 David Sancho
David Sancho Michel Enamorado
Michel Enamorado Johan Garaude
Johan Garaude- 1Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), Madrid, Spain
- 2Institute for Regenerative Medicine and Biotherapy, INSERM U1183, Montpellier, France
Sensing of microbe-associated molecular patterns or danger signals by innate immune receptors drives a complex exchange of information. Innate receptor signaling not only triggers transcriptional events but also induces profound changes in metabolic fluxes, redox balance, and metabolite abundance thereby influencing immune cell function. Mitochondria are at the core of metabolic adaptation to the changing environment. The close interaction between mitochondrial metabolism and immune signaling has emerged as a central regulator of innate sensing. Metabolic processes generate a constant flow of electrons that eventually end up in the mitochondrial electron transport chain (ETC). Two electron carriers and four respiratory complexes that can assemble as larger molecular supercomplexes compose the ETC in the mitochondrial inner membrane. While the meaning and biological relevance of such structural organization is a matter of passionate debates, recent data support that innate stimuli remodel the ETC. We will review the function of mitochondrial metabolism and ETC dynamics as innate rheostats that regulate signaling, transcription, and epigenetics to orchestrate innate immune responses.
Introduction: Myeloid Cells Reprogram Their Metabolism in Response to Environmental Cues
Ligation of pattern recognition receptors, cytokine receptors, and phagocytosis of dying or dead cells provoke key changes in myeloid cell metabolism that are only beginning to be explored (1). As an example, tissue damage-derived signals can modulate myeloid cell metabolic reprogramming since uptake of apoptotic cells increases the mitochondrial membrane potential to downregulate phagocytosis (2). Another relevant example of metabolic consequences induced by innate immune signal is the stimulation of mouse macrophages and bone marrow-derived dendritic cells differentiated with GM-CSF (GM-DCs) with agonists for toll-like receptors (TLRs) or for the β-glucan receptor Dectin-1, which result in aerobic glycolysis (the Warburg effect) (1, 3–8). Many recent reviews have already remarkably described the recent advances in our understanding of metabolic reprogramming from a “metabolic flux” point of view (1, 9–13). However, recent works have provided new elements on the mechanisms bridging innate immune recognition and mitochondrial metabolic functions, suggesting that mitochondria regulate their electron flow as adaptation to innate immune signals. Here, we will summarize some of these findings that point toward a key function of the mitochondrial respiratory chain in governing innate immune cell fate.
Mitochondria, a Metabolic Rheostat for Innate Immune Receptors
Metabolism As a Flow of Electrons
Metabolism is often seen as a complex arrangement of metabolic fluxes, redox signaling, and translational or posttranslational events that are mutually dependent. However, metabolism can also be seen as a flow of electrons through multiple parallel and alternative pathways where metabolites act as potential carriers of electrons. From this point of view, the main and ultimate acceptor of electrons is the mitochondrial electron transport chain (ETC). Many catabolic processes indeed supply electrons to the ETC in the form of reducing equivalents of nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH2), whose relative proportion depends on the nature of the fuel used (14). The capacity of the cell to use different fuels efficiently is thus critical for its ability to adapt to changing environmental cues (15). Mitochondria must therefore regulate their location, biogenesis, fusion or fission, structure, and internal metabolite fluxes in response to changes in fuel source or signals received by cell membrane receptors or intracellular sensors. In turn, mitochondria control cell metabolism by governing the balance of anabolism (lipogenesis and antioxidant defenses from citrate, gluconeogenesis, serine, and glycine biosynthesis from pyruvate, nucleotide biosynthesis) and catabolism (Krebs cycle, β-oxidation, oxidative phosphorylation) (16). Mitochondria are central for ATP synthesis, redox balance, reactive oxygen species (ROS) production, thermogenesis, and generation of metabolites, all of which impact cell function. Since the release of cytochrome c to the cytosol is a major trigger for apoptosis, mitochondria also regulate cell survival.
The ETC comprises two electron carriers [coenzyme Q (CoQ)/ubiquinone and cytochrome c] and four respiratory complexes [complexes I–IV (CI–CIV)], which, excluding CII, can assemble as larger molecular supercomplexes (SCs) in the mitochondrial inner membrane (16) (Figure 1). The assembly of SCs is dynamic and may adapt the electron flux to the available substrates (17, 18). The nature of the fuel conditions the proportion of electrons feeding into the ETC from NADH and FADH2, with a NADH/FADH2 electron ratio of 5 following full oxidation of glucose and a ratio of 2 following oxidation of the fatty acid palmitate (14, 17, 18). The H+ gradient generated by the ETC is then used by the H+-ATP synthase (CV) to generate ATP. Recent results support the notion that innate sensing of microbial features leads to adaptations in SC assembly and electron flow through the ETC in mouse macrophages (5, 19) suggesting that ETC exerts key functions in macrophage activation processes (Figure 1).
