 Pavlos Fanis1
Pavlos Fanis1 Nicos Skordis1,2,3
Nicos Skordis1,2,3 Meropi Toumba1,4
Meropi Toumba1,4 Michalis Picolos5
Michalis Picolos5 George A. Tanteles6
George A. Tanteles6 Vassos Neocleous1Leonidas A. Phylactou1*
Vassos Neocleous1Leonidas A. Phylactou1*- 1Department of Molecular Genetics, Function and Therapy, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
- 2Division of Paediatric Endocrinology, Paedi Center for specialized Paediatrics, Nicosia, Cyprus
- 3School of Medicine, University of Nicosia, Nicosia, Cyprus
- 4Department of Paediatrics, Paediatric Endocrinology Clinic, Aretaeio Hospital, Nicosia, Cyprus
- 5Department of Endocrinology, Alithias Endocrinology Center, Nicosia, Cyprus
- 6Department of Clinical Genetics, The Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus
Objective: The study aimed to identify the pathogenic status of p.Gln319Ter (NM_000500.7: c.955C>T) variant when inherited in a single CYP21A2 gene (bimodular RCCX haplotype) and to discriminate between a non-causing congenital adrenal hyperplasia (CAH) allele when inherited in a duplicated and functional CYP21A2 gene context (trimodular RCCX haplotype).
Methods: 38 females and 8 males with hyperandrogenemia, previously screened by sequencing and identified as carriers for the pathogenic p.Gln319Ter, were herein tested by multiplex ligation-dependent probe amplification (MLPA) and a real-time PCR Copy number Variation (CNV) assay.
Results: Both MLPA and real-time PCR CNV analyses confirmed a bimodular and pathogenic RCCX haplotype with a single CYP21A2 in 19/46 (41.30%) p.Gln319Ter carriers and who in parallel all shared elevated 17-OHP levels. The remaining 27 individuals that also carried the p.Gln319Ter exhibited low 17-OHP levels as a result of their carriership of a duplicated CYP21A2 with a trimodular RCCX haplotype. Interestingly, all of these individuals also carried in linkage disequilibrium with p.Gln319Ter two single nucleotide polymorphisms, the c.293-79G>A (rs114414746) in intron 2 and the c.*12C>T (rs150697472) in the 3’-UTR. Therefore, these variants can be used to distinguish between pathogenic and non-pathogenic genomic contexts of the c.955T (p.Gln319) in the genetic diagnosis of congenital adrenal hyperplasia (CAH).
Conclusion: The employed methodologies identified a considerable number of individuals with non-pathogenic p.Gln319Ter from the individuals that typically carry the pathogenic p.Gln319Ter in a single CYP21A2. Therefore, it is extremely important the detection of such haplotypes for the prenatal diagnosis, treatment and genetic counseling in patients with CAH.
Introduction
Congenital adrenal Hyperplasia (CAH) represents a group of autosomal recessive defects each of which is caused by the impairment of several enzymes that are involved in cortisol biosynthesis (1, 2). The most frequent form of CAH is 21-hydroxylase deficiency (21-OHD) (90–95% of cases) followed by 11β-hydroxylase deficiency (11β-OHD) (∼5% of cases) and other rarer forms (3). The Non Classic (NC)-CAH which is the mild form of the disorder is more frequently observed in females (4). Males with NC-CAH are diagnosed significantly less frequently than females due to less frequently presented and recognized signs of androgen excess (4, 5). The multiallelic and tandem RCCX (R=RP1 also known as STK19, C=C4 or complement Component C4, C=CYP21A2 or steroid 21-hydroxylase, and X=TNXB or tenascin-X) copy number variation (CNV) complex exists in the major histocompatibility complex (MHC) class III region on chromosome 6p21.3. The RCCX complex typically comprises of one (monomodular), two (bimodular) or three (trimodular) segments with an incidence of 15%, 75% and 10% in Europeans, respectively (6, 7). Each segment itself is also quite multipart since it contains a series of four genes; the serine/threonine kinase 19 (STK19), the complement 4 (C4), the steroid 21-hydroxylase (CYP21A2), the tenascin-X (TNX) and their corresponding pseudogenes (8). The majority of pathogenic variants in 21-OHD cases are transferred to the CYP21A2 gene by small conversions from the CYP21A1P pseudogene as a result of unequal crossover during meiosis (6, 9–13). The complex genetic structure of the RCCX CNV complicates further the genetic diagnosis of 21-hydroxylase deficiency, in particular when a haplotypic structure with the segment harboring two CYP21A2 gene copies and one CYP21A1P pseudogene copy is involved [6]. A very characteristic example of a haplotypic RCCX CNV structure containing three distinct segments (i.e the trimodular haplotype) with one of them carrying the deleterious p.Gln319Ter variant is quite often observed and confuses the genetic diagnosis of CAH (6, 14–17). Numerous studies in the Mediterranean region identified p.Gln319Ter pathogenic variant as quite frequent (18–20). Similarly, in a previous study by our group three hundred unrelated subjects (150 males and 150 females) from the general population of Cyprus were screened for pathogenic variants in the CYP21A2 gene and identified p.Gln319Ter variant among six others as the second most frequent (21). In this same study, an estimated CYP21A2 carrier frequency of 9.83% for the Cypriot population was also reported, with the mild p.Val282Leu being the most frequent (4.3%), followed by p.Gln319Ter (2.5%), p.Pro454Ser (1.33%), p.Val305Met (0.83%), p.Pro483Ser (0.67%) and p.Met284Val (0.17%) (21).
