 Vijay Kumar1,2*†
Vijay Kumar1,2*†- 1Children's Health Queensland Clinical Unit, Faculty of Medicine, School of Clinical Medicine, Mater Research, University of Queensland, Brisbane, QLD, Australia
- 2Faculty of Medicine, School of Biomedical Sciences, University of Queensland, Brisbane, QLD, Australia
The lung is a primary organ for gas exchange in mammals that represents the largest epithelial surface in direct contact with the external environment. It also serves as a crucial immune organ, which harbors both innate and adaptive immune cells to induce a potent immune response. Due to its direct contact with the outer environment, the lung serves as a primary target organ for many airborne pathogens, toxicants (aerosols), and allergens causing pneumonia, acute respiratory distress syndrome (ARDS), and acute lung injury or inflammation (ALI). The current review describes the immunological mechanisms responsible for bacterial pneumonia and sepsis-induced ALI. It highlights the immunological differences for the severity of bacterial sepsis-induced ALI as compared to the pneumonia-associated ALI. The immune-based differences between the Gram-positive and Gram-negative bacteria-induced pneumonia show different mechanisms to induce ALI. The role of pulmonary epithelial cells (PECs), alveolar macrophages (AMs), innate lymphoid cells (ILCs), and different pattern-recognition receptors (PRRs, including Toll-like receptors (TLRs) and inflammasome proteins) in neutrophil infiltration and ALI induction have been described during pneumonia and sepsis-induced ALI. Also, the resolution of inflammation is frequently observed during ALI associated with pneumonia, whereas sepsis-associated ALI lacks it. Hence, the review mainly describes the different immune mechanisms responsible for pneumonia and sepsis-induced ALI. The differences in immune response depending on the causal pathogen (Gram-positive or Gram-negative bacteria) associated pneumonia or sepsis-induced ALI should be taken in mind specific immune-based therapeutics.
Introduction
Lungs serve as vital organs for the gaseous exchange in the vertebrates. They have evolved from their very primitive stage (air sacs found in the very primitive and well-armored fossil placoderm fish Bothriolepis) to the most advanced form present in mammals depending on their habitat and the oxygen demand. Thus, due to continuous gaseous exchange function, lungs serve as a very easy target organ for the airborne pathogens, allergens, and other toxicants to cause pulmonary infections or inflammation. However, pulmonary damage may be acute, or chronic depending on the intensity and the duration of the exposure. For example, chronic obstructive pulmonary disease (COPD) and allergic asthma cause chronic inflammatory changes in the lungs. Whereas, acute microbial (bacterial or viral) infections responsible for pneumonia or sepsis cause severe inflammatory damage to the lungs, leading to the development of acute lung injury/inflammation (ALI) or acute respiratory distress syndrome (ARDS) in critically ill patients (1).
ALI in response to the severe pulmonary microbial infections occurs as a result of the immunological recognition of the pathogen responsible for inducing a pro-inflammatory immune response. The ALI causes severe tissue damage, and in severe cases, irreversible pulmonary damage may lead to death. For example, the protein-rich hydrostatic pulmonary edema characterizing ALI causes refractory hypoxemia, stiffening of the lungs, and difficulty to respire. Rene Laennec (invented the stethoscope in 1861) in 1821 first described the ARDS as an “idiopathic pulmonary edema” occurring without heart failure, which was further modified into “wet lung or shock lung” (2, 3). However, Ashbaugh et al. for the first time, coined the term ARDS to describe the rapid onset of tachypnoea, hypoxemia, and the loss of compliance after a variety of stimuli (4). Sepsis is a leading (6–42%) cause of the ALI (5). Depending on the ALI/ARDS cause, age, and sex of the host, the pulmonary innate immune system plays a very significant role in the ALI pathogenesis (6).
The innate immune system serves as the first line of defense against foreign pathogens via recognizing their pathogen-associated molecular patterns (PAMPs) or microbe-associated molecular patterns (MAMPs). Also, innate immune cells recognize the damage or danger-associated molecular patterns (DAMPs) generated during the pro-inflammatory conditions disturbing immune homeostasis (7). The recognition of PAMPs or MAMPs and DAMPs involves several pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and multiple germ line encoded receptors [NOD-like receptors (NLRs), retinoic acid inducible gene I (RIG-I)-like receptors (RLRs), C-type lectin receptors (CLRs) and multiple intracellular DNA sensors expressed (cGAS-STING signaling pathway, Aim 2 like receptors (ALRs)] (8–11). This induces the pro-inflammatory immune response generating different cytokines, chemokines, interferons (IFNs), and other molecules, including reactive oxygen or nitrogen species (ROS or RNS) for clearing the infection to maintain the immune homeostasis. However, the innate immune response dysregulation during infection may increase its severity via increasing the pathogen load due to the inefficient pathogen clearance or by causing increased and irreversible organ damage in patients succumbed to sepsis (12, 13). Hence, a regulated innate immune response during both acute and chronic infections is essential for clearing the infection.
The organ-specific innate immune response determines infection severity. For example, the potent innate immune response generation in the lungs during localized pulmonary infections (pneumonia) or its dysregulation as seen in the non-pulmonary sepsis (sepsis originating from other sources, including the peritoneum, urinary tract, various soft tissues, and skin)-associated acute ALI or ARDS plays a crucial role in the disease outcome (12). Thus, the major aim of the present article is to describe the pulmonary innate immune response responsible for the ALI observed during bacterial pneumonia and sepsis, as evidenced by both animal and human findings.
Lung as an Innate Immune Organ
Lungs are the vital organs designed not only for the gaseous exchange but also serve as a major immune organ to protect the host from diseases caused by the pathogen inhalation during respiration along with allergens and xenobiotics (allergic asthma, pneumonia, sepsis-associated ALI) (12, 14). In the early 1960s, Askonas and Humphrey showed upon intravenous injection of pneumococcal antigens lungs potentially contribute to developing more specific antibodies in comparison to the rest of the lymphoid organs (15). Later on, another study in rabbits showed that the local intranasal instillation of pneumococcal antigen-induced the specific immunity and pulmonary resistance to the infection without generating the antibody (Ab)-mediated systemic immunity (16). Furthermore, the pulmonary DNA vaccine-based immunization also induces the local CD8+T cell-based protective anti-viral (vaccinia and influenza virus) immunity without recruiting peripheral T cells (17). This pulmonary immune response during vaccination can further be enhanced by the nasal administration of the adjuvants (18). However, the pulmonary challenge with recombinant vaccines has the potential to generate local (lung) as well as systemic immunity against pathogens (19). Thus, lung can induce protective immunity against respiratory pathogens without the involvement or activation of peripheral or systemic immunity by working as a potent immune organ.
Lungs can be categorized into two components both from a physiological and immunological point of view, (1) Upper respiratory tract serving as mucosal (IgA serves as predominating class of antibody) and glandular component, and (2) peripheral airways without any mucosal tissue (dominated by IgG antibody). Furthermore, the peripheral airways on the luminal side constantly remain in contact with the Broncho-alveolar cells (BACs, 90% of which under normal homeostasis comprise of alveolar macrophages), and 10% of which is comprised of lymphocytes (Figure 1). Thus, the pulmonary immune system is separable into different compartments, which have the potential to interact (20). Similar to the epithelial lymphocyte compartment of the gut, a compartment of lymphocytes residing in the respiratory tract epithelium over the epithelial membrane and between the epithelial cells also exists. Thus, protecting the host from invasive pulmonary infections. The other compartment of the respiratory lymphoid cells (RLCs) comprises of the organized lymphoid tissues lying within the bronchial walls. This RLC compartment comprises of either solitary lymphoid follicles (SLFs) or their aggregates resembling the Peyer's Patches (PPs) of the intestine (21, 22). Thus, this bronchus-associated lymphoid tissue (BALT) is morphologically and functionally analogous to the gut-associated lymphoid tissue (GALT) of the intestine (23, 24). For example, receptor activator of nuclear factor-κB and its ligand (RANKL) is a common inducer of M cells in the lungs and gastrointestinal tracts (GITs) (25). M cells play a crucial role in respiratory diseases (26).
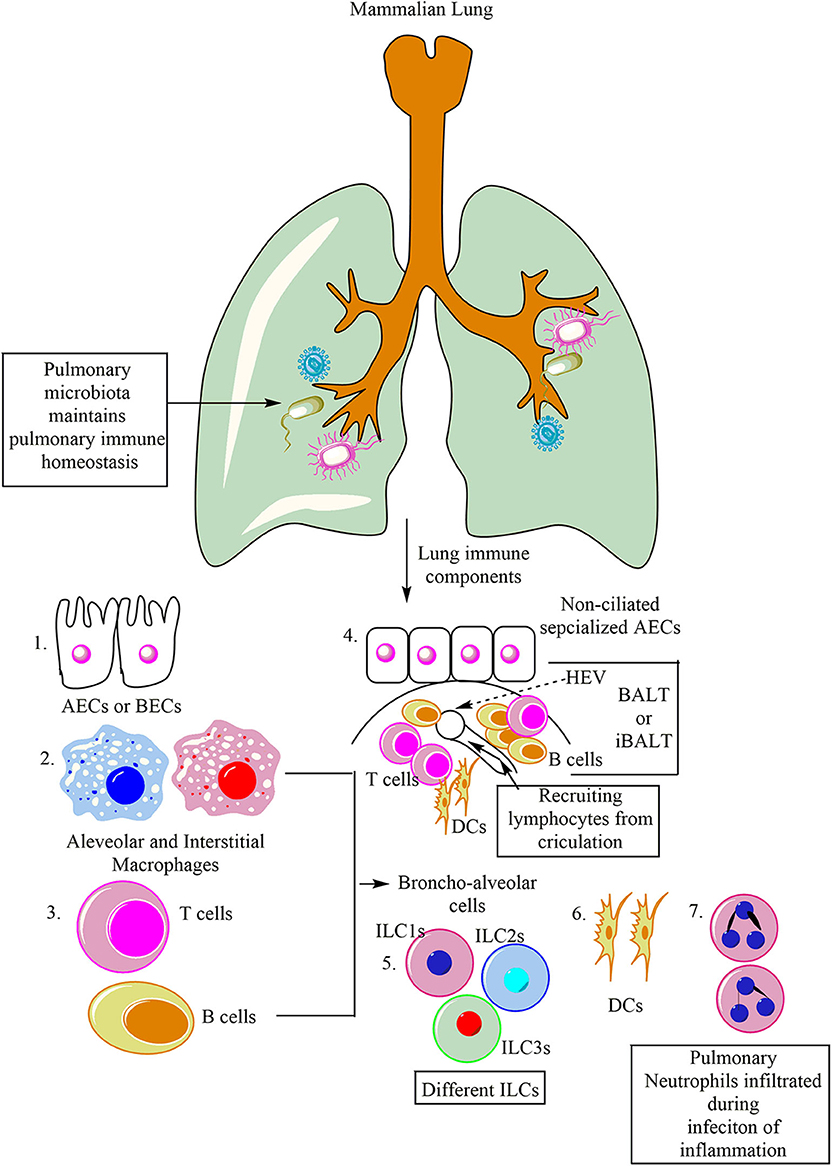
Figure 1. Major immune cells in the mammalian lung. Lungs are potent immune organs and contain macrophages, which may be divided into alveolar macrophages (AM) and interstitial macrophages (IMs), alveolar and bronchial epithelial cells (AECs and BECs), DCs, NK cells along with other ILCs (ILC1s, ILC2s, and ILC3s), and adaptive immune cells (different T and B cells). Neutrophils also migrate to the lungs in response to the infection or inflammatory insult. Additionally, like Peyer's patches (PPs) of gut-associated lymphoid tissue (GALT), lungs also have BALT. BALT contains T cells (T cell Zone), B cells (B cell zone), and DCs. The BALT induced in response to the infection is called iBALT.
The BALT is covered by a lymphoepithelium, and its follicle-associated epithelium selectively samples both soluble and particulate matter from the respiratory tract lumen (Figure 1) (27, 28). Of note, in humans, BALT is present only in the lungs of kids and adolescents, and adults show BALT only during chronic inflammatory diseases, where it is called inducible BALT (iBALT) (Figure 1) (29). On the other hand, BALT may present in the fetal and neonate's lungs, depending on the antigenic stimulation (30, 31). However, these RLCs comprising the lymphoid follicles of the BALT in humans expand or proliferate considerably in a group of patients suffering from recurrent respiratory tract infections (RTIs) of unknown etiology due to the occlusion of the bronchiolar or bronchial lumen (32). B cells are the major immune cell population of the BALT responsible to generate IgA (Figure 1) (20, 33). T cells comprising T cell zone are also present in BALT. T cell zones also have DCs. BALTs also have high endothelial venule (HEV), which serves to transport lymphocytes and antigens to and from the circulation (Figure 1). The IgA produced may bind to the lymphocytes to increase their Ab-dependent cytotoxic action. The secreted IgA also protects against viral and bacterial infections along with the allergy. The other compartment comprises of BACs, which can be obtained through broncho-alveolar lavage fluid (BALF) from the peripheral airways. BALF may contain alveolar macrophages (AMs), innate lymphoid cells (ILCs), and dendritic cells (DCs), providing protection against pathogens, toxicants, and allergens inhaled. These pulmonary innate immune cells serve as antigen-presenting cells (APCs) and secrete several cytokines and chemokines to regulate both the pulmonary innate and adaptive immunity. Under normal healthy conditions, BACs in BALF mainly comprises of AM (90%) and rest (10%) are lymphocytes (14). These lymphocytes, via lymph, circulate through the lung and patrol for potential antigen inhaled or entered into the lung through circulation.
The pulmonary immune system matures in the postnatal environment depending on the richness and the type of antigen exposure to the host (34). However, during in utero embryonic development lungs remain sterile, but during vaginal delivery, they acquire maternal microbiota (35, 36). The pulmonary microbiota helps in the pulmonary immune system development, tolerance induction, and its homeostasis (Figure 1) (37, 38). The pulmonary residential epithelial cells, ILCs, and AMs along with other pulmonary immune cells, are essential to maintain the steady-state in the lungs. However, their ability to recognize different airway pathogens and allergens also induces inflammatory changes in the lungs. Under some situations, these pulmonary inflammatory changes are mild and resolve by itself, but the ALI observed during bacterial pneumonia and sepsis may prove harmful to the host depending on the severity of the infection and the inflammatory innate immune response.
Pulmonary Innate Immune Response During Bacterial Pneumonia
According to the National Center for Health Statistics, bacterial pneumonia and influenza comprised eighth causes of mortality in the United States in 2014–2018 (39, 40). However, in children, among infectious diseases, pneumonia is the single most cause of death all over the world (41). Thus, pneumonia is a serious life-threatening infection among the children and older population. Pneumonia pathogenesis is a very complex process involving the microbial invasion of the lower respiratory tract through community or hospital spread. It may occur through inhalation of the causal pathogen. For example, S. pneumoniae is the most common pathogen responsible for community-acquired pneumonia (CAP). In addition to the S. pneumoniae, Legionella pneumophila, Mycoplasma pneumoniae, Chlamydophila pneumoniae, Chlamydophila psittaci, and Coxiella burnetii are several other common pathogens responsible for CAP (42, 43). Most hospital-acquired pneumonia are caused by Gram-negative pathogens (Klebsiella pneumoniae, Pseudomonas aeruginosa, etc.). The details of CAP and hospital-acquired pneumonia (HAP) are described somewhere else (41). The pulmonary innate immune response during pneumonia initiates with the activation of residential innate immune cells (AECs, AMs, etc.) inducing the neutrophil infiltration into the lungs. Toll-like receptor 4 (TLR4) activation induced immune response protects the experimental animals infected with Gram-positive (Streptococcus pneumoniae) or Gram-negative bacteria (Klebsiella pneumoniae) induced pneumonia (44).
Neutrophil Infiltration in Lungs During Pneumonia-Associated ALI
The mechanism of neutrophil infiltration in the lungs varies from the process involved in other organs and has been described in detail somewhere else (45, 46). The mechanism of neutrophil infiltration in the lungs during ALI varies during Gram-negative and -positive bacterial pneumonia (47). For example, during Gram-negative bacterial (E. coli or P. aeruginosa) pneumonia, alveolar neutrophil infiltration is mediated by CD18 or β2 integrin, whereas in Gram-positive bacterial (S. pneumoniae) pneumonia, it is mediated by the CD29 or β1 integrin (48). Additionally, patients with ALI show an elevated chemokine (CXCL8 or IL-8, CXCL1, or keratinocyte-cell derived chemokine (KC), CXCL5, or epithelial cell-derived neutrophil-activating peptide-78 (ENA-78), and CCL-2) levels in their BALF, which further regulate neutrophil infiltration into the lungs (49, 50).