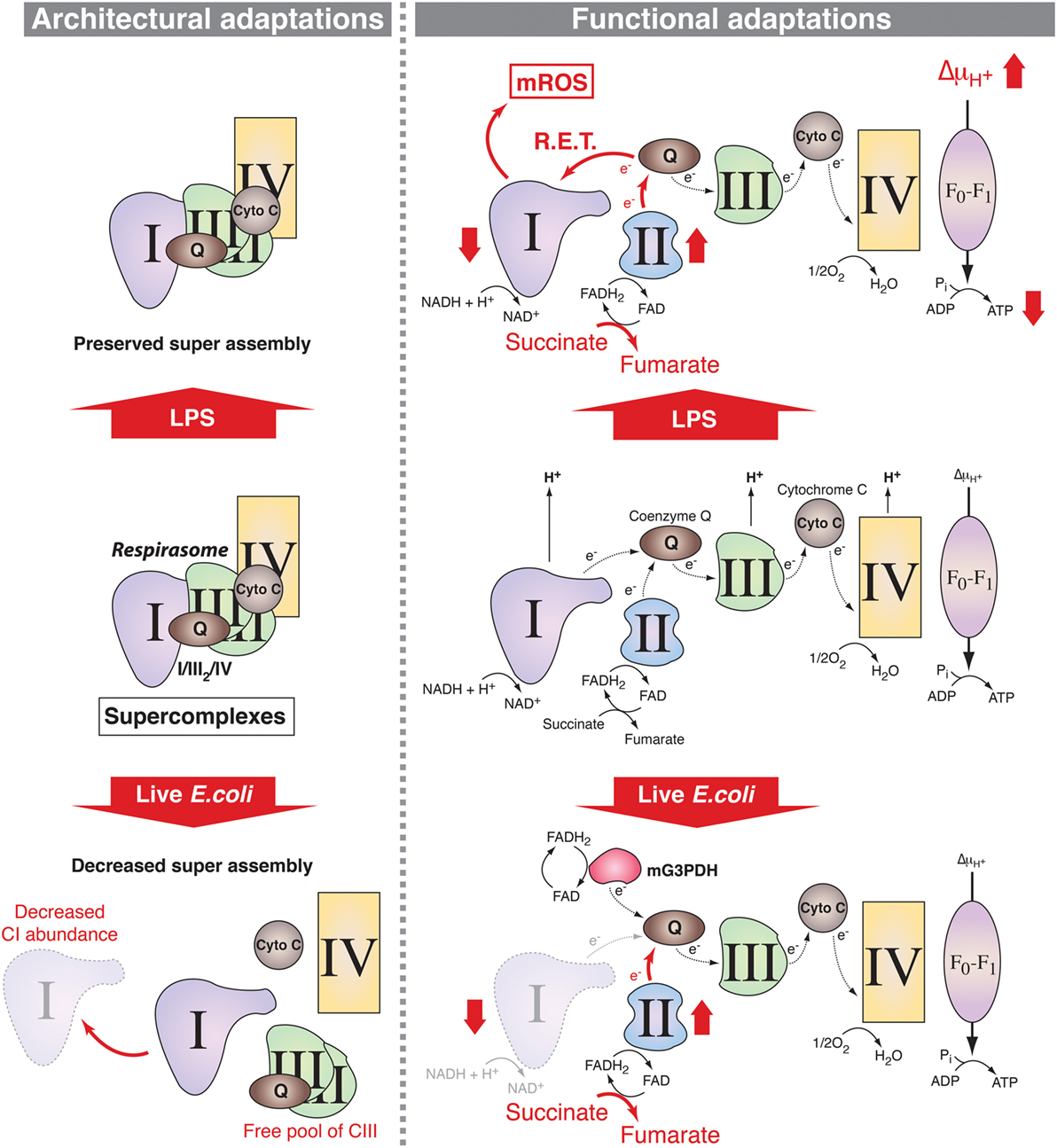
Figure 1. Mitochondrial respiratory chain adaptations following innate immune sensing. Mitochondrial respiratory complexes, except for CII, can associate into supercomplexes (SCs) including the respirasome, composed of CI + CIII2 + CIV (left panel). In lipopolysaccharide (LPS)-stimulated mouse macrophages, SC assembly is preserved (upper left) but there are functional adaptations such as increase in CII (succinate dehydrogenase) activity, which enhances succinate oxidation along with increased mitochondrial membrane potential and decreased mitochondrial ATP synthase-mediated production of ATP (upper right). This is accompanied by a decrease in the NAD+/NADH ratio, supporting a possible reverse electron transport (RET) from coenzyme Q to CI thereby inducing the production of mitochondrial reactive oxygen species (mROS). Upon detection of live Escherichia coli, increase in phagosomal reactive oxygen species mediates Fgr-dependent activation of CII and decrease in CI-containing SCs probably due to CI disassembly. This provokes a change in electron flow in the electron transport chain, with a drop in the entry of electron derived from NADH that is compensated by the induction of the activities of FADH2-consuming enzymes CII and mitochondrial glycerol-3-phosphate dehydrogenase (mG3PDH). The increase in CII activity generates fumarate that modulates macrophage function.
Mitochondria As Platforms for Innate Signaling
Mitochondria can both generate ligands and serve as signaling platform for innate sensing receptors (12). First, some mitochondrial-derived molecules trigger immune receptors. This is the case for mitochondrial N-formyl peptides, which are damage-associated molecular patterns that activate receptors such as formyl peptide receptor-1 to promote cytokine production (20). Furthermore, mitochondrial DNA (mtDNA) has hypomethylated CpG that can trigger TLR9 activation (20). mtDNA can access the cytosol through an altered permeability of the mitochondrial membrane to activate the NLRP3 inflammasome (21, 22). Later, it was discovered that glycolytic enzymes can directly contribute to innate sensing of microbes. Hexokinase is a glycolysis enzyme associated with the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (23). Following degradation of microbe-associated peptidoglycans in the phagosomes of mouse macrophages and DCs, peptidoglycan-derived N-acetylglucosamine binds to hexokinase causing its dissociation from the mitochondria outer membranes and VDAC (24). The NLRP3 inflammasome is subsequently activated, possibly relying on variations of the mitochondrial membrane permeability and the access of mtDNA to the cytosol. Thus, hexokinase moonlights as a regulator of NLRP3 activation and subsequent maturation of pro-IL-1β to promote an antibacterial pro-inflammatory response (24, 25). In addition, cytosolic location of mtDNA can also induce antiviral immunity by triggering the DNA sensor cGAS and the STING-IRF3-dependent pathway to promote IFN-I production (26).
