 Kinga K. Hosszu
Kinga K. Hosszu Alisa Valentino
Alisa Valentino Ellinor I. Peerschke
Ellinor I. Peerschke Berhane Ghebrehiwet
Berhane Ghebrehiwet- 1Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 2Department of Lab Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, United States
- 3The Department of Medicine, Stony Brook University, Stony Brook, NY, United States
Genetic deficiency in C1q is a strong susceptibility factor for systemic lupus erythematosus (SLE). There are two major hypotheses that potentially explain the role of C1q in SLE. The first postulates that C1q deficiency abrogates apoptotic cell clearance, leading to persistently high loads of potentially immunogenic self-antigens that trigger autoimmune responses. While C1q undoubtedly plays an important role in apoptotic clearance, an essential biological process such as removal of self- waste is so critical for host survival that multiple ligand-receptor combinations do fortunately exist to ensure that proper disposal of apoptotic debris is accomplished even in the absence of C1q. The second hypothesis is based on the observation that locally synthesized C1q plays a critical role in regulating the earliest stages of monocyte to dendritic cell (DC) differentiation and function. Indeed, circulating C1q has been shown to keep monocytes in a pre-dendritic state by silencing key molecular players and ensuring that unwarranted DC-driven immune responses do not occur. Monocytes are also able to display macromolecular C1 on their surface, representing a novel mechanism for the recognition of circulating “danger.” Translation of this danger signal in turn, provides the requisite “license” to trigger a differentiation pathway that leads to adaptive immune response. Based on this evidence, the second hypothesis proposes that deficiency in C1q dysregulates monocyte-to-DC differentiation and causes inefficient or defective maintenance of self-tolerance. The fact that C1q receptors (cC1qR and gC1qR) are also expressed on the surface of both monocytes and DCs, suggests that C1q/C1qR may regulate DC differentiation and function through specific cell-signaling pathways. While their primary ligand is C1q, C1qRs can also independently recognize a vast array of plasma proteins as well as pathogen-associated molecular ligands, indicating that these molecules may collaborate in antigen recognition and processing, and thus regulate DC-differentiation. This review will therefore focus on the role of C1q and C1qRs in SLE and explore the gC1qR/C1q axis as a potential target for therapy.
C1q: A Brief Overview
The first component of complement, C1, is a multimeric protein comprised of C1q and the Ca2+ – dependent tetramer C1r2–C1s2 (1–6). C1q itself is a 460 kDa collagen-like glycoprotein that is comprised of six globular “heads” (gC1q) linked to six collagen-like “stalks” (cC1q), and serves as the recognition signal triggering the classical pathway of complement (7–9). Each subunit of C1q is made up of three different, but highly conserved polypeptide chains – A, B, and C (10, 11). C1q belongs to the collectin (collagen containing lectin) family of molecules that contain collagen-like sequences contiguous with non-collagen-like stretches. Although it lacks a consensus carbohydrate recognition domain (which allows other collectins to recognize glycoconjugates containing mannose and fucose on microorganisms but not on self-proteins), C1q contains collagen sequences which allow it to bind to protein motifs in immunoglobulin (Ig)G or IgM. These motifs allow C1q to bind to immune complexes and engage in complement-mediated microbial killing and phagocytosis (12–14). While the majority of C1q circulates in plasma, it is also synthesized by many cell types including macrophages and dendritic cells (DCs), and secreted locally at sites of inflammation (15–24). Approximately 80% of circulating C1q is associated with the C1 complex, while the remaining portion is in its monomeric, “free” form (25).
In recent decades multiple groups have shown evidence that C1q plays a role in recognizing and clearing altered self and apoptotic cells by binding to the apoptotic cell surface and initiating phagocytic uptake by macrophages and DCs through interaction with C1q receptors expressed both on the phagocytic cell, (e.g., cC1qR/CD91) and the apoptotic cell (gC1qR and phosphatidylserine) (26–29). This clearance of immune complexes and apoptotic debris is crucial for maintaining homeostasis to avoid immune recognition of hidden epitopes – a critical immunopathogenic event leading to autoimmune disease.
C1q Receptors
C1q receptors mediate many immunologic functions involved in innate and adaptive immunity. There are at least two types of distinct, ubiquitously expressed cell surface molecules which bind human C1q: gC1qR, the receptor for the globular heads, and cC1qR, the receptor for the collagen tail (28, 30–35).
Predominantly found in the storage compartments of the endoplasmic reticulum, cC1qR (60 kDa), a homolog of calreticulin (CR) (sometimes also referred to as cC1qR/CR or the “collagen receptor”) fulfills a multiplicity of functions. It is a molecular chaperone, an extracellular compartment protein, an intracellular mediator of integrin function, an inhibitor of steroid hormone-regulated gene expression, and a receptor for C1q (36–43). However, studies have shown that C1q can only bind stably to cC1qR after it has been immobilized, heat-treated, or bound to IgG, suggesting that cC1qR is a receptor for an altered conformation of C1q (44, 45).
cC1qR does not contain a transmembrane domain or a GPI-anchor attachment site, and instead needs other adaptor molecules for signal transduction. One such molecule is CD91 (46), which binds to cC1qR and C1q on the surface of monocytes to initiate uptake of apoptotic cells (26). However, the uptake process cannot be completely inhibited by antibody blockade or genetic deficiency of CD91, indicating that it is not actually required for the C1q-mediated enhancement of phagocytosis (26, 47). Additional co-receptors of cC1qR are scavenger receptor A on antigen presenting cells (48), CD59 on neutrophils (49), α2β1 integrin and glycoprotein VI on resting platelets (50), MHC class I on T cells (51), and CD69 on human peripheral blood mononuclear cells (PBMCs) (52).
GC1qR (p32/p33/HABP1) is another well-described C1q receptor. It is a highly acidic homotrimer, comprised of three 33-kDa chains with a ubiquitous and multi-compartmental distribution including on the cell surface. As a result, gC1qR has a highly asymmetric surface charge with a negatively charged “solution face” exposed to plasma and a neutral or basic “membrane face” on the reverse side, suggesting that the two sides have different functions (53–56). It is present on the surface of human monocytes, DCs, macrophages, and many other cells (19, 33, 34, 57, 58). Additionally, gC1qR’s capacity to elicit biological responses and transduce intracellular signals affects a variety of cell types (32, 57, 59–64). Similar to cC1qR, it lacks a transmembrane segment, and requires a docking/signaling partner, some of which are β1-integrins on endothelial cells (32), vasopressin V2 receptor on the HEK 293 cell line, alpha(1B)-adrenergic receptor on the COS 7 cell line (65), DC-SIGN on DCs (66, 67) and LAIR-1 on DCs and T cells (68–71).
Due to gC1qR’s ability to recognize and bind to a plethora of ligands, many pathogens employ immune escape mechanisms to exploit the normal regulatory functions of C1q/gC1qR. Among the growing list of pathogenic microorganisms are HIV (67, 72–74), adenovirus (75, 76), Epstein-Barr virus (77), Herpesvirus Saimiri (78), rubella virus (79–81), hepatitis B virus (82), hepatitis C virus (HCV) (59, 63, 74), L. monocytogenes (83), S. aureus (84), and B. cereus (85). These microorganisms have a strong affinity for gC1qR, which further indicates that gC1qR plays an important role in immune regulation. For example, in vitro studies have shown that HCV, which binds gC1qR at the C1q binding site, employs gC1qR on monocyte-DC precursors to prevent DC immunogenic activity (57, 58).
C1q and SLE
The connection between C1q and autoimmune diseases such as rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) is well established. In RA, antibodies to C1q may cross-react with collagen type II and contribute to the disease process that leads to tissue destruction and inflammation (86, 87). In animal models of RA, C1q function is impaired by autoantibodies, indicating a regulatory role for C1q in suppressing immune activity (87, 88). Moreover, a synthetic decapeptide corresponding to the A-chain of C1q injected into DBA/1 mice delays disease onset and reduces the severity of collagen-induced arthritis (86, 89).
Hereditary homozygous C1q deficiency, while rare, is the strongest known susceptibility factor for SLE (90–93). The vast majority of patients (≥95%) develop clinical symptoms closely related to SLE, with rashes, glomerulonephritis, and central nervous system disease (91, 94). Additionally, about a third of SLE patients have high affinity autoantibodies to C1q directed to a neo-epitope in the A-chain (91, 94). In a subset of patients who are C1q sufficient, the SLE disease process itself causes consumption of C1q, therefore mimicking the genetic deficiency of C1q. This acquired partial deficiency of C1q, either due to complement activation or to the presence of anti-C1q autoantibodies, is even more commonly observed in lupus patients than genetic C1q deficiency (92, 95, 96). Multiple studies have shown associations between the presence of anti-C1q antibodies and active nephritis in SLE (97–100). There is, however, evidence that the presence of anti-C1q antibodies is not associated with active lupus nephritis, but rather with SLE global activity, indicating that although C1q’s main function is the clearance of immune complexes during apoptosis, it has other biologic functions with inhibitory/protective factors (30).
