
- 1 Institute for Microbiology, Eidgenössische Technische Hochschule Zurich, Zurich, Switzerland
- 2 Laboratory for Gastroenterology, DKF, University of Berne, Berne, Switzerland
- 3 Complex Traits Group, Department of Microbiology and Immunology, McGill University, Montreal, QC, Canada
- 4 Institute for Infectious Diseases, University of Berne, Berne, Switzerland
Intestinal bacteria outnumber our own human cells in conditions of both health and disease. It has long been recognized that secretory antibody, particularly IgA, is produced in response to these microbes and hypothesized that this must play an important role in defining the relationship between a host and its intestinal microbes. However, the exact role of IgA and the mechanisms by which IgA can act are only beginning to be understood. In this review we attempt to unravel the complex interaction between so-called “natural,” “primitive” (T-cell-independent), and “classical” IgA responses, the nature of the intestinal microbiota/intestinal pathogens and the highly flexible dynamic homeostasis of the mucosal immune system. Such an analysis reveals that low-affinity IgA is sufficient to protect the host from excess mucosal immune activation induced by harmless commensal microbes. However, affinity-maturation of “classical” IgA is essential to provide protection from more invasive commensal species such as segmented filamentous bacteria and from true pathogens such as Salmonella typhimurium. Thus a correlation is revealed between “sophistication” of the IgA response and aggressiveness of the challenge. A second emerging theme is that more-invasive species take advantage of host inflammatory mechanisms to more successfully compete with the resident microbiota. In many cases, the function of IgA may be to limit such inflammatory responses, either directly by coagulating or inhibiting virulence of bacteria before they can interact with the host or by modulating immune signaling induced by host recognition. Therefore IgA appears to provide an added layer of robustness in the intestinal ecosystem, promoting “commensal-like” behavior of its residents.
Introduction
Immunity at our mucosal surfaces at first appears to tread a very fine line between excessive pathological responsiveness to the abundant commensal microbes, and insufficient responsiveness to potentially pathogenic microbes. In reality, the composition of the microbiota varies with circadian rhythm (Caporaso et al., 2011), eating (Kiss et al., 2011; Li et al., 2011), chemical toxins (Veldhoen et al., 2008), etc. Further, a broad range of immunodeficiencies (Vaishnava et al., 2008; Slack et al., 2009) are known to be compatible with the formation of host–commensal mutualism at least with a specific-pathogen-free microbiota. Thus it is revealed that the host–microbiota relationship is a dynamic homeostasis that is both remarkably robust and mechanistically flexible. The observed inflammation and induction of high-affinity adaptive immune responses during pathogenic infection can be seen as a (preferably temporary) deviation of the bacterial–host relationship sharply away from its usual homeostatic set-point, with the corresponding decrease in function of the intestine that accompanies any such deviation.
The study of genetically manipulated mice has revealed many factors that contribute to the robustness of this homeostasis [including mucus production (Van der Sluis et al., 2006; Johansson et al., 2008), antimicrobial peptide production (Vaishnava et al., 2008, 2011), innate immunity (Slack et al., 2009)]. This robustness may also explain how the intestinal immune system is able to accommodate a wide range of microbes that have evolved a wide variety of intestinal lifestyles ranging from symbiotic to opportunistic pathogenic/“pathobiotic” (Chow et al., 2011).
For many years, the role of intestinal IgA in host–commensal, and host–pathogen, interactions has been actively pursued. The mild phenotype of IgA-deficiency both in mice and in humans [approx. 1 in 600 Caucasians fail to secrete measurable quantities of IgA into the intestine without any clear association to clinical disease (Hammarström et al., 2000)] may be easily explained by the necessary robustness of the intestinal immune system. Nevertheless, IgA is normally produced at a rate of 5 g/day, making it the most abundantly produced antibody isotype, and a considerable metabolic burden. This hints toward a strong evolutionary selective pressure for its maintenance. The purpose of this review is to discuss recently published progress in our understanding of the role and mechanisms of action of secretory IgA in mucosal homeostasis, mucosal vaccination, and responses to infection.
Microbiota-Driven IgA – Is it “Natural”?
“Primitive” and “Classical” IgA
Before we consider the function of IgA, it is first pertinent to discuss one major aspect of IgA biology: many IgA responses are considered to be “primitive” (Macpherson et al., 2000), but the exact meaning of this is easily confused with “natural” and/or “innate.”
Evidence of secretory antibodies can be seen in the genome of species as diverse as cartilaginous fish, amphibians, and mammals. Intriguingly, teleost fish, which branched from the mammalian and cartilaginous fish lineages during the Triassic period, produce only variants of IgM with no evidence of intestinal secretion (Tort et al., 2003). This suggests that mammalian IgA arose later than the divergence of teleosts and the rest of the Gnathostomata species, and is therefore considerably less ancient than the adaptive immune system itself.
Nevertheless, although “classical” adaptive immunity generates high-affinity antibodies via T-cell driven somatic hypermutation (Table 1), T-cell-deficient mice can still produce so-called “primitive” IgA (Table 1) of a sufficient affinity/avidity to bind commensal bacterial antigens on a western blot or ELISA. This suggests that commensal-induced IgA does not absolutely require classical T-cell help for its generation (Macpherson et al., 2000, 2001). The term “primitive” here refers to the seemingly simpler induction mechanism not involving T-cells. It is intriguing to note that the breadth of epitopes, particularly of carbohydrate epitopes never produced by mammalian cells, is much broader in bacteria than in commonly studied viruses such as LCMV (which necessarily rely upon host protein and carbohydrate synthesis machinery). This potentially allows for evolutionary selection of antibody V, D, and J segments capable of forming high-affinity interactions with common bacterial antigens even in germ-line conformation. Indeed, (monoclonal) IgA isolated from monocolonized mice commonly displays no evidence of somatic hypermutation (Bos et al., 1996; Hapfelmeier et al., 2010; Wei et al., 2011; Lindner et al., 2012) but nevertheless can be of sufficient affinity to bind to the bacterial surface of the colonizing species in a flow cytometry stain (where the typical affinity minimum cut-off is considered to be around 109 L/mol (Macey, 2007) as opposed to much lower affinity cut-offs that can be detected by Western blot or “dirty-plate” ELISA).

Table 1. Key features of “natural,” “primitive,” and “classical” intestinal IgA.