Another example of how the outer mitochondrial membrane acts as a scaffold structure involved in innate sensing is the mitochondrial antiviral-signaling protein (MAVS) that binds to mitochondrial-associated membranes connecting the endoplasmic reticulum to the outer mitochondrial membrane (27). RIG-I binds to and promotes MAVS aggregates in the outer mitochondrial membrane to trigger downstream signaling (28). Thus, signaling and adaptor proteins downstream of pattern recognition receptor (PRR) sensing pathways can localize to mitochondria raising the possibility that PRRs could modulate mitochondrial functions. Such a regulatory role has been already suggested for tumor necrosis factor receptor-associated factor 6 (TRAF6), which can interact with evolutionarily conserved signaling intermediate in toll pathways (ECSIT), a protein involved in CI assembly (29, 30).
Metabolic Reprogramming in Macrophages upon Innate Immune Receptor Engagement
The specificities of macrophage metabolism compared to other immune cells have been a subject of interest for a long time as exemplified by pioneer works from the 80s (31–33). Our current appreciation of metabolic reprogramming led us to postulate that most inflammatory agonists for PRRs engage similar metabolic adaptations, although some specificities on the outcomes for host defense may exist. The main characteristic of metabolic adaptations upon innate immune receptor engagement is a strong induction of glycolysis even in presence of substantial oxygen (1, 3–8). Indeed, mouse macrophage stimulation with the TLR4 agonist lipopolysaccharide (LPS), the main component of Gram-negative bacterial cell wall, induces the activation of transcription factor hypoxia-inducible factor-1α (HIF-1α) (8) that controls the expression of several enzymes implicated in glycolysis (34, 35). LPS-activated mouse macrophages express a highly active isoform of phosphofructokinase-2 that promotes glycolysis (36) and LPS induces pyruvate kinase M2, which associates with and stabilizes HIF-1α to further induce glycolysis and enhance proinflammatory cytokines (37, 38). Such induction of glycolytic flux was also found in phagocytic cells activated through TLR2 (4, 39), TLR3 (4, 5), TLR7/8 (4, 5), TLR9 (4, 40), or Dectin-1 (3, 41), indicating that enhanced glycolysis might be a common feature to PRR-activated cells. The detection of a number of microbes including Salmonella typhimurium (5, 37), Escherichia coli (5), or Mycobacterium tuberculosis (37, 42, 43) strongly induce glycolysis in mouse macrophages. However, inflammasome activation of caspase-1 mediates the cleavage of glycolytic enzymes, e.g., during NLRC4 sensing of S. typhimurium (44) highlighting the complex interplay between PRRs and myeloid cell metabolism. In fact, induction of glycolysis does not seem to solely contribute to metabolic reprogramming but might also directly sustain pathogen sensing and host defense.
Although glycolysis is thought to largely contribute to ATP production in activated myeloid cells (40, 45), it also provides metabolic intermediates that could feed other metabolic pathways to serve macromolecule synthesis (45). Along with glycolysis, the pentose phosphate pathway (PPP) is induced in LPS-activated mouse macrophages (7). The mechanisms engaged are not fully understood, but they likely involve the inhibition of the PPP inhibitor carbohydrate kinase-like protein (46).
Because the glucose catabolic product pyruvate is diverted from entry to mitochondria in activated macrophages and is rather metabolized to lactate, it was tempting to speculate that engagement of innate immune receptors could globally dampen mitochondrial respiration. However, innate stimulation also activates pathways such as glutaminolysis to replenish the tricarboxylic acid (TCA) cycle and maintain global metabolic flux (7, 8, 47) likely avoiding cell death. Thus, a significant activity of the mitochondrial respiratory chain and the mitochondrial membrane potential must be maintained. This is partially achieved by an increase in anaplerosis including glutaminolysis (7, 8), which feeds the TCA cycle at α-ketoglutarate, and the aspartate–argininosuccinate shunt, which feeds the TCA cycle at malate and fumarate (7). Glutaminolysis is also enhanced in human monocytes trained with β-glucan, which triggers the C-type lectin receptor Dectin-1 (41, 48), suggesting that induction of such metabolic feature is not limited to TLRs. Therefore, an important issue to address is whether different PRRs induce specific metabolic reprogramming signatures, which would allow specific manipulation of myeloid cell metabolism for therapy.
How innate immune receptors regulate lipid metabolism in macrophages is a field of active research. On the one hand, engagement of TLRs upon recognition of various bacteria enhances fatty acid uptake and incorporation to triglycerides for storage and simultaneously decrease fatty acid oxidation and lipolysis suggesting that lipids may serve other needs than energy production in activated mouse macrophages (6, 49, 50). In line with this, fatty acid catabolism is not induced in LPS-activated macrophages compared to alternatively activated macrophages (7). Consistently, mouse macrophage fatty acid synthase is induced during sepsis in a mechanism depending on the mitochondrial uncoupling protein 2 (UCP2) (51). On the other hand, fatty acid oxidation is increased in mouse and human macrophages primed in conditions that activate the NLRP3 inflammasome, a process that requires mitochondrial NADPH oxidase 4 (NOX4)-dependent ROS production (52). Therefore, it is clear that needs for lipid metabolism in activated macrophages is significantly increased but whether this fulfills mitochondrial energy production requirements or rather constitutes the primal building blocks for anabolism needs to be further investigated.
