 Christin Riess1,2,3
Christin Riess1,2,3 Björn Schneider4Hanna Kehnscherper3Julia Gesche3Nina Irmscher3Fatemeh Shokraie1Carl Friedrich Classen1Elisa Wirthgen1Grazyna Domanska5
Björn Schneider4Hanna Kehnscherper3Julia Gesche3Nina Irmscher3Fatemeh Shokraie1Carl Friedrich Classen1Elisa Wirthgen1Grazyna Domanska5 Annette Zimpfer4
Annette Zimpfer4 Daniel Strüder6Christian Junghanss3
Daniel Strüder6Christian Junghanss3 Claudia Maletzki3*
Claudia Maletzki3*- 1University Children's Hospital, Rostock University Medical Centre, Rostock, Germany
- 2Institute for Medical Microbiology, Virology, and Hygiene, Rostock University Medical Centre, Rostock, Germany
- 3Medical Clinic III - Hematology, Oncology, Palliative Care, Department of Internal Medicine, Rostock University Medical Center, Rostock, Germany
- 4Institute of Pathology, Rostock University Medical Center, University of Rostock, Rostock, Germany
- 5Institute of Immunology and Transfusion Medicine, University of Greifswald, Greifswald, Germany
- 6Department of Otorhinolaryngology, Head and Neck Surgery “Otto Koerner”, Rostock University Medical Center, Rostock, Germany
Indoleamine 2,3-dioxygenase (IDO) and tryptophan 2,3-dioxygenase (TDO2) are the key enzymes of tryptophan (TRP) metabolism in the kynurenine pathway (KP). Both enzymes function as indicators of immunosuppression and poor survival in cancer patients. Direct or indirect targeting of either of these substances seems thus reasonable to improve therapy options for patients. In this study, glioblastoma multiforme (GBM) as well as head and neck squamous cell carcinomas (HNSCC) were examined because of their different mechanisms of spontaneous and treatment-induced immune escape. Effects on gene expression and protein levels were examined. Accompanying assessment of TRP metabolites from treated GBM cell culture supernatants was conducted. Our results show a heterogeneous and inversely correlated expression profile of TRP-metabolizing genes among GBM and HNSCC cells, with low, but inducible IDO1 expression upon IFNγ treatment. TDO2 expression was higher in GBM cells, while genes encoding kynurenine aminotransferases were mainly confined to HNSCC cells. These data indicate that the KP is active in both entities, with however different enzymes involved in TRP catabolism. Upon treatment with Temozolomide, the standard of care for GBM patients, IDO1 was upregulated. Comparable, although less pronounced effects were seen in HNSCC upon Cetuximab and conventional drugs (i.e., 5-fluorouracil, Gemcitabine). Here, IDO1 and additional genes of the KP (KYAT1, KYAT2, and KMO) were induced. Vice versa, the novel yet experimental cyclin-dependent kinase inhibitor Dinaciclib suppressed KP in both entities. Our comprehensive data imply inhibition of the TRP catabolism by Dinaciclib, while conventional chemotherapeutics tend to activate this pathway. These data point to limitations of conventional therapy and highlight the potential of targeted therapies to interfere with the cells' metabolism more than anticipated.
Introduction
Tumor cells release immunosuppressive factors that shape a tolerogenic environment and enable progression and invasion. Indoleamine 2,3-dioxygenase (IDO1) is an intracellular monomeric, immune-checkpoint molecule that degrades the essential amino acid l-tryptophan along the kynurenine pathway (KP) (1, 2). Like other immune checkpoints, including programmed cell death protein 1 and cytotoxic T-lymphocyte-associated protein 4, IDO suppresses the hosts' antitumor immunity by inducing apoptosis in T- and natural killer cells (3). As a direct consequence of this, many cancer and cancer-associated cells express IDO1 (mesenchymal stromal cells, myeloid-derived suppressor cells, dendritic cells, endothelial cells, tumor-associated macrophages, and fibroblasts) (3–6). IDO1 is influenced by interferon-γ (IFNγ) (7–9), nitric oxide (10), pro- [interleukin (IL)-1β, tumor necrosis factor α] and anti-inflammatory (IL4, IL10, transforming growth factor β) cytokines. IDO1 activity inhibits T-cell activation and proliferation and even mediates regulatory T-cell recruitment to the tumor microenvironment, provoking local immune tolerance. In head and neck squamous cell carcinomas (HNSCCs), IDO1 inversely correlates with programmed cell death protein ligand 1, which constitutes an important prognostic biomarker for immune-checkpoint inhibition (11). The increased IDO1 activation decreases intratumoral TRP levels, resulting in tumor starvation and increase in kynurenine (KYN) metabolites (which are toxic to lymphocytes) (12). This immune exhaustion may be further boosted by conventional chemotherapeutics, leading to decreased efficacy. Therefore, IDO1 overexpression in the tumor microenvironment intimately impairs patients' outcome and may serve as a future prognostic predictor and drug target (13–18).
In the KP, most studies focused on IDO1 because this molecule is amenable to pharmacological intervention (19–22), and a couple of specific and global IDO inhibitors [including natural compounds (17, 23, 24)] already entered clinical trials, mostly reporting safe application and efficacy (stable disease at best outcome) (25). Current trials are evaluating the efficacy of IDO1 inhibitors in combination with chemotherapy, radiotherapy, and other immunotherapies including cytotoxic T-lymphocyte-associated protein 4 blockade (11, 22). The latter is based on the observation of an enhanced lytic ability of tumor-antigen-specific T cells upon IDO1 inhibition and decreased numbers of local immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells (20, 26). The efficacy and toxicity data from recent clinical trials with IDO1 inhibitors is reviewed in Yentz and Smith (27). In most cases, however, overall survival was not significantly improved, leaving the future role for this combination therapy in question (28). More key enzymes are involved in TRP metabolism: tryptophan 2.3-dioxygenase (TDO2), a member of the oxidoreductases family, catalyzes the same initial step of the KP as IDO1 (2). Thus, TDO2 has been shown to be constitutively and highly expressed in various cancer cells such as malignant glioma and HNSCC (29, 30). More importantly, TDO2 also has immunomodulatory functions by promoting immune tolerance. This, in turn, promotes survival, growth, invasion, and metastasis and decreases patients' survival (just like IDO1) (13, 22, 31, 32).
In this study, we performed a comprehensive analysis on the expression status of genes belonging to the KP. HNSCC and glioblastoma multiforme (GBM) were picked as prime examples for different spontaneous and treatment-induced immune escape mechanisms. Therefore, expression changes were determined under standard and targeted therapy, and results were compared among each other.
Materials and Methods
Tumor Cell Lines and Culture Conditions
Patient-derived GBM cell lines (N = 13; HROG02, HROG04, HROG05, HROG06, HROG10, HROG15, HROG24, HROG36, HROG38, HROG52, HROG63, HROG73, HROG75) and HNSCC cell lines (N = 6; FADU, Detroit-562, Cal-33, PE/CA/PJ-15, UT-SCC-14, UT-SCC-15) were either established and basically characterized in our lab or originally obtained from the German collection of cell cultures (DSMZ; Braunschweig, Germany). UT-SCC14 and UT-SCC15 cells were kindly provided by Prof. R. Grenman [University of Turku, Finland (33)]. All cells were routinely cultured in our lab and maintained in full medium: Dulbecco's modified Eagle Medium/HamsF12 supplemented with 10% fetal calf serum, glutamine (2 mmol/L), and antibiotics (medium and supplements were purchased from PAA, Cölbe, Germany). For functional analysis, cell lines from each tumor entity were chosen, and all subsequent experiments were performed with these lines only.
IFNγ Stimulation
Cells were cultured in six-well plates or ibidi chamber slides, incubated overnight and treated with IFNγ (50 ng/ml, Immunotools, Friesoythe, Germany) for 24 and 72 h, respectively. Thereafter, cells were harvested and further processed.
Cytostatic Drugs and Targeted Substance
Cytostatics used in this study included 5-fluorouracil (5-FU) (2.5 μM), Cisplatin (0.2 μM), Gemcitabine (0.0002 μM), and Cetuximab (0.34 μM) for HNSCC, as well as Temozolomide (10 μM, TMZ) for GBM (pharmacy of the University Hospital Rostock). CDKi Dinaciclib (10 or 100 nM) was used as experimental targeted drug. All substances were used in doses below the IC50 as determined before.
Apoptosis/Necrosis Assay
A Yo-Pro-1/PI-based assay for discriminating early apoptotic, late apoptotic, and necrotic cells was applied as described before (34).
Hemolysis Assay
Hemolytic activity of Dinaciclib was determined by hemoglobin release from whole blood cells after 2 h of incubation. Briefly, whole blood of healthy donors (N = 5) was seeded in 96-well plates and treated with increasing Dinaciclib doses (ranging from 1, 5, and 10 μM). Negative controls were left untreated, and positive controls (=maximum lysis) were treated with 1% sodium dodecyl sulfate. Following the incubation period, cell-free supernatants were transferred into a new 96-well plate, and absorption was measured on a plate reader at 560 nm (reference wave length, 750 nm). Hemolytic activity was quantified according to the following formula and corrected for spontaneous hemolysis (=untreated controls):
In addition, peripheral blood mononuclear cells' (PBMC) viability (N = 5) were determined by Calcein AM staining. This was done upon 24 h incubation at the above-mentioned doses. Fluorescence measurement and quantification were done as described (34).