C1q plays a critical role in recognizing harmful molecules, ranging from pathogen-associated molecular ligands (non-self) to damage-associated molecular targets (altered self) (29). Therefore, in this manner, C1q acts as a molecular bridge between the phagocytic cell and the apoptotic debris to be cleared. While many studies suggest that failure to properly clear apoptotic cells in the absence of C1q could result in an immunogenic state (91, 94, 101), many observations have challenged this idea. Disruption of other apoptotic uptake processes, such as those mediated by CD14 (102), β3 or β5 integrin (103), mannose-binding lectin (104), all result in the accumulation of apoptotic bodies without triggering autoimmunity. In fact, apoptotic cells can actively inhibit the inflammatory program. For example, preincubating macrophages with apoptotic cells can significantly reduce the inflammatory response induced by lipopolysaccharide (LPS) (105–107). During this process, anti-inflammatory cytokines, such as transforming growth factor (TGF)-β and interleukin (IL)-10, are released and act via paracrine or autocrine mechanisms to sustain an anti-inflammatory state (107). Administration or accumulation of apoptotic cells have been shown to ameliorate multiple inflammatory disorders, such as diabetes (108, 109), Experimental Autoimmune Encephalomyelitis (110, 111), arthritis (112), colitis (113), pulmonary fibrosis (114–116), fulminant hepatitis (117), contact hypersensitivity (118, 119), acute and chronic graft rejection (120–123), and hematopoietic cell engraftment (124–127). Data from these studies indicate that apoptotic cells modulate immune responses and can prevent the onset and/or establishment of inflammatory disease. Based on these observations, it is likely that processes other than the accumulation of apoptotic debris play a decisive role in SLE development.
In recent years, increasing evidence has emerged that aside from the recognition and triggering of the classical complement pathway, C1q also modulates the acquired immune response. In this context, C1q provides active protection from autoimmunity by silencing key molecular markers or regulating autoreactive immune cells.
Multiple studies have shown that C1q regulates cytokine secretion and polarizes antigen presenting cells (APCs) toward a tolerogenic phenotype (17, 128–135). Specifically, macrophages and DCs that have been exposed to C1q exhibit enhanced production of anti−inflammatory and reduced pro-inflammatory cytokines (129, 134, 135). Immature DCs (iDC) in the presence of immobilized C1q have reduced capacity to induce allogeneic Th1 and Th17 cells, and demonstrate a trend toward increased Treg proliferation (130, 136). Furthermore, C1q-primed macrophages have elevated PD-L1 and PD-L2 and suppressed surface CD40, and C1q-polarized DCs have higher surface PD-L2 and reduced CD86 (130). Plasmacytoid DCs (pDCs), a major interferon-α (IFN-α)-producing cell type, also play a pivotal role in SLE pathogenesis (137–139). In the presence of immune complexes, C1q interacts with pDCs and strongly inhibits IFN-α production (140–142), while in the absence of C1q, immune complexes can preferentially engage pDCs and increase IFN-α production (143). These data suggest that C1q provides a protective, anti-inflammatory function by regulating IFN-α production in pDCs.
Our lab was the first to show that monocytes are able to display macromolecular C1 on their surface with the globular heads of C1q displayed outwardly, toward the extracellular milieu (144). Thus, membrane associated C1q can potentially recognize and capture circulating immune complexes or pathogen-associated molecular patterns and signal monocytes to migrate into tissues, differentiate into macrophages or DCs, and initiate the process of antigen elimination. Unoccupied C1q, on the other hand, may silence key molecular players, ensuring that unwarranted DC-driven immune responses do not occur.
Using a C1q-deficient mouse model of SLE, Ling et al. showed that C1q ameliorates the response to self-antigens by modulating the mitochondrial metabolism of CD8+ T cells (145). Conversely, C1q deficiency can trigger an effector CD8+ T cell response to chronic viral infection leading to lethal immunopathology.
Taken together, these data suggest that upon interacting with APCs, C1q regulates the subsequent activation of T effector functions to modulate the adaptive immune response and prevent the initiation/propagation of autoimmunity.
C1q Receptors as an Immune Checkpoint
While the wide array of immunological processes exhibited by C1q appear to be the principal component of its immune-modulatory function, its underlying mechanisms remain poorly described. The unique structure of C1q, which allows it to interact with its primary receptors, gC1qR and cC1qR, via either its globular head or collagen tail domains, may shed light to this dilemma. The observation that C1q functions as a molecular switch during the narrow window of monocyte to DC transition (128, 133) is also reflected by the differential expression of gC1qR and cC1qR during this process (Figure 1) (128). While gC1qR is steadily expressed, the expression of cC1qR is low on monocytes and increases as the cells commit to the dendritic cell lineage. At the time corresponding to firm commitment to the DC lineage, there is an inverse correlation between gC1qR and cC1qR expression on the cell surface, which, in turn, may influence the nature and specificity of the cells’ response to C1q (128).
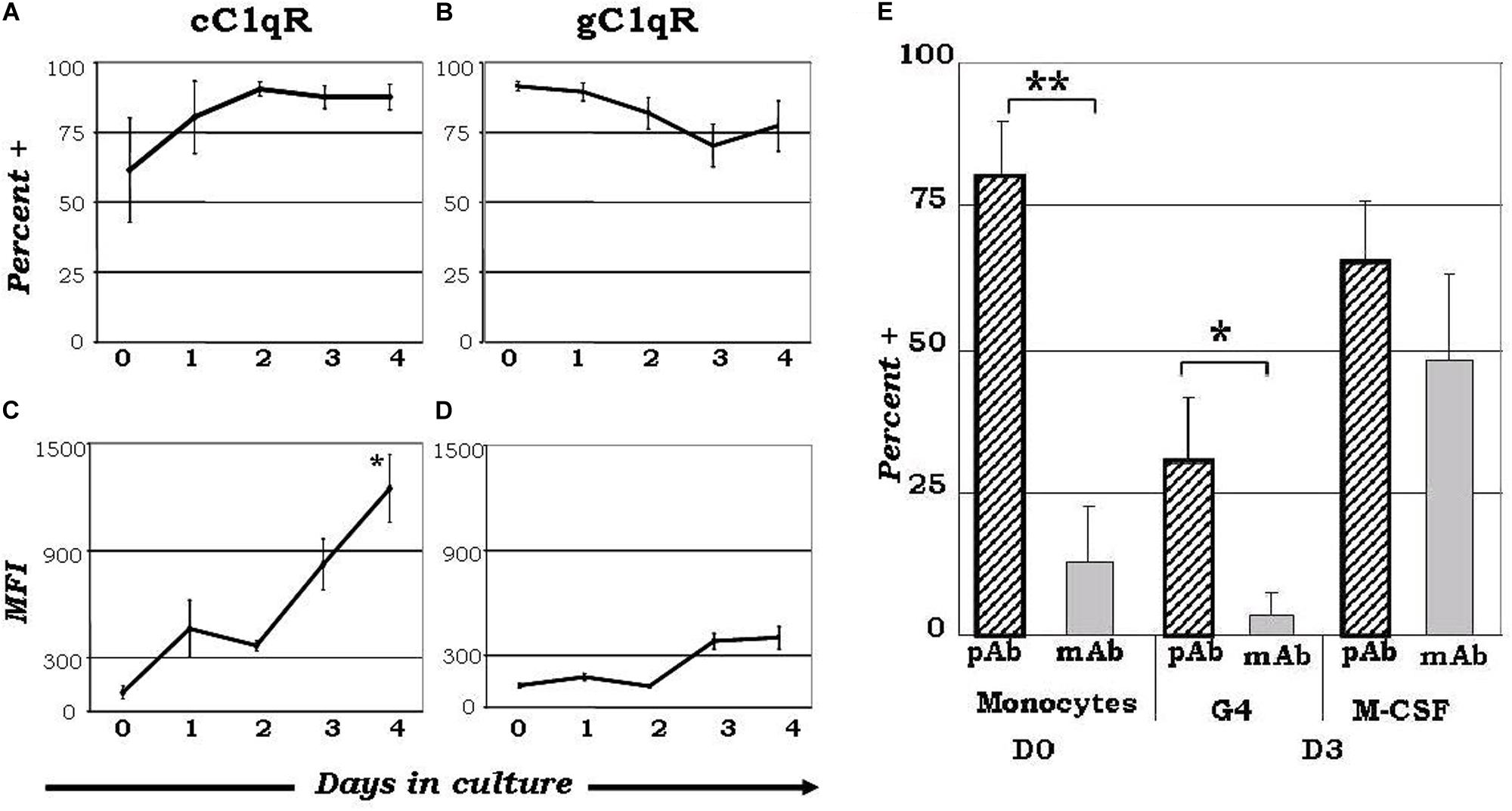
Figure 1. Varied expression of C1q receptors and specific binding orientation of surface bound C1q on monocyte-DC precursors may regulate DC differentiation events. Mononuclear cells cultured in the presence of GM-CSF+ IL-4 were analyzed for the expression of cC1qR (A,C) and gC1qR (B,D) expression, and C1q binding orientation (E). (A) The percentage of cC1qR expression was variable on monocytes, but by day 2 nearly all monocyte-DCs had the receptor on their surface (n = 4). (B) On day 0, gC1qR was present on almost all the cells, and its expression was only slightly reduced by day 4 (n = 4). (C) Mean fluorescence analysis revealed that cC1qR expression was dramatically amplified by days 3 and 4 (n = 4). (D) Mean fluorescence intensity of gC1qR remained at relatively steady levels throughout the days (n = 4). (E) C1q is bound to the monocyte and DC surface via its globular head regions, while on M-CSF treated monocyte-macrophages its orientation is reversed. Binding orientation of C1q was determined using monoclonal antibodies specific to the globular head regions of C1q as well as polyclonal antibodies to the whole protein, and assessed by flow cytometry (n = 3). Experiments were gated on HLA-DR+ cells. *P < 0.05, **P < 0.01. [Adapted from ref (128)]
Upon binding to C1qR, specific pathways get activated to trigger downstream signaling. Incubating C1q or a monoclonal antibody which recognizes the C1q binding site on gC1qR, with T cells, inhibits T cell proliferation, possibly through the activation of PI3K, NADPH oxidase and p190 RhoGAP (53, 146). Additionally, it causes the inactivation of TC10, and the translocation of NKp44L from the cytoplasm to the plasma membrane (147). Ligand engagement of gC1qR at the C1q binding site (by HCV core protein and mAb) in LPS-stimulated monocytes increases PI3K activation and Akt phosphorylation, and in macrophages it induces A20 expression via P38, JNK and NF-κB signaling pathways, in an ERK independent manner (57, 58, 148). Similarly, engagement of gC1qR by C1q activates the MAPK and PI3K/AKT signaling pathways in macrophages (148). Furthermore, binding of HCV core protein to gC1qR down-regulates many inflammatory cytokines in macrophages, including IL-6 and IL-1β, indicating that gC1qR relays an anti-inflammatory signal (148). Conversely, ligation of cC1qR by a mAb increases TNFα and IL6 secretion, as well as the expression and phosphorylation of STAT6 in macrophages, indicating that cC1qR is a pro-inflammatory receptor (149).
