 Shu-Ping Tseng1
Shu-Ping Tseng1 James K. Wetterer2
James K. Wetterer2 Andrew V. Suarez3
Andrew V. Suarez3 Chow-Yang Lee4Tsuyoshi Yoshimura1
Chow-Yang Lee4Tsuyoshi Yoshimura1 DeWayne Shoemaker5*
DeWayne Shoemaker5* Chin-Cheng Scotty Yang1*
Chin-Cheng Scotty Yang1*- 1Research Institute for Sustainable Humanosphere, Kyoto University, Kyoto, Japan
- 2Wilkes Honors College, Florida Atlantic University, Jupiter, FL, United States
- 3Department of Evolution, Ecology and Behavior and Department of Entomology, University of Illinois Urbana-Champaign, IL, United States
- 4Department of Entomology, University of California, Riverside, CA, United States
- 5Entomology and Plant Pathology, University of Tennessee, Knoxville, TN, United States
Understanding the phylogeographic history of an invasive species may facilitate reconstructing the history and routes of its invasion. The longhorn crazy ant, Paratrechina longicornis, is a ubiquitous agricultural and household pest throughout much of the tropics and subtropics, but little is known about the history of its spread. Here, we examine worldwide genetic variation in P. longicornis and its associated Wolbachia bacterial symbionts. Analyses of mtDNA sequences of 248 P. longicornis workers (one per colony) from 13 geographic regions reveal two highly diverged mtDNA clades that co-occur in most of the geographic regions. These two mtDNA clades are associated with different Wolbachia infection patterns, but are not congruent with patterns of nDNA (microsatellite) variation. Multilocus sequence typing reveals two distinct Wolbachia strains in P. longicornis, namely, wLonA and wLonF. The evolutionary histories of these two strains differ; wLonA appears to be primarily transmitted maternally, and patterns of mtDNA and nDNA variation and wLonA infection status are consistent with a relatively recent Wolbachia-induced selective sweep. In contrast, the observed patterns of mtDNA variation and wLonF infections suggest frequent horizontal transfer and losses of wLonF infections. The lack of nDNA structure among sampled geographic regions coupled with the finding that numerous mtDNA haplotypes are shared among regions implies that inadvertent long-distance movement through human commerce is common in P. longicornis and has shaped the genetic structure of this invasive ant worldwide.
Introduction
Globalized human commerce has facilitated and intensified the spread of alien species, and the number of invasive species threatening native biodiversity, natural resources, and the economy continues to increase (Pimentel et al., 2000; Leppc et al., 2002; Occhipinti-Ambrogi and Savini, 2003; Meyerson and Mooney, 2007). Knowledge of the invasion histories, routes, and subsequent spread of invasive species provides important information for developing practical management strategies (Estoup and Guillemaud, 2010). Population genetic analyses on invasive species may provide insights into the introduction pathways and help us understand the mechanisms underlying the invasion success. Such analyses also may help define management objectives and assist policy makers in developing management, prevention, and restoration strategies (Abdelkrim et al., 2005; Le Roux and Wieczorek, 2009; Chadès et al., 2011; Cristescu, 2015).
The longhorn crazy ant, Paratrechina longicornis (Latreille, 1802) (Hymenoptera: Formicidae), is a widespread agricultural and household pest found throughout much of the tropics and subtropics in both the Old World and New World (Wetterer, 2008). A previous study demonstrated the occurrence of an extraordinary, double-clonal reproduction system in a population of P. longicornis from Thailand. In this population, queens are produced clonally from their mother, males are produced clonally from their fathers, and workers are produced sexually and characterized by an excess of heterozygosity (Pearcy et al., 2011). High heterozygosity of workers, close association with humans, and high adaptability in disturbed environments of this species may help explain to some extent how this ant spread rapidly around the world even prior to the 20th century (Weber, 1939; Harris and Berry, 2005; Lester, 2005; Wetterer, 2008; Pearcy et al., 2011). While the precise native range of this ant has been a source of debate and remains uncertain, distribution records of P. longicornis and its closest relatives suggest either a Southeast Asian or African origin (Wetterer, 2008; LaPolla et al., 2010; LaPolla et al., 2013; LaPolla and Fisher, 2014). A comprehensive phylogeographic study of P. longicornis is needed to help identify more precisely where the species originated as well as its subsequent dispersal routes around the globe.
Researchers routinely analyze both mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) data to address questions in molecular ecology and invasion biology. Typically, low genetic variation within a focal population is interpreted as resulting from one or more population bottlenecks after colonization. However, low mtDNA variation also can result from a recent “selective sweep” of a single, highly successful mtDNA variant, a process which may have no discernable effect on nDNA variation (Nei et al., 1975; Aquadro, 1997). This pattern also can stem from indirect selection associated with a selectively-favored, maternally-inherited symbiont (Hurst and Jiggins, 2005). Such symbionts are common in many insect populations and play a major role in shaping host mtDNA evolutionary history (Hurst and Jiggins, 2005; Moran et al., 2008; Charlat et al., 2009; Feldhaar, 2011; Richardson et al., 2012; Bennett and Moran, 2015; Schuler et al., 2016; Schuler et al., 2018). If a maternally-inherited symbiont confers a sufficient selective advantage to spread within and among host populations, the mtDNA variant originally associated with this symbiont may spread with it, and result in a skewed frequency distribution of mtDNA alleles during the process (Caspari and Watson, 1959; Kambhampati et al., 1992; Turelli et al., 1992; Narita et al., 2006; Atyame et al., 2011; Schuler et al., 2016). Several genera of such bacterial symbionts are found in insects, including Wolbachia, Cardinium, Rickettsia, Spiroplasma, and Arsenophonus (Duron et al., 2008; Engelstädter and Hurst, 2009). Among these, Wolbachia appears to be the most widespread maternally-transmitted symbiont in insects (Zug and Hammerstein, 2012; Weinert et al., 2015). Wolbachia variants typically spread within host species by increasing the relative fitness of infected females, either by conferring direct fitness benefits, such as increased fecundity (Vavre et al., 1999; Weeks et al., 2007; Zélé et al., 2018) or providing nutrients (Hosokawa et al., 2010; Nikoh et al., 2014), or by manipulating host reproduction via cytoplasmic incompatibility (CI), male-killing, feminization of genetic males, or thelytokous parthenogenesis (Werren et al., 2008; Saridaki and Bourtzis, 2010; Ma and Schwander, 2017). A relatively high proportion of ant species harbor Wolbachia infections (34%; Russell, 2012; Russell et al., 2012). Thus, possible symbiont effects on mtDNA variation in ants cannot be ignored. Incorporation of data from nuclear genes is essential to verify results obtained for mtDNA data because Wolbachia selective sweeps often, but not always, have little to no effects on nuclear variation (Rokas et al., 2001).
In this study we attempt to understand worldwide genetic variation and prevalence of Wolbachia in P. longicornis. We also assessed the geographic patterns of mtDNA variation in P. longicornis, to see if phylogeographic structure can help track the routes of dispersal of this invasive ant species. Our combined results allow us to test whether Wolbachia have exerted some selective pressure on mtDNA variation in P. longicornis. Also, patterns of mtDNA and nDNA variation were compared for incongruence, which would be predicted if mtDNA variation has been affected by co-evolving reproductive parasite. Lastly, because mtDNA genomes and endosymbionts are maternally co-inherited, analyses of mtDNA structure and variation can shed light on historical transmission patterns (e.g., potential source and spread) of endosymbionts in P. longicornis.
Materials and Methods
mtDNA Sequencing and Phylogenetic Analyses
We obtained P. longicornis workers from field collections and from other researchers (Table S1). A total of 248 ant colonies were sampled across the current geographic distribution of P. longicornis, including 22 colonies from Northeast Asia, 81 colonies from East Asia, 71 colonies from South Asia, 9 colonies from Indian Subcontinent, 17 colonies from Oceania, 9 colonies from Polynesia, 9 colonies from North America, 2 colonies from South America, 19 colonies from Caribbean, 2 colonies from Arabia, 2 colonies from Southeastern Europe, 4 colonies from West Africa, and 1 colony from South Africa. To generate statistically unbiased samples, only a single worker ant was used from each colony for subsequent genetic analyses. DNA was extracted from individual P. longicornis workers using the Gentra Puregene cell and tissue kit (Qiagen, USA) following the manufacturer’s instructions, and stored at −20°C. Portions of the cytochrome oxidase subunit I (COI, 1,203 bp), an intergenic spacer (106 to 127 bp), tRNA–Leu (70 to 77 bp), and the cytochrome oxidase subunit II (COII, 547 bp) genes were amplified via polymerase chain reaction (PCR). PCR was performed using the primer pair C1-J-1745M-F/PLCOII-R2 for partial COI and PLCOII-F1/C2-N-3661R for CO1-tRNA-COII region follow the PCR conditions described below (Degnan et al., 2004; Table S2). PCR mixtures contained 1–2 µL of template DNA, 0.2 µM of each primer, Takara EmeraldAmp Max PCR Master Mix (Takara, Japan) and water (20 µL reactions). PCR conditions included an initial denaturation step at 98°C (3 min) followed by 35 cycles of 94°C (30 s), 52°C (30 s), 72°C (2 min), and a final extension phase at 72°C (7 min). All PCR products were sequenced in both directions by Genomics BioSci and Tech Corp. (Taipei, Taiwan) using an ABI3730 sequencer. Sequence data were assembled using Sequencher 4.9 (GeneCodes).
Sequences were aligned using MUSCLE as implemented in MEGA 6 with default settings (Tamura et al., 2013). The intergenic spacer and tRNA–Leu region were excluded from phylogenetic analyses due to its ambiguous alignment. We performed phylogenetic analyses using two mtDNA datasets, one including all 248 P. longicornis workers and a second containing only a single representative sequence for each of the 43 mitochondrial haplotypes and plus two outgroup taxa, P. zanjensis and P. ankarana (45 OTUs). PartitionFinder 1.0.1 software (Lanfear et al., 2012) was used to determine the best fit substitution model and partitioning scheme based on Akaike information criterion (AIC) scores. PartitionFinder for our full dataset indicated the best scheme had four partitions: first position of COI and COII, second position of COI, third position of COI and COII, and second position of COII. The preferred evolutionary model for these four partitions were GTR + G, HKY + I, GTR + G, and F81, respectively (GTR = General Time Reversible; G = gamma distribution; HKY = Hasegawa-Kishino-Yano; I = proportion of invariable sites; F81 = Felsenstein 1981). For the singleton haplotype dataset (45 OTU), PartitionFinder suggested the best scheme had five partitions: 1) first position of COI, 2) second position of COI, 3) third position of COI and COII, 4) first position of COII, and 5) second position of COII. The preferred evolutionary model for these five partitions was GTR + G, HKY + I, GTR + G, HKY + G, and F81, respectively. These best schemes were used as priors for Bayesian phylogeny inference. A Bayesian phylogeny was reconstructed using MrBayes 3.2.1 (Ronquist et al., 2012). Two independent runs of 107 generations with 4 MCMC (Markov Chain Monte Carlo) chains were conducted simultaneously, starting from random trees and resampling each tree every 1,000 generations. Posterior probabilities were obtained from the 50% majority-rule consensus of trees sampled after discarding the first 25% of sampled trees.
Network Analysis and Neutrality Tests
A median joining mtDNA haplotype network was constructed using POPART (Leigh and Bryant, 2015; software available at: www.popart.otago.ac.nz) to infer relationships among haplotypes. Net genetic divergence between and within groups (p-distance) was estimated using MEGA 6 (Tamura et al., 2013). Population genetic parameters, including number of segregating sites S (Watterson, 1975), number of haplotypes h, haplotype diversity Hd (Nei, 1987), and nucleotide diversity π/bp (Nei, 1987), were estimated using DNASP v5.10 (Librado and Rozas, 2009). This software also was used to perform neutrality tests including Tajima’s D (Tajima, 1989), Fu and Li’s D* and F* tests (Fu and Li, 1993) and McDonald and Kreitman test (McDonald and Kreitman, 1991). A mtDNA sequence from P. zanjensis was used as the outgroup for neutrality tests. Negative values of Tajima’s D, Fu and Li’s D* and F* may reflect a recent population expansion, purifying selection, or genetic hitchhiking, whereas positive values generally reflect a population bottleneck, genetic structure and/or balancing selection. The McDonald and Kreitman test (M–K test) compares the ratio of fixed and polymorphic synonymous and nonsynonymous changes (McDonald and Kreitman, 1991). Additionally, the DHEW test (Zeng et al., 2007b) was performed to detect the signatures of positive selection and hitchhiking on host mtDNA as implemented in the DH program (Zeng et al., 2007a; Zeng et al., 2007b). The DHEW test we used was a compound test of Tajima’s D (Tajima, 1989), Fay and Wu’s Hn (Fay and Wu, 2000), and Ewens–Watterson test (Watterson, 1978), and is thought to be more powerful in detecting positive selection and more robust to historical demographic changes. P-values of the DHEW test were estimated using 100,000 replications of coalescent simulation using DH package (available online: http://zeng-lab.group.shef.ac.uk/wordpress/?page_id=28). Normalized Fay and Wu’s Hn (Fay and Wu, 2000; Zeng et al., 2006) was also calculated using the same package.
