 Elisabeth Müller1,2*
Elisabeth Müller1,2* Martin Speth2
Martin Speth2 Panagiotis F. Christopoulos1
Panagiotis F. Christopoulos1 Anna Lunde1Ajna Avdagic1
Anna Lunde1Ajna Avdagic1 Inger Øynebråten1
Inger Øynebråten1 Alexandre Corthay1*
Alexandre Corthay1*- 1Tumor Immunology Lab, Department of Pathology, Rikshospitalet, Oslo University Hospital, University of Oslo, Oslo, Norway
- 2Department of Biosciences, University of Oslo, Oslo, Norway
Triggering or enhancing antitumor activity of tumor-associated macrophages is an attractive strategy for cancer treatment. We have previously shown that the cytokine interferon-γ (IFN-γ), a type II IFN, could synergize with toll-like receptor (TLR) agonists for induction of antitumor M1 macrophages. However, the toxicity of IFN-γ limits its clinical use. Here, we investigated whether the less toxic type I IFNs, IFN-α, and IFN-β, could potentially replace IFN-γ for induction of antitumor M1 macrophages. We measured in vitro the ability of type I and II IFNs to synergize with TLR agonists for transcription of inducible nitric oxide synthase (iNOS) mRNA and secretion of nitric oxide (NO) by mouse bone marrow-derived macrophages (BMDMs). An in vitro growth inhibition assay was used to measure both cytotoxic and cytostatic activity of activated macrophages against Lewis lung carcinoma (LLC) cancer cells. We found that both type I and II IFNs could synergize with TLR agonists in inducing macrophage-mediated inhibition of cancer cell growth, which was dependent on NO. The ability of high dose lipopolysaccharide (LPS) to induce tumoricidal activity in macrophages in the absence of IFN-γ was shown to depend on induction of autocrine type I IFNs. Antitumor M1 macrophages could also be generated in the absence of IFN-γ by a combination of two TLR ligands when using the TLR3 agonist poly(I:C) which induces autocrine type I IFNs. Finally, we show that encapsulation of poly(I:C) into nanoparticles improved its potency to induce M1 macrophages up to 100-fold. This study reveals the potential of type I IFNs for activation of antitumor macrophages and indicates new avenues for cancer immunotherapy based on type I IFN signaling, including combination of TLR agonists.
Introduction
During the last decade, breakthrough therapies aiming to unleash the body's own immune response against cancer have provided significant clinical benefits to patient groups previously faced with limited treatment options (1). It has become clear that not only cancer cells, but also the immune and stromal cells within tumors, can be targeted with great effects. One important type of immune cell in tumors is the macrophage (2, 3). Macrophage accumulation in tumors can be detrimental because cancer cells can exploit macrophages for their ability to promote growth, angiogenesis, and immune suppression (4). Nevertheless, macrophages can also acquire potent antitumor activities upon appropriate stimulation. Such M1 macrophages mediate both direct antitumor activity through killing of cancer cells and inhibition of angiogenesis, as well as indirect effects through stimulation of T cells and induction of Th1 immune responses (5–9).
Several interventions targeting macrophages have shown promising results in mouse models for cancer, including a histone deacetylase inhibitor (10), agonistic anti-CD40 antibodies (9, 11), TGF-β inhibition in combination with toll-like receptor 7 (TLR7) ligation (12), and attenuated Listeria monocytogenes (13). Several TLR agonists are currently in clinical trials (14), but so far no therapeutic strategies which directly induces antitumor macrophages have been approved. The best characterized and most established protocol for inducing antitumor macrophages in vitro is based on activation with the TLR4 agonist lipopolysaccharide (LPS), alone or in combination with interferon (IFN)-γ (15, 16). Unfortunately, LPS is highly toxic, and IFN-γ has also shown severe dose-dependent side effects, including influenza-like symptoms, nausea, dizziness, anorexia, depression and leukopenia (17, 18). We have previously shown that LPS can be replaced by other, potentially better tolerated TLR ligands such as the TLR1/2 agonist Pam3CSK4 (a lipopeptide that mimics the acylated amino terminus of bacterial lipoproteins), and the TLR7 agonist CL264 (a 9-benzyl-8 hydroxyadenine derivative) for induction of an antitumor macrophage phenotype in vitro (19). Both Pam3CSK4 and CL264 were able to synergize with IFN-γ to induce antitumor M1 macrophages, but, unlike LPS, had no effect alone (19).
Combinations of multiple TLR agonists have synergistic effects on the production of proinflammatory cytokines and nitric oxide (NO) by macrophages in vitro (20, 21) and on antitumor activity of the immune system in vivo (22). All TLRs (except TLR3) signal through the adapter protein MyD88 (myeloid differentiation primary response 88), leading to activation of nuclear factor-κB (NF-κB). A second, MyD88-independent signaling pathway, which results in the induction of type I IFNs, depends on the TRIF adapter molecule (TIR-domain-containing adapter-inducing IFN-β). The TRIF pathway is activated by LPS through TLR4 or poly(I:C) through TLR3 (23–26). We have recently shown that poly(I:C) encapsulated in nanoparticles strongly synergizes with the TLR2 agonist bacille Calmette-Guérin (BCG) in inducing cytokine and NO production in mouse bone-marrow derived macrophages (BMDM) via TRIF-mediated autocrine type I IFN signaling (21). Autocrine signaling through IFN-α/β has also been shown to be crucial for the expression of inducible NO synthase (iNOS) and NO production in response to LPS (27). Expression of iNOS is a well-established marker for mouse proinflammatory M1 macrophages, and NO production is required for macrophage-mediated inhibition of cancer cell growth in vitro (19). Therefore, type I IFNs emerge as an attractive mediator for inducing antitumor macrophages.
In this study, we found that autocrine production of type I IFNs was important for the ability of LPS to induce antitumor macrophages in the absence of IFN-γ. We further observed that both recombinant and endogenously produced type I IFNs could synergize with Pam3CSK4 for induction of antitumor macrophages in a similar fashion as IFN-γ. Finally, we could show that macrophage antitumor activity is ~100-fold more efficiently induced in Pam3CSK4/poly(I:C) co-treated macrophages by using poly(I:C)-encapsulated nanoparticles [poly(I:C)-NP] instead of soluble poly(I:C). Our data reveal the potential of type I IFNs in the activation of antitumor macrophages and suggest a potential strategy for macrophage-targeted immunotherapy utilizing combinations of TLR agonists and nanoparticle technology.
Methods
Mice
C57BL/6NRj mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France) and bred at the Department of Comparative Medicine, Oslo University Hospital, Rikshospitalet (Oslo, Norway) in specific pathogen free (SPF) conditions. Bones from mice deficient in the IFN alpha/beta receptor 1 (Ifnar1−/−) were obtained from the Helmholtz Centre for Infection Research, Braunschweig, Germany, and the TWINCORE, Centre for Experimental and Clinical Infection Research, Hannover, Germany (28, 29).
Cell Lines
Lewis lung carcinoma (LLC) is a cell line originating from a spontaneous lung carcinoma in a C57BL/6 mouse and was obtained from CLS Cell Lines Service (Eppelheim, Germany) (30). L929 is a fibroblast-like cell line originating from connective tissue of a C3H/An mouse and was obtained from ATCC (Manassas, VA, USA) (31). Both cell lines were negative for mycoplasma infection as tested by use of MycoSensor PCR Assay kit (#302109, Agilent, Santa Clara, CA, USA).
TLR Agonists and Cytokines
The following TLR agonists were used: TLR1/TLR2 agonist Pam3CSK4 (#tlrl-pms, InvivoGen, San Diego, CA, USA); TLR3 agonist polyinosinic:polycytidylic acid [poly(I:C)] of high molecular weight type (#tlrl-pic, InvivoGen); TLR4 agonist LPS from E. coli (#L4391, Sigma-Aldrich, St. Louis, MO, USA); and TLR7 agonist CL264 (#tlrl-c264e-5, InvivoGen). The TLR agonists were used alone or in combination with mouse recombinant IFN-γ (#315-05, Peprotech, Rocky Hill, NJ, USA), mouse recombinant IFN-β (#8234-MB, R&D Systems Inc., Minneapolis, MN USA), or mouse recombinant IFN-α type A (#12100-1, PBL Assay Sciences, Piscataway, NJ, USA).
Production of Poly(I:C)-Encapsulating Nanoparticles [Poly(I:C)-NPs]
Poly(I:C)-encapsulating nanoparticles were produced as described previously (21). Briefly, equal volumes of 1 mg/mL of low molecular weight poly(I:C) (#tlrl-picw, InvivoGen) in 0.9% NaCl and 2 mg/mL of chitosan (KiOmedine-CsU, #740063, Sigma-Aldrich) dissolved in MilliQ H2O were mixed under stirring at room temperature. Spontaneously formed poly(I:C)-NPs were then collected by centrifugation at 10,000 g for 20 min on a glycerol bed, before resuspension in 0.9% NaCl and dissociation by water bath sonication for 10 min. The content of poly(I:C) encapsulated in poly(I:C)-NPs was determined indirectly by quantification of the amount of non-encapsulated (free) poly(I:C) in the supernatant after the centrifugation step. Poly(I:C) was quantified by measuring the absorbance at 260 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Generation of Bone Marrow-Derived Macrophages (BMDMs)
Mouse macrophages were differentiated from bone marrow cells according to an established protocol using L929 cell-conditioned medium as source of macrophage colony-stimulating factor (M-CSF) (32). The L929 cell-conditioned medium was produced as follows: Confluent L929 cells were cultured in RPMI 1640 medium (#61870044, Thermo Fisher Scientific) containing 10% fetal bovine serum (FBS, #BCHRS0405, Biochrom GmbH, Berlin, Germany) for 10 days before the medium was centrifuged, filtered and stored at −20°C until use. Bone marrow from the femurs and tibiae of the hind legs of 8-12 weeks old male and female C57BL/6NRj mice was flushed with RPMI 1640 medium containing 10% FBS under sterile conditions, and passed through a 70-μm cell strainer (#CLS431751-50EA, Sigma-Aldrich). Red blood cells (RBC) were removed by incubation in RBC lysis buffer (150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM Na2 EDTA) for 10 min at room temperature. After centrifugation and washing, the remaining cells were cultured in 10 cm non-tissue culture treated dishes (#734-2359, VWR, Radnos, PA, USA) in RPMI 1640 medium containing 30% L929-cell-conditioned medium. Bone marrow cells were cultured for 5 days, after which non-adherent cells were washed off using phosphate buffered saline (PBS, #D8662, Sigma-Aldrich) and the adherent macrophages were cultured for an additional day. Differentiated BMDMs were detached by incubation in PBS without CaCl2 and MgCl2 (#D8537, Sigma-Aldrich) for 30 min at 4°C and by repeated gentle flushing of the dish, before the BMDMs were collected by centrifugation. BMDMs were then frozen in FBS with 10% dimethyl sulfoxide (DMSO, #0231-500 ml, VWR) and stored at −150°C for future experiments. The purity of the cells was ≥99% as routinely analyzed by flow cytometry using the macrophage markers CD11b (clone M1/70, #101219, BioLegend, San Diego, CA, USA) and F4/80 (clone BM8, #123116, BioLegend).