C1q also engages in molecular complexing at the cell surface. In monocyte-derived iDCs, C1q, DC-SIGN and gC1qR form a trimolecular complex on the plasma membrane, which is presumed to modulate DC differentiation and function through DC-SIGN-mediated signaling pathways [26]. Signaling through DC-SIGN has been shown to increase phosphorylation of Raf-1 on Ser338 and Tyr340/341 (150). Furthermore, stimulation of DC-SIGN with a mannose receptor-1 Ab activates the MEK/ERK kinase cascade (151). However, whether direct stimulation of C1q participates in these signaling pathways still remains to be investigated.
The leukocyte-associated immunoglobulin-like receptor 1 (LAIR-1) is another C1q-binding transmembrane receptor that can serve as a potential co-receptor to gC1qR. On T cells, LAIR-1 engagement by C1q inhibits TCR signaling by decreasing the phosphorylation of LCK, LYN, ZAP-70, extracellular signal-regulated kinase, c-Jun N-terminal kinase 1/2, and p38, indicating that LAIR-1 activation may be a strategy for controlling inflammation (70). Studies by Son et al. showed that C1q and HMGB1 can cooperate to terminate inflammation, and induce the differentiation of monocytes to anti-inflammatory M2-like macrophages through a complex with RAGE and LAIR-1 (71). In myelomonocytes, the globular head of C1q binds to CD33 and LAIR-1 and activates CD33/LAIR-1 inhibitory motifs (68). Binding of C1q to LAIR-1 on monocytes significantly up-regulates the expression of IL-8, IL-10, LAIR-1, and the phosphorylation of JNK, p38-MAPK, AKT, and NF-κB (152).
Taken together, these data suggest that the regulatory effects of C1q may depend on specific C1q/C1qR interactions; and these interactions may in turn control the transition from the tolerogenic state toward a pro-inflammatory state. Fundamental to this mechanism is the differential expression of the C1q/C1qR system, which, through the engagement of distinct receptors (gC1qR versus cC1qR), and the resulting binding orientation of C1q – heads versus tails – actively avoids self-directed adaptive immune responses to modified-self as well as non-self antigens.
As illustrated by Figure 2, this functional duality of the C1q/gC1qR axis is very similar to the role of the PD1/PDL1 checkpoint in cancer, which helps maintain the balance between immune surveillance and cancer cell proliferation (153). In this setting, the C1q/C1qR axis would serve as an immune checkpoint supporting a tolerogenic/anti-inflammatory signal by the interaction between membrane-associated C1q on the signaling cell or soluble C1q in the extracellular milieu, and the membrane associated C1q receptors on the target cell. Conversely, when this interaction is blocked by antigen binding to the soluble or membrane-associated C1q, a pro-inflammatory signal is relayed through cC1qR. These specific interactions ensure that the immune system is activated only at the appropriate time in order to minimize the possibility of chronic autoimmune inflammation.
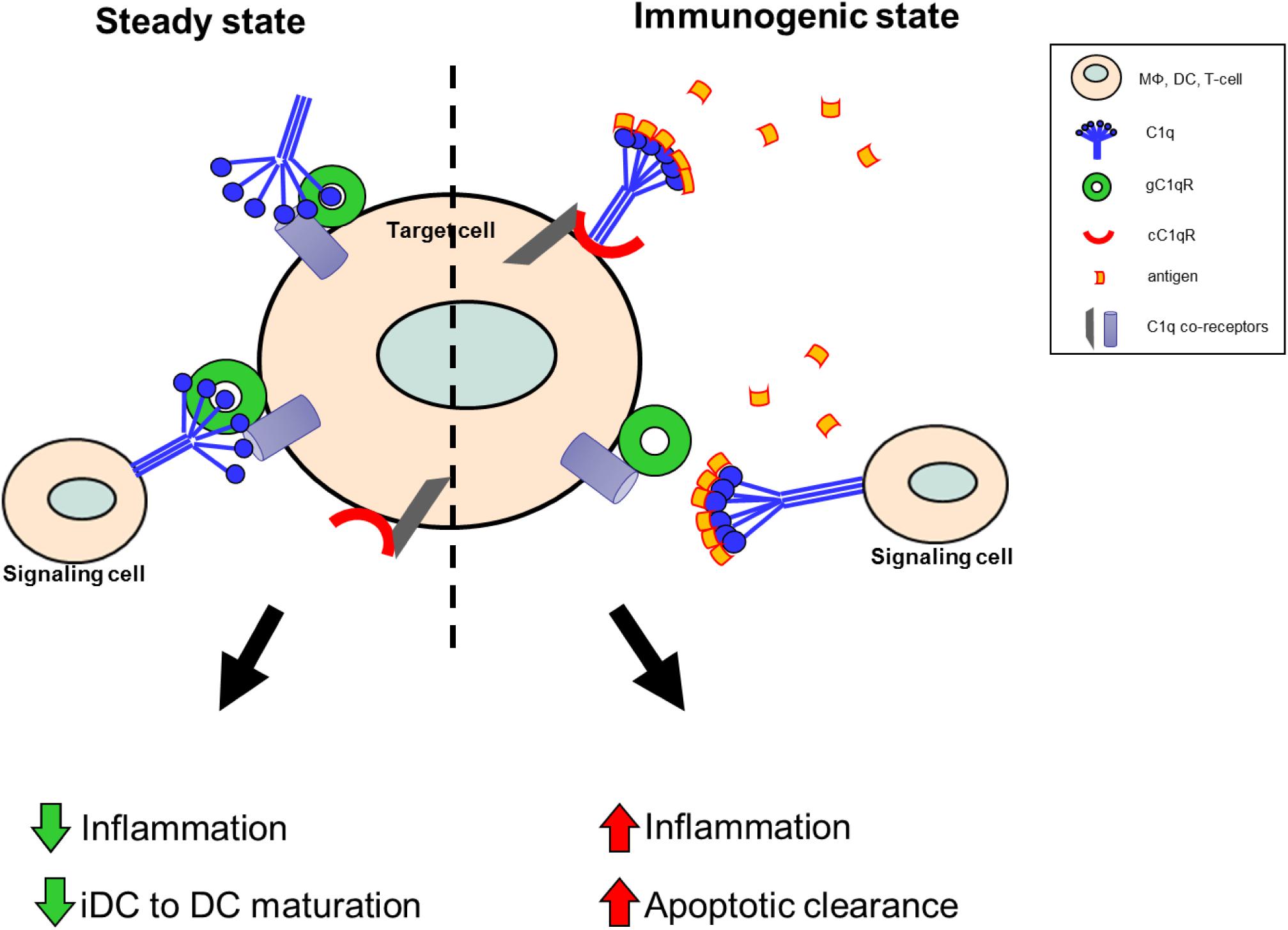
Figure 2. Theoretical model of the C1q/gC1qR immune checkpoint in inflammation and autoimmunity. Under steady state conditions, in the absence of danger signals (PAMPs, DAMPs, etc.), membrane-associated C1q on the signaling cell, or soluble C1q in the extracellular milieu, is available to bind to gC1qR on the target cell to support a tolerogenic state. During these conditions anti-inflammatory processes are dominant and DC maturation is decreased to keep cells in a tolerogenic/immature state. When C1q recognizes and captures circulating immune complexes or pathogen-associated molecular patterns, it undergoes a conformational change and only the collagen tail is available to bind. Thus, the resulting C1q/cC1qR interactions drive increased pro-inflammatory signals and signal monocytes to migrate into tissues, differentiate into macrophages or DCs, and initiate the process of antigen elimination.