Screening for Wolbachia Infection and MLST Sequencing
We screened the DNA samples for Wolbachia using three primer pairs that amplified part of the Wolbachia surface protein gene (wsp), 16S rRNA gene, and cell division protein (ftsZ) (Table S3). PCR primers are published elsewhere and listed in Table S3. PCRs were carried out for each of the three genes, and at least one Wolbachia-positive sample and deionized/distilled H2O were included as positive control and blank, respectively. Three workers per colony were used to determine the infection status for each colony. Our preliminary results indicated a high intra-colony infection rate of Wolbachia in both workers and queens (approximately 0.96–0.97, Tseng et al., unpublished data). All PCR amplicons that yielded a single band on agarose gels were sequenced by Genomics BioSci and Tech Corp. (Taipei, Taiwan) using an ABI3730 sequencer. Some workers appeared to be infected with multiple Wolbachia (see Results section for more details), and, in these cases, sequence data for individual Wolbachia was obtained by PCR using group- or strain-specific primers (Table S4).
Although Wolbachia surface protein (wsp) gene sequence data have been used for phylogenetic analyses in numerous studies, the phylogenetic relationships inferred using data based on a single gene may not be robust due to a high level of recombination among Wolbachia strains (Baldo and Werren, 2007). Therefore, we employed a multilocus sequence typing (MLST) approach developed by Baldo et al. (2006) in which a total of five MLST genes (gatB, coxA, hcpA, ftsZ, and fbpA) were sequenced following the methods of Baldo et al. (2006). Wolbachia strains were characterized by comparisons with other sequences in the Wolbachia MLST database (http://pubmlst.org/Wolbachia/) and NCBI Genbank database (https://www.ncbi.nlm.nih.gov/genbank/). ClonalFrame version 1.1 was used to construct a Wolbachia MLST genealogy (Didelot and Falush, 2007). ClonalFrame accounts for both substitutions and recombination events, providing more reliable clonal relationships based on multilocus data (Didelot and Falush, 2007). Two independent runs were performed, each with 1,000,000 MCMC burn-in iterations and 1,000,000 as sampling period and a sampling frequency of 1,000. A 50% majority rule consensus tree was built from combined data from the two independent runs.
Reconstruction of Ancestral States of Wolbachia Infection Status
The program BayesTraits was utilized to reconstruct the ancestral states of Wolbachia infections in P. longicornis mtDNA lineages (Pagel et al., 2004; Mark and Andrew, 2006). This software considers uncertainties in estimating a tree and its branch lengths when inferring ancestral states, and thus may provide different inference results from the parsimonious expectation. Two Wolbachia strains, wLonA and wLonF, were found in some of our P. longicornis samples (see Results section for details). We tested for a correlation between the occurrence/absence of wLonA and the occurrence/absence of wLonF by performing BayesTrait analyses using both dependent (i.e., the infection history of wLonA was correlated with wLonF) and independent models. The difference between the two models was assessed by Bayes Factor (BF) based on the final harmonic mean of the likelihoods model. A log BF value greater than two was interpreted as supporting the dependent model (i.e., correlated patterns of infections). Prior to the MCMC runs, maximum likelihood analyses were performed using the consensus tree obtained from MrBayes, and the derived results were used to set the priors for MCMC analyses. Considering the results of the likelihood analysis, all MCMC priors were set as uniform distribution for all rates, with different ranges used for each parameter. A total of 7,500 trees were generated by MrBayes (full dataset with 248 OTUs, discarded first 25% trees as burn-in) and used in the MCMC inferences to account for phylogenetic uncertainty. These input trees did not include an outgroup species because we focused only on infection histories of the two Wolbachia strains in P. longicornis. Terminal taxa were coded for presence (1) or absence (0) of Wolbachia infection. The rate deviation parameter was tuned automatically to achieve an average acceptance rate between 20% and 40% and ancestral states were reconstructed using the command “addnode”. The MCMC chains were run for 109 iterations, sampled every 105 iterations with a burn-in of 108 iterations.
We tested for associations between mitochondrial lineages and Wolbachia infection status by using the BaTS program (Bayesian tip-association significance testing) to compute the parsimony score statistic of clustering strength (PS), the association index statistic (AI), and the exclusive single-state clade size statistic (MC) (Parker et al., 2008). PS represents the most parsimonious number of character changes in the phylogeny. AI is an estimate of the frequency of the most common branch tip trait subtended by internal nodes. MC measures the size of the maximum monophyletic clade in which all tips share the same trait. Thus, a significantly lower value of PS, lower AI and higher MC would indicate a strong phylogeny–trait association. The association between mitochondrial lineage and Wolbachia infection is predicted to be strong if Wolbachia infections are transmitted vertically only, while frequent horizontal transfers of Wolbachia infections would erode this association. In BaTS analyses, 7,500 trees were generated by MrBayes as input and tested each parameter by generating a null distribution from 1,000 replicates.
nDNA Analyses
We genotyped a subset of P. longicornis workers from three well-sampled regions (41 colonies from East Asia, 21 colonies from Northeast Asia, and 71 colonies from South Asia) at 20 microsatellite loci (Tseng et al., 2019) (Table S5) to test for congruence (or incongruence) of mtDNA and nuclear DNA variation patterns. DNA of the same worker (one worker per colony) was used for both mtDNA and microsatellite analyses. Microsatellite loci were amplified by using a multiplex PCR method following procedures described by Blacket et al. (2012). The purified PCR products were analyzed on an ABI-3730 Genetic Analyzer (Applied Biosystems) by Genomics BioSci and Tech Co., Ltd (Taipei, Taiwan). GeneMarker (version 2.4.0, Softgenetics LLC) was employed to visualize and score alleles. Genetic variation at each microsatellite locus was characterized in terms of number of alleles (Na), effective number of alleles (Ne), observed (Ho) and expected (He) heterozygosity, Shannon’s information index (I), fixation index (F), and Hedrick’s standardized Gst for small number of populations (G’’ST), using the program GENALEX 6.502 (Peakall and Smouse, 2006).
Genetic structure was assessed using the Bayesian model-based clustering software STRUCTURE 2.3.4 (Pritchard et al., 2000; Hubisz et al., 2009). Five independent STRUCTURE runs were executed for each of K = 1–10 (K, the number of assumed genetic clusters) under the admixture model and allele frequencies correlated with 1,000,000 MCMC iterations and an initial burn-in of 100,000 generations. The optimal number of genetic clusters within the data was estimated by Evanno et al. (2005) in STRUCTURE HARVESTER v. 0.9.94 (Earl and vonHoldt, 2012) (available online: http://taylor0.biology.ucla.edu/structureHarvester/). STRUCTURE results were visualized using CLUMPAK server (Jakobsson and Rosenberg, 2007) (available online: http://clumpak.tau.ac.il/). Genetic relationships among populations were examined by applying a discriminant analysis of principal components (DAPC) (Jombart et al., 2010) available in the R (R Core Team, 2014) package adegenet (Jombart, 2008) on all microsatellite data. Population labels were input as the prior cluster information in DAPC. The first 20 principal components (PCs) accounted for 80% of the total microsatellite genetic variation and were retained from the analysis.
Results
mtDNA Analyses
We sequenced mtDNA of 248 P. longicornis workers (one per colony) from 13 geographic regions. A total of 43 different mtDNA haplotypes were found with 172 polymorphic sites present over the entire 1,750bp COI-COII region (Table S6; collection site information in Table S1). Nucleotide diversity was highest in samples from the Indian Subcontinent (0.041) (Figure 1; Table S6) genetic diversity values from Arabia, Southeastern Europe, West Africa, and South America are likely biased due to low sample size). Nevertheless, the populations across Old World regions exhibit similar levels of genetic diversity. Bayesian phylogenetic analyses indicated the presence of two mtDNA clades (Clade I and II), one of which (Clade II) was divided into three subclades (Clade II-1, -2 and -3) (Figure 2). The average genetic distance between Clades I and II was 0.057, suggesting deep divergence between the two clades. The average pairwise genetic distance among haplotypes was higher within Clade II (0.010) compared with Clade I (0.002). Average genetic distances among workers within the three subclades each had a mean value of 0.001. MtDNA variation was not strongly correlated with geographic location (Figure 3). Workers belonging to Clades I and II were found at 11 of the 13 sampled geographic regions (all except South and West Africa; Figure 3). Workers with haplotypes belonging to subclade II-1 were found in the Old World, but not in the New World (Figure 3). The median-joining network constructed for all 43 unique mtDNA haplotypes further revealed no clear spatial clustering of haplotypes from Clade I (Figure 4). In particular, haplotype Hap08 (Clade I) was common across the sampled ranges and was connected to several tip haplotypes with low frequency, implying that this haplotype may represent a putative ancestral haplotype within Clade I from which the latter are derived. Similar to Clade I, the haplotype network revealed negligible spatial clustering in Clade II (Figure 4), with approximately half of all haplotypes in this clade present in more than one geographic region.
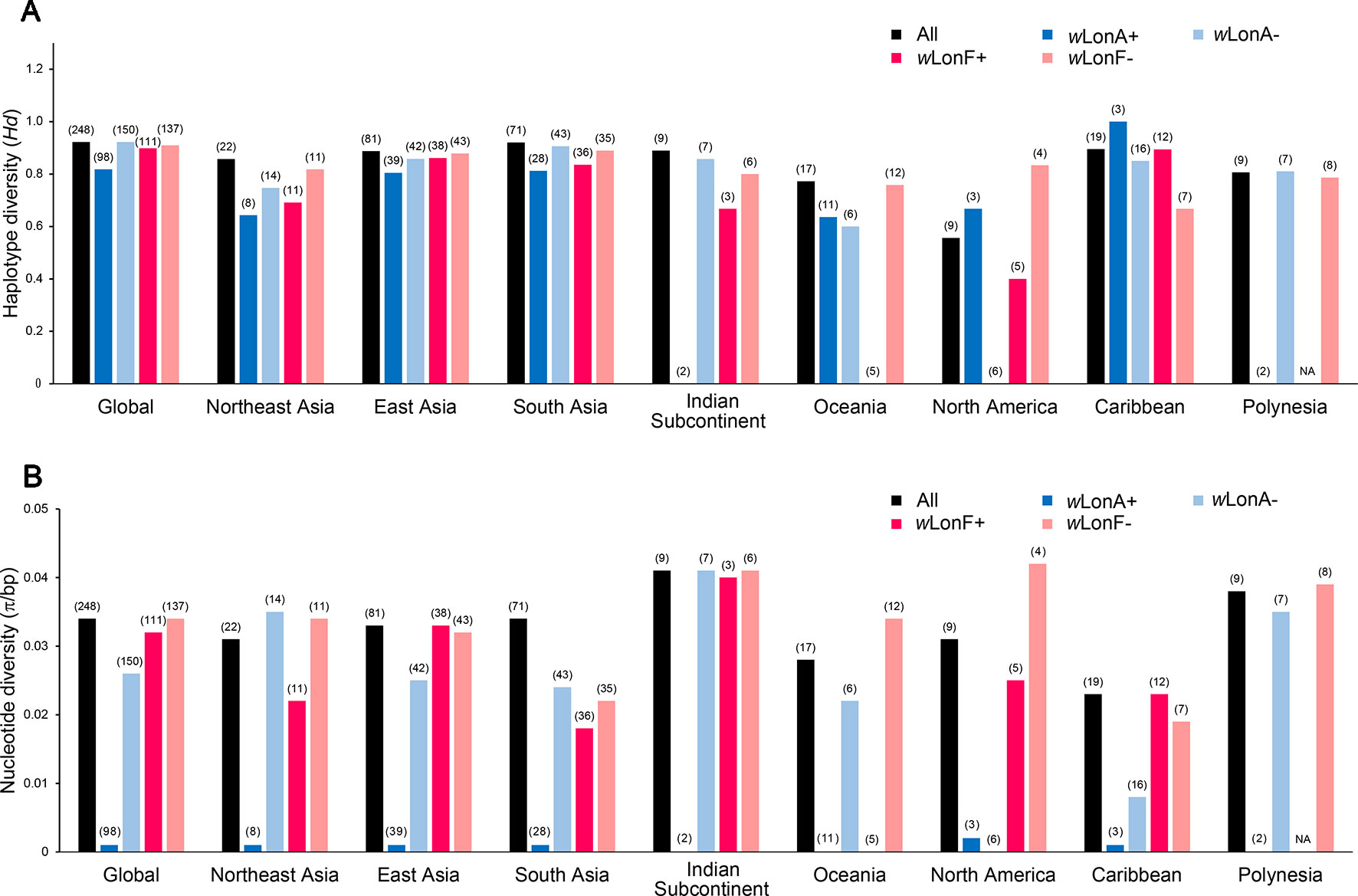
Figure 1 Regional mitochondrial genetic diversity of Paratrechina longicornis as expressed by (A) haplotype diversity and (B) nucleotide diversity with respect to their Wolbachia infection status. wLonA+, wLonA−, wLonF+, and wLonF− denote wLonA-infected, wLonA-uninfected, wLonF-infected, and wLonF-uninfected ants in a given region, respectively. Sample size of each region is indicated in parentheses.