The above observations all support the idea that IgA can be produced in a “primitive” fashion, independently from T-cell-mediated immunity. However, it should be noted that bacterial binding by IgA from T-cell-deficient mice is observed when total IgA concentrations are normalized between wild-type and T-cell-deficient mice, but total intestinal IgA levels are lower in T-cell-deficient mice than in wild-type mice, depending on the microbiota (Macpherson et al., 2000). Further, when T-cell-deficient or CD4 T-cell-depleted mice are challenged intragastrically with either a commensal or a pathogen, we cannot detect IgA of sufficient affinity to be measured by flow cytometry staining of the bacterial surface, even when total IgA concentrations are normalized (Slack et al., 2009; Slack and Hapfelmeier, unpublished observations). As well as T-cells, organized lymphoid structures in the intestine are required for a substantial proportion of IgA+ plasma cell generation as production of these cells is severely impaired in mice lacking RORγt which fail to develop any organized lymphoid structures in the intestine (Eberl and Littman, 2004), lymphotoxin-alpha deficient mice which lack all organized lymphoid structures in the intestine (Kang et al., 2002; Newberry et al., 2002), and RORγt-deficient mice reconstituted with bone marrow from lymphotoxin-alpha deficient mice which fail to generate follicular dendritic cells in the Peyer’s patches (PP; Tsuji et al., 2008). Further, germinal centers are always histologically observed in the mucosa-associated lymphoid tissues of SPF or recently bacterially colonized animals (Talham et al., 1999; Hapfelmeier et al., 2010). Therefore, although “primitive” bacterially induced IgA can be observed in the absence of “classical” T-cell-dependent antibody responses, the generation of abundant and high-affinity IgA still appears to require CD4 T-cell help and organized lymphoid structures.
“Natural” and “Innate” IgA
How these responses relate to what are defined as “natural” and “innate” antibody responses is also intriguing. Classically, “natural” responses are defined as pre-existing antibody responses that an animal produces in the absence of microbial or antigenic stimulation. Until recently, all antibodies in the serum and intestine of an unvaccinated uninfected SPF animal would be considered “natural.” However, at least 90% of the IgA in the intestine is induced by the microbiota (to be further discussed below, see section The Role of of IgA in Host–Commensal Mutualism), suggesting this term can only be classically applied to the antibody responses found in germ-free mice (Table 1). Germ-free animals produce reduced but still significant amounts of intestinal IgA. Recent next-generation sequencing of the IgA repertoire indicates that this “natural” repertoire contains a similar distribution of low frequency and high frequency plasma cell clones to that in SPF mice (Lindner et al., 2012). However, intestinal IgA from germ-free mice does not contain sufficient levels of bacterial-antigen-specific clones to bind bacterial antigens on western blots, even when total IgA concentrations are normalized (Macpherson et al., 2000, 2001).
In the fields of virology, antibody affinities of greater than 108L/mol are required to neutralize infection with extensively characterized viruses such as VSV (Bachmann et al., 1997). Such antibody responses are only induced after extensive somatic hypermutation during germinal center reactions, and in this situation, the distinction between natural antibody responses and induced antibody responses is simple to make. For anti-bacterial antibodies, it is almost impossible to define a bacterial equivalent of “neutralizing” antibody unless the response is bacterial toxin-specific, adding considerably to the complexity of discussion.
Thus, both “primitive” T-independent, and “classical” adaptive T-dependent IgA responses appear to operate in parallel in the intestine to provide both extremely rapid, and slower but higher-affinity responses to bacteria, as appropriate. How “natural” the IgA response is in both situations remains to be fully explored, but next-generation-sequencing-based approaches to the IgA repertoire are beginning to provide clear answers to this: there is evidence for the microbiota-induced rapid expansion of a near-germ-line “natural” IgA repertoire (pre-existent in new-borns and germ-free animals) combined with the subsequent gradual diversification of this repertoire by T-dependent somatic hypermutation in the murine intestine (Wei et al., 2011; Lindner et al., 2012). The relative contributions of the different types of responses to the various functions of IgA are now starting to be unraveled and will be discussed below.
Functions of Intestinal IgA
The commensal microbiota of a free-living mammal contains a range of species from those which barely interact with the host, via organisms that form truly mutualistic relationships (Bacteroides thetaiotaomicron, Lactococcus lactis) through to well known opportunistic pathogens (Staphylococcus aureus, Clostridium difficile) and “pathobionts” [e.g., “segmented filamentous bacteria” (SFB); Chow et al., 2011]. It should be noted that all such terms are an attempt to draw lines onto the continuous spectrum of bacterial behaviors that depend not only on the bacterial species, but also on the host. Indeed bacteria that have been defined as true commensals or “pathobionts” in wild-type mice can begin to display behavior of true opportunistic pathogens in immunocompromised mouse strains (Fagarasan et al., 2002; Elinav et al., 2011; Wei et al., 2011; Henao-Mejia et al., 2012) further indicating the flexibility of these definitions. A useful distinction is thus that commensalism represents a state of homeostasis that can be maintained long-term, whereas pathogenic infection represents a major deviation from this homeostasis. The function of IgA in both of these situations will be discussed below.
The Role of IgA in Host–Commensal Mutualism
It is well established that the majority (approx. >90%) of intestinal IgA production is microbiota-driven, since germ-free animals contain around 10-fold reduced numbers of mucosal IgA-producing cells (Crabbé et al., 1970; Moreau et al., 1978). This state is readily reversed by the re-association of such animals with a normal microbiota (Crabbé et al., 1970; Moreau et al., 1978). In line with this it was shown that most intestinal bacterial cells are IgA-coated in situ (van der Waaij et al., 1996), but it has also been noted that not all IgA that is induced by a specific commensal microbe has detectable reactivity to the inducing microbe (Bry et al., 1996; Talham et al., 1999; see also Stoel et al., 2005).
Compensatory immune reactions reveal IgA-mediated modulation of immune function
The study of severely innate immune deficient mice has shown that both innate and adaptive immunity are critically important for intestinal containment also of commensal bacteria (even the completely avirulent laboratory model organism E. coli K-12). Indeed, considerable functional complementarity of innate and adaptive immunity can compensate for partial immune deficiency (Slack et al., 2009). This flexibility almost certainly also underlies the lack of overt pathology in IgA−/− or even completely antibody-deficient animals (Shulzhenko et al., 2011). It also suggests that the functionality of IgA in host–commensal interaction is best measured indirectly by measuring upregulation of other, compensatory commensal-induced mucosal immune mechanisms. This has been successfully observed in a number of recent publications, which can be roughly divided into those showing an effect of non-specific IgA, those showing an effect of total IgA and those showing an effect of high-affinity IgA.
The work published by Petersen et al. (Peterson et al., 2007), demonstrates that a monoclonal antibody with high-affinity for the surface of Bacteroides thetaiotaomicron can specifically reduce intestinal innate immune activation in a B. thetaiotaomicron-monocolonized hybridoma back-pack RAG1-deficient (therefore T and B cell-deficient) mouse model. Increased innate immune activation is also observed in the epithelium of completely antibody-deficient mice (Shulzhenko et al., 2011). Previous studies have also reported spontaneous pathology and small intestinal bacterial overgrowth, predominantly of the “pathobiont” SFB, in AID-deficient mice, which have a defect in somatic hypermutation (Fagarasan et al., 2002; Suzuki et al., 2004; Wei et al., 2011), suggesting that affinity-matured IgA responses are necessary for the control of this species. In contrast to these studies on high-affinity antibodies, quasi-monoclonal IgA from Qm mice, and also the “natural” IgA from germ-free mice, was shown to be sufficient to prevent premature priming of antibody responses against the commensal microbiota in neonatal mice, despite presumably negligible affinity of this IgA for the intestinal microbiota (Harris et al., 2006). Elson and co-workers report another indirect IgA effect in host–commensal mutualism by showing that IgA-deficiency leads to increased commensal-driven generation of intestinal FoxP3+ regulatory T-cells (Cong et al., 2009), although no distinction could be made between low-affinity and high-affinity IgA responses in this system.
