 Jiatao Li
Jiatao Li Jean Tan3
Jean Tan3 Mikaël M. Martino
Mikaël M. Martino Kathy O. Lui
Kathy O. Lui- 1Department of Chemical Pathology, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, Hong Kong
- 2Li Ka Shing Institute of Health Sciences, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong, Hong Kong
- 3European Molecular Biology Laboratory Australia, Australian Regenerative Medicine Institute, Monash University, Melbourne, VIC, Australia
The identification of stem cells and growth factors as well as the development of biomaterials hold great promise for regenerative medicine applications. However, the therapeutic efficacy of regenerative therapies can be greatly influenced by the host immune system, which plays a pivotal role during tissue repair and regeneration. Therefore, understanding how the immune system modulates tissue healing is critical to design efficient regenerative strategies. While the innate immune system is well known to be involved in the tissue healing process, the adaptive immune system has recently emerged as a key player. T-cells, in particular, regulatory T-cells (Treg), have been shown to promote repair and regeneration of various organ systems. In this review, we discuss the mechanisms by which Treg participate in the repair and regeneration of skeletal and heart muscle, skin, lung, bone, and the central nervous system.
Introduction
The global number of individuals suffering from organ dysfunction as a result of acute injuries, chronic disorders, or aging has been on the rise and thus inadvertently places a high demand for organ transplantation. However, organ and tissue transplantation is obviously limited by the shortage of donors and side effects associated with the use of immunosuppressants (1), placing stress upon current methodology and creating a need for an alternative therapeutic avenue. By virtue of its self-renewal properties and capability in differentiating into multiple cell types, recent advances in human pluripotent stem cell research has offered a literally unlimited amount and varieties of therapeutic cells for transplantation (2, 3). Nevertheless, there is a lack of clinical evidence showing their long-term engraftment following transplantation possibly due to poor cell survival and chronic immune rejection (4, 5). Moreover, regenerative therapies stimulating endogenous regeneration such as growth factor-based strategies have shown mixed results in the clinic due to safety concerns and cost-effectiveness (6, 7). Therefore, it is necessary to find new ways to improve regenerative strategies and one of them is to control and utilize the host immune system. Nevertheless, in order to design immune-centric regenerative therapies, it is imperative to understand how the various immune components modulate tissue repair and regeneration.
Since decades, the immune system is well known to be implied in tissue repair and regeneration. For instance, inflammation following injury greatly contributes to tissue repair and scar formation, while excessive inflammation led by immune cells causes pathological fibrosis that debilitates tissue function and may lead to organ failure. Immune-mediated tissue healing processes are complex, yet, highly orchestrated. After injury, invading pathogens, necrotic debris, the clotting reaction, and tissue-resident immune cells trigger an inflammatory response, which result in the recruitment of various immune cells. The activity of immune cells during wound healing can be separated in three phases (8): First, pro-inflammatory cells are recruited to the site of injury for host defense and phagocytosis of necrotic tissues. Second, the pro-inflammatory response is dampened via immune cells such as anti-inflammatory macrophages, while immune cells also directly participate in stimulating angiogenesis, myofibroblast activation, and tissue progenitor cell proliferation. Last, most immune cells exit the site of injury or are eliminated by apoptosis and the tissue homeostasis is restored. Nonetheless, the role of the various immune cells and their subsets as well as the mechanisms by which they regulate tissue healing remain largely elusive. It is, therefore, imperative to understand how tissue healing is controlled by the immune system and harnessing the endogenous regenerative capacity has recently become an active area of research.
An interesting observation supporting the critical role of immunity in regeneration (as opposed to tissue repair and scarring) comes from the evolution of the immune system among species and during development. Compared to lower vertebrates such as amphibians and teleost fishes that are capable of completely regenerating many body parts, mammals have a limited regenerative potential. To explain this difference, it has been postulated that the loss of regenerative capacity in mammals is in part associated with maturation of their immune system compared to lower vertebrates (9, 10). The immune system also changes during development and throughout life. For example, some organs such as the mammalian heart is notorious for not being able to regenerate and the necrotic cardiac muscles are replaced by dysfunctional scar tissues after injury. However, accumulating evidence shows that the neonatal hearts of mammals including humans have a transient regenerative capacity compared to adults (11–13). Indeed, the mammalian adaptive immune system is relatively immature after birth, which coincides with the period of neonatal regeneration. In contrast to adults, neonates do not mount a robust fibrotic but a more angiogenic response that facilitates tissue regeneration after injury (10). Therefore, since immune cells regulate both fibrosis and angiogenesis during tissue healing, targeting the immune system to promote neoangiogenesis with minimal fibrosis would be an interesting approach to stimulate regeneration. Therefore, understanding how immunity regulates tissue fibrosis and neoangiogenesis would shed light on the development of potential therapeutics targeting endogenous tissue regeneration. During the last decade, innate immunity, in particular, macrophages and their various polarization states, has been considered as a central regulator of the tissue healing process. However, recent evidences suggest that the adaptive immune system is also a critical actor. In this review, we focus on the role of regulatory T-cells (Treg).
Overview of the Immune Functions of Treg During Tissue Healing
Treg are required for maintenance of self-tolerance, preventing excessive inflammation and autoimmune diseases. The most reliable cell-specific marker of Treg is Forkhead box P3 (FOXP3), which is essential for the maturation and function of Treg. Congenital deficiency in Treg, due to mutation of the Foxp3 gene, causes fatal autoimmunity in mice, the scurfy phenotype, and enteropathy, X-linked (IPEX) syndrome in human (14, 15). Treg are normally present in lymphoid organs but have been shown to accumulate in damaged tissues to some extent. Long recognized as potent suppressors of the immune system, Treg have been recently rediscovered as indirect and direct regulators of tissue healing, while the mechanisms are still largely unknown (16–18).
Uncontrolled inflammation after tissue injury can lead to impaired healing and tissue remodeling. In many tissues, Treg are recruited to the damaged site to facilitate inflammation resolution and to regulate immunity after injury (19). For instance, Treg can indirectly modulate regeneration by controlling neutrophils (20–22), inducing macrophage polarization (23, 24), and regulating helper T-cells (22, 25) (Figure 1) Moreover, Treg have been shown to directly facilitate regeneration via activating progenitor cells locally (16, 17).