IDO1 Immunofluorescence
Tumor cells were treated with 50 ng/ml of IFNγ (Immunotools), TMZ, Cetuximab, or Dinaciclib for 24 h in chamber slides, respectively. Cells were washed with phosphate-buffered saline, fixed in 4% paraformaldehyde w/o methanol (Thermo Scientific, Darmstadt, Germany) for 20 min, washed again, followed by cell permeabilization in 0.3% Triton X−100/5% normal bovine serum in phosphate-buffered saline for 60 min. Cells were then incubated overnight at 4°C in monoclonal rabbit IDO1 primary antibody (1:100; Cell Signaling Technology, Frankfurt/Main, Germany). Cells were washed, labeled with fluorochrome-conjugated secondary antibody using goat antirabbit secondary antibody (1:250, Boster Biological Technology, Pleasanton CA, USA), and incubated in the dark for 2 h. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and cells were analyzed with a Zeiss LSM-780 Confocal Laser Microscope (Zeiss, Jena, Germany). Quantification of staining intensity was done using the ImageJ software. Therefore, channels were split into red, green, and blue. Subsequently, integrated density profiles of the same size were measured in the green channel.
IDO1 Immunohistochemistry on Patients' Tumor Samples
Primary antibody against IDO1 (rabbit IgG, clone D5J4E, Cell Signaling Technology, dilution 1:200) was used. All samples were pretreated for 20 min at 97°C and pH 6.9. Standard immunoperoxidase technique was applied using an automated immunostainer (DAKO link) with diaminobenzidine as chromogen. IDO1 expression was defined as cytoplasmatic and membranous staining in >1% inflammatory cells.
Quantification of Tryptophan, Kynurenine, and Kynurenic Acid in Cell Culture Supernatant by Liquid Chromatography Tandem Mass Spectrometry System
The basis for the measurement was the method of Fuertig et al. which was adapted to the system used here (35).
Sample Preparation
Cell culture supernatant was mixed 1:1 with internal standards [10 μM D5-kynurenic acid (Buchem BV, Apeldoorn, Netherlands), 10 μM D5-phenylalanine (Cambridge Isotope Laboratories, Inc. Andover, MA, United States), 5 μM D4-kynurenine (Cambridge Isotope Laboratories), 10 μM D5-tryptophan (Sigma Aldrich, Hamburg, Germany), 10 μM D3-quinolinic acid (Buchem BV), 5.5 nM 15N5-8-hydroxy-2-deoxyguanosine (Cambridge Isotope Laboratories)], and with 10 μl of mobile phase (0.4% formic acid, 1% acetonitrile in water). Reagents were gently shaken on a mixer, and 150 μl of ice-cold methanol was added. Samples were incubated overnight at −20°C to allow protein precipitation. On the following day, samples were centrifugated at 0°C and 18,000×g for 15 min. Supernatants were transferred to a new tube, and the liquid phase was removed by evaporation at 30°C among vacuum. Solid samples were stored until measurement at −20°C. Afterwards, dried extracts were reconstituted in 100 μl of acidified mobile phase. Samples were incubated at 40°C (1 h), centrifuged (4°C, 18,000×g, 5 min), and clear supernatant (100 μl) was transferred onto a 96-well plate.
Liquid Chromatography Tandem Mass Spectrometry
Measurements were performed on an AB Sciex 5500 QTrap™ mass spectrometer (AB SCIEX, Darmstadt, Germany) with electrospray ionization in positive mode combined with a high-performance liquid chromatography system (Agilent 1260 Infinity Binary LC, Santa Clara, United States) including a degasser unit, column oven, autosampler, and a binary pump. Twenty microliters of the supernatant was injected and separated using a VisionHT C18 column (100 × 2.1 mm; particle size, 3 μm; Grace, MD, United States). To prevent contamination, a precolumn (VisionHT C18, Guard 5 × 2 mm) was used additionally. The temperature of the column oven was set at 15°C. The flowrate was set to 0.4 ml/min, and the sample was separated in a total run time of 11 min using solution A (water + 0.1% formic acid + 0.01% trifluoroacetic acid) and solution B (MeOH + 0.1% formic acid + 0.01% trifluoroacetic acid) with the following gradient: 0–2.8 min, 97% A, 3% B; 2.8–3.3 min, 70% A, 30% B; 3.3–4.4, 40% A, 60% B; 4.5–5.0 min, 40% A, 60% B; 5.0–5.5, 5% A, 95% B; 5.5–6.9 min, 5% A, 95% B; 6.9–7.0 min, 97% A, 3% B; 7.0–11.0 min, 97% A, 3% B.
The eluate between 0.5 and 9 min was introduced into the mass spectrometer and analyzed in MRM mode. The ion spray voltage (IS) was 4,000 V, the curtain gas flow was 40.0 psi, and the ion source temperature were set at 550°C.
Internal standards were used for metabolite quantification (Table 1). Data analysis, including peak integration and concentration determination, was performed with Analyst software (Version 1.5.1, AB Sciex, Darmstadt, Germany).
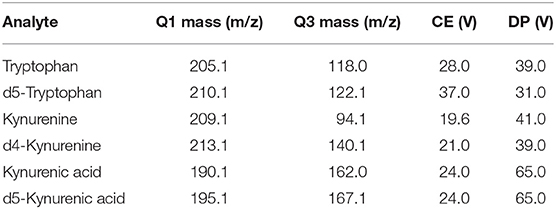
Table 1. Internal standards.
RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR
Total RNA was isolated with RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturers' instructions. RNA was reverse transcribed into complementary DNA (cDNA) from 1 μg RNA using 1 μl dNTP mix (10 mM), oligo (dT)15 primer (50 ng/μl), 1 μl reverse transcriptase (100 U), and 4 μl 5× reverse transcription buffer complete (all purchased from Bioron GmbH, Ludwigshafen, Germany). Final reaction volume was 20 μl (filled with RNAse free water). cDNA synthesis conditions were as follows: 70°C for 10 min, 45°C for 120 min, and 70°C for 10 min. Target cDNA levels of human cell lines were analyzed by quantitative real-time PCR using TaqMan Universal PCR Master Mix and self-designed TaqMan gene expression assays either labeled with 6-FAM-3′ BHQ-1 or 5′ HEX−3′ BHQ-1 to be used as duplex: IDO1, TDO2, KMO, HAAO, KYAT1/2/3/4, KYNU, QPRT, and GAPDH or ß-actin were used as housekeeping genes. Reaction was performed in the light cycler Viia7 (Applied Biosystems, Foster City, USA) with the following PCR conditions: 95°C for 10 min, 40 cycles of 15 s at 95°C, and 1 min at 60°C. All reactions were run in triplicates. The messenger RNA (mRNA) levels of target genes were normalized to GAPDH/β-actin. Reactions were performed in triplicate wells and repeated four times. The general expression level of each sample was considered by calculating 2−ΔCT (ΔCt = Cttarget – CtHousekeeping genes).
Statistical Analysis
All values are reported as mean ± SD. After proving the assumption of normality, differences between controls and treated cells were determined using the unpaired Student's t-test. If normality failed, the non-parametric Mann-Whitney U-test was applied. Statistical evaluation was performed using GraphPad PRISM software, version 5.02 (GraphPad Software, San Diego, CA, USA). In case of multiple comparisons, two- or one-way ANOVA on ranks (Bonferroni's multiple comparison test) was used. The criterion for significance was taken to be p < 0.05.
Results
Basal IDO1 and Related Genes in GBM and HNSCC Cell Lines
While IDO1 itself is not the only mechanism by which tumors can resist immune-mediated killing, we studied the expression status of different KP-related genes on a panel of human GBM and HNSCC cell lines. These experiments revealed not only differences between both entities but also a heterogeneous profile of all tested genes among cell lines (Figure 1). IDO1 was differently expressed by most glioma samples (11/13) analyzed. In general, IDO1 was only detectable at very low levels (Figure 1). TDO2, the other rate-limiting enzyme of the KP (36), was constitutively expressed by all glioma samples, and expression was even higher in comparison to IDO1. Generally, expression status for TDO2 and kynurenine hydrolase (KYNU) was higher in GBM, while HNSCC expressed more kynurenine aminotransferases (KYAT) (Figure 1). Hence, these data indicate that the KP is active in both entities, with however different enzymes being involved in TRP catabolism.