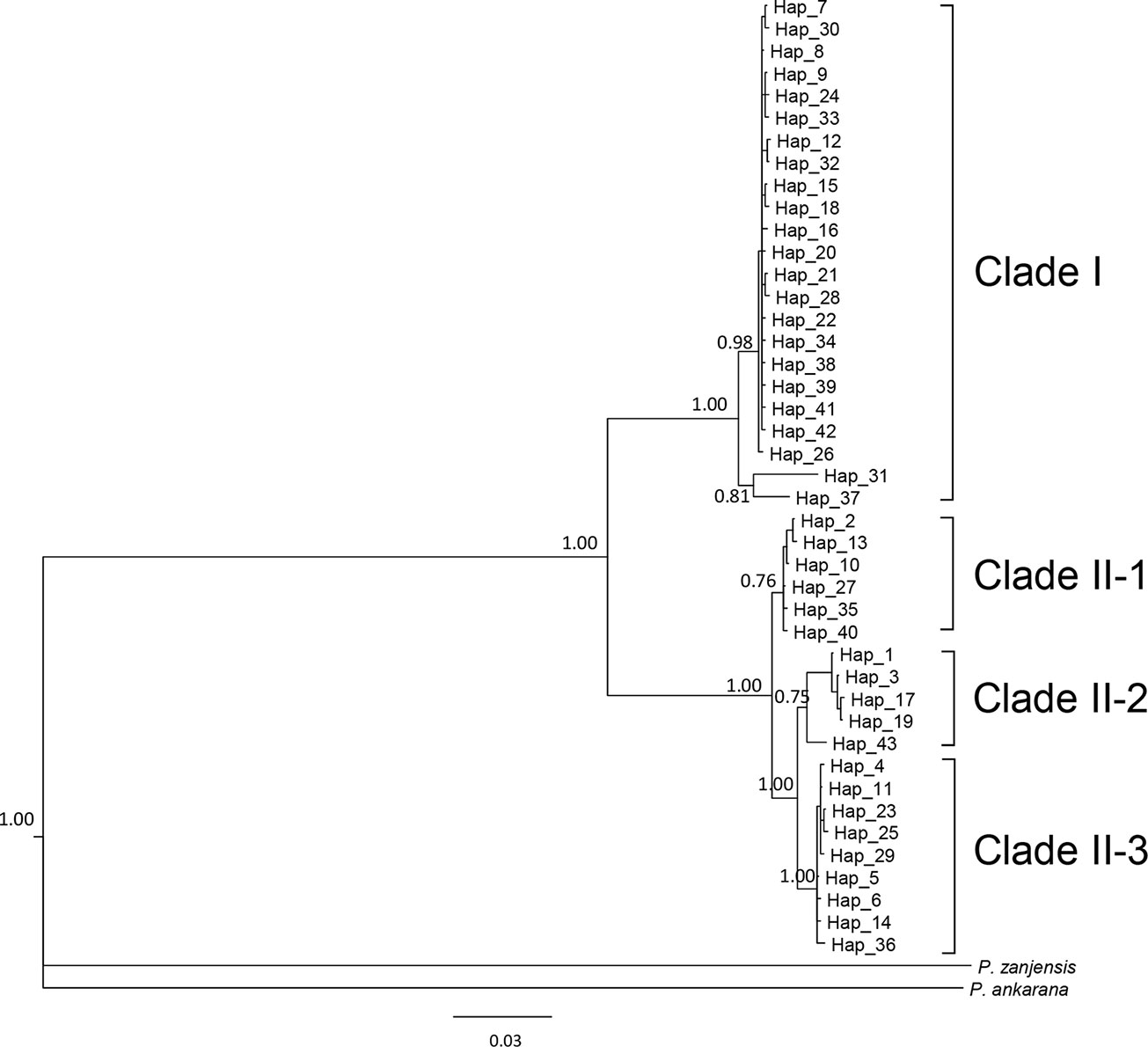
Figure 2 The 50% majority rule consensus tree for all sampled Paratrechina longicornis, inferred by Bayesian analysis. Numbers above branches indicate Bayesian posterior probability calculated by MrBayes. Refer to Table S1 for respective geographic information of each haplotype.
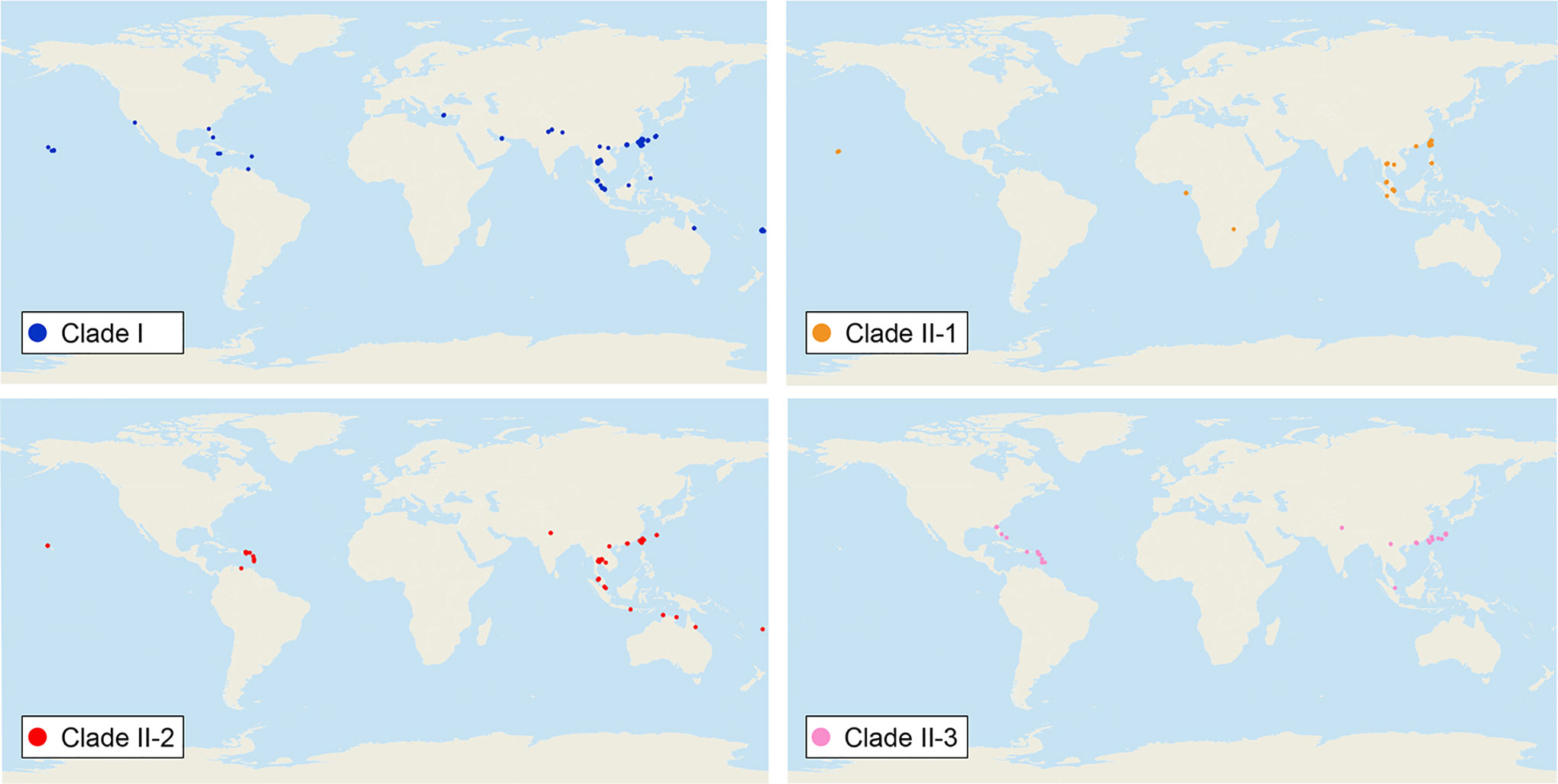
Figure 3 Distribution of all mitochondrial haplogroups in the study regions. Haplogroups are denoted by colors: Clade I (blue), Clade II-1 (orange), Clade II-2 (red), and Clade II-3 (pink).
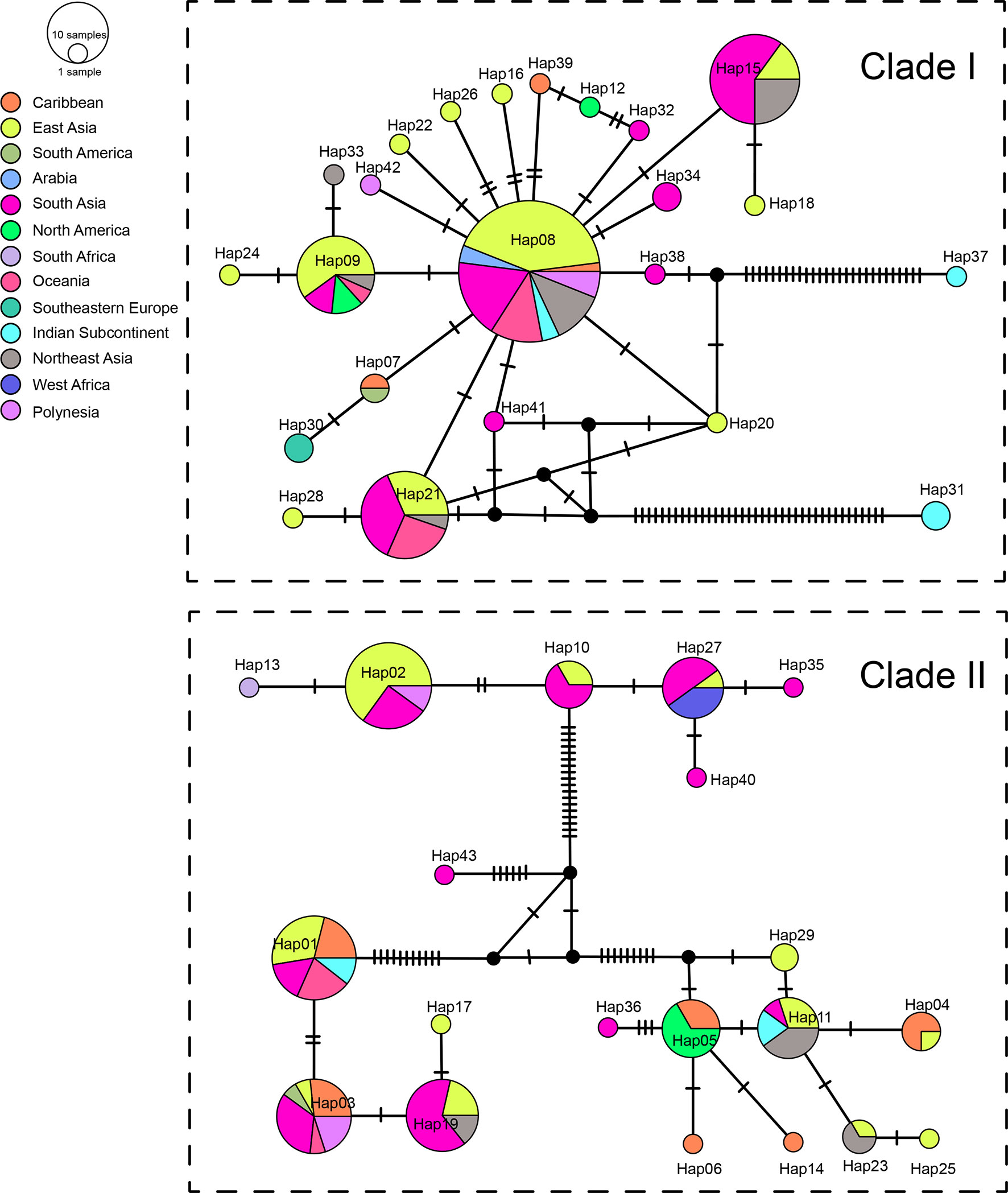
Figure 4 Haplotype networks of the mitochondrial genes. Circle sizes are proportional to the number of sequences per haplotype. Colors correspond to geographic regions.