The CXCR2 (a chemokine receptor) binding to different chemokines [CXCL1, CXCL8 (in humans), CXCL5, CXCL2, CXCL3, CXCL6, and CXCL7 (in humans)] regulates the neutrophil infiltration in the lungs (Figure 2A) (51). The lung epithelial cells (LECs) produce the CXCL5 during bacterial (E. coli) pneumonia that induces neutrophil infiltration in the lungs, whereas in naïve murine blood, platelets are a crucial source of CXCL5 (51). However, CXCL5 deficiency during E. coli pneumonia increases neutrophil influx in the lungs, accelerates the pathogen clearance, improves pulmonary edema, and protects the mice from severe pneumonia and, thus, the ALI (52). CXCL5−/− mice do not show much decrease in CXCR2 expression on bone marrow and blood neutrophils as compared to the wild type (WT) mice upon E. coli-induced pneumonia, but the CXCR2 expression on neutrophils remains unchanged during intranasal lipopolysaccharide (LPS) challenge (52). In the absence of CXCL5, CXCL1, and CXCL2 bind to the Duffy Antigen Receptor for Chemokines (DARC) to increase the neutrophil infiltration in the lungs, which enhances bacterial clearance, and protects the animal from severe pneumonia (52). The levels of these chemokines (CXCL8) affect the severity of ALI and the mortality among patients regulating neutrophil infiltration (53). The CXCL1 regulates neutrophil infiltration and the bacterial clearance during K. pneumoniae-induced pneumonia via regulating the CXCL2/MIP-2 and CXCL5, and NF-κB and MAPKs activation in the lungs (54). Thus, the pulmonary neutrophil infiltration is crucial in the ALI induction and its resolution (55, 56). The pulmonary residential innate immune cells [Airway epithelial cells (AECs), macrophages, dendritic cells (DCs), and innate lymphoid cells (ILCs)] are crucial in the pathogenesis of bacterial pneumonia and associated ALI and its outcome.
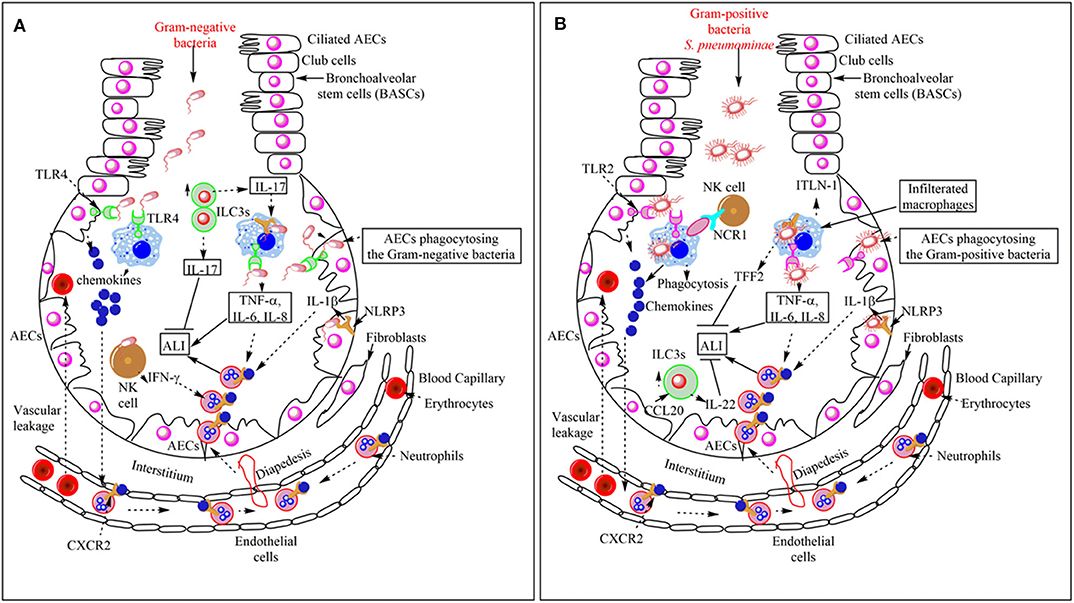
Figure 2. Overview of the bacterial pneumonia-associated innate immune response responsible for ALI. (A) Gram-negative bacterial pneumonia and ALI. The PRRs [TLRs (TLR4) and Inflammasome proteins (NLRP3)] expressed on the pulmonary innate immune cells (AECs, BECs, AMs, DCs, and NK cells) recognize Gram-negative bacteria in the lungs. This recognition induces pro-inflammatory cytokine (TNF-α, IL-6, IL-8, and IL-1β) and chemokine release. Chemokines and pro-inflammatory cytokines induce the neutrophil infiltration in the alveoli from the pulmonary blood capillaries through their vascular endothelium via diapedesis. The infiltrated neutrophils help in the pathogen clearance but also cause bystander inflammatory pulmonary tissue damage via damaging PECs or AECs. In the severe pneumonia-associated ALI, the vascular leakage of proteins and erythrocytes also occurs in the lungs. The pulmonary NK cells also release IFN-γ, which further enhances neutrophil infiltration and ALI. However, the increase in ILC3s at later stages increases the IL-17 level. This cytokine helps in the increased phagocytosis of the pathogen and the resolution of the Gram-negative bacterial pneumonia-induced ALI. (B) Gram-positive bacterial pneumonia-associated ALI. The recognition of the Gram-positive bacteria (S. aureus) induces the increased the production of chemokines and pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-8), which via binding to the corresponding receptors (CXCR2) on the neutrophils induce their diapedesis to the lung alveoli from the pulmonary vascular endothelium. Neutrophils, along with bacterial clearance, also cause ALI. Pulmonary vascular endothelium damage also causes vascular leakage. The Pulmonary NK cells via their NCR1 interact with AMs to further increase the pro-inflammatory cytokine release, which further aggravates the neutrophil infiltration, pathogen clearance, and ALI also. The NLRP3 also works independently of inflammasome activation via inducing the release of TFF2 and ITLN-1. TFF2 inhibits ALI and helps in its resolution, whereas ITLN-1 clears the infection via increasing the pathogen phagocytosis. The CCL20 released from AECs increases the pulmonary ILC3 numbers, which release IL-22 that inhibits ALI and helps in its resolution.
Airway Epithelial Cells (AECs) and PRRs (TLRs and Inflammasomes) During Pneumonia
AECs comprising of bronchial epithelial cells (BECs) and alveolar epithelial cells [categorized into (1) type I AECs, which are primarily involved in facilitating gaseous exchange and may also recognize pathogens, and (2) type II AECs, also called type II pneumocytes and serve as innate immune cells] serve as a protective mechanical barrier against inhaled pathogens responsible for pneumonia (57, 58). Type II pneumocytes also secrete surfactant proteins on their apical side. These surfactant proteins serve as mucins. The type II AECs secrete repair enzymes (fibrinogen or FBG) basolaterally, which respond to the change in osmotic pressure of the cell very quickly and sense pore-forming toxins secreted by pathogenic bacteria (59). The released FBG helps in the cellular response in response to the inflammatory cell damage (60). AECs also serve as potent regulators of the generation of the primary immune response against invading pathogens via releasing various immune mediators [antimicrobial peptides (AMPs), cytokines] and interacting directly with other immune cells (macrophages, neutrophils, and DCs) (61). The prolonged activation of AECs may prove harmful to the host due to the release of large quantities of pro-inflammatory cytokines, chemokines, and their increased cell death (necrosis or necroptosis).
During pneumonia, AECs recognize various pathogens due to the expression of different PRRs, including TLRs [TLR1, TLR2, TLR4, TLR5, TLR6 (Extracellular TLRs), and TLR3, TLR7, TLR8, and TLR9 (Intracellular TLRs)] and NLRs comprising inflammasome (58, 62–64). The recognition of pathogens by these PRRs serves as the first line of defense and helps in the pathogen clearance. The downstream adaptor molecules [myeloid differentiation primary response protein 88 (MyD88) and Toll/IL-1R domain-containing adaptor-inducing IFN-β (TRIF)] of TLR signaling play crucial roles in bacterial pneumonia. For example, MyD88−/− and TRIF−/− mice develop severe pneumonia due to the profound bacterial growth during Gram-negative pneumonia (K. pneumoniae, P. aeruginosa, and E. coli) in response to the impaired immune response, including the reduced generation of Th1 immune response (TNF-α, IL-6, and IL-8) and almost no neutrophil influx and regulated upon activation normal T Cells expressed and presumably Secreted (RANTES or CCL5) production (65–68).
The TRIF signaling in response to the TLR activation during K. pneumoniae-induced pneumonia also exerts antibacterial defense via inducing the interferon (IFN)-x03B3 in the lungs (69). However, Toll/IL-1R Domain-Containing Adaptor Protein (TIRAP) plays a critical role during K. pneumoniae-induced pneumonia but not during P. aeruginosa-induced pneumonia due to the attenuation of neutrophil sequestration, and MIP-2, TNF-α, IL-6, and LIX (lipopolysaccharide-induced CXC chemokine) production (70). The LIX production, neutrophil infiltration, and bacterial clearance during P. aeruginosa-induced pneumonia do not require TIRAP (70). The TLR2-induced MyD88 activation is not required for the S. aureus clearance during pneumonia and only exerts the potent inflammatory immune response, but it plays a crucial role in P. aeruginosa clearance (71). Thus, TLR2 activation does not have a significant role in the pathogen clearance and survival of the mice but is only required for the inflammatory immune response during Gram-positive bacterial (S. pneumoniae) pneumonia (Figure 2B) (72). The TLR2 signaling activation during Gram-positive bacterial (S. pneumoniae) pneumonia increases the non-small cell lung cancer cell (NSCLC) metastasis (73). Thus, Gram-positive bacterial pneumonia may increase the metastasis of cancer cells in cancer patients.
Inflammasomes and their component proteins also play a crucial role in pathogen detection and clearance during pneumonia. For example, NLRP1 (NLR Family Pyrin Domain Containing 1) enhances the host's resistance to pneumonia via detecting their virulence factors [Bacillus anthracis lethal factor (LF) protease] (74). The LF protease induces the proteasome-mediated degradation of amino-terminal domains of NLRPB1 to liberate the carboxyl-terminal fragment, a potent caspase-1 (CASP1) activator (75). Also, the NLR Family Pyrin Domain Containing 3 (NLRP3) activation in BECs during various pneumonia-causing bacterial (K. pneumoniae, S. pneumoniae, S. aureus, C. pneumoniae, and L. pneumophila) infections protects the host from infections (Figures 2A,B) (63, 76, 77). The human BECs also express NLRP3 inflammasome (78). The NLRP3-mediated control of K. pneumoniae-induced pneumonia involves the increased neutrophil infiltration, macrophage necrosis, and the release of high-mobility group box-1 protein (HMGB-1) (Figure 2A) (79).
The apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) or pyrin domain (PYD) (ASC, also known as PYCARD) is an inflammasome adapter protein required for the formation of the absent in melanoma 2 (AIM2) and NLRP3 inflammasomes. Inflammasome activation causes the ASC speck formation, which forms a platform to activate caspase-1 (CASP-1). However, NLRP3 and ASC maintain pulmonary innate immune homeostasis during S. pneumoniae-induced pneumonia through an inflammasome independent manner without activating the CASP1 and CASP11 (80). During this process, they (NLRP3 and ASC) stimulate the optimal expression of several mucosal innate immune proteins, including trefoil factor 2 (TFF2) and intelectin-1 (ITLN-1, a secretory galactofuranose-binding lectin) via expressing the SAM pointed domain-containing Ets transcription factor (SPDEF), which facilitates the mucosal defense factor genes (Figure 2B) (80). SPDEF activation involves STAT6 activation. TFF2 protects from increased inflammatory damage via inducing decreased neutrophil recruitment through inhibiting the endothelial vascular cell adhesion molecule 1 (VCAM1) expression and nitric oxide (NO.) release from macrophages via inhibiting inducible nitric oxide synthase (iNOS) (81, 82). TFF2 also antagonizes IL-12 (a cytokine required for inducing IFN-γ production and activating Th1 immune cells) secretion from dendritic cells (DCs) and macrophages (83). TFF2 also serves as mucosal healers via protecting mucosal damage, promoting cell motility, and alveolar type 2 cell proliferation, and restores pulmonary gas exchange after infection (84, 85). TFF2 also induces IL-25 and IL-33 after infection to induce type 2 immunity and repair (86). Pulmonary macrophages also utilize the TFF2/Wnt axis to induce pulmonary epithelial cell proliferation to repair the damage following ALI (87). ITLN-1 provides protection via directly binding to the S. pneumoniae and representing them to phagocytes for phagocytosis (Figure 2B) (80, 87). The BECs express ITLN-1 and may also clear the S. pneumoniae via phagocytosis (88–90).
Aged mice exhibit a reduced NLRP3 expression and function, which increases their susceptibility to developing pneumonia, ALI, and mortality (91). The lower expression and function of NLRP3 in aged immune cells (macrophages, epithelial cells, and DCs) attribute to the increased unfolded protein responses (UPRs), which causes a decreased inflammasome assembly and function increasing the severity of pneumonia caused by S. pneumoniae (92). The aging also increases the susceptibility of the host to secondary S. pneumoniae-induced pneumonia due to the decreased NLRP3 expression and function in the aged lung (93). The treatment of these aged mice with inflammasome activators [Nigericin, which promotes potassium (K+) efflux increases the synthesis and release of inflammasome activation-dependent cytokines (IL-1β and IL-18)] increase their survival and decreases their susceptibility toward pneumonia and ALI. Furthermore, the pre-treatment of aged mice with endoplasmic reticulum (ER) chaperone and the stress-reducing agent tauroursodeoxycholic acid (TUDCA) decreases the pneumonia-associated mortality among the aged mice due to the activation of the NLRP3 inflammasome, which increases the pathogen clearance, and lowers the infection-associated pneumonitis (92). The aged mice also express lower levels of TLR1, TLR6, and TLR9 in the lungs, which also increases their susceptibility to pneumonia (93).
Of note, during lethal pneumonia caused by a low dose of serotype 3 S. pneumoniae, NLRP3 increases the incidence of ALI and mortality due to the bacterial dissemination and the development of the sepsis (94). Also, during S. aureus-induced pneumonia, NLRP3 deficiency prevents the onset of severe necrotic pneumonia via promoting bacterial clearance (95). The NLRP3 activation by α-hemolysin during S. aureus pneumonia induces necrotic pulmonary injury or necrotizing pneumonia independent of IL-1β signaling (95, 96). The NLRP3 activation by α-hemolysin in innate immune cells depends on A Disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) expression and activity (97). ADAM10 binding with α-hemolysin increases NLRP3 activation and cell death due to the availability of ADAM10 on the cell surface. However, ADAM10 protease activity does not play a significant role in NLRP3 activation. Thus, the profound NLRP3 inflammasome activation depending on the severity of the infection proves harmful to the host. In addition to the NLRP3 inflammasome, S. aureus pneumonia also activates NLRC4 inflammasome to induce necroptosis through inhibiting the IL-17A-induced neutrophil accumulation in the lungs and IL-18 production (98). The deficiency of NLRC4 increases the pulmonary neutrophil infiltration, decreases the necroptosis, increases the pathogen clearance, and improves the host survival. Thus, the loss of NLRC4 in both hematopoietic and non-hematopoietic cells protects the host against S. aureus pneumonia (98).
The murine AECs also express NLRP6 inflammasome (63). NLRP6 activation during S. aureus pneumonia also increases the pyroptosis and necroptosis, causing necrotizing pneumonia, increases bacterial burden in the lungs, and decreases the pulmonary neutrophil infiltration (99). The neutrophils isolated from NLRP6 knockout (KO) animals exhibit an increased NADPH-dependent reactive oxygen species (ROS) production and increased bacterial killing. Thus, therapeutic targeting of NLRP3, NLRC4, and NLRP4 inflammasome during Gram-positive bacteria-induced severe pneumonia responsible for ALI may prove beneficial to the host. An experimental study has shown the beneficial effects (inhibition of ALI, decrease in pro-inflammatory cytokines levels, and decrease in the mortality) of resveratrol during K. pneumoniae-induced pneumonia through the NLRP3 inflammasome inhibition (100). NLRC4 activation during Gram-negative bacterial (K. pneumoniae, P. aeruginosa) pneumonia proves beneficial to the host via producing IL-1β, IL-17A, and neutrophil chemoattractants (keratinocyte cell-derived chemokines, MIP-2, and LPS-induced CXC chemokines) in the lungs (101). However, during P. aeruginosa pneumonia, NLRC4 activation-induces inflammatory lung damage, increases pulmonary bacterial burden, and necroptosis (102). Hence the inhibition of NLRC4 inflammasome activation during Gram-negative pneumonia remains a tricky scenario, and further studies will prove helpful in the direction.