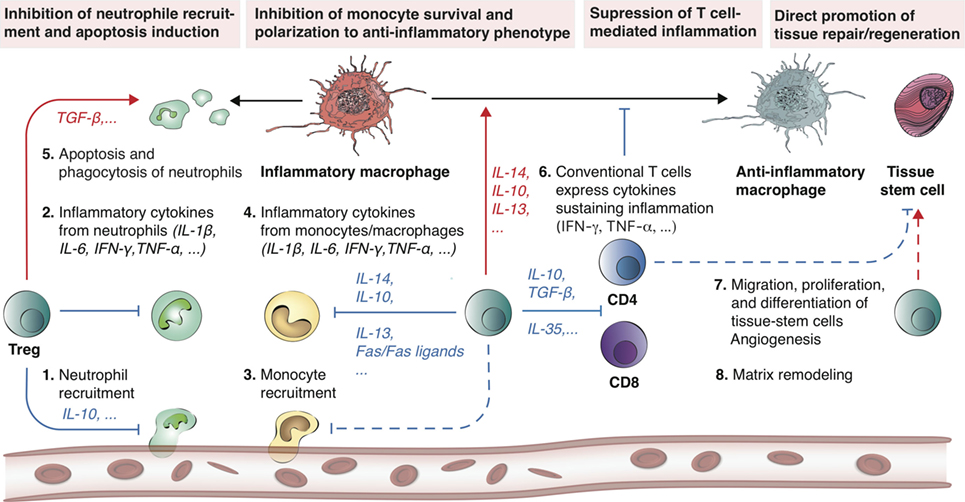
Figure 1. Treg promote tissue repair and regeneration by modulating inflammation. Treg have demonstrated the ability to promote tissue repair and regeneration by controlling both the innate and adaptive immune systems. Following tissue injury, a cascade of immune events is triggered (steps 1–6) until a new tissue is formed (steps 7–8). Treg are involved in all these different steps. At the onset of inflammation, Treg can neutralize inflammatory cytokine secretion (e.g., IL-6, IFN-γ, TNF-α, and IL-1β) by inhibiting neutrophil extravasation via IL-10. In addition, Treg are able to promote apoptosis of neutrophils and encourage phagocytosis of dead neutrophils by macrophages. Concomitantly, Treg further inhibit monocyte activity, survival, and stimulate macrophage polarization toward an anti-inflammatory phenotype (M2) via the release of anti-inflammatory cytokines (e.g., IL-4, IL-10, IL-13). Similarly, Treg have the natural ability to suppress CD4 and CD8 T cell-mediated inflammation (via IL-10, TGF-β, and IL-35). Overall, these Treg-mediated mechanisms result in the inhibition of neutrophil, inflammatory macrophage, as well as CD4 and CD8 T-cell activity, which is generally favorable for tissue repair and regeneration. Dashed lines indicate a hypothetical mechanism. Red arrows indicate induction, while blue arrows indicate inhibition.
Treg Interact with Innate Immune Cells to Control Inflammation After Tissue Injury
Treg are able to control the functions of neutrophils and macrophages, which have been widely shown to be involved in the tissue healing process. Neutrophils are among the first leukocytes recruited to the injury site, and they directly modulate tissue healing either positively or negatively. For instance, after skeletal muscle injury, it has been demonstrated that neutrophils impair restoration of muscle structures and function through the release of hypochlorous acid, NAPDH oxidase, and other cytokines (26, 27). A negative role of neutrophils has also been demonstrated in a lung ischemia-reperfusion model, where neutrophils enhance the injury (28). However, in an inflammatory lung disease model, mice treated with intratracheal LPS, which induces neutrophil transmigration show activated β-catenin signaling in lung epithelial cells, triggering repair of the lung epithelium (29). Therefore, it is likely that neutrophils modulate tissue healing in a context-specific manner.
Treg have shown ability to modulate tissue healing via controlling neutrophil behavior. For instance, an in vitro study has demonstrated that activated Treg promote neutrophils to secrete anti-inflammatory molecules including IL-10 and TGF-β, heme oxygenase-1, and indoleamine 2,3-dioxygenase (IDO). This is also preceded by inhibition of neutrophil’s IL-6 production, suggesting that Treg can modulate inflammation through inhibition of neutrophil activity (30). Concomitantly, Treg have been shown to induce neutrophil apoptosis and death both in vitro and in vivo (21, 31). For example, in an acute lung injury model, Treg mediate resolution of lung injury via TGF-β-induced neutrophil apoptosis (21). In addition, Treg can modulate neutrophil infiltration to the site of injury. For instance, deletion of Treg leads to increased infiltration of neutrophils after cardiac injury and subsequently results in impaired healing (20, 22). In a model of kidney ischemia reperfusion injury, Treg suppress infiltration of neutrophils and attenuate kidney injury via IL-10 secretion (32). These studies indicate that Treg-mediated modulation of neutrophil behavior and activity is an important step toward regulating tissue healing.
Asides from neutrophils, Treg interact with other key innate immune cells involved in the inflammatory response such as macrophages. In addition to being scavengers that phagocytose cellular debris including apoptotic neutrophils and other cells, macrophages have been shown to play a pivotal role in tissue repair and regeneration. As a remarkable example, salamanders are well-known to be able to regenerate limbs, but depletion of macrophages leads to failure of limb blastemal formation and regeneration (33). Similarly, genetic ablation of macrophages during blastemal proliferation leads to failure of tail fin regeneration in adult zebrafish (34). In mice, depletion of macrophages leads to excessive fibrosis and lack of neoangiogenesis, resulting in failure in neonatal heart regeneration after myocardial infarction (MI) or apex resection (11, 24). Likewise, macrophages are important for cardio protection driven by cardiosphere-derived cells (CDCs), a stem-like population derived from cardiac biopsies ex vivo. Indeed, systemic depletion of macrophages with clodronate abolishes CDC-mediated cardioprotection and inhibits their regenerative capability in adult hearts after MI (35).
Importantly, during tissue healing, there are at least two different subsets of monocyte-derived macrophages, namely M1 and M2 macrophages. M1 are pro-inflammatory macrophages usually induced by IFN-γ or TNF-α, while M2 are anti-inflammatory usually induced by IL-4/IL-13 or IL-10. In this context, Treg are important regulator of macrophage phenotypes and functions (36–38). For example, monocytes cocultured with Treg produce decreased level of TNF-α and IL-6 in response to LPS; and the inhibition is associated with secretion of IL-10, IL-4, and IL-13 by Treg (39). Additionally, coculture of monocytes with Treg induces macrophages to polarize toward a M2 phenotype characterized by the upregulation of CD206, CD163, and decreased expression of HLA-DR (40). Treg can attenuate tissue injury and help tissue repair also by modulating macrophage activity and survival. For example, in a chronic kidney disease model, Treg protect kidney injury through inhibition of macrophage activity, which is dependent on Treg-derived TGF-β (41). In this context, Treg also inhibit monocyte survival through the Fas/FasL pathway (41).
Treg Facilitate Tissue Healing via Regulation of Conventional T–Cell Activities
Mounting evidence suggest that conventional T-cells are most likely detrimental for tissue healing (42). For example, CD4- and CD8-deficient mice have improved renal function in renal ischemia reperfusion model. SCID mice, which lack lymphocytes have significantly decreased intestinal leakage of albumin compared to wild-type mice after mesenteric artery ischemia and reperfusion (43, 44). In a MI model, CD8+ cytotoxic T-cells can respond to cardiomyocytes after being exposed to autoantigen in vivo and kill healthy cardiomyocytes in vitro (45, 46). Moreover, Rag−/− mice, which lack T-cells have significantly smaller infarct size compared to control mice (47). In the context of bone, conventional T-cells may inhibit regeneration by promoting osteoclast differentiation (48). In addition, recruitment of CD8+ effector T-cells is correlated with delayed fracture healing and osteogenesis, due to secretion of IFN-γ and TNF-α (49). Deletion of CD8+ T-cells in mouse osteotomy model enhances fracture healing while adoptive transfer of CD8+ T-cells results in impaired healing (49).
The negative role of conventional T-cells in tissue injury is most likely mediated by the expression of inflammatory cytokines such as TNF-α and IFN-γ (50–52), but Treg can suppress conventional T-cells through various mechanisms including secretion of anti-inflammatory cytokines such as IL-10, TGF-β, and IL-35 (53–56). For example, it has been shown that deletion of Treg increase CD4+ and CD8+ cell number in the heart injury zone. In this context, both CD8+ and CD4+ T-cells show an increased secretion of IFN-γ and TNF-α, suggesting that Treg not only decrease the infiltration of conventional T-cells, but also attenuate their activity (22). Yet, the mechanisms by which Treg modulate tissue repair and regeneration are most likely tissue-dependent. In the next sections, we will discuss the role of Treg in the repair and regeneration of various tissues including skeletal and heart muscle, skin, lung, bone, and the central nervous system (CNS) (Figure 2).