Tumor Cell Growth Inhibition Assay
Frozen aliquots of BMDMs were thawed and cultured for 3 days in non-tissue culture treated dishes (#734-2359, VWR) in RPMI 1640 medium supplemented with 10% FBS (Biochrom) and 10% L929-cell conditioned medium. Next, BMDMs were harvested by scraping and incubated for 2 h at 37°C with 10 mg/mL mitomycin C (#M4287, Sigma-Aldrich) to block eventual proliferation of BMDMs. After washing twice with PBS, BMDMs were seeded in flat bottom 96 well plates (#734-1793, Costar, Washington DC, USA) at density 6 × 104 cells/well in 200 μL RPMI 1640 medium supplemented with 10% FBS (Biochrom) and 10% L929-cell conditioned medium. After 24 h of incubation, the culture medium was replaced with medium containing different combinations of TLR agonists and cytokines and the cells were stimulated for 24 h. Then, half of the cell supernatants (100 μL) were removed and used for quantification of and cytokines. Target LLC tumor cells were added to each well (3,000 cells in 100 μL), resulting in 20:1 ratio of effector to target cells. LLC cells were also added to wells without BMDMs and were either left untreated or treated with the same concentrations of TLR agonists and cytokines as the wells with co-cultured BMDMs and LLC cells. After 24 h of culturing, cells were pulsed with [3H]thymidine (#MT6032, Hartmann Analytic, Braunschweig, Germany) and harvested 18 h later by a freeze and thaw cycle. Quantification of radiolabeled DNA was performed on a 1450 MicroBeta Trilux Microplate Scintillation counter (Perkin Elmer, Waltham, MA, USA) as counts per minutes (cpm). For each condition, triplicate wells were analyzed.
Quantification of Nitrite () by the griess test
Supernatants of BMDM cultures were centrifuged at 410 g for 5 min to remove detached cells and immediately assayed for nitrite () as a measure for macrophage NO production. Fifty microliters of supernatant was added to 50 μL of a mixture of 1% sulphanilamide (#S9251, Sigma-Aldrich) and 5% phosphoric acid (#W290017, Sigma-Aldrich) in MilliQ water (Griess reagent A) and incubated in the dark at room temperature for 10 min. Finally, 50 μL of 0.1% N-(1-napthyl) ethylenediamine (#N9125, Sigma-Aldrich) in MilliQ water (Griess reagent B) was added and the absorbance at 540 nm was measured using a microplate reader (BioTek Instruments, Winooski, VT, USA). For each condition, triplicate wells were analyzed and a serial dilution of NaNO2 (#67398, Sigma-Aldrich) was performed to create a standard curve of in the range of 1.56 to 100 μM.
Inhibition of iNOS
N-[[3-(aminomethyl)phenyl]methyl]-ethanimidamide dihydrochloride (1400 w, #1415, Tocris/Bio-Techne, MN, USA) is a specific inhibitor of iNOS (33) and was used to block NO production by activated BMDMs.
Cytokine Quantification By Luminex Technology
BMDMs (6 × 104 cells/well) were cultured in 200 μL RPMI 1640 supplemented with 10% FBS (Biochrom) and 10% L929-cell conditioned medium and activated for 24 h with TLR agonists with or without IFN-γ. Supernatants were collected and centrifuged at 410 g for 5 min to remove cells and stored at −80°C for maximum 1 week. The concentrations of IFN-α and IFN-β were determined by a multiplex Luminex assay (#EPX020-22187-901, ProcartaPlex, ThermoFischer Scientific) according to the manufacturer's instructions. The detection limit was 4 pg/mL for IFN-α and 2 pg/mL for IFN-β. Samples were analyzed in duplicates, using a Bio-Plex MAGPIX Multiplex Reader and Bio-Plex Manager 6.1 software (Bio-Rad Laboratories).
Cell Viability/Proliferation Assay
LLC cells were seeded in 96 well plates at 10,000 cells/well in 200 μL RPMI medium without phenol red supplemented with 10% FBS. Recombinant IFN-β was added to the cells to achieve final concentrations ranging from 0.1 to 20 ng/mL. After 48 h of incubation, LLC cell growth was analyzed in triplicate wells per condition using the Cell Counting Kit 8 (CCK-8) (#96992, Sigma-Aldrich) according to the manufacturer's instructions. The CCK-8 assay is based on the formation of an orange formazan product from colorless WST-8 reagent by cellular dehydrogenases, which is strongly correlated with the number of cells. The absorbance was measured at 450 nm using a microplate reader (Perkin Elmer).
Determination of iNOS mRNA Levels by Real-Time Quantitative PCR
BMDMs were seeded in 12 well plates (#CLS3513, Sigma-Aldrich) at a density of 4.5 × 105 cells/well in 1 ml/well RPMI 1640 supplemented with 10% FBS and treated with either IFN-γ (40 ng/mL), IFN-β (40 ng/mL), poly(I:C) (100 μg/mL), LPS (1 or 1,000 ng/mL), Pam3CSK4 (100 ng/mL) or combinations thereof for 24 h. Total RNA was extracted using 300 μL/well TRI-Reagent (#T9424-100ML, Merck KGaA, Darmstadt, Germany) and Direct-zol RNA minipreps (#R2072, Zymo Research, Irvine, CA, USA) according to manufacturer's instructions. In total, 250 ng RNA of each sample were reverse transcribed to cDNA using the Primescript RT kit (#RR036A, Takara Bio Inc., Shiga, Japan) according to manufacturer's instructions. Real-time quantitative PCR (RT-qPCR) was performed with 50 ng of the obtained cDNA, using a Kapa SYBR fast qPCR kit (#KK4600, Kapa Biosystems, Wilmington, MA, USA) and 0.2 μM of mRNA specific primers for the mouse gene Nos2 which encodes iNOS (forward primer: TTCACCCAGTTGTGCATCGACCTA, reverse primer: TCCATGGTCACCTCCAACACAAGA) in selected cycling conditions (95°C for 3 min, 95°C for 3 s, 60°C for 30 s for 40 cycles). All samples were run in duplicates and the final values were averaged. Following melting curve analysis, the relative differences in iNOS mRNA levels were expressed using the -ΔCt values (Ctreference-Cttarget) and with 18s RNA (forward primer: CGCTTCCTTACCTGGTTGAT, reverse primer: GAGCGACCAAAGGAACCATA) as the endogenous control.
Statistical Analysis
Multiple groups were compared by using two-way ANOVA followed by a post-hoc Dunnett's and Sidak's test for multiple comparisons. P < 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001). Statistical analysis was performed using GraphPad Prism 7.02 software.
Results
LPS-Mediated Activation of Antitumor M1 Macrophages is Associated With Autocrine Production of Type I IFNs
We have recently reported that LPS, a classical M1 stimulus, was the only TLR agonist which could activate mouse BMDMs to an antitumor phenotype in the absence of IFN-γ (19). To further investigate this phenomenon, we used a previously established growth inhibition assay (19), in which the growth of cancer cells cultured alone or together with mitomycin C-treated BMDMs was analyzed by measuring the incorporation of radiolabeled thymidine as schematically depicted in Figure 1A. A main advantage of such a growth inhibition assay is that both cytotoxic and cytostatic activities of activated macrophages against cancer cells are measured. BMDMs were left untreated or activated with the indicated factors for 24 h, before NO production and type I IFN secretion were measured in cell culture supernatants (Figure 1A). LLC cancer cells were then added as target cells and co-cultured with BMDMs for another 24 h before radiolabeled [3H] thymidine was added for the remaining 18 h of the assay. Finally, [3H] thymidine incorporation by LLC cells was analyzed as a measure for cancer cell proliferation (Figure 1A).