The TLR2, TLR4, and MyD88 deficiency did not alter the host response during lethal pneumonia. Any abnormality in the AEC function may lead to the predisposition of the host toward pulmonary infections, including bacterial pneumonia due to the enhanced microbial colonization. For example, patients with allergic asthma are more prone to develop bacterial pneumonia due to the increased pathogenic bacterial colonization, including Staphylococcus aureus (S. aureus). It may be explained as the higher Th2 cytokines (IL-4 and IL-13) decrease the antimicrobial action of AECs via suppressing the synthesis of human β-defensins 2. However, during streptococcal or pneumococcal pneumonia, AECs also express secreted and transmembrane (Sectm) 1, Sectm1a, and Sectm1b genes due to the type 1 IFN signaling induction in AECs via signal transducer and activator of transcription 1 (STAT-1) activation (103). The Sectm1a binds to the neutrophils only in the presence of the infection and increases the CXCL2 expression. Thus, Sectm1 synthesis and release by AECs during pneumonia increase the neutrophil infiltration into the lungs and helps to clear the infection. However, its dysregulated synthesis may lead to the development of ALI or ARDS.
PECs protect from K. pneumoniae-induced pneumonia via ingesting and controlling their number through phagocytosing them via producing the complement component C3, which opsonizes them for phagocytosis (Figure 2A) (104). CD46 recognizes the C3 opsonized K. pneumoniae for the AEC-mediated phagocytosis or internalization (105). However, the complement resistant strains of K. pneumoniae have been emerged and are posing a potential threat to the host (106). The type 1 AECs also highly express epithelial membrane protein 2 (EMP2), a tetraspan protein, which promotes recruitment of different integrins (α6β1, αVβ3) and adhesion molecules (ICAM-1) to the lipid rafts (107). Both rodent and human type II AECs and AMs do not express EMP2 (108). The EMP2 expression of type 1 AECs plays a crucial role in the transepithelial neutrophil migration into the alveoli by regulating the expression of integrins and adhesion molecules (ICAM-1) and suppression of caveolins during bacterial pneumonia (109). Mice lacking EMP2 show a decreased neutrophil infiltration in the alveoli and lung injury during pneumonia. Thus, the activation of the residential PECs as innate immune cells during bacterial pneumonia plays a crucial role in the pathogenesis of pneumonia-associated ALI and its outcome depending on the pathogens (Gram-positive or Gram-negative bacteria) causing pneumonia and the associated immune response.
Pulmonary Innate Lymphoid Cells (ILCs) During Pneumonia and Associated ALI
ILCs serve as immunoregulatory innate immune cells at mucosal surfaces and play a crucial role in the pathogenesis of inflammation and inflammatory diseases (110, 111). The details of their development, classification, regulatory transcription factor (TFs), and function are described somewhere else (112–114). ILCs are divided into three major categories depending on their effector functions and transcriptional requirements: (1) Group 1 ILCs include type 1 ILCs, and Natural Killer (NK) cells, (2) Group 2 ILCs or ILC2s, and (3) Group 3 ILCs or ILC3s (114). Group 1 ILCs, including NK cells, are a rapid source of interferon-γ (IFN-γ), and mice deficient in IFN-γ develop more severe K. pneumoniae or L. pneumophila-induced pneumonia upon intratracheal inoculation of the pathogen due to impaired IL-1 and IL-6 production, and the defective clearance of the bacteria (115, 116).
NK cells in the lungs are present in its parenchyma in humans and comprise 10–20% of total lung lymphocytes, and in mice, they account for 10% of total lung lymphocytes (117, 118). Human lung NK cells are mostly CD16+CD56low, and KIR+CD57+NKG2A− highly differentiated NK cells are also found in the lungs (117, 119). However, the pulmonary resident NK cells express CD69, CD49a, and CD103, and most of them are CD56highCD16− and display a lesser mature form (120). In mice, pulmonary NK cells protect against K. pneumoniae-induced pneumonia via secreting IFN-γ and IL-22, which launch the bacterial growth-controlling interactions between alveolar macrophages and NK cells (Figure 2A) (121, 122). IFN-γ plays a crucial role in the bactericidal action of alveolar macrophages and the release of NK cell amplifying IL-12 and CXCL10 (122). The NCR1 (natural cytotoxicity receptor 1) on pulmonary NK cells controls their activation and the IFN-γ release during the early stages of S. pneumoniae-induced pneumonia without mediating the pathogen recognition (123). However, NCR1 ligands are expressed by pulmonary macrophages and DCs, which directly interact with NK cells during the early stages of S. pneumoniae-induced pneumonia (Figure 2B). This interaction increases their phagocytic activity required to clear the infection and mounting the effective immune response (Figure 2B).
Group 2 ILCs secrete Th2 cytokines [IL-4, IL-5, IL-6, IL-9, IL-13, and Amphiregulin (Arg)] and group 3 ILCs depending on the cytokine released, can be categorized into IL-17 secreting and IL-22 secreting ILC3s. In addition to these cytokines, ILC3s also secrete IL-26 (in humans), GM-CSF, and TNF-α (124). Lymphoid tissue inducer (LTi) cells also belong to group 3 ILCs and secrete IL-22 and IL-17. However, LTi cells have not been seen in the lungs in homeostasis and acute inflammation (125). In human lungs, they have also not been identified due to the lack of known human LTi markers (126). For example, CCR6 is a marker for mice LTi cells, but in humans, all ILC3s express CCR6, and therefore CCR6 does not serve as a marker for human LTi cells (127). Even tertiary lymphoid organs or follicles (TLOs or TLFs), such as iBALT form in the lung tissues of Rorc−/− and Id2−/− mice, which lack LTi cells, following influenza virus infection and inflammation (128). However, iBALT development depends on IL-17 secreted by Th17 cells, which triggers lymphotoxin-independent expression of CXCL13 and CCL19. Thus, LTi cells are dispensable for the aspect of lung immunity.
Lungs are the crucial sites for all the three groups of ILCs (125). Haemophilus influenza pulmonary infection increases the number of IFN-γ producing ILC1-like (Lin−IL-7Rα+IL-12Rβ2+IL-18Rα+Tbet+) cells and increases the pulmonary inflammatory immune response due to the plasticity among pulmonary ILC2s (129). AECs or PECs or pulmonary macrophages during pneumonia secrete IL-1β that governs the ILC2s plasticity (130). IL-1β impacts ILC2 plasticity via inducing the low expression of T-bet (TF) and inducing the IL-12Rβ2 expression, which converts these cells into ILC1s in the presence of IL-12 (131). The treatment with IL-12 during pneumonia exerts a protective action via increasing the infiltration of inflammatory cells (ILC1s, NK cells, and neutrophils) and inflammatory cytokines (IFN-γ) (132, 133). The transforming growth factor-β1 (TGF-β1) secreted by AECs or PECs primes pulmonary ILC2s (134). Pulmonary ILC2s express TGF-βRII. The CD127+CD90+CCR6+RORγt+ group 3 ILCs have been identified in the lung mucosa (135). Pulmonary ILC2s are unable to migrate efficiently within the lung tissue in the absence of TGF-β (134). IL-33 protects against pneumonia via enhancing bacterial clearance and improving the mortality via increasing the neutrophil infiltration and pulmonary ILC2s number (136). The ILC2s convert into ILC1s, which clear the pathogens. Also, these ILC2s are crucial for IL-13-dependent differentiation of pulmonary M2 macrophages, required for the resolution phase of inflammation and infection (137).
The S. pneumoniae pneumonia frequently induces the group 3ILCs accumulation in the lungs, which produce IL-22 to protect against severe pneumonia (Figure 2B). Furthermore, the administration of TLR5 agonist (flagellin) enhances the IL-22 production from group 3 ILCs during S. pneumoniae-induced pneumonia (Figure 2B). Studies have also shown earlier, the protective action of mucosal (including sublingual root) flagellin administration to mice infected with S. pneumoniae-induced pneumonia without the activation of NLRC4 inflammasome (138, 139). Also, the TLR5 agonist (flagellin) administration increases the efficacy of antibiotic treatment during pneumonia (140). The group 3 ILCs activation to produce IL-22 during S. pneumoniae-induced pneumonia also involves the activation of pulmonary dendritic cells (DC). Thus, the activation of pulmonary mucosal group 3 ILCs may prove beneficial to contain the pulmonary infection or pneumonia associated with severe lung inflammation and ALI. The number of group 3 ILCs producing IL-17 also increases during K. pneumoniae-induced pneumonia, which helps in the resolution of pulmonary inflammation at later stages to prevent the development of ALI during pneumonia (Figure 2A) (141). The release of TNF-α increases the pulmonary ILC3s number and also acts on AECs or PECs to synthesize CCL20. CCL20 chemoattracts ILC3s at the site of infection and inflammation (Figure 2B). Also, ILC3 produce IL-17A, which enhances the phagocytic uptake and killing of the bacteria by pulmonary macrophages to clear pneumonia (Figure 2A). Thus, ILC3s secrete IL-17A to clear infection during early stages and help in the resolution of the inflammation to prevent ALI during pneumonia.
The recruitment of IL-22 producing ILC3s into the lungs of neonate mice on exposure to commensal bacteria protects them from neonatal pneumonia (142, 143). This protection involves the intestinal mucosal DCs mediated sensing of commensal bacteria. Furthermore, the murine gut microbiota comprising segmented filamentous bacteria (Sfb) controls the resistance to the S. aureus pneumonia via enhancing the number of IL-22 and IL-17 producing innate immune cells (144). Hence pulmonary ILC3s protect the host from pneumonia and associated ALI during early childhood and later in adult life. Thus, pulmonary ILCs serve as crucial pulmonary innate immune cells to protect against pneumonia-induced ALI and in the resolution of the lung inflammation during pneumonia.
Pulmonary Macrophages During Bacterial Pneumonia and Associated ALI
Pulmonary macrophages account for 90–95% of lung immune cells at homeostasis (145). They are of two types: (1) Interstitial macrophages or IMs (reside in lung parenchyma and highly express CD11b but lower levels of CD11c), and (2) Alveolar macrophages or AMs (located in airway space, express high levels of CD11c and low levels of CD11b at their quiescent stage) in the healthy lung (146). Both AMs and IMs express the macrophage-specific markers [CD64 or Fc-gamma receptor 1 (FcγRI) and MER Proto-Oncogene or Tyro-Axl-MerTK (TAM) family of receptor Tyrosine Kinase (Mertk) is involved in efferocytosis] (147, 148). AMs are crucial for maintaining pulmonary immune homeostasis and host defense due to their unique location at the interface between the pulmonary mucosa and the external environment, and are inherently suppressive, whereas IMs exhibit the regulatory function in the lung (149). IMs produce high levels of IL-10 as compared to the AMs, which mainly produce non-specific antimicrobial molecules, including NO., TNF-α, and IFN-γ (146). The steady-state AMs express CD206 (a mannose receptor, which is a C-type lectin and serves as a PRR) and β-glucan specific receptor (Dectin-1), which are also expressed by alternatively-activated macrophages (AAMs) or M2 as their definitive markers (150, 151). The serum CD206 (sCD206) levels increases in the patients of community-acquired pneumonia (CAP) with the increase in its severity [pneumonia severity index (PSI)], which can be used for CAP prognosis (152). Also, the infiltration of CD206+ macrophages increases in the lungs of patients with fatal pneumonia.
AMs play a crucial role in the pathogenesis of bacterial pneumonia and associated ALI. For example, AMs during Gram-negative bacterial pneumonia produce TNF-α, which induces granulocyte-macrophage colony-stimulating factor (GM-CSF) in AECs that elicits proliferative signaling in AECs via autocrine stimulation contributing to the alveolar epithelial barrier restoration (153). However, during S. pneumoniae-induced pneumonia infiltrating peripheral macrophages replace the resident AMs and IMs. Also, the AM-mediated clearance of apoptotic cells decreases their potential to phagocytose the bacterial pathogens, which increases the bacterial burden in the lungs (Figure 2B) (154, 155). The efferocytosis induces the release of prostaglandin E2 (PGE2), which binds to the prostanoid receptors EP2-EP4 activating inhibitory cAMP and PKA pathway, which impairs the neutrophil infiltration and induces the IL-10 release to impair the pathogen clearance (155, 156). PGE2 also impairs the S. pneumoniae intracellular killing (ICK) by AMs via inhibiting the hydrogen peroxide (H2O2) production (157). IL-18 produced by AMs protects against pneumonia and ALI associated with S. pneumoniae infection via enhancing the bacterial clearance (158). However, IL-18 proves detrimental to P. aeruginosa-induced pneumonia and enhances its invasiveness to cause sepsis and ALI (159). Thus, type (Gram-positive or Gram-negative) of bacterial pathogens also determines the macrophage-mediated immune response, including the protective or detrimental action of IL-18 released.
The transition of M1 to M2 macrophages during the late stages of pneumonia mediates the inflammation resolution via producing IL-4 and IL-13, which promote STAT6 activation (160). Also, pulmonary macrophages secrete TNF-α stimulated gene-6 (TSG-6), which helps in the ALI resolution via promoting the M1 to M2 macrophage transition However, the efferocytosis of neutrophils by AMs during later stages of pneumonia helps in the resolution of lung inflammation due to expression of growth arrest-specific 6 (Gas6), a member of vitamin K-dependent family of proteins, which binds to its receptors Tyro3, Axl and Mer (TAM), or Mertk (148, 160). The Mertk activation causes ERK-mediated sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA2) expression to decrease the cytosolic Ca2+ levels, which suppresses the calcium//calmodulin-dependent protein kinase II (148). This process decreases the mitogen activating protein kinase (MAPK) and MK2 kinase activity to increase the abundance of non-phosphorylated cytosolic lipoxygenase (LOX), called 5-LOX, to enhance the production of specialized pro-resolving mediators (SPMs) mediating inflammation resolution (148). Thus, TSG-6 activates STAT6 to induce Gas6 expression in AMs for the ALI resolution during pneumonia.
Lipoxin A4 release by pulmonary endothelial cells, immigrated neutrophils, and pulmonary macrophages at later stages of pneumonia, inhibits neutrophil infiltration, promotes the efferocytosis of dead neutrophils by serving as a proapoptotic signal through downregulating Mac-1 (a β2 integrin) expression, to induce the pulmonary inflammation resolution (161–163). The lipoxin A4-induced neutrophil apoptosis involves the myeloperoxidase (MPO)-induced extracellular signal-regulated kinase (Erk) and Akt-mediated Bcl2-associated agonist of cell death (Bad) phosphorylation along with reducing the antiapoptotic protein myeloid cell leukemia-1 (Mcl-1) expression, which aggravates the mitochondrial dysfunction. This is because Mcl-1 promotes neutrophil survival through heterodimerization and neutralization of Bcl-2 interacting protein (Bim) or Bcl-2 homologous antagonist/killer (Bak) in the mitochondrial outer membrane (162, 164, 165). Lipoxin A4 also enhances the pathogen (E. coli) clearance by pulmonary macrophages through inducing the AMP expression (161). The Mac-1 binding to its ligands (ICAM-1, FBG, and MPO) suppresses the apoptosis (163). However, the Mac-1-dependent phagocytosis of complement-opsonized pathogens triggers rapid neutrophil apoptosis that depends on NADPH oxidase-generated reactive oxygen species (ROS) and caspase (CASP) activation (166). Lipoxin A4 also inhibits the CXCL8 or IL-8 release from pulmonary macrophages (167). Furthermore, Lipoxin A4 agonist, BML-111 induces autophagy in pulmonary macrophages through suppressing MAPK 1 and 8 signaling. The autophagy of pulmonary macrophages protects against ALI during Gram-negative bacterial pneumonia (168). Lipoxin A4-dependent autophagy among alveolar macrophages during pneumonia occurs independently of mTOR signaling. Hence pulmonary AMs play a crucial role in the induction of protective inflammatory immune during pneumonia and later on in the resolution of the inflammation.
This resolution process occurs at the expense of local pulmonary innate immunity comprising AMs (suppressing phagocytosis) to predisposes the recovering host to severe secondary pneumonia (169). This defective phagocytic function of AMs from pneumonia recovering animals stays for at least 28 days. Even the AMs transplanted intratracheally from normal mice to pneumonia recovered mice become paralyzed AMs, indicating the presence of long term inflammatory innate immune response suppression to make sure the complete resolution of the pulmonary inflammation (169). However, regulatory T cells (Tregs), cytokines (TGF-β1 and TNF-α), and DAMPs (HMGB1) do not play a significant role in the induction of paralyzed AMs during resolution of pulmonary inflammation following pneumonia. Also, these paralyzed AMs are not metabolically exhausted as they produce more lactate as compared to the control AMs and produce the same amount of TNF-α upon LPS challenge. Of note, the process of macrophage renewal in mice recovering from pneumonia is similar to normal mice. Thus, AMs of mice recovering from pneumonia are defective in phagocytosis and are unable to clear bacterial pathogens efficiently, increasing their susceptibility to secondary pneumonia. However, these defective or paralyzed AMs are derived from the local pulmonary macrophages in response to the increased expression of signal regulatory protein α (SIRPα), a regulator of tyrosine kinase-coupled signaling processes (phagocytosis) (170–172).