Since the first report by Globerman et al. (22) where a stop codon in exon 8 of the CYP21A2 gene at position 319 (p.Gln319Ter) was identified, numerous other followed (4, 23–25). Individuals homozygous for this mutation have the severe salt-wasting (SW) form of CAH and no enzymatic activity (22, 26). Several other reports have also demonstrated that heterozygosity for the p.Gln319Ter variant can be an issue especially in females and that exhibit the milder NC-CAH form (27–29).
In the present study, we used two different quantitative methods, a multiplex ligation-dependent probe amplification (MLPA) methodology and a real-time PCR assay to test for the presence of a duplicated CYP21A2 gene. We have re-examined using the above-mentioned two quantitative methodologies, 46 heterozygous and/or compound heterozygous individuals and that were initially screened by Sanger and were identified as carriers of the severe p.Gln319Ter mutation. Our findings demonstrated that a lower frequency with the duplicated CYP21A2 gene trimodular RCCX haplotype exists in the Cypriot cohort under investigation. Furthermore, in the same cohort a lower incidence of the pathogenic p.Gln319Ter mutation is carried in a RCCX haplotype with one copy of the CYP21A2 gene. This finding demonstrates the necessity of using in addition to the traditional Sanger sequencing also other contemporary methodologies that are needed for the more precise and accurate diagnosis of CAH.
Materials and methods
Study subjects
In the present study a sample collection of 46 (38 females and 8 males) Cypriot patients with clinical hyperandrogenism. According to age, patients were diagnosed with clinical signs of hyperandrogenemia. Pre-pubertal patients; girls younger than 8 years of age and boys younger than 9 years of age presented with premature adrenarche (pubic or/and underarm hair, body odor, acne spots), and/or bone age advancement, and/or hypertrophic clitoris. Patients in post-pubertal ages presented with severe acne, hirsutism, infrequent menstrual bleeding, voice hoarseness and/or male pattern balding. All patients underwent measurements of basal and/or stimulated 17-OH Progesterone (P) levels. Sanger sequencing was previously performed to all patients with clinical hyperandrogenaemia either with normal or mildly elevated basal or/and stimulated levels of 17-OH Progesterone (P) and irrespectively of the levels of the other androgens (testosterone, androstenedione and DHEA-S). All patients included, were previously identified with Sanger sequencing as carriers for the p.Gln319Ter in the CYP21A2 gene, were also subjected to further genetic analysis as described below using two other quantitative PCR based methodologies. Parental samples were not available for analysis. Informed consent was obtained from all adult patients and the parents or guardians of the minors. The project was approved by the Cyprus National Ethics Committee (EEBK/EΠ/2016/28) and all methods were performed in accordance with the relevant guidelines and regulations and the Declaration of Helsinki of 1964 and its later amendments.
Copy number variation analysis by MLPA and real-time PCR assay
All 46 samples were further re-examined using two quantitative PCR based methodologies as per the recommendation of the CAH Best Practice Guidelines (30). The two methodologies used, were the multiplex ligation-dependent probe amplification (MLPA) as previously described (12, 31) using the SALSA MLPA CAH P050-D1 Probemix (MRC-Holland, Amsterdam, the Netherlands) and the commercial CAH Real Fast CNV Assay real-time PCR according to manufacturer’s instructions (ViennaLab Diagnostics GmbH, Austria). The Real-time PCR was performed on QuantStudio 3 (Applied Biosystems, Foster City, CA, USA). Both methods intended to test for the presence of a duplicated CYP21A2 gene.
Sanger sequencing analysis
The complete CYP21A2 gene downstream of the TNXB gene was amplified for all 46 samples using the primers CYP779f (5’-AGGTGGGCTGTTTTCCTTTCA-3’) and Tena32F (5’-CTGTGCCTGGCTATAGCAAGC-3’) according to the protocol of Lee et al. (32), generating an 8.5 Kb product that was sequenced with specific internal primers. Specifically, the PCR reaction mixture was performed using the PrimeStar GXL DNA polymerase kit (Takara-Bio, Shiga, Japan) at a final volume of 50 μl and contained 10 μl of Primestar GLX buffer (5x), 4 μl dNTP mix (2.5 mM), 1 μl of each primer (10μM), 2 μl PrimeSTAR GXL DNA Polymerase (1.25 U/μl) and 250 ng of genomic DNA. Amplification was performed with an initial denaturing temperature at 98°C for 5min, followed by 32 cycles of denaturation (98°C, 10sec), annealing (60°C, 30sec), extension (68°C, 3min), with a final extension at 68°C for 7min. For the 27 samples identified by MLPA and Real-time PCR assay to have three copies of the CYP21A2 gene, we also amplified and sequenced the complete the CYP21A2 gene downstream of the TNXA gene. To achieve this we initially amplify the gene using the CYP779f and XA-36F (5’-GGACCCAGAAACTCCAGGTGG-3’) primers according to the protocol of Tsai et al. (17), generating a 6.1 Kb product. The PCR mixture was the same as above and the amplification was performed with an initial denaturing temperature at 98°C for 5min, followed by 28 cycles of denaturation (98°C, 10sec), extension (68°C, 3min), with a final extension at 68°C for 7min. Next, 20 μl of the PCR product were used for restriction enzyme digestion with TaqI at 65°C for 2 hrs. The whole digestion reaction was loaded on a 1% agarose gel and the 3.7 Kb fragment was extracted and purified using the NucleoSpin gel and PCR cleanup kit (Macherey-Nagel, Düren, Germany) and then sequenced with specific internal primers. The sequencing results were analysed using the Seqscape v3.0 software (Applied Biosystems, Foster City, CA).