Taken in parallel with the absence of intestinal inflammation in IgA or antibody-deficient mice in many animal facilities, this suggests that requirements of the IgA system for maintaining homeostasis are highly dependent on the nature of the intestinal microbiota. In the majority of SPF facilities, compensation by other immune mechanisms appears to prevent the appearance of clinically overt disease. Whilst affinity-matured antibodies appear to be necessary to protect against immune activation caused by bacteria like SFB, non-specific IgA such as in the QM mice, is sufficient to prevent early immune activation by a less invasive microbiota (Figure 1).
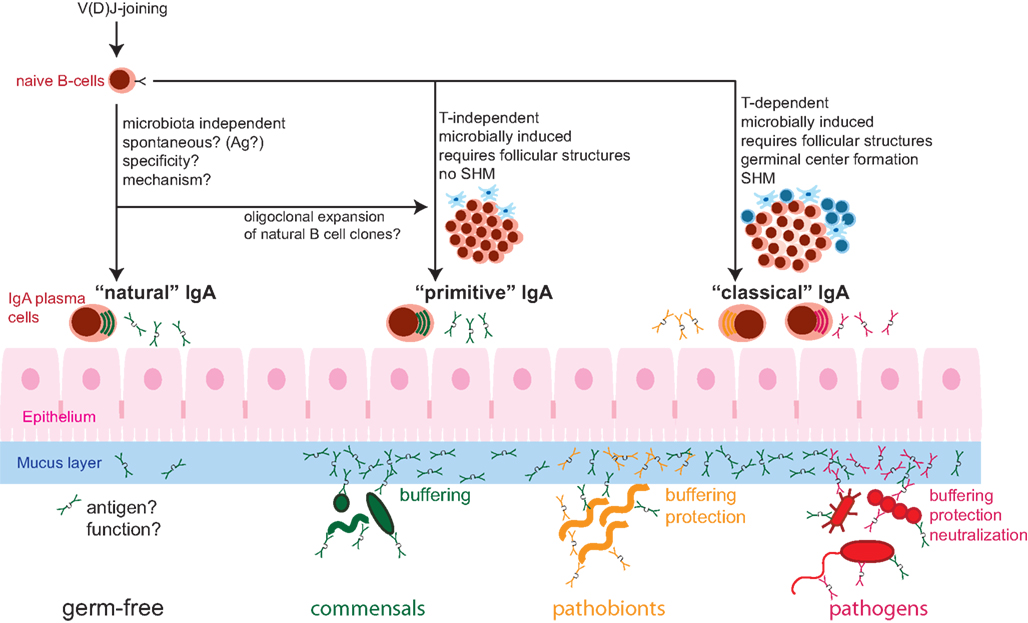
Figure 1. Schematic of the composite nature of the mucosal IgA system, consisting of “natural” (microbiota-independent), “primitive” (microbobially induced, T-cell independent), and “classical” (microbe/pathogen-induced, helper T-cell-dependent) IgA responses. Whether there is a specific immunogen inducing “natural” IgA is unknown. “Natural” IgA can be sufficient for controlling benign commensal bacteria. Commensal microbes induce “primitive” T-cell independent IgA with is sufficient for the management of commensal bacteria (Green), mainly by exclusion from the mucus layer and the resulting “buffering” of mucosal responses. The more invasive pathobionts (orange) and infection with pathogenic microbes (red) further induce “classical” T-cell-dependent IgA of high-affinity that is important for the control of these types of microorganisms via efficient mucosal exclusion and neutralization. SHM, somatic hypermutation.
Mechanisms of IgA-mediated immune modulation
The mechanism by which IgA can minimize induction of innate or adaptive immunity in the intestine remains to be definitely proven. This may be achieved by decreasing bacterial penetration of the intestinal epithelial barrier and minimizing access of bacterial antigen to gut associated lymphoid tissues (GALT), or by alteration of immune signaling induced by recognition of IgA-coated bacteria in the epithelium and lamina propria.
The first of these mechanisms could be called “innate immune exclusion”, in analogy of the classical term of “immune exclusion” (i.e., the effect of IgA in downregulating its own induction by excluding the cognate antigen from accessing IgA inductive sites [Stokes et al., 1975]). A prominent example of this is the monocolonization of germ-free animals with a strongly IgA-inducing commensal bacterium like Morganella morganii. Despite the persistent nature of bacterial intestinal colonization, this leads to a contained IgA response with only transient germinal center reactions (Shroff et al., 1995).
Recent progress in this field has led to a more detailed consideration of intestinal mucus in such an exclusion mechanism. The viscous mucous layer covering the intestinal epithelium physically impedes bacterial contact with the epithelium and concentrates secreted antimicrobials that kill mucus-invading bacteria, without affecting the commensal habitat as a whole. If the viscous mucus layer is missing, the micro-compartmentalized nature of the mucosal barrier literally collapses. This can precipitate excessive defense reactions toward commensal bacteria and chronic inflammatory disease, as can be observed in Muc2-deficient mice (Van der Sluis et al., 2006; Johansson et al., 2008). It has recently been demonstrated that the antimicrobial peptide RegIIIγ is concentrated in the mucus layer and deficiency for this peptide results in heavy bacterial colonization of the mucus layer (Vaishnava et al., 2011). Histologically it is also clear that IgA is concentrated in the intestinal mucus, and similar experiments examining mucus colonization in IgA-deficient and antibody-deficient animals would reveal whether IgA contributes to the sterility of the intestinal mucus.
A second possible mechanism is that recognition of IgA-coated bacteria induces altered signaling when compared to recognition of bacteria alone. Many different proteins have been suggested to bind IgA, in addition to the PolyIg receptor that mediates its transport across epithelia. These include the Fcα/μR (Shibuya et al., 2000), CD89 (reviewed in (Bakema and van Egmond, 2011)), asialoglycoprotein receptors (Stockert et al., 1982), transferrin receptors (CD71; Moura et al., 2001), and secretory component receptors (Lamkhioued et al., 1995). In humans the IgA receptor CD89 is expressed on myeloid cells (although absent from intestinal macrophages) and can signal in either an activatory or inhibitory manner depending on the degree of cross-linking (Reviewed in (Bakema and van Egmond, 2011)). Although mice lack a direct homolog of CD89, transgenic mice expressing human CD89 on myeloid cells and passively transferred with human IgA specific for mycobacterium tuberculosis were protected from infection (Balu et al., 2011). IgA can be shown to bind to the surface of murine macrophages, suggesting that a functional homolog of CD89 exists, but as yet the responsible receptor complex has not been identified (Reljic, 2006). Until further characterization of IgA receptors in the common murine models has been achieved, signaling induced by IgA in different contexts will remain hard to define.