Mitochondrial Respiratory Chain Adaptations in Macrophages upon Innate Immune Receptor Engagement
As stated above, most catabolic processes converge on the mitochondrial ETC by supplying electrons in the form of the reductive equivalents NADH and FADH2. The intramitochondrial NADH/FADH2 ratio depends on the nature of the fuels that feed the mitochondrial metabolism and the respiratory chain adapts to these fuel source fluctuations (16), particularly during PRR-mediated macrophage activation. Indeed, two recent studies provide evidence that changes in the ETC occur in activated mouse macrophages (5, 19). The phagocytosis of live Gram-negative bacteria by mouse macrophages trigger a profound change in ETC structural organization (5) (Figure 1). This is characterized by a decreased abundance of ETC SCs that contain CI and a relative increase in the free form of CIII and is accompanied by a substantial decrease in the activity of CI. By contrast, the activity of the glycerol 3-phosphate dehydrogenase and CII, two enzymes that use FADH2, are increased in response to live bacteria or to TLR-mediated sensing of bacterial RNA. CII activity is driven by the production of phagosomal ROS. This activates the ROS-dependent tyrosine kinase Fgr, which was previously found to phosphorylate CII (53). Consistently, CII was essential to mitochondrial respiration in bacteria-activated macrophages. This work thus suggests that adjustments of the organization of the ETC and of the activity of its components are required for metabolic reprogramming in macrophages. A study by O’Neill and colleagues has further precised the mechanism by which ETC integrates signals emerging from TLRs (19) (Figure 1). The combination of enhanced succinate dehydrogenase (SDH) activity and increased mitochondrial potential in LPS-activated mouse macrophages allows for the induction of mitochondrial ROS production at the level of CI thereby reprogramming mitochondria from ATP production to ROS production in order to establish an inflammatory state. This is consistent with a previous study showing that metformin-mediated inhibition of CI-dependent ROS production suppresses the inflammatory capacity of activated mouse macrophages (54). These data point out CI as a major regulator of inflammatory macrophages. This is backed up by genetical evidences demonstrating that mouse macrophages deficient for the CI subunit NDUFS4 exhibit an inflammatory phenotype (55). Interestingly, the deletion of TLR2/4 in NDUFS4-deficient mice attenuated the inflammatory phenotype (55), highlighting the close relationship between TLRs and the ETC. However, the contributions of mitochondrial respiratory chain to innate immune functions seems not limited to TLRs since the engagement of Dectin-1 in human monocytes induces accumulation of CII-product fumarate, which can induce an epigenetic program accounting for trained immunity by inhibiting KDM5 histone demethylases (41). Whether adaptations of the ETC reflect solely the metabolic fluctuations within activated macrophages or can be directly regulated by signals emerging from innate immune receptors remains to be clarified.
Functional Consequences of Mitochondrial Adaptations in Macrophages
Regulation of Macrophage Polarization and Cytokine Response
The finding that IL-4, a well-known inducer of alternatively activated mouse macrophages (M2), promotes a distinct metabolic reprogramming compared to pro-inflammatory (M1) mouse macrophage stimulating LPS/IFN-γ indicates that mitochondrial metabolism adjusts to the type of immune responses required to eliminate the threats encountered (1). M1 mouse macrophages produce high amount of pro-inflammatory cytokines and antimicrobial peptides and excel in phagocytosing and destroying microbes. Those macrophages exhibit a high glycolytic rate and present two breaks in the TCA cycle, one at isocitrate dehydrogenase, the enzyme that converts isocitrate to α-ketoglutarate, and another one at SDH that catalyzes the oxidation of succinate to fumarate (7). M2 mouse macrophages are essential at fighting against helminth infection, exert tissue repair functions, and conserve an intact TCA cycle (7). The specificities of metabolic fluxes in differentiated macrophages is a subject of intense research and has been reviewed elsewhere (1). However, whether mitochondrial ETC organization and its functional adjustments reflect the metabolic specificities of M1 and M2 macrophages is still poorly understood. A recent report nevertheless showed that LPS + IFN-γ treatment promotes NO that inhibits mitochondrial respiration thereby preventing the repolarization of M1 into M2 macrophages in mice and humans upon IL-4 treatment (56). In line with this, various reports suggest that manipulating the ETC modulates the balance between pro- and anti-inflammatory cytokine production. The use of the CI inhibitors metformin and rotenone decreased mitochondrial ROS production in LPS-activated mouse macrophages thereby reducing IL-1β production and boosting IL-10 production (54). In addition to their function as TLR7 agonists, imiquimod and CL097 also inhibited the quinone oxidoreductases NQO2 and CI, inducing ROS that contributed to NLRP3 activation (57). Conversely, the CII inhibitor dimethyl malonate enhanced the production of IL-1RA and IL-10 and promote an anti-inflammatory response by preventing mitochondrial ROS generation and succinate oxidation (19).