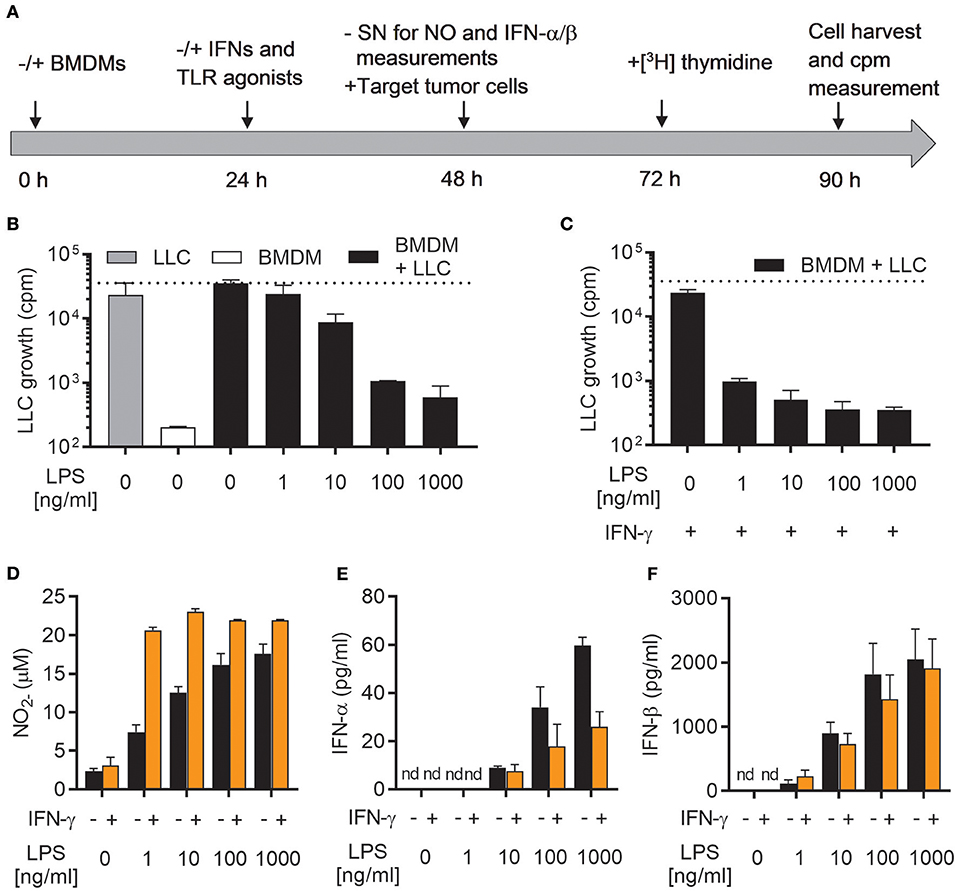
Figure 1. LPS induces growth inhibitory capacity of macrophages and production of both NO and type I IFNs. (A) Timeline for the growth inhibition assay used to assess cytotoxic and cytostatic activity of BMDMs toward cancer cells. Mitomycin C-treated BMDMs were seeded out (6 × 104 in 200 μL) and, after 24 h, stimulated with IFNs and/or TLR agonists. At 48 h, half of the cell culture supernatant (SN) was removed for analysis of nitric oxide (NO) and interferon (IFN)-α/β production, before LLC tumor cells (3 × 103) were added to the BMDM cultures, resulting in a 20:1 BMDM effector to LLC target cell ratio. Control wells with LLC cancer cells alone, were treated correspondingly. At 72 h, radiolabeled [3H]thymidine was added to all wells, and at 90 h, cells were harvested. Growth of LLC cancer cells was measured by the incorporation of [3H]thymidine (counts per minute; cpm). (B) Growth inhibition of LLC cells co-cultured with BMDMs, which had been left untreated or stimulated with various concentrations of LPS for 24 h. Cultures of untreated LLC cells alone and mitomycin C-treated BMDMs alone were used as controls. (C) Inhibition of growth of LLC cells in co-cultures with BMDMs stimulated for 24 h with 40 ng/mL IFN-γ alone or 40 ng/mL IFN-γ in combination with various concentrations of LPS for 24 h. (D) Production of NO by the BMDMs plated for use in analysis of LLC growth inhibition presented in (B,C). The Griess assay was used to measure NO indirectly as nitrite () in the supernatants of BMDMs. (E,F) Luminex technology was used to measure the levels of IFN-α (E) and IFN-β (F) in supernatants from BMDMs plated for analysis of LLC growth inhibition shown in (B,C). The BMDMs were stimulated 24 h with LPS alone or LPS together with IFN-γ. Data are presented as means ± SD of triplicate wells from one representative experiment out of three. nd, not detectable.
As previously reported, stimulation with LPS alone was able to activate BMDMs to inhibit the growth of LLC cancer cells (Figure 1B) (19). This effect was concentration-dependent, and only high concentrations of LPS (100 and 1,000 ng/mL) resulted in efficient LLC cell growth inhibition. Lower concentrations were only partially (10 ng/mL), or not at all (1 ng/mL), able to induce LLC growth inhibition (Figure 1B). Upon co-stimulation with LPS and IFN-γ, BMDMs were able to completely inhibit the growth of LLC cancer cells already at the lowest tested concentration of LPS (1 ng/mL) (Figure 1C). Mirroring the growth inhibition results, production of NO by BMDMs stimulated with LPS alone was concentration-dependent and the lowest concentration of LPS (1 ng/ml) failed to induce substantial levels of NO (Figure 1D). Co-stimulation of BMDMs with LPS/IFN-γ resulted in equally high NO levels for all tested LPS concentrations (1-1,000 ng/ml) (Figure 1D).
A characteristic feature of LPS stimulated leukocytes, such as macrophages and dendritic cells, is the induction of type I IFN production, in particular IFN-α and IFN-β, among other pro-inflammatory cytokines (24). Autocrine activation of the type I IFN signaling pathway has been shown to be essential for LPS-mediated induction of iNOS expression and NO production by macrophages (34, 35). Indeed, stimulation of BMDMs with LPS for 24 h resulted in significant amounts of both IFN-α and IFN-β, which varied with the concentration of LPS (Figures 1E,F). The levels of IFN-α and IFN-β were not further increased by co-stimulation with IFN-γ (Figures 1E,F). In fact, macrophages activated by LPS/IFN-γ appeared to secrete less IFN-α compared to LPS stimulation alone (Figure 1E). Thus, activation of antitumor M1 macrophages by LPS (alone or together with IFN-γ) is associated with autocrine production of the type I IFNs, IFN-α, and IFN-β.
Autocrine Type I IFN Signaling is Required For Lps-Induced No Production and Optimal Cancer Cell Growth Inhibition in the Absence of IFN-γ
We investigated the importance of autocrine type I IFN signaling for the induction of an antitumor phenotype in macrophages activated with LPS. For that purpose, we used BMDMs derived from Ifnar1−/−mice, which lack the common receptor for both IFN-α and IFN-β. WT and Ifnar1−/− BMDMs did not differ in their ability to produce IFN-β after treatment with 100 ng/mL LPS for 24 h (Figure 2A). In contrast, Ifnar1−/− BMDMs failed to produce NO in response to 100 ng/mL LPS alone (Figure 2B). NO production by Ifnar1−/− BMDMs was not reduced compared to WT BMDMs upon co-stimulation with LPS and IFN-γ (Figure 2B). Furthermore, Ifnar1−/− BMDMs stimulated with LPS alone were much less able to inhibit LLC cancer cell growth compared to WT BMDMs (Figures 2C,D). When co-stimulated with LPS/IFN-γ, Ifnar1−/−BMDMs did not differ from WT BMDMs in their ability to completely inhibit LLC cell growth (Figures 2C,D).
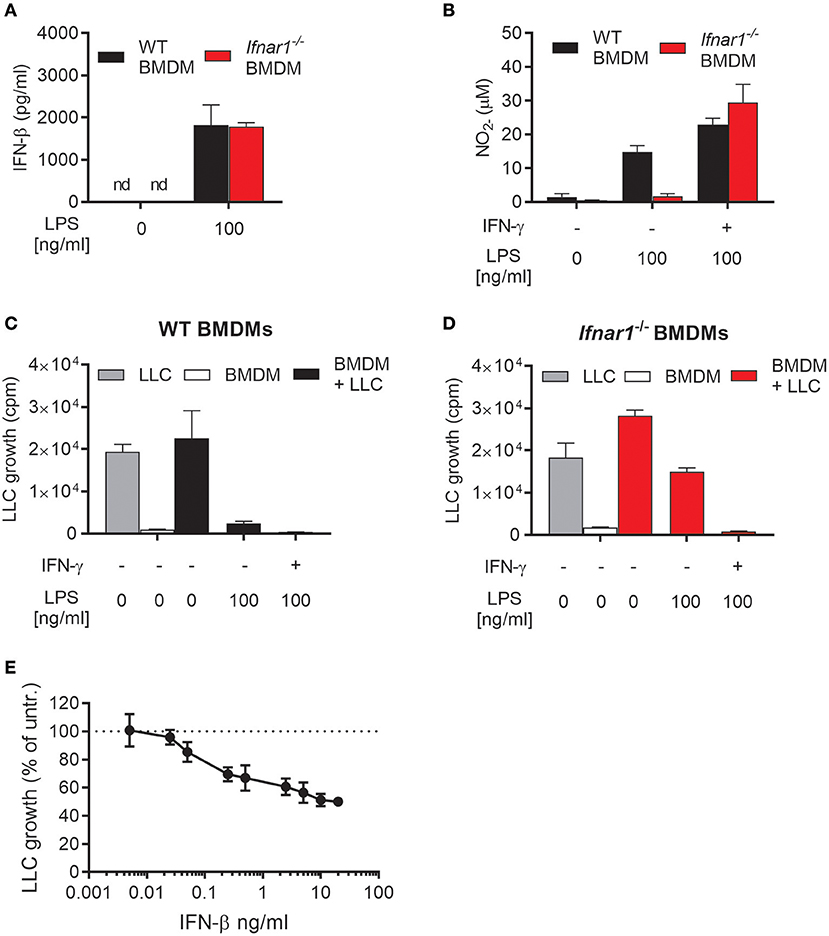
Figure 2. Role of autocrine type I IFNs for NO production and inhibitory activity of macrophages activated by LPS. (A) Levels of IFN-β in the supernatant of WT and IFN alpha/beta receptor 1 knockout (Ifnar1−/−) BMDMs left untreated or treated with 100 ng/mL LPS for 24 h. Data are presented as means ± SD of triplicate wells from a single experiment. (B) NO production, measured indirectly as nitrite (), by WT and Ifnar1−/− BMDMs (6 × 104 in 200 μL) in response to stimulation for 24 h with 100 ng/mL LPS alone or in combination with 40 ng/mL IFN-γ. (C,D) Growth inhibition of LLC cells co-cultured with WT (C) or Ifnar1−/− (D) BMDMs, which were stimulated as described in (B). Data in (B–D) are presented as means ± SD of triplicate wells from one representative experiment out of three. (E) Direct effect of exogenous IFN-β on the growth of LLC cells. The proportion of viable cells after 48 h of treatment with different concentrations of IFN-β was determined using the CCK-8 viability assay. Untreated cells were used as control and the results are presented in percentage of the untreated control group. Pooled data from four independent experiments are presented as means ± SD. nd, not detectable.
Ifnar1−/− BMDMs activated with LPS alone retained some inhibitory effect on LLC cells as compared with untreated Ifnar1−/− BMDMs (Figure 2D). Notably, Ifnar1−/− BMDMs produced normal levels of IFN-β upon LPS stimulation (Figure 2A). It is well-documented that type I IFNs, including IFN-β, can inhibit the growth of cancer cells in a direct manner (36–38). Therefore, we assessed the direct effect of IFN-β on the growth of LLC cells by measuring the LLC cell viability after incubation with different concentrations of recombinant mouse IFN-β for 48 h (Figure 2E). LLC cell growth was inhibited by relatively low concentrations of IFN-β, and a tendency was observed already at 0.05 ng/mL IFN-β. In contrast to IFN-β, a direct effect of IFN-α or IFN-γ on LLC growth was not observed as measured by the thymidine-based growth inhibition assay (Supplementary Figure 1). Thus, the ability of LPS to induce antitumor macrophages in the absence of IFN-γ depends on production of autocrine type I IFNs by the macrophages. Type I IFN signaling in BMDMs is required for LPS-induced NO production and complete inhibition of tumor cell growth. Additionally, LPS-induced IFN-β production is likely to directly contribute to the growth inhibition of LLC cancer cells.