A third proposed mechanism of action of IgA appears paradoxical to the first. It has been observed that IgA also plays a role in increasing the translocation of bacteria across PP follicle associated epithelia, where IgA-coated bacteria bind to the luminal side and are transcytosed into the PP (Kadaoui and Corthésy, 2007; reviewed in Corthésy, 2007, 2009). However, it is still not fully understood whether the IgA-coated antigen then feeds into a positive or negative feedback loop of antigen presentation for the regulation of bacterial-specific antibody responses. It is important to keep this bacterial-uptake mechanism in mind, as a common measure of mucosal containment defects is to quantify the load of live bacteria present in the PPs and MLNs. An absence of phenotype in IgA-deficient mice by this method may represent a composite of decreased active transport into the PP and increased passive translocation, masking the effect. This also suggests that the concept of IgA-mediated exclusion of bacteria from the lamina propria is over-simplified and the immune consequences of recognition of IgA-coated bacteria need to be fully investigated.
An entirely different mechanism by which IgA may function is a possible direct effect of IgA on commensal bacterial colonization of the intestine (such as by influencing growth rate, survival, bacterial responses, etc.). Despite enormous technical innovation and experimental creativity, the attempts to demonstrate effects of immune functions on microbiota composition have been a challenge and so far have only found subtle effects of mucosal immunity on the commensal bacterial fitness (Peterson et al., 2007; Goodman et al., 2009). Supporting functional data for the impact of bacteria-binding IgA on commensal bacterial fitness come from the Peterson at al. study (Peterson et al., 2007) where it could be demonstrated that phase variation of surface polysaccharide that leads to escape from IgA recognition confers a selective advantage for intestinal colonization. Interestingly, it is known that inflammatory responses, such as increased antimicrobial peptide production and upregulation of siderophore-sequestering proteins, negatively impact on commensal bacteria. Therefore beneficial effects of IgA on commensal colonization may also be indirect via modulation of the activation state of the mucosal immune system.
In conclusion, the immune set-points and anatomy of the mucosal barrier together with IgA may function primarily in robustly “protecting commensals from themselves,” to avoid unnecessary inflammatory reactions where the collateral damage would always outweigh the benefits and likely lead to a chronic state of exaggerated inflammation as in inflammatory bowel diseases. IgA is therefore a typical example of a mucosal immune specialization with the potential to actively exclude bacteria from host tissues without any concomitant induction of acute inflammation, thus broadening the window of immune system and physical barrier function compatible with stable host–microbiota homeostasis.
Functions of IgA in Bacterial Pathogen Defense
In considering the literature on the role of IgA in pathogen defense, it is also crucial to make the distinction between low-affinity pre-existing IgA responses found in animals never previously exposed to the pathogen, and high-affinity IgA responses formed during vaccination or by previous exposure to the pathogen.
Can “Natural” IgA protect against pathogens?
In contrast to our knowledge on the role of low-affinity IgA responses in host–commensal mutualism, only limited data supports a role of low-affinity antibodies in protecting from acute infection with bona fide obligate pathogens. Indeed PolyIg receptor (pIgR)-deficient mice, which fail to transport any polymeric immunoglobulin into the intestinal lumen, are equally as susceptible as wild-type mice in the typhoid model of Salmonella typhimurium infection (Uren et al., 2005), although pIgR-deficient mice have a reduced colonization resistance toward S. typhimurium (Wijburg et al., 2006). However, this may in part be a consequence of the mild non-specific protein losing enteropathy phenotype of this mouse strain in some hygiene conditions (Johansen et al., 1999). Our own data in the streptomycin-pretreatment model of non-typhoidal salmonellosis further confirm these observations (E. Slack and W.-D. Hardt, unpublished data). Similarly, naive IgA-knock-out mice show comparable susceptibility to Influenza infection, to naïve wild-type controls (Mbawuike et al., 1999). Importantly, given the recent progress in our understanding of the role of the microbiota in disease (Stecher and Hardt, 2011), it would be valuable for the field to revisit these observations in gnotobiotic settings. Further, the presence of possible masking of natural IgA effects by compensatory production of other isotypes, of increased innate immunity and increased innate lymphoid cell and T-cell function requires full exploration. However, although antibody-mediated-compensation for innate immunodeficiency in MyD88-deficient mice is sufficient to protect these animals from some commensal bacterial species (Slack et al., 2009), this was shown to be insufficient to protect from Salmonella infection (Ko et al., 2009).
High-affinity “classical” IgA induced by pathogens
Induced IgA responses with measurable affinity for particular pathogens have been extensively investigated in the context of mucosal vaccination. Early experiments with cholera toxin vaccination demonstrated strong induction of IgA after gastric administration of the toxin (Svennerholm et al., 1978; Fuhrman and Cebra, 1981). Svennerholm et al. further showed a strong correlation between the titer of IgA against cholera toxin and protection from murine cholera infection. Later, “back-pack” IgA-producing hybridoma experiments and adoptive transfer of monoclonal dimeric IgA revealed that Vibrio cholerae LPS-specific IgA was protective against murine cholera whereas anti-toxin IgA alone provided very limited protection (Winner et al., 1991; Apter et al., 1993), although the relative affinities of the IgA clones employed were not assessed. It could also be demonstrated that adoptive transfer with a monoclonal dimeric IgA specific for the outer membrane of Helicobacter felis could protect germ-free mice from Helicobacter infection (Czinn et al., 1993). Endt et al. (2010) recently elucidated that high-affinity surface (predominantly LPS O-antigen)-binding IgA protects from colitis upon re-infection with S. typhimurium in the non-typhoidal salmonellosis model. This appears to be mediated by antibody-mediated bacterial coagulation and “luminal trapping,” hindering innate immune recognition and bacterial delivery of proinflammatory effector molecules. It was recently found that pathogen-binding IgA can also protect from pathogens by direct modulation of bacterial motility (Forbes et al., 2008) or virulence organelles, such as type III secretion systems. For example, a monoclonal Shigella flexneri LPS-O-antigen specific IgA antibody when bound to the bacterial surface specifically inhibits the S. flexneri type III secretion system that is required for epithelial invasion and induction of pathology (Forbes et al., 2011). In analogy to the virus-neutralizing antibodies that directly protect from virus infection, these antibodies could be termed “bacteria-neutralizing.” Taken together, these studies indicate that high-affinity IgA responses can provide protection against a number of gastrointestinal pathogens in the stomach and intestines.