Mitochondrial metabolites, e.g., those related to α-ketoglutarate, such as succinate and fumarate, regulate transcription and influence the pattern of cytokine production that characterizes differentiated macrophages. Succinate is accumulated in M1 mouse macrophages and was originally thought to stabilize HIF-1α through inhibition of prolyl hydroxylases (58). However, new evidence shows that succinate rather drives CII activity, which in turn promotes mitochondrial reactive oxygen species (mROS) production to stabilize HIF-1α (19), thereby promoting pro-inflammatory cytokine IL-1β expression (8) (Figure 2). Succinate may also contribute to increased reverse electron transport and ROS production from CI (59), a process that is potentially important for macrophage function (60). Likewise, SDH (part of the CII) metabolizes succinate to fumarate in the TCA cycle, and fumarate inhibits histone demethylases thus regulating epigenetics (41). In this process, CII accepts the electrons from FADH2, and this function as electron carrier drives modulation of ROS signaling (60). These results establish how ROS drives expression of pro-inflammatory cytokines that characterize M1 macrophages but the mechanism by which succinate inhibits anti-inflammatory gene expression is still an open question. Taken together, those studies show the great potential of mitochondrial metabolism for regulation of transcription, cytokine production, and macrophage polarization that can be exploited for therapy.
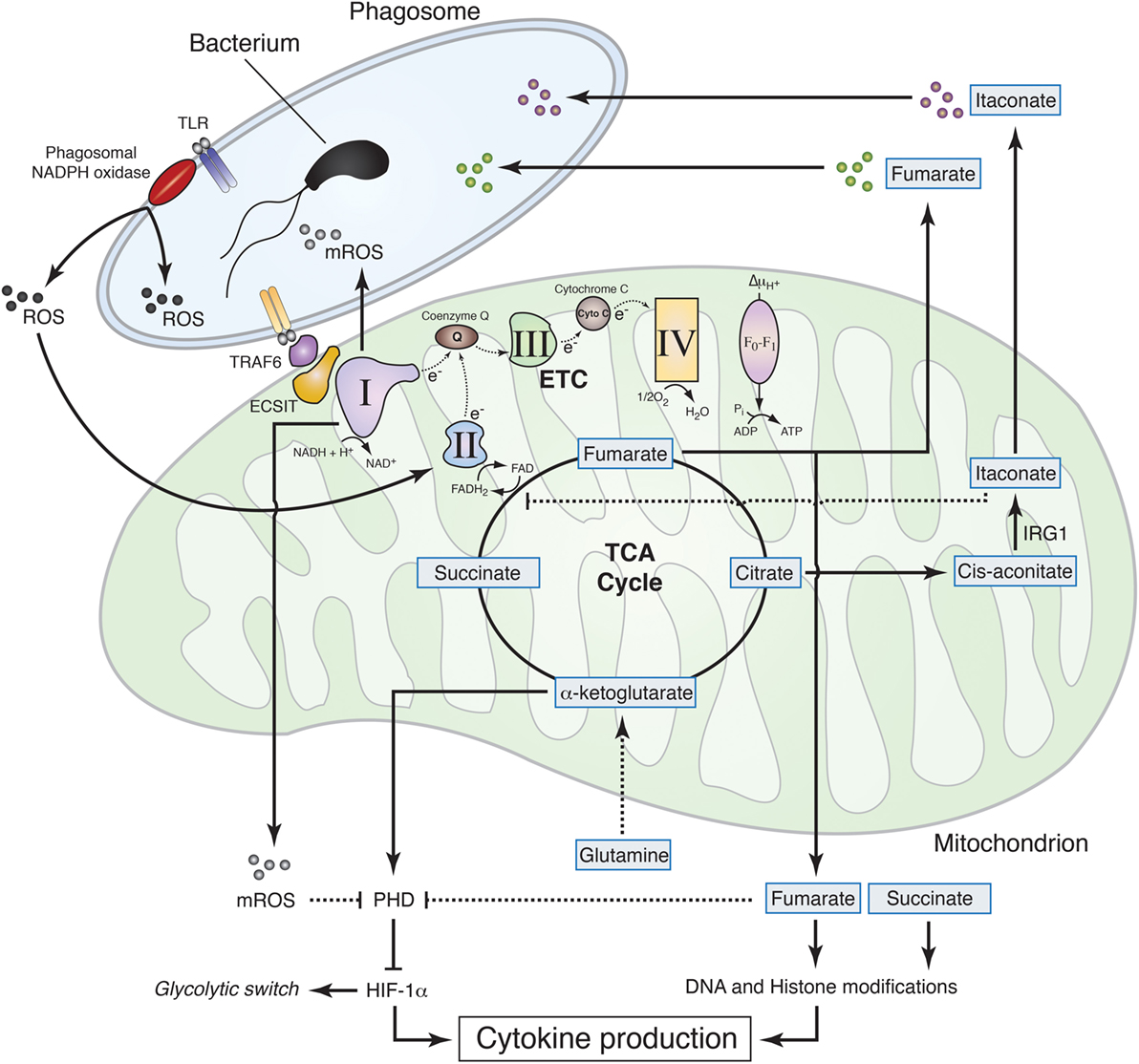
Figure 2. Mitochondrial metabolism contributions to macrophage effector functions. Sensing of live Gram-negative bacteria by toll-like receptors (TLR) induces the production of phagosomal reactive oxygen species (ROS) by phagosomal NADPH oxidase. Phagosomal ROS directly contribute to the killing of the bacteria inside the phagosome and induce CII activity thereby promoting fumarate accumulation, which exerts bactericidal properties and modulates histone posttranslational modifications to control cytokine production. The increase in CII activity is associated with a reduction in CI activity and a subsequent increase in mitochondrial ROS (mROS). TLR mediates recruitment of the mitochondria to the phagosome through tumor necrosis factor receptor-associated factor 6 (TRAF6)–evolutionarily conserved signaling intermediate in toll pathways (ECSIT) interaction and concomitantly increases mROS, which contribute to bacteria killing in the phagosome and inhibit prolyl hydroxylase (PHD), thus promoting hypoxia-inducible factor-1α (HIF-1α) stabilization. TLR signaling also induces accumulation of succinate, which, together with fumarate, inhibits α-ketoglutarate-dependent DNA hydroxylases and histone demethylases, regulating cytokine expression and glycolytic switch. Succinate oxidation, in turn, is required to induce mROS production at the level of CI, thereby inhibiting PHD, stabilizing HIF-1α, and further controlling cytokine production. In addition to fumarate, other tricarboxylic acid cycle-related metabolites exhibit antibactericidal properties. Citrate is used to produce itaconate through the decarboxylation of cis-aconitate by IRG1. Itaconate was found to have antimicrobial properties and to regulate succinate dehydrogenase (CII) activity in lipopolysaccharide-activated macrophages.