SIRPα increases during the resolution phase in response to the increased surfactant protein-D (SP-D) level. SP-D is an agonist for SIRPα and induces the immunosuppressive environment to produce trained but paralyzed AMs, which stay for weeks after infection to make sure the complete resolution of the inflammation. The increased Sirpα expression upregulates the Mir142 (a micro RNA regulating gene expression in mononuclear phagocytes) expression but down-regulates Setdb2 gene (encoding a histone methyltransferase, which controls the chemokine response during viral pneumonia) (173, 174). The Sedtb2 down-regulation may prevent neutrophil infiltration during the resolution phase to dampen the pneumonia-induced ALI, as indicated previously (173). Also, the Sedtb2 down-regulation alter the pro-inflammatory phenotype of macrophages to a reparative phenotype (175). Mir142 is also shown to regulate immunometabolic reprogramming and favors glycolysis via regulating fatty acid oxidation (FAO) through directly targeting carnitine palmitoyltransferase −1a (CPT1a), a key regulator of the FA pathway (176). Thus, pulmonary macrophages play a crucial role in the resolution of ALI associated with pneumonia.
The Interaction Between PECs and AMs During Pneumonia and Associated ALI
The AMs highly express CD200R (an OX2 glycoprotein of the superfamily of immunoglobulins) on their surface and its levels are maintained by epithelial expression of IL-10 and TGF-β (177). The PECs express ligand for CD200R called, CD200 on their apical side. The CD200R-CD200 interaction on AMs inhibits their pro-inflammatory action during pneumonia that prevents the induction of ALI (177). The CD200-CD200R interaction increases AAMs or M2a phenotype via cAMP-response element-binding protein-C/EBP-beta signaling and upregulates TGF-β expression (178). Also, M2a (anti-inflammatory or regulatory) macrophages generated in the presence of IL-4 and IL-13 also express CD200R in humans (179, 180). The CD200-CD200R interaction inhibits the downstream signaling pathway comprising of the ERK1/2 signaling pathway required for macrophage activation downstream of IFN-γ signaling through Janus-associated kinase (JAK)/STAT-1 activation (181). The CD200−/− mice develop ALI during pneumonia due to the increased pro-inflammatory function of macrophages and a decrease in the resolution of inflammation (177). Also, the AMs attached to the alveolar wall form connexin 43 (Cx43)-containing gap junction channels with the airway epithelium during bacterial pneumonia and intercommunicate through synchronized Ca2+ waves, through utilizing the epithelium as the conducting pathway (182, 183). This interaction further supports the anti-inflammatory role of PEC-AM interaction. As mice with AM-specific knockout of Connexin-43 show an increased neutrophil infiltration into the pulmonary alveoli and increased pro-inflammatory cytokine levels in BALF during Gram-negative bacterial (P. aeruginosa) pneumonia (182, 183). Thus, the interaction between PECs and AMs controls the inflammatory outcome of the pulmonary infections, including pneumonia leading to the development of ALI and its resolution.
Pulmonary Innate Immune Response During Bacterial Sepsis
Sepsis leads to the pulmonary inflammation that does not resolve and leads to the development of ALI or ARDS, causing irreversible damage to the lungs (12). Earlier studies have shown that the sepsis is responsible for more than 210,000 cases of ALI/ARDS in the US alone/annually, causing over 74,500 deaths (184, 185). The sepsis-associated ALI//ARDS has a higher mortality rate as compared to the ALI occurring due to other causes (186). The sepsis-associated ALI/ARDS may initiate on any side, including direct lung injury due to the pulmonary epithelial damage or indirect damage comprising the endothelial cell damage (187, 188). The neutrophil infiltration plays a crucial event in this outcome, and the recruitment of neutrophils into the lungs depends on the expression of E-selectin [CD62E or endothelial-leukocyte adhesion molecule 1 (ELAM-1), or leukocyte-endothelial cell adhesion molecule-2 (LECMA-2)]. E-selectin does not express on unstimulated endothelium, but its expression increases on pulmonary vascular endothelium due to the impact of pro-inflammatory cytokines and induces neutrophil infiltration in sepsis-induced ALI (Figure 3) (189).
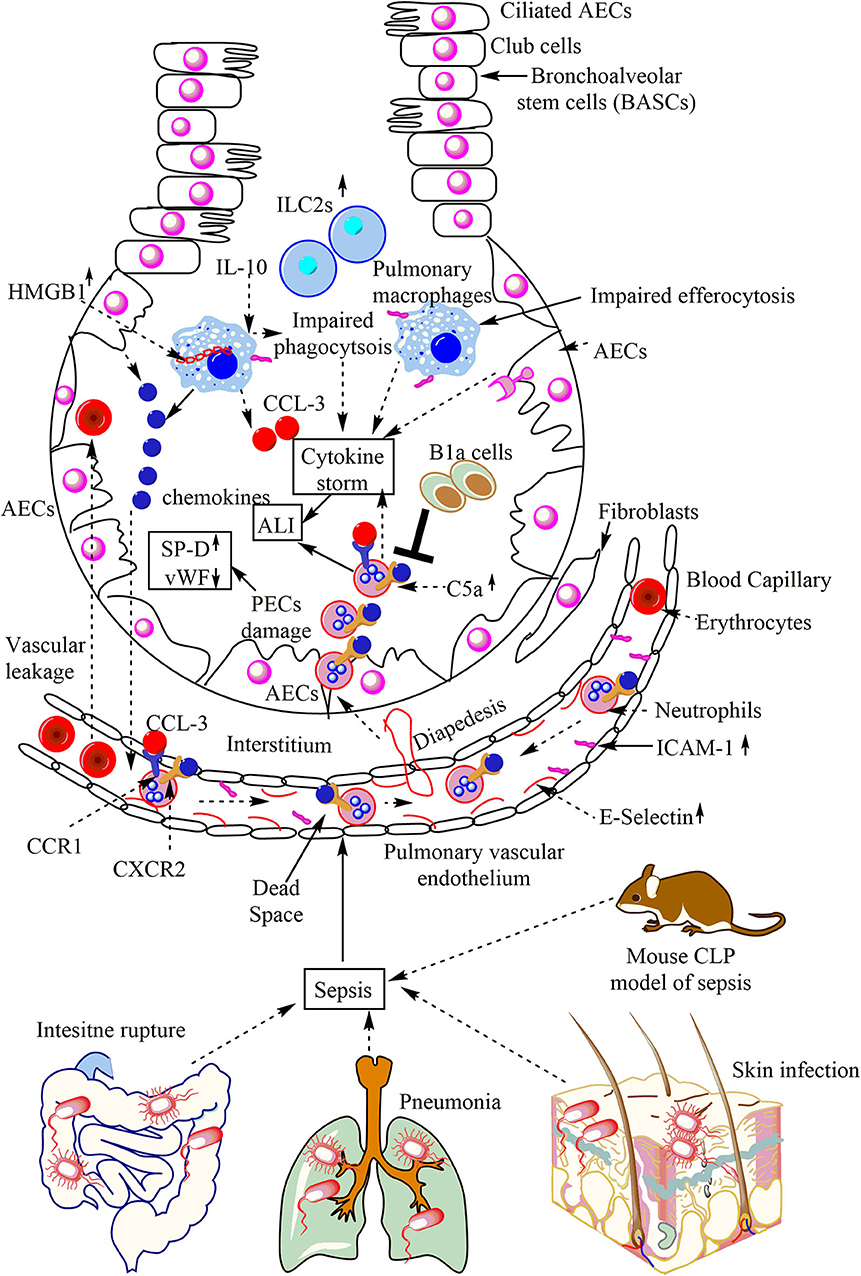
Figure 3. Overview of sepsis-induced ALI. Local infections of the skin (S. aureus), lungs (pneumonia), and intestinal commensal bacteria leak into the blood may lead to sepsis development. Sepsis leads to the neutrophil and monocyte infiltration in the lung alveoli via pulmonary transendothelial migration due to the profound release of the pro-inflammatory mediators (cytokine storm) damaging endothelial monolayer and inducing endothelial vascular leakage. These neutrophils and monocytes reach into the lung alveoli through crossing the pulmonary epithelial layer due to damage of PECs (AECs and BECs). These PECs express C5aR and C3aR receptors. The profound release of complement component C5a during sepsis induces the inflammatory damage, and the death of PECs during sepsis causes sepsis-associated ALI. The PECs death/damage increases the SP-D, but Vwf levels decrease. The increase in the IL-10 levels in the lungs at later stages of sepsis impairs the bactericidal action of AMs along with inducing a defective efferocytosis. The defective efferocytosis among AMs further increases the ALI. The necrotic death of AMs (indicated by the cytosolic HMG-B1) at later stages of sepsis further aggravates the ALI. The neutrophils infiltrated into the lung alveoli during sepsis are apoptosis-resistant and aggravate the ALI due to their increased pro-inflammatory action on lung tissues. B1a cells inhibit neutrophil infiltration and, thus, the sepsis-induced ALI. The increase in pulmonary ILC2s also occurs.
The immunohistochemical analysis has shown an increased expression of CD62E or E-selectin in the pulmonary microvasculature in sepsis-associated fatalities. The pulmonary intravascular, interstitial and intra-alveolar leukocytes strongly express very late antigen-4 or VLA-4 (CD49d/CD29) or α4β1integrin in sepsis-associated casualties. The ICAM-1 (CD54) is highly expressed on the pulmonary endothelial cells, pulmonary macrophages, and lymphocytes in sepsis-associated fatalities (Figure 3). The pulmonary epithelial damage during ALI/ARDS seen in patients with pneumonia-associated sepsis may be indicated by an elevation of surfactant protein-D (SP-D). However, but these patients have low levels of von Willebrand factor (vWF) and IL-6, and IL-8, which are the markers of endothelial damage (Figure 3) (190). The pulmonary epithelial damage seen during direct sepsis-associated ALI is more severe as compared to the indirect non-pneumonia-mediated sepsis (190). However, the damage to the endothelium during direct sepsis-associated ALI is less severe. The pulmonary B1a cells exert a protective role in cecal-ligation and puncture (CLP)-induced sepsis via inhibiting neutrophil infiltration and MPO production in the lungs (Figure 3) (191). The CXCR2−/− mice exposed to peritoneal sepsis show a decreased pulmonary damage due to the low neutrophil infiltration in the lungs, and the increased CXCL10 expression in the peritoneum (192). CCL-3 or macrophage inflammatory protein-1α (MIP-1α) also mediates sepsis-induced ALI via promoting neutrophil infiltration, pulmonary vascular leakage, and early mortality (Figure 3) (193). The following sections highlight the pulmonary innate immune response during sepsis-induced ALI//ARDS.
PECs During Sepsis and Associated ALI
The generation of pro-inflammatory molecules (cytokines, ROS, and RNS) and hypoxia damage the pulmonary epithelial barrier during sepsis-induced ALI (Figure 2) (194, 195). This damage to the pulmonary epithelium alters its barrier function and induces the fluid and protein leakage into the alveolar space. The injury to both type I and II AECs during sepsis can easily be assessed in both plasma and pulmonary lavage fluid by the presence of several biomarkers as described previously (194, 196). The pulmonary epithelial damage and increase in its permeability during sepsis involve the change in actin organization (197). The PECs damage due to actin reorganization during sepsis does not include MAPK signaling or the alterations in the tight junction (TJ) proteins. The PECs (BECs and AECs) of the septic lung show an increased αvβ3 integrin, but its inhibition during sepsis-associated ALI needs to study as it may increase the endothelial permeability and thus the sepsis-associated ALI (198, 199). The later (proliferative or fibroproliferative stage observed during the second week of sepsis) stages of ALI during sepsis involves the transformation of the damaged epithelial cells to fibroblast-like cells (epithelial-mesenchymal transition), which requires mitochondrial ROS and hypoxia-inducible factor-1α (HIF-1α) (200).
The PECs express a higher Fas level during sepsis-associated ALI, and the increased infiltration of FasL expressing inflammatory immune cells in the lungs occurs (201). The apoptotic death of PECs during non-pulmonary sepsis (sepsis originating outside the lungs or in the absence of pulmonary infection) involves Fas-FasL interaction, and the Fas inhibition protects their apoptotic cell death via diminishing lung tissue TNF-α, IL-6, IL-10, IFN-γ, IL-12, and CASP-3 activity (202). The BECs show an increased expression of both C3aR and C5aR during sepsis (203). An increased intrapulmonary or intra alveolar C5a level during sepsis may cause severe ALI via binding to the C5aR1 or C5aR, which induces an increased neutrophil infiltration into the septic lung and cytokine/chemokine storm (Figure 3) (204, 205). The infiltrated neutrophils in the lungs during ALI/ARDS have a distinctive phenotype and are resistant to apoptosis, and exhibit an enhanced phosphoinositide 3-kinase-dependent (PI3K)-dependent respiratory burst (206). A human study has also indicated the infiltration of less apoptotic neutrophils in the lungs of Sepsis-associated ALI/ARDS patients (207). Hence neutrophils migrated to the lungs during sepsis-associated ALI exert more damaging effects to the lungs as compared to bacterial pneumonia (Figure 3).
The apoptosis of neutrophils enhances the resolution of the inflammation that is lost in the sepsis-associated ALI. However, the cyclin-dependent kinase (CDK) inhibitor, called AT7519 enhances the apoptosis of infiltrated neutrophils during sepsis-associated ALI or ARDS, can be used as a mediator of initiating the resolution phase of inflammation during sepsis-associated ALI (208). Of note, mechanisms causing ALI and resolution of inflammation occur in parallel during sepsis-associated ALI/ARDS. The first resolution step involves the reestablishment of the alveolar-capillary barrier and the migration of AT-II epithelial cells to replace injured AT I epithelial cells, following the proliferation of tissue-resident progenitor cells (187). However, the uncontrolled inflammatory process causing severe ALI overpowers the resolution process, which causes irreversible damage during Gram-negative bacteria (Klebsiella pneumoniae)-induced sepsis as indicated by accumulation of the lungs with apoptosis-resistant neutrophils and elevation of pro-inflammatory cytokines (IL-1α, TNF-α) in BALF (12). Furthermore, keratinocyte growth factor (KGF) treatment induced the resolution in PECs in vitro and in vivo in mice, but it failed in phase II clinical trial and aggravated the ALI (209–211). Thus, due to severe PEC damage during sepsis-induced ALI, it is more damaging and irreversible as compared to the ALI observed during bacterial pneumonia only without the development of sepsis.
ILCs During ALI/ARDS Observed During Sepsis
There is a doubt regarding the presence of ILC1s in naïve lungs or during homeostasis. However, their number increases during Haemophilus influenzea infection. It occurs due to the phenotypic change in lung-resident ILC2s in response to the downregulation of T1/ST2, GATA3, IL-5, and IL-13 expression (125). ILC2s represent the majority of ILCs in both mouse and human (30% of all ILCs in adult human lungs) lungs (212, 213). Although Lungs have a low number of ILC2s in the steady-state, and it increases only during pulmonary allergic diseases. Lungs have both NCR+ and NCR− ILC3s during a steady state. Lin-CD127+RORγt+ILC3s comprise 30% of ILCs in mice, and the majority of them also co-express CCR6. These can be activated with IL-23 and IL-1β in vitro to produce IL-22 and IL-17A (135). The human lungs also have ILC3s (Lin−CD127+CRTh2−CD117+) expressing RORγt. During S. pneumoniae lung infection, the depletion of ILC3s protects the host from ALI due to inhibition of IL-22 and IL-17A production (135).
The systemic levels of ILCs (ILC1s and ILC3s) significantly decrease in patients with sepsis in comparison to the control group due to their increased apoptotic death (214). The HLA-DR expression increases in the ILCs of the septic patients without any effect on their capacity to produce TNF-α in response to the TLR agonists. The apoptotic cell death among ILCs (ILC1s, ILC2s, and ILC3s) occurs due to the increase in CASP3 level and activity within <24 h of sepsis diagnosis (214). However, no significant decrease in systemic ILC2s occurs during the early stages of sepsis despite the increase in CASP3 activity. It may be attributed to the sphingosine-1-phosphate (S1P)-dependent migration of ILC2s to distant organs, including lungs (215). The plasma S1P-1 level decreases with the severity of the sepsis (216). The ILC2 migration to the lungs in response to S1P occurs due to increased expression of S1P receptors (S1PRs, S1P1-SIP5) (217). A study in CLP-induced sepsis has shown the increase in ILC2s in the peritoneum and small intestine along with the increased IL-13 and IL-33 levels in the peritoneal lavage fluid (PLF) within 24 h post sepsis development (218).