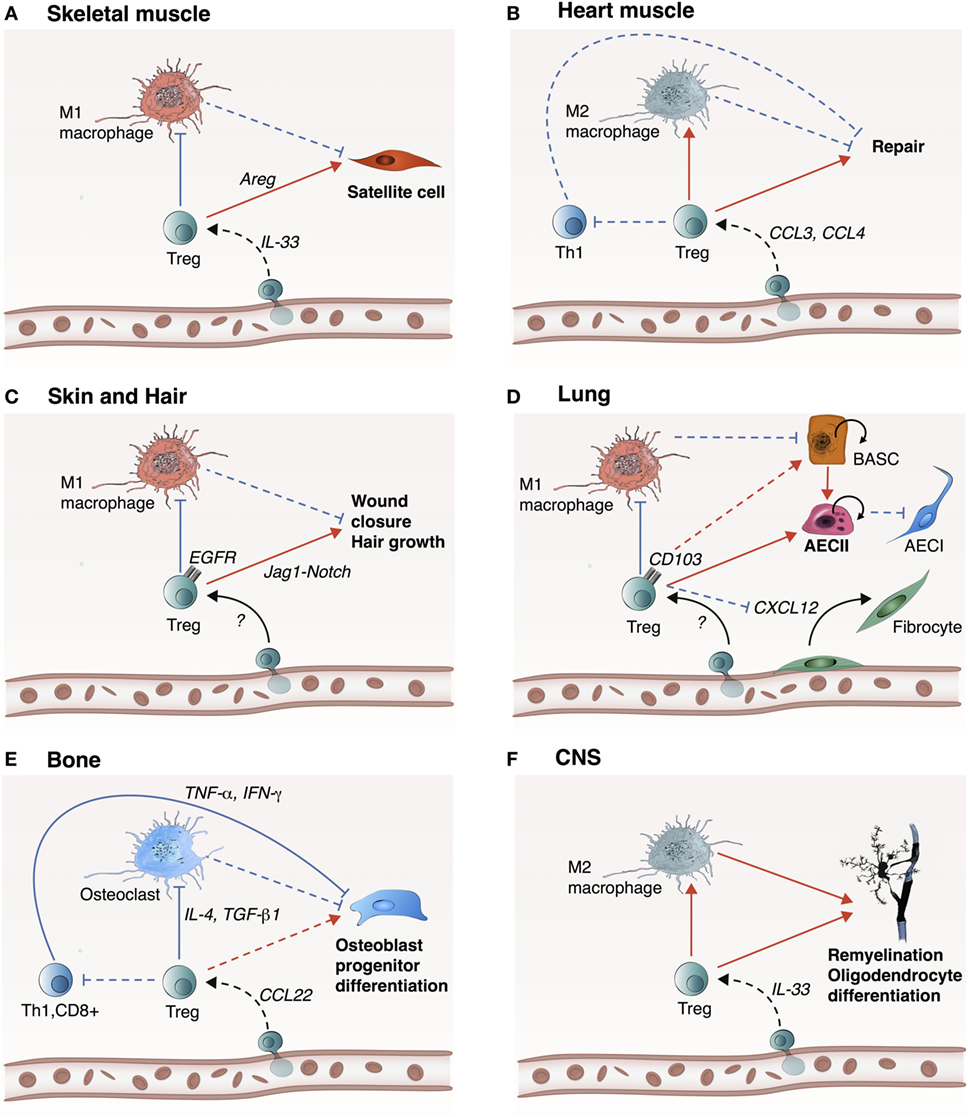
Figure 2. Treg likely promote tissue repair and regeneration in a tissue-specific manner. Treg play an important role in the repair and regeneration of skeletal muscle, heart muscle, skin and hair, lung, bone, and central nervous system (CNS). (A) In skeletal muscle, IL-33 participates to Treg recruitment into the site of injury. Treg inhibit M1 macrophage-mediated inflammation, which promote transition to the resolution phase. Treg also directly activate satellite cell proliferation and differentiation through Areg. (B) In the heart, Treg are recruited via CCR5 signaling (e.g., CCL3 and CCL4) allowing inhibition of Th1 cell activity and inhibition of M1 macrophages. (C) In skin and hair, mechanism of Treg recruitment is still unknown, but upon recruitment, Treg inhibit M1 macrophage inflammatory activity and promote wound closure and hair growth via the Jag1-Notch signaling pathway. (D) In the lung, Treg inhibit M1 macrophage inflammatory activity and encourage proliferation and differentiation of damaged alveolar type 2 epithelial cells (AECII) into AECIs. This step can be mediated by Areg or CD103 to E-cadherin ligand-receptor binding. Alternatively, Treg could potentially activate progenitor bronchioalveolar stem cells (BASCs) to differentiate into AECII cells. Concurrently, Treg prevent fibrosis by inhibiting fibrocyte recruitment and proliferation via CXCL12. (E) In the bone, Treg are most likely recruited via CCL22, which act on inhibiting Th1, CD8+, and M1 macrophages to support osteoblast progenitor differentiation. (F) In CNS, Treg are recruited by IL-33 and play a reparative role by encouraging M2 macrophage polarization to facilitate re-myelination and differentiation of oligodendrocytes. Treg may also directly act on oligodendrocytes via CCN3. Dashed lines indicate a hypothetical mechanism. Red arrows indicate induction, while blue arrows indicate inhibition.
The Role of Treg in Tissue-Specific Repair and Regeneration
Skeletal Muscle
It has been shown that Treg accumulate in the skeletal muscle of acutely injured mice or in mdx model of Duchenne muscular dystrophy (18, 57). Normal repair of skeletal muscle is found to require local expansion of Treg, since Treg ablation following treatment with diphtheria toxin in Foxp3DTR mice or following treatment with the depleting anti-CD25mAb targeting CD4+CD25hi Treg increases muscle damage in dystrophic mice (18, 57). Similarly, treatments that enhance Treg activities including complexes of recombinant IL-2 with anti-IL-2 mAb prevented muscle damage in dystrophic mice (18, 57). Comparing the transcriptome of Treg isolated from regenerating muscle and lymphoid tissues including spleen and lymph nodes, Treg from muscle, but not naïve Treg from lymphoid organ express the growth factor Amphiregulin that directly acts on muscle satellite cells in vitro and improves muscle repair in vivo (18). Depletion of Treg also leads to increased muscle inflammation characterized by an increased IFN-γ response and activation of M1 macrophages (57). Moreover, it has been shown in vitro that coculture of induced Treg with muscle satellite cells enhances muscle satellite expansion and inhibits their myogenic differentiation (16). Nevertheless, direct evidence of Treg converting satellite cells into muscle has yet to be demonstrated.
To date, factors, which contribute to the accumulation of Treg in damaged tissues remain elusive. Nevertheless, IL-33 has been shown to facilitate recovery after CNS injury (58) and to drive accumulation of Treg in visceral adipose tissue of lean mice (59) and damaged muscle in young mice (60). IL-33 acts on the suppression of tumorigenicity 2 (ST2) receptor of Treg. Treg devoid of ST2 due to Treg cell-specific ablation of the Il1rl1 gene show impaired recruitment to injured muscle, resulting in delayed muscle regeneration (60). Moreover, aged mice with more severely impaired muscle repair are found to have less IL-33-dependent accumulation of Treg after acute injury compared to young mice (60). Thus, IL-33 is important in mobilizing Treg in muscle and supplementation of IL-33 can reverse these effects and facilitate muscle regeneration in aged mice (16, 60).