Regulation of Macrophage Bactericidal Functions
Although phagosomal ROS are considered to be a main component of bacteria killing machinery within macrophages, mitochondria seem to significantly contribute to antibacterial functions by generating a substantial amount of ROS (Figure 2). Engagement of a cell membrane-associated TLRs (TLR1, TLR2, and TLR4) results in the recruitment of mitochondria to macrophage phagosomes and increases mROS production. This response involves translocation of the TLR signaling adaptor TRAF6 to mitochondria where it engages the mitochondrial respiratory complex I assembly adaptor ECSIT (30). Consistently, mouse macrophages that lack ECSIT or TRAF6 show decreased levels of TLR-induced ROS and impaired ability to kill intracellular bacteria. In addition, reducing mouse macrophage mROS levels by expressing catalase in mitochondria results in defective bacterial killing, confirming the role of mROS in bactericidal activity (30). Additional studies support the notion that suppressing mROS production regulates antibacterial innate immune responses. Mice deficient for UCP2 and infected with a lethal load of Toxoplasma gondii were significantly more resistant compared with wild-type (WT) animals. Consistently, UCP2-deficient macrophages generated more ROS in response to T. gondii underlying this enhanced parasite-killing capacity (61). A subsequent study detailed a similar phenotype, demonstrating that Ucp2−/− mice were more resistant to Listeria monocytogenes and display higher splenic ROS levels than WT mice (62). LPS stimulation also reduces macrophage UCP2 expression, indicating that the abundance of UCP2 in the cell regulates mROS generation following TLR4 engagement (63). Additionally, it was demonstrated that IFNγ induces an estrogen-related receptor-α (ERRα)- and a proliferator-activated receptor-γ co-activator (PGC1β)-dependent transcriptional program in mouse macrophages that induces mitochondrial function and mROS production upon bacterial infection (64). Consequently, ERRα- or PGC1β-deficient macrophages produce less ROS upon L. monocytogenes challenge and display a reduced bacteria killing capacity (64). In this context, ETC re-arrangement may also regulate mROS production by controlling electron leak within the respiratory complexes (16). Future studies will likely provide additional details on the role of ETC architecture and electron flow for antimicrobial function of mitochondria.
The notion that mitochondria are important players for the microbicidal properties of macrophage has recently gained further interest with the finding that mitochondrial metabolites directly contribute to macrophage bactericidal functions. The mitochondrial protein immune responsive gene 1 (IRG1) is highly expressed in mouse and human macrophages during inflammation. It metabolizes cis-aconitate into itaconate, which can drive the production of β-oxidation-dependent mitochondrial ROS (65). Importantly, itaconate inhibits citrate-lyase in different bacterial strains and thus shows direct antimicrobial activity (66) (Figure 2). IFNs induce IRG1, which in turn accumulates in mitochondria that closely associate with Legionella-containing vacuoles, as step that may be required for bacteria killing (67). In addition, itaconate has been postulated to modulate mouse macrophage metabolism and effector functions by inhibiting SDH. Indeed, Irg1−/− mice, which cannot produce itaconate, have decreased level of succinate and increased mitochondrial respiration and inflammatory cytokine production upon LPS stimulation (68). Reorganization of the ETC may also participate to such processes, since the SDH product fumarate can directly inhibit bacteria proliferation in response to ETC adjustments upon sensing of viable bacteria (5) (Figure 2). Notably, the substrate of SDH succinate did not show antimicrobial properties as compared to fumarate, revealing specific immune properties of closely related mitochondria-derived metabolites (5).
Concluding Remarks
Since mitochondria have emerged as key conductors of the metabolic adaptations to the innate sensing by modulating the electron flow in the ETC (5, 19), a key open question is whether targeting of mitochondrial metabolism can be used to modulate immune cell function with substantial improvement for therapeutic strategies. This would be particularly interesting for enhanced vaccination and modulation of immunity and tolerance when targeting DCs, or for the treatment of inflammatory diseases in the case of targeting macrophages. In the later, mitochondrial manipulation would constitute an approach of choice to facilitate macrophage repolarization (56). Because some mitochondrial metabolites and the use of ETC CI or CII inhibitors can modulate cytokine production (8, 19, 41, 54, 57), such approach seems very promising. Moreover, mitochondrial ROS and metabolites, such as fumarate or itaconate, show a direct microbicidal effect (5, 30, 66). The exciting findings in this rapidly moving field thus emphasize the great potential of targeting mitochondrial metabolism for modulation of transcription, polarization, cytokine production, and microbicidal capacity of macrophages with potential to offer new therapeutic approaches.
