With Pheochromocytoma and Paraganglioma in a Single Center in Korea. Front Endocrinol (Lausanne) (2020) 11:610746. doi: 10.3389/fendo.2020.610746
9. Havekes B, Romijn JA, Eisenhofer G, Adams K, Pacak K. Update on Pediatric Pheochromocytoma. Pediatr Nephrol (2009) 24(5):943–50. doi: 10.1007/s00467-008-0888-9
10. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell (2017) 31(2):181–93. doi: 10.1016/j.ccell.2017.01.001
11. Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, et al. Pheochromocytoma and Paraganglioma in Children: A Review of Medical and Surgical Management at a Tertiary Care Center. Pediatrics (2006) 118(3):1109–17. doi: 10.1542/peds.2005-2299
12. Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, et al. Pheochromocytomas in Von Hippel-Lindau Syndrome and Multiple Endocrine Neoplasia Type 2 Display Distinct Biochemical and Clinical Phenotypes. J Clin Endocrinol Metab (2001) 86(5):1999–2008. doi: 10.1210/jcem.86.5.7496
13. Eisenhofer G, Huynh TT, Elkahloun A, Morris JC, Bratslavsky G, Linehan WM, et al. Differential Expression of the Regulated Catecholamine Secretory Pathway in Different Hereditary Forms of Pheochromocytoma. Am J Physiol Endocrinol Metab (2008) 295(5):E1223–33. doi: 10.1152/ajpendo.90591.2008
14. Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, et al. A HIF1alpha Regulatory Loop Links Hypoxia and Mitochondrial Signals in Pheochromocytomas. PLoS Genet (2005) 1(1):72–80. doi: 10.1371/journal.pgen.0010008
15. Crona J, Taieb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev (2017) 38(6):489–515. doi: 10.1210/er.2017-00062
16. Pacak K. Preoperative Management of the Pheochromocytoma Patient. J Clin Endocrinol Metab (2007) 92(11):4069–79. doi: 10.1210/jc.2007-1720
17. Nazari MA, Rosenblum JS, Haigney MC, Rosing DR, Pacak K, et al. Pathophysiology and Acute Management of Tachyarrhythmias in Pheochromocytoma: JACC Review Topic of the Week. J Am Coll Cardiol (2020) 76(4):451–64. doi: 10.1016/j.jacc.2020.04.080
18. Gupta G, Pacak K, Committee AAS. Precision Medicine: An Update on Genotype/Biochemical Phenotype Relationships in Pheochromocytoma/Paraganglioma Patients. Endocr Pract (2017) 23(6):690–704. doi: 10.4158/EP161718.RA
19. Choat H, Derrevere K, Knight L, Brown W, Mack EH, et al. SDHB-Associated Paraganglioma in a Pediatric Patient and Literature Review on Hereditary Pheochromocytoma-Paraganglioma Syndromes. Case Rep Endocrinol (2014) 2014:502734. doi: 10.1155/2014/502734
20. Olson SW, Yoon S, Baker T, Prince LK, Oliver D, Abbott KC, et al. Longitudinal Plasma Metanephrines Preceding Pheochromocytoma Diagnosis: A Retrospective Case-Control Serum Repository Study. Eur J Endocrinol (2016) 174(3):289–95. doi: 10.1530/EJE-15-0651
21. Hamidi O. Metastatic Pheochromocytoma and Paraganglioma: Recent Advances in Prognosis and Management. Curr Opin Endocrinol Diabetes Obes (2019) 26(3):146–54. doi: 10.1097/MED.0000000000000476
22. Barontini M, Levin G, Sanso G. Characteristics of Pheochromocytoma in a 4- to 20-Year-Old Population. Ann N Y Acad Sci (2006) 1073:30–7. doi: 10.1196/annals.1353.003
23. Jochmanova I, Abcede AMT, Guerrero RJ S, Malong CLP, Wesley R, Huynh T, et al. Clinical Characteristics and Outcomes of SDHB-Related Pheochromocytoma and Paraganglioma in Children and Adolescents. J Cancer Res Clin Oncol (2020) 146(4):1051–63. doi: 10.1007/s00432-020-03138-5
24. Zhang JJ, Liu T, Peng F. Primary Paraganglioma of the Lung: A Case Report and Literature Review. J Int Med Res (2012) 40(4):1617–26. doi: 10.1177/147323001204000442