Results
CYP21A2 gene p.Gln319Ter pathogenic variant
In the course of Sanger sequencing for the CYP21A2 gene downstream of the TNXB gene, the 46 patients with hyperandrogenemia were previously identified by our group as carriers for the p.Gln319Ter pathogenic variant (12, 31). Thirty-five of the above patients carried p.Gln319Ter in heterozygosity and the remaining eleven in compound heterozygosity with another CYP21A2 pathogenic causing variant (Table 1). As described below the employment of MLPA and the quantitative Real-time PCR CNV assay delineated the pathogenic p.Gln319Ter variants.
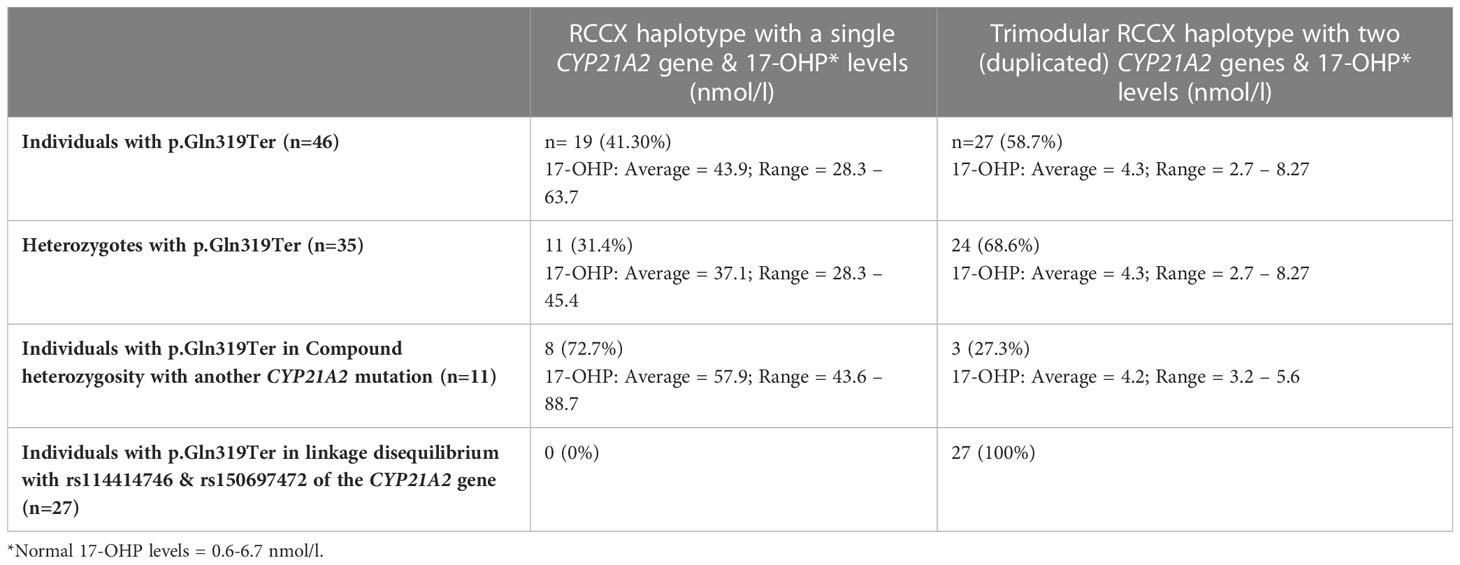
Table 1 RCCX copy number variation haplotypes in 46 individuals with hyperandrogenemia all found to carry the p.Gln319Ter pathogenic variant in the CYP21A2 gene.
MLPA analysis for the detection of CYP21A2 gene duplication
Further analysis by MLPA of the 46 patients initially identified by Sanger sequencing as carriers for the p.Gln319Ter, revealed the presence of four haplotypes that were combined in different genotypes. The four haplotypes were H1: bimodular RCCX module with one CYP21A2 gene, H2: monomodular RCCX module with one CYP21A2 gene, H3: trimodular RCCX module with one CYP21A2 gene and H4: trimodular RCCX module with two CYP21A2 genes (Figure 1A). Interestingly, only 19 (41.30%) of the patients were found to carry two haplotypes containing one CYP21A2 gene each (Table 1, Supplementary Table 1, Figure 1A, B(i)). Eleven of these 19 hyperandrogenic patients carried p.Gln319Ter in the heterozygous state with the remaining eight to carry it in compound heterozygosity with another CYP21A2 causing variant. The remaining 27 patients that carried the p.Gln319Ter variant in the heterozygous form shared the trimodular H4 haplotype with two CYP21A2 genes and a single CYP21A1P pseudogene [Table 1, Supplementary Table 1, Figure 1A, B(ii)].