Author Contributions
DS, ME, and JG conceived and wrote the manuscript. JG did the figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
DS’s laboratory is funded by the CNIC and grants from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO) and European Fund for Regional Development (FEDER) (SAF-2016-79040-R), the European Commission (635122-PROCROP H2020), the Fondation ACTERIA, and the European Research Council. The CNIC is supported by the MINECO and the Pro-CNIC Foundation, and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505). ME is the recipient of a CNIC International PhD Programme fellowship “la Caixa”—Severo Ochoa, 2013 Call (OSLC-CNIC-2013-04). JG’s laboratory is funded by INSERM and a European FP7-Marie Curie Career Integration Grant (332881).
References
1. O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213(1):15–23. doi:10.1084/jem.20151570
2. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature (2011) 477(7363):220–4. doi:10.1038/nature10340
3. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi:10.1126/science.1250684
4. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKK? supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15(4):323–32. doi:10.1038/ni.2833
5. Garaude J, Acin-Perez R, Martinez-Cano S, Enamorado M, Ugolini M, Nistal-Villan E, et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat Immunol (2016) 17(9):1037–45. doi:10.1038/ni.3509
6. Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol (2014) 15(9):846–55. doi:10.1038/ni.2956
7. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi:10.1016/j.immuni.2015.02.005
8. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496(7444):238–42. doi:10.1038/nature11986
9. Mills EL, O’Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol (2016) 46(1):13–21. doi:10.1002/eji.201445427
10. Gleeson LE, Sheedy FJ. Metabolic reprogramming and inflammation: fuelling the host response to pathogens. Semin Immunol (2016) 28(5):450–68. doi:10.1016/j.smim.2016.10.007
11. Pearce EJ, Everts B. Dendritic cell metabolism. Nat Rev Immunol (2015) 15(1):18–29. doi:10.1038/nri3771
12. Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity (2015) 42(3):406–17. doi:10.1016/j.immuni.2015.02.002
13. Arts RJ, Joosten LA, Netea MG. Immunometabolic circuits in trained immunity. Semin Immunol (2016) 28(5):425–30. doi:10.1016/j.smim.2016.09.002
14. Speijer D. Oxygen radicals shaping evolution: why fatty acid catabolism leads to peroxisomes while neurons do without it: FADH(2)/NADH flux ratios determining mitochondrial radical formation were crucial for the eukaryotic invention of peroxisomes and catabolic tissue differentiation. Bioessays (2011) 33(2):88–94. doi:10.1002/bies.201000097
15. Stanley IA, Ribeiro SM, Gimenez-Cassina A, Norberg E, Danial NN. Changing appetites: the adaptive advantages of fuel choice. Trends Cell Biol (2014) 24(2):118–27. doi:10.1016/j.tcb.2013.07.010
16. Enriquez JA. Supramolecular organization of respiratory complexes. Annu Rev Physiol (2016) 78:533–61. doi:10.1146/annurev-physiol-021115-105031
17. Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, Latorre-Pellicer A, Colas C, Balsa E, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science (2013) 340(6140):1567–70. doi:10.1126/science.1230381
18. Guaras A, Perales-Clemente E, Calvo E, Acin-Perez R, Loureiro-Lopez M, Pujol C, et al. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep (2016) 15(1):197–209. doi:10.1016/j.celrep.2016.03.009
19. Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell (2016) 167(2):457–70 e413. doi:10.1016/j.cell.2016.08.064
20. Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature (2010) 464(7285):104–7. doi:10.1038/nature08780
21. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12(3):222–30. doi:10.1038/ni.1980
22. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36(3):401–14. doi:10.1016/j.immuni.2012.01.009
23. Pastorino JG, Hoek JB. Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr (2008) 40(3):171–82. doi:10.1007/s10863-008-9148-8
24. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell (2016) 166(3):624–36. doi:10.1016/j.cell.2016.05.076
25. Moon J-S, Hisata S, Park M-A, DeNicola GM, Ryter SW, Nakahira K, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep (2015) 12(1):102–15. doi:10.1016/j.celrep.2015.05.046
26. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature (2015) 520(7548):553–7. doi:10.1038/nature14156
27. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A (2011) 108(35):14590–5. doi:10.1073/pnas.1110133108
28. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell (2011) 146(3):448–61. doi:10.1016/j.cell.2011.06.041
29. Vogel RO, Janssen RJ, van den Brand MA, Dieteren CE, Verkaart S, Koopman WJ, et al. Cytosolic signaling protein ECSIT also localizes to mitochondria where it interacts with chaperone NDUFAF1 and functions in complex I assembly. Genes Dev (2007) 21(5):615–24. doi:10.1101/gad.408407
30. West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature (2011) 472(7344):476–80. doi:10.1038/nature09973
31. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect Immun (1996) 64(1):108–12.