25. Liao W, Ding ZY, Zhang B, Chen L, Li G X, Wu JJ, et al. Primary Functioning Hepatic Paraganglioma Mimicking Hepatocellular Carcinoma: A Case Report and Literature Review. Medicine (Baltimore) (2018) 97(17):e0293. doi: 10.1097/MD.0000000000010293
26. Nolting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized Management of Pheochromocytoma and Paraganglioma. Endocr Rev (2022) 43(2):199–239. doi: 10.1210/endrev/bnab019
27. Berends AMA, Buitenwerf E, de Krijger RR, Veeger N, van der Horst-Schrivers ANA, Links TP, et al. Incidence of Pheochromocytoma and Sympathetic Paraganglioma in the Netherlands: A Nationwide Study and Systematic Review. Eur J Intern Med (2018) 51:68–73. doi: 10.1016/j.ejim.2018.01.015
28. Flynn JT, Kaelber DC, Baker-Smith CM, Blowey D, Carroll AE, Daniels SR, et al. Clinical Practice Guideline for Screening and Management of High Blood Pressure in Children and Adolescents. Pediatrics (2017) 140(3):7–18 7–18. doi: 10.1542/peds.2017-3035
29. Cascon A, Inglada-Perez L, Comino-Mendez I, de Cubas AA, Leton R, Mora J, et al. Genetics of Pheochromocytoma and Paraganglioma in Spanish Pediatric Patients. Endocr Relat Cancer (2013) 20(3):L1–6. doi: 10.1530/ERC-12-0339
30. Ciftci AO, Tanyel FC, Senocak ME, Buyukpamukcu N. Pheochromocytoma in Children. J Pediatr Surg (2001) 36(3):447–52. doi: 10.1053/jpsu.2001.21612
31. Chen Y, Hodin RA, Pandolfi C, Ruan DT, McKenzie TJ. Hypoglycemia After Resection of Pheochromocytoma. Surgery (2014) 156(6):1404–8; discussion 1408-9. doi: 10.1016/j.surg.2014.08.020
32. Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, et al. A Current Review of the Etiology, Diagnosis, and Treatment of Pediatric Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab (2010) 95(5):2023–37. doi: 10.1210/jc.2009-2830
33. Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of Plasma Free Metanephrines for Detecting Childhood Pheochromocytoma. J Clin Endocrinol Metab (2002) 87(5):1955–60. doi: 10.1210/jcem.87.5.8446
34. Eisenhofer G, Peitzsch M, Kaden D, Langton K, Mangelis A, Pamporaki C, et al. Reference Intervals for LC-MS/MS Measurements of Plasma Free, Urinary Free and Urinary Acid-Hydrolyzed Deconjugated Normetanephrine, Metanephrine and Methoxytyramine. Clin Chim Acta (2019) 490:46–54. doi: 10.1016/j.cca.2018.12.019
35. Eisenhofer G, Prejbisz A, Peitzsch M, Pamporaki C, Masjkur J, Rogowski-Lehmann N, et al. Biochemical Diagnosis of Chromaffin Cell Tumors in Patients at High and Low Risk of Disease: Plasma Versus Urinary Free or Deconjugated O-Methylated Catecholamine Metabolites. Clin Chem (2018) 64(11):1646–56. doi: 10.1373/clinchem.2018.291369
36. Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein F, et al. Plasma Methoxytyramine: Clinical Utility With Metanephrines for Diagnosis of Pheochromocytoma and Paraganglioma. Eur J Endocrinol (2017) 177(2):103–13. doi: 10.1530/EJE-17-0077
37. Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma Methoxytyramine: A Novel Biomarker of Metastatic Pheochromocytoma and Paraganglioma in Relation to Established Risk Factors of Tumour Size, Location and SDHB Mutation Status. Eur J Cancer (2012) 48(11):1739–49. doi: 10.1016/j.ejca.2011.07.016
38. Zuber S, Wesley R, Prodanov T, Eisenhofer G, Pacak K, Kantorovich V, et al. Clinical Utility of Chromogranin A in SDHx-Related Paragangliomas. Eur J Clin Invest (2014) 44(4):365–71. doi: 10.1111/eci.12245
39. Hannah-Shmouni F, Pacak K, Stratakis CA. Metanephrines for Evaluating Palpitations and Flushing. JAMA (2017) 318(4):385–6. doi: 10.1001/jama.2017.5926