Mechanisms of IgA “immune diplomacy” – dampening inflammation wherever it can be avoided
Although counterintuitive at first, an important protective mechanism of high-affinity secretory IgA appears to be also the dampening of bactericidal immune responses provoked by certain mucosal pathogens, for the benefit of the commensal microbiota and for intestinal homeostasis. Seminal work by Stecher et al. and others (Lupp et al., 2007; Stecher et al., 2007; Ackermann et al., 2008; Brown et al., 2008) introduced the concept that mucosal pathogens such as Salmonella and Citrobacter cause pathology not with the primary goal of colonizing extra-intestinal sites (which may eventually kill the host), but rather to modify the intestinal microenvironment to improve their competitive edge. Commensal species are continuously selected by conditions found within the non-inflamed intestine; i.e., competition for space and resources and resistance to constitutive antimicrobial activity. The majority of mechanisms required to resist full-blown host inflammatory immune responses are energy-demanding and therefore often absent from commensal species. On the other hand, obligate pathogens require such resistance mechanisms to survive and propagate. Although it at first seems counterintuitive, for many intestinal bacterial pathogens induction of inflammation actually provides them with a significant advantage over the commensal microbiota, that are more susceptible to induced immunity. The role of intestinal IgA may therefore be two-fold: Firstly directly inhibiting adhesion, effacement and invasion by intestinal pathogens and secondly, limiting the induction of inflammation by such pathogens. This hypothesis is nicely supported by the observation that LPS O-antigen-specific IgA can prevent Salmonella from interacting with the intestinal epithelium, thus preventing induction of inflammation and allowing the commensal microbiota to re-grow (Endt et al., 2010). The specific role of anti-bacterial IgA in dampening the bacterial elicitation of inflammatory responses has also been worked out for Shigella [in a rabbit ligated ileal loop infection model (Boullier et al., 2009)] coming to similar conclusions.
TNFα/iNOS-producing IgA plasma cells – more than just IgA producers
A recent finding now indicates that IgA-producing cells in the intestine may have crucial immune functions beyond IgA production. Local intestinal production of nitric oxide catalyzed by the inducible nitric oxide synthase (iNOS) is important for class switch recombination to IgA and generation of small intestinal IgA plasma cells (Tezuka et al., 2007). A recent study identified the IgA plasma cells themselves as a dominant iNOS and TNFα-producing cell population of the intestinal lamina propria, and B-lineage-specific TNFα/iNOS deficiency led to a an intestinal IgA-deficiency equivalent to a full TNFα/iNOS double knockout (Fritz et al., 2012). Intriguingly, the generation of these TNFα/iNOS-producing IgA plasma cells required microbial exposure, since the baseline lamina propria IgA plasma cells of germ-free animals consisted only of TNFα/iNOS-negative cells (Fritz et al., 2012). Colonization and infection experiments using mice with a B cell specific TNFα/iNOS deficiency further suggested that B-lineage-derived TNFα/iNOS contributed to the control of Citrobacter rodentium infection and intestinal colonization with the pathobiont SFB (Fritz et al., 2012). These observations document a novel immune regulatory function of IgA plasma cells and suggest that intestinal microbes induce plasma cells with a more “myeloid” cell-like phenotype.
The challenge of inducing protective IgA by oral vaccination
A major challenge in mucosal immunology currently is the development of vaccines that protect against mucosal infections such as typhoidal and non-typhoidal salmonellosis, cholera, shigellosis, enterotoxigenic E. coli, and tuberculosis. Currently many vaccines tested against these organisms show low efficacy when compared to vaccination against viruses (measles, influenza, rubella). In part, this reflects the complexity of the pathogens, although novel vaccines against papilloma viruses and Varicella zoster combat organisms approaching bacterial levels of complexity (recently reviewed in Stanley, 2008; Abendroth et al., 2010). Intracellular bacterial pathogens appear to be particularly challenging for the immune system, often residing within macrophages or dendritic cells which they modify to best evade immune detection (Bedoui et al., 2010). However, a vaccine that induces a long-lasting high-affinity IgA response can potentially inhibit the initial infection step before bacteria become resident intracellular pathogens. This poses two challenges for vaccine developers: Firstly, to induce IgA that has a sufficiently high-affinity for a sufficient number of relevant strains. Secondly, to produce a long-lasting IgA response safely.
Recent experiments using reversible colonization of germ-free mice indicate that both of these aims are harder to achieve in the mucosal system than in systemic IgG-based vaccination (Hapfelmeier et al., 2010). Firstly, the mucosal immune system is rather insensitive to bacteria. Whereas as few as 103 bacteria given systemically can induce a robust IgG response, over 109 live bacteria need to be delivered orally in order to see a measurable IgA response in the intestine (Hapfelmeier et al., 2010). Therefore any oral vaccination needs to introduce very high numbers of the vaccine strain into the system, either by using a live strain that can colonize to over this density, or by giving extremely high numbers of non-replicating bacteria. Alternatively, methods need to be employed that deliver sufficient quantities of antigen across the mucosal epithelium, or deliver strong enough adjuvanticity that these numbers can be reduced. Killed bacteria seem to be at least 100-fold less effective at inducing IgA responses than live bacteria, presumably due to their inactivity and the digestion of dead bacteria during transit through the stomach and intestine (Macpherson and Uhr, 2004; Hapfelmeier et al., 2010). To make matters worse, there is no clear prime-boost effect of intestinal immunization. Rather with each oral dose of bacteria there is a step-wise increase in specific IgA production that is independent of the interval between doses. A further challenge is that in the normally colonized intestine, the rate of attrition of the IgA repertoire is extremely high (Hapfelmeier et al., 2010). Thus it seems that any successful IgA-inducing vaccine must contain high numbers of bacteria, in a format that survives passage through the stomach, and which provides persistent antigen at a high enough concentration to avoid attrition of the response. Intriguingly, older works on the immune response to cholera toxin in the intestine demonstrate robust prime-boost effects of the IgA response and the presence of memory B cells up to 16 weeks post vaccination, suggesting that the nature of the antigen or the adjuvant may be critical in determining longevity of the IgA response (Pierce, 1978).
Remarkably, it could be demonstrated that intestinal infection with virulent Salmonella produces more and higher-affinity IgA than an equally densely colonizing avirulent Salmonella mutant, and only the IgA induced by virulent bacteria conferred robust protection from colitis upon re-challenge (Martinoli et al., 2007; Endt et al., 2010). More needs to be learned about the underlying qualitative and quantitative differences between pathogen and non-pathogen-induced mucosal immunity to understand this difference. Presumably, like pathogenic and commensal bacteria, also virulent and avirulent variants of a pathogen necessitate different degrees and kinds of immune protection. This is clearly relevant for the design of mucosal vaccines intended to be protective against mucosal infections: it is absolutely possible that the optimization of completely attenuated bacterial strains as live vaccines turns out to be counterproductive. Such a strategy could lead to agents that potently induce anti-commensal immunity but not the intended anti-pathogen immune protection.
IgA-independent protective mucosal immunity
Importantly, whilst IgA can be sufficient for protection from pathogen infection, it can be demonstrated that immunity induced by exposure of mice to a live mucosal vaccine or pathogen is highly redundant and in many cases IgA is dispensable. This has been shown both for Influenza virus infection (Mbawuike et al., 1999), and for typhoidal salmonellosis (Uren et al., 2005). In the non-typhoidal model of salmonellosis, Endt et al. (2010) demonstrated robust protection from colitis by specific secretory IgA but an absence of sterilizing immunity – indeed clearance of Salmonella from the gut lumen critically required re-growth of the microbiota, rather than active immune mechanisms. It has further been observed in humans that there is no correlation between any anti-typhoid antibody titer measured in serum or saliva and rate of recovery from typhoid (Dham and Thompson, 1982). Further, specifically induced CD4 effector T-cells can confer antibody-independent protection from mucosal infection. This was shown in an intranasal Klebsiella pneumoniae infection model, where Th17 cells conferred cross-protection between multiple serotypes of the same pathogen, independent of antibodies (Chen et al., 2011).
