 Kristen N. Morrow
Kristen N. Morrow Craig M. Coopersmith
Craig M. Coopersmith Mandy L. Ford
Mandy L. Ford- 1Immunology and Molecular Pathogenesis Program, Laney Graduate School, Emory University, Atlanta, GA, United States
- 2Department of Surgery, Emory University School of Medicine, Atlanta, GA, United States
- 3Emory Critical Care Center, Emory University School of Medicine, Atlanta, GA, United States
- 4Emory Transplant Center, Emory University School of Medicine, Atlanta, GA, United States
Sepsis is a major cause of morbidity and mortality worldwide despite numerous attempts to identify effective therapeutics. While some sepsis deaths are attributable to tissue damage caused by inflammation, most mortality is the result of prolonged immunosuppression. Ex vivo, immunosuppression during sepsis is evidenced by a sharp decrease in the production of pro-inflammatory cytokines by T cells and other leukocytes and increased lymphocyte apoptosis. This allows suppressive cytokines to exert a greater inhibitory effect on lymphocytes upon antigen exposure. While some pre-clinical and clinical trials have demonstrated utility in targeting cytokines that promote lymphocyte survival, this has not led to the approval of any therapies for clinical use. As cytokines with a more global impact on the immune system are also altered by sepsis, they represent novel and potentially valuable therapeutic targets. Recent evidence links interleukin (IL)-17, IL-27, and IL-33 to alterations in the immune response during sepsis using patient serum and murine models of peritonitis and pneumonia. Elevated levels of IL-17 and IL-27 are found in the serum of pediatric and adult septic patients early after sepsis onset and have been proposed as diagnostic biomarkers. In contrast, IL-33 levels increase in patient serum during the immunosuppressive stage of sepsis and remain high for more than 5 months after recovery. All three cytokines contribute to immunological dysfunction during sepsis by disrupting the balance between type 1, 2, and 17 immune responses. This review will describe how IL-17, IL-27, and IL-33 exert these effects during sepsis and their potential as therapeutic targets.
Introduction to Sepsis and the IL-17/IL-27/IL-33 Axis
Although it was first described centuries ago, sepsis remains a leading cause of morbidity and mortality. While the infectious agent and the organ system(s) impacted can vary between patients, sepsis is characterized by immune dysfunction linked to alterations in systemic cytokine levels and lymphocyte apoptosis (1). The immune response during sepsis was originally thought to proceed through two distinct phases through which an initially hyper-inflammatory immune response shifted toward profound immunosuppression caused by lymphocyte impairment (2). However, this only reflects the phenotype of circulating lymphocytes in some immunocompetent patients (3–6) and does not reflect the immune response in immunocompromised patients (7). In addition, evidence now exists that both pro- and anti- inflammatory cytokines are released shortly after sepsis onset (8, 9) and continue to be released in tandem throughout the course of the illness (10–12).
While there have been many positive animal studies demonstrating the beneficial effect of targeting cytokines during sepsis, this has not translated into improvements in clinical treatment; no clinical trials so far have led to an approved therapeutic. The reasons behind this are multifactorial and partially stem from a failure to consider the interaction between individual cytokines and the larger cytokine milieu. In addition, the cytokine milieu varies between septic patients, making it difficult to distinguish any benefit in large studies of heterogenous patients. Although the failure of cytokine-based therapy in septic patients has been disappointing, recent phase I clinical trials (such as IL-7 infusion) have demonstrated the potential benefit of immunomodulation (13). However, before additional cytokines can be considered as therapeutic targets for sepsis, further work needs to be done to define the alterations that occur across the cytokine milieu during sepsis and distinguish how individual cytokines interact and modulate the effects of one another.
Recent work on the cytokines IL-17, IL-27, and IL-33 suggest the presence of a novel cytokine axis during sepsis. IL-17 primarily acts to promote the inflammatory response in mucosal tissue. In humans, serum levels of IL-17 are predictive of the development of sepsis and mortality in poly-trauma patients (14) and mutations in the IL-17A gene are associated with increased susceptibility to infection caused by gram positive bacteria and mortality (15). The immunosuppressive cytokine IL-27 increases in the plasma of many septic patients (16–27) and has been shown to inhibit the differentiation of Th17 cells (16, 18, 19, 28–33). These results have been recapitulated in diverse models of sepsis in mice (34–38). The blockade of the p28 subunit of IL-27 (38) or depletion of IL-27 using a soluble and recombinant IL-27Rα (34) significantly reduces mortality in the cecal ligation and puncture (CLP) model of sepsis and is associated with reduced bacterial burden in the tissues and blood. IL-33 is a member of the IL-1 family of cytokines that modulates Th2 responses and decreases the differentiation of T cells into Th17 cells (39). IL-33 signals through the cytokine receptor ST2 and plays an anti-inflammatory role during sepsis, improving survival during the early stages of sepsis but ultimately leading to long lasting immunosuppression through the induction of regulatory T cells (Tregs) (40–42). In addition to its interactions with IL-17, IL-33 has also been reported to interact with IL-27, with both modulating the activity of ILC2 cells (43–46). As the importance of ILC2 cells during sepsis has recently been described (47–49), these interactions may become increasingly significant for the development of effective therapeutics.
In this review, we will further discuss the individual and combined roles of IL-17, IL-27, and IL-33 during sepsis and how this axis might be therapeutically targeted.
The Role of IL-17 in Sepsis Pathophysiology
The IL-17 family of cytokines is composed of the structurally similar IL-17A-F. Apart from IL-17A (classically referred to as IL-17) and IL-17F, all the cytokines in this family are encoded separately, although they share conserved sequences. The earliest studies addressing the role of IL-17A during sepsis in animal models reported that they induced significant pathology and that eliminating IL-17A resulted in significantly improved survival (50, 51). However, subsequent studies using mice deficient in the IL-17 receptor found opposite results (52), and the literature now contains numerous studies demonstrating the mixed effects of IL-17A blockade in sepsis.
