 Lauren H. Fairley
Lauren H. Fairley Jia Hui Wong
Jia Hui Wong Anna M. Barron
Anna M. Barron- Neurobiology of Aging and Disease Laboratory, Lee Kong Chian School of Medicine, Nanyang Technological University Singapore, Singapore, Singapore
Alzheimer’s disease (AD) is an age-associated terminal neurodegenerative disease with no effective treatments. Dysfunction of innate immunity is implicated in the pathogenesis of AD, with genetic studies supporting a causative role in the disease. Microglia, the effector cells of innate immunity in the brain, are highly plastic and perform a diverse range of specialist functions in AD, including phagocytosing and removing toxic aggregates of beta amyloid and tau that drive neurodegeneration. These immune functions require high energy demand, which is regulated by mitochondria. Reflecting this, microglia have been shown to be highly metabolically flexible, reprogramming their mitochondrial function upon inflammatory activation to meet their energy demands. However, AD-associated genetic risk factors and pathology impair microglial metabolic programming, and metabolic derailment has been shown to cause innate immune dysfunction in AD. These findings suggest that immunity and metabolic function are intricately linked processes, and targeting microglial metabolism offers a window of opportunity for therapeutic treatment of AD. Here, we review evidence for the role of metabolic programming in inflammatory functions in AD, and discuss mitochondrial-targeted immunotherapeutics for treatment of the disease.
Introduction
Alzheimer’s disease (AD) is an age-associated terminal neurodegenerative disease characterized by the presence of two hallmark proteinopathies: extracellular aggregates of toxic beta amyloid (Aβ) and intracellular neuronal accumulation of misfolded tau. Inflammation is also an early feature of the neurodegenerative cascade (1, 2), identified as a key driver in pathogenesis and a promising target for AD therapeutic development (3). Although once viewed as a downstream consequence of AD pathogenesis, genome-wide association studies have implicated innate immunity as causative in the disease process, with variants of microglial regulators identified as key AD risk factors such as triggering receptor myeloid 2 (TREM2) and its downstream signaling molecule, Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1 (SHIP1) (4–8). Whether microglial overactivation or insufficiency contributes to neurodegeneration in AD is still a matter of controversy. However, in order to develop potential immunotherapeutics for AD, identification of mechanisms governing the switch between protective and dysfunctional microglial states is critical.
Emerging evidence indicates that metabolic function plays a critical role in the regulation of microglial immune function in AD, with several genetic risk factors for AD identified as important regulators of microglial metabolic fitness (3, 9, 10). Microglia are highly metabolically flexible and metabolic programming is a key regulator of functional plasticity, an emerging field known as immunometabolism (11, 12). Here we discuss the role of mitochondria as metabolic hubs and intracellular signaling platforms coordinating microglial immune functions in AD, and potential targets for the development of immunometabolic therapies for disease treatment.
Innate Immunity and Metabolism in AD
Microglia perform a diverse range of specialist functions in the AD brain. They can mitigate neurodegeneration through phagocytic clearance of pathological Aβ (2, 13) and tau (14), by removing dying neurons thus preventing “bystander” neuronal death (15, 16) and releasing neurotrophic factors promoting neuronal support including nerve growth factor, brain-derived neurotrophic factor, and insulin-like growth factor-I (17–20). On the other hand, they can exacerbate neurodegeneration by mediating the spread of misfolded forms of tau, phagocytosing healthy or functional neurons, releasing neurotoxic cytokines and increasing oxidative stress through the generation of reactive oxygen species (ROS) (21–23).
Although activated microglia have previously been roughly divided into two categories—classically activated M1 host defence responses, with pro-inflammatory and cytotoxic properties, and alternatively activated M2 regeneration and repair responses—recent single cell transcriptomic analyses have revealed a high degree of heterogeneity and complexity within microglial states and populations that change with aging and disease (24–29). This microglial diversity is distinguished not only by unique immune signatures but also by altered metabolic phenotypes (3, 26, 29).
Diversity of mitochondrial structure, localization and function within and between cells has been identified as an important factor determining cell-to-cell heterogeneity, as well as contributing to heterogeneous outcomes in aging (30). In microglia, mitochondria coordinate energy supply, generation of ROS, and production of substrates for membrane biosynthesis, immune signaling molecules and growth factors (31). However, mitochondrial quality and activity declines in aging and age-related diseases, such as AD (32). Microglia have very low mitochondrial turnover (33) and are severely affected by mitochondrial impairment (34). Further, chronic exposure to pathogens such as Aβ and tau also induces mitochondrial toxicity and metabolic dysfunction in microglia (35). It is therefore important to understand the effect of aging and disease on microglial metabolic programming in AD.
Microglial Metabolism in AD
Microglial metabolism is tightly controlled in response to environmental cues, including nutrient availability, cytokines, and damage- or pathogen-associated molecular patterns (DAMPs and PAMPs), including Aβ and tauopathy. A recent large-scale proteomics analysis of AD brain demonstrated early metabolic changes associated with microglial activation (3). Similarly, proteomics analysis of microglia isolated from AD mice identified enrichment in proteins involved in energy metabolism and mitochondrial processes (36). Here we will examine how the AD microenvironment impacts microglial metabolism and mitochondrial function.
Nutrient Availability and Metabolic Stress
The AD brain is under metabolic stress, with impairments in nutrient availability including glucose occurring early in the disease due to impaired blood-brain glucose transfer (34, 37). Glucose is the primary substrate used by microglia for energy production and is taken into the cell via various glucose transporters (GLUTs) (38–40) where it can metabolise glucose via glycolysis in the cytoplasm, enzymatically converting glucose into pyruvate then lactate to generate energy in the form of adenosine triphosphate (ATP). Alternatively, pyruvate can be shuttled into the mitochondria where it is converted into acetyl coenzyme A (Acetyl CoA) and consumed by the tricarboxylic acid (TCA) cycle, generating substrates for oxidative phosphorylation (OXPHOS). Glycolysis is less efficient than OXPHOS, requiring nearly 20-fold more glucose to yield equivalent quantities of ATP, but much faster, making it ideal under conditions in which rapid energy production is required. Microglia express genes required for both glycolytic and oxidative metabolism (41, 42) and can switch between these metabolic programmes in response to inflammatory stimuli (35, 43–46).
Glucose can also be metabolized by microglia via the pentose phosphate pathway (PPP) to generate nicotinamide adenine dinucleotide phosphate (NADPH), which fuels NADPH oxidase to produce ROS as well as providing the building blocks for nucleotide synthesis (47). However, long-term reductions in glucose availability in aging and AD necessitate the use of alternative energy sources in microglia, or they risk metabolic derailment and dysfunction.