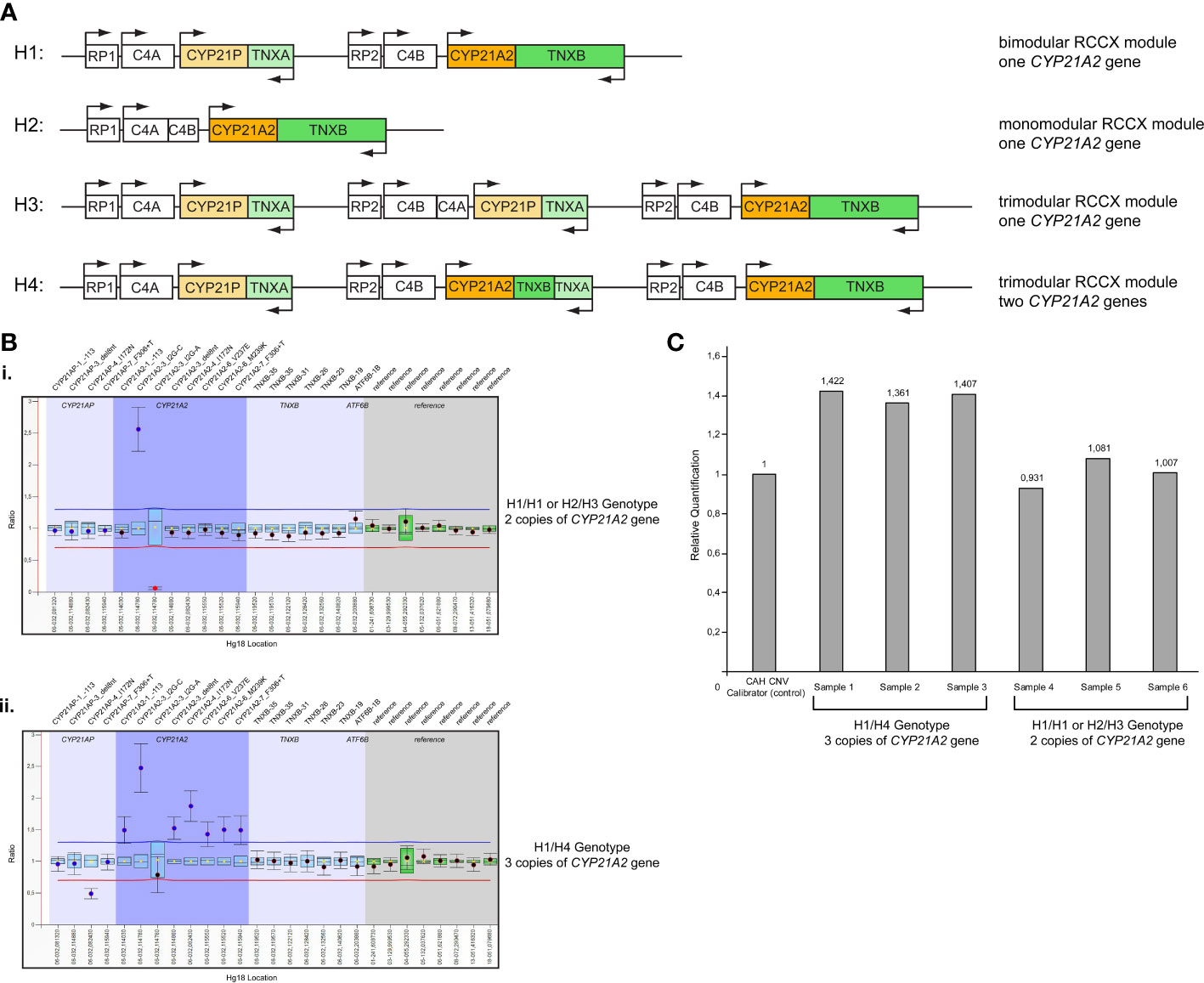
Figure 1 (A) Schematic representation of the four different haplotypes identified in the present study. H1: Haplotype 1, bimodular RCCX module with one CYP21A2 gene; H2: Haplotype 2, monomodular RCCX module with one CYP21A2 gene; H3: Haplotype 3, trimodular RCCX module with one CYP21A2 gene; H4: Haplotype 4, trimodular RCCX module with two CYP21A2 genes. (B) MLPA representative ratio charts of patients identified as carriers for the pathogenic variant p.Gln319Ter showing the i) H1/H1 or H2/H3 genotype and the ii) H1/H4 genotype on chromosome 6p21.3. (C) Representative CAH Real Fast CNV Assay of patients identified as carriers for the pathogenic variant p.Gln319Ter showing the H1/H4 genotype (samples 1-3) and the H1/H1 or H2/H3 genotype (samples 4-6). CAH CNV calibrator (Vienna Lab) used as control with ratio=1.
Real-time PCR CNV assay for the detection of CYP21A2 gene duplication
All 46 samples from the unrelated patients with hyperandrogenemia were further re-analyzed using the commercial real-time PCR CAH Real Fast CNV Assay (Vienna Lab) so as to re-confirm the presence or absence of the duplicated CYP21A2 genes obtained by MLPA. Likewise, the real-time PCR assay confirmed the presence of a duplicated CYP21A2 in the same 19 patients that were detected with MLPA (Figure 1C).
Molecular characterization of the trimodular H4 haplotype with two CYP21A2 genes and a single CYP21A1P pseudogene
All 27 subjects who shared the trimodular H4 haplotype were also found to carry in complete linkage disequilibrium (LD) with the p.Gln319Ter the SNPs rs114414746 (NM_000500.9: c.293-79G>A) in intron 2 and the rs150697472 (NM_000500.9: c.*12C>T) in the 3’-UTR. To investigate this further, we specifically sequence the CYP21A2 gene, which is located downstream of the TNXA gene. Sequencing revealed that the CYP21A2 gene downstream of the TNXA gene contains the SNP rs150697472 but not p.Gln319Ter and SNP rs114414746 (Figure 2A). Combined with the results of sequencing of the CYP21A2 gene downstream of the TNXB gene, the middle segment of the H4 trimodular haplotype harbors the SNP c.*12T (rs150697472), while the third segment harbors the two variants c.293-79A (rs114414746) and the c.955T (p.Gln319) (Figure 2B).