The C1q/C1qR Axis: A Functional Example
The role of C1q in the regulation of DC differentiation and function has been greatly studied in recent years. A significant portion of the work has centered, around the potential regulatory role of C1q during DC maturation, once the cells have fully committed to the DC lineage. These data show that C1q treatment of LPS-primed human iDCs decreases the cell surface expression of CD80, CD83 and CD86, the secretion of IL-6, TNF-α, and IL-10, as well as the ability of the cells to stimulate T helper (TH) 1 cell proliferation in a mixed leukocyte reaction (154). These results suggest that C1q treated iDCs may be resistant to LPS-induced maturation. Yamada and colleagues showed that C1q treatment after LPS-stimulation or CpG oligodeoxynucleotide induction suppresses IL-12p40 production in bone marrow-derived DCs, reduces NF-κB activity and delays the phosphorylation of p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase (155). These data further indicate that C1q may function by suppressing pro-inflammatory responses after DC activation. As ligation of gC1qR results in decreased secretion of pro-inflammatory cytokines like IL-6 and TNFα, soluble C1q in these experiments putatively acts through a gC1qR-mediated pathway.
However, in order to imitate the role of C1q as an opsonin in vitro, some studies employed immobilized C1q. Nauta and colleagues found that the uptake of C1q-opsonized apoptotic cells by iDCs stimulated the production of IL-6, IL-10, and TNF-α, without an effect on IL-12p70 (156). Additionally, iDCs placed on immobilized C1q, gC1q or cC1q, showed enhanced maturation, translocation of NF-κB to the nucleus and enhanced secretion of IL-12 and TNF-α, in addition to elevated TH1-stimulating capacity (157). The increased secretion of pro-inflammatory cytokines in these studies suggest that fixation of C1q supports DC maturation and acts in a cC1qR-mediated pathway.
So far, very little data is available on how soluble C1q that is present in the plasma and interstitial tissues under steady state conditions might regulate DC differentiation during the earliest stages of mono-DC growth. These yet unexplored functions would provide important details of how C1q regulates adaptive immune functions via iDCs in the absence of infection or inflammation. Studies from our lab (158) and others (159) have shown that C1q acts as a chemoattractant to iDCs, but not mature DCs. C1q-induced migration is mediated through ligation of both gC1qR and cC1qR and activation of Akt and MAPK pathways. C1q treatment during DC differentiation was also shown to give rise to CD1a+DC-SIGN+ iDCs with high phagocytic capacity, and low expression of CD80, CD83, and CD86 (154). Because this narrow window of differentiation represents the important interface between innate and adaptive immunity, more work is needed to explore this crucial stage.
Implications for Therapy and Concluding Remarks
Since C1q and C1qRs are involved in a multitude of inflammatory processes that accompany various disease conditions, including infection, cancer, and autoimmune diseases, understanding the underlying mechanism is important to identify new targets for the design of therapeutic strategies. While the role of C1q in apoptotic clearance has been well described and is supported by a plethora of evidence, it is still not clear how deficiency of C1q contributes to the loss of tolerance. This review is aimed to provide new insights and stimulate discussion around the topic. Understanding how the interactions between C1q and C1qRs control the transition from steady state to a pro-inflammatory response, will not only give us insight into how the C1q/C1qR system regulates the immune response, but may also provide us with alternative approaches for designing better therapeutic options. Molecules or peptides that inhibit the interaction between antigen-bound C1q and cC1qR, or those that can mimic the interaction between C1q and gC1qR, can potentially be used as templates for the development of therapeutic interventions to reduce C1q-mediated pro-inflammatory responses. One potential target for an inhibitory-drug design is the N-terminal region (residues 160–283) on the collagen tail of C1q, which binds to cC1qR, and contains several short (7–10 amino acids) CH2-like motifs (ExKxKx) similar to the C1q binding motif found in the CH2 domain of IgG (160). For gC1qR, some therapeutic molecules already exist. One example is the use of mAb 74.5.2, which inhibits the binding of kininogen to gC1qR, thus blocking the generation of bradykinin and other vasoactive molecules that have been shown to contribute to inflammation (161). Another example of a therapeutic molecule is mAb 60.11, which is specific to the C1q binding site on gC1qR (aa 76–93). This antibody has been shown to reduce cell proliferation, decrease tumor growth, increase apoptosis, and impair angiogenesis (162). In summary, the data reviewed in this article supports the idea that the C1q/C1qR system is an ideal molecular target for the design of antibody- or peptide-based therapy to attenuate acute and chronic inflammation associated with autoimmune diseases, SLE in particular.
Author Contributions
BG supervised the work. All authors contributed to the article and approved the submitted version.
Funding
The work included in this article was supported in part by grants from the National Institutes of Allergy and Infectious Diseases R01 AI 060866, R01 AI-084178, and R56-AI 1223476 (to BG) and the NIH/NCI cancer support grant P30 CA008748 (to MSKCC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
gC1q, the globular heads of C1q; cC1q, the collagen domain of C1q; gC1qR, receptor for gC1q; cC1qR, receptor for cC1q; CR, calreticulin (another name for cC1qR); ghA, ghB and ghC, globular heads (gh) of the AB and C chains of C1q.
References
1. Perkins SJ. Molecular modelling of human complement subcomponent C1q and its complex with C1r2C1s2 derived from neutron-scattering curves and hydrodynamic properties. Biochem J. (1985) 228:13–26. doi: 10.1042/bj2280013
2. Siegel RC, Schumaker VN. Measurement of the association constants of the complexes formed between intact C1q or pepsin-treated C1q stalks and the unactivated or activated C1r2C1s2 tetramers. Mol Immunol. (1983) 20:53–66. doi: 10.1016/0161-5890(83)90105-0
3. Weiss V, Fauser C, Engel J. Functional model of subcomponent C1 of human complement. J Mol Biol. (1986) 189:573–81. doi: 10.1016/0022-2836(86)90325-6
4. Calcott MA, Müller-Eberhard HJ. C1q protein of human complement. Biochemistry. (1972) 11:3443–50. doi: 10.1021/bi00768a018
5. Lepow IH, Naff GB, Todd EW, Pensky J, Hinz CF. Chromatographic resolution of the first component of human complement into three activities. J Exp Med. (1963) 117:983–1008. doi: 10.1084/jem.117.6.983
6. Muller-Eberhard HJ, Kunkel HG. Isolation of a thermolabile serum protein which precipitates gamma-globulin aggregates and participates in immune hemolysis. Proc Soc Exp Biol Med. (1961) 106:291–5. doi: 10.3181/00379727-106-26313
7. Brodsky-Doyle B, Leonard KR, Reid KB. Circular-dichroism and electron-microscopy studies of human subcomponent C1q before and after limited proteolysis by pepsin. Biochem J. (1976) 159:279–86. doi: 10.1042/bj1590279
8. Reid KB, Porter RR. Subunit composition and structure of subcomponent C1q of the first component of human complement. Biochem J. (1976) 155:19–23. doi: 10.1042/bj1550019
10. Sasaki T, Yonemasu K. Chemical studies on the isolated collagen-like and globular fragment of complement component C1q. Comparative studies on bovine and human C1q. Biochim Biophys Acta. (1983) 742:122–8. doi: 10.1016/0167-4838(83)90367-9
11. Kishore U, Leigh LE, Eggleton P, Strong P, Perdikoulis MV, Willis AC, et al. Functional characterization of a recombinant form of the C-terminal, globular head region of the B-chain of human serum complement protein C1q. Biochem J. (1998) 333(Pt 1):27–32. doi: 10.1042/bj3330027
12. Lu J, Wiedemann H, Timpl R, Reid KB. Similarity in structure between C1q and the collectins as judged by electron microscopy. Behring Inst Mitt. (1993) 93:6–16.
13. Bobak DA, Gaither TA, Frank MM, Tenner AJ. Modulation of FcR function by complement: subcomponent C1q enhances the phagocytosis of IgG-opsonized targets by human monocytes and culture-derived macrophages. J Immunol. (1987) 138:1150–6.
14. Leist-Welsh P, Bjornson AB. Immunoglobulin-independent utilization of the classical complement pathway in opsonophagocytosis of Escherichia coli by human peripheral leukocytes. J Immunol. (1982) 128:2643–51. doi: 10.1016/0161-5890(82)90091-8
15. Schwaeble W, Schäfer MK, Petry F, Fink T, Knebel D, Weihe E, et al. Follicular dendritic cells, interdigitating cells, and cells of the monocyte-macrophage lineage are the C1q-producing sources in the spleen. Identification of specific cell types by in situ hybridization and immunohistochemical analysis. J Immunol. (1995) 155:4971–8.
16. Bensa JC, Reboul A, Colomb MG. Biosynthesis in vitro of complement subcomponents C1q, C1s and C1 inhibitor by resting and stimulated human monocytes. Biochem J. (1983) 216:385–92. doi: 10.1042/bj2160385
17. Castellano G, Woltman AM, Nauta AJ, Roos A, Trouw LA, Seelen MA, et al. Maturation of dendritic cells abrogates C1q production in vivo and in vitro. Blood. (2004) 103:3813–20. doi: 10.1182/blood-2003-09-3046
18. Kaul M, Loos M. Expression of membrane C1q in human monocyte-derived macrophages is developmentally regulated and enhanced by interferon-gamma. FEBS Lett. (2001) 500:91–8. doi: 10.1016/s0014-5793(01)02592-3
19. Vegh Z, Goyarts EC, Rozengarten K, Mazumder A, Ghebrehiwet B. Maturation-dependent expression of C1q-binding proteins on the cell surface of human monocyte-derived dendritic cells. Int Immunopharmacol. (2003) 3:345–57. doi: 10.1016/S1567-5769(02)00234-5
20. Dillon SP, D’Souza A, Kurien BT, Scofield RH. Systemic lupus erythematosus and C1q: a quantitative ELISA for determining C1q levels in serum. Biotechnol J. (2009) 4:1210–4. doi: 10.1002/biot.200800273
21. Hughes-Jones NC. Functional affinity constants of the reaction between 125I-labelled C1q and C1q binders and their use in the measurement of plasma C1q concentrations. Immunology. (1977) 32:191–8.