In 2012, Ogiku et al. reported that mice lacking IL-17A had significantly increased mortality following CLP that correlated with higher bacteremia at 12 h (53). Similarly, a more recent paper using the CLP model concluded that IL-17 has a partially protective role during sepsis: wild type mice had significantly increased survival and IgA production after CLP when compared to IL-17−/− mice (54). Interestingly, this study found that non-canonical signaling through NF-κB was responsible for much of the IL-17A production, as mice lacking RANKL and NF-κB inducing kinase (NIK) signaling in their intestinal epithelium cells had significantly reduced IL-17A and mortality similar to IL-17−/− mice (54).
Other studies have found that the impact of IL-17A on sepsis mortality depends on the microbe that initiated the infection. Using a bacterial pneumonia model, Ritchie et al. found that the role of IL-17A in sepsis is highly dependent on the encapsulation status of the infecting bacterium (55). IL-17A was beneficial during infections caused by minimally encapsulated bacteria, but significantly increased lung pathology and mortality if the infectious organism was heavily encapsulated (55). The authors concluded that this was due to the accumulation of neutrophils unable to phagocytose the bacteria (55). In conjunction with IL-23 signaling, IL-17A increases the recruitment of neutrophils and their accumulation in the lung following CLP, partially explaining the inflammation seen in the lung following polymicrobial sepsis originating in other tissues (56). IL-17 has also been linked to the development of acute kidney injury in septic patients and animal models (57). Given these findings, it is not surprising that multiple groups have reported that the neutralization of IL-17A or IL-17F improves survival (58, 59).
As the results of these sepsis studies conflict, it is important to note that IL-17A can induce the production of other IL-17 family cytokines, especially IL-17C (60). Although it is a distinct cytokine, it plays a similar role in neutrophil recruitment and the inflammatory process to IL-17A (60). In a mouse model of pneumonia induced by Pseudomonas aeruginosa, mice lacking IL-17C had 100% survival at 48 h, whereas wild type mice had only 25% survival at this time point (60). In contrast, another recent paper reported that IL-17C induction provides protection against LPS-induced endotoxemia (61). As IL-17C has been reported promote the production of IL-17A by Th17 lymphocytes in inflammatory conditions (such as autoimmune disease) (62), the authors concluded that these effects may be due more to the promotion of IL-17A than to IL-17C alone.
The Role of IL-27 in Sepsis Pathophysiology
Originally thought to be pro-inflammatory, there is now consensus that IL-27 is a potent immunosuppressant. It is composed of an alpha subunit (IL-27p28, also known as IL-30) and EBI3 (shared with IL-35) (63). IL-27 binds to the IL-27 receptor alpha (IL-27Rα, also known as WSX-1) and gp130 and is primarily produced by dendritic cells (DCs), monocytes and macrophages (63). The lymphocyte populations that respond to the presence of IL-27 or one of its subunits are T cells, natural killer (NK) cells, natural killer T (NKT) cells, and DCs (64–69). This allows IL-27 to have wide ranging effects on cells of both the innate and adaptive immune response in addition to autocrine effects.
In septic patients and in murine models of sepsis, the plasma concentration of IL-27 significantly increases (34, 35, 70, 71), briefly causing it to be considered as a potential diagnostic biomarker in adults (22–24) and children (20, 26). However, these results have not been consistently replicated in humans, limiting its current therapeutic potential. In mice, the results are more consistent and indicate a clear role for IL-27 in the pathology of sepsis and critical illness. When the p28 subunit is neutralized or the IL-27Rα is blocked, mortality is significantly decreased in both CLP and endotoxemia (34, 37, 38).
In a study by Cao et al., mice lacking the IL-27Rα were resistant to a secondary bacterial infection caused by Pseudomonas aeruginosa following CLP in a manner dependent on alveolar macrophages and neutrophils (37). Specifically, the neutrophils and alveolar macrophages in these mice had a significantly improved ability to kill P. aeruginosa upon phagocytosis (37). Similarly, Bosmann et al. observed that the oxidative burst of macrophages was improved upon the elimination of IL-27 signaling, and determined that IL-10 limits the production of IL-27p28 in vivo following CLP (38). In addition, this study found that the cells primarily responsible for the production of IL-27p28 in the CLP model of sepsis are splenic macrophages (38). However, a more recent study has found conflicting evidence that indicates a protective role for p28 during sepsis (72). In this study, the administration of the p28 subunit or its overproduction through genetic therapy led to a reduction in mortality during sepsis directly linked to the reduction in NKT cell production of inflammatory cytokines (72).
In addition to its modulation of innate cells, IL-27 has a significant impact on T cells. IL-27 can promote the differentiation of Th1 cells and it is also a potent inducer of type 1 Treg (Tr1) cells (73). While Tr1 cells produce IFN-γ, they also produce large quantities of IL-10 and have potent suppressive functions (74). In addition to the induction of this cell population, IL-27 signaling leads to an increase in co-inhibitory molecule expression on T cells following chronic antigen exposure and during cancer (75). As T cell dysfunction and exhaustion is associated with the development of immunosuppression during sepsis and ultimately worsened survival (76–79), IL-27 could be an effective therapeutic target. However, mice can produce IL-27p28 in the absence of EBI3, so it is unclear if the reported effects of IL-27 during sepsis are actually due to the full heterodimeric cytokine or merely to its alpha subunit. A group has recently reported the development of transgenic B57L/6J mice in which the IL-27p28 subunit cannot be produced independently of EBI3 (80). This animal model will be necessary to truly distinguish the effects of IL-27p28 from those of IL-27.