IFN-β Synergizes With TLR Agonists for Induction of Antitumor M1 Macrophages
We next wanted to examine whether IFN-β could, similarly to IFN-γ, synergize with TLR ligands for induction of antitumor macrophages. The TLR ligands Pam3CSK4 (a TLR1/2 agonist) and CL264 (a TLR7 agonist) were selected because we have previously shown that these TLR agonists cannot alone, but in combination with IFN-γ, induce an antitumor macrophage phenotype (19). First, we tested whether these TLR ligands stimulate macrophages to produce type I IFNs, and measured the levels of IFN-α and IFN-β in the supernatant of BMDMs stimulated for 24 h with Pam3CSK4 or CL264 alone or in combination with IFN-γ. None of the conditions induced detectable levels of IFN-α (Figure 3A). IFN-β could be detected, but at levels ~100-fold lower than upon stimulation with LPS alone or with LPS in combination with IFN-γ (Figure 3B). The data presented in Figures 1E,F, 3A,B are from the same experiment, and the results of stimulation with 1 μg/ml LPS alone or in combination with IFN-γ (Figures 1E,F) are also included in Figures 3A,B but now as positive control.
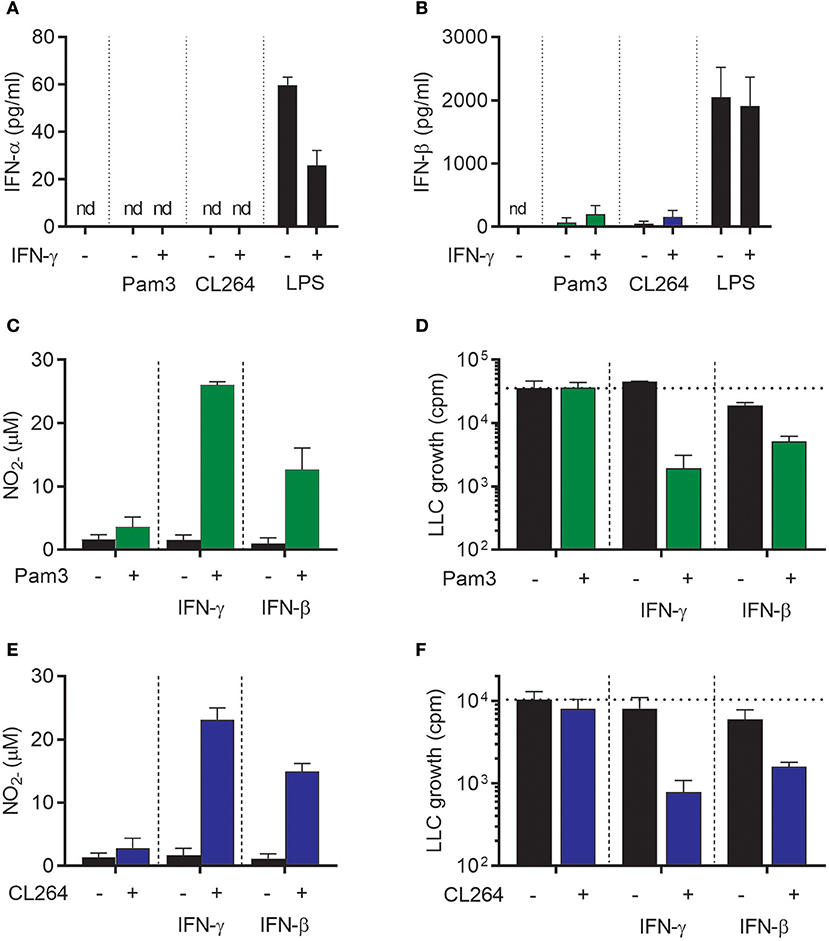
Figure 3. IFN-β synergizes with TLR agonists for activation of antitumor M1 macrophages in a similar fashion as IFN-γ. (A,B) Luminex analysis of the levels of IFN-α (A) and IFN-β (B) produced by BMDMs (6 × 104 in 200 μL) left untreated or stimulated with 100 ng/mL Pam3CSK4, 1 μg/mL CL264, or 1 μg/mL LPS for 24 h in the presence or absence of 40 ng/mL IFN-γ. Note that the results were obtained from the same experiments as the results presented in Figures 1E,F and therefore show the same data for stimulation with 1 μg/ml LPS, which represents the positive control in (A,B). (C) NO production, measured indirectly as nitrite (), by BMDMs stimulated for 24 h with 100 ng/mL Pam3CSK4, 40 ng/mL IFN-γ, 40 ng/mL IFN-β or the indicated combinations. (D) Inhibition of LLC cell growth in BMDM-LLC co-cultures. BMDMs were stimulated as described in (C) before addition of LLC cells. (E) NO production by BMDMs treated for 24 h with 1 μg/mL CL264, 40 ng/mL IFN-γ, 40 ng/mL IFN-β or the indicated combinations. (F) Growth inhibition of LLC cells in co-cultures with BMDMs. The BMDMs were stimulated as described in (E) before addition of LLC cells. Data are presented as means ± SD of triplicate wells from one representative experiment out of three. nd, not detectable; Pam3, Pam3CSK4.
Stimulation of BMDMs with Pam3CSK4, IFN-γ, or IFN-β for 24 h did not activate BMDMs to produce NO (Figure 3C). However, combined treatment of BMDMs with Pam3CSK4/IFN-γ or with Pam3CSK4/IFN-β induced strong NO production in a synergistic manner (Figure 3C). Furthermore, growth-inhibition of LLC cells was only induced when BMDMs were treated with Pam3CSK4/IFN-γ or Pam3CSK4/IFN-β, whereas Pam3CSK4 alone had no effect (Figure 3D). The effect of CL264 was similar to that of Pam3CSK4 as combined activation with CL264/IFN-γ or with CL264/IFN-β induced strong NO production. Treatment with CL264 alone did not result in NO production (Figure 3E). Co-stimulation of BMDMs with CL264/IFN-γ or CL264/IFN-β was also required for BMDM-mediated growth-inhibition of LLC cells, since CL264 alone had no effect (Figure 3F). In accordance with the data in Figure 2D and Supplementary Figure 1, treatment with exogenous, recombinant IFN-β alone had a limited inhibitory effect on LLC cell growth (Figures 3D,F). Thus, IFN-β synergizes with TLR ligands for induction of NO production and inhibitory activity of macrophages toward cancer cells, in a similar fashion as IFN-γ.
Both IFN-α and IFN-β Synergize With Pam3CSK4 For Induction of M1 Antitumor Macrophages
The ability of IFN-α, IFN-β, and IFN-γ to induce antitumor M1 macrophages was investigated in more detail. We stimulated BMDMs with Pam3CSK4 in combination with different concentrations of either IFN-γ, IFN-β, or the other major type I IFN, IFN-α (type A), for 24 h before analyzing NO production and growth inhibition of LLC cancer cells. In accordance with a previous report (39), recombinant IFN-γ synergized with Pam3CSK4 in inducing NO production and BMDM-mediated growth inhibition of cancer cells in a dose-dependent manner starting from the lowest IFN-γ concentration, 0.04 ng/mL (Figures 4A,B). Likewise, recombinant IFN-β as well as IFN-α were able to synergize in a dose-dependent manner with Pam3CSK4 in inducing NO production (Figures 4C,E). Both IFN-β and IFN-α induced BMDM-mediated growth inhibition of cancer cells in combination with Pam3CSK4, although only at relatively high concentrations compared to IFN-γ: at 4 ng/mL and at 100 ng/mL for IFN-β and IFN-α, respectively (Figures 4D,F). We therefore conclude that, similarly to type II IFN, both of the tested type I IFNs can synergize with Pam3CSK4 to induce an antitumor M1 macrophage phenotype.
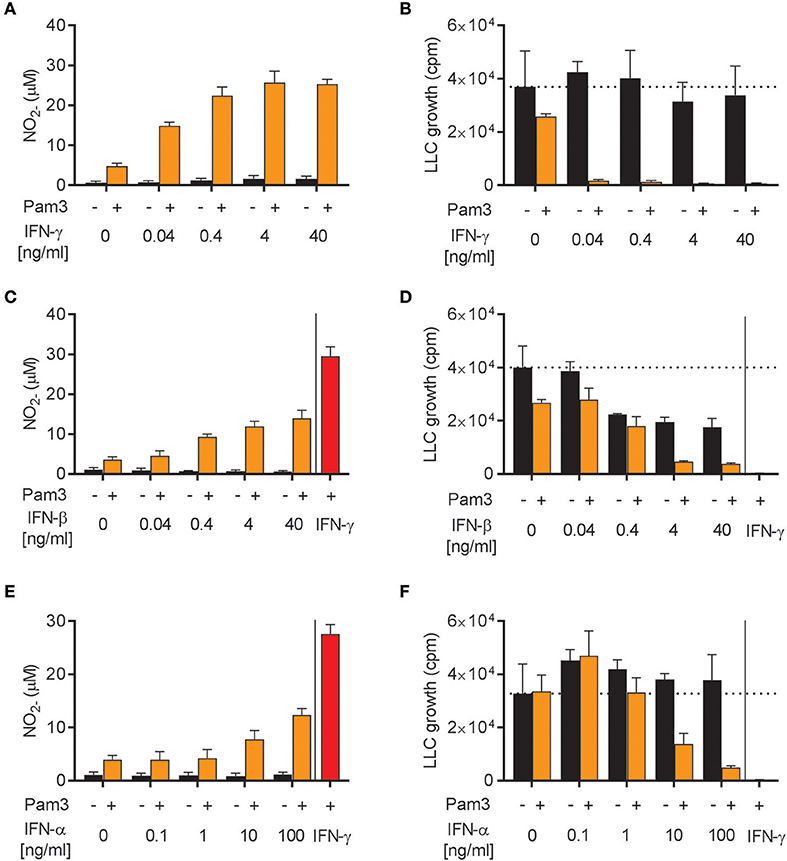
Figure 4. Both IFN-α and IFN-β synergize with Pam3CSK4 in inducing NO production by BMDMs and inhibition of tumor cell growth. (A) NO production, measured indirectly as nitrite (), by BMDMs (6 × 104 in 200 μL) stimulated for 24 h with the indicated concentrations of IFN-γ in the absence or presence of 100 ng/mL Pam3CSK4. (B) BMDMs were stimulated as described in (A) before LLC cells (3 × 103) were added to measure inhibition of LLC cell growth in co-cultures. (C) NO production by BMDMs stimulated with the indicated concentrations of IFN-β in the absence or presence of 100 ng/mL Pam3CSK4 for 24 h. (D) Inhibition of LLC cell growth in co-cultures with BMDMs stimulated as described in (C). (E) NO production by BMDMs stimulated with the indicated concentrations of IFN-α in the absence or presence of 100 ng/mL Pam3CSK4 for 24 h. (F) Inhibition of LLC cell growth in co-cultures with BMDMs stimulated as described in (E). In (C–F) 40 ng/mL IFN-γ was used as positive control. Data are presented as means ± SD of triplicate wells from one representative experiment out of three (A-F). Pam3, Pam3CSK4.