22. Schuller E, Helary M. Determination in the nanogram range of C1q in serum and unconcentrated CSF by electro-immunodiffusion. J Immunol Methods. (1983) 56:159–65. doi: 10.1016/0022-1759(83)90407-6
23. Breitner S, Störkel S, Reichel W, Loos M. Complement components C1q, C1r/C1s, and C1INH in rheumatoid arthritis. Correlation of in situ hybridization and northern blot results with function and protein concentration in synovium and primary cell cultures. Arthritis Rheum. (1995) 38:492–8. doi: 10.1002/art.1780380406
24. Soda K, Ando M, Sakata T, Sugimoto M, Nakashima H, Araki S. C1q and C3 in bronchoalveolar lavage fluid from patients with summer-type hypersensitivity pneumonitis. Chest. (1988) 93:76–80. doi: 10.1378/chest.93.1.76
25. Sjöholm AG, Mårtensson U, Laurell AB. C1 dissociation in serum: estimation of free C1q by electroimmunoassay. Acta Pathol Microbiol Immunol Scand C. (1985) 93:161–8. doi: 10.1111/j.1699-0463.1985.tb02939.x
26. Vandivier RW, Ogden CA, Fadok VA, Hoffmann PR, Brown KK, Botto M, et al. Role of surfactant proteins A, D, and C1q in the clearance of apoptotic cells in vivo and in vitro: calreticulin and CD91 as a common collectin receptor complex. J Immunol. (2002) 169:3978–86. doi: 10.4049/jimmunol.169.7.3978
27. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. (1997) 158:4525–8.
28. Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. (2001) 194:781–95. doi: 10.1084/jem.194.6.781
29. Païdassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. (2008) 180:2329–38. doi: 10.4049/jimmunol.180.4.2329
30. Ghebrehiwet B, Lim BL, Peerschke EI, Willis AC, Reid KB. Isolation, cDNA cloning, and overexpression of a 33-kD cell surface glycoprotein that binds to the globular “heads” of C1q. J Exp Med. (1994) 179:1809–21. doi: 10.1084/jem.179.6.1809
31. Peterson KL, Zhang W, Lu PD, Keilbaugh SA, Peerschke EI, Ghebrehiwet B. The C1q-binding cell membrane proteins cC1q-R and gC1q-R are released from activated cells: subcellular distribution and immunochemical characterization. Clin Immunol Immunopathol. (1997) 84:17–26. doi: 10.1006/clin.1997.4374
32. Feng X, Tonnesen MG, Peerschke EIB, Ghebrehiwet B. Cooperation of C1q receptors and integrins in C1q-mediated endothelial cell adhesion and spreading. J Immunol. (2002) 168:2441–8. doi: 10.4049/jimmunol.168.5.2441
33. Herwald H, Dedio J, Kellner R, Loos M, Müller-Esterl W. Isolation and characterization of the kininogen-binding protein p33 from endothelial cells. Identity with the gC1q receptor. J Biol Chem. (1996) 271:13040–7. doi: 10.1074/jbc.271.22.13040
34. Peerschke EI, Reid KB, Ghebrehiwet B. Identification of a novel 33-kDa C1q-binding site on human blood platelets. J Immunol. (1994) 152:5896–901.
35. Sim RB, Moestrup SK, Stuart GR, Lynch NJ, Lu J, Schwaeble WJ, et al. Interaction of C1q and the collectins with the potential receptors calreticulin (cC1qR/collectin receptor) and megalin. Immunobiology. (1998) 199:208–24. doi: 10.1016/s0171-2985(98)80028-4
36. Malhotra R. Collectin receptor (C1q receptor): structure and function. Behring Inst Mitt. (1993) 93:254–61.
37. Malhotra R, Sim RB, Reid KB. Interaction of C1q, and other proteins containing collagen-like domains, with the C1q receptor. Biochem Soc Trans. (1990) 18:1145–8. doi: 10.1042/bst0181145
38. Nauseef WM, McCormick SJ, Clark RA. Calreticulin functions as a molecular chaperone in the biosynthesis of myeloperoxidase. J Biol Chem. (1995) 270:4741–7. doi: 10.1074/jbc.270.9.4741
39. Wada I, Imai S, Kai M, Sakane F, Kanoh H. Chaperone function of calreticulin when expressed in the endoplasmic reticulum as the membrane-anchored and soluble forms. J Biol Chem. (1995) 270:20298–304. doi: 10.1074/jbc.270.35.20298
40. Somogyi E, Petersson U, Hultenby K, Wendel M. Calreticulin–an endoplasmic reticulum protein with calcium-binding activity is also found in the extracellular matrix. Matrix Biol. (2003) 22:179–91. doi: 10.1016/S0945-053X(02)00117-8
41. Dedhar S. Novel functions for calreticulin: interaction with integrins and modulation of gene expression? Trends Biochem Sci. (1994) 19:269–71. doi: 10.1016/0968-0004(94)90001-9
42. Burns K, Duggan B, Atkinson EA, Famulski KS, Nemer M, Bleackley RC, et al. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature. (1994) 367:476–80. doi: 10.1038/367476a0
43. Platet N, Cunat S, Chalbos D, Rochefort H, Garcia M. Unliganded and liganded estrogen receptors protect against cancer invasion via different mechanisms. Mol Endocrinol. (2000) 14:999–1009. doi: 10.1210/mend.14.7.0492
44. Vandenberg RJ, Easterbrook-Smith SB. Conformational changes in C1q upon binding to IgG oligomers. FEBS Lett. (1986) 207:276–9. doi: 10.1016/0014-5793(86)81504-6
45. Steinø A, Jørgensen CS, Laursen I, Houen G. Interaction of C1q with the receptor calreticulin requires a conformational change in C1q. Scand J Immunol. (2004) 59:485–95. doi: 10.1111/j.0300-9475.2004.01425.x
46. Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. (2001) 14:303–13. doi: 10.1016/s1074-7613(01)00111-x
47. Lillis AP, Greenlee MC, Mikhailenko I, Pizzo SV, Tenner AJ, Strickland DK, et al. Murine low-density lipoprotein receptor-related protein 1 (LRP) is required for phagocytosis of targets bearing LRP ligands but is not required for C1q-triggered enhancement of phagocytosis. J Immunol. (2008) 181:364–73. doi: 10.4049/jimmunol.181.1.364
48. Berwin B, Hart JP, Rice S, Gass C, Pizzo SV, Post SR, et al. Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. EMBO J. (2003) 22:6127–36. doi: 10.1093/emboj/cdg572
49. Ghiran I, Klickstein LB, Nicholson-Weller A. Calreticulin is at the surface of circulating neutrophils and uses CD59 as an adaptor molecule. J Biol Chem. (2003) 278:21024–31. doi: 10.1074/jbc.M302306200
50. Elton CM, Smethurst PA, Eggleton P, Farndale RW. Physical and functional interaction between cell-surface calreticulin and the collagen receptors integrin alpha2beta1 and glycoprotein VI in human platelets. Thromb Haemost. (2002) 88:648–54. doi: 10.1267/th02100648
51. Santos SG, Powis SJ, Arosa FA. Misfolding of major histocompatibility complex class I molecules in activated T cells allows cis-interactions with receptors and signaling molecules and is associated with tyrosine phosphorylation. J Biol Chem. (2004) 279:53062–70. doi: 10.1074/jbc.M408794200
52. Vance BA, Harley PH, Backlund PS, Ward Y, Phelps TL, Gress RE. Human CD69 associates with an N-terminal fragment of calreticulin at the cell surface. Arch Biochem Biophys. (2005) 438:11–20. doi: 10.1016/j.abb.2005.04.009
53. Ghebrehiwet B, Lu PD, Zhang W, Lim BL, Eggleton P, Leigh LE, et al. Identification of functional domains on gC1Q-R, a cell surface protein that binds to the globular “heads” of C1Q, using monoclonal antibodies and synthetic peptides. Hybridoma. (1996) 15:333–42. doi: 10.1089/hyb.1996.15.333
54. Ghebrehiwet B, Tantral L, Titmus MA, Panessa-Warren BJ, Tortora GT, Wong SS, et al. The exosporium of B. cereus contains a binding site for gC1qR/p33: implication in spore attachment and/or entry. Adv Exp Med Biol. (2007) 598:181–97. doi: 10.1007/978-0-387-71767-8_13