The Role of IL-33 in Sepsis Pathophysiology
ST2 was an orphan receptor until 2005, when Schmitz et al. reported their discovery of IL-33 (81). A member of the IL-1 family, IL-33 is constitutively expressed by endothelial and epithelial cells in barrier tissues and is also found at high levels under inflammatory conditions in other tissues (82, 83). When T cells, mast cells, eosinophils, and ILC2s receive IL-33 signaling, the immune response shifts toward a type 2 response (81, 84).
The first paper to describe the role of IL-33 (rather than its receptor ST2) in sepsis was published by Alves-Filho et al. (40). The authors found that survival significantly increased when IL-33 was administered to mice following CLP (40). Another 2010 study found that IL-33 is protective against LPS induced endotoxemia (85). The ability of IL-33 to improve survival during sepsis is linked to the rescue of neutrophil migration to the site of infection (40), to improvements in bacterial clearance, and to a reduction in lymphocyte apoptosis (41). IL-33 also suppresses the inflammatory response by a variety of innate lymphocytes (86) and modulates the activity of ILC2 cells (47–49). In addition to direct effects on other lymphocytes, IL-33 impacts the activity of other cytokines, including IL-17 (41, 87). While IL-33 can bind to a soluble form of ST2 (sST2), the effects of IL-33 during sepsis appear to be dependent on signaling through membrane bound ST2; in one study, patients who had did not survive sepsis had higher levels of sST2 than patients that went on to survive their infections (40).
Despite being linked to improvements in survival early after sepsis onset, IL-33 signaling may not always be beneficial. IL-33 is implicated in the induction and maintenance of immunosuppression during sepsis through the induction of Tregs (42). Nascimento et al. found that this occurs through the production of IL-4 and IL-13 by ILC2s that receive IL-33 signaling (42). The IL-4 and IL-13 then drives the proliferation of IL-10 producing macrophages and ultimately an expansion in Treg numbers (42). When they examined the blood of a small number of patients who had been diagnosed with sepsis 5–10 months prior, they found that sepsis survivors had significantly higher concentrations of both IL-10 and IL-33 and higher circulating Treg numbers compared to previously healthy patients (42). While these findings need to be replicated, they suggest that the impact of IL-33 signaling may depend on the stage of disease.
Interactions Between IL-17, IL-27, and IL-33
The ability for lymphocytes to recognize and respond to slight changes in their environment makes the immune system very adaptable and ensures that the balance between inflammatory and immunosuppressive responses is fine-tuned. While the ability of lymphocytes to respond so readily to their surroundings is beneficial from an evolutionary point of view, it makes it significantly harder to elucidate the role of individual cytokines. The individual and combined actions of the cytokines in the IL-17, IL-27, and IL-33 axis are summarized in Figure 1.
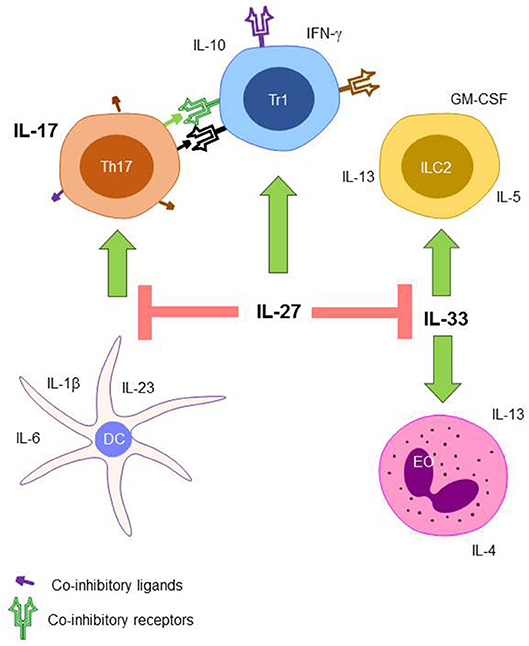
Figure 1. The proposed role of the IL-17, IL-27, and IL-33 axis during sepsis. Th17 cells that receive co-inhibitory signaling from IL-27 induced Tr1 cells have inhibited production of IL-17. Differentiation of naïve T cells into Th17 cells is also inhibited by IL-27 through the modulation of DC cytokine production. ILC2 cells and EOs expansion are also inhibited through the action of IL-27 signaling on IL-33. Th17, T helper type 17 cell; Tr1, T regulatory type 1 cell; ILC2, innate lymphocyte type 2 cell; DC, dendritic cell; EO, eosinophil.
IL-17 and IL-27
IL-17 plays a harmful role in many autoimmune diseases, particularly experimental autoimmune encephalomyelitis (EAE) and rheumatoid arthritis (RA). By limiting the differentiation of naïve CD4+ T cells into Th17 cells, IL-27 is able to attenuate these diseases (16, 19, 28, 31, 32, 88). Similarly, IL-27 signaling prevents the development of neurological damage during chronic Toxoplasma gondii infection (18) and reduces tissue damage during RSV infection (89). Further research has shown that STAT1 signaling (which IL-27 induces) inhibits the expression of the transcription factor RORγt, necessary for Th17 differentiation, while promoting the induction of the protein suppressor of cytokine signaling 1 (SOCS1) (16, 19, 30, 90). This leads to the suppression of IL-22 production by Th17 cells, impairing antimicrobial defenses in the epithelium (30, 33). In addition to its direct effects on T cells, IL-27 can also inhibit Th17 differentiation by inhibiting the production of the Th17-polarizing cytokines IL-1β, IL-6, and IL-23 by DCs (29). In contrast, T cells that have already committed to the Th17 lineage are not directly inhibited by IL-27 signaling (28, 91). Instead, inhibition occurs indirectly through the induction of Tr1 (29) and the expression of co-inhibitory receptors and their ligands (32, 88).