Microglia are also capable of using a range of non-glucose based energy sources, a potentially important adaptive function in the hypoglycemic AD brain. Fatty acids (FA), which are released following the degradation of lipid droplets, are transported into mitochondria and used to fuel mitochondrial OXPHOS, a process known as fatty acid β-oxidation (FAO). A number of genes involved in FAO are expressed in microglia (48), and FAs have been shown to fuel macrophage functions under glucose deprivation (49, 50). Microglia have also been shown to take up ketones and lactate, through the monocarboxylic transporters MCT1 and MCT2 (51). Glutamine is another alternative energy source consumed by microglia via glutaminolysis under hyperglycemic conditions, which feeds into the TCA cycle (52). Although the extent to which these non-glucose substrates fuel microglial function in vivo is largely unknown, a recent study used fluorescence lifetime imaging (FLIM) to indirectly measure glycolysis and OXPHOS in the normal mouse brain under insulin-induced hypoglycaemia, demonstrating microglia shift to glutaminolysis in the absence of glucose (52). Positron emission tomography (PET) can also be used to visualize glycolysis and OXPHOS in both animals and the humans. Glycolysis can be measured using the glucose analogue, 18F-fluoro-2-deoxy-D-glucose-PET (FDG-PET) (53, 54) in combination with cerebral oxygen consumption to measure glycolysis (55, 56), while OXPHOS can be measured using mitochondrial complex-I-PET (57). These metabolic imaging approaches will enable investigation of microglial metabolism in the living brain under both normal and AD conditions and may provide biomarkers of microglial metabolism.
Aβ and Tau Alter Microglial Metabolism
AD-associated proteinopathy, Aβ and tau, induce mitochondrial toxicity and metabolic dysfunction. Exposure to Aβ alone or in combination with other inflammatory stimuli has been shown to induce a shift in metabolic programming from OXPHOS to glycolysis, impairing ATP production, increasing the generation of ROS, and inducing mitochondrial fission, fragmentation and extracellular release (35, 45, 46, 58). Fragmented mitochondria released from microglia have been shown to impair not only microglia, but also nearby neurons and astrocytes (58). Likewise, microglia isolated from AD mice are characterized by dependence on glycolytic metabolism as well as impaired mitochondrial quality control due to inhibition of mitophagy (59). Consistent with this, PET studies have shown increased reliance on aerobic glycolysis in areas spatially correlated with Aβ deposition in the AD brain (55, 56). Similarly, pathological forms of tau can bind mitochondria and impair OXPHOS and ATP synthesis (60); and Aβ and tau act synergistically to induce defects in OXPHOS and ATP synthesis, while increasing ROS production in AD mice (61).
Aβ can influence mitochondrial metabolism through direct binding and accumulation within mitochondria (62–64), inducing toxicity (65–67) and mitochondrial bioenergetic impairments (68–70). Mechanistically, Aβ inhibits OXPHOS by targeting ATP synthase, the enzyme that catalyses ATP production in the final step of OXPHOS (71). Others have implicated the nutrient sensor, mammalian target of rapamycin (mTOR), in Aβ-induced microglial metabolic reprogramming. Exposure to Aβ increases phosphorylation of mTOR via serine/threonine protein kinase B (Akt), which increases the expression of HIF-1a, the master transcriptional regulator of glycolysis (35).
Additionally, Aβ and tau can indirectly affect metabolism through upregulation of cytokines classically associated with M1, proinflammatory responses, such as interleukin-1β (IL-1β) and interferon-γ (IFNγ). Inflammatory cytokines induce glycolytic programming accompanied by breaks in the TCA cycle and uncoupling of OXPHOS (43, 45, 46). OXPHOS uncoupling impairs ATP production from mitochondrial respiratory chain activity, causing an increase in ROS generation (72). To support glycolysis in response to pro-inflammatory cytokines, microglia upregulate expression of GLUTs to facilitate increased glucose (40). A metabolic break in citrate metabolism fragments the TCA cycle, increases citrate availability for FA synthesis (FAS) and lipogenesis (73). Further, Aβ has been implicated in the suppression of mitochondrial succinate dehydrogenase (74), which underlies a second TCA cycle break widely observed in pro-inflammatory microglia, leading to accumulation of succinate. These findings suggest that AD-associated stimuli directly alter metabolic processes in microglia in a variety of ways. However, the precise signaling mechanisms involved remain to be fully elucidated.
AD Genetic Risk Factors and Microglial Metabolism
A number of immune-related genetic risk factors for AD have been shown to modulate microglial metabolism. The most prevalent genetic risk factor for AD, apolipoprotein E4 (ApoE4), is primarily expressed by glia in the brain and has been shown to play a role in mitochondrial energy production (9). In human iPSC-derived microglia, the AD-associated E4 variant of ApoE severely impaired metabolism, inhibiting both glycolysis and OXPHOS (75). Further, cognitively normal ApoE4 carriers demonstrate abnormally low cerebral metabolic rates for glucose (76). Additionally, ApoE binds and transports lipoproteins, and ApoE knockdown has been shown to alter FA levels and lipid metabolism in the brain (77). Interestingly, ApoE is a ligand for TREM2, which is a microglial surface receptor required for diverse microglial responses in neurodegeneration, including proliferation, survival, clustering and phagocytosis (46, 78–80). Increased risk of developing AD is associated with loss-of-function variants of TREM2, and TREM2 has been shown to induce ApoE signaling in microglia (27, 81).
Recently, TREM2 has also been identified as a regulator of mitochondrial metabolic fitness in microglia and macrophages (10). TREM2-deficient microglia exhibit decreased expression of genes encoding glucose transporters, glycolytic enzymes, as well as decreased expression of the metabolic coordinator, mTOR (10). Combined transcriptomic and metabolic analysis of TREM2 deficient macrophages revealed reduced ATP production and defects in metabolites and enzymes involved in glycolysis, the TCA cycle and PPP (10). Furthermore, TREM2 knockout mice exhibit cerebral hypometabolism measured by FDG-PET (82, 83). TREM2 also plays an important role in lipid metabolism, with TREM2 deficiency causing pathogenic lipid accumulation in microglia (84).