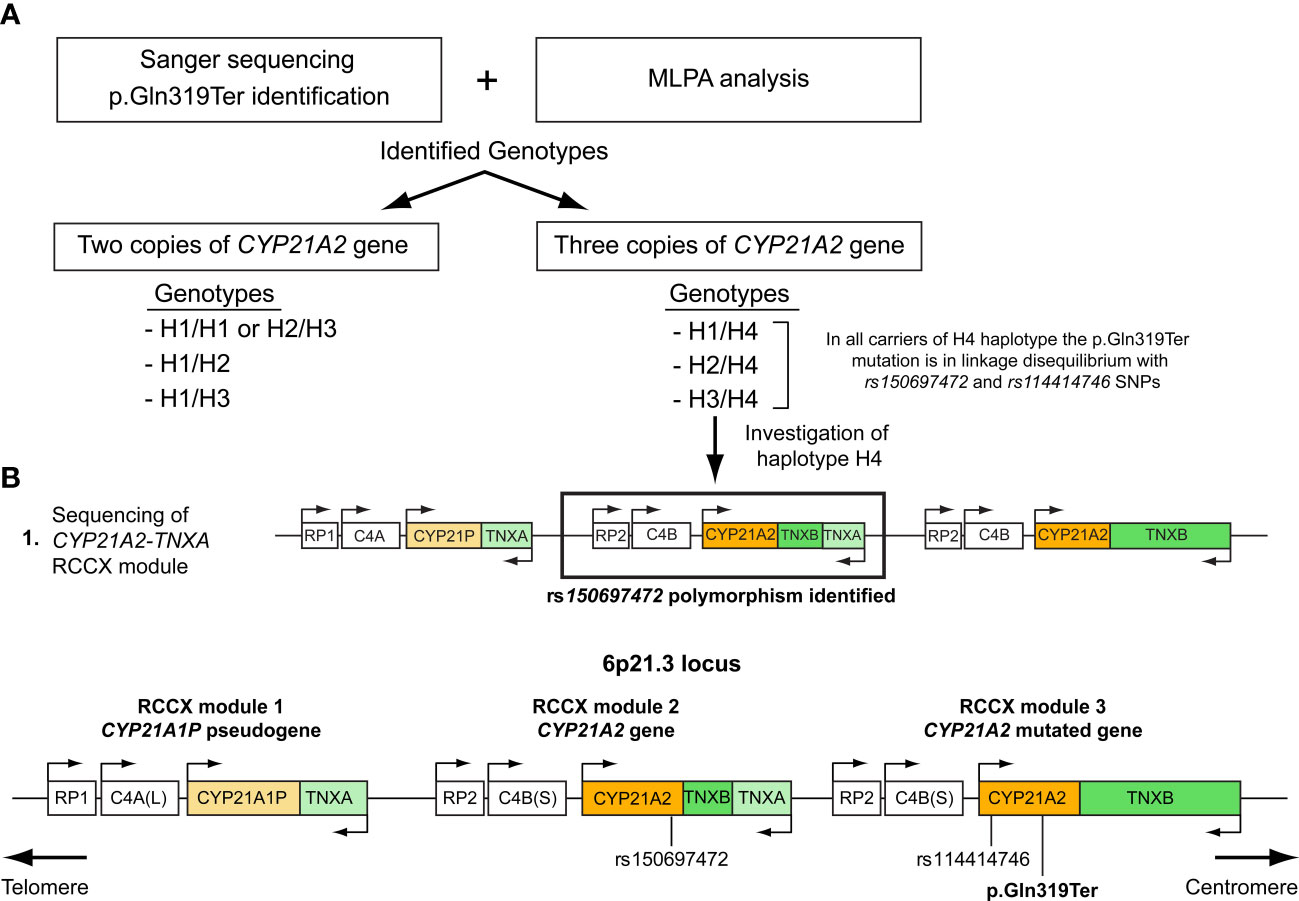
Figure 2 (A) Illustration of the procedure followed in this study to genotype the samples carrying the p.Gln319Ter mutation. (B) Schematic representation of the trimodular RCCX haplotype on chromosome 6p21.3 consisting of three segments. RCCX module 1 containing the CYP21A1P pseudogene and RCCX modules 2 and 3 containing the CYP21A2 gene. Both the p.Gln319Ter mutation and the rs114414746 (NM_000500.9: c.293-79G>A) polymorphism in intron 2 of the CYP21A2 gene are located in the third non-functional segment, while the rs150697472 (NM_000500.9: c.*12C>T) is located in the 3’UTR of the second functional segment.