Patients with sepsis show increased plasma IL-33 levels (218). Many investigators have suggested different roles of pulmonary ILC2s during sepsis, depending on the experimental model. For example, increased IL-33 levels (released by epithelial cells of the lungs) in CLP-induced sepsis in mice cause sepsis-induced ALI, and IL-33 inhibition causes a decrease in sepsis-associated ALI due to the decreased neutrophil and monocyte infiltration into the lungs (219). This IL-33 dependent ALI during sepsis also occurs via IL-5 upregulation in pulmonary ILC2s, and the IL-5 neutralization decreases the neutrophil infiltration, and ALI during sepsis (219). Thus, an increased activation of pulmonary ILC2s during CLP-induced sepsis may contribute to the sepsis-associated ALI. However, another study has shown the protective effect of the pulmonary ILC2s during sepsis-induced ALI via preventing the endothelial cell damage in response to the IL-33 released, which via binding to the ST2 receptor, mediates the ILC2 expansion (220).
The pulmonary ILC2s produce IL-9, which prevents CASP1 activation and the pyroptosis of pulmonary endothelial cells. It reduces the sepsis-associated ALI severity. The pulmonary ILC2s increase within the first 12 h of the sepsis development along with an increase in the peritoneal ILC2s (220). However, in another study, the ILC2s (as measured by the production of IL-5 and IL-13) pre-activation via intra-tracheal IL-33 administration before the lethal S. aureus sepsis protects the host from ALI and death via pulmonary eosinophilia induction, which clears the pathogen from the lungs and suppresses neutrophilia (221). However, without IL-33 pre-treatment, S. aureus is unable to induce ILC2 proliferation and function. Hence ILC2s play both beneficial and detrimental roles in ALI and sepsis-associated mortality depending on their activation stage. It will be interesting to investigate the impact of sepsis-associated ALI in people previously affected with parasitic infections causing a rise in pulmonary ILC2s and eosinophilia. Thus, pulmonary ILCs are crucial innate immune cells of the lungs, but their relevance to the sepsis-induced ALI/ARDS is a topic for the research and future immunomodulatory therapeutics. However, a decrease in the systemic ILC population is well-described even during the early phase of the sepsis.
Alveolar Macrophages (AMs) and Sepsis-Induced ALI//ARDS
The pro-inflammatory mediators released from AMs play a crucial role in the sepsis-induced ALI via inducing neutrophil infiltration into the lungs (Figure 3). The interstitial-to-vascular chemotactic gradient establishment facilitates the emigration of the vascular neutrophils in the lung alveoli (Figure 3) (222, 223). The bacterial peritonitis-induced sepsis activates AMs and neutrophil infiltration in the lung alveoli (Figure 3). The neutrophil infiltration in the lung alveoli occurs via pulmonary transendothelium in response to the AM activation during sepsis (Figure 3) (224). The NADPH oxidase activation in the pulmonary endothelium generates superoxide anion in response to the AM activation that plays a crucial role in the transendothelial neutrophil migration during sepsis-associated ALI (224). These neutrophils are less prone to apoptotic cell death and play a significant role in the sepsis-induced severe ALI (Figure 3). Furthermore, these infiltrated neutrophils block pulmonary microcirculation due to their prolonged entrapment in the capillaries inducing the dead space, which further aggravates the sepsis-induced ALI (Figure 3) (225). These neutrophils express Mac-1 (CD11b/CD18), and the Mac-1 inhibitor decreases the incidence of disturbing pulmonary microcirculation and the sepsis-induced ALI. Also, the impaired phagocytic activity of AMs during late stages in response to the released IL-10 during abdominal sepsis further enhances the incidence and the severity of sepsis-induced ALI (Figure 3) (226).
The impaired efferocytosis by AMs during the late stages of sepsis further increases the severity of ALI/ARDS due to the accumulation of dead neutrophils and other pulmonary cells (Figure 3) (227). However, the IFN-β treatment reverses the impaired AM function in response to the IL-10 at the late stage of sepsis and decreases the severity of sepsis-associated ALI/ARDS and the associated mortality (228). The HMG-B1 release in the cytosol of AMs during the late sepsis indicates their necrotic cell death, which further increases the ALI severity (Figure 3) (229). The macrophages endocytose HMG-B1 during sepsis (230). The HMG-B1 promotes pyroptosis of macrophages and endothelial cells by delivering the LPS via the receptor for advanced glycation end products (RAGE) into the cytosol, which destabilizes phagolysosome and induces CASP11 activation during lethal sepsis (231).
The CASP11 is the important inflammasome component, and HMG-B1 is known to activate NLRP3 inflammasome and the IL-1β release. The CASP11 activation causes pyroptosis via cleaving gasdermin D (GSDMD) into amino-terminal GSDMD (N-GSDMD) and carboxy-terminal GSDMD (C-GSDMD) (232, 233). The N-GSDMD is responsible for the pyroptosis. The increased lipid peroxidation (LPO) in the sepsis-associated ALI has been observed (12). This increased LPO further activates CASP11, and thus, the GSDMD to cause the pyroptosis of AMs and infiltrated monocytes and macrophages in a phospholipase C gamma 1 (PLCG1)-dependent manner (234). Also, the inflammatory IL-1β reduces the cyclic adenosine monophosphate (cAMP) and transcription factor cAMP response element-binding (CREB) in lung endothelial cells (235). This CREB blockage inhibits the VE-cadherin transcription, which induces pulmonary vascular endothelial damage to aggravate pulmonary vascular leakage and sepsis-associated ALI. Also, the treatment with rolipram (a drug inhibiting the type 4 cyclic nucleotide phosphodiesterase–mediated (PDE4-mediated) hydrolysis of cAMP) prevents sepsis-induced pulmonary vascular injury and thus the ALI via preserving the CREB-mediated VE-cadherin expression (235). Of note, the deficiency of neutrophils before sepsis also impairs the monocyte and macrophage infiltration in the lungs during both early and late stages and thus inflammatory process (236).
The iNOS induction in AMs during sepsis also causes protein leakage in the lungs and sepsis-induced ALI. The AM depletion attenuates the sepsis-induced increase in pulmonary microvascular protein leak and MPO activity that depends on the activation of iNOS (237). The increased nitric oxide (NO.) and MPO levels in BALF and lung homogenate of mice subjected to K. pneumoniae B5055-induced sepsis has been reported on all days in an experimental study (12). Furthermore, microRNA-199a (miR-199a) upregulation in AMs during Gram-negative bacterial sepsis also aggravates the sepsis-induced ALI, which can be prevented by the activation of sirtuin 1 [SIRT1 (Silent information regulator 2 homolog 1), an NAD+-dependent class III protein deacetylase or histone deacetylase regulating cell growth, differentiation, stress resistance, oxidative damage, and metabolism] (238, 239). The induction of miR-199a in AMs during sepsis increases the release of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α), the MPO activity, ALI, and the high levels of CASP3, Bax and lowers the Bcl-2 levels (238). The miR-199a inhibition during sepsis decreases the release of pro-inflammatory cytokines from AMs, MPO activity, the incidence of vascular leakage from pulmonary endothelium. The SIRT1 activation during sepsis also prevents the sepsis-induced ALI via inhibiting the NLRP3 inflammasomes in AMs and pulmonary vascular endothelial cells, which prevents the release of pro-inflammatory mediators (ICAM-1 and HMG-B1), disruption of tight and adherens junctions as indicated by the reduced lung claudin-1 and vascular endothelial-cadherin expression (240, 241).
The sepsis-associated altered AM function predisposes these mice to severe pulmonary infections and increases their mortality when challenged with Gram-negative bacteria (P. aeruginosa) due to the IL-1 receptor-associated kinase–M (IRAK-M) upregulation, which causes sepsis-associated immunosuppression at later stages (242). IRAK-M-mediated impaired TNF-α and iNOS expression in AMs is associated with the reduced acetylation and methylation of specific histones (AcH4 and H3K4me3) and reduced binding of RNA polymerase II to the promoters of these genes (243). However, the TLR2 and TLR4 levels remain the same in septic lungs as compared to the control group (242). Of note, diabetic rats show milder sepsis-associated ALI due to the impaired activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), increased suppressor of cytokine signaling 1 (SOCS1), and decreased MyD88 mRNA, and thus the decreased MyD88 downstream signaling in response to the TLR stimulation on AMs (244). The decreased AM activation in diabetic rats inhibits neutrophil infiltration, cyclo-oxygenase II (COX-II) expression and activity, and the pulmonary edema. The low incidence of sepsis-induced ALI has been also observed in patients with diabetes developing sepsis (245–248). However, a recent clinical study indicates that diabetes does not have any impact on sepsis-associated mortality and the 60-days mortality of ALI/ARDS (249). Diabetes may reduce the incidence of the sepsis-induce ALI/ARDS but not the associated mortality. Further studies in the direction and the establishment of AMs role in clinical patients of sepsis with diabetes about the ALI may prove helpful in a patient-specific therapeutic approach.
Hence sepsis leads to the severe ALI as compared to the ALI seen in pneumonia only patients. Furthermore, sepsis causes prolonged immunosuppressive stage in the lungs, which increases the chances of developing severe secondary pulmonary infections (hospital-acquired or community-acquired). For example, the pulmonary alveolar macrophages decrease in number in patients recovered from sepsis and show defective phagocytic function against bacterial pathogens (E. coli and S. aureus), which are frequently responsible for hospital-acquired pneumonia (169). This defective number of pulmonary alveolar macrophages stays at least for 6 months. These clinical findings (severely compromised phagocytic activity of AMs) have further been confirmed in mice subjected to secondary pneumonia caused by E. coli or S. aureus (169). These macrophages from patients recovering from sepsis also showed increased SIRPα expression. However, the outcome of sepsis-associated ALI may also depend on several other chronic inflammatory conditions, including type 2 diabetes mellitus (T2DM). Further studies are urgently required in the field due to the high mortality of sepsis patients due to the Sepsis-induced ALI/ARDS.
Conclusion
Pneumonia and sepsis, both are associated with the onset of ALI/ARDS. However, the pneumonia-associated ALI is less severe and often resolves once the infection has cleared. But this resolution of ALI during sepsis has not been observed. However, the pneumonia-associated ALI resolution leaves a long-lasting impact on the host immune response to future infection. This resolution of ALI involves the transforming growth factor-β (TGF-β) generation and the activation of pulmonary regulatory T cells (Tregs) inducing the immunosuppressive environment in the lungs (169). It causes the induction of paralyzed pulmonary macrophages and DCs, which are defective in the phagocytosis of the pathogen but further secrete TGF-β responsible for the Tregs accumulation. These paralyzed DCs express an increased amount of transcription repressor called B lymphocyte-induced maturation protein-1 (Blimp-1) but a lower amount of interferon regulatory factor 4 (IRF4). Blimp-1 is essential for tolerogenic DCs. Thus, the increased expression of Blimp-1 induces a tolerogenic phenotype of DCs. Whereas, IRF-4 is for expressing the molecules required for the antigen presentation, and its lower level in paralyzed DCs decrease their antigen presentation potential.
The immunosuppressive environment in the lung following the resolution of ALI-associated with pneumonia further disposes the host to acquire secondary pulmonary infection. However, this can be avoided by following the instruction, like keeping the recovered patient in the pathogen-free environment and keeping him/her on the immune-boosting diet. For example, the study has shown that these paralyzed macrophages and DCs generated following the resolution of pneumonia-associated ALI remains active for at least 21 days post clearance of the pathogen (169). Hence the impairment in the expression of Blimp-1 and IRF-4 in other immune cells, including PECs, T, and B cells post, pneumonia should also be studied to explore the unknowns associated with the cost of resolution of ALI associated with pneumonia and the patients recovered from sepsis. ALI during sepsis proves detrimental to the host. Even the neutrophils and monocytes infiltrated into the septic lungs show the apoptosis-resistant phenotype that proves harmful to the host by further aggravating the sepsis-induced ALI. Thus, therapeutics able to induce their phagocytosis later in the sepsis will prove beneficial to prevent the sepsis-induced ALI. Keeping in mind the difference in the action of the pulmonary innate immune response during sepsis and pneumonia-induced ALI, different therapeutics should be designed as the drug or molecule worth for one may not be useful for the other. Future studies are required in the direction to prevent the sepsis or pneumonia-induced ALI by studying the pulmonary, innate immunity. For example, the discovery of ILCs in the lungs and further research in their functional role in pneumonia and sepsis-induced ALI has changed their pathogenesis and opened the door to design better and new therapeutics, including the vaccines for pneumonia.
Author Contributions
VK has developed the idea, searched the literature, compiled, and wrote the article.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rezoagli E, Fumagalli R, Bellani G. Definition G, and epidemiology of acute respiratory distress syndrome. Ann Transl Med. (2017) 5:282. doi: 10.21037/atm.2017.06.62
3. Bernard G. Acute lung failure — our evolving understanding of ARDS. N Engl J Med. (2017) 377:507–9. doi: 10.1056/NEJMp1706595
4. Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. (1967) 2:319–23. doi: 10.1016/S0140-6736(67)90168-7
5. Sessler CN, Bloomfield GL, Fowler AA. Current concepts of sepsis and acute lung injury. Clin Chest Med. (1996) 17:213–35. doi: 10.1016/S0272-5231(05)70310-5
6. Pham T, Rubenfeld GD. Fifty years of research in ARDS. The epidemiology of acute respiratory distress syndrome. A 50th birthday review. Am J Respir Crit Care Med. (2017) 195:860–70. doi: 10.1164/rccm.201609-1773CP
7. Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem. (2014) 289:35237–45. doi: 10.1074/jbc.R114.619304
8. Kumar V. A STING to inflammation and autoimmunity. J leukoc Biol. (2019) 106:171–85. doi: 10.1002/JLB.4MIR1018-397RR
9. Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. (2018) 59:391–412. doi: 10.1016/j.intimp.2018.03.002
10. Hoving JC, Wilson GJ, Brown GD. Signalling C-type lectin receptors, microbial recognition and immunity. Cell Microbiol. (2014) 16:185–94. doi: 10.1111/cmi.12249
11. Franchi L, Warner N, Viani K, Nuñez G. Function of Nod-like receptors in microbial recognition and host defense. Immunol Rev. (2009) 227:106–28. doi: 10.1111/j.1600-065X.2008.00734.x
12. Kumar V, Chhibber S. Acute lung inflammation in Klebsiella pneumoniae B5055-induced pneumonia and sepsis in BALB/c mice: a comparative study. Inflammation. (2011) 34:452–62. doi: 10.1007/s10753-010-9253-9
13. Guirgis FW, Khadpe JD, Kuntz GM, Wears RL, Kalynych CJ, Jones AE. Persistent organ dysfunction after severe sepsis: a systematic review. J Crit Care. (2014) 29:320–6. doi: 10.1016/j.jcrc.2013.10.020
14. Bienenstock J. The lung as an immunologic organ. Ann Rev Med. (1984) 35:49–62. doi: 10.1146/annurev.me.35.020184.000405
15. Askonas BA, Humphrey JH. Formation of specific antibodies and gamma-globulin in vitro. A study of the synthetic ability of various tissues from rabbits immunized by different methods. Biochem J. (1958) 68:252–61. doi: 10.1042/bj0680252
16. Bull CG, McKee CM. Respiratory immunity in rabbits. vii. Resistance to intranasal infection in the absence of demonstrable antibodies. Am J Epidemiol. (1929) 9:490–9. doi: 10.1093/oxfordjournals.aje.a121662
17. Bivas-Benita M, Gillard GO, Bar L, White KA, Webby RJ, Hovav AH, et al. Airway CD8(+) T cells induced by pulmonary DNA immunization mediate protective anti-viral immunity. Mucosal Immunol. (2013) 6:156–66. doi: 10.1038/mi.2012.59
18. Takaki H, Ichimiya S, Matsumoto M, Seya T. Mucosal immune response in nasal-associated lymphoid tissue upon intranasal administration by adjuvants. J Innate Immun. (2018) 10:515–21. doi: 10.1159/000489405
19. Song K, Bolton DL, Wei C-J, Wilson RL, Camp JV, Bao S, et al. Genetic immunization in the lung induces potent local and systemic immune responses. Proc Natl Acad Sci. (2010) 107:22213–8. doi: 10.1073/pnas.1015536108
20. McDermott MR, Befus AD, Bienenstock J. The structural basis for immunity in the respiratory tract. Int Rev Exp Pathol. (1982) 23:47–112.
21. Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. I. Morphologic characteristics. Lab Invest. (1973) 28:686–92.
22. Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. II. Functional characterisitics. Lab Invest. (1973) 28:693–8.