The TREM2/ApoE axis has also been identified as a regulatory checkpoint in the differentiation of specialized microglial phenotypes associated with age and disease, coined disease-associated microglia (DAMs) (26). DAMs express high levels of lipid metabolism genes, including APOE, Cst7 and lipoprotein lipase (Lpl), which catalyse the release of FAs for FAO (50). Other studies have identified disease and injury associated microglial signatures that share overlap with some key DAM-associated genes, including APOE and Lpl (24, 25, 27). A recent study of microglial proteomic changes in AD mice demonstrated upregulation of the TREM2/ApoE axis and increased proteins involved in FA metabolism (85).
These findings indicate that AD-associated pathology and genetic risk factors are intricately associated with mitochondrial metabolic functions in microglia (summarized in Figure 1). However, further research is needed to elucidate the mechanistic pathways involved in metabolic regulation in microglia, which may in turn aid the identification of druggable targets to modulate immunometabolic impairment.
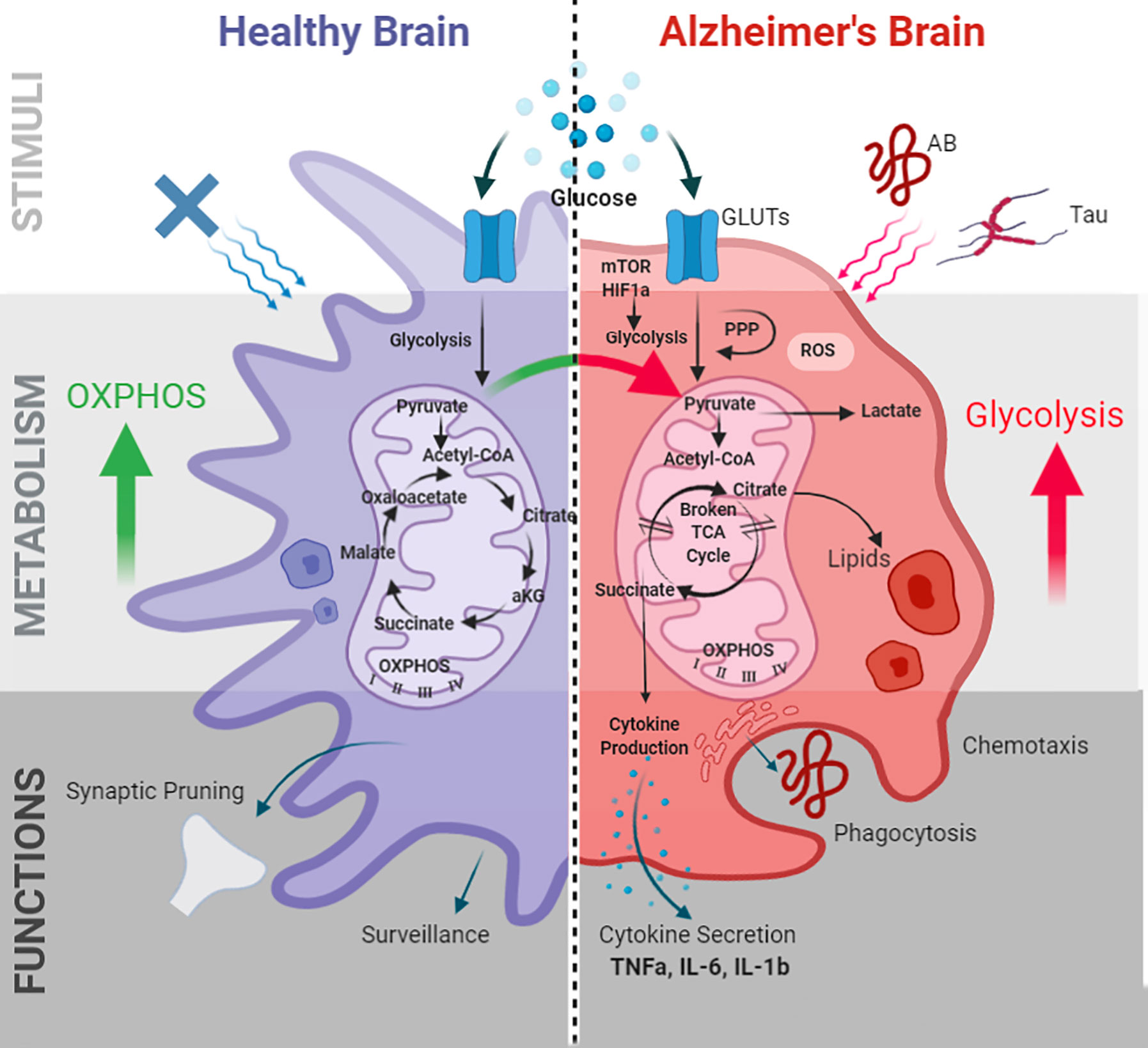
Figure 1 Microglial metabolic programming and immune functions in AD. Alzheimer’s pathogenic stimuli Aβ and tau induce microglial metabolic alterations. Metabolic alterations are mediated by the mTOR-HIF1a pathway and characterized by decreased OXPHOS, increased glycolysis, impaired ATP production, a “broken” TCA cycle, increased ROS, and lipid droplet accumulation. These alterations in turn effect microglial immune functions including phagocytosis, chemotaxis, cytokine production, membrane biogenesis, and antigen presentation. GLUTs, glucose transporters; PPP, pentose phosphate pathway; TCA, tricarboxylic acid; ROS, reactive oxygen species; OXPHOS, oxidative phosphorylation; TNF-α, tumor necrosis factor- α; IL-6, interleukin-6; IL-1β, interleukin-1β; HIF-1α, hypoxia inducible factor-1α; mTOR, mammalian target of rapamycin.
Aging and Microglial Metabolism
Aging is associated with a glycolytic metabolic shift in both human and mouse microglia, coupled with increased expression of markers of cellular senescence (86, 87). Aging also leads to accumulation of lipid droplets in microglia in the mouse and human brain, named lipid droplet accumulating microglia (LDAM) (29). LDAMs are characterized by upregulation of genes involved in lipogenesis, TCA cycle and FAO, and exhibit increased ROS production. Meanwhile, key enzymes involved in lipid degradation are downregulated. A recent study has demonstrated that physical coupling between the mitochondria and lipid droplets plays an important role in the regulation of glycolytic metabolic reprogramming in immune cells (88). Whether lipid droplet-mitochondrial interactions regulate metabolic programming in microglia remains to be addressed, but may provide important new insights into how microglial metabolism is coordinated in aging and AD.