Conclusion
The intestinal IgA system acts at the interface between body and one the densest bacterial consortia known. The challenge of bacterial containment and discrimination between non-pathogens and pathogens has shaped the evolution of an intestinal immune system that robustly maintains intestinal health due to highly redundant and functionally flexible barrier functions. The current literature suggests that IgA adds robustness to host–microbiota homeostasis in the face of this flexibility, by acting as an immunological “buffer”: i.e., that combines protection from invasion by pathogenic microbes with suppression of excessive inflammation induced by non-pathogenic commensal microbes. The affinity of microbe-IgA interaction required to achieve this buffering appears to be highly tailored to the invasiveness of the microbial species in question. How exactly IgA functions in each situation remains only patchily understood with mechanisms proposed that directly affect the microorganism or that modulate host immune signaling. Inducing long-lived, effective mucosal vaccination will require an in-depth understanding of the interaction of different specificities and affinities of IgA with the pathogen in question, as well as the effects of pre-existing IgA and continuous repertoire re-shaping. We propose that great advances in this field will come from a new approach, made possible by modern technologies, aiming to examine the functioning of IgA within the mucosal ecosystem as a whole, simultaneously assessing the effects on microbiota structure and mucosal activation, and subsequently dissecting the crucial relationship between these two entities.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors report no potential conflicts of interests. Siegfried Hapfelmeier is supported by an European Research Council Starting Grant. Emma Slack is supported an Ambizione Fellowship of the Swiss National Science Foundation. Research in the laboratory of Jörg H. Fritz is funded by the Canadian Institutes of Health Research (CIHR, Operating Grant MOP#114972) and the Natural Sciences and Engineering Research Council of Canada (NSERC, Discovery Grant).
References
Abendroth, A., Kinchington, P. R., and Slobedman, B. (2010). Varicella zoster virus immune evasion strategies. Curr. Top. Microbiol. Immunol. 342, 155–171.
Ackermann, M., Stecher, B., Freed, N. E., Songhet, P., Hardt, W.-D., and Doebeli, M. (2008). Self-destructive cooperation mediated by phenotypic noise. Nature 454, 987–990.
Apter, F. M., Michetti, P., Winner, L. S., Mack, J. A., Mekalanos, J. J., and Neutra, M. R. (1993). Analysis of the roles of antilipopolysaccharide and anti-cholera toxin immunoglobulin A (IgA) antibodies in protection against Vibrio cholerae and cholera toxin by use of monoclonal IgA antibodies in vivo. Infect. Immun. 61, 5279–5285.
Bachmann, M. F., Kalinke, U., Althage, A., Freer, G., Burkhart, C., Roost, H., Aguet, M., Hengartner, H., and Zinkernagel, R. M. (1997). The role of antibody concentration and avidity in antiviral protection. Science 276, 2024–2027.
Bakema, J. E., and van Egmond, M. (2011). The human immunoglobulin A Fc receptor FcαRI: a multifaceted regulator of mucosal immunity. Mucosal Immunol. 4, 612–624.
Balu, S., Reljic, R., Lewis, M. J., Pleass, R. J., McIntosh, R., van Kooten, C., van Egmond, M., Challacombe, S., Woof, J. M., and Ivanyi, J. (2011). A novel human IgA monoclonal antibody protects against tuberculosis. J. Immunol. 186, 3113–3119.
Bedoui, S., Kupz, A., Wijburg, O. L., Walduck, A. K., Rescigno, M., and Strugnell, R. A. (2010). Different bacterial pathogens, different strategies, yet the aim is the same: evasion of intestinal dendritic cell recognition. J. Immunol. 184, 2237–2242.
Bos, N. A., Bun, J. C., Popma, S. H., Cebra, E. R., Deenen, G. J., van der Cammen, M. J., Kroese, F. G., and Cebra, J. J. (1996). Monoclonal immunoglobulin A derived from peritoneal B cells is encoded by both germ line and somatically mutated VH genes and is reactive with commensal bacteria. Infect. Immun. 64, 616–623.
Boullier, S., Tanguy, M., Kadaoui, K. A., Caubet, C., Sansonetti, P., Corthésy, B., and Phalipon, A. (2009). Secretory IgA-mediated neutralization of Shigella flexneri prevents intestinal tissue destruction by down-regulating inflammatory circuits. J. Immunol. 183, 5879–5885.
Brown, S. P., Le Chat, L., and Taddei, F. (2008). Evolution of virulence: triggering host inflammation allows invading pathogens to exclude competitors. Ecol. Lett. 11, 44–51.
Bry, L., Falk, P. G., Midtvedt, T., and Gordon, J. I. (1996). A model of host-microbial interactions in an open mammalian ecosystem. Science 273, 1380–1383.
Caporaso, J. G., Lauber, C. L., Costello, E. K., Berg-Lyons, D., González, A., Stombaugh, J., Knights, D., Gajer, P., Ravel, J., Fierer, N., Gordon, J. I., and Knight, R. (2011). Moving pictures of the human microbiome. Genome Biol. 12, R50.
Chen, K., McAleer, J. P., Lin, Y., Paterson, D. L., Zheng, M., Alcorn, J. F., Weaver, C. T., and Kolls, J. K. (2011). Th17 cells mediate clade-specific, serotype-independent mucosal immunity. Immunity 35, 997–1009.
Chow, J., Tang, H., and Mazmanian, S. K. (2011). Pathobionts of the gastrointestinal microbiota and inflammatory disease. Curr. Opin. Immunol. 23, 473–480.
Cong, Y., Feng, T., Fujihashi, K., Schoeb, T. R., and Elson, C. O. (2009). A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl. Acad. Sci. U.S.A. 106, 19256–19261.
Corthésy, B. (2007). Roundtrip ticket for secretory IgA: role in mucosal homeostasis? J. Immunol. 178, 27–32.
Corthésy, B. (2009). Secretory immunoglobulin A: well beyond immune exclusion at mucosal surfaces. Immunopharmacol. Immunotoxicol. 31, 174–179.
Crabbé, P. A., Nash, D. R., Bazin, H., Eyssen, H., and Heremans, J. F. (1970). Immunohistochemical observations on lymphoid tissues from conventional and germ-free mice. Lab. Invest. 22, 448–457.
Czinn, S. J., Cai, A., and Nedrud, J. G. (1993). Protection of germ-free mice from infection by Helicobacter felis after active oral or passive IgA immunization. Vaccine 11, 637–642.
Dham, S. K., and Thompson, R. A. (1982). Studies of cellular and humoral immunity in typhoid fever and TAB vaccinated subjects. Clin. Exp. Immunol. 48, 389–395.
Eberl, G., and Littman, D. (2004). Thymic origin of intestinal alpha beta T cells revealed by fate mapping of ROR gamma t(+) cells. Science 305, 248–251.