IL-17 and IL-33
Similar to IL-27, IL-33 has been reported to attenuate EAE through the suppression of Th17 responses (92). While IL-33 attenuates sepsis mortality, it is less clear if this is due to any effect on the Th17 response. One group reported that the administration of IL-33 actually enhanced the production of IL-17 while decreasing the levels of IL-6, IL-10, and IFNγ following CLP (41). Similarly, another group found that the deletion of the IL-33 receptor ST2 led to a reduction in the frequency and number of IL-17 producing NK cells after CLP (86). However, a recent study of human patients with Staphylococcus aureus bacteremia revealed that a higher ratio of Th17 to Th1 cytokines early after sepsis onset was associated with increased mortality (49). As there was a trend toward increased Th2 cells in surviving patients, the authors did a follow up study using a mouse model of S. aureus bacteremia (49). IL-33 provided a survival benefit in this model that was dependent on functional ILC2s and EOs, suggesting that IL-33 is protective in part because it re-balances type 1, 2, and 17 responses during sepsis (49).
IL-27 and IL-33
While IL-27 signaling promotes type 1 immune responses and directly limits type 17 immunity, it also serves as a negative regulator of type 2 responses by interfering with IL-33 signaling. The first paper to describe this phenomenon utilized in vitro experiments which showed that IL-27 reduced type 2 cytokine production in bone marrow cells exposed to IL-33, including IL-5, IL-13, and GM-CSF (43). For IL-5, this effect was dependent on STAT1 signaling, as STAT1 knockout bone marrow cells were not impacted by the presence of IL-27 (43). Moro et al. confirmed these findings in vivo using STAT1 knockout mice, and revealed that while IL-27 reduces type 2 cytokine production by ILC2 cells, it does not affect cytokine production in Th2 cells (44). Another recent study reported that the administration of IL-27 limits IL-33 induced ILC2 accumulation and activation in the lungs, liver, spleen, and mesenteric lymph node in vivo (45). The administration of IL-27 also led to the overrepresentation of IL-27Rα−/− cells in chimeric mice (45). While not specifically addressing IL-27, another murine study found that STAT1 signaling induced by infection with respiratory syncytial virus is sufficient to reduce the production of IL-33 (46). These studies collectively show that a major function of IL-27 is to negatively regulate the type 2 immune response, specifically ILC2 cells, in a manner that is dependent on STAT1 signaling.
Exploiting the IL-17, IL-27, and IL-33 Axis During Sepsis
While many reviews have discussed the therapeutic potential for targeting IL-17, IL-27, and IL-33 during sepsis (93–95), none have considered the effect that treatment targeting only one of these cytokines may have on the others. In addition, the compartmentalization of the immune response during sepsis means that cytokine therapies that restore the function of circulating lymphocytes could cause excessive stimulation and ultimately programmed cell death in the more normally responsive tissue lymphocytes. To aid the specificity of these therapies, binding should be targeted to cells expressing the circulatory chemokine receptor molecule CCR7 or the integrin CD62L (required for lymphocyte extravasation into the lymphatic system).
As IL-17 can have either beneficial or detrimental roles during sepsis depending on the murine model used, it is currently unclear what course of action would be most beneficial for human patients. However, anything that significantly increases IL-17 levels for a long period of time raises the risk of auto-immune disease formation and increased tissue damage. It seems more tenable to target IL-27 and IL-33, with an eye to keeping a balance between these cytokines and IL-17.
Neutralizing IL-27 seems likely to provide a survival benefit in septic patients if administered early after disease onset. IL-27 signaling shifts the balance too far toward a type 1 regulatory response, but its neutralization would balance type 1 and type 2 responses through the increase in activity of the type 2 promoting cytokine IL-33. IL-33 signaling has been shown to improve sepsis survival in the short term in murine models, although one report suggests that IL-33 is linked to the development of immunosuppression during sepsis (42). In this study, mice lacking the IL-33 receptor had attenuated immunosuppression associated with a reduction in type 2 cytokines, ILC2 cells, and Tregs (42). It is currently unknown how much IL-33 signaling changes during sepsis in the absence of IL-27 signaling and therefore might lower the efficacy of IL-27 blockade in improving long term survival in sepsis patients who receive no further interventions.
Ultimately, targeting any of these cytokines in an indiscriminate fashion is unlikely to be clinically beneficial. However, understanding the complex interplay between IL-17, IL-27, and IL-33—including the timing in which cytokine augmentation or blockade may potentially be beneficial—suggests this axis may potentially be manipulatable for therapeutic gain as part of a precision medicine approach toward sepsis treatment.
Author Contributions
KM wrote the initial draft of the manuscript. The final manuscript includes equal contribution from MF and CC.
Funding
This work was supported by funding from the National Institutes of Health (GM113228, GM104323, AA027396, GM072808).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gogos C, Kotsaki A, Pelekanou A, Giannikopoulos G, Vaki I, Maravitsa P, et al. Early alterations of the innate and adaptive immune statuses in sepsis according to the type of underlying infection. Crit Care. (2010) 14:R96. doi: 10.1186/cc9031
2. Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. (2001) 16:83–96. doi: 10.1097/00024382-200116020-00001
3. Cavaillon JM, Adib-Conquy M, Cloez-Tayarani I, Fitting C. Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res. (2001) 7:85–93. doi: 10.1177/09680519010070020201
4. Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH. Differential induction of apoptosis in lymphoid tissues during sepsis: variation in onset, frequency, and the nature of the mediators. Blood. (1996) 87:4261–75.