Microglial Metabolism and the Innate Immune Response in AD
Microglial metabolism and immune function are reciprocally regulated. Microglia not only undergo adaptive metabolic reprogramming in response to inflammatory stimuli, but immune responses are dependent upon these metabolic shifts. Changes in cell morphology, chemotaxis, and phagocytosis all require reorganization of the actin cytoskeleton, which is dependent on the coordinated supply of ATP (89). Phagocytic degradation of engulfed materials, which plays an important role in the clearance of Aβ and tau, also relies upon the coordinated production and delivery of mitochondrial ROS to the phagolysosome (90). Similarly, the production of cytokines and growth factors requires resources such as amino acids, nucleotides, and fatty acids, which are supplied by metabolites generated during energy production. Mitochondrial metabolites are also utilized for lipogenesis to support membrane biogenesis for filopodia formation, antigen presentation and organelle biogenesis during proliferation and growth (73, 91). As such, changes in microglial adaptive metabolic reprogramming underpins immune function. Here we will discuss how age and disease associated changes in microglial metabolic programming modulates critical microglial functions in AD.
In microglia, initiation of the classic pro-inflammatory response is dependent upon glycolytic metabolic reprogramming, with immune responses including phagocytosis and pro-inflammatory cytokine production blocked by inhibition of glycolysis (25, 40, 92–94). In addition to rapidly generating ATP for energetically demanding chemotaxis and phagocytosis, upregulation of glycolysis and its branched pathway, the PPP, has been shown to be essential for the production of ROS-dependent phagosome degradation (95). In the AD brain, proteomics analysis has demonstrated a strong association between microglia and glycolytic metabolism (3). Upregulation of markers enriched in the AD brain was observed in microglia undergoing active Aβ phagocytosis in AD mouse brain, suggesting a protective function of hyperglycolytic microglia in AD (3). Markers identified in this study overlapped with markers of the TREM2-dependent, protective, phagocytic microglial subpopulation, DAMs (26). Knockout studies have shown that TREM2 promotes a metabolic programme fuelled by glycolysis, PPP and the TCA cycle to support phagocytosis (10). TREM2 deletion impairs microglial chemotaxis and phagocytosis in AD mice, resulting in inability of microglia to cluster around and clear aggregates of Aβ (10). These microglial deficits were mitigated with dietary cyclocreatine, a creatine analog that can supply ATP, indicating metabolic impairments caused the immune function impairments following TREM2 deletion (10).
Microglial hyperglycolysis is also observed in the aging brain but, in contrast is associated with compromised chemotaxis, phagocytosis and Aβ engulfment, and elevated secretion of pro-inflammatory cytokines (86, 87). Likewise, Aβ-induced glycolysis is associated with impaired chemotaxis and phagocytosis of Aβ in cultured microglia, a phenomena also observed in microglia isolated from AD mice (59). Further, multiple studies have shown that microglial Aβ phagocytosis is enhanced by promoting OXPHOS, rather than glycolysis (96, 97). One potential explanation for these differences may come from the chronic versus acute effects of Aβ on microglial function. Because glycolysis is metabolically inefficient, persistent reliance on glycolysis in microglia may lead to impaired immune function and reduced capacity to perform immune functions over time (97). In line with this, acute exposure to Aβ increased glycolysis and enhanced immune functions in microglia, whereas chronic exposure induced metabolic dysregulation and diminished immune functions, including phagocytosis and cytokine secretion (35). Further, Aβ-induced mitochondrial toxicity may disrupt coordinated immunometabolic programming. Consistent with this, in AD mice pharmacological induction of mitophagy to restore mitochondrial quality control enhanced microglial phagocytosis of Aβ and decreased the production of pro-inflammatory cytokines TNF-α and IL-6 (98).
Lipid metabolism has also been identified as important in microglial phagocytic functions in aging and AD. Lpl, the major enzyme responsible for liberating FAs from lipid droplets, is upregulated by Aβ in microglia, and silencing Lpl has been shown to impair Aβ phagocytosis (99). In macrophages, phagocytosis has been shown to be dependent on the availability of FAs following the degradation of lipid droplets, linking effective lysis of lipid droplets with successful phagocytosis (49). Supporting this, Lpl has been identified as a key marker of DAMs. In contrast, LDAMs, which are characterized by lipid droplet accumulation, lipogenesis and reduced FAO, also exhibit impaired phagocytosis (29). Lipid accumulation is a key feature observed in immune dysfunction, for example foamy macrophages observed in atherosclerotic lesions and lipid droplets have been identified as potential structural markers of inflammation (100). Interestingly, inhibition of FA synthesis restored phagocytic function in these microglia, again highlighting the potential to restore microglial immune function by restoring metabolic function in aging and AD.
Microglial Metabolic Reprogramming for the Treatment of AD
AD is one of the leading causes of death worldwide with no effective treatments available, leading to urgent calls for the development of disease modifying-agents (101). Given the pivotal role of inflammation in AD pathophysiology, here we discuss potential therapeutic strategies that improve microglial function through regulation of metabolism.
Ketone Body Therapeutics
Microglia can utilize ketone bodies as an alternative energy substrate to glucose, and ketosis has been shown to modulate a range of microglial inflammatory processes and reduce Aβ and tau accumulation in AD mice (101–106). Ketosis can be induced through several methods, including dietary modification, ketone body supplements, and pharmacological inhibitors of glycolysis. High-fat, low-carbohydrate ketogenic diets are thought to trigger a shift from glucose metabolism towards fatty acid metabolism, which in turn yields increased ketone body concentrations. Ketogenic diet decreased microglia activation and pro-inflammatory cytokine IL-6, IL-1β and TNF-α levels (107). Similarly, oral administration of ketone body metabolites such as β-hydroxybutyrate (β-OHB) have been shown to reduce microglial inflammation (108), reduce expression of pro-inflammatory cytokines IL-1b, IL-6, CCL2/MCP-1 (109), and inhibit NLRP3 inflammasome activation (110). Competitive inhibitors of glycolysis, such as 2-deoxy-D-glucose (2-DG) have been shown to induce compensatory metabolic processes and promote ketosis. Transgenic AD mice fed a diet supplemented with 2-DG exhibited increased serum ketone body levels and brain expression of enzymes required for ketone body metabolism, as well as decreased oxidative stress and reduced levels of Aβ oligomers (111). Further, pharmacological treatment with 2-DG has been shown to reduce markers of microglial activation following LPS treatment (41), reduce expression of inducible nitric oxide synthase (iNOS) (112), and decrease IL-6, IL-1β levels (113) in BV2 and primary microglia. These findings suggest that treatments targeted towards increasing microglial ketosis in AD may have therapeutic benefits, however ketogenic diet, β-OHB, and 2-DG are all known to exert non-microglial specific effects in a range of cell types in the brain. Further research aimed at identifying microglia-specific promoters of ketosis may be of benefit in the treatment of neuroinflammatory diseases such as AD.