Wolbachia Infections in P. longicornis
Both sequence data and phylogenetic analyses of concatenated MLST data suggest that two Wolbachia strains, wLonA and wLonF, occur in P. longicornis, with the former belonging to supergroup A and the latter to supergroup F (Figure S1). Forty-two of the 248 P. longicornis workers were infected with wLonA only (17%), 55 workers were infected with wLonF only (22%), 56 workers were co-infected with wLonA and wLonF (23%), and 95 workers were uninfected (38%) (Table 1). Wolbachia infection status was strongly associated with mtDNA variation. Specifically, the majority of ants belonging to Clade I were either infected with wLonA only (33%) or co-infected with wLonA and wLonF (44%), whereas none of workers belonging to Clade II was infected with wLonA (Table 1). We did not observe a significant association between Wolbachia infection status and host geographic range (Figure S2).
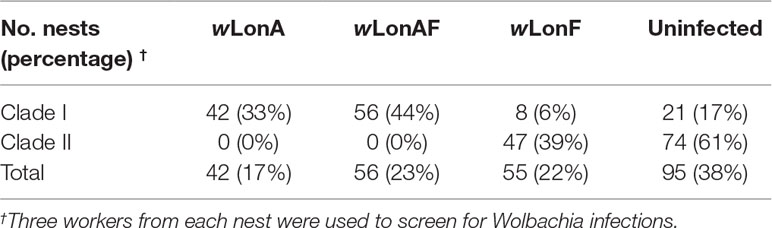
Table 1 Prevalence of WolbachiawLonA and wLonF infections in Paratrechina longicornis.
The MLST allelic profile for wLonA was identical to a sequence type in the Wolbachia MLST database, whereas wLonF represented a new sequence type that has not been reported in the database. wLonA allelic profiles for gatB, coxA, hcpA, ftsZ, fbpA and wsp were 7, 6, 7, 3, 8, and 18, respectively (Table S7). wLonA belongs to sequence type 19 (ST-19), and is similar to Wolbachia variants detected in a moth (Ephestia kuehniella), several ants (Technomyrmex albipes, Leptomyrmex sp., Pheidole plagiara, Ph. sauberi, and Leptogenys sp.) and two butterflies (Ornipholidotos peucetia and Aricia artaxerxes) (Table S7). wLonA shared an identical sequence type with Wolbachia Ekue_A (ID 13) detected from Ephestia kuehniella, a transinfected A group Wolbachia from Cadra cautella (wCauA) to E. kuehniella. WolbachiawCauA has been reported to cause cytoplasmic incompatibility (CI) in C. cautella, and male killing in E. kuehniella (Sasaki et al., 2002; Sasaki et al., 2005). wLonF allelic profiles for gatB, coxA, hcpA, ftsZ, fbpA and wsp were 168, 147, 262, 132, 226, and 708, respectively (Table S7), and were unique to consider wLonF a new sequence type (denoted as ST-471). Similar sequence types included ST-239, ST-242, and ST-243, all of which were detected from two dragonflies (Brachythemis contaminata and Orthetrum sabina). The most similar wsp sequence to wLonF in GenBank was from a Wolbachia variant infecting bat flies Cyclopodia dubia (KT751165; 99% similarity) (Wilkinson et al., 2016).
Wolbachia Infection History in P. longicornis
The BayesTraits analyses indicated the dependent model was not significantly better than the independent model (log Bayes factors = 0.707), suggesting no correlation between wLonA infection status and wLonF infection status. Therefore, infection history of wLonA and wLonF was inferred separately (Figure 5). BayesTraits analyses suggested a single ancestral wLonA infection (on the common ancestor of node 2 or node 3, Figure 5) occurred in P. longicornis that subsequently has been characterized by vertical Wolbachia transmission in the populations of Clade I with only occasional losses of infections (Figure 5). On the other hand, the history wLonF infections in P. longicornis appears to be characterized by frequent gains of wLonF through horizontal transmission as well as frequent losses of wLonF over time. Although the association between wLonF infection status and host mtDNA phylogeny was weaker than that of wLonA (both AI and PS values of wLonF were higher than those of wLonA), the BaTS results indicated that both wLonA and wLonF are significantly associated with the host mtDNA phylogeny (Table 2). These results suggest that wLonF infection within P. longicornis has been shaped by both horizontal and vertical transmission. For example, individuals bearing haplotype 2 (Hap 2) likely obtained wLonF via horizontal transmissions whereas the high prevalence of wLonF in Clade II-3 is consistent with vertical transmission of wLonF over time (Figure 5).
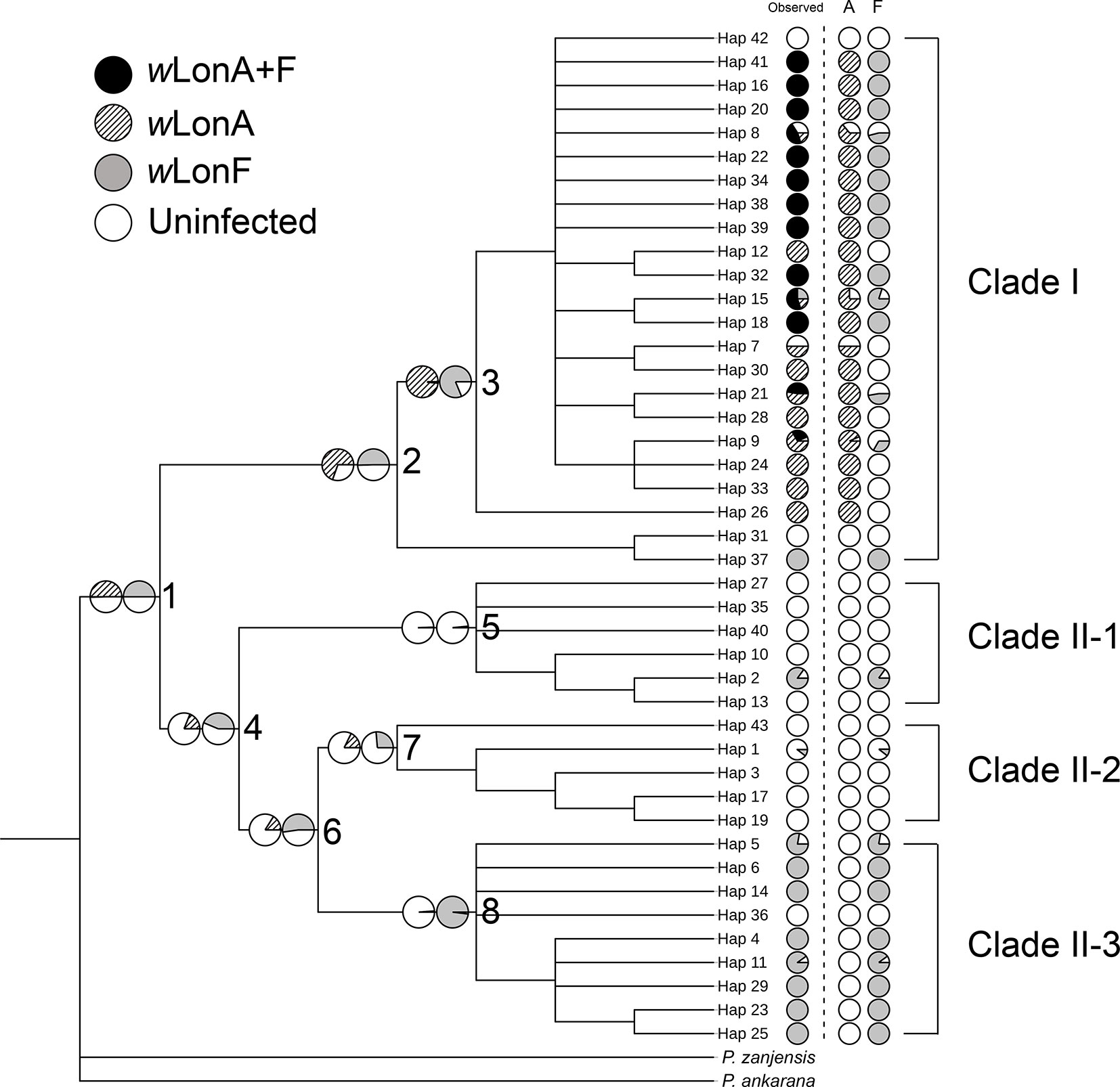
Figure 5 Simulated ancestral states of Wolbachia infection in Paratrechina longicornis inferred by BayesTraits. For each haplotype, pie charts following the haplotype name indicate observed Wolbachia infection status combined, wLonA only and wLonF only (wLonA+F co-infection: black; wLonA infected: upward diagonal; wLonF infected: grey; lack of infection: white). Pie charts on branches indicate simulated probabilities of Wolbachia infected status (left: wLonA; right: wLonF) for each numbered node.
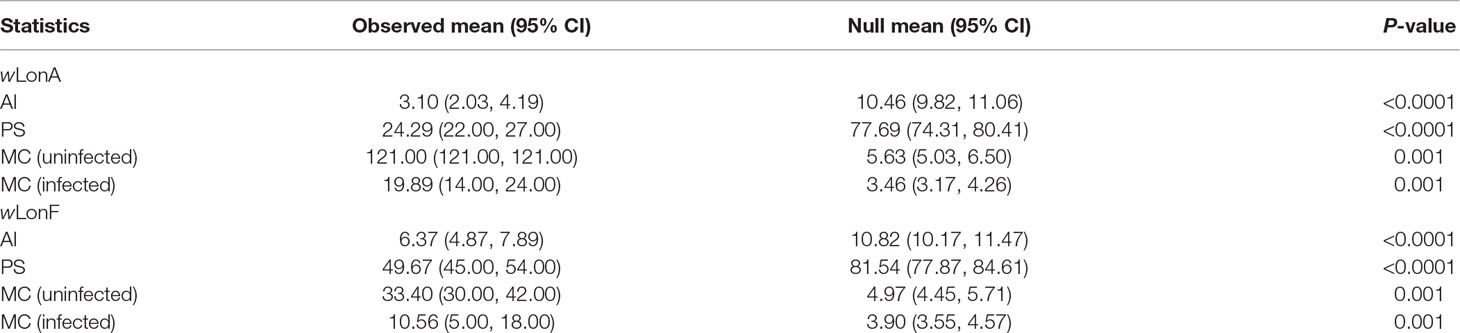
Table 2 Significance of correlations between Paratrechina longicornis mtDNA phylogeny and Wolbachia infection status as identified by BaTS. Association index statistic (AI) and parsimony score (PS) statistic of clustering strength, and exclusive single-state clade size (MC) statistic.
Patterns of mtDNA Variation Within Wolbachia-Infected and -Uninfected Ants
wLonA and wLonF may have undergone independent invasion histories in P. longicornis (Figure 5), therefore wLonA (or F)-positive individuals (including individuals with co-infection) are grouped together when mtDNA variation was analyzed to show the effect of a single strain on the mtDNA evolution. Analyses of mtDNA variation revealed that nucleotide diversity and numbers of segregating sites are much lower in wLonA-infected workers than those in wLonA-uninfected workers within all sampled regions, except North America (Figure 1; Table S6). Estimates of nucleotide diversity were more than 8-fold lower for wLonA-infected workers than in wLonA-uninfected workers despite limited differences in mtDNA variation between wLonF-infected workers and wLonF-uninfected workers.
Statistical tests of departures from neutral expectations are presented in Table S8 and Figure 6. Estimates of Tajima’s D, Fu and Li’s D*, Fu and Li’s F*, and Fay and Wu’s Hn were negative and statistically significant when all wLonA-infected workers were combined (Global group; Table S8; Figure 6). The result of the DHEW test on the combined wLonA-infected workers supported the hypothesis of selection influencing mtDNA variation (Table S8). The neutrality index (NI) of the McDonald–Kreitman (M–K) test was greater than one and deviated from neutral expectations (Table S8). In contrast, Tajima’s D, Fu and Li’s D*, Fu and Li’s F* tests performed on combined samples in other three groups (wLonA-uninfected, wLonF-infected and wLonF-uninfected workers) were all positive, but only five of these estimates were significant (Table S8; Figure 6). The estimates of Fay and Wu’s Hn in wLonA-uninfected and wLonF-uninfected workers were negative and significantly less than zero. However, the results of DHEW tests failed to support the presence of positive selection of these two groups. The results of M–K tests indicated that the NI of these three groups were not significantly different from one.