5. Cavaillon JM, Annane D. Compartmentalization of the inflammatory response in sepsis and SIRS. J Endotoxin Res. (2006) 12:151–70. doi: 10.1179/096805106X102246
6. Cavaillon JM, Adib-Conquy M. Determining the degree of immunodysregulation in sepsis. Contrib Nephrol. (2007) 156:101–11. doi: 10.1159/000102075
7. Kalil AC, Opal SM. Sepsis in the severely immunocompromised patient. Curr Infect Dis Rep. (2015) 17:487. doi: 10.1007/s11908-015-0487-4
8. Tamayo E, Fernandez A, Almansa R, Carrasco E, Heredia M, Lajo C, et al. Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw. (2011) 22:82–7. doi: 10.1684/ecn.2011.0281
9. Cazalis MA, Lepape A, Venet F, Frager F, Mougin B, Vallin H, et al. Early and dynamic changes in gene expression in septic shock patients: a genome-wide approach. Intensive Care Med Exp. (2014) 2:20. doi: 10.1186/s40635-014-0020-3
10. Frencken JF, van Vught LA, Peelen LM, Ong DSY, Klein Klouwenberg PMC, Horn J, et al. An unbalanced inflammatory cytokine response is not associated with mortality following sepsis: a prospective cohort study. Crit Care Med. (2017) 45:e493–e9. doi: 10.1097/CCM.0000000000002292
11. Osuchowski MF, Craciun F, Weixelbaumer KM, Duffy ER, Remick DG. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude, or phase of sepsis. J Immunol. (2012) 189:4648–56. doi: 10.4049/jimmunol.1201806
12. Andaluz-Ojeda D, Bobillo F, Iglesias V, Almansa R, Rico L, Gandia F, et al. A combined score of pro- and anti-inflammatory interleukins improves mortality prediction in severe sepsis. Cytokine. (2012) 57:332–6. doi: 10.1016/j.cyto.2011.12.002
13. Francois B, Jeannet R, Daix T, Walton AH, Shotwell MS, Unsinger J, et al. Interleukin-7 restores lymphocytes in septic shock: the IRIS-7 randomized clinical trial. JCI Insight. (2018) 3:98960. doi: 10.1172/jci.insight.98960
14. Ahmed Ali M, Mikhael ES, Abdelkader A, Mansour L, El Essawy R, El Sayed R, et al. Interleukin-17 as a predictor of sepsis in polytrauma patients: a prospective cohort study. Eur J Trauma Emerg Surg. (2018) 44:621–6. doi: 10.1007/s00068-017-0841-3
15. Nakada TA, Russell JA, Boyd JH, Walley KR. IL17A genetic variation is associated with altered susceptibility to Gram-positive infection and mortality of severe sepsis. Crit Care. (2011) 15:R254. doi: 10.1186/cc10515
16. Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. (2009) 182:5748–56. doi: 10.4049/jimmunol.0801162
17. Colgan J, Rothman P. All in the family: IL-27 suppression of T(H)-17 cells. Nat Immunol. (2006) 7:899–901. doi: 10.1038/ni0906-899
18. Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, et al. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. (2006) 7:937–45. doi: 10.1038/ni1376
19. Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, et al. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. (2006) 7:929–36. doi: 10.1038/ni1375
20. Wong HR, Cvijanovich NZ, Hall M, Allen GL, Thomas NJ, Freishtat RJ, et al. Interleukin-27 is a novel candidate diagnostic biomarker for bacterial infection in critically ill children. Crit Care. (2012) 16:R213. doi: 10.1186/cc11847
21. Scicluna BP, van der Poll T. Interleukin-27: a potential new sepsis biomarker exposed through genome-wide transcriptional profiling. Crit Care. (2012) 16:188. doi: 10.1186/cc11893
22. Wong HR, Lindsell CJ, Lahni P, Hart KW, Gibot S. Interleukin 27 as a sepsis diagnostic biomarker in critically ill adults. Shock. (2013) 40:382–6. doi: 10.1097/SHK.0b013e3182a67632
23. Wong HR, Liu KD, Kangelaris KN, Lahni P, Calfee CS. Performance of interleukin-27 as a sepsis diagnostic biomarker in critically ill adults. J Crit Care. (2014) 29:718–22. doi: 10.1016/j.jcrc.2014.04.004
24. Hanna WJ, Berrens Z, Langner T, Lahni P, Wong HR. Interleukin-27: a novel biomarker in predicting bacterial infection among the critically ill. Crit Care. (2015) 19:378. doi: 10.1186/s13054-015-1095-2
25. Gao F, Yang YZ, Feng XY, Fan TT, Jiang L, Guo R, et al. Interleukin-27 is elevated in sepsis-induced myocardial dysfunction and mediates inflammation. Cytokine. (2016) 88:1–11. doi: 10.1016/j.cyto.2016.08.006
26. He Y, Du WX, Jiang HY, Ai Q, Feng J, Liu Z, et al. Multiplex cytokine profiling identifies interleukin-27 as a novel biomarker for neonatal early onset sepsis. Shock. (2017) 47:140–7. doi: 10.1097/SHK.0000000000000753
27. He J, Zhang Q, Zhang W, Chen F, Zhao T, Lin Y, et al. The interleukin-27−964A>G polymorphism enhances sepsis-induced inflammatory responses and confers susceptibility to the development of sepsis. Crit Care. (2018) 22:248. doi: 10.1186/s13054-018-2180-0
28. El-behi M, Ciric B, Yu S, Zhang GX, Fitzgerald DC, Rostami A. Differential effect of IL-27 on developing versus committed Th17 cells. J Immunol. (2009) 183:4957–67. doi: 10.4049/jimmunol.0900735