Exercise
Exercise is consistently associated with improvement in cognitive and neuronal function in aged animals (114, 115) and reductions in Aβ and tau pathology in AD mice (116–118). The mechanism underlying exercise-related benefits in the brain is not well understood, however decreased pro-inflammatory cytokine expression has been proposed as one potential mechanism (119, 120). Recently, metabolic reprogramming has been identified as a mediator of exercise-relate changes in cognition and immune functions, as exercise attenuated age-dependent inflammatory cytokine expression and cognitive decline in mice, while decreasing glycolytic enzymes and increasing phagocytosis in isolated microglia (86). However, whether exercise exerts beneficial effects by promoting OXPHOS or FAO, or alternative substrate use, remains to be elucidated. Although no study to date has investigated whether metabolic reprogramming underlies exercise-related changes in inflammation and pathology in an AD-specific context, these findings suggest that exercise is a promising avenue for therapeutic investigation.
mTOR Targeted Therapeutics
The mTOR-HIF-1α pathway is a central mediator of inflammation in the brain and has been implicated in the regulation of microglial metabolic reprogramming in AD (10, 35). Two compounds that target the mTOR pathway and are currently being trialled for clinical efficacy in AD are rapamycin and metformin. Rapamycin directly inhibits mTOR via binding of mTOR Complex 1 (mTORC1), whereas metformin acts upstream of mTOR by targeting the glycolytic inhibitor AMP-activated protein kinase (AMPK). Both compounds have been shown to reduce glycolytic metabolism in favor of increased OXPHOS in immune cells (121, 122) and decrease the production of proinflammatory cytokines by microglia and macrophages (123, 124). As the mTOR pathway is found ubiquitously in all cell types, targeting microglial-specific mTOR signaling is challenging and both rapamycin and metformin are known to exert non-immune related effects in neurons and astrocytes. TREM2 has recently been identified as a microglial-specific target that regulates mTOR in AD (10), however there is currently a paucity of druggable targets identified in the TREM2-mTOR signaling pathway. SHIP1, which is primarily expressed in microglia, is one of the only therapeutic targets in the TREM2-specific mTOR signaling pathway that is under investigation for the treatment of AD. SHIP1 is known to inhibit TREM2 signaling (125) and pan-SHIP1/2 inhibitors have been shown to increase microglial phagocytosis of Aβ1-42 in vitro via mTOR regulation (126). These findings suggest that modulating TREM2-dependent mTOR signaling could provide neuroprotective effects in AD, however further research is needed to identify additional targets for therapeutic modulation in this pathway.
TSPO Targeted Therapeutics
The translocator protein (TSPO) is an outer mitochondrial membrane protein that is predominantly expressed in microglia in the brain and is upregulated in AD (127, 128). Consequently, TSPO is widely regarded as a biomarker of neuroinflammation, and TSPO ligands have been shown to exert a range of protective effects in mouse models of neurodegeneration (129, 130). In particular, ligands targeting TSPO have been shown to decrease Aβ deposition and reduce markers of inflammation in mouse models of AD (131). TSPO has also been implicated in microglial metabolic programming, as TSPO deficiency suppressed both OXPHOS and glycolysis, resulting in overall metabolic deficits in primary microglia (132) and increased fatty acid oxidation in steroidogenic cells (133). In contrast, treatment with TSPO ligands improved mitochondrial respiration, decreased oxidative stress-induced cell death by reducing ROS, and lowered Aβ levels in H1299 cells (134). These findings suggest that TSPO may be a marker of beneficial microglial phenotypes, and treatments aimed at increasing TSPO expression may confer neuroprotective effects in AD. However, further research investigating the metabolic modulatory effects of TSPO ligands in microglia specifically is required.
Discussion
Innate immunity plays a causative role in the pathogenesis of AD, and coordinated microglial immune responses have the potential to either ameliorate or exacerbate AD pathology, depending on microglial phenotypes. However, efforts to elucidate the cellular mechanisms and molecular signals mediating neuroinflammatory responses in AD have been hampered by the large heterogeneity in immune cell types and responses within the brain. Emerging evidence indicates that metabolic function plays a critical role in the regulation of microglial function in AD, with metabolic programming underlying diverse microglial immune functions and phenotypes. Further, therapeutic strategies modulating microglial metabolic programming have shown neuroprotective effects, by reducing amyloid and tau load, and improving cognitive deficits. These findings suggest that microglial function and metabolism are intricately associated processes in AD, however, further research is needed to elucidate the mechanistic pathways involved in metabolic regulation in microglia, which may in turn aid in the identification of druggable targets to modulate immunometabolic impairment.
Author Contributions
LF, JW, and AB wrote and reviewed the manuscript. LF: prepared the figure. All authors contributed to the article and approved the submitted version.
Funding
AB acknowledges funding support from the Singapore Ministry of Education under its Singapore Ministry of Education Academic Research Fund Tier 1 (RG42/18), Nanyang Assistant Professorship from Nanyang Technological University Singapore, and the Alzheimer’s Association (AARG-18-566427).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figure created with BioRender.com.