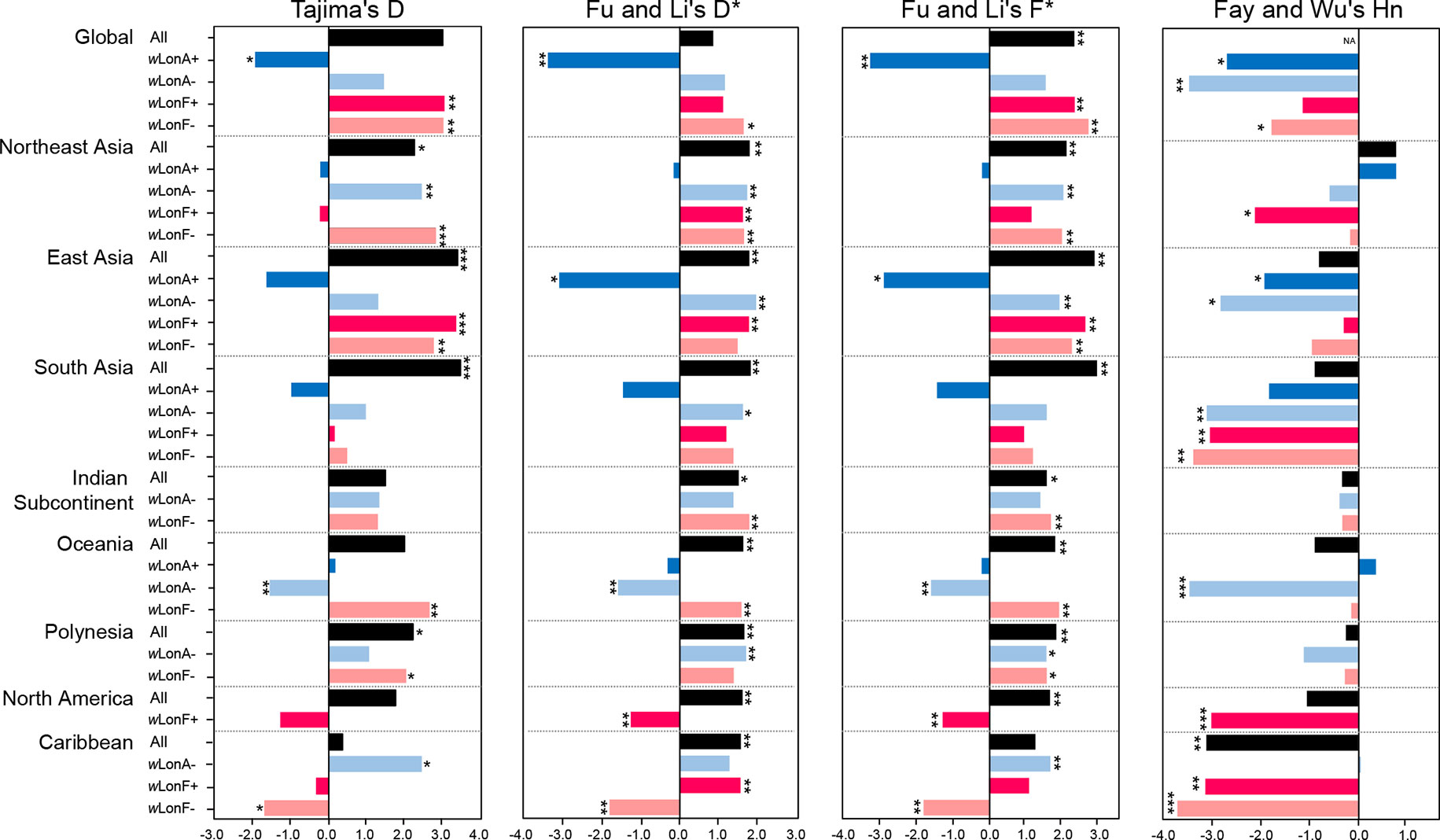
Figure 6 Tests for departure from neutrality for mtDNA sequence variation in Paratrechina longicornis. wLonA+, wLonA−, wLonF+, and wLonF− denote wLonA-infected, wLonA-uninfected, wLonF-infected, and wLonF-uninfected ants in a given region, respectively. *P < 0.05; **P < 0.01; ***P < 0.0001; statistics significantly deviated from expectations under neutrality.
Similar trends were found for estimates of Tajima’s D, Fu and Li’s D*, and Fu and Li’s F*statistic for all groups harboring wLonA (i.e., generally less than zero). However, the normalized Hn statistics of Fay and Wu test and results of DHEW varied among regions (Table S8). We obtained a negative yet significant estimate of Hn only for wLonA-infected workers from East Asia, and the results of DHEW tests for wLonA-infected groups from East Asia and South Asia regions were significant. The NI estimates from M–K test were larger than one for all groups harboring wLonA, and were significant in groups from Northeast and East Asia.
Tajima’s D, Fu and Li’s D*, Fu and Li’s F* generally were positive for wLonA-uninfected, wLonF-infected and wLonF-uninfected groups for each region with few exceptions. The significant negative estimates of D, D*, F*, Hn and significant results of DHEW test were observed in three groups, wLonA-uninfected workers from Oceania, wLonF-infected workers from North America, and wLonF-uninfected workers from Caribbean.
Nuclear DNA Variation and Population Genetic Structure
We compared the extent of mtDNA and nuclear (microsatellite) differentiation in the three selected Asia regions. A total of 191 alleles were observed across all loci for the 134 sampled workers. The average number of alleles in each sampled region ranged from 4.85 to 8.10 (Table S9). Shannon’s information index was used to assess gene diversity (Figure 7; Table S9), and, when incorporating infection status into analysis, genetic diversities among Wolbachia-infected workers are similar to those for uninfected workers across the three selected regions.
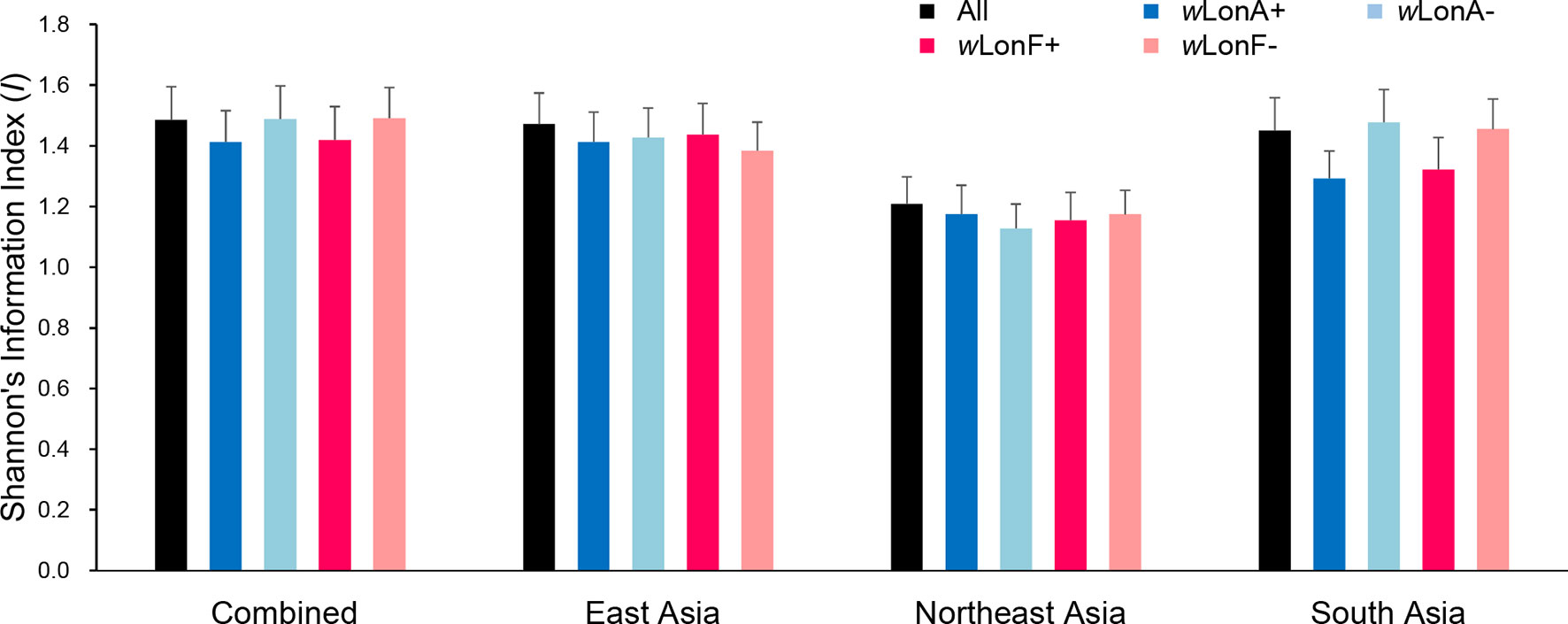
Figure 7 Genetic diversity, as expressed by Shannon’s information index, of Paratrechina longicornis in selected regions based on 20 microsatellite markers. wLonA+, wLonA−, wLonF+, and wLonF− denote wLonA-infected, wLonA-uninfected, wLonF-infected, and wLonF-uninfected ants in a given region, respectively. Error bars indicate standard errors.
Bayesian cluster analysis performed using STRUCTURE revealed six distinct genetic clusters for the entire data set (K = 3 based on ∆K statistic; Table S10). Most workers were admixed (i.e., had membership in more than one cluster; Figure S3), and genetic differentiation among geographic regions or mtDNA clades was not observed. DAPC analysis indicated the lack of differentiation among groups in each of the three selected regions as well as between the two mtDNA clades (Figure S4). The estimate of G’’ST between the two mtDNA clades was 0.049 (P = 0.001), suggesting a low level of nuclear differentiation between workers from the two mtDNA clades.
Discussion
Our results showed that global patterns of mtDNA and nDNA variation among populations of P. longicornis were discordant, characterized by two highly divergent mtDNA clades with no parallel pattern of nuclear genetic divergence (based on microsatellite loci) between workers from the two mtDNA clades. Several evolutionary scenarios possibly explaining such mitochondrial-nuclear discordance include sex-biased dispersal, local adaptation, historical demography, incomplete lineage sorting, and endosymbiont-driven hitchhiking effects (reviewed in Toews and Brelsford, 2012). Male-biased dispersal (López-Uribe et al., 2014) and local adaptation of mtDNA haplotypes (Cheviron and Brumfield, 2009; Ribeiro et al., 2011; Spottiswoode et al., 2011) are unlikely for at least two reasons: 1) the geographical distributions of ants from the two mtDNA clades overlap considerably and coexist in virtually all geographic regions we sampled and 2) all ant samples were collected from human-modified habitats (e.g., roadsides, parks or near buildings), suggesting negligible habitat preference between ants from the two clades. The strong association between Wolbachia infection status and host mtDNA lineage, as well as reduced mtDNA diversity associated with wLonA in P. longicornis, are consistent with Wolbachia influencing patterns of host mtDNA structure and variation. Levels of nuclear variation were nearly identical for ants from the wLonA-infected workers (mtDNA Clade I) and uninfected groups (mtDNA Clade II), which is consistent with the prediction that Wolbachia endosymbionts have minimal or no effects on nuclear genetic variation and divergence (assuming host reproduces sexually) due to biparental inheritance (Rokas et al., 2001).
Results from additional analyses also were largely consistent with the predicted effects of Wolbachia on mtDNA variation. Tajima’s D, Fu and Li’s D*, and Fu and Li’s F* tests for departures from neutral evolution were negative for all groups harboring wLonA and for global datasets. Moreover, the results of Fay and Wu’s Hn and DHEW were also consistent with expected patterns for a relatively recent Wolbachia-driven selective sweep occurring in some, but not all, geographic regions, such as East and South Asia, despite the fact that mtDNA variation is low in almost all populations harboring wLonA in every geographic region. One potential explanation for these inconsistencies is that selective sweeps of Wolbachia in some of these populations may have occurred far enough in the distant past such that any signature of selection on the mtDNA may have been eroded. The results of M–K tests also imply mtDNA substitution patterns may have been influenced by wLonA, but surprisingly that a signature of purifying selection is registered. The NI values of M–K test for groups harboring wLonA ranged between 5,074 to 12,888, and were significantly larger than values for groups from Northeast and East Asia. One possible explanation is that hitchhiking events associated with wLonA resulted in accumulation of slightly deleterious mutations (Shoemaker et al., 2000; Fay and Wu, 2000) followed by negative selection as Wolbachia-driven haplotype replacements cease (Bazykin and Kondrashov, 2011).