23. Richmond I, Pritchard GE, Ashcroft T, Avery A, Corris PA, Walters EH. Bronchus associated lymphoid tissue (BALT) in human lung: its distribution in smokers and non-smokers. Thorax. (1993) 48:1130–4. doi: 10.1136/thx.48.11.1130
24. Bienenstock J. Gut and bronchus associated lymphoid tissue: an overview. Adv Exp Med Biol. (1982) 149:471–7. doi: 10.1007/978-1-4684-9066-4_66
25. Kimura S, Mutoh M, Hisamoto M, Saito H, Takahashi S, Asakura T, et al. Airway M cells arise in the lower airway due to RANKL signaling and reside in the bronchiolar epithelium associated with iBALT in murine models of respiratory disease. Front Immunol. (2019) 10:1323. doi: 10.3389/fimmu.2019.01323
26. Corr SC, Gahan CCGM, Hill C. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol Med Microbiol. (2008) 52:2–12. doi: 10.1111/j.1574-695X.2007.00359.x
27. Bockman DE, Cooper MD. Pinocytosis by epithelium associated with lymphoid follicles in the bursa of Fabricius, appendix, Peyer's patches. An electron microscopic study. Am J Anat. (1973) 136:455–77. doi: 10.1002/aja.1001360406
28. Rácz P, Tenner-Rácz K, Myrvik QN, Fainter LK. Functional architecture of bronchial associated lymphoid tissue and lymphoepithelium in pulmonary cell-mediated reactions in the rabbit. J Reticuloendothel Soc. (1977) 22:59–83.
29. Tschernig T, Pabst R. Bronchus-associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology. (2000) 68:1–8. doi: 10.1159/000028109
30. Gould SJ, Isaacson PG. Bronchus-associated lymphoid tissue (BALT) in human fetal and infant lung. J Pathol. (1993) 169:229–34. doi: 10.1002/path.1711690209
31. Ersch J, Tschernig T, Stallmach T. Frequency and potential cause of bronchus-associated lymphoid tissue in fetal lungs. Pediatr Allergy Immunol. (2005) 16:295–8. doi: 10.1111/j.1399-3038.2005.00269.x
32. Meuwissen HJ, Hussain M. Bronchus-associated lymphoid tissue in human lung: correlation of hyperplasia with chronic pulmonary disease. Clin Immunol Immunopathol. (1982) 23:548–61. doi: 10.1016/0090-1229(82)90139-8
33. Sato A. Basic and clinical aspects of bronchus-associated lymphoid tissue. Nihon Kokyuki Gakkai Zasshi. (2000) 38:3–11.
34. Lloyd CM, Marsland BJ. Lung homeostasis: influence of age, microbes, and the immune system. (2017) 46:549–61. doi: 10.1016/j.immuni.2017.04.005
35. Gallacher DJ, Kotecha S. Respiratory microbiome of new-born infants. Front Pediatr. (2016) 4:10. doi: 10.3389/fped.2016.00010
36. Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. (2010) 107:11971–5. doi: 10.1073/pnas.1002601107
37. Gollwitzer ES, Saglani S, Trompette A, Yadava K, Sherburn R, McCoy KD, et al. Lung microbiota promotes tolerance to allergens in neonates via PD-L1. Nat Med. (2014) 20:642–7. doi: 10.1038/nm.3568
38. Wang J, Li F, Sun R, Gao X, Wei H, Li LJ, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat Commun. (2013) 4:2106. doi: 10.1038/ncomms3106
39. Murphy SL, Kochanek KD, Xu J, Arias E. Mortality in the United States, 2014. NCHS Data Brief . (2015) 1–8. doi: 10.1093/gao/9781884446054.article.T2271585
40. Murphy SL, Xu J, Kochanek KD, Arias E. (2018) Mortality in the United States, 2017. NCHS Data Brief. 328:1–8.
41. Cilloniz C, Martin-Loeches I, Garcia-Vidal C, San Jose A, Torres A. Microbial etiology of pneumonia: epidemiology, diagnosis and resistance patterns. Int J Mol Sci. (2016) 17:2120. doi: 10.3390/ijms17122120
42. Arnold FW, Summersgill JT, Ramirez JA. Role of atypical pathogens in the etiology of community-acquired pneumonia. Semin Respir Crit Care Medi. (2016) 37:819–28. doi: 10.1055/s-0036-1592121
43. Cilloniz C, Torres A, Niederman M, van der Eerden M, Chalmers J, Welte T, et al. Community-acquired pneumonia related to intracellular pathogens. Intensive Care Med. (2016) 42:1374–86. doi: 10.1007/s00134-016-4394-4
44. Branger J, Knapp S, Weijer S, Leemans JC, Pater JM, Speelman P, et al. Role of toll-like receptor 4 in gram-positive and gram-negative pneumonia in mice. Infect Immun. (2004) 72:788–94. doi: 10.1128/IAI.72.2.788-794.2004
45. Rossaint J, Zarbock A. Tissue-specific neutrophil recruitment into the lung, liver, and kidney. J Innate Immun. (2013) 5:348–57. doi: 10.1159/000345943
46. Maas SL, Soehnlein O, Viola JR. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front Immunol. (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
47. Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol. (2009) 40:519–35. doi: 10.1165/rcmb.2008-0348TR
48. Mizgerd JP. Molecular mechanisms of neutrophil recruitment elicited by bacteria in the lungs. Semin Immunol. (2002) 14:123–32. doi: 10.1006/smim.2001.0349
49. Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, et al. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. (1996) 154:602–11. doi: 10.1164/ajrccm.154.3.8810593
50. Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, et al. Interleukin-8 C, and development of adult respiratory distress syndrome in at-risk patient groups. Lancet. (1993) 341:643–7. doi: 10.1016/0140-6736(93)90416-E
51. Koltsova EK, Ley K. The mysterious ways of the chemokine CXCL5. Immunity. (2010) 33:7–9. doi: 10.1016/j.immuni.2010.07.012
52. Mei J, Liu Y, Dai N, Favara M, Greene T, Jeyaseelan S, et al. CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity. (2010) 33:106–17. doi: 10.1016/j.immuni.2010.07.009
53. Miller EJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, et al. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis. (1992) 146:427–32. doi: 10.1164/ajrccm/146.2.427
54. Cai S, Batra S, Lira SA, Kolls JK, Jeyaseelan S. CXCL1 regulates pulmonary host defense to Klebsiella infection via CXCL2, CXCL5, NF-κB, and MAPKs. J Immunol. (2010) 185:6214–25. doi: 10.4049/jimmunol.0903843
55. Zhou X, Dai Q, Huang X. Neutrophils in acute lung injury. Front Biosci. (2012) 17:2278–83. doi: 10.2741/4051
56. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol. (2014) 306:L217–30. doi: 10.1152/ajplung.00311.2013
57. Eisele NA, Anderson DM. Host defense and the airway epithelium: frontline responses that protect against bacterial invasion and pneumonia. J Pathog. (2011) 2011:249802. doi: 10.4061/2011/249802
58. Leiva-Juárez MM, Kolls JK, Evans SE. Lung epithelial cells: therapeutically inducible effectors of antimicrobial defense. Mucosal Immunology. (2018) 11:21–34. doi: 10.1038/mi.2017.71
59. Guadiz G, Sporn LA, Goss RA, Lawrence SO, Marder VJ, Simpson-Haidaris PJ. Polarized secretion of fibrinogen by lung epithelial cells. Am J Respir Cell Mol Biol. (1997) 17:60–9. doi: 10.1165/ajrcmb.17.1.2730
60. Haidaris PJ. Induction of fibrinogen biosynthesis and secretion from cultured pulmonary epithelial cells. Blood. (1997) 89:873–82. doi: 10.1182/blood.V89.3.873
61. Hippenstiel S, Opitz B, Schmeck B, Suttorp N. Lung epithelium as a sentinel and effector system in pneumonia – molecular mechanisms of pathogen recognition and signal transduction. Respir Res. (2006) 7:97. doi: 10.1186/1465-9921-7-97
62. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. (2014) 14:546–58. doi: 10.1038/nri3713
63. Chaput C, Sander LE, Suttorp N, Opitz B. NOD-like receptors in lung diseases. Front Immunol. (2013) 4:393. doi: 10.3389/fimmu.2013.00393
64. Lee S, Suh GY, Ryter SW, Choi AM. Regulation and function of the nucleotide binding domain leucine-rich repeat-containing receptor, pyrin domain-containing-3 inflammasome in lung disease. Am J Respir Cell Mol Biol. (2016) 54:151–60. doi: 10.1165/rcmb.2015-0231TR
65. Cai S, Batra S, Shen L, Wakamatsu N, Jeyaseelan S. Both TRIF- and MyD88-dependent signaling contribute to host defense against pulmonary Klebsiella infection. J Immunol. (2009) 183:6629–38. doi: 10.4049/jimmunol.0901033
66. van Lieshout MH, Blok DC, Wieland CW, de Vos AF, van't Veer C, van der Poll T. Differential roles of MyD88 and TRIF in hematopoietic and resident cells during murine gram-negative pneumonia. J Infect Dis. (2012) 206:1415–23. doi: 10.1093/infdis/jis505
67. Power MR, Li B, Yamamoto M, Akira S, Lin TJ. A role of toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J Immunol. (2007) 178:3170–6. doi: 10.4049/jimmunol.178.5.3170
68. Jeyaseelan S, Young SK, Fessler MB, Liu Y, Malcolm KC, Yamamoto M, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF)-mediated signaling contributes to innate immune responses in the lung during Escherichia coli pneumonia. J Immunol. (2007) 178:3153–60. doi: 10.4049/jimmunol.178.5.3153
69. van Lieshout MH, Florquin S, Van't Veer C, de Vos AF, van der Poll T. TIR-domain-containing adaptor-inducing interferon-β (TRIF) mediates antibacterial defense during gram-negative pneumonia by inducing interferon-x03B3. J Innate Immun. (2015) 7:637–46. doi: 10.1159/000430913
70. Jeyaseelan S, Young SK, Yamamoto M, Arndt PG, Akira S, Kolls JK, et al. Toll/IL-1R domain-containing adaptor protein (TIRAP) is a critical mediator of antibacterial defense in the lung against Klebsiella pneumoniae but not Pseudomonas aeruginosa. J Immunol. (2006) 177:538–47. doi: 10.4049/jimmunol.177.1.538
71. Skerrett SJ, Liggitt HD, Hajjar AM, Wilson CB. Cutting edge: myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J Immunol. (2004) 172:3377–81. doi: 10.4049/jimmunol.172.6.3377
72. Knapp S, Wieland CW, van't Veer C, Takeuchi O, Akira S, Florquin S, et al. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. (2004) 172:3132–8. doi: 10.4049/jimmunol.172.5.3132
73. Gowing SD, Chow SC, Cools-Lartigue JJ, Chen CB, Najmeh S, Jiang HY, et al. Gram-positive pneumonia augments non-small cell lung cancer metastasis via host toll-like receptor 2 activation. Int J Cancer. (2017) 141:561–71. doi: 10.1002/ijc.30734
74. Chavarria-Smith J, Vance RE. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. (2013) 9:e1003452. doi: 10.1371/journal.ppat.1003452
75. Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 364:e aau1330. doi: 10.1126/science.aau1330
76. de Nardo D, de Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol. (2014) 184:42–54. doi: 10.1016/j.ajpath.2013.09.007
77. Leissinger M, Kulkarni R, Zemans RL, Downey GP, Jeyaseelan S. Investigating the role of nucleotide-binding oligomerization domain-like receptors in bacterial lung infection. Am J Respir Crit Care Med. (2014) 189:1461–8. doi: 10.1164/rccm.201311-2103PP
78. Hirota JA, Hirota SA, Warner SM, Stefanowicz D, Shaheen F, Beck PL, et al. The airway epithelium nucleotide-binding domain and leucine-rich repeat protein 3 inflammasome is activated by urban particulate matter. J Allergy Clin Immunol. (2012) 129:1116–25.e6. doi: 10.1016/j.jaci.2011.11.033
79. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol. (2009) 183:2008–15. doi: 10.4049/jimmunol.0900138
80. Fang R, Uchiyama R, Sakai S, Hara H, Tsutsui H, Suda T, et al. ASC and NLRP3 maintain innate immune homeostasis in the airway through an inflammasome-independent mechanism. Mucosal Immunol. (2019) 12:1092–103. doi: 10.1038/s41385-019-0181-1
81. Giraud AS, Pereira PM, Thim L, Parker LM, Judd LM. TFF-2 inhibits iNOS/NO in monocytes, and nitrated protein in healing colon after colitis. Peptides. (2004) 25:803–9. doi: 10.1016/j.peptides.2004.01.019
82. Soriano-Izquierdo A, Gironella M, Massaguer A, May FE, Salas A, Sans M, et al. Trefoil peptide TFF2 treatment reduces VCAM-1 expression and leukocyte recruitment in experimental intestinal inflammation. J Leukoc Biol. (2004) 75:214–23. doi: 10.1189/jlb.0803396
83. McBerry C, Egan CE, Rani R, Yang Y, Wu D, Boespflug N, et al. Trefoil factor 2 negatively regulates type 1 immunity against Toxoplasma gondii. J Immunol. (2012) 189:3078–84. doi: 10.4049/jimmunol.1103374
84. Taupin D, Podolsky DK. Trefoil factors: initiators of mucosal healing. Nat Rev Mol Cell Biol. (2003) 4:721–32. doi: 10.1038/nrm1203
85. Hung L-Y, Oniskey TK, Sen D, Krummel MF, Vaughan AE, Cohen NA, et al. Trefoil factor 2 promotes type 2 immunity and lung repair through intrinsic roles in hematopoietic and nonhematopoietic cells. Am J Pathol. (2018) 188:1161–70. doi: 10.1016/j.ajpath.2018.01.020
86. Wills-Karp M, Rani R, Dienger K, Lewkowich I, Fox JG, Perkins C, et al. Trefoil factor 2 rapidly induces interleukin 33 to promote type 2 immunity during allergic asthma and hookworm infection. J Exp Med. (2012) 209:607–22. doi: 10.1084/jem.20110079
87. Tsuji S, Uehori J, Matsumoto M, Suzuki Y, Matsuhisa A, Toyoshima K, et al. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. J Biol Chem. (2001) 276:23456–63. doi: 10.1074/jbc.M103162200
88. Watanabe T, Chibana K, Shiobara T, Tei R, Koike R, Nakamura Y, et al. Expression of intelectin-1 in bronchial epithelial cells of asthma is correlated with T-helper 2 (Type-2) related parameters and its function. Allergy Asthma Clin Immunol. (2017) 13:35. doi: 10.1186/s13223-017-0207-8
89. Günther J, Seyfert H-M. The first line of defence: insights into mechanisms and relevance of phagocytosis in epithelial cells. Semin Immunopathol. (2018) 40:555–65. doi: 10.1007/s00281-018-0701-1
90. Kumar V. Phagocytosis: phenotypically simple yet a mechanistically complex process. Int Rev Immunol. (2020) 39:118–50. doi: 10.1080/08830185.2020.1732958
91. Stout-Delgado HW, Vaughan SE, Shirali AC, Jaramillo RJ, Harrod KS. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J Immunol. (2012) 188:2815–24. doi: 10.4049/jimmunol.1103051
92. Cho SJ, Rooney K, Choi AMK, Stout-Delgado HW. NLRP3 inflammasome activation in aged macrophages is diminished during Streptococcus pneumoniae infection. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L372–87. doi: 10.1152/ajplung.00393.2017
93. Cho SJ, Plataki M, Mitzel D, Lowry G, Rooney K, Stout-Delgado H. Decreased NLRP3 inflammasome expression in aged lung may contribute to increased susceptibility to secondary Streptococcus pneumoniae infection. Exp Gerontol. (2018) 105:40–6. doi: 10.1016/j.exger.2017.11.010
94. van Lieshout MHP, de Vos AF, Dessing MC, de Porto APNA, de Boer OJ, de Beer R, et al. ASC and NLRP3 impair host defense during lethal pneumonia caused by serotype 3 Streptococcus pneumoniae in mice. Eur J Immunol. (2018) 48:66–79. doi: 10.1002/eji.201646554
95. Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, et al. Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis. (2012) 205:807–17. doi: 10.1093/infdis/jir846
96. Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, et al. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE. (2009) 4:e7446. doi: 10.1371/journal.pone.0007446
97. Ezekwe EA Jr, Weng C, Duncan JA. ADAM10 cell surface expression but not activity is critical for staphylococcus aureus alpha-hemolysin-mediated activation of the NLRP3 inflammasome in human monocytes. Toxins. (2016) 8:95. doi: 10.3390/toxins8040095
98. Paudel S, Ghimire L, Jin L, Baral P, Cai S, Jeyaseelan S. NLRC4 suppresses IL-17A-mediated neutrophil-dependent host defense through upregulation of IL-18 and induction of necroptosis during Gram-positive pneumonia. Mucosal Immunol. (2019) 12:247–57. doi: 10.1038/s41385-018-0088-2
99. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog. (2018) 14:e1007308. doi: 10.1371/journal.ppat.1007308
100. Wu S HJ. Resveratrol alleviates Staphylococcus aureus pneumonia by inhibition of the NLRP3 inflammasome. Exp Ther Med. (2017) 14:6099–14. doi: 10.3892/etm.2017.5337
101. Cai S, Batra S, Wakamatsu N, Pacher P, Jeyaseelan S. NLRC4 inflammasome-mediated production of il-1β modulates mucosal immunity in the lung against gram-negative bacterial infection. J Immunol. (2012) 188:5623–35. doi: 10.4049/jimmunol.1200195
102. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. (2013) 123:1630-7. doi: 10.1172/JCI66142
103. Kamata H, Yamamoto K, Wasserman GA, Zabinski MC, Yuen CK, Lung WY, et al. Epithelial cell–derived secreted and transmembrane 1a signals to activated neutrophils during pneumococcal pneumonia. Am J Respir Cell Mol Biol. (2016) 55:407–18. doi: 10.1165/rcmb.2015-0261OC
104. Cortes G, Alvarez D, Saus C, Alberti S. Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun. (2002) 70:1075–80. doi: 10.1128/IAI.70.3.1075-1080.2002
105. de Astorza B, Cortes G, Crespi C, Saus C, Rojo JM, Alberti S. C3 promotes clearance of Klebsiella pneumoniae by A549 epithelial cells. Infect Immun. (2004) 72:1767–74. doi: 10.1128/IAI.72.3.1767-1774.2004
106. Doorduijn DJ, Rooijakkers SH, van Schaik W, Bardoel BW. Complement resistance mechanisms of Klebsiella pneumoniae. Immunobiology. (2016) 221:1102–9. doi: 10.1016/j.imbio.2016.06.014