NO is Required for Macrophage-Mediated Growth Inhibition of LLC Cells Induced by Pam3CSK4 in Combination With Both Type I and Type II IFNs
Next, we examined whether the cancer cell growth inhibition induced by Pam3CSK4 in combination with IFN-β or IFN-α was mediated by NO, as it is the case for Pam3CSK4 in combination with IFN-γ (19). NO production was successfully blocked by the iNOS inhibitor 1400 w when macrophages were activated by Pam3CSK4 in combination with any of the IFNs tested (Figure 5A). The presence of 1400 w also abolished the macrophage-mediated inhibition of cancer cell growth induced by Pam3CSK4 and IFNs (Figure 5B). Therefore, we conclude that both type I and type II IFN can be combined with Pam3CSK4 to induce macrophages to inhibit LLC cell growth in an NO-dependent manner.
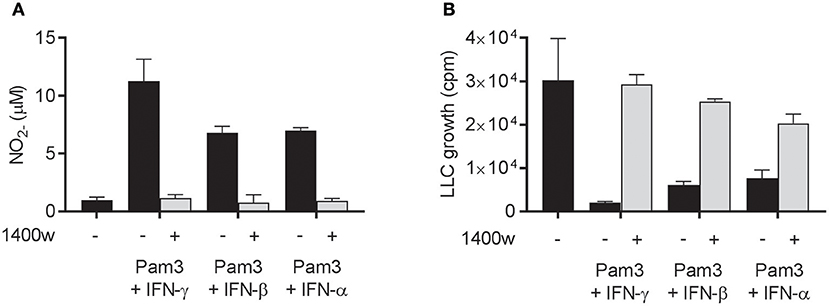
Figure 5. NO is required for macrophage-mediated growth inhibition of LLC cells induced by Pam3CSK4 in combination with both type I and type II IFNs. (A,B) BMDMs (6 × 104 in 200 μL) were left untreated or activated for 24 h with Pam3CSK4 (100 ng/mL) and the indicated IFNs (40 ng/mL IFN-γ, 40 ng/mL IFN-β, or 100 ng/mL IFN-α) in the presence or absence of 50 μM of the iNOS inhibitor 1400 w. (A) NO production was measured indirectly as nitrite () in the supernatant of untreated or activated BMDMs, and (B) inhibition of LLC growth in co-cultures of untreated or activated BMDMs was measured by thymidine incorporation. Data are presented as means ± SD of triplicate wells from one representative experiment out of two. Pam3, Pam3CSK4.
Combination of Two TLR Ligands Can Be Used to Activate Antitumor M1 Macrophages Through Induction of Autocrine Type I IFNs
In addition to LPS, the TLR3 agonist poly(I:C), which mimics viral double stranded RNA, stimulates macrophages to produce type I IFN via the TRIF signaling pathway (26). We have previously shown that type I IFNs produced upon stimulation with poly(I:C) activated BMDMs in an autocrine manner and that type I IFNs could synergize with TLR ligands, such as the TLR2 agonist Lipomannan and the TLR1/2 agonist Pam3CSK4, in inducing pro-inflammatory macrophages that produce high levels of NO (21). Therefore, we hypothesized that autocrine type I IFN production triggered by poly(I:C) in BMDMs could potentially synergize with Pam3CSK4 in inducing antitumor activity, in a similar manner as recombinant IFN-α or IFN-β. As expected, poly(I:C) induced production of both IFN-α and IFN-β, although the level of IFN-β was much lower than after stimulation with LPS (Figures 6A,B). Stimulation of BMDMs with poly(I:C) alone for 24 h did not result in high NO levels, but the NO production was markedly increased by co-stimulation with poly(I:C) and Pam3CSK4 (Figure 6C). In accordance with these data, we found that BMDMs stimulated with poly(I:C) alone failed to inhibit cancer cell growth, whereas BMDMs stimulated with Pam3CSK4/poly(I:C) inhibited LLC growth to a similar degree as the Pam3CSK4/IFN-γ treated, control BMDMs (Figure 6D).
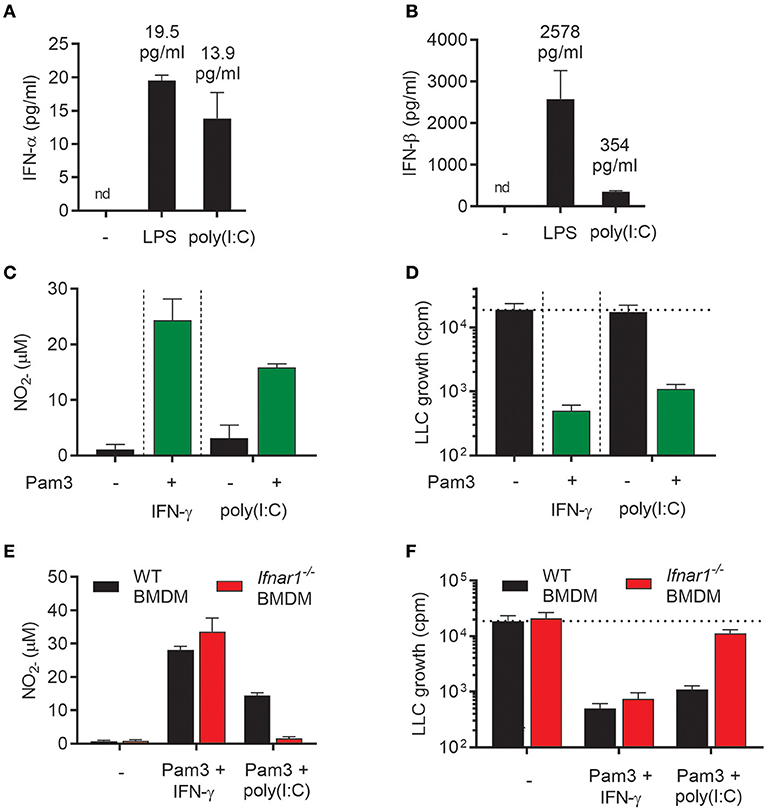
Figure 6. Poly(I:C) synergizes with Pam3CSK4 for activation of antitumor M1 macrophages through induction of type I IFNs. (A,B) Production of IFN-α (A) and IFN-β (B) by BMDMs (6 × 104 in 200 μL) left untreated, activated with 1 μg/mL LPS or 100 μg/mL poly(I:C) for 24 h. (C) Production of NO by untreated BMDMs or BMDMs stimulated for 24 h with 100 ng/mL Pam3CSK4 in combination with 40 ng/mL IFN-γ, 100 μg/mL poly(I:C) alone or 100 μg/mL poly(I:C) in combination with 100 ng/mL Pam3CSK4. (D) Inhibition of growth of LLC cells (3 × 103 per well) co-cultured with BMDMs (6 × 104), which were stimulated as described in (C). (E) NO production by WT and Ifnar1−/− BMDMs in response to co-stimulation with Pam3CSK4/IFN-γ or Pam3CSK4/poly(I:C) for 24 h. (F) Growth inhibition of LLC cells co-cultured with WT or Ifnar1−/− BMDMs treated as described in (E). Data in (A–F) are presented as the means ± SD of triplicate wells from one representative experiment out of three. nd, not detectable; Pam3, Pam3CSK4.
To study the role of autocrine type I IFN in the induction of antitumor BMDMs by poly(I:C)/Pam3CSK4, Ifnar1−/− BMDMs were compared with WT BMDMs in the growth inhibition assay. Ifnar1−/− BMDMs co-stimulated with poly(I:C)/Pam3CSK4 completely failed to produce NO (Figure 6E) and showed a strongly reduced ability to inhibit cancer cell growth (Figure 6F). In contrast, neither NO production nor BMDM-mediated growth inhibition was affected when Ifnar1−/− BMDMs had been stimulated with Pam3CSK4/IFN-γ (Figures 6E,F). Thus, the combination of two TLR ligands, poly(I:C) and Pam3CSK4, efficiently activated BMDMs to an antitumor phenotype via poly(I:C)-induced autocrine type I IFN signaling.
Both Type I and Type II IFNs Synergize With TLR Agonists For Induction of Nos2 Gene Expression
Our results showed that TLR agonists can synergize with both type I IFNs (recombinant or endogenously induced by poly(I:C) stimulation) and type II IFN for induction of NO production by macrophages (Figures 4, 6). In accordance with previous reports, NO was found to be critical for macrophage-mediated cancer cell growth inhibition (Figure 5) (19). To clarify the underlying mechanism, we analyzed the expression of Nos2, which is the gene coding for iNOS, by RT-qPCR. BMDMs were treated for 24 h with TLR agonists alone, IFNs alone, or combinations of both types of ligands. We found that stimulation with IFN-γ alone induced significant Nos2 expression in BMDMs compared with the untreated control (Figure 7A). LPS also induced significant Nos2 expression, but only when it was used at high concentrations (1,000 ng/mL) (Figure 7A). Stimulation with IFN-γ and LPS had a synergistic effect and resulted in significant Nos2 expression also at low LPS concentrations (1 ng/mL). IFN-γ did not further increase the expression of Nos2 induced by high concentrations of LPS (Figure 7A).