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., Peaper, D. R., Bertin, J., Eisenbarth, S. C., Gordon, J. I., and Flavell, R. A. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757.
Endt, K., Stecher, B., Chaffron, S., Slack, E., Tchitchek, N., Benecke, A., Van Maele, L., Sirard, J.-C., Mueller, A. J., Heikenwalder, M., Macpherson, A. J., Strugnell, R., von Mering, C., and Hardt, W. D. (2010). The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog. 6, e1001097. doi:10.1371/journal.ppat.1001097
Fagarasan, S., Muramatsu, M., Suzuki, K., Nagaoka, H., Hiai, H., and Honjo, T. (2002). Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 298, 1424–1427.
Forbes, S. J., Bumpus, T., McCarthy, E. A., Corthésy, B., and Mantis, N. J. (2011). Transient suppression of Shigella flexneri type 3 secretion by a protective O-antigen-specific monoclonal IgA. MBio 2, e00042–11.
Forbes, S. J., Eschmann, M., and Mantis, N. J. (2008). Inhibition of Salmonella enterica serovar typhimurium motility and entry into epithelial cells by a protective antilipopolysaccharide monoclonal immunoglobulin A antibody. Infect. Immun. 76, 4137–4144.
Fritz, J. H., Rojas, O. L., Simard, N., McCarthy, D. D., Hapfelmeier, S., Rubino, S., Robertson, S. J., Larijani, M., Gosselin, J., Ivanov, I. I., Martin, A., Casellas, R., Philpott, D. J., Girardin, S. E., McCoy, K. D., Macpherson, A. J., Paige, C. J., and Gommerman, J. L. (2012). Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481, 199–203.
Fuhrman, J. A., and Cebra, J. J. (1981). Special features of the priming process for a secretory IgA response. B cell priming with cholera toxin. J. Exp. Med. 153, 534–544.
Goodman, A. L., McNulty, N. P., Zhao, Y., Leip, D., Mitra, R. D., Lozupone, C. A., Knight, R., and Gordon, J. I. (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289.
Hammarström, L., Vorechovsky, I., and Webster, D. (2000). Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin. Exp. Immunol. 120, 225–231.
Hapfelmeier, S., Lawson, M. A. E., Slack, E., Kirundi, J. K., Stoel, M., Heikenwalder, M., Cahenzli, J., Velykoredko, Y., Balmer, M. L., Endt, K., Geuking, M. B., Curtiss, R., McCoy, K. D., and Macpherson, A. J. (2010). Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science 328, 1705–1709.
Harris, N. L., Spoerri, I., Schopfer, J. F., Nembrini, C., Merky, P., Massacand, J., Urban, J. F., Lamarre, A., Burki, K., Odermatt, B., Zinkernagel, R. M., and Macpherson, A. J. (2006). Mechanisms of neonatal mucosal antibody protection. J. Immunol. 177, 6256–6262.
Henao-Mejia, J., Elinav, E., Jin, C., Hao, L., Mehal, W. Z., Strowig, T., Thaiss, C. A., Kau, A. L., Eisenbarth, S. C., Jurczak, M. J., Camporez, J. P., Shulman, G. I., Gordon, J. I., Hoffman, H. M., and Flavell, R. A. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185.
Johansen, F.-E., Pekna, M., Norderhaug, I. N., Haneberg, B., Hietala, M. A., Krajci, P., Betsholtz, C., and Brandtzaeg, P. (1999). Absence of epithelial immunoglobulin A transport, with increased mucosal leakiness, in polymeric immunoglobulin receptor/secretory component-deficient mice. J. Exp. Med. 190, 915–922.
Johansson, M. E. V., Phillipson, M., Petersson, J., Velcich, A., Holm, L., and Hansson, G. C. (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. U.S.A. 105, 15064–15069.
Kadaoui, K. A., and Corthésy, B. (2007). Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J. Immunol. 179, 7751–7757.
Kang, H.-S., Chin, R. K., Wang, Y., Yu, P., Wang, J., Newell, K. A., and Fu, Y.-X. (2002). Signaling via LTbetaR on the lamina propria stromal cells of the gut is required for IgA production. Nat. Immunol. 3, 576–582.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., and Diefenbach, A. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565.
Ko, H.-J., Yang, J.-Y., Shim, D.-H., Yang, H., Park, S.-M., Curtiss, R., and Kweon, M.-N. (2009). Innate immunity mediated by MyD88 signal is not essential for induction of lipopolysaccharide-specific B cell responses but is indispensable for protection against Salmonella enterica serovar Typhimurium infection. J. Immunol. 182, 2305–2312.
Lamkhioued, B., Gounni, A. S., Gruart, V., Pierce, A., Capron, A., and Capron, M. (1995). Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur. J. Immunol. 25, 117–125.
Li, Y., Innocentin, S., Withers, D. R., Roberts, N. A., Gallagher, A. R., Grigorieva, E. F., Wilhelm, C., and Veldhoen, M. (2011). Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640.
Lindner, C., Wahl, B., Föhse, L., Suerbaum, S., Macpherson, A. J., Prinz, I., and Pabst, O. (2012). Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 209, 365–377.
Lupp, C., Robertson, M. L., Wickham, M. E., Sekirov, I., Champion, O. L., Gaynor, E. C., and Finlay, B. B. (2007). Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2, 119–129.
Macpherson, A. J., Gatto, D., Sainsbury, E., Harriman, G. R., Hengartner, H., and Zinkernagel, R. M. (2000). A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science 288, 2222–2226.
Macpherson, A. J., Lamarre, A., McCoy, K., Harriman, G. R., Odermatt, B., Dougan, G., Hengartner, H., and Zinkernagel, R. M. (2001). IgA production without mu or delta chain expression in developing B cells. Nat. Immunol. 2, 625–631.
Macpherson, A. J., and Uhr, T. (2004). Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 303, 1662–1665.
Martinoli, C., Chiavelli, A., and Rescigno, M. (2007). Entry route of Salmonella typhimurium directs the type of induced immune response. Immunity 27, 975–984.
Mbawuike, I. N., Pacheco, S., Acuna, C. L., Switzer, K. C., Zhang, Y., and Harriman, G. R. (1999). Mucosal immunity to influenza without IgA: an IgA knockout mouse model. J. Immunol. 162, 2530–2537.
Moreau, M. C., Ducluzeau, R., Guy-Grand, D., and Muller, M. C. (1978). Increase in the population of duodenal immunoglobulin A plasmocytes in axenic mice associated with different living or dead bacterial strains of intestinal origin. Infect. Immun. 21, 532–539.
Moura, I. C., Centelles, M. N., Arcos-Fajardo, M., Malheiros, D. M., Collawn, J. F., Cooper, M. D., and Monteiro, R. C. (2001). Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 194, 417–425.
Newberry, R. D., McDonough, J. S., McDonald, K. G., and Lorenz, R. G. (2002). Postgestational lymphotoxin/lymphotoxin beta receptor interactions are essential for the presence of intestinal B lymphocytes. J. Immunol. 168, 4988–4997.