107. Wadehra M, Iyer R, Goodglick L, Braun J. The tetraspan protein epithelial membrane protein-2 interacts with beta1 integrins and regulates adhesion. J Biol Chem. (2002) 277:41094–100. doi: 10.1074/jbc.M206868200
108. Dahlin K, Mager EM, Allen L, Tigue Z, Goodglick L, Wadehra M, et al. Identification of genes differentially expressed in rat alveolar type I cells. Am J Respir Cell Mol Biol. (2004) 31:309–16. doi: 10.1165/rcmb.2003-0423OC
109. Lin W-C, Gowdy KM, Madenspacher JH, Zemans RL, Yamamoto K, Lyons-Cohen M, et al. Epithelial membrane protein 2 governs transepithelial migration of neutrophils into the airspace. J Clin Invest. (2020) 130:157–70. doi: 10.1172/JCI127144
110. Kumar V. Innate lymphoid cells: immunoregulatory cells of mucosal inflammation. Eur J Inflamm. (2014) 12:11–20. doi: 10.1177/1721727X1401200102
111. Kumar V. Innate lymphoid cells: new paradigm in immunology of inflammation. Immunol Lett. (2014) 157:23–37. doi: 10.1016/j.imlet.2013.11.003
112. Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
113. Serafini N, Vosshenrich CA, Di Santo JP. Transcriptional regulation of innate lymphoid cell fate. Nat Rev Immunol. (2015) 15:415–28. doi: 10.1038/nri3855
114. Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. (2014) 41:354–65. doi: 10.1016/j.immuni.2014.09.005
115. Moore TA, Perry M, Getsoian AG, Newstead MW, Standiford TJ. Divergent role of gamma interferon in a murine model of pulmonary versus systemic Klebsiella pneumoniae. Infect Immun. (2002) 70:6310–8. doi: 10.1128/IAI.70.11.6310-6318.2002
116. Shinozawa Y, Matsumoto T, Uchida K, Tsujimoto S, Iwakura Y, Yamaguchi K. Role of interferon-gamma in inflammatory responses in murine pulmonary infection with Legionella pneumophila. J Med Microbiol. (2002) 51:225–30. doi: 10.1099/0022-1317-51-3-225
117. Marquardt N, Kekalainen E, Chen P, Kvedaraite E, Wilson JN, Ivarsson MA, et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional CD69(-)CD56(dim) cells. J Allergy Clin Immunol. (2017) 139:1321–30.e4. doi: 10.1016/j.jaci.2016.07.043
118. Wang J, Li F, Zheng M, Sun R, Wei H, Tian Z. Lung natural killer cells in mice: phenotype and response to respiratory infection. Immunology. (2012) 137:37–47. doi: 10.1111/j.1365-2567.2012.03607.x
119. Cong J, Wei H. Natural killer cells in the lungs. Front Immunol. (2019) 10:1416. doi: 10.3389/fimmu.2019.01416
120. Hervier B, Russick J, Cremer I, Vieillard V. NK cells in the human lungs. Front Immunol. (2019) 10:1263. doi: 10.3389/fimmu.2019.01263
121. Xu X, Weiss ID, Zhang HH, Singh SP, Wynn TA, Wilson MS, et al. Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. J Immunol. (2014) 192:1778–86. doi: 10.4049/jimmunol.1300039
122. Ivin M, Dumigan A, de Vasconcelos FN, Ebner F, Borroni M, Kavirayani A, et al. Natural killer cell-intrinsic type I IFN signaling controls Klebsiella pneumoniae growth during lung infection. PLoS Pathog. (2017) 13:e1006696. doi: 10.1371/journal.ppat.1006696
123. Elhaik-Goldman S, Kafka D, Yossef R, Hadad U, Elkabets M, Vallon-Eberhard A, et al. The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS ONE. (2011) 6:e23472. doi: 10.1371/journal.pone.0023472
124. Panda SK, Colonna M. Innate lymphoid cells in mucosal immunity. Front Immunol. (2019) 10:861. doi: 10.3389/fimmu.2019.00861
125. Barlow JL, McKenzie ANJ. Innate lymphoid cells of the lung. Annu Rev Physiol. (2019) 81:429-52. doi: 10.1146/annurev-physiol-020518-114630
126. Shikhagaie MM, Björklund ÅK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, et al. Neuropilin-1 is expressed on lymphoid tissue residing LTi-like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep. (2017) 18:1761–3. doi: 10.1016/j.celrep.2017.01.063
127. Lügering A, Ross M, Sieker M, Heidemann J, Williams IR, Domschke W, et al. CCR6 identifies lymphoid tissue inducer cells within cryptopatches. Clin Exp Immunol. (2010) 160:440–9. doi: 10.1111/j.1365-2249.2010.04103.x
128. Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, et al. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. (2011) 12:639–46. doi: 10.1038/ni.2053
129. Silver JS, Kearley J, Copenhaver AM, Sanden C, Mori M, Yu L, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. (2016) 17:626–35. doi: 10.1038/ni.3443
130. Ohne Y, Silver JS, Thompson-Snipes L, Collet MA, Blanck JP, Cantarel BL, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. (2016) 17:646–55. doi: 10.1038/ni.3447
131. Bal SM, Bernink JH, Nagasawa M, Groot J, Shikhagaie MM, Golebski K, et al. IL-1beta XR, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat immunol. (2016) 17:636–45. doi: 10.1038/ni.3444
132. Ruan S, McKinley L, Zheng M, Rudner X, D'Souza A, Kolls JK, et al. Interleukin-12 and host defense against murine pneumocystis pneumonia. Infect Immun. (2008) 76:2130–7. doi: 10.1128/IAI.00065-08
133. Nguyen Q-T, Furuya Y, Roberts S, Metzger DW. Role of interleukin-12 in protection against pulmonary infection with methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. (2015) 59:6308–16. doi: 10.1128/AAC.00968-15
134. Denney L, Byrne AJ, Shea TJ, Buckley JS, Pease JE, Herledan GM, et al. Pulmonary epithelial cell-derived cytokine TGF-beta1 is a critical cofactor for enhanced innate lymphoid cell function. Immunity. (2015) 43:945–58. doi: 10.1016/j.immuni.2015.10.012
135. van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis. (2014) 210:493–503. doi: 10.1093/infdis/jiu106
136. Robinson KM, Ramanan K, Clay ME, McHugh KJ, Rich HE, Alcorn JF. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. (2018) 11:199–208. doi: 10.1038/mi.2017.32
137. Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, et al. First-breath-induced type 2 pathways shape the lung immune environment. Cell Rep. (2017) 18:1893–905. doi: 10.1016/j.celrep.2017.01.071
138. Munoz N, Van Maele L, Marques JM, Rial A, Sirard JC, Chabalgoity JA. Mucosal administration of flagellin protects mice from Streptococcus pneumoniae lung infection. Infect Immun. (2010) 78:4226–33. doi: 10.1128/IAI.00224-10
139. Munoz-Wolf N, Rial A, Fougeron D, Tabareau J, Sirard JC, Chabalgoity JA. Sublingual flagellin protects against acute pneumococcal pneumonia in a TLR5-dependent and NLRC4-independent fashion. Future Microbiol. (2016) 11:1167–77. doi: 10.2217/fmb-2016-0045
140. Porte R, Fougeron D, Munoz-Wolf N, Tabareau J, Georgel AF, Wallet F, et al. A toll-like receptor 5 agonist improves the efficacy of antibiotics in treatment of primary and influenza virus-associated pneumococcal mouse infections. Antimicrob Agents Chemother. (2015) 59:6064–72. doi: 10.1128/AAC.01210-15
141. Xiong H, Keith JW, Samilo DW, Carter RA, Leiner IM, Pamer EG. Innate lymphocyte/Ly6C(hi) monocyte crosstalk promotes Klebsiella pneumoniae clearance. Cell. (2016) 165:679–89. doi: 10.1016/j.cell.2016.03.017
142. Gray J, Oehrle K, Worthen G, Alenghat T, Whitsett J, Deshmukh H. Intestinal commensal bacteria mediate lung mucosal immunity and promote resistance of newborn mice to infection. Sci Transl Med. (2017) 9:aaf9412. doi: 10.1126/scitranslmed.aaf9412
143. Tamburini S, Clemente JC. Gut microbiota: neonatal gut microbiota induces lung immunity against pneumonia. Nat Rev Gastroenterol Hepatol. (2017) 14:263–4. doi: 10.1038/nrgastro.2017.34
144. Gauguet S, D'Ortona S, Ahnger-Pier K, Duan B, Surana NK, Lu R, et al. Intestinal microbiota of mice influences resistance to Staphylococcus aureus pneumonia. Infect Immun. (2015) 83:4003–14. doi: 10.1128/IAI.00037-15
145. Kopf M SS, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol. (2015) 16:36–44. doi: 10.1038/ni.3052
146. Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
147. Puttur F, Gregory LG, Lloyd CM. Airway macrophages as the guardians of tissue repair in the lung. Immunol Cell Biol. (2019) 97:246–57. doi: 10.1111/imcb.12235
148. Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, Tabas I. MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal. (2018) 11:eaar3721. doi: 10.1126/scisignal.aar3721
149. Byrne AJ, Maher TM, Lloyd CM. Pulmonary macrophages: a new therapeutic pathway in fibrosing lung disease? Trends Mol Med. (2016) 22:303–16. doi: 10.1016/j.molmed.2016.02.004
150. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
151. Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Macrophage receptors and immune recognition. Annu Rev Immunol. (2005) 23:901–44. doi: 10.1146/annurev.immunol.23.021704.115816
152. Kazuo T, Suzuki Y, Yoshimura K, Yasui H, Karayama M, Hozumi H, et al. Macrophage mannose receptor CD206 predicts prognosis in community-acquired pneumonia. Sci Rep. (2019) 9:18750. doi: 10.1038/s41598-019-55289-2
153. Cakarova L, Marsh LM, Wilhelm J, Mayer K, Grimminger F, Seeger W, et al. Macrophage tumor necrosis factor-alpha induces epithelial expression of granulocyte-macrophage colony-stimulating factor: impact on alveolar epithelial repair. Am J Respir Crit Care Med. (2009) 180:521–32. doi: 10.1164/rccm.200812-1837OC
154. Taut K, Winter C, Briles DE, Paton JC, Christman JW, Maus R, et al. Macrophage turnover kinetics in the lungs of mice infected with streptococcus pneumoniae. Am J Respir Cell Mol Biol. (2008) 38:105–13. doi: 10.1165/rcmb.2007-0132OC
155. Medeiros AI, Serezani CH, Lee SP, Peters-Golden M. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med. (2009) 206:61–8. doi: 10.1084/jem.20082058
156. Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. (2004) 173:559–65. doi: 10.4049/jimmunol.173.1.559
157. Salina AC, Souza TP, Serezani CH, Medeiros AI. Efferocytosis-induced prostaglandin E2 production impairs alveolar macrophage effector functions during Streptococcus pneumoniae infection. Innate immun. (2017) 23:219–27. doi: 10.1177/1753425916684934
158. Lauw FN, Branger J, Florquin S, Speelman P, van Deventer SJH, Akira S, et al. IL-18 improves the early antimicrobial host response to pneumococcal pneumonia. J Immunol. (2002) 168:372–8. doi: 10.4049/jimmunol.168.1.372
159. Schultz MJ, Knapp S, Florquin S, Pater J, Takeda K, Akira S, et al. Interleukin-18 impairs the pulmonary host response to Pseudomonas aeruginosa. Infect Immun. (2003) 71:1630–4. doi: 10.1128/IAI.71.4.1630-1634.2003
160. Nepal S, Tiruppathi C, Tsukasaki Y, Farahany J, Mittal M, Rehman J, et al. STAT6 induces expression of Gas6 in macrophages to clear apoptotic neutrophils and resolve inflammation. Proc Natl Acad Sci USA. (2019) 116:16513–8. doi: 10.1073/pnas.1821601116
161. Sham HP, Walker KH, Abdulnour RE, Krishnamoorthy N, Douda DN, Norris PC, et al. 15-epi-Lipoxin A4, Resolvin D2, and Resolvin D3 induce NF-kappaB regulators in bacterial pneumonia. J Immunol. (2018) 200:2757–66. doi: 10.4049/jimmunol.1602090
162. El Kebir D, József L, Pan W, Wang L, Petasis NA, Serhan CN, et al. 15-epi-lipoxin A4 inhibits myeloperoxidase signaling and enhances resolution of acute lung injury. Am J Respir Crit Care Med. (2009) 180:311–9. doi: 10.1164/rccm.200810-1601OC
163. El Kebir D, Filep JG. Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Front Immunol. (2013) 4:60. doi: 10.3389/fimmu.2013.00060
164. Reed JC. Proapoptotic multidomain Bcl-2/Bax-family proteins: mechanisms, physiological roles, therapeutic opportunities. Cell Death Differ. (2006) 13:1378–86. doi: 10.1038/sj.cdd.4401975
165. Brenner D, Mak TW. Mitochondrial cell death effectors. Curr opin Cell Biol. (2009) 21:871–7. doi: 10.1016/j.ceb.2009.09.004
166. Mayadas TN, Cullere X. Neutrophil beta2 integrins: moderators of life or death decisions. Trends Immunol. (2005) 26:388–95. doi: 10.1016/j.it.2005.05.002
167. Jozsef L, Zouki C, Petasis NA, Serhan CN, Filep JG. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit peroxynitrite formation, NF-kappa B and AP-1 activation, and IL-8 gene expression in human leukocytes. Proc Natl Acad Sci USA. (2002) 99:13266–71. doi: 10.1073/pnas.202296999
168. Liu H, Zhou K, Liao L, Zhang T, Yang M, Sun C. Lipoxin A4 receptor agonist BML-111 induces autophagy in alveolar macrophages and protects from acute lung injury by activating MAPK signaling. Respir Res. (2018) 19:243. doi: 10.1186/s12931-018-0937-2
169. Roquilly A, McWilliam HEG, Jacqueline C, Tian Z, Cinotti R, Rimbert M, et al. Local modulation of antigen-presenting cell development after resolution of pneumonia induces long-term susceptibility to secondary infections. Immunity. (2017) 47:135–47.e5. doi: 10.1016/j.immuni.2017.06.021
170. Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. (2006) 6:457–64. doi: 10.1038/nri1859
171. Veillette A, Chen J. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. (2018) 39:173–84. doi: 10.1016/j.it.2017.12.005
172. Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, et al. Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol. (2020) 21:636–48. doi: 10.1038/s41590-020-0673-x
173. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. (2015) 16:67–74. doi: 10.1038/ni.3046
174. Mildner A, Chapnik E, Manor O, Yona S, Kim KW, Aychek T, et al. Mononuclear phagocyte miRNome analysis identifies miR-142 as critical regulator of murine dendritic cell homeostasis. Blood. (2013) 121:1016–27. doi: 10.1182/blood-2012-07-445999
175. Kimball AS, Davis FM, denDekker A, Joshi AD, Schaller MA, Bermick J, et al. The histone methyltransferase Setdb2 modulates macrophage phenotype and uric acid production in diabetic wound repair. Immunity. (2019) 51:258–71.e5. doi: 10.1016/j.immuni.2019.06.015
176. Sun Y, Oravecz-Wilson K, Bridges S, McEachin R, Wu J, Kim SH, et al. miR-142 controls metabolic reprogramming that regulates dendritic cell activation. J Clin Invest. (2019) 129:2029–42. doi: 10.1172/JCI123839
177. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol. (2011) 2:65. doi: 10.3389/fimmu.2011.00065
178. Hayakawa K, Wang X, Lo EH. CD200 increases alternatively activated macrophages through cAMP-response element binding protein - C/EBP-beta signaling. J Neurochem. (2016) 136:900–6. doi: 10.1111/jnc.13492
179. Koning N, van Eijk M, Pouwels W, Brouwer MS, Voehringer D, Huitinga I, et al. Expression of the inhibitory CD200 receptor is associated with alternative macrophage activation. J Innate Immun. (2010) 2:195–200. doi: 10.1159/000252803
180. Wright GJ, Puklavec MJ, Willis AC, Hoek RM, Sedgwick JD, Brown MH, et al. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity. (2000) 13:233–42. doi: 10.1016/S1074-7613(00)00023-6
181. Ocaña-Guzman R, Vázquez-Bolaños L, Sada-Ovalle I. Receptors that inhibit macrophage activation: mechanisms and signals of regulation and tolerance. J Immunol Res. (2018) 2018:8695157. doi: 10.1155/2018/8695157
182. Westphalen K, Gusarova GA, Islam MN, Subramanian M, Cohen TS, Prince AS, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. (2014) 506:503–6. doi: 10.1038/nature12902
183. Sarieddine MZ, Scheckenbach KE, Foglia B, Maass K, Garcia I, Kwak BR, et al. Connexin43 modulates neutrophil recruitment to the lung. J Cell Mol Med. (2009) 13:4560–70. doi: 10.1111/j.1582-4934.2008.00654.x
184. Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, et al. Incidence LD, and outcomes of acute lung injury. N Engl J Med. (2005) 353:1685–93. doi: 10.1056/NEJMoa050333
185. Rubenfeld Herridge GD. Epidemiology MS, and outcomes of acute lung injury. Chest. (2007) 131:554–62. doi: 10.1378/chest.06-1976
186. Stapleton RD, Wang BM, Hudson LD, Rubenfeld GD, Caldwell ES, Steinberg, et al. Causes KP, timing of death in patients with ARDS. Chest. 128:525–32. doi: 10.1378/chest.128.2.525
187. Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight. (2019) 4:e124061. doi: 10.1172/jci.insight.124061
188. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. (2019) 40:31–9. doi: 10.1055/s-0039-1683996
189. Tsokos M. Immunohistochemical detection of sepsis-induced lung injury in human autopsy material. Leg Med (Tokyo). (2003) 5:73–86. doi: 10.1016/S1344-6223(03)00010-5
190. Calfee CS, Janz DR, Bernard GR, May AK, Kangelaris KN, Matthay MA, et al. Distinct molecular phenotypes of direct vs indirect ARDS in single-center and multicenter studies. Chest. (2015) 147:1539–48. doi: 10.1378/chest.14-2454
191. Aziz M, Ode Y, Zhou M, Ochani M, Holodick NE, Rothstein TL, et al. B-1a cells protect mice from sepsis-induced acute lung injury. Mol Med. (2018) 24:26. doi: 10.1186/s10020-018-0029-2
192. Ness TL, Hogaboam CM, Strieter RM, Kunkel SL. Immunomodulatory role of CXCR2 during experimental septic peritonitis. J Immunol. (2003) 171:3775–84. doi: 10.4049/jimmunol.171.7.3775
193. Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, et al. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. (1995) 155:1515–24.