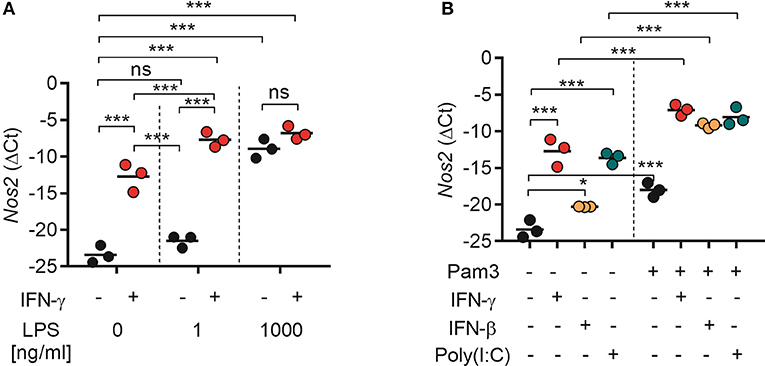
Figure 7. Both type I and II IFNs synergize with TLR agonists for induction of Nos2 gene expression. BMDMs (4.5 × 105) were stimulated for 24 h. Total RNA was reverse transcribed into cDNA and analyzed by RT-qPCR using primers specific for mouse Nos2 mRNA. (A) Nos2 expression in BMDMs treated with IFN-γ (40 ng/mL), or LPS at the indicated concentrations, or a combination of both. (B) Nos2 expression in BMDMs treated with IFN-γ (40 ng/mL), IFN-β (40 ng/mL), poly(I:C) (100 μg/mL), Pam3CSK4 (100 ng/mL), or the indicated combinations. Data are presented as means (ΔCt) of duplicates for each of the three independent experiments. Each circle in the graphs represents the mean value from one experiment. P-values from a two-way ANOVA multiple comparison test is displayed as follows: *p < 0.05, ***p < 0.001; ns, not significant; Pam3, Pam3CSK4.
Stimulation with Pam3CSK4, recombinant IFN-β or poly(I:C) alone induced significant Nos2 expression in BMDMs (Figure 7B). Furthermore, the Nos2 expression induced by treatment with IFN-γ, IFN-β, or poly(I:C) was significantly increased when the ligands were combined with Pam3CSK4 (Figure 7B). The relative levels of Nos2 expression correlated with the levels of NO induced by single or combined activation of BMDMs with TLR agonists and IFNs as observed in the previous experiments. Taken together, these results indicate that activation of BMDMs with TLR agonists and type I or type II IFNs results in downstream signaling which converge to the induction of Nos2 gene expression.
Encapsulation of Poly(I:C) in Nanoparticles Potentiates the Synergistic Interaction With Pam3CSK4 for Inducing Antitumor Macrophages Up to 100-fold
We have previously shown that the potency of poly(I:C) can be enhanced manifold by encapsulation into chitosan-based nanoparticles (poly(I:C)-NPs) (21). Encapsulation can protect poly(I:C) against enzymatic degradation and enhance the uptake of poly(I:C) by macrophages (21). When tested in combination with TLR ligands such as Lipomannan or BCG bacteria, poly(I:C)-NPs were up to 100-fold more potent than soluble poly(I:C) at activating BMDMs in respect to production of NO and pro-inflammatory cytokines (21). We therefore compared the ability of poly(I:C)-NPs to that of soluble poly(I:C) to induce antitumor BMDMs in the presence of Pam3CSK4. BMDMs stimulated with poly(I:C)-NPs for 24 h produced higher levels of IFN-β compared to BMDMs stimulated with the same concentrations of poly(I:C) in soluble form (Figure 8A). BMDMs stimulated with poly(I:C)-NPs alone displayed a modest growth inhibitory effect on LLC cells, possibly through the potent induction of IFN-β production (Figure 8B).
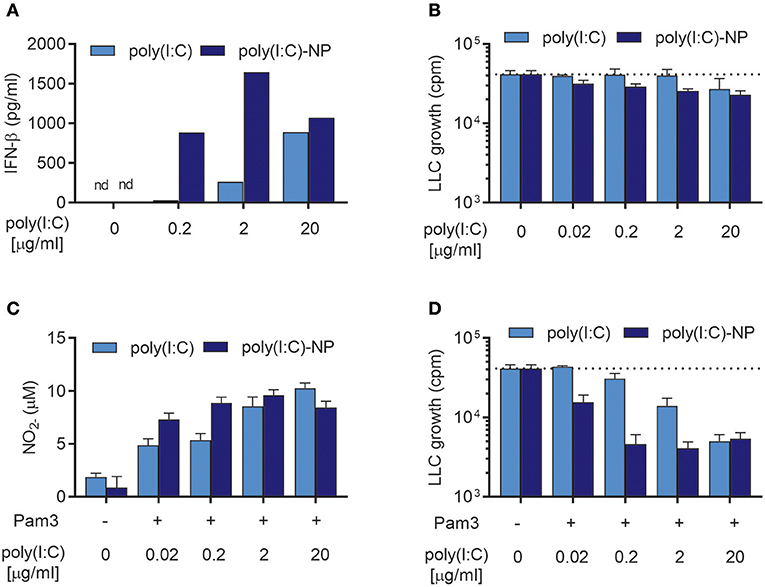
Figure 8. Encapsulation of poly(I:C) into nanoparticles (poly(I:C)-NPs) potentiates the synergy of combined stimulation with Pam3CSK4 and poly(I:C) in the induction of antitumor BMDMs. (A) Production of IFN-β by BMDMs (6 × 104 in 200 μL) stimulated with the indicated concentrations of poly(I:C) either in soluble form or encapsulated in nanoparticles (poly(I:C)-NPs) for 24 h. (B) Growth inhibition of LLC-cells (3 × 103) in co-culture with BMDMs (6 × 104), stimulated for 24 h with the indicated concentrations of either soluble poly(I:C) or poly(I:C)-NPs. (C) Production of NO by BMDMs (6 × 104) treated for 24 h with the indicated concentrations of either soluble poly(I:C) or poly(I:C)-NPs alone or in combination with 100 ng/mL Pam3CSK4. (D) Growth inhibition of LLC cells in co-culture with BMDMs, stimulated as described in (C). Data represent the means ± SD of triplicate wells of one representative experiment out of two. nd, not detectable; Pam3, Pam3CSK4.
Both soluble poly(I:C) and poly(I:C)-NPs synergized with Pam3CSK4 resulting in dose-dependent NO production (Figure 8C) but failed to induce NO production by BMDMs when used alone (Supplementary Figure 2). At low concentrations of poly(I:C) there was a tendency toward higher NO production by poly(I:C)-NP compared to soluble poly(I:C) (Figure 8C). This improved potency of poly(I:C)-NP was more apparent in the growth inhibition assay, however, when combined stimulation with Pam3CSK4 of the BMDMs was necessary. Whereas, maximal inhibition of LLC growth by BMDMs was achieved already at 0.2 μg/mL of poly(I:C)-NP, a concentration of 20 μg/mL was required to achieve the same effect for soluble poly(I:C) (Figure 8D). Taken together, encapsulation of poly(I:C) into poly(I:C)-NPs increased the ability of poly(I:C) to induce IFN-β production by BMDMs and to synergize with Pam3CSK4 in inducing antitumor macrophages. In consequence, the concentration of poly(I:C) required to induce almost complete growth inhibition of LLC cells was reduced from 20 to 0.2 μg/ml by using poly(I:C)-NPs as compared to soluble poly(I:C).
Discussion
In this study, we describe a common function of type I and type II IFNs in the activation of antitumor macrophages in vitro. It is well-established that LPS, a commonly used agonist for in vitro activation of macrophages, induces production of type I IFN through triggering of the TRIF-dependent signaling pathway (24, 25). Autocrine type I IFN signaling through STAT1 was previously found to be critical for LPS-induced iNOS expression by mouse macrophages in vitro (27). An early study by Vadiveloo et al. showed that Ifnar1−/− BMDMs did not produce NO in response to LPS alone. However, these macrophages responded strongly to LPS in combination with recombinant IFN-γ, and produced NO (40). We and others have previously shown that macrophage-mediated cancer cell growth inhibition depends on NO production by mouse macrophages (19, 41, 42). Here, we found that the ability of LPS to induce macrophage-mediated cancer cell growth inhibition in the absence of IFN-γ depended on the induction of autocrine type I IFN signaling, which in turn was essential for the induction of NO production by BMDMs. These results are in agreement with the study by Vadiveloo et al. (40), as is the finding that in the presence of IFN-γ, NO production, and antitumor activation of BMDMs by LPS did not depend on autocrine type I IFN signaling. In addition, we found that the type I IFN, IFN-β had a direct inhibitory effect on the growth of the LLC cancer cell line, whereas IFN-γ or IFN-α type A did not reduce LLC growth. The observed growth inhibitory effect of IFN-β on LLC cells is in agreement with previous studies (38, 43) and can explain the partially retained ability of LPS-stimulated Ifnar1−/− BMDMs to inhibit cancer cell growth, despite their inability to respond to type I IFNs and to produce NO.
A study from 1987 by Koestler et al. demonstrated that LPS can be combined with IFN-γ, or alternatively with IFN-α/β for induction of antitumor macrophages (44). However, to the best of our knowledge, no other study has investigated the ability of type I IFNs to activate macrophages to an antitumor phenotype in combination with other TLR agonists. We therefore tested whether recombinant IFN-β could synergize with two other TLR agonists, Pam3CSK4 (TLR1/2) and CL264 (TLR7) in inducing NO production and macrophage-mediated cancer cell growth inhibition by BMDMs. Both TLR agonists, by themselves, failed to induce production of IFN-α and IFN-β, and the macrophages required a second stimulus in form of IFN-γ to achieve an antitumor phenotype. Recombinant IFN-β and IFN-α type A were able to synergize with both of the TLR agonists for induction of NO production and macrophage-mediated inhibition of cancer cell growth. Both IFN-α and IFN-β were found to be able to replace IFN-γ as a second signal for antitumor macrophage activation, even though the type I IFNs were less effective than IFN-γ when compared at the same concentrations. However, BMDMs activated with TLR agonists in combination with any of the IFNs inhibited growth of LLC cells through a NO-dependent mechanism.
The synergistic effect of LPS and IFN-γ on NO production by macrophages has been well-described, and investigations into the underlying mechanisms have revealed that the two factors synergize to induce transcription of Nos2 (45). Nos2 is encoding the enzyme iNOS which is responsible for NO production by macrophages (46). By RT-qPCR analysis of macrophage total RNA, we found that both IFN-γ and IFN-β (exogenous or endogenous) synergized with TLR signaling for induction of Nos2 mRNA. Previously, several factors have been described to contribute to the synergistic effect of TLR and IFN signaling on iNOS expression. The promoter regions of iNOS contain binding sites for both NF-κB, IFN-γ-activating sites (GASs) and IFN-stimulated responsive elements (ISREs) (47, 48). Phosphorylated STAT1, which is induced by both type I and type II IFNs, has been shown to be able to bind GASs and ISREs and induce iNOS expression (49). Furthermore, TLR-induced activation of NF-κB and IFN-induced STAT1 and STAT2 activation has been reported to result in transcription of the Nos2 gene in macrophages through RNA polymerase II recruitment to the Nos2 promoter (50).