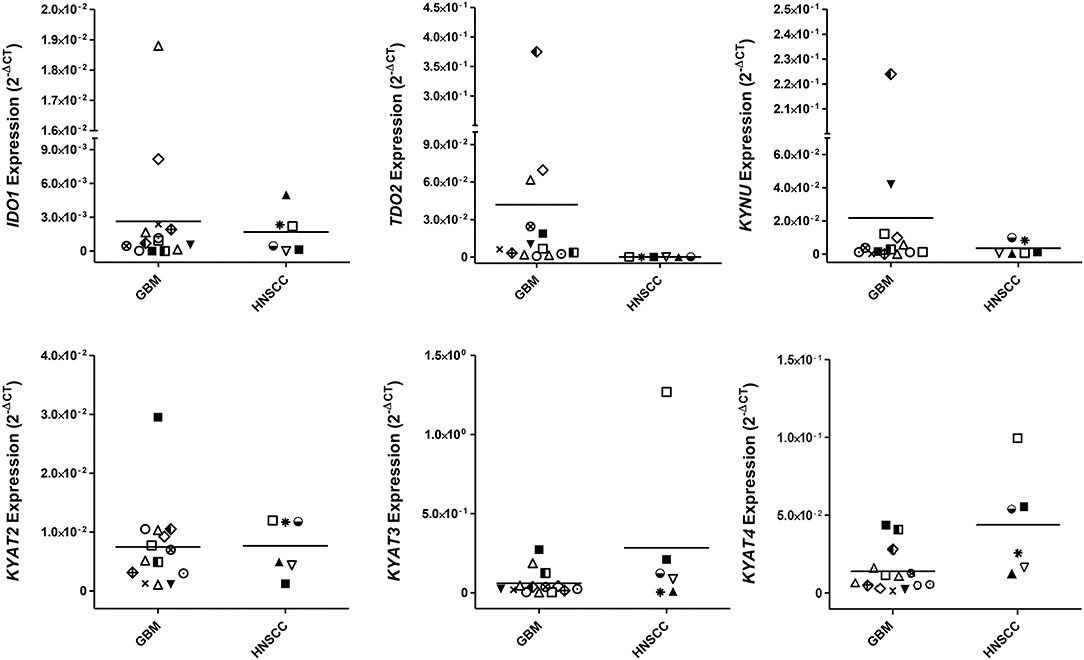
Figure 1. Relative messenger RNA (mRNA) expression of IDO1, TDO2, KYNU, KYAT2, KYAT3, and KYAT4 in glioblastoma multiforme (GBM) and head and neck squamous cell carcinoma cells (HNSCC). The graphs indicate the mRNA expression normalized to the housekeeping genes (2−ΔCT). GBM (N = 13, HROG02, HROG04, HROG05, HROG06, HROG10, HROG15, HROG24, HROG36, HROG38, HROG52, HROG63, HROG73, HROG75) and HNSCC cell lines [N = 6; FADU, Detroit-562, Cal-33, PE/CA/PJ-15, UT-SCC14, UT-SCC-15 (33)].
Still, tumor cell lines grown in vitro not necessarily represent the in vivo situation; we therefore analyzed the IDO1 abundance in clinical resection specimens (Figure 2). In GBM, IDO1 was detectable in one of three cases (representative images are shown in Figure 2). By contrast, HNSCC samples presented with IDO1 but only on a small fraction of tumor-infiltrating lymphocytes (Figure 2). Although not analyzed systematically, the only HPVpositive case in this small cohort showed highest IDO1 abundance, nicely reflecting the tumors' immunogenicity (11, 37).
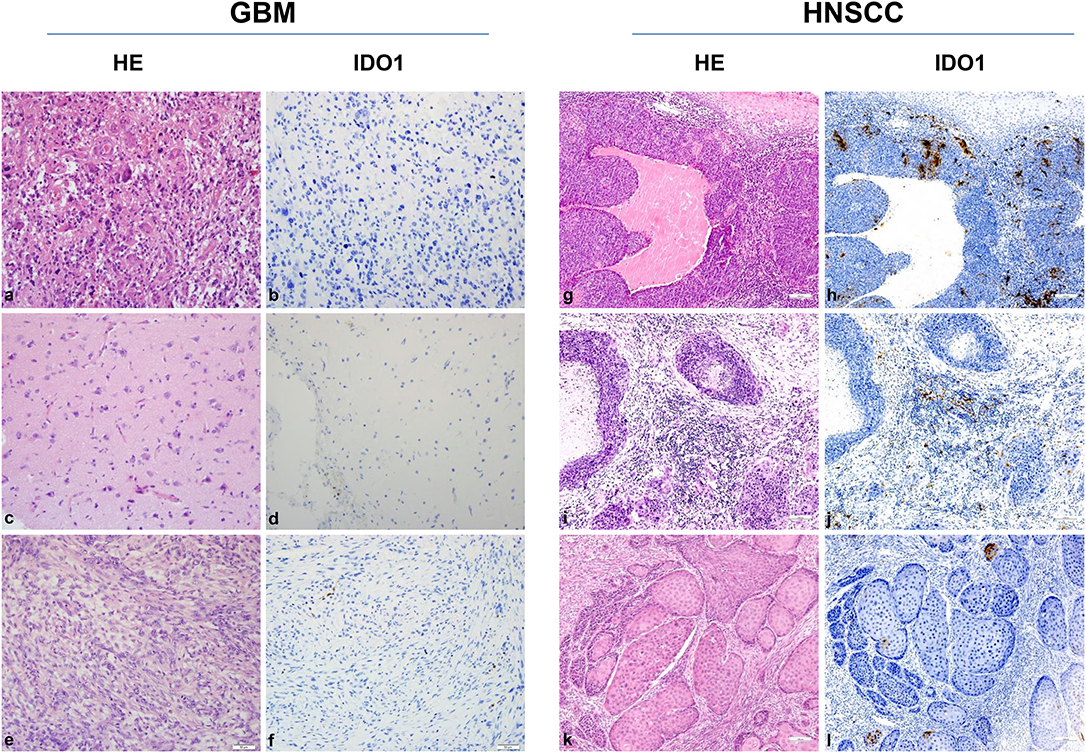
Figure 2. IDO1 immunohistochemistry. Representative images of primary glioblastoma multiforme (GBM) [HROG36 (a,b), HROG63 (c,d), and HROG73 (e,f)] and head and neck squamous cell carcinoma (HNSCC) [HNSCC06 (g,h), HNSCC02 (i,j), HNSCC01 (k,l)] samples. Left panel: Routine HE staining. Right panel: Note the focal IDO1 expression on tumor-infiltrating lymphocytes exclusively in HNSCC cases. HNSCC case 1 (g,h): tonsil (HPVpositive); case 2 (i,j): mouth base (HPVnegative, relapse); case 3 (k,l): larynx (HPVnegative). Pictures were taken at 20× and 10× magnification, respectively.
Gene Expression and Protein Changes Upon IFNγ Stimulation
IDO1 is an IFNγ-inducible enzyme. Upon stimulation, the KP is activated to induce immunosuppression. In vitro stimulation with IFNγ mimics the in vivo situation of an inflammatory microenvironment. Hence, upon immune-mediated inflammation, IDO1-negative tumor cells may upregulate IDO1 as resistance mechanism.
Using five individual GBM cell lines, IDO1 expression was inducible in all cases (Figure 3A). Upregulation of IDO1 was high on protein levels in HROG05 cells and marginal in HROG63 (Figure 3B). TDO2 and KYAT3 were suppressed upon IFNγ stimulation in three of five samples and hardly detectable in one cell line, supporting data from a recent publication (38). KYNU was not affected by IFNγ stimulation (Figure 3A).
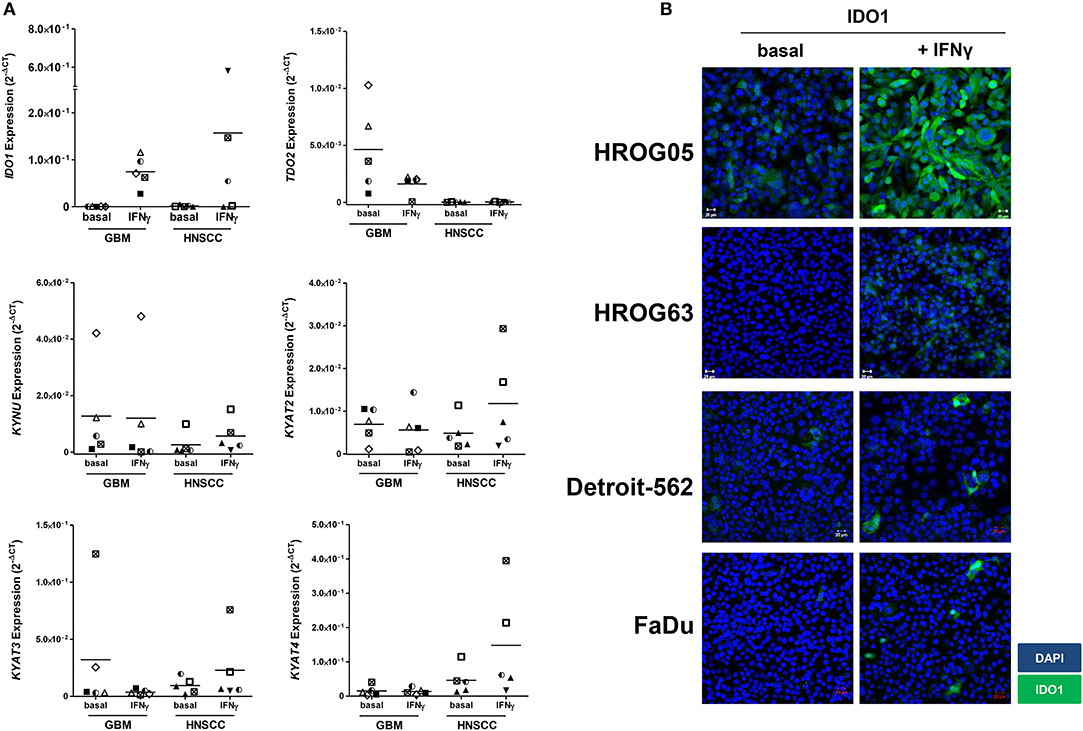
Figure 3. Relative messenger RNA (mRNA) expression of IDO1, TDO2, KYNU, KYAT2, KYAT3, and KYAT4 as well as IDO1 protein abundance following interferon-γ (IFNγ) stimulation in glioblastoma multiforme (GBM) and head and neck squamous cell carcinoma (HNSCC) cells. The cell cultures were either untreated or treated with IFNγ (50 ng/ml) for 24 h. (A) The graphs indicate the mRNA expression normalized to the housekeeping genes (2−ΔCT). GBM N = 5 cell lines (HROG02, HROG05, HROG52, HROG63, HROG75); HNSCC, N = 4 cell lines (FADU, Detroit-562, Cal-33, PE/CA/PJ-15). (B) IDO1 immunofluorescence in selected cell lines. Cell nuclei were stained with DAPI, and IDO1 was depicted by monoclonal rabbit IDO1 primary antibody (1:100; Cell Signaling Technology), followed by secondary antibody (1:250, Boster Biological Technology Pleasanton, CA, United States) labeling. Cells were analyzed with a Zeiss LSM-780 Confocal Laser Microscope. Original magnification 20×.