The virtual absence of genetic structure across a large geographic area in P. longicornis and the co-occurrence of divergent mtDNA haplotypes in almost every geographic region suggest that human-mediated, long-distance movement of this species is common. The time of divergence between any random pair of wLonA-uninfected groups (i.e., mtDNA subclades II-1, 2, and 3) is roughly 34,000 years ago (estimated based on average of pairwise genetic distances assuming a substitution rate of 1.455% per site per million years, the estimated rate for the COI gene of ants; Resende et al., 2010), which apparently predates potential human dispersal. However, this divergence, along with the deep divergence between Clades I and II, may stem from accelerated mtDNA substitution rates due to recurrent Wolbachia sweeps (Shoemaker et al., 2000), with the assumption that the former had infected ancestor at some point in time in the past. A likely explanation for the specific genetic patterns observed in P. longicornis is the ant has experienced multiple human-mediated dispersal events from genetically distinct source populations followed by global dispersal (i.e., high propagule pressure as a result of rampant migration/movement). The genetic patterns we describe are similar to those found in several other globally distributed insects, especially those that are common in human-modified landscapes [e.g., German cockroach (Blattella germanica), American cockroach (Periplaneta americana); Vargo et al., 2014; von Beeren et al., 2015], further highlighting the role of human-mediated dispersal in shaping population structure of insect species closely associated with humans.
Reduction in genetic variation as a result of a population bottleneck is a common feature observed in the introduced ranges of numerous invasive species (Nei et al., 1975; Allendorf and Lundquist, 2003; Dlugosch and Parker, 2008). The general consensus is that P. longicornis originated in the Old World tropics, but to narrow this down further to a specific sub-region has been somewhat controversial (Wetterer, 2008). We did not find evidence for reduced mtDNA diversity in any sampled subregions of the Old World. Also, mtDNA structure within Clade II appears to be less associated with Wolbachia infections, and inferring the origin of this ant using mtDNA patterns of variation in Clade II remains challenging primarily due to insufficient sampling in certain areas (Table S11). Identification of the native range of P. longicornis on a finer geographic scale is further obscured by presumed frequent human-mediated dispersals, a multi-century old invasion history, and the potential effects of Wolbachia infections on mtDNA variation. However, it is interesting to note that haplotypes from the northern part of India and Nepal (Himalayan region) (Hap31 and Hap37) are divergent from other haplotypes in Clade I and form a clade sister to all other haplotypes in this clade (Figure 2), implying the populations in Himalayan region might be the source of invasive populations of Clade I. More comprehensive sampling and additional nuclear data from queens and males may help efforts to identify the likely origin and to reconstruct with more confidence the invasion history of this ant.
The loss of Wolbachia in invasive ranges is common, but not universal, in invasive insects possibly due to founder effects or altered selection pressures in the new habitats (Shoemaker et al., 2000; Tsutsui et al., 2003; Reuter et al., 2004; Yang et al., 2010; Nguyen et al., 2016). For example, both the red imported fire ant, Solenopsis invicta, and the Argentine ant, Linepithema humile, had higher Wolbachia infection prevalences in their native populations compared with introduced populations where the symbionts are nearly absent (Shoemaker et al., 2000; Tsutsui et al., 2003; Reuter et al., 2004; Yang et al., 2010). However, we did not observe a similar phenomenon in P. longicornis. Both wLonA and wLonF are found throughout geographic range of P. longicornis, including known invasive areas such as North America and Caribbean (Figure S2). Nevertheless, a few individuals in clade I (wLonA lost) and subclade II-3 (wLonF lost) have lost Wolbachia and this loss appears to be stochastic. The loss of Wolbachia likely is attributable to imperfect maternal transmission or natural curing events (Stevens and Wicklow, 1992; Hoffmann and Turelli, 1997; Clancy and Hoffmann, 1998).
Our simulation results suggest wLonA was acquired by the common ancestor of Clade I, and the fitness advantage associated with harboring wLonA infections compared with uninfected ants may have facilitated the spread of Wolbachia and the associated mtDNA haplotype. One possible fitness advantage of harboring wLonA infections could be Wolbachia-induced cytoplasmic incompatibility (CI). Although spread of Wolbachia inducing CI in haplodiploid species appears to be less efficient than in diploid species (Vavre et al., 2000), limited movement of P. lonigicornis (Trager, 1984; Harris and Berry, 2005) could have enabled Wolbachia to increase in frequency within small local populations through genetic drift, allowing the bacterium to exceed the threshold frequency for spread in a host population (Vavre et al., 2003). While this possibility remains to be tested, a survey of Wolbachia prevalence across numerous ant species appears supportive that Wolbachia infections generally are more prevalent in ant species that have limited mobility (i.e. reproducing by budding or fusion) (Wenseleers et al., 1998).
In contrast, wLonF appears to have been gained and lost multiple times in P. longicornis over evolutionary time and has had little or no significant effect on host mtDNA variation. One possible explanation for this pattern is that wLonF is simply a passive passenger in longhorn crazy ant (e.g., having negligible fitness effects), and persists because the rate of wLonF loss occurs roughly at the same rate as horizontal transmission (Hoffmann et al., 1996; Charlat et al., 2004; Bouwma and Shoemaker, 2011). Invasive species may acquire new Wolbachia in their new environments (Rocha et al., 2005; Himler et al., 2011; Schuler et al., 2013). For example, the North American fruit fly, Rhagoletis cingulata (Diptera: Tephritidae) acquired a new Wolbachia strain through interspecific horizontal transmission from the Eurasian endemic R. cerasi (Schuler et al., 2013). However, supergroup F Wolbachia, to the best of our knowledge, has only been discovered in a single ant species, Ocymyrmex picardi (Russell et al., 2009), suggesting that the prevalence of this variant is very low in ants, and that acquisition of this variant from other sympatric ant species in the introduced range of P. longicornis appears unlikely. Effective and efficient horizontal transmission of Wolbachia depends on intimate ecological associations that provide opportunities to bring Wolbachia into close contact with novel hosts (Sintupachee et al., 2006; Stahlhut et al., 2010; Boivin et al., 2014). Host-parasitoid associations have been commonly suggested as a route of the Wolbachia horizontal transmission, and evidence this has occurred includes between host and parasitoid (Heath et al., 1999), between hosts (Ahmed et al., 2015), and between parasitoids sharing the same host (Huigens et al., 2000; Huigens et al., 2004). Other putative ecological associations for successful Wolbachia horizontal transmission are prey-predator and parasite-host associations (Le Clec’h et al., 2013; Brown and Lloyd, 2015). One well-known case for the latter involves an inquiline social parasite (Solenopsis daguerrei) and its ant host (S. invicta) (Dedeine et al., 2005). While no social parasites have been reported in P. longicornis to date, colonies of this ant often host a variety of arthropods, such as the ant cricket Myrmecophilus americanus, the beetle Coluocera maderae and the ant mite Macrodinychus multispinosus (Wollaston, 1854; Wetterer, 2008; Werren et al., 2008; Lachaud et al., 2016). These arthropods, termed “myrmecophiles,” represent good candidates for Wolbachia transfer because they all have intimate ecological associations and interactions with their ant hosts (Kronauer and Pierce, 2011). A future study of Wolbachia in the organisms ecologically associated with P. longicornis may uncover the routes and mechanisms underlying Wolbachia horizontal transmission in this ant.
Data Availability
MtDNA sequences (accession numbers KY769964-KY770018) are deposited at GenBank. The sequences of the five MLST genes and wsp gene of two Wolbachia strains are deposited at PubMLST database (ID:1827 and 1828).
Ethics Statement
We thank Future Development Funding Program of the Kyoto University Research Coordination Alliance for financial support.
Author Contributions
CCSY, S-PT, and DS originally conceived the ideas. JW, AS, and C-YL helped develop an overarching research programme and collect the samples. TY and CCSY applied the grant. SPT performed the experiments. S-PT, DS, and CCSY carried out the analyses and drafted the manuscript. All authors contributed substantially to editing the manuscript.
Funding
We thank Future Development Funding Program of the Kyoto University Research Coordination Alliance for financial support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Amy Low, Ni-Chen Chang, Chung-Wei You, Chùn-Têng Coody Chiu, Chun-Yi Lin, Ching-Chen Lee, En-Cheng Yang, Han-Chih Ho, Hui-Siang Tee, Hugo Darras, Hung-Wei Hsu, Jia-Wei Tay, Kean Teik Koay, Li Yan Gan, Mark Ooi, Nellie Wong, Peter G. Hawkes, Ping-Chih Lin, Po-Wei Hsu, Singham Veera, Su-Chart Lee, Sylvain Hugel, Yi-Ming Weng, Yueh-Hua Wu, Yu-Fang Tseng, and Zhengwei Jong, for assistance in field collection. We also thank the Future Development Funding Program of the Kyoto University Research Coordination Alliance for financial support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2019.00838/full#supplementary-material
References
Abdelkrim, J., Pascal, M., Calmet, C., Samadi, S. (2005). Importance of assessing population genetic structure before eradication of invasive species: examples from insular Norway rat populations. Conserv. Biol. 19, 1509–1518. doi: 10.1111/j.1523-1739.2005.00206.x
Ahmed, M. Z., Li, S. J., Xue, X., Yin, X. J., Ren, S. X., Jiggins, F. M., et al. (2015). The intracellular bacterium Wolbachia uses parasitoid wasps as phoretic vectors for efficient horizontal transmission. PLoS Pathogens. 11, e1004672. doi: 10.1371/journal.ppat.1004672
Allendorf, F. W., Lundquist, L. L. (2003). Introduction: population biology, evolution, and control of invasive species. Conserv. Biol. 17, 24–30. doi: 10.1046/j.1523-1739.2003.02365.x
Aquadro, C. F. (1997). Insights into the evolutionary process from patterns of DNA sequence variability. Curr. Opin. Genet. Dev. 7, 835–840. doi: 10.1016/S0959-437X(97)80048-2
Atyame, C. M., Delsuc, F., Pasteur, N., Weill, M., Duron, O. (2011). Diversification of Wolbachia endosymbiont in the Culex pipiens mosquito. Mol. Bio. Evol. 28, 2761–2772. doi: 10.1093/molbev/msr083
Baldo, L., Dunning Hotopp, J. C., Jolley, K. A., Bordenstein, S. R., Biber, S. A., Choudhury, R. R., et al. (2006). Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl. Environ. Microbiol. 72, 7098–7110. doi: 10.1128/AEM.00731-06
Baldo, L., Werren, J. H. (2007). Revisiting Wolbachia supergroup typing based on WSP: spurious lineages and discordance with MLST. Curr. Microbiol. 55, 81–87. doi: 10.1007/s00284-007-0055-8
Bazykin, G. A., Kondrashov, A. S. (2011). Detecting past positive selection through ongoing negative selection. Genome Biol. Evol. 3, 1006-1013. doi: 10.1093/gbe/evr086
Bennett, G. M., Moran, N. A. (2015). Heritable symbiosis: the advantages and perils of an evolutionary rabbit hole. Proc. Natl. Acad. Sci. U.S.A. 112, 10169– 10176. doi: 10.1073/pnas.1421388112
Blacket, M. J., Robin, C., Good, R. T., Lee, S. F., Miller, A. D. (2012). Universal primers for fluorescent labelling of PCR fragments-an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 12, 456– 463. doi: 10.1111/j.1755-0998.2011.03104.x
Boivin, T., Henri, H., Vavre, F., Gidoin, C., Veber, P., Auger-Rozenberg, M. A. (2014). Epidemiology of asexuality induced by the endosymbiotic Wolbachia across phytophagous wasp species: host plant specialization matters. Mol. Ecol. 23, 2362–2375. doi: 10.1111/mec.12737
Brown, A. N., Lloyd, V. K. (2015). Evidence for horizontal transfer of Wolbachia by a Drosophila mite. Exp. Appl. Acarol. 66, 301–311. doi: 10.1007/s10493-015-9918-z
Bouwma, A. M., Shoemaker, D. (2011). Wolbachia wSinvictaA infections in natural populations of the fire ant Solenopsis invicta: testing for phenotypic effects. J. Insect Sci. 11. doi: 10.1673/031.011.0111
Caspari, E., Watson, G. S. (1959). On the evolutionary importance of cytoplasmic sterility in mosquitoes. Evolution 13, 568–570. doi: 10.1111/j.1558-5646.1959.tb03045.x
Chadès, I., Martin, T. G., Nicol, S., Burgman, M. A., Possingham, H. P., Buckley, Y. M. (2011). General rules for managing and surveying networks of pests, diseases, and endangered species. Proc. Natl. Acad. Sci. U.S.A. 108, 8323–8328. doi: 10.1073/pnas.1016846108
Cheviron, Z. A., Brumfield, R. T. (2009). Migration-selection balance and local adaptation of mitochondrial haplotypes in rufous-collared sparrows (Zonotrichia capensis) along an elevational gradient. Evolution 63, 1593–1605. doi: 10.1111/j.1558-5646.2009.00644.x
Charlat, S., Ballard, J. W. O., Mercot, H. (2004). What maintains noncytoplasmic incompatibility inducing Wolbachia in their hosts: a case study from a natural Drosophila yakuba population. J. Evol. Biol. 17, 322–330. doi: 10.1046/j.1420-9101.2003.00676.x
Charlat, S., Duplouy, A., Hornett, E. A., Dyson, E. A., Davies, N., Roderick, G. K., et al. (2009). The joint evolutionary histories of Wolbachia and mitochondria in Hypolimnas bolina. BMC Evol. Biol. 9, 64. doi: 10.1186/1471-2148-9-64
Clancy, D. J., Hoffmann, A. A. (1998). Environmental effects on cytoplasmic incompatibility and bacterial load in Wolbachia-infected Drosophila simulans. Entomol. Exp. Appl. 86, 13–24. doi: 10.1046/j.1570-7458.1998.00261.x
Cristescu, M. E. (2015). Genetic reconstructions of invasion history. Mol. Ecol. 24, 2212–2225. doi: 10.1111/mec.13117
Dedeine, F., Ahrens, M., Calcaterra, L., Shoemaker, D. D. (2005). Social parasitism in fire ants (Solenopsis spp.): a potential mechanism for interspecies transfer of Wolbachia. Mol. Ecol. 14, 1543–1548. doi: 10.1111/j.1365-294X.2005.02499.x
Degnan, P. H., Lazarus, A. B., Brock, C. D., Wernegreen, J. J. (2004). Host–symbiont stability and fast evolutionary rates in an ant–bacterium association: cospeciation of Camponotus species and their endosymbionts, Candidatus Blochmannia. Syst. Biol. 53, 95–110. doi: 10.1080/10635150490264842
Didelot, X., Falush, D. (2007). Inference of bacterial microevolution using multilocus sequence data. Genetics 175, 1251–1266. doi: 10.1534/genetics.106.063305
Dlugosch, K. M., Parker, I. M. (2008). Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449. doi: 10.1111/j.1365-294X.2007.03538.x
Duron, O., Bouchon, D., Boutin, S., Bellamy, L., Zhou, L., Engelstädter, J., et al. (2008). The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 6, 27. doi: 10.1186/1741-7007-6-27
Earl, D. A., vonHoldt, B. M. (2012). Structure harvester: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Engelstädter, J., Hurst, G. D. (2009). The ecology and evolution of microbes that manipulate host reproduction. Annu. Rev. Ecol. Syst. 40, 127–149. doi: 10.1146/annurev.ecolsys.110308.120206
Estoup, A., Guillemaud, T. (2010). Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19, 4113–4130. doi: 10.1111/j.1365-294X.2010.04773.x
Evanno, G., Regnaut, S., Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14; 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Fay, J. C., Wu, C.-I. (2000). Hitchhiking under positive Darwinian selection. Genetics 155, 1405–1413.