In vivo, IFN-γ has been shown to play a key role in mediating tumor surveillance and tumoricidal activity of the immune system by exerting direct antitumor effects such as inhibiting angiogenesis and cancer cell growth, sensitizing cancer cells to apoptosis or by activating tumoricidal activity in T cells and macrophages (51). Despite these important functions in anti-tumor immune responses, clinical use of IFN-γ for cancer treatment in general and for antitumor activation of TAMs in particular has so far been hampered by significant dose-limiting toxicities and the complex pleiotropic effects of IFN-γ on a large number of different cell types (52). These challenges could potentially be met by more targeted therapies, such as IFN-γ gene delivery to tumors by oncolytic viruses (39). The finding that type I IFNs, in a similar way as IFN-γ, can synergize with TLR agonists to induce antitumor macrophages opens up for further alternative treatment strategies targeting TAMs. Induction of endogenous type I IFN production by the TLR3 agonist poly(I:C) was previously utilized as a strategy to improve the immunogenicity of BCG, either by coating live BCG mycobacteria with poly(I:C) (53) or by combined activation with BCG and poly(I:C)-encapsulating chitosan nanoparticles (21). In both studies, poly(I:C) was shown to synergize with the TLR2 agonist BCG for production of NO and proinflammatory cytokines by macrophages, and this synergy was dependent on activation of the TRIF-dependent pathway and autocrine type I IFN signaling (21). Interestingly, Cardif-KO macrophages showed no difference to WT macrophages in induction of NO, proinflammatory cytokine or IFN-β production in response to activation with the combination of BCG and poly(I:C)-encapsulating nanoparticles (21). This demonstrates that the effect of poly(I:C) is mediated through activation of the TLR pathway rather than the Rig-1/Mda5-Cardif pathway. Multiple other studies have described similar synergy between MyD88-dependent and TRIF-dependent TLR agonists in immune cell cytokine production (54–57). Poly(I:C) is currently being investigated as a potential cancer vaccine adjuvant, with at least two poly(I:C)-based drugs in clinical development (Hiltonol® and Ampligen®) (58). In our study, we show that poly(I:C) induces a robust type I IFN production by BMDMs, which allows poly(I:C) to synergize with Pam3CSK4 for induction of antitumor macrophages. Furthermore, the potency of poly(I:C) could be improved ~100-fold by encapsulation into nanoparticles that we have recently developed (21). Nanoparticles are able to protect poly(I:C) against enzymatic degradation and improve uptake of poly(I:C) by macrophages, resulting in improved stability and potency of poly(I:C)(21, 59). The increased efficacy of poly(I:C)-NPs, compared to soluble poly(I:C) in inducing antitumor macrophages is likely due to a combination of (i) a modest increase in NO production and (ii) a strong increase in production of IFN-β which mediates direct and indirect antitumor effects. Further studies are required to verify that Pam3CSK4 and soluble or nanoparticle-encapsulated poly(I:C) can target and activate tumor-associated macrophages in vivo.
Our results suggest that macrophages can be activated to an antitumor phenotype, characterized by high iNOS expression, NO production, and the ability to inhibit cancer cell growth by three different scenarios summarized in Figure 9. The possible therapeutic potential of activating antitumor macrophages in these ways will need to be explored in vivo. Finally, there are differences between mouse and human macrophages in the induction of iNOS expression as well as disagreement regarding the importance of this pathway for macrophage functions in humans (60), and it remains to be investigated whether human macrophages can be activated by similar combinations of TLR agonists and IFNs.
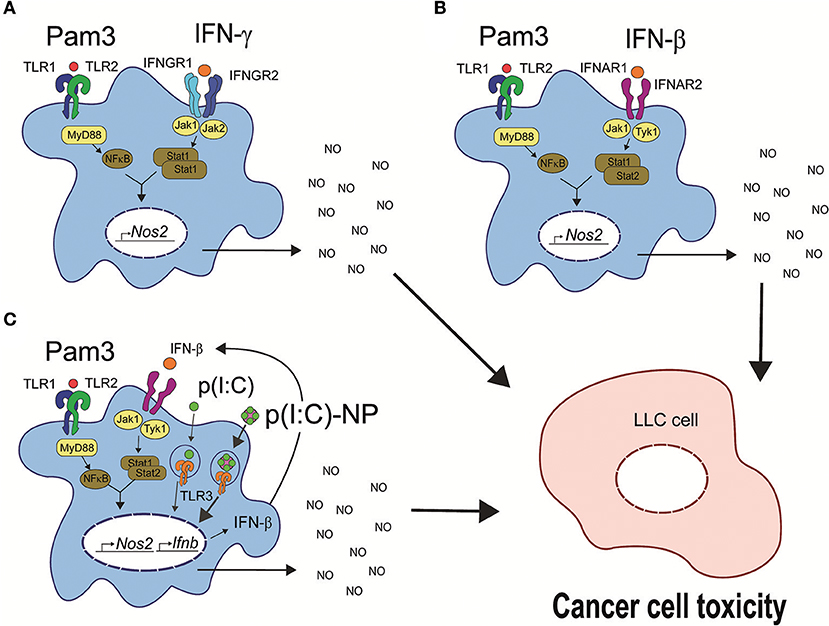
Figure 9. Three possible pathways for induction of antitumor macrophages. (A) TLR agonists such as LPS or Pam3CSK4 synergize with IFN-γ for induction of antitumor macrophages. TLR signaling through MyD88 leads to activation of NF-κB, and IFN-γ signaling through the tyrosine kinases Jak1 and Jak2 leads to STAT1 homodimer formation (61). The two pathways synergize to induce transcription of Nos2 and NO production resulting in cancer cell growth inhibition and cell toxicity. (B) Synergy between a TLR agonist such as Pam3CSK4 with recombinant IFN-β for induction of antitumor macrophages. IFN-β signals through the tyrosine kinases Jak1 and Tyk2 resulting in STAT1/STAT2 heterodimer formation (62), which synergize with TLR-induced NF-κB for induction of Nos2 gene transcription similar to (A). (C) The TLR agonists Pam3CSK4 and poly(I:C) synergize through endogenous type I IFN production for induction of antitumor macrophages. Poly(I:C) signals through TLR3 and induces endogenous IFN-β production, which mediates the second signal and synergizes with TLR signaling as described for exogenous IFN-β in (B). Pam3, Pam3CSK4.
Author Contributions
EM contributed to the conception and design of the experiments. Performed experimental work, prepared all figures and wrote the manuscript. MS contributed to the conception and design of the experiments in collaboration with EM, prepared poly(I:C)- encapsulating nanoparticles, performed experimental work, analyzed results, and contributed to writing the manuscript. PC performed experimental work and contributed to the writing of the manuscript. AL performed experimental work and analyzed results. AA performed early, preliminary experimental work. IØ provided supervision and experimental help, discussed the results, and contributed to writing the final version of the manuscript. AC designed, supervised, and evaluated the experiments and contributed to writing the manuscript. All authors read and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from The Research Council of Norway, the South-Eastern Norway Regional Health Authority, The Norwegian Cancer Society, Henrik Homans Minde fund, Ella and Kristian Nyerrøds fund, and S. G. Sønneland Foundation fund.
Acknowledgments
The authors would like to thank Prof Dr. Ulrich Kalinke at the Helmholtz Centre for Infection Research, Braunschweig, Germany, and the TWINCORE, Centre for Experimental and Clinical Infection Research, Hannover, Germany, for kindly providing bones from mice deficient in the IFN alpha/beta receptor 1 (Ifnar1−/−).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02520/full#supplementary-material
Abbreviations
BCG, bacille Calmette-Guérin; BMDM, bone marrow-derived macrophage; CCK-8, cell counting kit-8; cpm, counts per minute; FBS, fetal bovine serum; IFN, interferón; IFNAR1, interferon alpha/beta receptor 1; iNOS, inducible nitric oxide synthase; LLC, Lewis lung carcinoma; LPS, lipopolysaccharide; M-CSF, macrophage colony-stimulating factor; MyD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NO, nitric oxide; , nitrite; Pam3CSK4, tripalmitoylated lipopeptide CysSerLys4; PBS, phosphate buffered saline; Poly(I:C), polyinosinic:polycytidylic acid; poly(I:C)-NP, poly(I:C)-encapsulating nanoparticles; RT-qPCR, real-time quantitative polymerase chain reaction; TAM, tumor-associated macrophage; TLR, toll-like receptor; TRIF, toll/interleukin-1 receptor homologous region (TIR)-domain-containing adapter-inducing interfere on-β.
References
1. Emens LA, Ascierto PA, Darcy PK, Demaria S, Eggermont AMM, Redmond WL, et al. Cancer immunotherapy: opportunities and challenges in the rapidly evolving clinical landscape. Eur J Cancer (2017) 81:116–29. doi: 10.1016/j.ejca.2017.01.035
2. Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. J Transl Med. (2011) 9:216. doi: 10.1186/1479-5876-9-216
3. Bottazzi B, Polentarutti N, Acero R, Balsari A, Boraschi D, Ghezzi P, et al. Regulation of the macrophage content of neoplasms by chemoattractants. Science (1983) 220:210–2.
4. Noy R, Pollard JW Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
5. Haabeth OA, Bogen B, Corthay A. A model for cancer-suppressive inflammation. Oncoimmunology (2012) 1:1146–55. doi: 10.4161/onci.21542
6. Haabeth OA, Lorvik KB, Hammarström C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. (2011) 2:240. doi: 10.1038/ncomms1239
7. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
8. Corthay A, Skovseth DK, Lundin KU, Røsjø E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity (2005) 22:371–83. doi: 10.1016/j.immuni.2005.02.003
9. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (2011) 331:1612–6. doi: 10.1126/science.1198443
10. Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature (2017) 543:428–32. doi: 10.1038/nature21409
11. Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, et al. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology (2011) 132:226–39. doi: 10.1111/j.1365-2567.2010.03357.x
12. Peng J, Tsang JY, Li D, Niu N, Ho DH, Lau KF, et al. Inhibition of TGF-β signaling in combination with TLR7 ligation re-programs a tumoricidal phenotype in tumor-associated macrophages. Cancer Lett. (2013) 331:239–49. doi: 10.1016/j.canlet.2013.01.001
13. Lizotte PH, Baird JR, Stevens CA, Lauer P, Green WR, Brockstedt DG, et al. Attenuated Listeria monocytogenes reprograms M2-polarized tumor-associated macrophages in ovarian cancer leading to iNOS-mediated tumor cell lysis. Oncoimmunology (2014) 3:e28926. doi: 10.4161/onci.28926
14. Iribarren K, Bloy N, Buqué A, Cremer I, Eggermont A, Fridman WH, et al. Trial watch: immunostimulation with toll-like receptor agonists in cancer therapy. Oncoimmunology (2016) 5:e1088631. doi: 10.1080/2162402X.2015.1088631
15. Cameron DJ, Churchill WH. Cytotoxicity of human macrophages for tumor cells: enhancement by bacterial lipopolysaccharides (LPS). J Immunol. (1980) 124:708–12.