Just as in GBM, IDO1 was inducible in HNSCC cells (Figure 3A). Immunofluorescence revealed focal expression of singular cells with different intensity (Figure 3B). Of note, IFNγ stimulation even induced upregulation of KYAT1, KYAT2, KYAT3, and KYAT4 (Figure 3A and data not shown), most likely constituting a compensatory mechanism as described before in experimental autochthonous tumor models (39).
Interference With the KP of Cytostatic and Targeted Therapies
Next, we examined whether cytostatic and targeted drugs have an influence on the KP. For GBM, TMZ was chosen, and for HNSCC, 5-FU, Cisplatin, Gemcitabine, as well as Cetuximab were used. As a targeted yet still experimental agent, the potent and specific CDKi Dinaciclib was applied to cells of both entities.
Before this experiment, drug doses were carefully tested in dose-response analyses (data not shown) along with discrimination of apoptosis and necrosis. Generally, drugs used in this study tended to induce necrosis, while apoptosis, if present, was only detectable at early time points. Exemplary results for the HNSCC cell line Detroit-562 are given in Supplementary Figures 1A,B. While cytostatics are well-known to affect normal cells' viability, the impact of the CDKi Dinaciclib on immune and red blood cells is less clear. We therefore performed a hemolysis and leukocyte viability assay. In this experiment, no toxicity was seen against normal cells (Supplementary Figure 1C). Even at high concentrations, Dinaciclib impaired cellular viability/integrity only marginally (Supplementary Figure 1C).
TMZ is an oral alkylating agent that methylates DNA at the O6 position of guanine causing cell cycle arrest at G2/M. It is used as standard of care for GBM. However, acquired resistance, a process not fully understood, leads to major limitations in treatment. Here, TMZ downregulated IDO1 in three of five GBM cell lines but led to increased expression in HROG52 and HROG63—a paired GBM cell line established from the very same patient (primary lesion and upon relapse) (Figure 4). Gene expression of KYAT2, KYAT4, and KMO was heterogeneous. Generally, there was a trend toward higher expression of those genes but with cell-line-specific differences (e.g., KYAT3: p < 0.05 vs. control in HROG05 cells; Figure 4). KYNU expression was not affected by TMZ (data not shown). Interestingly, the combination of IFNγ and TMZ that mimics the in vivo situation led to similar or even stronger IDO1 upregulation compared to IFNγ alone in two out of four glioma samples (Figure 5). Adding Dinaciclib to either IFNγ or TMZ lowered the mRNA expression of IDO1 massively. Other KP-related genes like TDO2 and KYAT1-4 were similarly downregulated (Figure 5). Supplementary Table for Figure 5 provides a detailed statistical analysis of each cell line in relation to the individual treatment regimens.
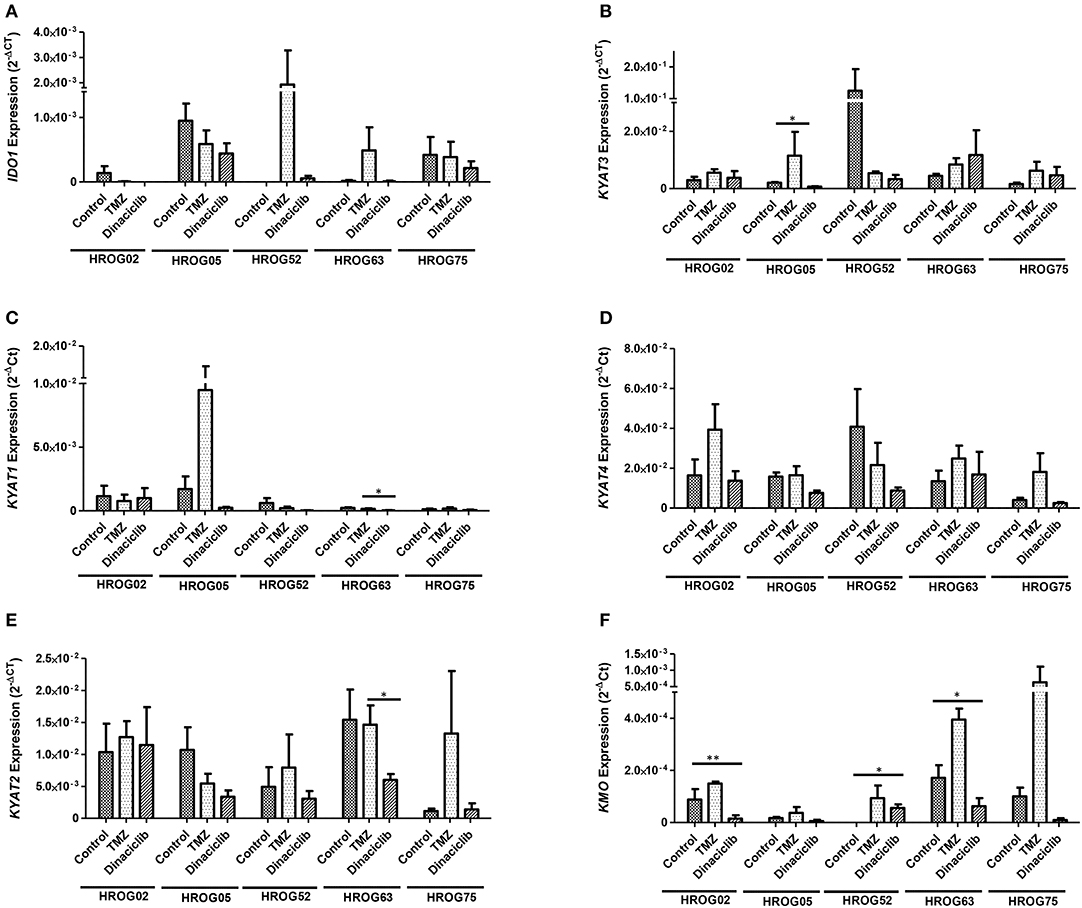
Figure 4. Relative messenger RNA (mRNA) expression of IDO1, KYAT1, KYAT2, KYAT3, KYAT4, and KMO upon cytostatic drugs and targeted therapy in glioblastoma multiforme (GBM) cells. Graphs indicate the mRNA expression of selected kynurenine pathway (KP)-related genes normalized to the housekeeping genes (2−ΔCT). Results show data of three independent experiments. *p < 0.05; **p < 0.01 vs. control. Two-way ANOVA.
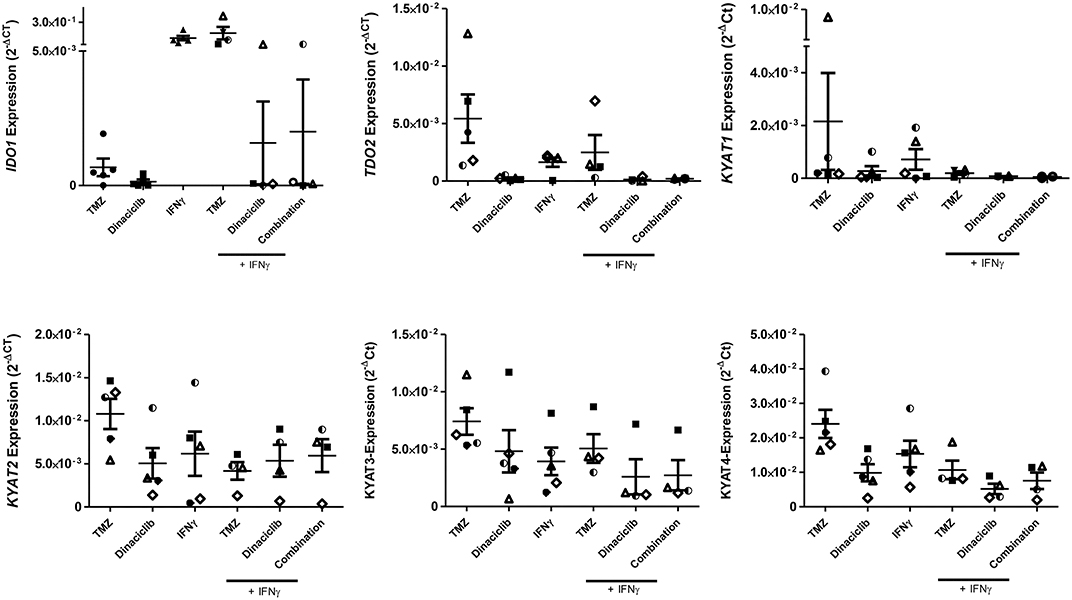
Figure 5. Relative messenger RNA (mRNA) expression of IDO1, TDO2, KYAT1, KYAT2, KYAT3, and KYAT4 upon concomitant treatment with interferon-γ (IFNγ) and cytostatic drugs in glioblastoma multiforme (GBM) cells. The cell cultures (N = 5, HROG02, HROG05, HROG52, HROG63, HROG75) were either left untreated or treated with IFNγ (50 ng/ml) for 24 h. Treatments were performed simultaneously, i.e., IFNγ ± TMZ, and/or Dinaciclib. Graphs indicate the mRNA expression of selected KP-related genes normalized to the housekeeping genes (2−ΔCT). Results show data of three independent experiments. A statistical report is given in Supplementary Table for Figure 5.