Biochemical and genotypic correlation of the identified patients with the p.Gln319Ter variant
Nineteen (12 females and 7 males) patients who were found to have two copies of the CYP21A2 gene and carry the pathogenic p.Gln319Ter variant either in the heterozygous or compound heterozygous state and had elevated 17-OHP levels (Average = 43.9, Range = 28.3 – 63.7). It is noted that normal levels of 17-OHP are 0.6 - 6.7 nmol/l. The remaining 27 subjects that carried in the trimodular H4 haplotype the p.Gln319Ter variant shared lower 17-OHP levels (Average = 4.3, Range = 2.7 – 8.27) (Table 1). As predicted, the heterozygote patients with the bimodular RCCX haplotype carrying the pathogenic p.Gln319Ter exhibited lower 17-OHP values (Average = 37.1, Range = 28.3 – 45.4) when compared to the values of patients (n=8) that shared p.Gln319Ter in compound heterozygosity with another CYP21A2 pathogenic variant (Average = 57.9, Range = 43.6 – 88.7) (Table 1). It should be noted that six out of eight of the above patients that carried p.Gln319Ter in a bimodular RCCX haplotype also carried in compound heterozygosity the p.Val282Leu mutation. All six (five females and one male) of them were classified as non-classic (NC) CAH and their 17-OHP levels ranged between 43.6-61.9 nmol/l. The remaining two female patients of the above cohort were respectively found to carry in their second allele the severe IVS-13A/C>G and the milder p.Pro454Ser mutations. As a result of the severe IVS-13A/C>G/p.Gln319Ter genotype the neonate female exhibited higher 17-OHP levels (88.7 nmol/l) and was born with ambiguous genitalia, therefore was classified with the severe Salt-Wasting (SW) CAH form. Lastly, the girl identified with the p.Gln319Ter/p.Pro454Ser genotype exhibited lower 17-OHP levels (52.4 nmol/l) and phenotypically was classified with the NC-CAH form.
Discussion
Duplications of the CYP21A2 gene have been reported as a result of the less frequent RCCX CNV trimodular haplotype that harbors one copy of the CYP21A1P pseudogene and two copies of the CYP21A2 gene, with the latter to also harbor the p.Gln319Ter variant in 2-7% of the general population (15, 16, 33, 34). In the present study genotyping of 46 individuals with p.Gln319Ter variant using the commercially available MLPA and the Real-time PCR CNV Assay, demonstrated that the majority of these individuals (27/46; 58.7%) carried the p.Gln319Ter aberration on the allele that harbors the duplicated CYP21A2 gene. It should be noted, that all 46 individuals of the present study were referred to our laboratory for genetic investigation since they were identified with clinical signs of hyperandrogenemia. As evidenced by the extended haplotype analysis from the MLPA and the real time PCR analyses of the present study, the p.Gln319Ter aberration was found in a genomic non-pathogenic context in 58.7% of the tested individuals as a result of the presence of the trimodular RCCX haplotype. Consequently, this haplotype format is not accountable for causing their clinical manifestations of hyperandrogenemia. Despite the fact that several recent studies likewise identified p.Gln319Ter often to be associated with an RCCX trimodular and duplicated CYP21A2 haplotype (33, 35), the unavailability of parental samples for more explicit haplotype analyses would be more appropriate to hypothesize this as described previously (36) (37). A future study aiming to determine the actual frequency of the trimodular non-pathogenic haplotype harboring the p.Gln319Ter variant in a cohort of clinically unaffected Cypriot individuals will undoubtedly contribute towards the avoidance of false-positive genotyping and assist for the more precise CAH diagnosis in the island.
It’s worth mentioning that for the more accurate CAH diagnosis the quantitative based methodologies such as the MLPA and the real-time PCR assay are absolutely essential and in the present study were proven so. The usage of both of these methodologies enabled us to clarify the carrier status of the p.Gln319Ter in the trimodular non-CAH causing allele from the bimodular CAH allele. Moreover, we have also noticed that the majority (8/11; 72.7%) of the compound heterozygote patients for the p.Gln319Ter indeed shared the bimodular and pathogenic RCCX haplotype with a single CYP21A2 gene (Table 1). On the contrary, the majority (24/35; 68.6%) of the non-pathogenic heterozygotes for the p.Gln319Ter variant shared the trimodular RCCX non-pathogenic haplotype as a result of the presence of two CYP21A2 gene copies (Table 1, Figure 2B).
Lastly, our observation that all carriers of the duplicated CYP21A2 gene with the p.Gln319Ter variant were in linkage disequilibrium with two infrequent SNPs, the c.293-79A (rs114414746) in intron 2 and the c.*12T (rs150697472) in the 3’-UTR (Table 1), and the fact that was in agreement with an earlier report by Kleinle et al. (33) reveals the importance of analyzing the CYP21A2 untranslated regions (Figure 2B). In a recent report by Doleschall et al., the sequences of the duplicated CYP21A2 genes (trimodular RCCX haplotype) were defined (8). More precisely, the CYP21A2 gene in the middle segment of the trimodular haplotype harbored the c.*12T (rs150697472) SNP, whereas the CYP21A2 gene in the 3’ segment harbored both the c.293-79A (rs114414746) SNP and the c.955T (p.Gln319) mutation. This finding in combination with earlier studies in healthy subjects of European descent (Spanish and Italian) (15, 35), and the evidenced by the present study (Figure 2) can be used to distinguish between pathogenic and non-pathogenic genomic contexts of the c.955C>T (p.Gln319Ter) in the genetic diagnosis of CAH.
Conclusions
In conclusion, the absence of solid correlation between genotype and phenotype observed in many individuals with clinical signs of hyperandrogenemia and CAH can largely be explained with the identification of the rare RCCX CNV haplotypes, therefore the need of their assessment is vital for the precise molecular diagnosis of 21-hydroxylase deficiency.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by Cyprus National Ethics Committee (EEBK/EΠ/2016/28). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
PF conceptualized and designed the study, interpreted data and performed the laboratory experiments. NS, MT, MP and GT performed clinical investigation and enrolled patients. VN conceptualized the study, interpreted data, drafted and revised the manuscript. LP conceptualized the study and revised the final version of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the A.G Leventis Foundation. The Department of Molecular Genetics, Function and Therapy of the Cyprus Institute of Neurology and Genetics is member of the European Reference Network on Rare Endocrine Conditions: Project ID N0 739543 (https://endo-ern.eu/about/reference-centres/).