Heart Muscle
Scarring of cardiac tissue after MI is likely the most deadly injury in humans (61). MI leads to a loss of large number of cardiomyocytes that are unable to regenerate, which ultimately progresses to cardiac failure. Cardiomyocyte death results in replacement by scar tissues and ventricular remodeling that further compromises heart function. Early cardiac wound healing is characterized by infiltration of both innate (62) and adaptive (63) immune cells into the myocardium. In patients with acute MI, increased systemic markers of inflammation correlate with higher mortality (64). Moreover, infiltration of activated CD4+CD25+ T-cells has been observed in the infarcted and remote regions of myocardium and heart-draining lymph nodes in patients with MI (63, 65). It has also been found that T-cells become activated in patients with coronary artery disease or a history of MI (65, 66). Nevertheless, the role of T-cells during pathogenesis or healing of the human heart is yet to be identified.
In mouse models of CD4+ T-cell deficiency, including CD4 or MHC-II knockout mice, or TCR specific for an irrelevant ovalbumin-derived peptide in transgenic OTII mice, CD4+ T cell-deficient mice show increased cardiac inflammation, impaired wound healing, left ventricular remodeling, and impaired survival (63). Although myocardial antigens are minimally accessible by the immune system, both MhcII−/− and OT-II mice have a higher rate of myocardial ruptures and mortality than wild-type mice. This suggest that CD4+ T cells are activated after MI driven by recognition of cardiac autoantigens on MHC-II and facilitate healing of the myocardium in an antigen-specific manner (63).
Indeed, T-cells specific for myocardial proteins exist in mice. Both immunization with troponin or myosin-derived peptides, and adoptive transfer of myosin-derived peptides loaded dendritic cells induce myocarditis in susceptible mice (67, 68). CD4+ T-cells are also found to be reactive to troponin, a complex comprised of three regulatory proteins troponin-C, -I, and -T, which are integral to cardiac muscle contractility (69). Moreover, the cardiomyocyte-specific protein, α-myosin heavy chain (α-MHC), is not expressed within the thymus of mice and human, and it resembles a non-self protein that activates CD4+ T-cells after MI (70). Thus, tolerance to α-MHC reactive T-cells is probably maintained by Treg to prevent autoimmunity after MI. The detrimental role of conventional CD4+ T-cells in MI healing most likely involves the adenosine receptor, since adenosine receptor depleted CD4+ T-cells are not able to recapitulate the injurious action of CD4+ T cells (47). In vitro activated Treg cells attenuate myocardial injury through expression of CD39, which promote extracellular degradation of nucleotides to form adenosine (71). Therefore, Treg in heart may function through CD39-mediated adenosine formation.
Treg also improve healing after MI by modulating monocytes and macrophages (22). Treg depletion in Foxp3DTR mice or following treatment with anti-CD25 mAb show increased infiltration of M1 macrophages, reduced cardiac function, and pronounced left ventricular dilation after MI (22). On the other hand, preferential induction of Treg via treatment with superagonistic anti-CD28 mAb leads to increased Treg infiltration into the infarcted myocardium after MI. The higher number of Treg promotes macrophages to polarize toward a M2 phenotype in the healing myocardium and reduce ventricular ruptures, which lead to better survival (22). Mechanistically, CCR5 is associated with Treg recruitment as CCR5 knockout mice show impaired Treg infiltration as well as adverse remodeling and cardiac deterioration after MI (72). Therefore, CCR5-mediated Treg recruitment restrains inflammation, excessive matrix degradation, and adverse remodeling after MI (72).
Skin and Hair
Layers of murine and human skin contain a large number of resident Treg (73–75). During a short-defined window of postnatal development, Treg migration to neonatal skin is important for the establishment of immune tolerance to commensal microbes (73). Upon entry to the skin through CCL20/CCR6 mediated migration, Treg localize and accumulate in hair follicles (76). Marked accumulation of CD4+ T-cells are observed in wounded skin with peaked infiltration at day 7 following injury. Interestingly, the majority of CD4+ T-cells are highly activated Treg characterized by increased expression of CD25, CTLA-4, and ICOS (77). Treg depletion following treatment with diphtheria toxin in Foxp3DTR mice after skin wounding results in significant attenuation of wound closure accompanied by increased tissue granulation and overlying eschar, indicating that Treg facilitate skin wound healing (77). Treg depletion also leads to an increased number of IFN-γ producing T-cells with augmented accumulation of proinflammatory macrophages in wounded skin. Moreover, ablation of epidermal growth factor receptor (EGFR) signaling in Treg using the Foxp3-Cre;EGFRfl/fl mouse model results in reduced Treg infiltration in the wounded skin and significantly delayed wound closure, indicating that the EGFR pathway plays a role in Treg activation and function during skin wound healing (77).
In alopecia areata disease displaying phenotype of hair follicle regeneration, genome-wide association studies have revealed single nucleotide polymorphisms in genes including Cd25, Ctla4, Eos, and Foxp3, which are important in differentiation and function of Treg (78–80). Treg in skin preferentially localize to hair follicles (81, 82) and are more abundant in the resting telogen than growing anagen phase during hair follicle cycling (17). More importantly, in the telogen phase, Treg display a highly activated phenotype. Transient or constant ablation of Treg in Foxp3DTR mice following treatment with diphtheria toxin leads to markedly reduced anagen induction in skin and subsequently reduced hair regrowth, indicating the important role of Treg in facilitating hair follicle regeneration by promoting the telogen-to-anagen transition (17). Immunofluorescence microscopy on dorsal skin derived from Foxp3GFP reporter mice further revealed that Treg preferentially localize to hair follicular stem cell niche, promoting proliferation and differentiation of hair follicular stem cells (17). By comparing the transcriptome of Treg derived from telogen skin and skin-draining lymph nodes, Treg of the skin expressed more Jag1 lymph node-Treg. Conditional ablation of Jag1 in Treg of Foxp3-Cre+/+;Jag1fl/fl mice significantly attenuates hair follicular stem cell proliferation, suggesting that the Jag1–Notch signaling pathway is essential in facilitating Treg-mediated hair follicle regeneration (17).
Lung
As microbes and other airborne materials can be frequently aspirated into the lung, pulmonary disease is easy to develop when the pulmonary immunity fails to protect the lungs during infections (83). However, the role of Treg during lung infections has been investigated in mouse models and has resulted in contradictory findings. It has been shown that patients with acute respiratory distress syndrome have increased Treg in their bronchoalveolar lavage fluid, suggesting that Treg play a role in the disease (21). In preclinical models of lung injury, T-cell deficient mice (Rag-1−/−) showed delayed lung resolution governed by high lung permeability as well as elevated number of neutrophils and macrophages, indicating that T-cells may play a reparative role during lung injury resolution (21). Moreover, further analysis has shown that infiltration of CD4+CD25+FOXP3+ Treg in the alveolar compartment increases upon LPS instillation (21). Furthermore, adoptive transfer of wild-type CD4+CD25+ splenocytes following intratracheal LPS instillation into Rag-1−/− mice successfully facilitates lung injury resolution, suggesting that Treg could serve as a rescue therapy after acute lung injury (21). Indeed, Treg play a central role in lung resolution, since adoptive transfer of Treg from wild-type into Rag-1−/− mice has shown to decrease the number of fibrocytes in LPS-treated lungs. Mechanistically, Treg reduce lung epithelial CXCL12 concentration which is responsible for CXCR4+ fibrocyte recruitment (84). In addition, Treg mediate resolution by stifling pro-inflammatory macrophage response and ultimately promote bronchioalveolar stem cells (BASCs) proliferation (84).