194. Walter JM, Wilson J, Ware LB. Biomarkers in acute respiratory distress syndrome: from pathobiology to improving patient care. Expert Rev Respir Med. (2014) 8:573–86. doi: 10.1586/17476348.2014.924073
195. Kellner M, Noonepalle S, Lu Q, Srivastava A, Zemskov E, Black SM. ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol. (2017) 967:105–37. doi: 10.1007/978-3-319-63245-2_8
196. Chawla LS, Fink M, Goldstein SL, Opal S, Gómez A, Murray P, et al. The epithelium as a target in sepsis. Shock. (2016) 45:249–58. doi: 10.1097/SHK.0000000000000518
197. Cohen TS, DiPaolo BC, Gray Lawrence G, Margulies SS. Sepsis enhances epithelial permeability with stretch in an actin dependent manner. PLoS ONE. (2012) 7:e38748. doi: 10.1371/journal.pone.0038748
198. Sheppard D. Modulation of acute lung injury by integrins. Proc Am Thorac Soc. (2012) 9:126–9. doi: 10.1513/pats.201112-052AW
199. Su G, Atakilit A, Li JT, Wu N, Bhattacharya M, Zhu J, et al. Absence of integrin αvβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med. (2012) 185:58–66. doi: 10.1164/rccm.201108-1381OC
200. Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, et al. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. (2009) 297:L1120–30. doi: 10.1152/ajplung.00007.2009
201. Kitamura Y, Hashimoto S, Mizuta N, Kobayashi A, Kooguchi K, Fujiwara I, et al. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med. (2001) 163:762–9. doi: 10.1164/ajrccm.163.3.2003065
202. Perl M, Chung C-S, Lomas-Neira J, Rachel T-M, Biffl WL, Cioffi WG, et al. Silencing of fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol. (2005) 167:1545–59. doi: 10.1016/S0002-9440(10)61240-0
203. Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, McCray PB, et al. Expression of the complement Anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. (2001) 166:2025–32. doi: 10.4049/jimmunol.166.3.2025
204. Bosmann M, Ward PA. Role of C3, C5 and anaphylatoxin receptors in acute lung injury and in sepsis. Adv Exp Med Biol. (2012) 946:147–59. doi: 10.1007/978-1-4614-0106-3_9
205. Russkamp NF, Ruemmler R, Roewe J, Moore BB, Ward PA, Bosmann M. Experimental design of complement component 5a-induced acute lung injury (C5a-ALI): a role of CC-chemokine receptor type 5 during immune activation by anaphylatoxin. FASEB J. (2015) 29:3762–72. doi: 10.1096/fj.15-271635
206. Juss JK, House D, Amour A, Begg M, Herre J, Storisteanu DML, et al. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3-kinase inhibition. Am J Respir Crit Care Med. (2016) 194:961–73. doi: 10.1164/rccm.201509-1818OC
207. Fialkow L FFL, Bozzetti MC, Milani AR, Rodrigues Filho EM, Ladniuk RM, Pierozan P, et al. Neutrophil apoptosis: a marker of disease severity in sepsis and sepsis-induced acute respiratory distress syndrome. Crit Care. (2006) 10:R155. doi: 10.1186/cc5090
208. Dorward DA, Felton JM, Robb CT, Craven T, Kipari T, Walsh TS, et al. The cyclin-dependent kinase inhibitor AT7519 accelerates neutrophil apoptosis in sepsis-related acute respiratory distress syndrome. Thorax. (2017) 72:182–85. doi: 10.1136/thoraxjnl-2016-209229
209. Galiacy S, Planus E, Lepetit H, Féréol S, Laurent V, Ware L, et al. Keratinocyte growth factor promotes cell motility during alveolar epithelial repair in vitro. Exp Cell Res. (2003) 283:215–29. doi: 10.1016/S0014-4827(02)00049-6
210. McAuley DF, Cross LM, Hamid U, Gardner E, Elborn JS, Cullen KM, et al. Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Respir Med. (2017) 5:484–91. doi: 10.1016/S2213-2600(17)30171-6
211. Ray P, Devaux Y, Stolz DB, Yarlagadda M, Watkins SC, Lu Y, et al. Inducible expression of keratinocyte growth factor (KGF) in mice inhibits lung epithelial cell death induced by hyperoxia. Proc Natl Acad Sci USA. (2003) 100:6098–103. doi: 10.1073/pnas.1031851100
212. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
213. de Grove KC, Provoost S, Verhamme FM, Bracke KR, Joos GF, Maes T, et al. characterization and quantification of innate lymphoid cell subsets in human lung. PLoS ONE. (2016) 11:e0145961. doi: 10.1371/journal.pone.0145961
214. Cruz-Zárate D, Cabrera-Rivera GL, Ruiz-Sánchez BP, Serafín-López J, Chacón-Salinas R, López-Macías C, et al. Innate lymphoid cells have decreased HLA-DR expression but retain their responsiveness to TLR ligands during sepsis. J Immunol. (2018) 201:3401–10. doi: 10.4049/jimmunol.1800735
215. Huang Y, Mao K, Chen X, Sun M-A, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (New York, N.Y.). (2018) 359:114–119. doi: 10.1126/science.aam5809
216. Winkler MS, Märtz KB, Nierhaus A, Daum G, Schwedhelm E, Kluge S, et al. Loss of sphingosine 1-phosphate (S1P) in septic shock is predominantly caused by decreased levels of high-density lipoproteins (HDL). J Intensive Care. (2019) 7:23. doi: 10.1186/s40560-019-0376-2
217. Germain RN, Huang Y. ILC2s - resident lymphocytes pre-adapted to a specific tissue or migratory effectors that adapt to where they move? Curr Opin Immunol. (2019) 56:76–81. doi: 10.1016/j.coi.2018.11.001
218. Chun TT, Chung C-S, Fallon EA, Hutchins NA, Clarke E, Rossi A-L, et al. Group 2 innate lymphoid cells (ILC2s) are key mediators of the inflammatory response in polymicrobial sepsis. Am J Pathol. (2018) 188:2097–108. doi: 10.1016/j.ajpath.2018.05.009
219. Xu H, Xu J, Xu L, Jin S, Turnquist HR, Hoffman R, et al. Interleukin-33 contributes to ILC2 activation and early inflammation-associated lung injury during abdominal sepsis. Immunol Cell Biol. (2018) 96:935–47. doi: 10.1111/imcb.12159
220. Lai D, Tang J, Chen L, Fan EK, Scott MJ, Li Y, et al. Group 2 innate lymphoid cells protect lung endothelial cells from pyroptosis in sepsis. Cell Death Dis. (2018) 9:369. doi: 10.1038/s41419-018-0412-5
221. Krishack PA, Louviere TJ, Decker TS, Kuzel TG, Greenberg JA, Camacho DF, et al. Protection against Staphylococcus aureus bacteremia-induced mortality depends on ILC2s and eosinophils. JCI Insight. (2019) 4:e124168. doi: 10.1172/jci.insight.124168
222. Puneet P, Moochhala S, Bhatia M. Chemokines in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. (2005) 288:L3–15. doi: 10.1152/ajplung.00405.2003
223. Wagner JG, Driscoll KE, Roth RA. Inhibition of pulmonary neutrophil trafficking during endotoxemia is dependent on the stimulus for migration. Am J Respir Cell Mol Biol. (1999) 20:769–76. doi: 10.1165/ajrcmb.20.4.3481
224. Wang Z, Rui T, Yang M, Valiyeva F, Kvietys PR. Alveolar macrophages from septic mice promote polymorphonuclear leukocyte transendothelial migration via an endothelial cell Src kinase/NADPH oxidase pathway. J Immunol. (2008) 181:8735–44. doi: 10.4049/jimmunol.181.12.8735
225. Park I, Kim M, Choe K, Song E, Seo H, Hwang Y, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J. (2019) 53:1800786. doi: 10.1183/13993003.00786-2018
226. Reddy RC, Chen GH, Newstead MW, Moore T, Zeng X, Tateda K, et al. Alveolar macrophage deactivation in murine septic peritonitis: role of interleukin 10. Infect Immun. (2001) 69:1394–401. doi: 10.1128/IAI.69.3.1394-1401.2001
227. Mahida R, Thickett D. Impaired alveolar macrophage efferocytosis in ARDS causes accumulation of apoptotic neutrophils & prolonged inflammation. Eur Respir J. (2018) 52:PA4283. doi: 10.1183/13993003.congress-2018.PA4283
228. Hiruma T, Tsuyuzaki H, Uchida K, Trapnell BC, Yamamura Y, Kusakabe Y, et al. IFN-β improves sepsis-related alveolar macrophage dysfunction and postseptic acute respiratory distress syndrome-related mortality. Am J Respir Cell Mol Biol. (2018) 59:45–55. doi: 10.1165/rcmb.2017-0261OC
229. Pahuja M, Tran C, Wang H, Yin K. Alveolar macrophage suppression in sepsis is associated with high mobility group box 1 transmigration. Shock. (2008) 29:754–60. doi: 10.1097/shk.0b013e31815d0c8f
230. Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ. (2014) 21:1229–39. doi: 10.1038/cdd.2014.40
231. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. (2018) 49:740–53.e7. doi: 10.1016/j.immuni.2018.08.016
232. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
233. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
234. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. (2018) 24:97–108.e4. doi: 10.1016/j.chom.2018.05.009
235. Xiong S, Hong Z, Huang LS, Tsukasaki Y, Nepal S, Di A, et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest. (2020) 130:3684–98. doi: 10.1172/JCI136908
236. Janardhan KS, Sandhu SK, Singh B. Neutrophil depletion inhibits early and late monocyte/macrophage increase in lung inflammation. Front Biosci. (2006) 11:1569–76. doi: 10.2741/1904
237. Farley KS, Wang LF, Razavi HM, Law C, Rohan M, McCormack DG, et al. Effects of macrophage inducible nitric oxide synthase in murine septic lung injury. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L1164–72. doi: 10.1152/ajplung.00248.2005
238. Liu Y, Guan H, Zhang J-L, Zheng Z, Wang T, Tao K, et al. Acute downregulation of miR-199a attenuates sepsis-induced acute lung injury by targeting SIRT1. Am J Physiol Cell Physiol. (2018) 314:C449–55. doi: 10.1152/ajpcell.00173.2017
239. Finkel T, Deng C-X, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. (2009) 460:587–91. doi: 10.1038/nature08197
240. Gao R, Ma Z, Hu Y, Chen J, Shetty S, Fu J. Sirt1 restrains lung inflammasome activation in a murine model of sepsis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L847–53. doi: 10.1152/ajplung.00274.2014
241. Li Y, Yang X, He Y, Wang W, Zhang J, Zhang W, et al. Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology. (2017) 222:552–61. doi: 10.1016/j.imbio.2016.11.002
242. Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, Flavell RA, et al. Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest. (2006) 116:2532–42. doi: 10.1172/JCI28054
243. Lyn-Kew K, Rich E, Zeng X, Wen H, Kunkel SL, Newstead MW, et al. IRAK-M regulates chromatin remodeling in lung macrophages during experimental sepsis. PLoS ONE. (2010) 5:e11145. doi: 10.1371/journal.pone.0011145
244. Filgueiras JLR, Martins JO, Serezani CH, Capelozzi VL, Montes MBA, Jancar S. Sepsis-induced acute lung injury (ALI) is milder in diabetic rats and correlates with impaired NFkB activation. PLoS ONE. (2012) 7:e44987. doi: 10.1371/journal.pone.0044987
245. Honiden S, Gong MN. Diabetes, insulin, and development of acute lung injury. Crit Care Med. (2009) 37:2455–64. doi: 10.1097/CCM.0b013e3181a0fea5
246. Esper AM, Moss M, Martin GS. The effect of diabetes mellitus on organ dysfunction with sepsis: an epidemiological study. Crit Care. (2009) 13:R18. doi: 10.1186/cc7717
247. Gu W-J, Wan Y-D, Tie H-T, Kan C, Sun T-W. Risk of acute lung injury/acute respiratory distress syndrome in critically ill adult patients with pre-existing diabetes: a meta-analysis. PLoS ONE. (2014) 9:e90426. doi: 10.1371/journal.pone.0090426
248. Yu S, Christiani DC, Thompson BT, Bajwa EK, Gong MN. Role of diabetes in the development of acute respiratory distress syndrome. Crit Care Med. (2013) 41:2720–32. doi: 10.1097/CCM.0b013e318298a2eb
Keywords: pneumonia, sepsis, ALI, ILCs, neutrophils, macrophages
Citation: Kumar V (2020) Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 11:1722. doi: 10.3389/fimmu.2020.01722
Received: 19 February 2020; Accepted: 29 June 2020;
Published: 04 August 2020.
Edited by:
Murugaiyan Gopal, Harvard Medical School, United StatesReviewed by:
Samithamby Jey Jeyaseelan, Louisiana State University, United StatesJames Harris, Monash University, Australia
Copyright © 2020 Kumar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vijay Kumar, vij_tox@yahoo.com
†ORCID: Vijay Kumar orcid.org/0000-0001-9741-3597