Acknowledgments
The authors thank all the participants of the study. The department of Molecular Genetics, Function and Therapy of the Cyprus Institute of Neurology and Genetics would also like to thank the European Reference Network on Rare Endocrine Conditions: Project ID N0 739543 (https://endo-ern.eu/about/reference-centres/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1156616/full#supplementary-material
References
1. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev (2022) 43(1):91–159. doi: 10.1210/endrev/bnab016
2. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
3. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med (2020) 383(13):1248–61. doi: 10.1056/NEJMra1909786
4. Auer MK, Nordenstrom A, Lajic S, Reisch N. Congenital adrenal hyperplasia. Lancet (2022) 401(10372):227-244. doi: 10.1016/S0140-6736(22)01330-7
5. Macut D, Zdravkovic V, Bjekic-Macut J, Mastorakos G, Pignatelli D. Metabolic perspectives for non-classical congenital adrenal hyperplasia with relation to the classical form of the disease. Front Endocrinol (2019) 10:681. doi: 10.3389/fendo.2019.00681
6. Carrozza C, Foca L, De Paolis E, Concolino P. Genes and pseudogenes: complexity of the RCCX locus and disease. Front Endocrinol (2021) 12:709758. doi: 10.3389/fendo.2021.709758
7. Concolino P. A rare CYP21A2 haplotype clarifies the phenotype-genotype discrepancy in an Italian patient with non classical congenital adrenal hyperplasia (NC-CAH). Mol Biol Rep (2020) 47(4):3049–52. doi: 10.1007/s11033-020-05379-6
8. Doleschall M, Luczay A, Koncz K, Hadzsiev K, Erhardt E, Szilagyi A, et al. A unique haplotype of RCCX copy number variation: from the clinics of congenital adrenal hyperplasia to evolutionary genetics. Eur J Hum Genet (2017) 25(6):702–10. doi: 10.1038/ejhg.2017.38
9. Morissette R, Chen W, Perritt AF, Dreiling JL, Arai AE, Sachdev V, et al. Broadening the spectrum of ehlers danlos syndrome in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2015) 100(8):E1143–52. doi: 10.1210/jc.2015-2232
10. Concolino P, Costella A. Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency: a comprehensive focus on 233 pathogenic variants of CYP21A2 gene. Mol diagnosis Ther (2018) 22(3):261–80. doi: 10.1007/s40291-018-0319-y
11. Neocleous V, Fanis P, Phylactou LA, Skordis N. Genotype is associated to the degree of virilization in patients with classic congenital adrenal hyperplasia. Front Endocrinol (2018) 9:733. doi: 10.3389/fendo.2018.00733
12. Neocleous V, Fanis P, Toumba M, Phedonos AAP, Picolos M, Andreou E, et al. Variations in the 3'UTR of the CYP21A2 gene in heterozygous females with hyperandrogenaemia. Int J Endocrinol (2017) 2017:8984365. doi: 10.1155/2017/8984365
13. Fanis P, Skordis N, Phylactou LA, Neocleous V. Salt-wasting congenital adrenal hyperplasia phenotype as a result of the TNXA/TNXB chimera 1 (CAH-X CH-1) and the pathogenic IVS2-13A/C > G in CYP21A2 gene. Hormones (Athens) (2022) 22(1): 71–77.. doi: 10.1007/s42000-022-00410-w
14. Kharrat M, Riahi A, Maazoul F, M'Rad R, Chaabouni H. Detection of a frequent duplicated CYP21A2 gene carrying a Q318X mutation in a general population with quantitative PCR methods. Diagn Mol Pathol (2011) 20(2):123–7. doi: 10.1097/PDM.0b013e3181f24807
15. Parajes S, Quinteiro C, Dominguez F, Loidi L. High frequency of copy number variations and sequence variants at CYP21A2 locus: implication for the genetic diagnosis of 21-hydroxylase deficiency. PloS One (2008) 3(5):e2138. doi: 10.1371/journal.pone.0002138
16. Wedell A, Stengler B, Luthman H. Characterization of mutations on the rare duplicated C4/CYP21 haplotype in steroid 21-hydroxylase deficiency. Hum Genet (1994) 94(1):50–4. doi: 10.1007/BF02272841
17. Tsai LP, Cheng CF, Chuang SH, Lee HH. Analysis of the CYP21A1P pseudogene: indication of mutational diversity and CYP21A2-like and duplicated CYP21A2 genes. Anal Biochem (2011) 413(2):133–41. doi: 10.1016/j.ab.2011.02.016
18. Loidi L, Quinteiro C, Parajes S, Barreiro J, Leston DG, Cabezas-Agricola JM, et al. High variability in CYP21A2 mutated alleles in Spanish 21-hydroxylase deficiency patients, six novel mutations and a founder effect. Clin Endocrinol (Oxf) (2006) 64(3):330–6. doi: 10.1111/j.1365-2265.2006.02465.x
19. Abid F, Tardy V, Gaouzi A, El Hessni A, Morel Y, Chabraoui L. CYP21A2 gene mutation analysis in Moroccan patients with classic form of 21-hydroxylase deficiency: high regional prevalence of p.Q318X mutation and identification of a novel p.L353R mutation. Clin Chem Lab Med (2008) 46(12):1707–13. doi: 10.1515/CCLM.2008.339
20. Dracopoulou-Vabouli M, Maniati-Christidi M, Dacou-Voutetakis C. The spectrum of molecular defects of the CYP21 gene in the Hellenic population: variable concordance between genotype and phenotype in the different forms of congenital adrenal hyperplasia. J Clin Endocrinol Metab (2001) 86(6):2845–8. doi: 10.1210/jcem.86.6.7574
21. Phedonos AA, Shammas C, Skordis N, Kyriakides TC, Neocleous V, Phylactou LA. High carrier frequency of 21-hydroxylase deficiency in Cyprus. Clin Genet (2013) 84(6):585–8. doi: 10.1111/cge.12153
22. Globerman H, Amor M, Parker KL, New MI, White PC. Nonsense mutation causing steroid 21-hydroxylase deficiency. J Clin Invest (1988) 82(1):139–44. doi: 10.1172/JCI113562
23. Adriaansen BPH, Schroder MAM, Span PN, Sweep F, van Herwaarden AE, Claahsen-van der Grinten HL. Challenges in treatment of patients with non-classic congenital adrenal hyperplasia. Front Endocrinol (2022) 13:1064024. doi: 10.3389/fendo.2022.1064024
24. Yau M, Khattab A, Yuen T, New M. Congenital adrenal hyperplasia. Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. South Dartmouth (MA: Endotext (2000).