40. Rosas AL, Kasperlik-Zaluska AA, Papierska L, Bass BL, Pacak K, Eisenhofer G, et al. Pheochromocytoma Crisis Induced by Glucocorticoids: A Report of Four Cases and Review of the Literature. Eur J Endocrinol (2008) 158(3):423–9. doi: 10.1530/EJE-07-0778
41. Bechmann N, Eisenhofer G. Hypoxia-Inducible Factor 2alpha: A Key Player in Tumorigenesis and Metastasis of Pheochromocytoma and Paraganglioma? Exp Clin Endocrinol Diabetes (2021) 130: 282–289. doi: 10.1055/a-1526-5263
42. Carrasquillo JA, Chen CC, Jha A, Ling A, Lin FI, Pryma DA, et al. Imaging of Pheochromocytoma and Paraganglioma. J Nucl Med (2021) 62(8):1033–42. doi: 10.2967/jnumed.120.259689
43. Pang Y, Lu Y, Caisova V, Liu Y, Bullova P, Huynh TT, et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin Cancer Res (2018) 24(14):3423–32. doi: 10.1158/1078-0432.CCR-17-3406
44. Read AD, Bentley RE, Archer SL, Dunham-Snary KJ. Mitochondrial Iron-Sulfur Clusters: Structure, Function, and an Emerging Role in Vascular Biology. Redox Biol (2021) 47:102164. doi: 10.1016/j.redox.2021.102164
45. Richter S, Klink B, Nacke B, de Cubas AA, Mangelis A, Rapizzi E, et al. Epigenetic Mutation of the Succinate Dehydrogenase C Promoter in a Patient With Two Paragangliomas. J Clin Endocrinol Metab (2016) 101(2):359–63. doi: 10.1210/jc.2015-3856
46. Bayley JP, Bausch B, Rijken JA, van Hulsteijn LT, Jansen JC, Ascher D, et al. Variant Type Is Associated With Disease Characteristics in SDHB, SDHC and SDHD-Linked Phaeochromocytoma-Paraganglioma. J Med Genet (2020) 57(2):96–103. doi: 10.1136/jmedgenet-2019-106214
47. Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-Inducible Factor Signaling in Pheochromocytoma: Turning the Rudder in the Right Direction. J Natl Cancer Inst (2013) 105(17):1270–83. doi: 10.1093/jnci/djt201
48. Amar L, Pacak K, Steichen O, Akker SA, Aylwin SJB, Baudin E, et al. International Consensus on Initial Screening and Follow-Up of Asymptomatic SDHx Mutation Carriers. Nat Rev Endocrinol (2021) 17(7):435–44. doi: 10.1038/s41574-021-00492-3
49. King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, et al. Metastatic Pheochromocytoma/Paraganglioma Related to Primary Tumor Development in Childhood or Adolescence: Significant Link to SDHB Mutations. J Clin Oncol (2011) 29(31):4137–42. doi: 10.1200/JCO.2011.34.6353
50. Rijken JA, Niemeijer ND, Jonker MA, Eijkelenkamp K, Jansen JC, van Berkel A, et al. The Penetrance of Paraganglioma and Pheochromocytoma in SDHB Germline Mutation Carriers. Clin Genet (2018) 93(1):60–6. doi: 10.1111/cge.13055
51. Burnichon N, Mazzella JM, Drui D, Amar L, Bertherat J, Coupier I, et al. Risk Assessment of Maternally Inherited SDHD Paraganglioma and Phaeochromocytoma. J Med Genet (2017) 54(2):125–33. doi: 10.1136/jmedgenet-2016-104297
52. Benn DE, Zhu Y, Andrews KA, Wilding M, Duncan EL, Dwight T, et al. Bayesian Approach to Determining Penetrance of Pathogenic SDH Variants. J Med Genet (2018) 55(11):729–34. doi: 10.1136/jmedgenet-2018-105427
53. Maher ER, Neumann HP, Richard S. Von Hippel-Lindau Disease: A Clinical and Scientific Review. Eur J Hum Genet (2011) 19(6):617–23. doi: 10.1038/ejhg.2010.175
54. Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, et al. Von Hippel-Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res (2017) 23(12):e68–75. doi: 10.1158/1078-0432.CCR-17-0547
55. Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A Gain-of-Function Mutations in Paraganglioma With Polycythemia. N Engl J Med (2012) 367(10):922–30. doi: 10.1056/NEJMoa1205119