16. Pace JL, Russell SW. Activation of mouse macrophages for tumor cell killing. I. Quantitative analysis of interactions between lymphokine and lipopolysaccharide. J Immunol. (1981) 126:1863–7.
17. Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer (2016) 16:131. doi: 10.1038/nrc.2016.14
18. Lundin JI, Checkoway H. Endotoxin and cancer. Environ Health Perspect. (2009) 117:1344–50. doi: 10.1289/ehp.0800439
19. Müller E, Christopoulos PF, Halder S, Lunde A, Beraki K, Speth M, et al. Toll-like receptor ligands and interferon-γ synergize for induction of antitumor M1 macrophages. Front Immunol. (2017) 8:1383. doi: 10.3389/fimmu.2017.01383
20. Mäkelä SM, Strengell M, Pietilä TE, Osterlund P, Julkunen I. Multiple signaling pathways contribute to synergistic TLR ligand-dependent cytokine gene expression in human monocyte-derived macrophages and dendritic cells. J Leukoc Biol. (2009) 85:664–72. doi: 10.1189/jlb.0808503
21. Speth MT, Repnik U, Müller E, Spanier J, Kalinke U, Corthay A, Griffiths G. Poly(I:C)–Encapsulating nanoparticles enhance innate immune responses to the tuberculosis vaccine Bacille Calmette-Guérin (BCG) via synergistic activation of innate immune receptors. Mol Pharmaceut. (2017) 14:4098–112. doi: 10.1021/acs.molpharmaceut.7b00795
22. Whitmore MM, DeVeer MJ, Edling A, Oates RK, Simons B, Lindner D, et al. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes enhanced antitumor activity. Cancer Res. (2004) 64:5850–60. doi: 10.1158/0008-5472.CAN-04-0063
23. Horng T, Barton GM, Flavell RA, Medzhitov R. The adaptor molecule TIRAP provides signalling specificity for toll-like receptors. Nature (2002) 420:329–33. doi: 10.1038/nature01180
24. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science (2003) 301:640–3. doi: 10.1126/science.1087262
25. O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat Rev Immunol. (2007) 7:353–64. doi: 10.1038/nri2079
26. Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C). Adv Drug Deliv Rev. (2008) 60:805–12. doi: 10.1016/j.addr.2007.11.005
27. Gao JJ, Filla MB, Fultz MJ, Vogel SN, Russell SW, Murphy WJ. Autocrine/paracrine IFN-alphabeta mediates the lipopolysaccharide-induced activation of transcription factor Stat1alpha in mouse macrophages: pivotal role of Stat1alpha in induction of the inducible nitric oxide synthase gene. J Immunol. (1998) 161:4803–10.
28. Spanier J, Lienenklaus S, Paijo J, Kessler A, Borst K, Heindorf S, et al. Concomitant TLR/RLH signaling of radioresistant and radiosensitive cells is essential for protection against vesicular stomatitis virus infection. J Immunol. (2014) 193:3045–54. doi: 10.4049/jimmunol.1400959
29. Müller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, et al. Functional role of type I and type II interferons in antiviral defense. Science (1994) 264:1918–21.
30. Bertram JS, Janik P. Establishment of a cloned line of lewis lung carcinoma cells adapted to cell culture. Cancer Lett. (1980) 11:63–73.
31. Sanford KK, Earle WR, Likely GD. The growth in vitro of single isolated tissue cells. J Natl Cancer Inst. (1948) 9:229–46.
32. Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. (2008) 2008:pdb.prot5080. doi: 10.1101/pdb.prot5080
33. Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, et al. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem. (1997) 272:4959–63. doi: 10.1074/jbc.272.8.4959
34. Zhang X, Alley EW, Russell SW, Morrison DC. Necessity and sufficiency of beta interferon for nitric oxide production in mouse peritoneal macrophages. Infect Immun. (1994) 62:33–40.
35. Jacobs AT, Ignarro LJ, Jacobs AT, Ignarro LJ. Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. J Biol Chem. (2001) 276:47950–7. doi: 10.1074/jbc.M106639200
36. Garrison JI, Berens ME, Shapiro JR, Treasurywala S, Floyd-Smith G. Interferon-beta inhibits proliferation and progression through S phase of the cell cycle in five glioma cell lines. J Neurooncol. (1996) 30:213–23. doi: 10.1007/BF00177272
37. Kusano H, Akiba J, Ogasawara S, Sanada S, Yasumoto M, Nakayama M, et al. Pegylated interferon-alpha2a inhibits proliferation of human liver cancer cells in vitro and in vivo. PLoS ONE (2013) 8:e83195. doi: 10.1371/journal.pone.0083195
38. Ogasawara S, Yano H, Momosaki S, Akiba J, Nishida N, Kojiro S, et al. Growth inhibitory effects of IFN-beta on human liver cancer cells in vitro and in vivo. J Interferon Cytokine Res. (2007) 27:507–16. doi: 10.1089/jir.2007.0183
39. Kurena B, Müller E, Christopoulos PF, Johnsen IB, Stankovic B, Øynebråten I, et al. Generation and functional in vitro analysis of semliki forest virus vectors encoding TNF-α and IFN-γ. Front Immunol. (2017) 8:1667. doi: 10.3389/fimmu.2017.01667
40. Vadiveloo PK, Vairo G, Hertzog P, Kola I, Hamilton JA. Role of type I interferons during macrophage activation by lipopolysaccharide. Cytokine (2000) 12:1639–46. doi: 10.1006/cyto.2000.0766
41. Stuehr DJ, Nathan CF. Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J Exp Med. (1989) 169:1543–55.
42. Hajri A, Metzger E, Vallat F, Coffy S, Flatter E, Evrard S, et al. Role of nitric oxide in pancreatic tumour growth: in vivo and in vitro studies. Br J Cancer (1998) 78:841–9.
43. Wong VL, Rieman DJ, Aronson L, Dalton BJ, Greig R, Anzano MA. Growth-inhibitory activity of interferon-beta against human colorectal carcinoma cell lines. Int J Cancer (1989) 43:526–30. doi: 10.1002/ijc.2910430331
44. Koestler TP, Johnson WJ, Rieman D, Dalton BJ, Greig RG, Poste G. Differential expression of murine macrophage-mediated tumor cytotoxicity induced by interferons. Cancer Res. (1987) 47:2804–8.
45. Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH, Russell SW. Expression of the nitric oxide synthase gene in mouse macrophages activated for tumor cell killing. Molecular basis for the synergy between interferon-gamma and lipopolysaccharide. J Biol Chem. (1993) 268:1908–13.
46. Bogdan C Nitric oxide synthase in innate and adaptive immunity: an update. Trends Immunol. (2015) 36:161–78. doi: 10.1016/j.it.2015.01.003
47. Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, et al. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc Natl Acad Sci USA. (1993) 90:9730–4.
48. Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. (2004) 500:255–66. doi: 10.1016/j.ejphar.2004.07.030
49. Gao J, Morrison DC, Parmely TJ, Russell SW, Murphy WJ. An interferon-γ-activated site (GAS) is necessary for full expression of the mouse iNOS Gene in response to interferon-γ and lipopolysaccharide. J Biol Chem. (1997) 272:1226–30. doi: 10.1074/jbc.272.2.1226
50. Farlik M, Reutterer B, Schindler C, Greten F, Vogl C, Müller M, et al. Nonconventional initiation complex assembly by STAT and NF-kappaB transcription factors regulates nitric oxide synthase expression. Immunity (2010) 33:25–34. doi: 10.1016/j.immuni.2010.07.001
51. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature (2001) 410:1107–11. doi: 10.1038/35074122
52. Miller CH, Maher SG, Young HA. Clinical use of interferon-gamma. Ann NY Acad Sci. (2009) 1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x
53. Speth MT, Repnik U, Griffiths G. Layer-by-layer nanocoating of live Bacille-Calmette-Guerin mycobacteria with poly(I:C) and chitosan enhances pro-inflammatory activation and bactericidal capacity in murine macrophages. Biomaterials (2016) 111:1–12. doi: 10.1016/j.biomaterials.2016.09.027
54. Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. (2005) 201:1435–46. doi: 10.1084/jem.20041964
55. Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol. (2007) 178:1164–71. doi: 10.4049/jimmunol.178.2.1164
56. Ouyang X, Negishi H, Takeda R, Fujita Y, Taniguchi T, Honda K. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activation. Biochem Biophys Res Commun. (2007) 354:1045–51. doi: 10.1016/j.bbrc.2007.01.090
57. Tan RS, Ho B, Leung BP, Ding JL. TLR cross-talk confers specificity to innate immunity. Int Rev Immunol. (2014) 33:443–53. doi: 10.3109/08830185.2014.921164
58. Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly(I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther. (2015) 146:120–31. doi: 10.1016/j.pharmthera.2014.09.010
59. Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H. Nanoparticle uptake: the phagocyte problem. Nano Today (2015) 10:487–510. doi: 10.1016/j.nantod.2015.06.006
60. Thomas AC, Mattila JT. “Of mice and men”: arginine metabolism in macrophages. Front Immunol. (2014) 5:479. doi: 10.3389/fimmu.2014.00479
61. Horvath CM. The Jak-STAT pathway stimulated by interferon gamma. Sci STKE (2004) 2004:tr8. doi: 10.1126/stke.2602004tr8
Keywords: macrophages, toll-like receptors, interferon-γ, interferon-α, interferon-β, cancer, nitric oxide, immunotherapy
Citation: Müller E, Speth M, Christopoulos PF, Lunde A, Avdagic A, Øynebråten I and Corthay A (2018) Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 9:2520. doi: 10.3389/fimmu.2018.02520
Received: 22 June 2018; Accepted: 12 October 2018;
Published: 02 November 2018.
Edited by:
Liwu Li, Virginia Tech, United StatesReviewed by:
Robert Adam Harris, Karolinska Institutet (KI), SwedenAshley Mansell, Hudson Institute of Medical Research, Australia
Copyright © 2018 Müller, Speth, Christopoulos, Lunde, Avdagic, Øynebråten and Corthay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabeth Müller, elisabethskn@googlemail.com
Alexandre Corthay, alexandre.corthay@ibv.uio.no