Feldhaar, H. (2011). Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol. Entomol. 36, 533–543. doi: 10.1111/j.1365-2311.2011.01318.x
Harris, R., Berry, J. (2005). “Paratrechina longicornis (LATREILLE),” in Invasive ant threat information sheet 20. Landcare Research contract report to Biosecurity New Zealand (New Zealand: Lincoln), 61.
Heath, B. D., Butcher, R. D. J., Whitfield, W. G. F., Hubbard, S. F. (1999). Horizontal transfer of Wolbachia between phylogenetically distant insect species by a naturally occurring mechanism. Curr. Biol. 9, 313–316. doi: 10.1016/S0960-9822(99)80139-0
Himler, A. G., Adachi-Hagimori, T., Bergen, J. E., Kozuch, A., Kelly, S. E., Tabashnik, B. E., et al. (2011). Rapid spread of a bacterial symbiont in an invasive whitefly is driven by fitness benefits and female bias. Science 332, 254– 256. doi: 10.1126/science.1199410
Hosokawa, T., Koga, R., Kikuchi, Y., Meng, X.-Y., Fukatsu, T. (2010). Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. U.S.A. 107, 769–774. doi: 10.1073/pnas.0911476107
Hoffmann, A. A., Clancy, D. J., Duncan, J. (1996). Naturally occurring Wolbachia infection in Drosophila simulans that does not cause cytoplasmic incompatibility. Heredity 76, 1–8. doi: 10.1038/hdy.1996.1
Hoffmann, A. A., Turelli, M., (1997). “Cytoplasmic incompatibility in insects,” in Influential passengers inherited microorganisms and arthropod reproduction. Eds. O’Neill, S. L., Hoffmann, A. A., Werren, J. H. (Oxford: Oxford University Press), 42–80.
Hubisz, M. J., Falush, D., Stephens, M., Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x
Huigens, M. E., de Almeida, R. P., Boons, P. A. H., Luck, R. F., Stouthamer, R. (2004). Natural interspecific and intraspecific horizontal transfer of parthenogenesis-inducing Wolbachia in Trichogramma wasps. Proc. R. Soc. Lond. B. Biol. Sci. 271, 509–515. doi: 10.1098/rspb.2003.2640
Huigens, M. E., Luck, R. F., Klaassen, R. H. G., Maas, M. F. P. M., Timmermans, M. J. T. N., Stouthamer, R. (2000). Infectious parthenogenesis. Nature 405, 178. doi: 10.1038/35012066
Hurst, G. D. D., Jiggins, F. M. (2005). Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. Lond. B. Biol. Sci. 272, 1525–1534. doi: 10.1098/rspb.2005.3056
Jakobsson, M., Rosenberg, N. A. (2007). CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Jombart, T. (2008). Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., Devillard, S., Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94. doi: 10.1186/1471-2156-11-94
Kambhampati, S., Rai, K. S., Verleye, D. M. (1992). Frequencies of mitochondrial DNA haplotypes in laboratory cage populations of the mosquito, Aedes albopictus. Genetics 132, 205–209.
Kronauer, D. J. C., Pierce, N. E. (2011). Myrmecophiles. Curr. Biol. 21, R208–R209. doi: 10.1016/j.cub.2011.01.050
Lachaud, J. P., Klompen, H., Pérez-Lachaud, G. (2016). Macrodinychus mites as parasitoids of invasive ants: an overlooked parasitic association. Sci. Rep. 6, 29995. doi: 10.1038/srep29995
Lanfear, R., Calcott, B., Ho, S. Y. W., Guindon, S. (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 29, 1695–1701. doi: 10.1093/molbev/mss020
LaPolla, J., Hawkes, P., Fisher, J. N. (2013). Taxonomic review of the ant genus Paratrechina, with a description of a new species from Africa. J. Hymenopt. Res. 35, 71–82. doi: 10.3897/jhr.35.5628
LaPolla, J. S., Brady, S. G., Shattuck, S. O. (2010). Phylogeny and taxonomy of the Prenolepis genus-group of ants (Hymenoptera: Formicidae). Syst. Entomol. 35, 118–131. doi: 10.1111/j.1365-3113.2009.00492.x
LaPolla, J. S., Fisher, B. L. (2014). Then there were five: a reexamination of the ant genus Paratrechina (Hymenoptera, Formicidae). ZooKeys 422, 35–48. doi: 10.3897/zookeys.422.7779
Latreille, P. A. (1802). Histoire naturelle des fourmis, et recueil de memoires et d’observations sur les abeilles, les araignees, les faucheurs, et autres insectes. Paris: De Crapelet, 445. doi: 10.5962/bhl.title.65810
Le Clec’h, W., Chevalier, F. D., Genty, L., Bertaux, J., Bouchon, D., Sicard, M. (2013). Cannibalism and predation as paths for horizontal passage of Wolbachia between terrestrial isopods. PloS one 8, e60232. doi: 10.1371/journal.pone.0060232
Leigh, J. W., Bryant, D. (2015). POPART: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Le Roux, J., Wieczorek, A. M. (2009). Molecular systematics and population genetics of biological invasions: towards a better understanding of invasive species management. Ann. Appl. Biol. 154, 1–17. doi: 10.1111/j.1744-7348.2008.00280.x
Leppc, E., Gollasch, S., Olenin, S. (2002). “Alien freshwater fishes of Europe,” in Invasive Aquatic Species of Europe: Distribution, Impacts and Management. Eds. Leppa¨koski, E., Gollasch, S., Olenin, S. (Dordrecht: Kluwer Academic Publishers), 153–161.
Lester, P. J. (2005). Determinants for the successful establishment of exotic ants in New Zealand. Divers. Distrib. 11, 279–288. doi: 10.1111/j.1366-9516.2005.00169.x
Librado, P., Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
López-Uribe, M. M., Zamudio, K. R., Cardoso, C. F., Danforth, B. N. (2014). Climate, physiological tolerance and sex-biased dispersal shape genetic structure of Neotropical orchid bees. Mol. Ecol. 23, 1874–1890. doi: 10.1111/mec.12689
Ma, W. J., Schwander, T. (2017). Patterns and mechanisms in instances of endosymbiont-induced parthenogenesis. J. Evol. Biol. 30, 868–888. doi: 10.1111/jeb.13069
Mark, P., Andrew, M. (2006). Bayesian analysis of correlated evolution of discrete characters by reversible-jump Markov chain Monte carlo. Am. Nat. 167, 808–825. doi: 10.1086/503444
McDonald, J. H., Kreitman, M. (1991). Adaptive protein evolution at the Adh locus in Drosophila. Nature 351, 652–654. doi: 10.1038/351652a0
Meyerson, L. A., Mooney, H. A. (2007). Invasive alien species in an era of globalization. Front. Ecol. Environ. 5, 199–208. doi: 10.1890/1540-9295(2007)5[199:IASIAE]2.0.CO;2
Moran, N. A., McCutcheon, J. P., Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Narita, S., Nomura, M., Kato, Y., Fukatsu, T. (2006). Genetic structure of sibling butterfly species affected by Wolbachia infection sweep: evolutionary and biogeographical implications. Mol. Eco. 15, 1095–1108. doi: 10.1111/j.1365-294X.2006.02857.x
Nei, M. (1987). Molecular evolutionary genetics. New York: Columbia University Press. doi: 10.7312/nei-92038
Nei, M., Maruyama, T., Chakraborty, R. (1975). The bottlenect effect and genetic variability in populations. Evolution 29, 1–10. doi: 10.1111/j.1558-5646.1975.tb00807.x
Nguyen, D. T., Spooner-Hart, R. N., Riegler, M. (2016). Loss of Wolbachia but not Cardinium in the invasive range of the Australian thrips species, Pezothrips kellyanus. Biol. Invasions 18, 197–214. doi: 10.1007/s10530-015-1002-4
Nikoh, N., Hosokawa, T., Moriyama, M., Oshima, K., Hattori, M., Fukatsu, T. (2014). Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc. Natl. Acad. Sci. U.S.A. 111, 10257–10262. doi: 10.1073/pnas.1409284111
Occhipinti-Ambrogi, A., Savini, D. (2003). Biological invasions as a component of global change in stressed marine ecosystems. Mar. Pollut. Bull. 46, 542–551. doi: 10.1016/S0025-326X(02)00363-6
Pagel, M., Meade, A., Crandall, K. (2004). A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Syst. Biol. 53, 571–581. doi: 10.1080/10635150490468675
Parker, J., Rambaut, A., Pybus, O. G. (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246. doi: 10.1016/j.meegid.2007.08.001
Pearcy, M., Goodisman, M. A. D., Keller, L. (2011). Sib mating without inbreeding in the longhorn crazy ant. Proc. R. Soc. Lond. B. Biol. Sci. 278, 2677– 2681. doi: 10.1098/rspb.2010.2562
Peakall, R. O. D., Smouse, P. E. (2006). Genelex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288– 295. doi: 10.1111/j.1471-8286.2005.01155.x
Pimentel, D., Lach, L., Zuniga, R., Morrison, D. (2000). Environmental and economic costs of nonindigenous species in the United States. BioScience 50, 53–65. doi: 10.1641/0006-3568(2000)050[0053:EAECON]2.3.CO;2
Pritchard, J. K., Stephens, M., Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
R Core Team, 2014. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. ISBN 3-900051-07-0. Retrieved from: http://www.R-project.org.