Peterson, D. A., McNulty, N. P., Guruge, J. L., and Gordon, J. I. (2007). IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2, 328–339.
Pierce, N. F. (1978). The role of antigen form and function in the primary and secondary intestinal immune responses to cholera toxin and toxoid in rats. J. Exp. Med. 148, 195–206.
Reljic, R. (2006). In search of the elusive mouse macrophage Fc-alpha receptor. Immunol. Lett. 107, 80–81.
Shibuya, A., Sakamoto, N., Shimizu, Y., Shibuya, K., Osawa, M., Hiroyama, T., Eyre, H. J., Sutherland, G. R., Endo, Y., Fujita, T., Miyabayashi, T., Sakano, S., Tsuji, T., Nakayama, E., Phillips, J. H., Lanier, L. L., and Nakauchi, H. (2000). Fc alpha/mu receptor mediates endocytosis of IgM-coated microbes. Nat. Immunol. 1, 441–446.
Shroff, K. E., Meslin, K., and Cebra, J. J. (1995). Commensal enteric bacteria engender a self-limiting humoral mucosal immune response while permanently colonizing the gut. Infect. Immun. 63, 3904–3913.
Shulzhenko, N., Morgun, A., Hsiao, W., Battle, M., Yao, M., Gavrilova, O., Orandle, M., Mayer, L., Macpherson, A. J., McCoy, K. D., Fraser-Liggett, C., and Matzinger, P. (2011). Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat. Med. 17, 1585–1593.
Slack, E., Hapfelmeier, S., Stecher, B., Velykoredko, Y., Stoel, M., Lawson, M. A. E., Geuking, M. B., Beutler, B., Tedder, T. F., Hardt, W.-D., Bercik, P., Verdu, E. F., McCoy, K. D., and Macpherson, A. J. (2009). Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science 325, 617–620.
Stecher, B., and Hardt, W.-D. (2011). Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 14, 82–91.
Stecher, B., Robbiani, R., Walker, A. W., Westendorf, A. M., Barthel, M., Kremer, M., Chaffron, S., Macpherson, A. J., Buer, J., Parkhill, J., Dougan, G., von Mering, C., and Hardt, W. D. (2007). Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5, 2177–2189. doi:10.1371/journal.pbio.0050244
Stockert, R. J., Kressner, M. S., Collins, J. C., Sternlieb, I., and Morell, A. G. (1982). IgA interaction with the asialoglycoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 79, 6229–6231.
Stoel, M., Jiang, H.-Q., van Diemen, C. C., Bun, J. C. A. M., Dammers, P. M., Thurnheer, M. C., Kroese, F. G. M., Cebra, J. J., and Bos, N. A. (2005). Restricted IgA repertoire in both B-1 and B-2 cell-derived gut plasmablasts. J. Immunol. 174, 1046–1054.
Stokes, C. R., Soothill, J. F., and Turner, M. W. (1975). Immune exclusion is a function of IgA. Nature 255, 745–746.
Suzuki, K., Meek, B., Doi, Y., Muramatsu, M., Chiba, T., Honjo, T., and Fagarasan, S. (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. U.S.A. 101, 1981–1986.
Svennerholm, A., Lange, S., and Holmgren, J. (1978). Correlation between intestinal synthesis of specific immunoglobulin A and protection against experimental cholera in mice. Infect. Immun. 21, 1–6.
Talham, G. L., Jiang, H. Q., Bos, N. A., and Cebra, J. J. (1999). Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect. Immun. 67, 1992–2000.
Tezuka, H., Abe, Y., Iwata, M., Takeuchi, H., Ishikawa, H., Matsushita, M., Shiohara, T., Akira, S., and Ohteki, T. (2007). Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature 448, 929–933.
Tort, L., Balasch, J. C., and Mackenzie, S. (2003). Fish immune system. A crossroads between innate and adaptive responses. Immunologia 22, 277–286.
Tsuji, M., Suzuki, K., Kitamura, H., Maruya, M., Kinoshita, K., Ivanov, I. I., Itoh, K., Littman, D. R., and Fagarasan, S. (2008). Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity 29, 261–271.
Uren, T. K., Wijburg, O. L. C., Simmons, C., Johansen, F.-E., Brandtzaeg, P., and Strugnell, R. A. (2005). Vaccine-induced protection against gastrointestinal bacterial infections in the absence of secretory antibodies. Eur. J. Immunol. 35, 180–188.
Vaishnava, S., Behrendt, C. L., Ismail, A. S., Eckmann, L., and Hooper, L. V. (2008). Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. U.S.A. 105, 20858–20863.
Vaishnava, S., Yamamoto, M., Severson, K. M., Ruhn, K. A., Yu, X., Koren, O., Ley, R., Wakeland, E. K., and Hooper, L. V. (2011). The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 334, 255–258.
Van der Sluis, M., De Koning, B. A. E., De Bruijn, A. C. J. M., Velcich, A., Meijerink, J. P. P., Van Goudoever, J. B., Büller, H. A., Dekker, J., Van Seuningen, I., Renes, I. B., and Einerhand, A. W. (2006). Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131, 117–129.
van der Waaij, L. A., Limburg, P. C., Mesander, G., and van der Waaij, D. (1996). In vivo IgA coating of anaerobic bacteria in human faeces. Gut 38, 348–354.
Veldhoen, M., Hirota, K., Westendorf, A. M., Buer, J., Dumoutier, L., Renauld, J.-C., and Stockinger, B. (2008). The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453, 106–109.
Wei, M., Shinkura, R., Doi, Y., Maruya, M., Fagarasan, S., and Honjo, T. (2011). Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nat. Immunol. 12, 264–270.
Wijburg, O. L. C., Uren, T. K., Simpfendorfer, K., Johansen, F.-E., Brandtzaeg, P., and Strugnell, R. A. (2006). Innate secretory antibodies protect against natural Salmonella typhimurium infection. J. Exp. Med. 203, 21–26.
Keywords: mucosal immunology, IgA, microbiota, mucosal infection, natural antibody, adaptive immunity, innate immunity
Citation: Slack E, Balmer ML, Fritz JH and Hapfelmeier S (2012) Functional flexibility of intestinal IgA – broadening the fine line. Front. Immun. 3:100. doi: 10.3389/fimmu.2012.00100
Received: 31 January 2012; Paper pending published: 23 February 2012;
Accepted: 15 April 2012; Published online: 03 May 2012.
Edited by:
Nils Yngve Lycke, University of Gothenburg, SwedenReviewed by:
Dennis Metzger, Albany Medical College, USANicholas J. Mantis, New York State Department of Health, USA
Copyright: © 2012 Slack, Balmer, Fritz and Hapfelmeier. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Siegfried Hapfelmeier, Institute for Infectious Diseases, University of Berne, Friedbühlstrasse 51, CH-3010 Bern, Switzerland. e-mail: hapfelmeier@ifik.unibe.ch
†Present address: Maria Luisa Balmer, Medizinische Poliklinik, Universitätsspital Basel, Petersgraben 4, CH-4031 Basel, Switzerland.