In HNSCC cells, Cetuximab was the only IDO1-inducing substance (exemplary results for Detroit-562 cells are given in Figure 6). Beyond that, the cytostatics as well as Cetuximab induced at least one of the KP-related genes (p < 0.05 vs. control), implicating activation of this pathway via different effectors. By adding Dinaciclib to cytostatic drugs, this effect was abrogated, even in the presence of IFNγ (Figure 6 and data not shown). Of note, Dinaciclib alone as well as in combination with other substances effectively suppressed all KP-related genes, implying inhibition of the TRP catabolism by this CDKi.
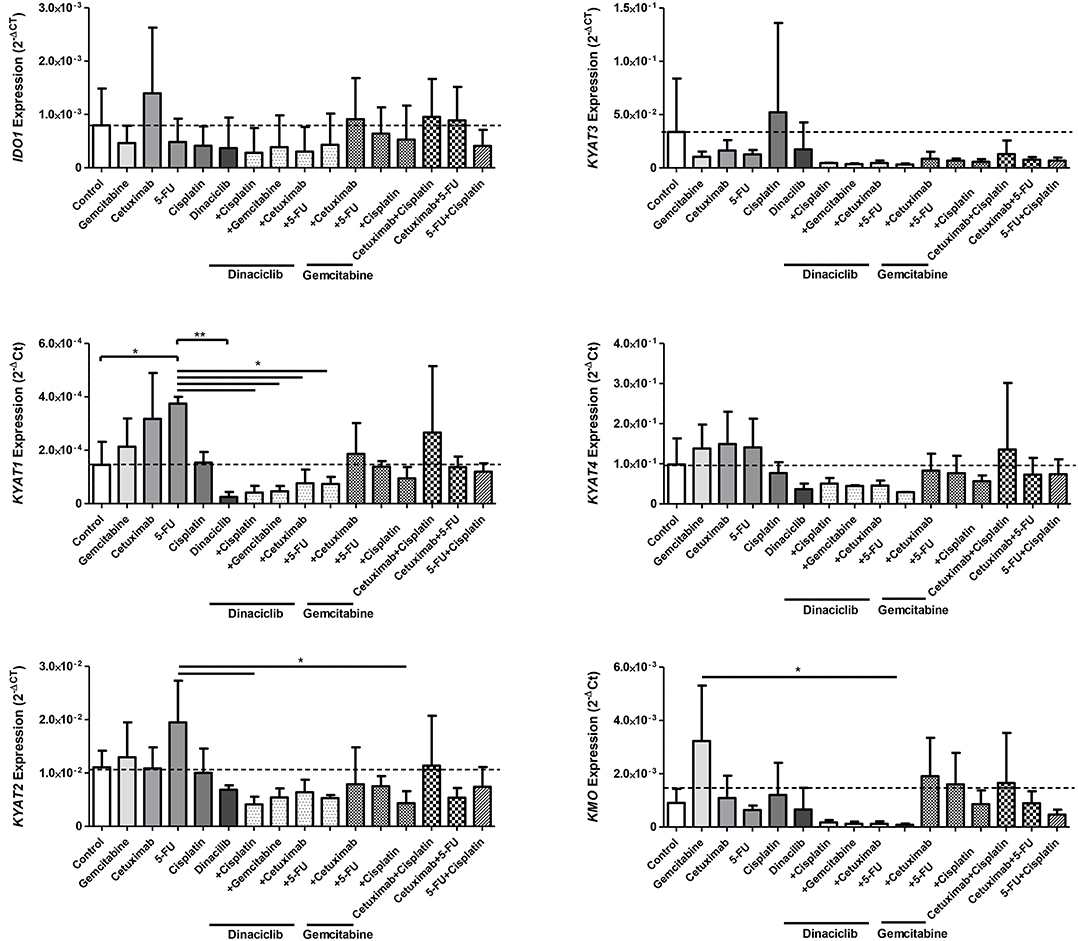
Figure 6. Relative messenger RNA (mRNA) expression of IDO1, KYAT1, KYAT2, KYAT3, KYAT4, and KMO upon cytostatic drugs and targeted therapy in head and neck squamous cell carcinoma (HNSCC) cells. Graphs indicate the mRNA expression of selected KP-related genes normalized to the housekeeping genes (2−ΔCT). Results show data of three independent experiments using HNSCC cell line Detroit-562. *p < 0.05; **p < 0.01. One-way ANOVA (Bonferroni's multiple comparison test).
Dinaciclib Blocks IFNγ-Induced IDO1 Expression in GBM and HNSCC Cells
Considering the active downregulation of KP-related genes by Dinaciclib, we investigated whether this CDKi is able to inhibit or reverse IFNγ-induced IDO1 upregulation in GBM and HNSCC cells on a protein level. TMZ and Cetuximab were included as active inductors of IDO1 and associated KP-related genes.
IFNγ and selected drugs were added simultaneously for 72 h. Dinaciclib effectively blocked IFNγ-induced IDO1 protein in both entities, while TMZ alone as well as the combination with IFNγ strongly enhanced IDO1 protein level (Figure 7). Hence, mRNA expression data were nicely confirmed.
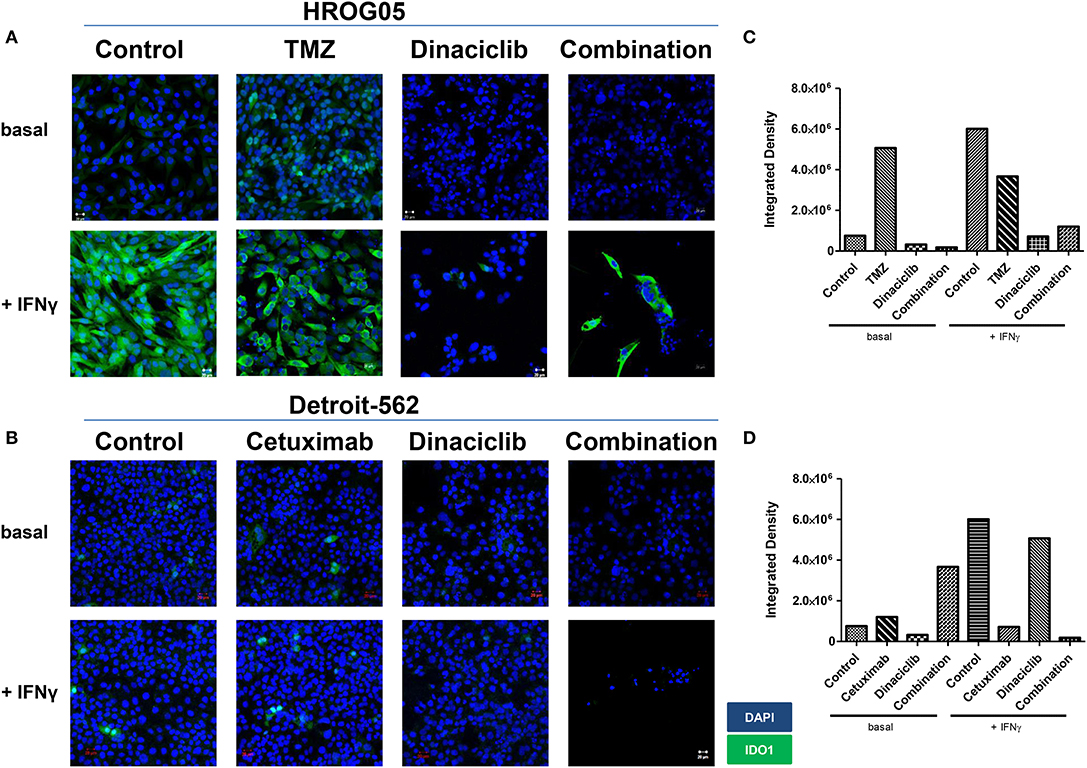
Figure 7. Indoleamine 2,3-dioxygenase (IDO1) protein abundance in selected glioblastoma multiforme (GBM) and head and neck squamous cell carcinoma (HNSCC) cells upon cytostatic drugs and targeted therapy. The cell cultures [(A) GBM: HROG05; (B) HNSCC: Detroit-562] were either left untreated or treated with IFNγ (50 ng/ml) for 24 h. Treatments were performed simultaneously, i.e., IFNγ ± TMZ, Cetuximab, and/or Dinaciclib. Cell nuclei were stained with DAPI. Original magnification 20×. (C,D) Quantification was done to score staining intensity in untreated and treated HROG05 and Detroit-562 cells. This was carried out using ImageJ software as described in Material and Methods.