Resende, H. C., Yotoko, K. S., Delabie, J. H., Costa, M. A., Campiolo, S., Tavares, M. G., et al. (2010). Pliocene and Pleistocene events shaping the genetic diversity within the central corridor of the Brazilian Atlantic Forest. Biol. J. Linn. Soc. Lond. 101, 949–960. doi: 10.1111/j.1095-8312.2010.01534.x
Reuter, M., Pedersen, J. S., Keller, L. (2004). Loss of Wolbachia infection during colonisation in the invasive Argentine ant Linepithema humile. Heredity 94, 364–369. doi: 10.1038/sj.hdy.6800601
Ribeiro, Â. M., Lloyd, P., Bowie, R. C. K. (2011). A tight balance between natural selection and gene flow in a southern African arid-zone endemic bird. Evolution 65, 3499–3514. doi: 10.1111/j.1558-5646.2011.01397.x
Richardson, M. F., Weinert, L. A., Welch, J. J., Linheiro, R. S., Magwire, M. M., Jiggins, F. M., et al. (2012). Population genomics of the Wolbachia endosymbiont in Drosophila melanogaster. PLoS Genetics 8, e1003129. doi: 10.1371/journal.pgen.1003129
Rocha, L., Mascarenhas, R., Perondini, A., Selivon, D. (2005). Occurrence of Wolbachia in Brazilian samples of Ceratitis capitata. Neotrop. Entomol. 34, 1013–1015. doi: 10.1590/S1519-566X2005000600020
Rokas, A., Atkinson, R. J., Brown, G. S., West, S. A., Stone, G. N. (2001). Understanding patterns of genetic diversity in the oak gallwasp Biorhiza pallida: demographic history or a Wolbachia selective sweep? Heredity 87, 294–304. doi: 10.1046/j.1365-2540.2001.00872.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Russell, J. A. (2012). The ants (Hymenoptera: Formicidae) are unique and enigmatic hosts of prevalent Wolbachia (Alphaproteobacteria) symbionts. Myrmecol. News 16, 7–23.
Russell, J. A., Funaro, C. F., Giraldo, Y. M., Goldman-Huertas, B., Suh, D., Kronauer, D. J. C., et al. (2012). A veritable menagerie of heritable bacteria from ants, butterflies, and beyond: broad molecular surveys and a systematic review. PLoS One 7, e51027. doi: 10.1371/journal.pone.0051027
Russell, J. A., Goldman-Huertas, B., Moreau, C. S., Baldo, L., Stahlhut, J. K., Werren, J. H., et al. (2009). Specialization and geographic isolation among Wolbachia symbionts from ants and lycaenid butterflies. Evolution 63, 624–640. doi: 10.1111/j.1558-5646.2008.00579.x
Saridaki, A., Bourtzis, K. (2010). Wolbachia: more than just a bug in insects genitals. Curr. Opin. Microbiol. 13, 67–72. doi: 10.1016/j.mib.2009.11.005
Sasaki, T., Kubo, T., Ishikawa, H. (2002). Interspecific transfer of Wolbachia between two Lepidopteran insects expressing cytoplasmic incompatibility: a Wolbachia variant naturally infecting Cadra Cautella causes male killing in Ephestia kuehniella. Genetics 162, 1313–1319.
Sasaki, T., Massaki, N., Kubo, T. (2005). Wolbachia variant that induces two distinct reproductive phenotypes in different hosts. Heredity 95, 389–393. doi: 10.1038/sj.hdy.6800737
Schuler, H., Bertheau, C., Egan, S. P., Feder, J. L., Riegler, M., Schlick-Steiner, B. C., et al. (2013). Evidence for a recent horizontal transmission and spatial spread of Wolbachia from endemic Rhagoletis cerasi (Diptera: Tephritidae) to invasive Rhagoletis cingulata in Europe. Mol. Ecol. 22, 4101–4111. doi: 10.1111/mec.12362
Schuler, H., Egan, S. P., Hood, G. R., Busbee, R. W., Driscoe, A. L., Ott, J. R. (2018). Diversity and distribution of Wolbachia in relation to geography, host plant affiliation and life cycle of a heterogonic gall wasp. BMC Evol. Biol. 18 (1), 37. doi: 10.1186/s12862-018-1151-z
Schuler, H., Köppler, K., Daxböck-Horvath, S., Rasool, B., Krumböck, S., Schwarz, D., et al. (2016). The hitchhiker’s guide to Europe: the infection dynamics of an ongoing Wolbachia invasion and mitochondrial selective sweep in Rhagoletis cerasi.. Mol. Ecol. 25, 1595–1609. doi: 10.1111/mec.13571
Sintupachee, S., Milne, J. R., Poonchaisri, S., Baimai, V., Kittayapong, P. (2006). Closely related Wolbachia strains within the pumpkin arthropod community and the potential for horizontal transmission via the plant. Microb. Ecol. 51, 294–301. doi: 10.1007/s00248-006-9036-x
Shoemaker, D. D., Ross, K. G., Keller, L., Vargo, E. L., Werren, J. H. (2000). Wolbachia infections in native and introduced populations of fire ants (Solenopsis spp.). Insect Mol. Biol. 9, 661–673. doi: 10.1046/j.1365-2583.2000.00233.x
Spottiswoode, C. N., Stryjewski, K. F., Quader, S., Colebrook-Robjent, J. F. R., Sorenson, M. D. (2011). Ancient host specificity within a single species of brood parasitic bird. Proc. Natl. Acad. Sci. U.S.A. 108, 17738–17742. doi: 10.1073/pnas.1109630108
Stahlhut, J. K., Desjardins, C. A., Clark, M. E., Baldo, L., Russell, J. A., Werren, J. H., et al. (2010). The Mushroom habitat as an ecological arena for global exchange of Wolbachia. Mol. Ecol. 19, 1940–1952. doi: 10.1111/j.1365-294X.2010.04572.x
Stevens, L., Wicklow, D. T. (1992). Multispecies interactions affect cytoplasmic incompatibility in Tribolium flour beetles. Am. Nat. 140, 642–653. doi: 10.1086/285432
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Toews, D. P. L., Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x
Trager, J. C. (1984). A revision of the genus Paratrechina (Hymenoptera: Formicidae) of the continental United States. Sociobiology 9, 49–162.
Turelli, M., Hoffmann, A. A., McKechnie, S. W. (1992). Dynamics of cytoplasmic incompatibility and mtDNA variation in natural Drosophila simulans populations. Genetics 132, 713–723.
Tseng, S. P., Darras, H., Lee, C. Y., Yoshimura, T., Keller, L., Yang, C. C. S. (2019). Isolation and characterization of novel microsatellite markers for a globally distributed invasive ant Paratrechina longicornis (Hymenoptera: Formicidae). Eur. J. Entomol. 116, 253-257. doi: 10.14411/eje.2019.029
Tsutsui, N. D., Kauppinen, S. N., Oyafuso, A. F., Grosberg, R. K. (2003). The distribution and evolutionary history of Wolbachia infection in native and introduced populations of the invasive argentine ant (Linepithema humile). Mol. Ecol. 12, 3057–3068. doi: 10.1046/j.1365-294X.2003.01979.x
Vargo, E. L., Crissman, J. R., Booth, W., Santangelo, R. G., Mukha, D. V., Schal, C. (2014). Hierarchical genetic analysis of German cockroach (Blattella germanica) populations from within buildings to across continents. PLoS One 9, e102321. doi: 10.1371/journal.pone.0102321
Vavre, F., Fleury, F., Varaldi, J., Fouillet, P., Boulétreau, M. (2000). Evidence for female mortality in Wolbachia-mediated cytoplasmic incompatibility in haplodiploid insects: epidemiologic and evolutionary consequences. Evolution 54, 191–200. doi: 10.1554/0014-3820(2000)054[0191:EFFMIW]2.0.CO;2
Vavre, F., Fouillet, P., Fleury, F. (2003). Between- and within-host species selection on cytoplasmic incompatibility-inducing Wolbachia in haplodiploids. Evolution 57, 421–427. doi: 10.1111/j.0014-3820.2003.tb00275.x
Vavre, F., Girin, C., Boulétreau, M. (1999). Phylogenetic status of a fecundity-enhancing Wolbachia that does not induce thelytoky in Trichogramma. Insect Mol. Biol. 8, 67–72. doi: 10.1046/j.1365-2583.1999.810067.x
von Beeren, C., Stoeckle, M. Y., Xia, J., Burke, G., Kronauer, D. J. C. (2015). Interbreeding among deeply divergent mitochondrial lineages in the American cockroach (Periplaneta americana). Sci. Rep. 5, 8297. doi: 10.1038/srep08297
Watterson, G. A. (1975). On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276. doi: 10.1016/0040-5809(75)90020-9
Weeks, A. R., Turelli, M., Harcombe, W. R., Reynolds, K. T., Hoffmann, A. A. (2007). From parasite to mutualist: rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 5, e114. doi: 10.1371/journal.pbio.0050114
Weinert, L. A., Araujo-Jnr, E. V., Ahmed, M. Z., Welch, J. J. (2015). The incidence of bacterial endosymbionts in terrestrial arthropods. Proc. R. Soc. Lond. B. Biol. Sci. 282, 20150249. doi: 10.1098/rspb.2015.0249
Wenseleers, T., Ito, F., Van Borm, S., Huybrechts, R., Volckaert, F., Billen, J. (1998). Widespread occurrence of the microorganism Wolbachia in ants. Proc. R. Soc. Lond. B. Biol. Sci. 265, 1447–1452. doi: 10.1098/rspb.1998.0456
Werren, J. H., Baldo, L., Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Wetterer, J. K. (2008). Worldwide spread of the longhorn crazy ant, Paratrechina longicornis (Hymenoptera: Formicidae). Myrmecol. News 11, 137–149.
Wilkinson, D. A., Duron, O., Cordonin, C., Gomard, Y., Ramasindrazana, B., Mavingui, P., et al. (2016). The bacteriome of bat flies (Nycteribiidae) from the Malagasy region: a community shaped by host ecology, bacterial transmission mode, and host-vector specificity. Appl. Environ. Microbiol. 82, 1778–1788. doi: 10.1128/AEM.03505-15
Wollaston, T. V. (1854). Insecta maderensia; being an account of the insects of the islands of the Madeiran group. London: J Van Voorst, 634. doi: 10.5962/bhl.title.9060
Yang, C. C., Yu, Y. C., Valles, S. M., Oi, D. H., Chen, Y. C., Shoemaker, D. D., et al. (2010). Loss of microbial (pathogen) infections associated with recent invasions of the red imported fire ant Solenopsis invicta. Biol. Invasions 12, 3307–3318. doi: 10.1007/s10530-010-9724-9
Zeng, K., Fu, Y.-X., Shi, S., Wu, C.-I. (2006). Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 174, 1431– 1439. doi: 10.1534/genetics.106.061432
Zeng, K., Mano, S., Shi, S., Wu, C.-I. (2007a). Comparisons of site- and haplotype-frequency methods for detecting positive selection. Mol. Biol. Evol. 24, 1562–1574. doi: 10.1093/molbev/msm078
Zeng, K., Shi, S., Wu, C.-I. (2007b). Compound tests for the detection of hitchhiking under positive selection. Mol. Biol. Evol. 24, 1898–1908. doi: 10.1093/molbev/msm119
Zélé, F., Denoyelle, J., Duron, O., Rivero, A. (2018). Can Wolbachia modulate the fecundity costs of Plasmodium in mosquitoes? Parasitology 145, 775–782. doi: 10.1017/S0031182017001330
Keywords: horizontal transfer, invasive species, phylogeography, selective sweep, Wolbachia
Citation: Tseng S-P, Wetterer JK, Suarez AV, Lee C-Y, Yoshimura T, Shoemaker DW and Yang C-CS (2019) Genetic Diversity and Wolbachia Infection Patterns in a Globally Distributed Invasive Ant. Front. Genet. 10:838. doi: 10.3389/fgene.2019.00838
Received: 27 April 2019; Accepted: 13 August 2019;
Published: 17 September 2019.
Edited by:
Horacio Naveira, University of A Coruña, SpainReviewed by:
Hannes Schuler, Free University of Bozen-Bolzano, ItalyDimitrios N. Avtzis, Hellenic Agricultural, Organization – ELGO, Greece
Copyright © 2019 Tseng, Wetterer, Suarez, Lee, Yoshimura, Shoemaker and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: DeWayne Shoemaker, dewayne.shoemaker@utk.edu; Chin-Cheng Scotty Yang, ccyang@rish.kyoto-u.ac.jp