55. Jiang J, Zhang Y, Krainer AR, Xu RM. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc Natl Acad Sci USA. (1999) 96:3572–7. doi: 10.1073/pnas.96.7.3572
56. Ghebrehiwet B, Peerschke EIB. cC1q-R (calreticulin) and gC1q-R/p33: ubiquitously expressed multi-ligand binding cellular proteins involved in inflammation and infection. Mol Immunol. (2004) 41:173–83. doi: 10.1016/j.molimm.2004.03.014
57. Waggoner SN, Cruise MW, Kassel R, Hahn YS. gC1q receptor ligation selectively down-regulates human IL-12 production through activation of the phosphoinositide 3-kinase pathway. J Immunol. (2005) 175:4706–14. doi: 10.4049/jimmunol.175.7.4706
58. Waggoner SN, Hall CHT, Hahn YS. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J Leukoc Biol. (2007) 82:1407–19. doi: 10.1189/jlb.0507268
59. Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. (2000) 106:1239–49. doi: 10.1172/JCI10323
60. Meenakshi J, Anupama, Goswami SK, Datta K. Constitutive expression of hyaluronan binding protein 1 (HABP1/p32/gC1qR) in normal fibroblast cells perturbs its growth characteristics and induces apoptosis. Biochem Biophys Res Commun. (2003) 300:686–93. doi: 10.1016/S0006-291X(02)02788-2
61. Moorman JP, Fitzgerald SM, Prayther DC, Lee SA, Chi DS, Krishnaswamy G. Induction of p38- and gC1qR-dependent IL-8 expression in pulmonary fibroblasts by soluble hepatitis C core protein. Respir Res. (2005) 6:105. doi: 10.1186/1465-9921-6-105
62. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement-dependent regulatory pathway. J Immunol. (2001) 167:5264–72. doi: 10.4049/jimmunol.167.9.5264
63. Yao ZQ, Eisen-Vandervelde A, Ray S, Hahn YS. HCV core/gC1qR interaction arrests T cell cycle progression through stabilization of the cell cycle inhibitor p27Kip1. Virology. (2003) 314:271–82. doi: 10.1016/S0042-6822(03)00419-7
64. Yao ZQ, Waggoner SN, Cruise MW, Hall C, Xie X, Oldach DW, et al. SOCS1 and SOCS3 are targeted by hepatitis C virus core/gC1qR ligation to inhibit T-cell function. J Virol. (2005) 79:15417–29. doi: 10.1128/JVI.79.24.15417-15429.2005
65. Xu Z, Hirasawa A, Shinoura H, Tsujimoto G. Interaction of the alpha(1B)-adrenergic receptor with gC1q-R, a multifunctional protein. J Biol Chem. (1999) 274:21149–54. doi: 10.1074/jbc.274.30.21149
66. Hosszu KK, Valentino A, Vinayagasundaram U, Vinayagasundaram R, Joyce MG, Ji Y, et al. C1q, and gC1qR form a trimolecular receptor complex on the surface of monocyte-derived immature dendritic cells. Blood. (2012) 120:1228–36. doi: 10.1182/blood-2011-07-369728
67. Pednekar L, Pandit H, Paudyal B, Kaur A, Al-Mozaini MA, Kouser L, et al. Complement protein C1q interacts with DC-SIGN via its globular domain and thus may interfere with HIV-1 transmission. Front Immunol. (2016) 7:600. doi: 10.3389/fimmu.2016.00600
68. Son M, Diamond B, Volpe BT, Aranow CB, Mackay MC, Santiago-Schwarz F. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci Rep. (2017) 7:270. doi: 10.1038/s41598-017-00290-w
69. Son M, Diamond B. C1q-mediated repression of human monocytes is regulated by leukocyte-associated Ig-like receptor 1 (LAIR-1). Mol Med. (2015) 20:559–68. doi: 10.2119/molmed.2014.00185
70. Park J-E, Brand DD, Rosloniec EF, Yi A-K, Stuart JM, Kang AH, et al. Leukocyte-associated immunoglobulin-like receptor 1 inhibits T-cell signaling by decreasing protein phosphorylation in the T-cell signaling pathway. J Biol Chem. (2020) 295:2239–47. doi: 10.1074/jbc.RA119.011150
71. Son M, Porat A, He M, Suurmond J, Santiago-Schwarz F, Andersson U, et al. C1q and HMGB1 reciprocally regulate human macrophage polarization. Blood. (2016) 128:2218–28. doi: 10.1182/blood-2016-05-719757
72. Berro R, Kehn K, de la Fuente C, Pumfery A, Adair R, Wade J, et al. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J Virol. (2006) 80:3189–204. doi: 10.1128/JVI.80.7.3189-3204.2006
73. Pednekar L, Valentino A, Ji Y, Tumma N, Valentino C, Kadoor A, et al. Identification of the gC1qR sites for the HIV-1 viral envelope protein gp41 and the HCV core protein: implications in viral-specific pathogenesis and therapy. Mol Immunol. (2016) 74:18–26. doi: 10.1016/j.molimm.2016.03.016
74. Ghebrehiwet B, Jesty J, Vinayagasundaram R, Vinayagasundaram U, Ji Y, Valentino A, et al. Targeting gC1qR domains for therapy against infection and inflammation. Adv Exp Med Biol. (2013) 735:97–110. doi: 10.1007/978-1-4614-4118-2_6
75. Matthews DA, Russell WC. Adenovirus core protein V interacts with p32–a protein which is associated with both the mitochondria and the nucleus. J Gen Virol. (1998) 79(Pt 7):1677–85. doi: 10.1099/0022-1317-79-7-1677
76. Ohrmalm C, Akusjärvi G. Cellular splicing and transcription regulatory protein p32 represses adenovirus major late transcription and causes hyperphosphorylation of RNA polymerase II. J Virol. (2006) 80:5010–20. doi: 10.1128/JVI.80.10.5010-5020.2006
77. Wang Y, Finan JE, Middeldorp JM, Hayward SD. P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein-Barr virus. Virology. (1997) 236:18–29. doi: 10.1006/viro.1997.8739
78. Hall KT, Giles MS, Calderwood MA, Goodwin DJ, Matthews DA, Whitehouse A. The Herpesvirus saimiri open reading frame 73 gene product interacts with the cellular protein p32. J Virol. (2002) 76:11612–22. doi: 10.1128/JVI.76.22.11612-11622.2002
79. Beatch MD, Everitt JC, Law LJ, Hobman TC. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J Virol. (2005) 79:10807–20. doi: 10.1128/JVI.79.16.10807-10820.2005
80. Beatch MD, Hobman TC. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J Virol. (2000) 74:5569–76. doi: 10.1128/JVI.74.12.5569-5576.2000
81. Mohan KVK, Ghebrehiwet B, Atreya CD. The N-terminal conserved domain of rubella virus capsid interacts with the C-terminal region of cellular p32 and overexpression of p32 enhances the viral infectivity. Virus Res. (2002) 85:151–61. doi: 10.1016/S0168-1702(02)00030-8
82. Lainé S, Thouard A, Derancourt J, Kress M, Sitterlin D, Rossignol JM. In vitro and in vivo interactions between the hepatitis B virus protein P22 and the cellular protein gC1qR. J Virol. (2003) 77:12875–80. doi: 10.1128/jvi.77.23.12875-12880.2003
83. Braun L, Ghebrehiwet B, Cossart P. gC1q-R/p32, a C1q-binding protein, is a receptor for the InlB invasion protein of Listeria monocytogenes. EMBO J. (2000) 19:1458–66. doi: 10.1093/emboj/19.7.1458
84. Peerschke EIB, Bayer AS, Ghebrehiwet B, Xiong YQ. gC1qR/p33 blockade reduces Staphylococcus aureus colonization of target tissues in an animal model of infective endocarditis. Infect Immun. (2006) 74:4418–23. doi: 10.1128/IAI.01794-05
85. Peerschke EIB, Ghebrehiwet B. The contribution of gC1qR/p33 in infection and inflammation. Immunobiology. (2007) 212:333–42. doi: 10.1016/j.imbio.2006.11.011
86. Maeurer MJ, Trinder PK, Störkel S, Loos M. C1q in autoimmune diseases: rheumatoid arthritis. Behring Inst Mitt. (1993) 93:262–78.