The adult lung has a remarkable regenerative potential after injury (85, 86). The alveolar compartment comprises largely (90–95%) of alveolar type I cells (ATI) involved in gas exchange and to a lesser amount (7%) of type II cells (ATII), which are involved in immune regulation, repair, and recovery (87). These cell types derive from BASCs found at the bronchioalveolar duct junction (88). In a left unilateral pneumonectomy mouse model, surgical removal of the left lung induces mass expansion in the intact lobes of the remaining right lung (89). This extravagant alveologenesis process is shown to be dependent on lung epithelial proliferation, specifically through ATII cells responsible for maintaining ATI number through differentiation (89). In acute lung injury or partial pneumonectomy models, it has been shown that epithelial proliferation during lung recovery is significantly impaired after specific elimination of Treg in Foxp3DTR mice following diphtheria toxin treatment (90). Lung epithelial proliferation is strongly correlated with Treg number after injury and Treg promote ATII proliferation through binding of their surface integrin CD103 to E-Cadherin expressed by epithelial cells (90). Furthermore, the growth factor amphiregulin (Areg) expression by Treg also seems to play a non-redundant role in lung repair (18). Rapid increase in expression level of Areg in lung tissues is observed at day 3 postviral infection. Using Foxp3-Cre;Aregfl/fl mice in which Areg is specifically ablated in Treg, it has been shown that the immunosuppressive function of Treg is preserved during antiviral immune responses. However, in the absence of Areg production by Treg, impaired recovery of lung function has been found in Foxp3-Cre;Aregfl/fl mice. Interestingly, by coculturing Treg with IL-18 or IL-33, it has been shown that Areg is induced by activation of IL-18R or ST2, instead of being activated through the TCR signaling pathway (91).
Bone
The adaptive immune system has been shown to play an important role in bone regeneration. Compared to most tissues, bone is capable of healing without scar tissue formation. The homeostasis of bone is mediated mainly by the interaction between osteoblasts which form bone and osteoclasts which resorb bone. Osteoblasts are principally differentiated from progenitors such as mesenchymal stem cells (MSC), while osteoclasts originate from bone marrow-derived monocytes. Interestingly, MSC can induce Treg from naïve T-cells and promote Treg proliferation through HO-1 (92). While CD3+ T-cells support peripheral blood mononuclear cell differentiation into osteoclasts in vitro, Treg inhibit such a differentiation through paracrine signaling of TGF-β and IL-4 (93, 94). Moreover, the number of Treg in peripheral blood is inversely correlated to serum marker of osteoclastogenesis in normal human and rheumatoid arthritis patients. In vivo experiments have also shown that Treg protect TNF-α-induced bone destruction and ovariectomy-induced bone loss (95, 96). Treatment with superagonistic anti-CD28 mAb ameliorates TNF-α-induced arthritis and increases bone mass in wild-type mice. The protective role of Treg in bone loss is most likely the result of impaired osteoclast differentiation and bone resorption via inflammatory cytokines. For example, in an actinobacillus actinomycetemcomitans-induced canine model, Treg are recruited to the site of injury by CCL22 and decrease bone resorption through reducing inflammation (97). Interestingly, Treg may also directly promote osteoblast differentiation from progenitor cells. For instance, it has been demonstrated that Treg facilitate MSC-based bone regeneration by inhibiting CD4+ conventional T-cells, which secrete IFN-γ and TNF-α (52, 98).
Central Nervous System
In mice deficient for CD4+ or CD8+ T-cells, remyelination is inhibited after lysolecithin injection, suggesting that CD4+ and CD8+ T-cells are required in remyelination of the CNS (99). In a myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis mouse model, Treg are found to expand in peripheral lymphoid compartment and accumulate in CNS (100). Even though infiltrating Treg fail to control autoimmune inflammation, it has been demonstrated that they promote myelin regeneration (25). Moreover, Treg-deficient mice show impaired remyelinataion and oligodendrocyte differentiation that can be rescued by adoptive transfer of Treg. IL-33 has been found to promote Treg recruitment into injured tissues, facilitating recovery after CNS injury. In addition, mice lacking IL-33 have impaired recovery after CNS injury, which is associated with reduced myeloid cell infiltrates and decreased induction of M2-assiociated genes at the injury site (58). Treg also promote oligodendrocyte progenitor cell differentiation and myelination in vitro and ex vivo. Interestingly, through proteome profiling of Treg conditioned media, nephroblastoma overexpressed, also known as CCN3, has been found to mediate Treg-driven oligodendrocyte progenitor cell differentiation and CNS myelination (25).
Future Perspectives
Recently, exploring the function of the immune system during tissue repair and regeneration has gained a lot of interest in regenerative medicine. Nevertheless, we still have sparse knowledge on how immunity—in particular the adaptive immune system—controls the tissue healing process. For instances, what are the neo antigens, if any, released to initiate adaptive immunity-mediated tissue healing? Similarly, is adaptive immunity-mediated tissue healing an antigen-specific process? If so, how are T-cells recruited, activated and function in response to self-antigens during injury that are different from responding to non-self antigens? Currently, the development of treatments targeting the immune system is hindered by the lack of markers that specifically define distinct subsets of immune cells. Recent advances in single-cell genomics could offer unprecedented delineation of lineage-specific markers and function of various subsets of immune cells operating during tissue repair and regeneration.
Furthermore, it is still unclear why scars are absent in some tissues such as in bone, but are forming in others such as in heart. Understanding how both innate and adaptive immune cells interact with tissue-resident progenitor cells and myofibroblasts would shed light on developing therapeutic strategies for improving healing and regeneration in the clinic. Moreover, accumulating evidence has shown that the function of the immune system declines with age (101, 102). Given that the immune system plays a crucial role in tissue repair and regeneration, whether the reduced tissue repair capacity is related to a degenerated immune system during aging awaits further investigations.
Overall, studies investigating the role of Treg during tissue regeneration have been largely based on the use of Foxp3DTR mouse model (17, 18, 22, 25, 57, 77, 90). However, one caveat of using such model is that the mice develop spontaneous systemic autoimmunity when Treg are depleted for long term (103). Careful data analysis should also include gain-of-function experiments such as adoptive transfer of purified Treg into Rag1−/− mice to determine the role of Treg in tissue repair and regeneration.
From a regenerative point of view, one could control tissue Treg to promote regeneration. Treg of adipose tissues, skeletal muscle, and colonic lamina propria are the best characterized tissue Treg that maintain organismal homeostasis (104). Similar to regenerative Treg as aforementioned, IL-33 has been reported to expand tissue Treg in colonic lamina propria (105) that are well equipped to participate in local repair responses with expression of the tissue repair factor, amphiregulin. The exact role of these tissue Treg in intestinal regeneration awaits further investigations. Nevertheless, manipulating Treg for alleviating inflammatory diseases has been tested in several clinical trials. For instances, the use of low dosage of IL-2 in selective expansion of Treg in human patients (106) as well as ex vivo expansion and adoptive transfer of Treg to treat Type 1 diabetes have been reported and tested in the clinic (107–109). Further studies are needed to investigate if these Treg-mediated strategies can also be utilized for inducing tissue regeneration.
Although it has been reported that superagonistic anti-CD28 mAb increases Treg infiltration or activities in mice, the use of humanized superagonisticanti-CD28 antibody TGF1412 caused cytokine storm, leading to organ failure in a previous trial (110). Furthermore, even though IL-33 plays an important role in recruitment and function of Treg in mice (60), IL-33 is dispensable in humans as individuals lacking IL-33 have no obvious health problems such as autoimmunity, indicating that the pro-regenerative function of IL-33 on Treg could also be different between mice and humans (111). Therefore, novel strategies in empowering Treg-mediated tissue regeneration for potential clinical uses would be needed in the future.