32. Hard GC. Some biochemical aspects of the immune macrophage. Br J Exp Pathol (1970) 51(1):97–105.
33. Newsholme P, Curi R, Gordon S, Newsholme EA. Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J (1986) 239(1):121–5. doi:10.1042/bj2390121
34. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi:10.1016/j.cmet.2006.02.002
35. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem (1996) 271(51):32529–37. doi:10.1074/jbc.271.51.32529
36. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185(1):605–14. doi:10.4049/jimmunol.0901698
37. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab (2015) 21(1):65–80. doi:10.1016/j.cmet.2014.12.005
38. Yang L, Xie M, Yang M, Yu Y, Zhu S, Hou W, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun (2014) 5:4436. doi:10.1038/ncomms5436
39. Lachmandas E, Beigier-Bompadre M, Cheng SC, Kumar V, van Laarhoven A, Wang X, et al. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur J Immunol (2016) 46(11):2574–86. doi:10.1002/eji.201546259
40. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115(23):4742–9. doi:10.1182/blood-2009-10-249540
41. Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab (2016) 24(6):807–19. doi:10.1016/j.cmet.2016.10.008
42. Gleeson LE, Sheedy FJ, Palsson-McDermott EM, Triglia D, O’Leary SM, O’Sullivan MP, et al. Cutting edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J Immunol (2016) 196(6):2444–9. doi:10.4049/jimmunol.1501612
43. Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, et al. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog (2014) 10(7):e1004265. doi:10.1371/journal.ppat.1004265
44. Shao W, Yeretssian G, Doiron K, Hussain SN, Saleh M. The caspase-1 digestome identifies the glycolysis pathway as a target during infection and septic shock. J Biol Chem (2007) 282(50):36321–9. doi:10.1074/jbc.M708182200
45. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood (2012) 120(7):1422–31. doi:10.1182/blood-2012-03-419747
46. Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab (2012) 15(6):813–26. doi:10.1016/j.cmet.2012.04.023
47. Meiser J, Kramer L, Sapcariu SC, Battello N, Ghelfi J, D’Herouel AF, et al. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J Biol Chem (2016) 291(8):3932–46. doi:10.1074/jbc.M115.676817
48. Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe (2012) 12(2):223–32. doi:10.1016/j.chom.2012.06.006
49. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol (2012) 92(4):829–39. doi:10.1189/jlb.1111537
50. Nicolaou G, Goodall AH, Erridge C. Diverse bacteria promote macrophage foam cell formation via toll-like receptor-dependent lipid body biosynthesis. J Atheroscler Thromb (2012) 19(2):137–48. doi:10.5551/jat.10249
51. Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity (2015) 44(1):155–66. doi:10.1016/j.immuni.2015.11.004
52. Moon J-S, Nakahira K, Chung K-P, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med (2016) 22(9):1002–12. doi:10.1038/nm.4153
53. Acin-Perez R, Carrascoso I, Baixauli F, Roche-Molina M, Latorre-Pellicer A, Fernandez-Silva P, et al. ROS-triggered phosphorylation of complex II by Fgr kinase regulates cellular adaptation to fuel use. Cell Metab (2014) 19(6):1020–33. doi:10.1016/j.cmet.2014.04.015
54. Kelly B, Tannahill GM, Murphy MP, O’Neill LA. Metformin inhibits the production of reactive oxygen species from NADH: ubiquinone oxidoreductase to limit induction of interleukin-1beta (IL-1beta) and boosts interleukin-10 (IL-10) in lipopolysaccharide (LPS)-activated macrophages. J Biol Chem (2015) 290(33):20348–59. doi:10.1074/jbc.M115.662114
55. Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab (2014) 20(3):483–98. doi:10.1016/j.cmet.2014.07.011
56. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep (2016) 17(3):684–96. doi:10.1016/j.celrep.2016.09.008
57. Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, et al. K+ Efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity (2016) 45(4):1–14. doi:10.1016/j.immuni.2016.08.010
58. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell (2005) 7(1):77–85. doi:10.1016/j.ccr.2004.11.022
59. Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature (2014) 515(7527):431–5. doi:10.1038/nature13909
60. Mills E, O’Neill LA. Succinate: a metabolic signal in inflammation. Trends Cell Biol (2014) 24(5):313–20. doi:10.1016/j.tcb.2013.11.008
61. Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet (2000) 26(4):435–9. doi:10.1038/82565
62. Rousset S, Emre Y, Join-Lambert O, Hurtaud C, Ricquier D, Cassard-Doulcier AM. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine (2006) 35(3–4):135–42. doi:10.1016/j.cyto.2006.07.012
63. Emre Y, Hurtaud C, Nubel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. Biochem J (2007) 402(2):271–8. doi:10.1042/BJ20061430
64. Sonoda J, Laganiere J, Mehl IR, Barish GD, Chong LW, Li X, et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev (2007) 21(15):1909–20. doi:10.1101/gad.1553007
65. Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating β-oxidation-dependent mitochondrial ROS production. Cell Metab (2013) 18(2):265–78. doi:10.1016/j.cmet.2013.06.018
66. Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci U S A (2013) 110(19):7820–5. doi:10.1073/pnas.1218599110
67. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog (2016) 12(2):e1005408. doi:10.1371/journal.ppat.1005408
Keywords: innate immune response, immunometabolism, electron transport chain, mitochondria, macrophages, dendritic cells, cytokines, inflammation
Citation: Sancho D, Enamorado M and Garaude J (2017) Innate Immune Function of Mitochondrial Metabolism. Front. Immunol. 8:527. doi: 10.3389/fimmu.2017.00527
Received: 22 December 2016; Accepted: 19 April 2017;
Published: 08 May 2017
Edited by:
Ping-Chih Ho, University of Lausanne, SwitzerlandReviewed by:
Tiffany Horng, Harvard University, USAPu-Ste Liu, Kaohsiung Medical University, Taiwan
Copyright: © 2017 Sancho, Enamorado and Garaude. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Sancho, dsancho@cnic.es;
Johan Garaude, johan.garaude@inserm.fr