87. Trinder PK, Maeurer MJ, Stoerkel SS, Loos M. Altered (oxidized) C1q induces a rheumatoid arthritis-like destructive and chronic inflammation in joint structures in arthritis-susceptible rats. Clin Immunol Immunopathol. (1997) 82:149–56. doi: 10.1006/clin.1996.4293
88. Trinder PK, Maeurer MJ, Brackertz D, Loos M. The collagen-like component of the complement system, C1q, is recognized by 7 S autoantibodies and is functionally impaired in synovial fluids of patients with rheumatoid arthritis. Immunology. (1996) 87:355–61. doi: 10.1046/j.1365-2567.1996.495559.x
89. Maeurer MJ, Trinder PK, Störkel S, Loos M. Modulation of type II collagen-induced arthritis in DBA/1 mice by intravenous application of a peptide from the C1q-A chain. Immunobiology. (1992) 185:103–20. doi: 10.1016/S0171-2985(11)80321-9
90. Ghebrehiwet B, Peerschke EI. Role of C1q and C1q receptors in the pathogenesis of systemic lupus erythematosus. Curr Dir Autoimmun. (2004) 7:87–97. doi: 10.1159/000075688
91. Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. (1998) 199:265–85. doi: 10.1016/S0171-2985(98)80032-6
92. Frémeaux-Bacchi V, Weiss L, Demouchy C, Blouin J, Kazatchkine MD. Autoantibodies to the collagen-like region of C1q are strongly associated with classical pathway-mediated hypocomplementemia in systemic lupus erythematosus. Lupus. (1996) 5:216–20. doi: 10.1177/096120339600500309
93. Goulielmos GN, Zervou MI, Vazgiourakis VM, Ghodke-Puranik Y, Garyfallos A, Niewold TB. The genetics and molecular pathogenesis of systemic lupus erythematosus (SLE) in populations of different ancestry. Gene. (2018) 668:59–72. doi: 10.1016/j.gene.2018.05.041
94. Walport MJ. Complement and systemic lupus erythematosus. Arthritis Res. (2002) 4(Suppl. 3):S279–93. doi: 10.1186/ar586
95. Sharma M, Vignesh P, Tiewsoh K, Rawat A. Revisiting the complement system in systemic lupus erythematosus. Expert Rev Clin Immunol. (2020) 16:397–408. doi: 10.1080/1744666X.2020.1745063
96. Schur PH, Sandson J. Immunologic factors and clinical activity in systemic lupus erythematosus. N Engl J Med. (1968) 278:533–8. doi: 10.1056/NEJM196803072781004
97. Siegert C, Daha M, Westedt ML, van der Voort E, Breedveld F. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J Rheumatol. (1991) 18:230–4.
98. Trendelenburg M, Lopez-Trascasa M, Potlukova E, Moll S, Regenass S, Frémeaux-Bacchi V, et al. High prevalence of anti-C1q antibodies in biopsy-proven active lupus nephritis. Nephrol Dial Transplant. (2006) 21:3115–21. doi: 10.1093/ndt/gfl436
99. Gargiulo MDLÁ, Gómez G, Khoury M, Collado MV, Suárez L, Álvarez C, et al. Association between the presence of anti-C1q antibodies and active nephritis in patients with systemic lupus erythematosus. Medicina. (2015) 75:23–8.
100. Marto N, Bertolaccini ML, Calabuig E, Hughes GRV, Khamashta MA. Anti-C1q antibodies in nephritis: correlation between titres and renal disease activity and positive predictive value in systemic lupus erythematosus. Ann Rheum Dis. (2005) 64:444–8. doi: 10.1136/ard.2004.024943
101. Hurst NP, Nuki G, Wallington T. Evidence for intrinsic cellular defects of “complement” receptor-mediated phagocytosis in patients with systemic lupus erythematosus (SLE). Clin Exp Immunol. (1984) 55:303–12.
102. Devitt A, Parker KG, Ogden CA, Oldreive C, Clay MF, Melville LA, et al. Persistence of apoptotic cells without autoimmune disease or inflammation in CD14-/- mice. J Cell Biol. (2004) 167:1161–70. doi: 10.1083/jcb.200410057
103. Lucas M, Stuart LM, Zhang A, Hodivala-Dilke K, Febbraio M, Silverstein R, et al. Requirements for apoptotic cell contact in regulation of macrophage responses. J Immunol. (2006) 177:4047–54. doi: 10.4049/jimmunol.177.6.4047
104. Stuart LM, Ezekowitz RA. Phagocytosis: elegant complexity. Immunity. (2005) 22:539–50. doi: 10.1016/j.immuni.2005.05.002
105. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. (1997) 390:350–1. doi: 10.1038/37022
106. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. (1998) 101:890–8. doi: 10.1172/JCI1112
107. Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. (2004) 172:880–9. doi: 10.4049/jimmunol.172.2.880
108. Xia C-Q, Peng R, Qiu Y, Annamalai M, Gordon D, Clare-Salzler MJ. Transfusion of apoptotic beta-cells induces immune tolerance to beta-cell antigens and prevents type 1 diabetes in NOD mice. Diabetes. (2007) 56:2116–23. doi: 10.2337/db06-0825
109. Xia C-Q, Qiu Y, Peng R-H, Lo-Dauer J, Clare-Salzler MJ. Infusion of UVB-treated splenic stromal cells induces suppression of beta cell antigen-specific T cell responses in NOD mice. J Autoimmun. (2008) 30:283–92. doi: 10.1016/j.jaut.2007.11.017
110. Qiu C-H, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel subset of CD8{alpha}+ dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J Immunol. (2009) 182:4127–36. doi: 10.4049/jimmunol.0803364
111. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest. (2007) 117:2268–78. doi: 10.1172/JCI31990
112. Gray M, Miles K, Salter D, Gray D, Savill J. Apoptotic cells protect mice from autoimmune inflammation by the induction of regulatory B cells. Proc Natl Acad Sci USA. (2007) 104:14080–5. doi: 10.1073/pnas.0700326104
113. Grau A, Tabib A, Grau I, Reiner I, Mevorach D. Apoptotic cells induce NF-κB and inflammasome negative signaling. PLoS One. (2015) 10:e0122440. doi: 10.1371/journal.pone.0122440
114. Yoon Y-S, Lee Y-J, Choi J-Y, Cho M-S, Kang JL. Coordinated induction of cyclooxygenase-2/prostaglandin E2 and hepatocyte growth factor by apoptotic cells prevents lung fibrosis. J Leukoc Biol. (2013) 94:1037–49. doi: 10.1189/jlb.0513255
115. Lee Y-J, Moon C, Lee SH, Park H-J, Seoh J-Y, Cho M-S, et al. Apoptotic cell instillation after bleomycin attenuates lung injury through hepatocyte growth factor induction. Eur Respir J. (2012) 40:424–35. doi: 10.1183/09031936.00096711
116. Yoon YS, Kim SY, Kim MJ, Lim JH, Cho MS, Kang JL. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol. (2015) 8:1031–46. doi: 10.1038/mi.2014.130
117. Zhang M, Xu S, Han Y, Cao X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-β. Hepatology. (2011) 53:306–16. doi: 10.1002/hep.24029
118. Ferguson TA, Herndon J, Elzey B, Griffith TS, Schoenberger S, Green DR. Uptake of apoptotic antigen-coupled cells by lymphoid dendritic cells and cross-priming of CD8(+) T cells produce active immune unresponsiveness. J Immunol. (2002) 168:5589–95. doi: 10.4049/jimmunol.168.11.5589
119. Griffith TS, Kazama H, VanOosten RL, Earle JK, Herndon JM, Green DR, et al. Apoptotic cells induce tolerance by generating helpless CD8+ T cells that produce TRAIL. J Immunol. (2007) 178:2679–87. doi: 10.4049/jimmunol.178.5.2679
120. Wu C, Zhang Y, Jiang Y, Wang Q, Long Y, Wang C, et al. Apoptotic cell administration enhances pancreatic islet engraftment by induction of regulatory T cells and tolerogenic dendritic cells. Cell Mol Immunol. (2013) 10:393–402. doi: 10.1038/cmi.2013.16
121. Mougel F, Bonnefoy F, Kury-Paulin S, Borot S, Perruche S, Kantelip B, et al. Intravenous infusion of donor apoptotic leukocytes before transplantation delays allogeneic islet graft rejection through regulatory T cells. Diabetes Metab. (2012) 38:531–7. doi: 10.1016/j.diabet.2012.08.008
122. Wang Z, Shufesky WJ, Montecalvo A, Divito SJ, Larregina AT, Morelli AE. In situ-targeting of dendritic cells with donor-derived apoptotic cells restrains indirect allorecognition and ameliorates allograft vasculopathy. PLoS One. (2009) 4:e4940. doi: 10.1371/journal.pone.0004940
123. Sun E, Gao Y, Chen J, Roberts AI, Wang X, Chen Z, et al. Allograft tolerance induced by donor apoptotic lymphocytes requires phagocytosis in the recipient. Cell Death Differ. (2004) 11:1258–64. doi: 10.1038/sj.cdd.4401500
124. Bonnefoy F, Perruche S, Couturier M, Sedrati A, Sun Y, Tiberghien P, et al. Plasmacytoid dendritic cells play a major role in apoptotic leukocyte-induced immune modulation. J Immunol. (2011) 186:5696–705. doi: 10.4049/jimmunol.1001523
125. Bonnefoy F, Masson E, Perruche S, Marandin A, Borg C, Radlovic A, et al. Sirolimus enhances the effect of apoptotic cell infusion on hematopoietic engraftment and tolerance induction. Leukemia. (2008) 22:1430–4. doi: 10.1038/sj.leu.2405061
126. Kleinclauss F, Perruche S, Masson E, de Carvalho Bittencourt M, Biichle S, Remy-Martin JP, et al. Intravenous apoptotic spleen cell infusion induces a TGF-beta-dependent regulatory T-cell expansion. Cell Death Differ. (2006) 13:41–52. doi: 10.1038/sj.cdd.4401699
127. Perruche S, Kleinclauss F, de Bittencourt M, Paris D, Tiberghien P, Saas P. Intravenous infusion of apoptotic cells simultaneously with allogeneic hematopoietic grafts alters anti-donor humoral immune responses. Am J Transplant. (2004) 4:1361–5. doi: 10.1111/j.1600-6143.2004.00509.x
128. Hosszu KK, Santiago-Schwarz F, Peerschke EIB, Ghebrehiwet B. Evidence that a C1q/C1qR system regulates monocyte-derived dendritic cell differentiation at the interface of innate and acquired immunity. Innate Immun. (2010) 16:115–27. doi: 10.1177/1753425909339815
129. Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol. (2009) 183:6175–85. doi: 10.4049/jimmunol.0902232
130. Clarke EV, Weist BM, Walsh CM, Tenner AJ. Complement protein C1q bound to apoptotic cells suppresses human macrophage and dendritic cell-mediated Th17 and Th1 T cell subset proliferation. J Leukoc Biol. (2015) 97:147–60. doi: 10.1189/jlb.3A0614-278R
131. Spivia W, Magno PS, Le P, Fraser DA. Complement protein C1q promotes macrophage anti-inflammatory M2-like polarization during the clearance of atherogenic lipoproteins. Inflamm Res. (2014) 63:885–93. doi: 10.1007/s00011-014-0762-0
132. Hosszu KK, Santiago-Schwarz F, Peerschke EI, Ghebrehiwet B. C1q is a molecular switch dictating the monocyte to dendritic cell (DC) transition and arrests DCs in an immature phenotype. FASEB J. (2008) 22:6731.