29. Murugaiyan G, Mittal A, Lopez-Diego R, Maier LM, Anderson DE, Weiner HL. IL-27 is a key regulator of IL-10 and IL-17 production by human CD4+ T cells. J Immunol. (2009) 183:2435–43. doi: 10.4049/jimmunol.0900568
30. Liu H, Rohowsky-Kochan C. Interleukin-27-mediated suppression of human Th17 cells is associated with activation of STAT1 and suppressor of cytokine signaling protein 1. J Interferon Cytokine Res. (2011) 31:459–69. doi: 10.1089/jir.2010.0115
31. Fitzgerald DC, Fonseca-Kelly Z, Cullimore ML, Safabakhsh P, Saris CJ, Zhang GX, et al. Independent and interdependent immunoregulatory effects of IL-27, IFN-beta, and IL-10 in the suppression of human Th17 cells and murine experimental autoimmune encephalomyelitis. J Immunol. (2013) 190:3225–34. doi: 10.4049/jimmunol.1200141
32. Moon SJ, Park JS, Heo YJ, Kang CM, Kim EK, Lim MA, et al. In vivo action of IL-27: reciprocal regulation of Th17 and Treg cells in collagen-induced arthritis. Exp Mol Med. (2013) 45:e46. doi: 10.1038/emm.2013.89
33. Wang H, Li Z, Yang B, Yu S, Wu C. IL-27 suppresses the production of IL-22 in human CD4(+) T cells by inducing the expression of SOCS1. Immunol Lett. (2013) 152:96–103. doi: 10.1016/j.imlet.2013.05.001
34. Wirtz S, Tubbe I, Galle PR, Schild HJ, Birkenbach M, Blumberg RS, et al. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J Exp Med. (2006) 203:1875–81. doi: 10.1084/jem.20060471
35. Nelson DA, Tolbert MD, Clemens MG, Bost KL. Interleukin-27 expression following infection with the murine gammaherpesvirus 68. Cytokine. (2010) 51:184–94. doi: 10.1016/j.cyto.2010.04.015
36. Gwyer Findlay E, Villegas-Mendez A, O'Regan N, de Souza JB, Grady LM, Saris CJ, et al. IL-27 receptor signaling regulates memory CD4+ T cell populations and suppresses rapid inflammatory responses during secondary malaria infection. Infect Immun. (2014) 82:10–20. doi: 10.1128/IAI.01091-13
37. Cao J, Xu F, Lin S, Song Z, Zhang L, Luo P, et al. IL-27 controls sepsis-induced impairment of lung antibacterial host defence. Thorax. (2014) 69:926–37. doi: 10.1136/thoraxjnl-2014-205777
38. Bosmann M, Russkamp NF, Strobl B, Roewe J, Balouzian L, Pache F, et al. Interruption of macrophage-derived IL-27(p28) production by IL-10 during sepsis requires STAT3 but not SOCS3. J Immunol. (2014) 193:5668–77. doi: 10.4049/jimmunol.1302280
39. Liew FY. IL-33: a Janus cytokine. Ann Rheum Dis. (2012) 71 (Suppl. 2):i101–4. doi: 10.1136/annrheumdis-2011-200589
40. Alves-Filho JC, Sonego F, Souto FO, Freitas A, Verri WA Jr, Auxiliadora-Martins M, et al. Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat Med. (2010) 16:708–12. doi: 10.1038/nm.2156
41. Li S, Zhu FX, Zhao XJ, An YZ. The immunoprotective activity of interleukin-33 in mouse model of cecal ligation and puncture-induced sepsis. Immunol Lett. (2016) 169:1–7. doi: 10.1016/j.imlet.2015.11.009
42. Nascimento DC, Melo PH, Pineros AR, Ferreira RG, Colon DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. (2017) 8:14919. doi: 10.1038/ncomms14919
43. Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol. (2016) 17:65–75. doi: 10.1038/ni.3308
44. Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol. (2016) 17:76–86. doi: 10.1038/ni.3309
45. McHedlidze T, Kindermann M, Neves AT, Voehringer D, Neurath MF, Wirtz S. IL-27 suppresses type 2 immune responses in vivo via direct effects on group 2 innate lymphoid cells. Mucosal Immunol. (2016) 9:1384–94. doi: 10.1038/mi.2016.20
46. Stier MT, Goleniewska K, Cephus JY, Newcomb DC, Sherrill TP, Boyd KL, et al. STAT1 represses cytokine-producing group 2 and group 3 innate lymphoid cells during viral infection. J Immunol. (2017) 199:510–9. doi: 10.4049/jimmunol.1601984
47. Xu H, Xu J, Xu L, Jin S, Turnquist HR, Hoffman R, et al. Interleukin-33 contributes to ILC2 activation and early inflammation-associated lung injury during abdominal sepsis. Immunol Cell Biol. (2018) 96:935–47. doi: 10.1111/imcb.12159
48. Chun TT, Chung CS, Fallon EA, Hutchins NA, Clarke E, Rossi AL, et al. Group 2 innate lymphoid cells (ILC2s) are key mediators of the inflammatory response in polymicrobial sepsis. Am J Pathol. (2018) 188:2097–108. doi: 10.1016/j.ajpath.2018.05.009
49. Krishack PA, Louviere TJ, Decker TS, Kuzel TG, Greenberg JA, Camacho DF, et al. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight. (2019) 4:e124168. doi: 10.1172/jci.insight.124168
50. Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. (2003) 170:1958–63. doi: 10.4049/jimmunol.170.4.1958
51. Flierl MA, Rittirsch D, Gao H, Hoesel LM, Nadeau BA, Day DE, et al. Adverse functions of IL-17A in experimental sepsis. FASEB J. (2008) 22:2198–205. doi: 10.1096/fj.07-105221
52. Freitas A, Alves-Filho JC, Victoni T, Secher T, Lemos HP, Sonego F, et al. IL-17 receptor signaling is required to control polymicrobial sepsis. J Immunol. (2009) 182:7846–54. doi: 10.4049/jimmunol.0803039