When Dinaciclib was combined with IFNγ and TMZ, the IDO1-inducing stimulus of these latter substances was far too strong to be suppressed (Figure 7). However, the low number of residual cells in this combination hints toward additive or even synergistic effects independent from IDO1 (Figures 7A,B).
While IDO1 was highly inducible in GBM cells only, we then determined protein level upon IFNγ-prestimulation approaching the in vivo situation. The cytotoxic effect of Dinaciclib was preserved; however, levels of IDO1 enzyme were not significantly altered (Supplementary Figures 2A,B). Comparable results were obtained for TMZ. Virtually, all residual cells showed positive staining; still there was a trend toward lower intensity in monotherapy and in combination (Supplementary Figure 2B).
Taken together, the CDKi Dinaciclib is able to block IFNγ-mediated and thus most likely even chemotherapy-induced IDO1 upregulation in GBM and HNSCC cells. However, blunt interference with this TRP-metabolizing enzyme is unlikely.
Treatment Induced Influence on KP-Related Metabolites
Our data revealed IDO1 induction by TMZ, which is reversible by Dinaciclib. Thus, we examined the influence on KP-related metabolites in GBM cell lines.
TRP, KYN, and the downstream metabolite kynurenic acid (KYNA) were quantified by MS using cell culture supernatants of GBM cell lines (Figures 8A,B). TRP was catabolized after 24 h from all cell lines among all treatment regimens. Adding TMZ or Dinaciclib in monotherapy marginally affected TRP consumption as well as KYN and KYNA production. Stimulation with either IFNγ or a combination of TMZ resulted in greatly enhanced TRP depletion and increased KYN levels, although to varying degrees in the different cell lines (Figure 8A). Small amounts of KYNA were produced constitutively and to a greater extent after IFNγ mono- and TMZ combination in all cell lines (Figure 8A). In contrast, KYNA level remained unchanged upon Dinaciclib in combination with IFNγ, confirming immunofluorescence results (please see Figure 7 for details). The same was true for the KYN/TRP ratio, being only affected in samples treated with IFNγ as well as the combination of IFNγ and TMZ (Figure 8B).
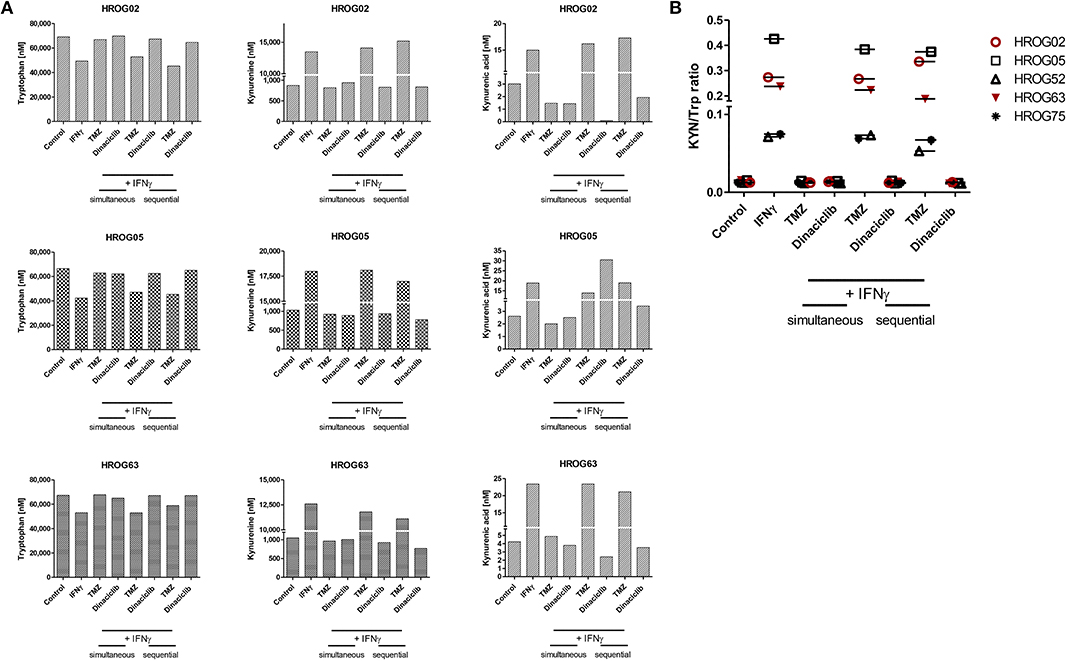
Figure 8. Indoleamine 2,3-dioxygenase (IDO) protein abundance in glioblastoma multiforme (GBM) and head and neck squamous cell carcinoma (HNSCC) cells as well as kynurenine pathway (KP) metabolite levels in GBM cells upon cytostatic drugs and targeted therapy. Treatments were performed sequentially, i.e., interferon-γ (IFNγ) pretreatment for IDO1 induction, followed by Temozolomide (TMZ) and/or Dinaciclib. (A) KP metabolites changed upon TMZ but not Dinaciclib treatment. The combination of TMZ and IFNγ accelerates tryptophan (TRP) consumption accompanied by kynurenine (KYN) and kynurenic acid (KYNA) production (B). KYN/TRP ratios in GBM cells were determined dividing KYN values by TRP values. Results show data of a single measurement.
These data underline our gene and protein expression data. The CDKi Dinaciclib is directly or indirectly capable of blocking the KP. TMZ particularly in combination with the proinflammatory cytokine IFNγ accelerates TRP consumption accompanied by KYN and KYNA production in GBM cells.
Discussion
The finding that high IDO1 expression is associated with shorter survival in cancer patients made IDO1 a promising target either by specific inhibitors or indirectly by immunomodulation. A recent study described dramatically suppressed tumor growth upon IDO1 knockdown by increasing the number of CD4+ and CD8+ T cells in murine GBM models (9). However, the exact mechanisms underlying IDO1 and thus TRP metabolism along the KP remain unclear. Therefore, we focused on the expression of IDO1 and IDO-related KP genes and their potential involvement in immune evasion in experimental models of HNSCC and GBM.
We were able to show that the KP is active in both entities, with different enzymes involved in TRP catabolism. Of note, basal IDO1 expression was low and inversely correlated with TDO2. In the only prior study on primary GBM cultures, similar results were described with constitutive TDO2 expression in most GBM cell cultures (29). In here, TDO2 likely promotes tumor growth by suppressing antitumor immune responses (2, 31). KP products are considered as therapeutic targets because IDO1 and other genes of the TRP metabolism are not expressed in healthy brain tissue, but gradually increase with GBM dedifferentiation (low vs. high grade GBM). In HNSCC, different results on IDO1 are documented, and expression is heterogeneous among different HNSCC cell lines. Of note, IDO1 abundance of primary resection specimen and cultured cells seems to be independent from anatomical site and HPV status (40). Still, IDO1 is a useful marker for progression of in oral squamous cell carcinoma (41). In esophageal squamous cell carcinoma, progression and metastasis correlates with strong inflammation at the tumors' invasive front and disturbed TRP metabolism (42). These cumulative data highlight the biological relevance of the KP in malignancies and may explain why IDO1 is barely detectable upon long-term in vitro culture. By mimicking the inflamed microenvironment and thus taking a step closer to the in vivo situation, IFNγ was added as strong IDO1 inductor (43). While GBM cells responded with the expected IDO1 upregulation on mRNA expression and protein level as well as accelerated TRP consumption, this molecule was barely inducible in HNSCC cells. It is conceivable that this is due to the duration of in vitro culture. GBM cells were established recently and thus used in defined low passages (<P40), whereas half of the HNSCC cell lines were long-term cultures with more or less unknown passage [Detroit-562 as well as UT-SCC14 and UT-SCC15 (44) are the only exceptions; <P40]. Cell lines may acquire additional mutations overtime changing their protein expression. Another in vitro limitation is that experiments were conducted without immunological pressure. In vivo studies are desirable to verify the results.
Indirect effects of TRP metabolism include interference with other biological functions like migration, angiogenesis, and cell growth regulation (18, 40). To investigate the influence of anticancer drugs on TRP catabolism, we performed a comprehensive analysis using conventional chemotherapy (TMZ, 5-FU, Cisplatin, Gemcitabine) and targeted drugs (Cetuximab, Dinaciclib). The KP-related gene expression and metabolites were determined in residual cells. In GBM, the standard of care drug TMZ was applied either with or without IFNγ stimulation. While this substance affected IDO1 on the expression level, the amount of the resulting protein increased. This may be explained by either increased protein's half-life due to a reduced rate of degradation or the preferential translation during cellular stress. In previous studies, exposure of several cultured human malignant glioma cell lines, primary neurons, and a neuroblastoma cell line to IFNγ reduced TRP levels in culture medium accompanied by increased IDO1 expression and KYN production (29, 45). Our results confirm these data, and in addition, we were able to demonstrate that IFNγ stimulation in combination with TMZ stimulated KYN and KYNA production and TRP catabolism in GBM cell cultures. The increase in TRP catabolism and KYN production (KYN/TRP ratio) is widely used as indirect indicator of the cumulative activities of TDO2, IDO1, and IDO-2 (38, 46). The KP in brain tumors is likely triggered by IFNγ from immediate surrounding tissue (29, 47, 48). Thus, IDO1 expression in brain tumor cells is likely to be triggered when IFNγ is produced from activated T cells and/or microglia and neurons. Furthermore, gliomas and glioneuronal tumors have an elevated tryptophan uptake and catabolism in vivo (49). Given our observation on a further enhanced KP activity upon TMZ treatment, this might provide an explanation of (acquired) drug resistance and final relapse. Hence, IDO1 blocking agents should be investigated in TMZ-tailored therapeutic approaches.