133. Hosszu KK, Santiago-Schwarz F, Peerschke EI, Ghebrehiwet B. C1q is a molecular switch that regulates dendritic cell maturation at the monocyte-to-dendritic cell transition. FASEB J. (2008) 45:4142–3. doi: 10.1016/j.molimm.2008.08.143
134. Lu J, Wu X, Teh BK. The regulatory roles of C1q. Immunobiology. (2007) 212:245–52. doi: 10.1016/j.imbio.2006.11.008
135. Espericueta V, Manughian-Peter AO, Bally I, Thielens NM, Fraser DA. Recombinant C1q variants modulate macrophage responses but do not activate the classical complement pathway. Mol Immunol. (2020) 117:65–72. doi: 10.1016/j.molimm.2019.10.008
136. Castellano G, Woltman AM, Schena FP, Roos A, Daha MR, van Kooten C. Dendritic cells and complement: at the cross road of innate and adaptive immunity. Mol Immunol. (2004) 41:133–40. doi: 10.1016/j.molimm.2004.03.018
137. Di Domizio J, Cao W. Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev Clin Immunol. (2013) 9:201–10. doi: 10.1586/eci.12.106
138. Rönnblom L, Alm GV. A pivotal role for the natural interferon alpha-producing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus. J Exp Med. (2001) 194:F59–63. doi: 10.1084/jem.194.12.f59
139. Cao W. Pivotal functions of plasmacytoid dendritic cells in systemic autoimmune pathogenesis. J Clin Cell Immunol. (2014) 5:212. doi: 10.4172/2155-9899.1000212
140. Lood C, Gullstrand B, Truedsson L, Olin AI, Alm GV, Rönnblom L, et al. C1q inhibits immune complex-induced interferon-alpha production in plasmacytoid dendritic cells: a novel link between C1q deficiency and systemic lupus erythematosus pathogenesis. Arthritis Rheum. (2009) 60:3081–90. doi: 10.1002/art.24852
141. Santer DM, Hall BE, George TC, Tangsombatvisit S, Liu CL, Arkwright PD, et al. C1q deficiency leads to the defective suppression of IFN-alpha in response to nucleoprotein containing immune complexes. J Immunol. (2010) 185:4738–49. doi: 10.4049/jimmunol.1001731
142. Mascarell L, Airouche S, Berjont N, Gary C, Gueguen C, Fourcade G, et al. The regulatory dendritic cell marker C1q is a potent inhibitor of allergic inflammation. Mucosal Immunol. (2017) 10:695–704. doi: 10.1038/mi.2016.87
143. Santer DM, Wiedeman AE, Teal TH, Ghosh P, Elkon KB. Plasmacytoid dendritic cells and C1q differentially regulate inflammatory gene induction by lupus immune complexes. J Immunol. (2012) 188:902–15. doi: 10.4049/jimmunol.1102797
144. Hosszu KK, Valentino A, Ji Y, Matkovic M, Pednekar L, Rehage N, et al. Cell surface expression and function of the macromolecular c1 complex on the surface of human monocytes. Front Immunol. (2012) 3:38. doi: 10.3389/fimmu.2012.00038
145. Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, et al. C1q restrains autoimmunity and viral infection by regulating CD8+ T cell metabolism. Science. (2018) 360:558–63. doi: 10.1126/science.aao4555
146. Chen A, Gaddipati S, Hong Y, Volkman DJ, Peerschke EI, Ghebrehiwet B. Human T cells express specific binding sites for C1q. Role in T cell activation and proliferation. J Immunol. (1994) 153:1430–40.
147. Fausther-Bovendo H, Vieillard V, Sagan S, Bismuth G, Debré P. HIV gp41 engages gC1qR on CD4+ T cells to induce the expression of an NK ligand through the PIP3/H2O2 pathway. PLoS Pathog. (2010) 6:e1000975. doi: 10.1371/journal.ppat.1000975
148. Song X, Yao Z, Yang J, Zhang Z, Deng Y, Li M, et al. HCV core protein binds to gC1qR to induce A20 expression and inhibit cytokine production through MAPKs and NF-κB signaling pathways. Oncotarget. (2016) 7:33796–808. doi: 10.18632/oncotarget.9304
149. Jiang Z, Chen Z, Hu L, Qiu L, Zhu L. Calreticulin blockade attenuates murine acute lung injury by inducing polarization of M2 subtype macrophages. Front Immunol. (2020) 11:11. doi: 10.3389/fimmu.2020.00011
150. Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TBH. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. (2007) 26:605–16. doi: 10.1016/j.immuni.2007.03.012
151. Caparrós E, Munoz P, Sierra-Filardi E, Serrano-Gómez D, Puig-Kröger A, Rodríguez-Fernández JL, et al. DC-SIGN ligation on dendritic cells results in ERK and PI3K activation and modulates cytokine production. Blood. (2006) 107:3950–8. doi: 10.1182/blood-2005-03-1252
152. Zhang Y, Li J, Rong Q, Xu Z, Ding Y, Cao Q, et al. The regulatory role of C1q on Helicobacter pylori-induced inflammatory cytokines secretion in THP-1 cells. Microb Pathog. (2019) 131:234–8. doi: 10.1016/j.micpath.2019.04.017
153. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
154. Castellano G, Woltman AM, Schlagwein N, Xu W, Schena FP, Daha MR, et al. Immune modulation of human dendritic cells by complement. Eur J Immunol. (2007) 37:2803–11. doi: 10.1002/eji.200636845
155. Yamada M, Oritani K, Kaisho T, Ishikawa J, Yoshida H, Takahashi I, et al. Complement C1q regulates LPS-induced cytokine production in bone marrow-derived dendritic cells. Eur J Immunol. (2004) 34:221–30. doi: 10.1002/eji.200324026
156. Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, et al. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol. (2004) 173:3044–50.
157. Csomor E, Bajtay Z, Sándor N, Kristóf K, Arlaud GJ, Thiel S, et al. Complement protein C1q induces maturation of human dendritic cells. Mol Immunol. (2007) 44:3389–97. doi: 10.1016/j.molimm.2007.02.014
158. Vegh Z, Kew RR, Gruber BL, Ghebrehiwet B. Chemotaxis of human monocyte-derived dendritic cells to complement component C1q is mediated by the receptors gC1qR and cC1qR. Mol Immunol. (2006) 43:1402–7. doi: 10.1016/j.molimm.2005.07.030
159. Liu S, Wu J, Zhang T, Qian B, Wu P, Li L, et al. Complement C1q chemoattracts human dendritic cells and enhances migration of mature dendritic cells to CCL19 via activation of AKT and MAPK pathways. Mol Immunol. (2008) 46:242–9. doi: 10.1016/j.molimm.2008.08.279
160. Kovacs H, Campbell ID, Strong P, Johnson S, Ward FJ, Reid KB, et al. Evidence that C1q binds specifically to CH2-like immunoglobulin gamma motifs present in the autoantigen calreticulin and interferes with complement activation. Biochemistry. (1998) 37:17865–74. doi: 10.1021/bi973197p
161. Ghebrehiwet B, Jesty J, Xu S, Vinayagasundaram R, Vinayagasundaram U, Ji Y, et al. Structure-function studies using deletion mutants identify domains of gC1qR/p33 as potential therapeutic targets for vascular permeability and inflammation. Front Immunol. (2011) 2:58. doi: 10.3389/fimmu.2011.00058
Keywords: c1q, gC1qR, cC1qR, complement, SLE, novel hypothesis
Citation: Hosszu KK, Valentino A, Peerschke EI and Ghebrehiwet B (2020) SLE: Novel Postulates for Therapeutic Options. Front. Immunol. 11:583853. doi: 10.3389/fimmu.2020.583853
Received: 15 July 2020; Accepted: 10 September 2020;
Published: 07 October 2020.
Edited by:
Elena Volokhina, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Mihaela Gadjeva, Harvard Medical School, United StatesKenneth Reid, University of Oxford, United Kingdom
Copyright © 2020 Hosszu, Valentino, Peerschke and Ghebrehiwet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Berhane Ghebrehiwet, Berhane.Ghebrehiwet@stonybrook.edu; Berhane.Ghebrehiwet@stonybrookmedicine.edu
†These authors have contributed equally to this work