56. Lorenzo FR, Yang C, Ng Tang Fui M, Vankayalapati H, Zhuang Z, Huynh T, et al. A Novel EPAS1/HIF2A Germline Mutation in a Congenital Polycythemia With Paraganglioma. J Mol Med (Berl) (2013) 91(4):507–12. doi: 10.1007/s00109-012-0967-z
57. Darr R, Nambuba J, Del Rivero J, Janssen I, Merino M, Todorovic M, et al. Novel Insights Into the Polycythemia-Paraganglioma-Somatostatinoma Syndrome. Endocr Relat Cancer (2016) 23(12):899–908. doi: 10.1530/ERC-16-0231
58. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ, et al. Neurofibromatosis Type 1. Nat Rev Dis Primers (2017) 3:17004. doi: 10.1038/nrdp.2017.4
59. Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM, et al. Von Recklinghausen's Disease and Pheochromocytomas. J Urol (1999) 162(5):1582–6. doi: 10.1016/S0022-5347(05)68171-2
60. Kepenekian L, Mognetti T, Lifante JC, Giraudet AL, Houzard C, Pinson S, et al. Interest of Systematic Screening of Pheochromocytoma in Patients With Neurofibromatosis Type 1. Eur J Endocrinol (2016) 175(4):335–44. doi: 10.1530/EJE-16-0233
61. Al-Sharefi A, Javaid U, Perros P, Ealing J, Truran P, Nag S, et al. Clinical Presentation and Outcomes of Phaeochromocytomas/Paragangliomas in Neurofibromatosis Type 1. Eur Endocrinol (2019) 15(2):95–100. doi: 10.17925/EE.2019.15.2.95
62. Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V, et al. Defects in the Kidney and Enteric Nervous System of Mice Lacking the Tyrosine Kinase Receptor Ret. Nature (1994) 367(6461):380–3. doi: 10.1038/367380a0
63. Wells SA Jr., Asa SL, Dralle H, Elisei R, Evans DB, Gagel RF, et al. Revised American Thyroid Association Guidelines for the Management of Medullary Thyroid Carcinoma. Thyroid (2015) 25(6):567–610. doi: 10.1089/thy.2014.0335
64. Moraitis AG, Martucci VL, Pacak K. Genetics, Diagnosis, and Management of Medullary Thyroid Carcinoma and Pheochromocytoma/Paraganglioma. Endocr Pract (2014) 20(2):176–87. doi: 10.4158/EP13268.RA
65. Bholah R, Bunchman TE. Review of Pediatric Pheochromocytoma and Paraganglioma. Front Pediatr (2017) 5:155. doi: 10.3389/fped.2017.00155
66. Eisenhofer G., Rivers G., Rosas A. L., Quezado Z., Manger W. M., Pacak K., et al. Adverse Drug Reactions in Patients With Phaeochromocytoma: Incidence, Prevention and Management. Drug Saf (2007) 30(11):1031–62. doi: 10.2165/00002018-200730110-00004
67. Taieb D, Jha A, Treglia G, Pacak K. Molecular Imaging and Radionuclide Therapy of Pheochromocytoma and Paraganglioma in the Era of Genomic Characterization of Disease Subgroups. Endocr Relat Cancer (2019) 26(11):R627–52. doi: 10.1530/ERC-19-0165
68. Shulkin BL, Shapiro B. Current Concepts on the Diagnostic Use of MIBG in Children. J Nucl Med (1998) 39(4):679–88.
69. Carrasquillo JA, Chen CC, Jha A, Pacak K, Pryma DA, Lin FI. Systemic Radiopharmaceutical Therapy of Pheochromocytoma and Paraganglioma. J Nucl Med (2021) 62(9):1192–9. doi: 10.2967/jnumed.120.259697
70. Jha A, Ling A, Millo C, Gupta G, Viana B, Lin FI, et al. Superiority of (68)Ga-DOTATATE Over (18)F-FDG and Anatomic Imaging in the Detection of Succinate Dehydrogenase Mutation (SDHx )-Related Pheochromocytoma and Paraganglioma in the Pediatric Population. Eur J Nucl Med Mol Imaging (2018) 45(5):787–97. doi: 10.1007/s00259-017-3896-9
71. States LJ, Reid JR. Whole-Body PET/MRI Applications in Pediatric Oncology. AJR Am J Roentgenol (2020) 215(3):713–25. doi: 10.2214/AJR.19.22677
72. Han S, Suh CH, Woo S, Kim YJ, Lee JJ. Performance of (68)Ga-DOTA-Conjugated Somatostatin Receptor-Targeting Peptide PET in Detection of Pheochromocytoma and Paraganglioma: A Systematic Review and Metaanalysis. J Nucl Med (2019) 60(3):369–76. doi: 10.2967/jnumed.118.211706