53. Ogiku M, Kono H, Hara M, Tsuchiya M, Fujii H. Interleukin-17A plays a pivotal role in polymicrobial sepsis according to studies using IL-17A knockout mice. J Surg Res. (2012) 174:142–9. doi: 10.1016/j.jss.2010.11.901
54. Ramakrishnan SK, Zhang H, Ma X, Jung I, Schwartz AJ, Triner D, et al. Intestinal non-canonical NFkappaB signaling shapes the local and systemic immune response. Nat Commun. (2019) 10:660. doi: 10.1038/s41467-019-08581-8
55. Ritchie ND, Ritchie R, Bayes HK, Mitchell TJ, Evans TJ. IL-17 can be protective or deleterious in murine pneumococcal pneumonia. PLoS Pathog. (2018) 14:e1007099. doi: 10.1371/journal.ppat.1007099
56. Cauvi DM, Williams MR, Bermudez JA, Armijo G, De Maio A. Elevated expression of IL-23/IL-17 pathway-related mediators correlates with exacerbation of pulmonary inflammation during polymicrobial sepsis. Shock. (2014) 42:246–55. doi: 10.1097/SHK.0000000000000207
57. Maravitsa P, Adamopoulou M, Pistiki A, Netea MG, Louis K, Giamarellos-Bourboulis EJ. Systemic over-release of interleukin-17 in acute kidney injury after septic shock: clinical and experimental evidence. Immunol Lett. (2016) 178:68–76. doi: 10.1016/j.imlet.2016.08.002
58. Li J, Zhang Y, Lou J, Zhu J, He M, Deng X, et al. Neutralisation of peritoneal IL-17A markedly improves the prognosis of severe septic mice by decreasing neutrophil infiltration and proinflammatory cytokines. PLoS ONE. (2012) 7:e46506. doi: 10.1371/journal.pone.0046506
59. Wynn JL, Wilson CS, Hawiger J, Scumpia PO, Marshall AF, Liu JH, et al. Targeting IL-17A attenuates neonatal sepsis mortality induced by IL-18. Proc Natl Acad Sci USA. (2016) 113:E2627–35. doi: 10.1073/pnas.1515793113
60. Wolf L, Sapich S, Honecker A, Jungnickel C, Seiler F, Bischoff M, et al. IL-17A-mediated expression of epithelial IL-17C promotes inflammation during acute Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol. (2016) 311:L1015–L22. doi: 10.1152/ajplung.00158.2016
61. Yamaguchi S, Nambu A, Numata T, Yoshizaki T, Narushima S, Shimura E, et al. The roles of IL-17C in T cell-dependent and -independent inflammatory diseases. Sci Rep. (2018) 8:15750. doi: 10.1038/s41598-018-34054-x
62. Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. (2011) 35:611–21. doi: 10.1016/j.immuni.2011.09.010
63. Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. (2002) 16:779–90. doi: 10.1016/S1074-7613(02)00324-2
64. Villarino AV, Larkin J III, Saris CJ, Caton AJ, Lucas S, Wong T, et al. Positive and negative regulation of the IL-27 receptor during lymphoid cell activation. J Immunol. (2005) 174:7684–91. doi: 10.4049/jimmunol.174.12.7684
65. Matta BM, Raimondi G, Rosborough BR, Sumpter TL, Thomson AW. IL-27 production and STAT3-dependent upregulation of B7-H1 mediate immune regulatory functions of liver plasmacytoid dendritic cells. J Immunol. (2012) 188:5227–37. doi: 10.4049/jimmunol.1103382
66. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. (2013) 14:1054–63. doi: 10.1038/ni.2695
67. Sowrirajan B, Saito Y, Poudyal D, Chen Q, Sui H, DeRavin SS, et al. Interleukin-27 enhances the potential of reactive oxygen species generation from monocyte-derived macrophages and dendritic cells by induction of p47(phox). Sci Rep. (2017) 7:43441. doi: 10.1038/srep43441
68. Harker JA, Wong KA, Dallari S, Bao P, Dolgoter A, Jo Y, et al. Interleukin-27R signaling mediates early viral containment and impacts innate and adaptive immunity after chronic lymphocytic choriomeningitis virus infection. J Virol. (2018) 92:e02196-17. doi: 10.1128/JVI.02196-17
69. Wehrens EJ, Wong KA, Gupta A, Khan A, Benedict CA, Zuniga EI. IL-27 regulates the number, function and cytotoxic program of antiviral CD4 T cells and promotes cytomegalovirus persistence. PLoS ONE. (2018) 13:e0201249. doi: 10.1371/journal.pone.0201249
70. O'Dwyer MJ, Mankan AK, White M, Lawless MW, Stordeur P, O'Connell B, et al. The human response to infection is associated with distinct patterns of interleukin 23 and interleukin 27 expression. Intensive Care Med. (2008) 34:683–91. doi: 10.1007/s00134-007-0968-5
71. Rinchai D, Khaenam P, Kewcharoenwong C, Buddhisa S, Pankla R, Chaussabel D, et al. Production of interleukin-27 by human neutrophils regulates their function during bacterial infection. Eur J Immunol. (2012) 42:3280–90. doi: 10.1002/eji.201242526
72. Yan J, Mitra A, Hu J, Cutrera JJ, Xia X, Doetschman T, et al. Interleukin-30 (IL27p28) alleviates experimental sepsis by modulating cytokine profile in NKT cells. J Hepatol. (2016) 64:1128–36. doi: 10.1016/j.jhep.2015.12.020
73. Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. (2007) 8:1380–9. doi: 10.1038/ni1541
74. Battaglia M, Gregori S, Bacchetta R, Roncarolo MG. Tr1 cells: from discovery to their clinical application. Semin Immunol. (2006) 18:120–7. doi: 10.1016/j.smim.2006.01.007
75. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature. (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
76. Xie J, Crepeau RL, Chen CW, Zhang W, Otani S, Coopersmith CM, et al. Sepsis erodes CD8(+) memory T cell-protective immunity against an EBV homolog in a 2B4-dependent manner. J Leukoc Biol. (2019) 105:565–75. doi: 10.1002/JLB.4A0718-292R
77. Ammer-Herrmenau C, Kulkarni U, Andreas N, Ungelenk M, Ravens S, Hubner C, et al. Sepsis induces long-lasting impairments in CD4+ T-cell responses despite rapid numerical recovery of T-lymphocyte populations. PLoS ONE. (2019) 14:e0211716. doi: 10.1371/journal.pone.0211716
78. Thampy LK, Remy KE, Walton AH, Hong Z, Liu K, Liu R, et al. Restoration of T cell function in multi-drug resistant bacterial sepsis after interleukin-7, anti-PD-L1, and OX-40 administration. PLoS ONE. (2018) 13:e0199497. doi: 10.1371/journal.pone.0199497
79. Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-Induced T cell immunoparalysis: the ins and outs of impaired T cell immunity. J Immunol. (2018) 200:1543–53. doi: 10.4049/jimmunol.1701618
80. Muller SI, Friedl A, Aschenbrenner I, Esser-von Bieren J, Zacharias M, Devergne O, et al. A folding switch regulates interleukin 27 biogenesis and secretion of its alpha-subunit as a cytokine. Proc Natl Acad Sci USA. (2019) 116:1585–90. doi: 10.1073/pnas.1816698116
81. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
82. Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin'? PLoS ONE. (2008) 3:e3331. doi: 10.1371/journal.pone.0003331
83. Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J Immunol. (2012) 188:3488–95. doi: 10.4049/jimmunol.1101977
84. Louten J, Rankin AL, Li Y, Murphy EE, Beaumont M, Moon C, et al. Endogenous IL-33 enhances Th2 cytokine production and T-cell responses during allergic airway inflammation. Int Immunol. (2011) 23:307–15. doi: 10.1093/intimm/dxr006
85. Oboki K, Ohno T, Kajiwara N, Arae K, Morita H, Ishii A, et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA. (2010) 107:18581–6. doi: 10.1073/pnas.1003059107
86. Babic ZM, Zunic FZ, Pantic JM, Radosavljevic GD, Jovanovic IP, Arsenijevic NN, et al. IL-33 receptor (ST2) deficiency downregulates myeloid precursors, inflammatory NK and dendritic cells in early phase of sepsis. J Biomed Sci. (2018) 25:56. doi: 10.1186/s12929-018-0455-z
87. Lv R, Zhao J, Lei M, Xiao D, Yu Y, Xie J. IL-33 attenuates sepsis by inhibiting IL-17 receptor signaling through upregulation of SOCS3. Cell Physiol Biochem. (2017) 42:1961–72. doi: 10.1159/000479836
88. Hirahara K, Ghoreschi K, Yang XP, Takahashi H, Laurence A, Vahedi G, et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity. (2012) 36:1017–30. doi: 10.1016/j.immuni.2012.03.024
89. de Almeida Nagata DE, Demoor T, Ptaschinski C, Ting HA, Jang S, Reed M, et al. IL-27R-mediated regulation of IL-17 controls the development of respiratory syncytial virus-associated pathogenesis. Am J Pathol. (2014) 184:1807–18. doi: 10.1016/j.ajpath.2014.02.004
90. Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, Pot C. IL-27 induces Th17 differentiation in the absence of STAT1 signaling. J Immunol. (2015) 195:4144–53. doi: 10.4049/jimmunol.1302246
91. El-Behi M, Dai H, Magalhaes JG, Hwang D, Zhang GX, Rostami A, et al. Committed Tc17 cells are phenotypically and functionally resistant to the effects of IL-27. Eur J Immunol. (2014) 44:3003–14. doi: 10.1002/eji.201344360
92. Jiang HR, Milovanovic M, Allan D, Niedbala W, Besnard AG, Fukada SY, et al. IL-33 attenuates EAE by suppressing IL-17 and IFN-gamma production and inducing alternatively activated macrophages. Eur J Immunol. (2012) 42:1804–14. doi: 10.1002/eji.201141947
93. Neurath MF. New therapies for sepsis: focus on the interleukin (IL)12 family member IL27. Ann Rheum Dis. (2007) 66 (Suppl. 3):iii29–31. doi: 10.1136/ard.2007.078337
94. Bosmann M, Ward PA. Therapeutic potential of targeting IL-17 and IL-23 in sepsis. Clin Transl Med. (2012) 1:4. doi: 10.1186/2001-1326-1-4
Keywords: sepsis, IL-17, IL-33, critical illness, cytokine, immunological dysfunction, IL-27
Citation: Morrow KN, Coopersmith CM and Ford ML (2019) IL-17, IL-27, and IL-33: A Novel Axis Linked to Immunological Dysfunction During Sepsis. Front. Immunol. 10:1982. doi: 10.3389/fimmu.2019.01982
Received: 15 June 2019; Accepted: 05 August 2019;
Published: 22 August 2019.
Edited by:
Christoph Thiemermann, Queen Mary University of London, United KingdomReviewed by:
Sian M. Henson, Queen Mary University of London, United KingdomMarcin Filip Osuchowski, Ludwig Boltzmann Institute for Experimental and Clinical Traumatology, Austria
Copyright © 2019 Morrow, Coopersmith and Ford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mandy L. Ford, mandy.ford@emory.edu