25. Fardella CE, Poggi H, Pineda P, Soto J, Torrealba I, Cattani A, et al. Salt-wasting congenital adrenal hyperplasia: detection of mutations in CYP21B gene in a Chilean population. J Clin Endocrinol Metab (1998) 83(9):3357–60. doi: 10.1210/jcem.83.9.5071
26. Pignatelli D, Carvalho BL, Palmeiro A, Barros A, Guerreiro SG, Macut D. The complexities in genotyping of congenital adrenal hyperplasia: 21-hydroxylase deficiency. Front Endocrinol (2019) 10:432. doi: 10.3389/fendo.2019.00432
27. Neocleous V, Shammas C, Phedonos AA, Phylactou LA, Skordis N. Phenotypic variability of hyperandrogenemia in females heterozygous for CYP21A2 mutations. Indian J Endocrinol Metab (2014) 18(Suppl 1):S72–9. doi: 10.4103/2230-8210.145077
28. Guarnotta V, Niceta M, Bono M, Marchese S, Fabiano C, Indelicato S, et al. Clinical and hormonal characteristics in heterozygote carriers of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol (2020) 198:105554. doi: 10.1016/j.jsbmb.2019.105554
29. Soveizi M, Mahdieh N, Setoodeh A, Sayarifard F, Abbasi F, Bose HS, et al. p.Gln318X and p.Val281Leu as the major variants of CYP21A2 gene in children with idiopathic premature pubarche. Int J Endocrinol (2020) 2020:4329791. doi: 10.1155/2020/4329791
30. Baumgartner-Parzer S, Witsch-Baumgartner M, Hoeppner W. EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur J Hum Genet (2020) 28(10):1341–67. doi: 10.1038/s41431-020-0653-5
31. Neocleous V, Fanis P, Toumba M, Stylianou C, Picolos M, Andreou E, et al. The spectrum of genetic defects in congenital adrenal hyperplasia in the population of Cyprus: a retrospective analysis. Horm Metab Res (2019) 51(9):586–94. doi: 10.1055/a-0957-3297
32. Lee HH, Lee YJ, Lin CY. PCR-based detection of the CYP21 deletion and TNXA/TNXB hybrid in the RCCX module. Genomics (2004) 83(5):944–50. doi: 10.1016/j.ygeno.2003.11.006
33. Kleinle S, Lang R, Fischer GF, Vierhapper H, Waldhauser F, Fodinger M, et al. Duplications of the functional CYP21A2 gene are primarily restricted to Q318X alleles: evidence for a founder effect. J Clin Endocrinol Metab (2009) 94(10):3954–8. doi: 10.1210/jc.2009-0487
34. Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. the load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med (2000) 191(12):2183–96. doi: 10.1084/jem.191.12.2183
35. Concolino P, Mello E, Minucci A, Giardina B, Capoluongo E. Genes, pseudogenes and like genes: the case of 21-hydroxylase in Italian population. Clin Chim Acta (2013) 424:85–9. doi: 10.1016/j.cca.2013.05.019
36. Lao Q, Jardin MD, Jayakrishnan R, Ernst M, Merke DP. Complement component 4 variations may influence psychopathology risk in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Genet (2018) 137(11-12):955–60. doi: 10.1007/s00439-018-1959-z
Keywords: CYP21A2, 21-hyrdroxylase deficiency, RCCX, gene duplications, CAH
Citation: Fanis P, Skordis N, Toumba M, Picolos M, Tanteles GA, Neocleous V and Phylactou LA (2023) The pathogenic p.Gln319Ter variant is not causing congenital adrenal hyperplasia when inherited in one of the duplicated CYP21A2 genes. Front. Endocrinol. 14:1156616. doi: 10.3389/fendo.2023.1156616
Received: 01 February 2023; Accepted: 04 May 2023;
Published: 31 May 2023.
Edited by:
Agnieszka Pazderska, St. James’s Hospital, IrelandReviewed by:
Qizong Lao, National Institutes of Health (NIH), United StatesPaola Concolino, Agostino Gemelli University Polyclinic (IRCCS), Italy
Copyright © 2023 Fanis, Skordis, Toumba, Picolos, Tanteles, Neocleous and Phylactou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonidas A. Phylactou, laphylac@cing.ac.cy