73. Jha A, Patel M, Carrasquillo JA, Ling A, Millo C, Saboury B, et al. Sporadic Primary Pheochromocytoma: A Prospective Intraindividual Comparison of Six Imaging Tests (CT, MRI, and PET/CT Using (68)Ga-DOTATATE, FDG, (18)F-FDOPA, and (18)F-FDA). AJR Am J Roentgenol (2022) 218(2):342–50. doi: 10.2214/AJR.21.26071
74. Hadi M, Chen CC, Whatley M, Pacak K, Carrasquillo JA. Brown Fat Imaging With (18)F-6-Fluorodopamine PET/CT, (18)F-FDG PET/CT, and (123)I-MIBG SPECT: A Study of Patients Being Evaluated for Pheochromocytoma. J Nucl Med (2007) 48(7):1077–83. doi: 10.2967/jnumed.106.035915
75. Aloj L, Giger O, Mendichovszky IA, Challis BG, Ronel M, Harper I, et al. The Role of [(68) Ga]Ga-DOTATATE PET/CT in Wild-Type KIT/PDGFRA Gastrointestinal Stromal Tumours (GIST). EJNMMI Res (2021) 11(1):5. doi: 10.1186/s13550-021-00747-0
76. Albano D, Mattia B, Giubbini R, Bertagna F. Role of 18F-FDG PET/CT in Restaging and Follow-Up of Patients With GIST. Abdom Radiol (NY) (2020) 45(3):644–51. doi: 10.1007/s00261-019-02274-y
77. Seamon ML, Yamaguchi I. Hypertension in Pheochromocytoma and Paraganglioma: Evaluation and Management in Pediatric Patients. Curr Hypertens Rep (2021) 23(5):32. doi: 10.1007/s11906-021-01150-9
78. Ilanchezhian M, Jha A, Pacak K, Del Rivero J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr Treat Options Oncol (2020) 21(11):85. doi: 10.1007/s11864-020-00787-z
79. Jha A, Taieb D, Carrasquillo JA, Pryma DA, Patel M, Millo C, et al. High-Specific-Activity-(131)I-MIBG Versus (177)Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin Cancer Res (2021) 27(11):2989–95. doi: 10.1158/1078-0432.CCR-20-3703
80. Jha A, Patel M, Baker E, Gonzales MK, Ling A, Millo C. Role of (68)Ga-DOTATATE PET/CT in a Case of SDHB-Related Pterygopalatine Fossa Paraganglioma Successfully Controlled With Octreotide. Nucl Med Mol Imaging (2020) 54(1):48–52. doi: 10.1007/s13139-019-00629-3
81. Kamihara J, Hamilton KV, Pollard JA, Clinton CM, Madden JA, Lin J, et al. Belzutifan, a Potent HIF2alpha Inhibitor, in the Pacak-Zhuang Syndrome. N Engl J Med (2021) 385(22):2059–65. doi: 10.1056/NEJMoa2110051
82. Raygada M, King KS, Adams KT, Stratakis CA, Pacak K, et al. Counseling Patients With Succinate Dehydrogenase Subunit Defects: Genetics, Preventive Guidelines, and Dealing With Uncertainty. J Pediatr Endocrinol Metab (2014) 27(9-10):837–44. doi: 10.1515/jpem-2013-0369
83. Mweempwa A, Xu H, Vissers JHA, Tothill RW, Pattison AD, Fellowes AP, et al. Novel RET Fusion RET-SEPTIN9 Predicts Response to Selective RET Inhibition With Selpercatinib in Malignant Pheochromocytoma. JCO Precis Oncol (2021) 5:1160–5. doi: 10.1200/PO.21.00127
Keywords: pediatric, metastatic, pheochromocytoma, paraganglioma, clinical presentation, diagnosis, genetics, therapeutic approach
Citation: Kuo MJM, Nazari MA, Jha A and Pacak K (2022) Pediatric Metastatic Pheochromocytoma and Paraganglioma: Clinical Presentation and Diagnosis, Genetics, and Therapeutic Approaches. Front. Endocrinol. 13:936178. doi: 10.3389/fendo.2022.936178
Received: 04 May 2022; Accepted: 13 June 2022;
Published: 12 July 2022.
Edited by:
Regis Coutant, Centre Hospitalier Universitaire d’Angers, FranceReviewed by:
Miklós Tóth, Semmelweis University, HungaryClaudio Casella, University of Brescia, Italy
Copyright © 2022 Kuo, Nazari, Jha and Pacak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karel Pacak, karel@mail.nih.gov