Author Contributions
JL and KL wrote the manuscript; JT and MM made the figures; MM and KL revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AA and handling Editor declared their shared affiliation.
Funding
This work was supported by Research Grants Council (04110515, 14111916, C4024-16W, C4026-17WF to KL); Health and Medical Research Fund (03140346, 04152566 to KL); Croucher Foundation Innovation Award (BL16380 to KL) and Startup Award (KL); Lui Chi Woo Innovative Medicine Seed Fund (MD14541 to KL), Direct Grant (MD15986 to KL), Faculty Innovation Award (BL16438 to KL), Postgraduate Studentship from the Chinese University of Hong Kong (to JL), EMBL Australia (to MM), and the Australian Research Council (DE170100398 to MM).
References
1. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the global burden of disease study 2010. Lancet (2012) 380(9859):2095–128. doi:10.1016/S0140-6736(12)61728-0
2. Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet (2012) 379(9817):713–20. doi:10.1016/S0140-6736(12)60028-2
3. Schwartz SD, Regillo CD, Lam BL, Eliott D, Rosenfeld PJ, Gregori NZ, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet (2015) 385(9967):509–16. doi:10.1016/S0140-6736(14)61376-3
4. Lui KO, Fairchild PJ, Waldmann H. Prospects for ensuring acceptance of ES cell-derived tissues. StemBook. Cambridge, MA (2008).
5. Lui KO, Waldmann H, Fairchild PJ. Embryonic stem cells: overcoming the immunological barriers to cell replacement therapy. Curr Stem Cell Res Ther (2009) 4(1):70–80. doi:10.2174/157488809787169093
6. Martino MM, Briquez PS, Maruyama K, Hubbell JA. Extracellular matrix-inspired growth factor delivery systems for bone regeneration. Adv Drug Deliv Rev (2015) 94:41–52. doi:10.1016/j.addr.2015.04.007
7. Briquez PS, Hubbell JA, Martino MM. Extracellular matrix-inspired growth factor delivery systems for skin wound healing. Adv Wound Care (2015) 4(8):479–89. doi:10.1089/wound.2014.0603
8. Eming SA, Wynn TA, Martin P. Inflammation and metabolism in tissue repair and regeneration. Science (2017) 356(6342):1026–30. doi:10.1126/science.aam7928
9. Julier Z, Park AJ, Briquez PS, Martino MM. Promoting tissue regeneration by modulating the immune system. Acta Biomater (2017) 53:13–28. doi:10.1016/j.actbio.2017.01.056
10. Uygur A, Lee RT. Mechanisms of cardiac regeneration. Dev Cell (2016) 36(4):362–74. doi:10.1016/j.devcel.2016.01.018
11. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science (2011) 331(6020):1078–80. doi:10.1126/science.1200708
12. Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, et al. Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res (2016) 118(2):216–21. doi:10.1161/CIRCRESAHA.115.307017
13. Aurora AB, Olson EN. Immune modulation of stem cells and regeneration. Cell Stem Cell (2014) 15(1):14–25. doi:10.1016/j.stem.2014.06.009
14. Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet (2001) 27(1):20–1. doi:10.1038/83713
15. Clark LB, Appleby MW, Brunkow ME, Wilkinson JE, Ziegler SF, Ramsdell F. Cellular and molecular characterization of the scurfy mouse mutant. J Immunol (1999) 162(5):2546–54.
16. Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, et al. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS One (2015) 10(6):e0128094. doi:10.1371/journal.pone.0128094
17. Ali N, Zirak B, Rodriguez RS, Pauli ML, Truong HA, Lai K, et al. Regulatory T cells in skin facilitate epithelial stem cell differentiation. Cell (2017) 169(6):1119–1129.e11. doi:10.1016/j.cell.2017.05.002
18. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell (2013) 155(6):1282–95. doi:10.1016/j.cell.2013.10.054
19. Murphy TJ, Ni Choileain N, Zang Y, Mannick JA, Lederer JA. CD4+CD25+ regulatory T cells control innate immune reactivity after injury. J Immunol (2005) 174(5):2957–63. doi:10.4049/jimmunol.174.5.2957
20. Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Pathophysiological role of neutrophils in acute myocardial infarction. Thromb Haemost (2013) 110(3):501–14. doi:10.1160/TH13-03-0211
21. D’Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, et al. CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest (2009) 119(10):2898–913. doi:10.1172/JCI36498
22. Weirather J, Hofmann UD, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res (2014) 115(1):55–67. doi:10.1161/CIRCRESAHA.115.303895
23. Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A (2014) 111(45):16029–34. doi:10.1073/pnas.1406508111
24. Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest (2014) 124(3):1382–92. doi:10.1172/JCI72181
25. Dombrowski Y, O’Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci (2017) 20(5):674–80. doi:10.1038/nn.4528
26. Toumi H, F’Guyer S, Best TM. The role of neutrophils in injury and repair following muscle stretch. J Anat (2006) 208(4):459–70. doi:10.1111/j.1469-7580.2006.00543.x
27. Pizza FX, Peterson JM, Baas JH, Koh TJ. Neutrophils contribute to muscle injury and impair its resolution after lengthening contractions in mice. J Physiol (2005) 562(Pt 3):899–913. doi:10.1113/jphysiol.2004.073965
28. Yang Z, Sharma AK, Linden J, Kron IL, Laubach VE. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J Thorac Cardiovasc Surg (2009) 137(3):695–702. doi:10.1016/j.jtcvs.2008.10.044
29. Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, et al. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci U S A (2011) 108(38):15990–5. doi:10.1073/pnas.1110144108
30. Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. Neutrophil – CD4+CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology (2013) 218(4):455–64. doi:10.1016/j.imbio.2012.05.029
31. Lewkowicz P, Lewkowicz N, Sasiak A, Tchórzewski H. Lipopolysaccharide-activated CD4+CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol (2006) 177(10):7155–63. doi:10.4049/jimmunol.177.10.7155
32. Kinsey GR, Sharma R, Huang L, Li L, Vergis AL, Ye H, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol (2009) 20(8):1744–53. doi:10.1681/ASN.2008111160
33. Godwin JW, Pinto AR, Rosenthal NA. Macrophages are required for adult salamander limb regeneration. Proc Natl Acad Sci U S A (2013) 110(23):9415–20. doi:10.1073/pnas.1300290110
34. Petrie TA, Strand NS, Yang CT, Rabinowitz JS, Moon RT. Macrophages modulate adult zebrafish tail fin regeneration. Development (2014) 141(13):2581–91. doi:10.1242/dev.098459
35. de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest (2015) 125(8):3147–62. doi:10.1172/JCI81321
36. Kubo T, Hatton RD, Oliver J, Liu X, Elson CO, Weaver CT. Regulatory T cell suppression and anergy are differentially regulated by proinflammatory cytokines produced by TLR-activated dendritic cells. J Immunol (2004) 173(12):7249–58. doi:10.4049/jimmunol.173.12.7249
37. Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev (2006) 212:314–29. doi:10.1111/j.0105-2896.2006.00422.x
38. Ghiringhelli F, Ménard C, Martin F, Zitvogel L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev (2006) 214:229–38. doi:10.1111/j.1600-065X.2006.00445.x
39. Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, et al. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol (2005) 66(3):222–30. doi:10.1016/j.humimm.2004.12.006
40. Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A (2007) 104(49):19446–51. doi:10.1073/pnas.0706832104
41. Venet F, Pachot A, Debard AL, Bohe J, Bienvenu J, Lepape A, et al. Human CD4+CD25+ regulatory T lymphocytes inhibit lipopolysaccharide-induced monocyte survival through a Fas/Fas ligand-dependent mechanism. J Immunol (2006) 177(9):6540–7. doi:10.4049/jimmunol.177.9.6540
42. Linfert D, Chowdhry T, Rabb H. Lymphocytes and ischemia-reperfusion injury. Transplant Rev (Orlando) (2009) 23(1):1–10. doi:10.1016/j.trre.2008.08.003
43. Veeneman JM, de Jong PE, Huisman RM, Reijngoud DJ. Re: Adey et al. Reduced synthesis of muscle proteins in chronic renal failure. Am J Physiol Endocrinol Metab 278: E219-E225, 2000. Am J Physiol Endocrinol Metab (2001) 280(1):E197–8. doi:10.1152/ajpendo.2001.280.1.E197
44. Shigematsu T, Wolf RE, Granger DN. T-lymphocytes modulate the microvascular and inflammatory responses to intestinal ischemia-reperfusion. Microcirculation (2002) 9(2):99–109. doi:10.1080/mic.9.2.99.109
45. Maisel A, Cesario D, Baird S, Rehman J, Haghighi P, Carter S. Experimental autoimmune myocarditis produced by adoptive transfer of splenocytes after myocardial infarction. Circ Res (1998) 82(4):458–63. doi:10.1161/01.RES.82.4.458
46. Varda-Bloom N, Leor J, Ohad DG, Hasin Y, Amar M, Fixler R, et al. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J Mol Cell Cardiol (2000) 32(12):2141–9. doi:10.1006/jmcc.2000.1261
47. Yang Z, Day YJ, Toufektsian MC, Xu Y, Ramos SI, Marshall MA, et al. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation (2006) 114(19):2056–64. doi:10.1161/CIRCULATIONAHA.106.649244
48. Brunetti G, Colucci S, Pignataro P, Coricciati M, Mori G, Cirulli N, et al. T cells support osteoclastogenesis in an in vitro model derived from human periodontitis patients. J Periodontol (2005) 76(10):1675–80. doi:10.1902/jop.2005.76.10.1675
49. Reinke S, Geissler S, Taylor WR, Schmidt-Bleek K, Juelke K, Schwachmeyer V, et al. Terminally differentiated CD8(+) T cells negatively affect bone regeneration in humans. Sci Transl Med (2013) 5(177):177ra36. doi:10.1126/scitranslmed.3004754
50. Krishnadasan B, Naidu B, Rosengart M, Farr AL, Barnes A, Verrier ED, et al. Decreased lung ischemia-reperfusion injury in rats after preoperative administration of cyclosporine and tacrolimus. J Thorac Cardiovasc Surg (2002) 123(4):756–67. doi:10.1067/mtc.2002.120351
51. Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol (2012) 107(1):232. doi:10.1007/s00395-011-0232-6
52. Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X, et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat Med (2011) 17(12):1594–601. doi:10.1038/nm.2542
53. Corthay A. How do regulatory T cells work? Scand J Immunol (2009) 70(4):326–36. doi:10.1111/j.1365-3083.2009.02308.x
54. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450(7169):566–9. doi:10.1038/nature06306
55. Hawrylowicz CM, O’Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol (2005) 5(4):271–83. doi:10.1038/nri1589
56. Joetham A, Takeda K, Taube C, Miyahara N, Matsubara S, Koya T, et al. Naturally occurring lung CD4(+)CD25(+) T cell regulation of airway allergic responses depends on IL-10 induction of TGF-beta. J Immunol (2007) 178(3):1433–42. doi:10.4049/jimmunol.178.8.5400
57. Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, et al. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci Transl Med (2014) 6(258):258ra142. doi:10.1126/scitranslmed.3009925
58. Gadani SP, Walsh JT, Smirnov I, Zheng J, Kipnis J. The glia-derived alarmin IL-33 orchestrates the immune response and promotes recovery following CNS injury. Neuron (2015) 85(4):703–9. doi:10.1016/j.neuron.2015.01.013
59. Kolodin D, van Panhuys N, Li C, Magnuson AM, Cipolletta D, Miller CM, et al. Antigen- and cytokine-driven accumulation of regulatory T cells in visceral adipose tissue of lean mice. Cell Metab (2015) 21(4):543–57. doi:10.1016/j.cmet.2015.03.005
60. Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, et al. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity (2016) 44(2):355–67. doi:10.1016/j.immuni.2016.01.009
61. Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res (2005) 66(1):22–32. doi:10.1016/j.cardiores.2005.01.011
62. Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation (2010) 121(22):2437–45. doi:10.1161/CIRCULATIONAHA.109.916346
63. Hofmann U, Beyersdorf N, Weirather J, Podolskaya A, Bauersachs J, Ertl G, et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation (2012) 125(13):1652–63. doi:10.1161/CIRCULATIONAHA.111.044164
64. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science (2013) 339(6116):161–6. doi:10.1126/science.1230719
65. Abbate A, Bonanno E, Mauriello A, Bussani R, Biondi-Zoccai GG, Liuzzo G, et al. Widespread myocardial inflammation and infarct-related artery patency. Circulation (2004) 110(1):46–50. doi:10.1161/01.CIR.0000133316.92316.81
66. Dangas G, Konstadoulakis MM, Epstein SE, Stefanadis CI, Kymionis GD, Toutouza MG, et al. Prevalence of autoantibodies against contractile proteins in coronary artery disease and their clinical implications. Am J Cardiol (2000) 85(7):870–2,A6,A9. doi:10.1016/S0002-9149(99)00883-8
67. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, et al. Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med (2003) 9(12):1484–90. doi:10.1038/nm960
68. Kaya Z, Katus HA, Rose NR. Cardiac troponins and autoimmunity: their role in the pathogenesis of myocarditis and of heart failure. Clin Immunol (2010) 134(1):80–8. doi:10.1016/j.clim.2009.04.008
69. Frenkel D, Pachori AS, Zhang L, Dembinsky-Vaknin A, Farfara D, Petrovic-Stojkovic S, et al. Nasal vaccination with troponin reduces troponin specific T-cell responses and improves heart function in myocardial ischemia-reperfusion injury. Int Immunol (2009) 21(7):817–29. doi:10.1093/intimm/dxp051
70. Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest (2011) 121(4):1561–73. doi:10.1172/JCI44583
71. Xia N, Jiao J, Tang TT, Lv BJ, Lu YZ, Wang KJ, et al. Activated regulatory T-cells attenuate myocardial ischaemia/reperfusion injury through a CD39-dependent mechanism. Clin Sci (Lond) (2015) 128(10):679–93. doi:10.1042/CS20140672
72. Dobaczewski M, Xia Y, Bujak M, Gonzalez-Quesada C, Frangogiannis NG. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory T cells. Am J Pathol (2010) 176(5):2177–87. doi:10.2353/ajpath.2010.090759
73. Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, Nosbaum A, et al. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity (2015) 43(5):1011–21. doi:10.1016/j.immuni.2015.10.016
74. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest (2014) 124(3):1027–36. doi:10.1172/JCI72932
75. Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol (2006) 176(7):4431–9. doi:10.4049/jimmunol.176.7.4431