In HNSCC cells, KP activation was different. KP-related genes were exclusively induced by standard drugs, and only Cetuximab induced IDO1. Additional upregulated genes involved kynurenine aminotransferases, responsible for synthesizing a neuroprotectant, and KMO. While the specific biochemical activity of these molecules and biological relevance in cancer is barely examined, we interpret this result as one possible mechanism of resistance upon therapy—a finding quite common after conventional chemotherapy and usually also being associated with poor response toward neoadjuvant therapy in other entities (50).
Mechanistically, this can be attributed to the secretion of proinflammatory substances, such as prostaglandin E2 or high-mobility group protein B1 by dying tumor cells, secondary contributing to KP activation. By accumulating TRP, toxic metabolites of tumor cells actively shape an immunosuppressive microenvironment. Breaking down this shield is one of the main objectives in pharmacological inhibition of KP. Questions remain why most inhibitors failed in clinical trials, and mechanisms are only just beginning to become clear. A fact worth mentioning is the functional redundancy of IDO1, IDO-2, and TDO2 (51), augmenting the risk of mechanistic bypass.
Dinaciclib is a potent and specific CDK inhibitor of CDK1, CDK2, CDK5, and CDK9. Preclinical studies showed that this inhibitor is capable of decelerating tumor growth in numerous cancer entities via cell cycle arrest and apoptosis induction (52, 53). In our study, Dinaciclib was the only KP-inhibiting substance tested here. Of note, impairment of the KP was independent from the combination partner, and this CDKi effectively suppressed IFNγ-induced IDO1 upregulation after simultaneous treatment. While this result was completely unexpected and has—to the best of our knowledge—not been described previously, our data do not support the idea of blunt interference with the KP. GBM cells with strong IDO1 expression showed only marginally reduced IDO1 protein level after Dinaciclib treatment. This effect might be boosted after long or repeated treatment cycles. In line with these findings, several preclinical studies already proposed synergistic effects of selective and unselective IDO1 inhibitors when administered in conjunction with chemo- and/or radiotherapy (4). This may finally have impact for second- or third-line immunotherapeutic approaches. Therefore, the late KYN/TRP index is indeed a relevant clinical benchmark providing prognostic value for GBM patients (54).
Summarizing our findings, we provide evidence for the relevance of TRP catabolism in malignancies especially in the context of standard therapy. The CDKi Dinaciclib was identified as indirect KP inhibitor. Lastly, specific KP inhibition may increase the efficacy of standard drugs by restoring immune function and thus improve patients' outcome.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
CR performed experiments, analyzed data, and participated in manuscript writing. BS and AZ performed immunohistochemistry and analysis, and provided images. HK, JG, NI, and FS performed experiments and analyzed data. GD performed LC-MS analyses. CC and CJ participated in paper finalization and critically revised the manuscript. DS and EW critically revised the manuscript. CM designed study, the outline of the manuscript, performed data interpretation, and wrote the manuscript.
Funding
CM was supported by grants from the Deutsche Forschungsgemeinschaft (MA5799/2-1 and MA5799/2-2).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are also grateful to Susanne Neumeister and Dr. Ann-Kristin Henning from the Institute of Clinical Chemistry and Laboratory Medicine, University of Greifswald, for assisting with the quantification of TRP metabolites.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00055/full#supplementary-material
Supplementary Figure 1. Quantitative analysis of cell death in Detroit-562 HNSCC cells upon cytostatic drugs and targeted therapy. The cells were treated with the given substances for 24 and 72 h. Thereafter, cells were harvested and stained with Yo-Pro-1 to detect early and late apoptotic cells, as well as propidium iodide for necrosis determination. Apoptosis/necrosis discrimination was done on a flow cytometer (BD FACSVerseTM) as described in material and methods. (A) Quantitative analysis of cell death after 24 and 72 h, respectively. *p < 0.05 vs. control; **p < 0.01 vs. control. t-test. (B) Representative dot plots showing elevated numbers of necrotic cells upon treatment. (C) Hemolysis and viability of PBMC upon treatment with Dinaciclib. Therefore, whole blood and PBMC were cultured in the presence of increasing Dinaciclib concentrations (1, 5, and 10 μM) for 2 and 24 h, respectively. Hemolytic activity was determined from cell-free supernatants (red blood cell lysis), Calcein AM was used for quantifying viability of PBMC. Mean + SD, N = 5 individual donors.
Supplementary Figure 2. IDO1 protein abundance in HROG05 GBM cells upon cytostatic drugs and targeted therapy. The cells were pretreated with IFNγ (50 ng/ml) for 24 h. Thereafter TMZ, Dinaciclib and the combination of both substances was added to see whether IFNγ-induced upregulation of IDO1 is reversible. (A) None of these substances downregulated IDO1 in the sequential setting. Cell nuclei were stained with DAPI. Original magnification 20x. (B) Quantification was done to score staining intensity in untreated and treated HROG05 cells. This was carried out by using ImageJ software as described in material and methods.
Supplementary Table for Figure 5. Statistical analysis of individual treatment regimens, depicted for each cell line, and genes analyzed.
Abbreviations
CDKi, cyclin-dependent kinase inhibitor; GBM, glioblastoma multiforme; HNSCC, head and neck squamous cell carcinoma; IDO1, indoleamine 2,3-dioxygenase; IFN, interferon; KYAT, kynurenine aminotransferase; KP, kynurenine pathway; PBMC, peripheral blood mononuclear cells; SCC, squamous cell carcinoma; TDO2, tryptophan 2,3-dioxygenase.
References
1. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. (2018) 11:100. doi: 10.1186/s13045-018-0644-y
2. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
3. Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
4. Muller AJ, DuHadaway JB, Donover PS, Sutanto-Ward E, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. (2005) 11:312–9. doi: 10.1038/nm1196
5. Moon YW, Hajjar J, Hwu P, Naing A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J Immunother Cancer. (2015) 3:51. doi: 10.1186/s40425-015-0094-9
6. Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. (2016) 37:193–207. doi: 10.1016/j.it.2016.01.002
7. Avril T, Saikali S, Vauleon E, Jary A, Hamlat A, De Tayrac M, et al. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. J Neuroimmunol. (2010) 225:22–33. doi: 10.1016/j.jneuroim.2010.04.003
8. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
9. Hanihara M, Kawataki T, Oh-Oka K, Mitsuka K, Nakao A, Kinouchi H. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg. (2016) 124:1594–601. doi: 10.3171/2015.5.JNS141901
10. Badawy AAB. Kynurenine pathway of tryptophan metabolism: regulatory and functional aspects. Int J Tryptophan Res. (2017) 10:1178646917691938. doi: 10.1177/1178646917691938
11. Solomon B, Young RJ, Rischin D. Head and neck squamous cell carcinoma: genomics and emerging biomarkers for immunomodulatory cancer treatments. Semin Cancer Biol. (2018) 52:228–40. doi: 10.1016/j.semcancer.2018.01.008
12. Acovic A, Gazdic M, Jovicic N, Harrell CR, Fellabaum C, Arsenijevic N, et al. Role of indoleamine 2,3-dioxygenase in pathology of the gastrointestinal tract. Therap Adv Gastroenterol. (2018) 11:1756284818815334. doi: 10.1177/1756284818815334
13. Sordillo PP, Sordillo LA, Helson L. The kynurenine pathway: a primary resistance mechanism in patients with glioblastoma. Anticancer Res. (2017) 37:2159–71. doi: 10.21873/anticanres.11551
14. Zamanakou M, Germenis AE, Karanikas V. Tumor immune escape mediated by indoleamine 2,3-dioxygenase. Immunol Lett. (2007) 111:69–75. doi: 10.1016/j.imlet.2007.06.001
15. Ma W-J, Wang X, Yan W-T, Zhou Z-G, Pan Z-Z, Chen G, et al. Indoleamine-2,3-dioxygenase 1/cyclooxygenase 2 expression prediction for adverse prognosis in colorectal cancer. World J Gastroenterol. (2018) 24:2181–90. doi: 10.3748/wjg.v24.i20.2181
16. Brandacher G, Perathoner A, Ladurner R, Schneeberger S, Obrist P, Winkler C, et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: effect on tumor-infiltrating T cells. Clin Cancer Res. (2006) 12:1144–51. doi: 10.1158/1078-0432.CCR-05-1966
17. Jeong Y-I, Kim SW, Jung ID, Lee JS, Chang JH, Lee C-M, et al. Curcumin suppresses the induction of indoleamine 2,3-dioxygenase by blocking the Janus-activated kinase-protein kinase Cdelta-STAT1 signaling pathway in interferon-gamma-stimulated murine dendritic cells. J Biol Chem. (2009) 284:3700–8. doi: 10.1074/jbc.M807328200
18. Platten M, von Knebel Doeberitz N, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. (2014) 5:673. doi: 10.3389/fimmu.2014.00673
19. Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. (2003) 9:1269–74. doi: 10.1038/nm934
20. Fox E, Oliver T, Rowe M, Thomas S, Zakharia Y, Gilman PB, et al. Indoximod: an immunometabolic adjuvant that empowers T cell activity in cancer. Front Oncol. (2018) 8:370. doi: 10.3389/fonc.2018.00370
21. Beatty GL, O'Dwyer PJ, Clark J, Shi JG, Bowman KJ, Scherle PA, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. (2017) 23:3269–76. doi: 10.1158/1078-0432.CCR-16-2272
22. Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. (2019) 18:379–401. doi: 10.1038/s41573-019-0016-5
23. Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, Matsumoto S, et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. (2011) 68:157–64. doi: 10.1007/s00280-010-1470-2
24. Maletzki C, Scheinpflug P, Witt A, Klar E, Linnebacher M. Targeting immune-related molecules in cancer therapy: a comprehensive in vitro analysis on patient-derived tumor models. Biomed Res Int. (2019) 2019:4938285. doi: 10.1155/2019/4938285
25. Vacchelli E, Aranda F, Eggermont A, Sautes-Fridman C, Tartour E, Kennedy EP, et al. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology. (2014) 3:e957994. doi: 10.4161/21624011.2014.957994
26. Schafer CC, Wang Y, Hough KP, Sawant A, Grant SC, Thannickal VJ, et al. Indoleamine 2,3-dioxygenase regulates anti-tumor immunity in lung cancer by metabolic reprogramming of immune cells in the tumor microenvironment. Oncotarget. (2016) 7:75407–24. doi: 10.18632/oncotarget.12249
27. Yentz S, Smith D. Indoleamine 2,3-dioxygenase (IDO) inhibition as a strategy to augment cancer immunotherapy. BioDrugs. (2018) 32:311–7. doi: 10.1007/s40259-018-0291-4
28. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
29. Adams S, Teo C, McDonald KL, Zinger A, Bustamante S, Lim CK, et al. Involvement of the kynurenine pathway in human glioma pathophysiology. PLoS ONE. (2014) 9:e112945. doi: 10.1371/journal.pone.0112945
30. Ball HJ, Jusof FF, Bakmiwewa SM, Hunt NH, Yuasa HJ. Tryptophan-catabolizing enzymes—party of three. Front Immunol. (2014) 5:485. doi: 10.3389/fimmu.2014.00485
31. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frederick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase. Proc Natl Acad Sci USA. (2012) 109:2497–502. doi: 10.1073/pnas.1113873109
32. Abdel-Magid AF. Targeting the inhibition of tryptophan 2,3-dioxygenase (TDO-2) for cancer treatment. ACS Med Chem Lett. (2017) 8:11–3. doi: 10.1021/acsmedchemlett.6b00458
33. Jamieson SM, Tsai P, Kondratyev MK, Budhani P, Liu A, Senzer NN, et al. Evofosfamide for the treatment of human papillomavirus-negative head and neck squamous cell carcinoma. JCI Insight. (2018) 3:122204. doi: 10.1172/jci.insight.122204
34. Maletzki C, Klier U, Marinkovic S, Klar E, Andrä J, Linnebacher M. Host defense peptides for treatment of colorectal carcinoma-a comparative in vitro and in vivo analysis. Oncotarget. (2014) 5:4467–79. doi: 10.18632/oncotarget.2039
35. Fuertig R, Ceci A, Camus SM, Bezard E, Luippold AH, Hengerer B. LC-MS/MS-based quantification of kynurenine metabolites, tryptophan, monoamines and neopterin in plasma, cerebrospinal fluid, and brain. Bioanalysis. (2016) 8:1903–17. doi: 10.4155/bio-2016-0111
36. Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. (2011) 478:197–203. doi: 10.1038/nature10491
37. Guo T, Califano JA. Molecular biology and immunology of head & neck cancer. Surg Oncol Clin N Am. (2015) 24:397–407. doi: 10.1016/j.soc.2015.03.002
38. Adam I, Dewi DL, Mooiweer J, Sadik A, Mohapatra SR, Berdel B, et al. Upregulation of tryptophanyl-tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. Oncoimmunology. (2018) 7:e1486353. doi: 10.1080/2162402X.2018.1486353
39. Smith C, Chang MY, Parker KH, Beury DW, DuHadaway JB, Flick HE, et al. IDO is a nodal pathogenic driver of lung cancer and metastasis development. Cancer Discov. (2012) 2:722–35. doi: 10.1158/2159-8290.CD-12-0014
40. Bates AM, Gomez Hernandez MP, Lanzel EA, Qian F, Brogden KA. Matrix metalloproteinase (MMP) and immunosuppressive biomarker profiles of seven head and neck squamous cell carcinoma (HNSCC) cell lines. Transl Cancer Res. (2018) 7:533–42. doi: 10.21037/tcr.2018.05.09
41. Seppala M, Halme E, Tiilikainen L, Luukkainen A, Laranne J, Rautiainen M, et al. The expression and prognostic relevance of indoleamine 2,3-dioxygenase in tongue squamous cell carcinoma. Acta Otolaryngol. (2016) 136:729–35. doi: 10.3109/00016489.2016.1152631
42. Cheng J, Jin H, Hou X, Lv J, Gao X, Zheng G. Disturbed tryptophan metabolism correlating to progression and metastasis of esophageal squamous cell carcinoma. Biochem Biophys Res Commun. (2017) 486:781–7. doi: 10.1016/j.bbrc.2017.03.120
43. Grant RS, Naif H, Espinosa M, Kapoor V. IDO induction in IFN-gamma activated astroglia: a role in improving cell viability during oxidative stress. Redox Rep. (2000) 5:101–4. doi: 10.1179/135100000101535357
44. Tonlaar N, Galoforo S, Thibodeau BJ, Ahmed S, Wilson TG, Yumpo Cardenas P, et al. Antitumor activity of the dual PI3K/MTOR inhibitor, PF-04691502, in combination with radiation in head and neck cancer. Radiother Oncol. (2017) 124:504–12. doi: 10.1016/j.radonc.2017.08.001
45. Miyazaki T, Moritake K, Yamada K, Hara N, Osago H, Shibata T, et al. Indoleamine 2,3-dioxygenase as a new target for malignant glioma therapy. Laboratory investigation. J Neurosurg. (2009) 111:230–7. doi: 10.3171/2008.10.JNS081141
46. Suzuki Y, Suda T, Asada K, Miwa S, Suzuki M, Fujie M, et al. Serum indoleamine 2,3-dioxygenase activity predicts prognosis of pulmonary tuberculosis. Clin Vaccine Immunol. (2012) 19:436–42. doi: 10.1128/CVI.05402-11
47. Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia. (2005) 49:15–23. doi: 10.1002/glia.20090
48. Adams S, Braidy N, Bessede A, Brew BJ, Grant R, Teo C, et al. The kynurenine pathway in brain tumor pathogenesis. Cancer Res. (2012) 72:5649–57. doi: 10.1158/0008-5472.CAN-12-0549
49. Juhasz C, Chugani DC, Muzik O, Wu D, Sloan AE, Barger G, et al. In vivo uptake and metabolism of alpha-[11C]methyl-L-tryptophan in human brain tumors. J Cereb Blood Flow Metab. (2006) 26:345–57. doi: 10.1038/sj.jcbfm.9600199
50. Li F, Wei L, Li S, Liu J. Indoleamine-2,3-dioxygenase and Interleukin-6 associated with tumor response to neoadjuvant chemotherapy in breast cancer. Oncotarget. (2017) 8:107844–58. doi: 10.18632/oncotarget.22253
51. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO-301 trial and beyond. Semin Immunopathol. (2019) 41:41–8. doi: 10.1007/s00281-018-0702-0
52. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. (2010) 9:2344–53. doi: 10.1158/1535-7163.MCT-10-0324
53. Lin SF, Lin JD, Hsueh C, Chou TC, Wong RJ. A cyclin-dependent kinase inhibitor, dinaciclib in preclinical treatment models of thyroid cancer. PLoS ONE. (2017) 12:e172315. doi: 10.1371/journal.pone.0172315
Keywords: targeted therapy, solid tumor models, tryptophan metabolites, IDO1, chemotherapy
Citation: Riess C, Schneider B, Kehnscherper H, Gesche J, Irmscher N, Shokraie F, Classen CF, Wirthgen E, Domanska G, Zimpfer A, Strüder D, Junghanss C and Maletzki C (2020) Activation of the Kynurenine Pathway in Human Malignancies Can Be Suppressed by the Cyclin-Dependent Kinase Inhibitor Dinaciclib. Front. Immunol. 11:55. doi: 10.3389/fimmu.2020.00055
Received: 16 September 2019; Accepted: 09 January 2020;
Published: 14 February 2020.
Edited by:
Ignacio Melero, University of Navarra, SpainReviewed by:
Benjamin Heng, Macquarie University, AustraliaMichael Platten, German Cancer Research Center (DKFZ), Germany
Copyright © 2020 Riess, Schneider, Kehnscherper, Gesche, Irmscher, Shokraie, Classen, Wirthgen, Domanska, Zimpfer, Strüder, Junghanss and Maletzki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Maletzki, claudia.maletzki@med.uni-rostock.de