76. Scharschmidt TC, Vasquez KS, Pauli ML, Leitner EG, Chu K, Truong HA, et al. Commensal microbes and hair follicle morphogenesis coordinately drive Treg migration into neonatal skin. Cell Host Microbe (2017) 21(4):467–77.e5. doi:10.1016/j.chom.2017.03.001
77. Nosbaum A, Prevel N, Truong HA, Mehta P, Ettinger M, Scharschmidt TC, et al. Cutting edge: regulatory T cells facilitate cutaneous wound healing. J Immunol (2016) 196(5):2010–4. doi:10.4049/jimmunol.1502139
78. Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature (2010) 466(7302):113–7. doi:10.1038/nature09114
79. Castela E, Le Duff F, Butori C, Ticchioni M, Hofman P, Bahadoran P, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol (2014) 150(7):748–51. doi:10.1001/jamadermatol.2014.504
80. Shin BS, Furuhashi T, Nakamura M, Torii K, Morita A. Impaired inhibitory function of circulating CD4+CD25+ regulatory T cells in alopecia areata. J Dermatol Sci (2013) 70(2):141–3. doi:10.1016/j.jdermsci.2013.01.006
81. Chow Z, Mueller SN, Deane JA, Hickey MJ. Dermal regulatory T cells display distinct migratory behavior that is modulated during adaptive and innate inflammation. J Immunol (2013) 191(6):3049–56. doi:10.4049/jimmunol.1203205
82. Gratz IK, Truong HA, Yang SH, Maurano MM, Lee K, Abbas AK, et al. Cutting edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol (2013) 190(9):4483–7. doi:10.4049/jimmunol.1300212
83. Mizgerd JP. Respiratory infection and the impact of pulmonary immunity on lung health and disease. Am J Respir Crit Care Med (2012) 186(9):824–9. doi:10.1164/rccm.201206-1063PP
84. Garibaldi BT, D’Alessio FR, Mock JR, Files DC, Chau E, Eto Y, et al. Regulatory T cells reduce acute lung injury fibroproliferation by decreasing fibrocyte recruitment. Am J Respir Cell Mol Biol (2013) 48(1):35–43. doi:10.1165/rcmb.2012-0198OC
85. Wansleeben C, Barkauskas CE, Rock JR, Hogan BL. Stem cells of the adult lung: their development and role in homeostasis, regeneration, and disease. Wiley Interdiscip Rev Dev Biol (2013) 2(1):131–48. doi:10.1002/wdev.58
86. Beers MF, Morrisey EE. The three R’s of lung health and disease: repair, remodeling, and regeneration. J Clin Invest (2011) 121(6):2065–73. doi:10.1172/JCI45961
87. Wong MH, Johnson MD. Differential response of primary alveolar type I and type II cells to LPS stimulation. PLoS One (2013) 8(1):e55545. doi:10.1371/journal.pone.0055545
88. Kim CF. Paving the road for lung stem cell biology: bronchioalveolar stem cells and other putative distal lung stem cells. Am J Physiol Lung Cell Mol Physiol (2007) 293(5):L1092–8. doi:10.1152/ajplung.00015.2007
89. Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, et al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell (2011) 147(3):539–53. doi:10.1016/j.cell.2011.10.003
90. Mock JR, Garibaldi BT, Aggarwal NR, Jenkins J, Limjunyawong N, Singer BD, et al. Foxp3+ regulatory T cells promote lung epithelial proliferation. Mucosal Immunol (2014) 7(6):1440–51. doi:10.1038/mi.2014.33
91. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell (2015) 162(5):1078–89. doi:10.1016/j.cell.2015.08.021
92. Mougiakakos D, Jitschin R, Johansson CC, Okita R, Kiessling R, Le Blanc K. The impact of inflammatory licensing on heme oxygenase-1-mediated induction of regulatory T cells by human mesenchymal stem cells. Blood (2011) 117(18):4826–35. doi:10.1182/blood-2010-12-324038
93. Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, Moon HB. Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem Biophys Res Commun (2007) 357(4):1046–52. doi:10.1016/j.bbrc.2007.04.042
94. Horwood NJ, Kartsogiannis V, Quinn JM, Romas E, Martin TJ, Gillespie MT. Activated T lymphocytes support osteoclast formation in vitro. Biochem Biophys Res Commun (1999) 265(1):144–50. doi:10.1006/bbrc.1999.1623
95. Zaiss MM, Frey B, Hess A, Zwerina J, Luther J, Nimmerjahn F, et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J Immunol (2010) 184(12):7238–46. doi:10.4049/jimmunol.0903841
96. Zaiss MM, Sarter K, Hess A, Engelke K, Böhm C, Nimmerjahn F, et al. Increased bone density and resistance to ovariectomy-induced bone loss in FoxP3-transgenic mice based on impaired osteoclast differentiation. Arthritis Rheum (2010) 62(8):2328–38. doi:10.1002/art.27535
97. Glowacki AJ, Yoshizawa S, Jhunjhunwala S, Vieira AE, Garlet GP, Sfeir C, et al. Prevention of inflammation-mediated bone loss in murine and canine periodontal disease via recruitment of regulatory lymphocytes. Proc Natl Acad Sci U S A (2013) 110(46):18525–30. doi:10.1073/pnas.1302829110
98. Liu Y, Yang R, Shi S. Systemic infusion of mesenchymal stem cells improves cell-based bone regeneration via upregulation of regulatory T cells. Tissue Eng A (2015) 21(3–4):498–509. doi:10.1089/ten.TEA.2013.0673
99. Bieber AJ, Kerr S, Rodriguez M. Efficient central nervous system remyelination requires T cells. Ann Neurol (2003) 53(5):680–4. doi:10.1002/ana.10578
100. Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med (2007) 13(4):423–31. doi:10.1038/nm1564
101. Montecino-Rodriguez E, Berent-Maoz B, Dorshkind K. Causes, consequences, and reversal of immune system aging. J Clin Invest (2013) 123(3):958–65. doi:10.1172/JCI64096
102. Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci U S A (2017) 114(12):E2420–9. doi:10.1073/pnas.1621047114
103. Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol (2007) 8(2):191–7. doi:10.1038/ni1428
104. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol (2016) 34:609–33. doi:10.1146/annurev-immunol-032712-095948
105. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513(7519):564–8. doi:10.1038/nature13577
106. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365(22):2055–66. doi:10.1056/NEJMoa1108188
107. Bayry J, Gautier JF. Regulatory T cell immunotherapy for type 1 diabetes: a step closer to success? Cell Metab (2016) 23(2):231–3. doi:10.1016/j.cmet.2016.01.010
108. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi:10.1126/scitranslmed.aad4134
109. Yi S, Ji M, Wu J, Ma X, Phillips P, Hawthorne WJ, et al. Adoptive transfer with in vitro expanded human regulatory T cells protects against porcine islet xenograft rejection via interleukin-10 in humanized mice. Diabetes (2012) 61(5):1180–91. doi:10.2337/db11-1306
110. Schneider CK, Kalinke U, Lower J. TGN1412 – a regulator’s perspective. Nat Biotechnol (2006) 24(5):493–6. doi:10.1038/nbt0506-493
Keywords: CD4+ regulatory T-cells, tissue repair and regeneration, stem cells, macrophages, heart regeneration
Citation: Li J, Tan J, Martino MM and Lui KO (2018) Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 9:585. doi: 10.3389/fimmu.2018.00585
Received: 09 September 2017; Accepted: 08 March 2018;
Published: 23 March 2018
Edited by:
Luis Graca, Universidade de Lisboa, PortugalReviewed by:
Afonso Almeida, Universidade de Lisboa, PortugalAwen Gallimore, Cardiff University, United Kingdom
Copyright: © 2018 Li, Tan, Martino and Lui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathy O. Lui, kathyolui@cuhk.edu.hk