References
1. Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun (2013) 4:2030. doi: 10.1038/ncomms3030
2. Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci (2008) 28(33):8354–60. doi: 10.1523/JNEUROSCI.0616-08.2008
3. Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med (2020) 26(5):769–80. doi: 10.1038/s41591-020-0815-6
4. Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener (2018) 13(1):66. doi: 10.1186/s13024-018-0298-9
5. Guerreiro R, Hardy J. Genetics of Alzheimer’s disease. Neurotherapeutics (2014) 11(4):732–7. doi: 10.1007/s13311-014-0295-9
6. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med (2013) 368(2):107–16. doi: 10.1056/NEJMoa1211103
7. Malik M, Parikh I, Vasquez JB, Smith C, Tai L, Bu G, et al. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol Neurodegener (2015) 10(1):52. doi: 10.1186/s13024-015-0048-1
8. Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med (2012) 2(10):1–10. doi: 10.1101/cshperspect.a006296
9. Liao F, Yoon H, Kim J. Apolipoprotein E metabolism and functions in brain and its role in Alzheimer’s disease. Curr Opin Lipidol (2017) 28(1):60. doi: 10.1097/MOL.0000000000000383
10. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell (2017) 170(4):649–63.e13. doi: 10.1016/j.cell.2017.07.023
11. Angajala A, Lim S, Phillips JB, Kim J-H, Yates C, You Z, et al. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front Immunol (2018) 9:1605. doi: 10.3389/fimmu.2018.01605
12. Lauro C, Limatola C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front Immunol (2020) 11(493):1–8. doi: 10.3389/fimmu.2020.00493
13. Lee CD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (2010) 117(8):949–60. doi: 10.1007/s00702-010-0433-4
14. Luo W, Liu W, Hu X, Hanna M, Caravaca A, Paul SM. Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep (2015) 5(1):11161. doi: 10.1038/srep11161
15. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem (2009) 109(4):1144–56. doi: 10.1111/j.1471-4159.2009.06042.x
16. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med (2005) 201(4):647–57. doi: 10.1084/jem.20041611
17. Sampaio TB, Savall AS, Gutierrez MEZ, Pinton S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: implications for pathogenesis and therapy. Neural Regen Res (2017) 12(4):549. doi: 10.4103/1673-5374.205084
18. Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR II, Lafaille JJ, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell (2013) 155(7):1596–609. doi: 10.1016/j.cell.2013.11.030
19. Rajendran L, Paolicelli RC. Microglia-mediated synapse loss in Alzheimer’s disease. J Neurosci (2018) 38(12):2911–9. doi: 10.1523/JNEUROSCI.1136-17.2017
20. Suh H-S, Zhao M-L, Derico L, Choi N, Lee SC. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: differential regulation by inflammatory mediators. J Neuroinflamm (2013) 10(1):805. doi: 10.1186/1742-2094-10-37
21. Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci (2015) 18(11):1584. doi: 10.1038/nn.413
22. Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, et al. Microglial derived tumor necrosis factor-alpha drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol Dis (2014) 62:273–85. doi: 10.1016/j.nbd.2013.10.007
23. Wilkinson BL, Landreth GE. The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer’s disease. J Neuroinflamm (2006) 3:30. doi: 10.1186/1742-2094-3-30
24. Frigerio CS, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep (2019) 27(4):1293–306. e6. doi: 10.1016/j.celrep.2019.03.099
25. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity (2019) 50(1):253–71.e6. doi: 10.1016/j.immuni.2018.11.004
26. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell (2017) 169(7):1276–90.e17. doi: 10.1016/j.cell.2017.05.018
27. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47(3):566–81.e9. doi: 10.1016/j.immuni.2017.08.008
28. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol (2018) 18(4):225–42. doi: 10.1038/nri.2017.125
29. Marschallinger J, Iram T, Zardeneta M, Lee SE, Lehallier B, Haney MS, et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci (2020) 23(2):194–208. doi: 10.1038/s41593-019-0566-1
30. Aryaman J, Johnston IG, Jones NS. Mitochondrial Heterogeneity. Front Genet (2019) 9:718. doi: 10.3389/fgene.2018.00718
31. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol (2011) 11(6):389–402. doi: 10.1038/nri2975
32. Sun N, Youle RJ, Finkel T. The Mitochondrial Basis of Aging. Mol Cell (2016) 61(5):654–66. doi: 10.1016/j.molcel.2016.01.028
33. Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, et al. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci (2008) 28(34):8624–34. doi: 10.1523/jneurosci.1957-08.2008
34. Ryu JK, Nagai A, Kim J, Lee MC, McLarnon JG, Kim SU. Microglial activation and cell death induced by the mitochondrial toxin 3-nitropropionic acid: in vitro and in vivo studies. Neurobiol Dis (2003) 12(2):121–32. doi: 10.1016/s0969-9961(03)00002-0
35. Baik SH, Kang S, Lee W, Choi H, Chung S, Kim J, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab (2019) 30(3):493–507.e6. doi: 10.1016/j.cmet.2019.06.005
36. Rangaraju S, Dammer EB, Raza SA, Gao T, Xiao H, Betarbet R, et al. Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol Neurodegener (2018) 13(1):34. doi: 10.1186/s13024-018-0266-4
37. Backes H, Walberer M, Ladwig A, Rueger MA, Neumaier B, Endepols H, et al. Glucose consumption of inflammatory cells masks metabolic deficits in the brain. NeuroImage (2016) 128:54–62. doi: 10.1016/j.neuroimage.2015.12.044
38. Kalsbeek MJ, Mulder L, Yi C-X. Microglia energy metabolism in metabolic disorder. Mol Cell Endocrinol (2016) 438:27–35. doi: 10.1016/j.mce.2016.09.028
39. Sasaki A, Yamaguchi H, Horikoshi Y, Tanaka G, Nakazato Y. Expression of glucose transporter 5 by microglia in human gliomas. Neuropathol Appl Neurobiol (2004) 30(5):447–55. doi: 10.1111/j.1365-2990.2004.00556.x
40. Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener (2019) 14(1):2. doi: 10.1186/s13024-019-0305-9
41. Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia (2018) 66(6):1200–12. doi: 10.1002/glia.23271
42. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci (2014) 34(36):11929–47. doi: 10.1523/jneurosci.1860-14.2014
43. Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, et al. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun (2018) 68:183–96. doi: 10.1016/j.bbi.2017.10.017
44. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia (2019) 67(6):1047–61. doi: 10.1002/glia.23587
45. Voloboueva LA, Emery JF, Sun X, Giffard RG. Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett (2013) 587(6):756–62. doi: 10.1016/j.febslet.2013.01.067
46. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med (2016) 213(5):667–75. doi: 10.1084/jem.20151948
47. Gimeno-Bayón J, López-López A, Rodríguez M, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res (2014) 92(6):723–31. doi: 10.1002/jnr.23356
48. Mauerer R, Ebert S, Langmann T. High glucose, unsaturated and saturated fatty acids differentially regulate expression of ATP-binding cassette transporters ABCA1 and ABCG1 in human macrophages. Exp Mol Med (2009) 41(2):126–32. doi: 10.3858/emm.2009.41.2.015
49. Chandak PG, Radovic B, Aflaki E, Kolb D, Buchebner M, Frohlich E, et al. Efficient phagocytosis requires triacylglycerol hydrolysis by adipose triglyceride lipase. J Biol Chem (2010) 285(26):20192–201. doi: 10.1074/jbc.M110.107854
50. Yin B, Loike JD, Kako Y, Weinstock PH, Breslow JL, Silverstein SC, et al. Lipoprotein lipase regulates Fc receptor-mediated phagocytosis by macrophages maintained in glucose-deficient medium. J Clin Invest (1997) 100(3):649–57. doi: 10.1172/JCI119576
51. Moreira TJ, Pierre K, Maekawa F, Repond C, Cebere A, Liljequist S, et al. Enhanced cerebral expression of MCT1 and MCT2 in a rat ischemia model occurs in activated microglial cells. J Cereb Blood Flow Metab (2009) 29(7):1273–83. doi: 10.1038/jcbfm.2009.50
52. Bernier L-P, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun (2020) 11(1):1–17. doi: 10.1038/s41467-020-15267-z
53. Barron AM, Tokunaga M, Zhang MR, Ji B, Suhara T, Higuchi M. Assessment of neuroinflammation in a mouse model of obesity and beta-amyloidosis using PET. J Neuroinflamm (2016) 13(1):221. doi: 10.1186/s12974-016-0700-x
54. Tsukada H, Nishiyama S, Fukumoto D, Kanazawa M, Harada N. Novel PET probes 18F-BCPP-EF and 18F-BCPP-BF for mitochondrial complex I: a PET study in comparison with 18F-BMS-747158-02 in rat brain. J Nucl Med (2014) 55(3):473–80. doi: 10.2967/jnumed.113.125328
55. Vaishnavi SN, Vlassenko AG, Rundle MM, Snyder AZ, Mintun MA, Raichle ME. Regional aerobic glycolysis in the human brain. Proc Natl Acad Sci U S A (2010) 107(41):17757–62. doi: 10.1073/pnas.1010459107
56. Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, et al. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta ) deposition. Proc Natl Acad Sci U S A (2010) 107(41):17763–7. doi: 10.1073/pnas.1010461107
57. Barron AM, Ji B, Fujinaga M, Zhang M-R, Suhara T, Sahara N, et al. In vivo positron emission tomography imaging of mitochondrial abnormalities in a mouse model of tauopathy. Neurobiol Aging (2020) 94:140–8. doi: 10.1016/j.neurobiolaging.2020.05.003
58. Joshi AU, Minhas PS, Liddelow SA, Haileselassie B, Andreasson KI, Dorn G 2nd, et al. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat Neurosci (2019) 22(10):1635–48. doi: 10.1038/s41593-019-0486-0
59. McIntosh A, Mela V, Harty C, Minogue AM, Costello DA, Kerskens C, et al. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol (2019) 29(5):606–21. doi: 10.1111/bpa.12704
60. Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, et al. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta (2008) 1777(10):1289–300. doi: 10.1016/j.bbabio.2008.07.004
61. Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, et al. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A (2009) 106(47):20057–62. doi: 10.1073/pnas.0905529106
62. Cenini G, Rüb C, Bruderek M, Voos W. Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol Biol Cell (2016) 27(21):3257–72. doi: 10.1091/mbc.E16-05-0313
63. Mamada N, Tanokashira D, Ishii K, Tamaoka A, Araki W. Mitochondria are devoid of amyloid β-protein (Aβ)-producing secretases: Evidence for unlikely occurrence within mitochondria of Aβ generation from amyloid precursor protein. Biochem Biophys Res Commun (2017) 486(2):321–8. doi: 10.1016/j.bbrc.2017.03.035
64. Schaefer PM, von Einem B, Walther P, Calzia E, von Arnim CA. Metabolic Characterization of Intact Cells Reveals Intracellular Amyloid Beta but Not Its Precursor Protein to Reduce Mitochondrial Respiration. PLoS One (2016) 11(12):e0168157. doi: 10.1371/journal.pone.0168157
65. Aleardi A, Benard G, Augereau O, Malgat M, Talbot J, Mazat J, et al. Gradual alteration of mitochondrial structure and function by β-amyloids: importance of membrane viscosity changes, energy deprivation, reactive oxygen species production, and cytochrome c release. J Bioenerg Biomembr (2005) 37(4):207–25. doi: 10.1007/s10863-005-6631-3
66. Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet (2006) 15(9):1437–49. doi: 10.1093/hmg/ddl066
67. Petersen CAH, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, et al. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci (2008) 105(35):13145–50. doi: 10.1073/pnas.0806192105
68. Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med (2008) 14(10):1097–105. doi: 10.1038/nm.1868
69. Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science (2004) 304(5669):448–52. doi: 10.1126/science.1091230
70. Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet (2011) 20(13):2495–509. doi: 10.1093/hmg/ddr139
71. Chiozzi P, Sarti AC, Sanz JM, Giuliani AL, Adinolfi E, Vultaggio-Poma V, et al. Amyloid beta-dependent mitochondrial toxicity in mouse microglia requires P2X7 receptor expression and is prevented by nimodipine. Sci Rep (2019) 9(1):6475. doi: 10.1038/s41598-019-42931-2
72. Zuo H, Wan Y. Metabolic Reprogramming in Mitochondria of Myeloid Cells. Cells (2019) 9(1):1–17. doi: 10.3390/cells9010005
73. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42(3):419–30. doi: 10.1016/j.immuni.2015.02.005
74. Kaneko I, Yamada N, Sakuraba Y, Kamenosono M, Tutumi S. Suppression of Mitochondrial Succinate Dehydrogenase, a Primary Target of β-Amyloid, and Its Derivative Racemized at Ser Residue. J Neurochem (1995) 65(6):2585–93. doi: 10.1046/j.1471-4159.1995.65062585.x
75. Konttinen H, Ohtonen S, Wojciechowski S, Shakirzyanova A, Caligola S, Giugno R, et al. PSEN1ΔE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-derived microglia. Stem Cell Rep (2019) 13(4):669–83. doi: 10.1016/j.stemcr.2019.08.004
76. Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A (2001) 98(6):3334–9. doi: 10.1073/pnas.061509598
77. Lee J, Choi J, Wong GW, Wolfgang MJ. Neurometabolic roles of ApoE and Ldl-R in mouse brain. J Bioenerg Biomembr (2016) 48(1):13–21. doi: 10.1007/s10863-015-9636-6
78. Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener (2014) 9:20. doi: 10.1186/1750-1326-9-20
79. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron (2016) 91(2):328–40. doi: 10.1016/j.neuron.2016.06.015
80. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron (2016) 90(4):724–39. doi: 10.1016/j.neuron.2016.05.003
81. Atagi Y, Liu C-C, Painter MM, Chen X-F, Verbeeck C, Zheng H, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem (2015) 290(43):26043–50. doi: 10.1074/jbc.M115.679043
82. Götzl JK, Brendel M, Werner G, Parhizkar S, Sebastian Monasor L, Kleinberger G, et al. Opposite microglial activation stages upon loss of PGRN or TREM 2 result in reduced cerebral glucose metabolism. EMBO Mol Med (2019) 11(6):e9711. doi: 10.1523/JNEUROSCI.2070-18.2019
83. Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, et al. The FTD-like syndrome causing TREM 2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J (2017) 36(13):1837–53. doi: 10.15252/embj.201796516
84. Nugent AA, Lin K, Van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron (2020) 105(5):837–54.e9. doi: 10.1016/j.neuron.2019.12.007
85. Monasor LS, Müller SA, Colombo AV, Tanrioever G, König J, Roth S, et al. Fibrillar Aβ triggers microglial proteome alterations and dysfunction in Alzheimer mouse models. Elife (2020) 9:e54083. doi: 10.7554/eLife.54083
86. Mela V, Mota BC, Milner M, McGinley A, Mills KH, Kelly ÁM, et al. Exercise-induced re-programming of age-related metabolic changes in microglia is accompanied by a reduction in senescent cells. Brain Behav Immun (2020) 87:413–28. doi: 10.1016/j.bbi.2020.01.012
87. Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun (2018) 9(1):539. doi: 10.1038/s41467-018-02926-5
88. Bosch M, Sánchez-Álvarez M, Fajardo A, Kapetanovic R, Steiner B, Dutra F, et al. Mammalian lipid droplets are innate immune hubs integrating cell metabolism and host defense. Science (2020) 370(6514):1–12. doi: 10.1126/science.aay8085
89. Brusco J, Haas K. Interactions between mitochondria and the transcription factor myocyte enhancer factor 2 (MEF2) regulate neuronal structural and functional plasticity and metaplasticity. J Physiol (2015) 593(16):3471–81. doi: 10.1113/jphysiol.2014.282459
90. Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H, et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol (2015) 16(11):1142–52. doi: 10.1038/ni.3268
91. Feingold KR, Shigenaga JK, Kazemi MR, McDonald CM, Patzek SM, Cross AS, et al. Mechanisms of triglyceride accumulation in activated macrophages. J Leukoc Biol (2012) 92(4):829–39. doi: 10.1189/jlb.1111537
92. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289(11):7884–96. doi: 10.1074/jbc.M113.522037
93. Michl J, Ohlbaum DJ, Silverstein SC. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages II. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and ATP generation. J Exp Med (1976) 144(6):1484–93. doi: 10.1084/jem.144.6.1484
94. Pavlou S, Wang L, Xu H, Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J Inflamm (2017) 14(1):4. doi: 10.1186/s12950-017-0151-x
95. Koo SJ, Szczesny B, Wan X, Putluri N, Garg NJ. Pentose Phosphate Shunt Modulates Reactive Oxygen Species and Nitric Oxide Production Controlling Trypanosoma cruzi in Macrophages. Front Immunol (2018) 9:202. doi: 10.3389/fimmu.2018.00202
96. Pan RY, Ma J, Kong XX, Wang XF, Li SS, Qi XL, et al. Sodium rutin ameliorates Alzheimer’s disease-like pathology by enhancing microglial amyloid-beta clearance. Sci Adv (2019) 5(2):eaau6328. doi: 10.1126/sciadv.aau6328
97. Rubio-Araiz A, Finucane OM, Keogh S, Lynch MA. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of beta-amyloid. J Neuroinflamm (2018) 15(1):247. doi: 10.1186/s12974-018-1281-7
98. Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci (2019) 22(3):401–12. doi: 10.1038/s41593-018-0332-9
99. Ma Y, Bao J, Zhao X, Shen H, Lv J, Ma S, et al. Activated cyclin-dependent kinase 5 promotes microglial phagocytosis of fibrillar beta-amyloid by up-regulating lipoprotein lipase expression. Mol Cell Proteomics (2013) 12(10):2833–44. doi: 10.1074/mcp.M112.026864
100. Gibson MS, Domingues N, Vieira OV. Lipid and Non-lipid Factors Affecting Macrophage Dysfunction and Inflammation in Atherosclerosis. Front Physiol (2018) 9:654. doi: 10.3389/fphys.2018.00654
101. Prince M, Wimo AGM, Ali GC, Wu YT, Prina M. World Alzheimer Report 2015: the global impact of dementia: an analysis of prevalence, incidence, cost and trends. London: Alzheimer’s Disease International (2015). p. 1–87. https://www.alzint.org/u/WorldAlzheimerReport2015.pdf.
102. Broom GM, Shaw IC, Rucklidge JJ. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (2019) 60:118–21. doi: 10.1016/j.nut.2018.10.003
103. Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) (2009) 6:31. doi: 10.1186/1743-7075-6-31
104. Kashiwaya Y, Bergman C, Lee JH, Wan R, King MT, Mughal MR, et al. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol Aging (2013) 34(6):1530–9. doi: 10.1016/j.neurobiolaging.2012.11.023
105. Ota M, Matsuo J, Ishida I, Takano H, Yokoi Y, Hori H, et al. Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci Lett (2019) 690:232–6. doi: 10.1016/j.neulet.2018.10.048
106. Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement (N Y) (2018) 4:28–36. doi: 10.1016/j.trci.2017.11.002
107. Yang X, Cheng B. Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J Mol Neurosci (2010) 42(2):145–53. doi: 10.1007/s12031-010-9336-y
108. Huang C, Wang P, Xu X, Zhang Y, Gong Y, Hu W, et al. The ketone body metabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia (2018) 66(2):256–78. doi: 10.1002/glia.23241
109. Dupuis N, Curatolo N, Benoist JF, Auvin S. Ketogenic diet exhibits anti-inflammatory properties. Epilepsia (2015) 56(7):e95–8. doi: 10.1111/epi.13038
110. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med (2015) 21(3):263–9. doi: 10.1038/nm.3804
111. Yao J, Chen S, Mao Z, Cadenas E, Brinton RD. 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer’s disease. PLoS One (2011) 6(7):e21788. doi: 10.1371/journal.pone.0021788
112. Forstermann U, Kleinert H. Nitric oxide synthase: expression and expressional control of the three isoforms. Naunyn Schmiedebergs Arch Pharmacol (1995) 352(4):351–64. doi: 10.1007/BF00172772
113. Fodelianaki G, Lansing F, Bhattarai P, Troullinaki M, Zeballos MA, Charalampopoulos I, et al. Nerve Growth Factor modulates LPS - induced microglial glycolysis and inflammatory responses. Exp Cell Res (2019) 377(1-2):10–6. doi: 10.1016/j.yexcr.2019.02.023
114. Birch AM, Kelly ÁM. Lifelong environmental enrichment in the absence of exercise protects the brain from age-related cognitive decline. Neuropharmacology (2019) 145:59–74. doi: 10.1016/j.neuropharm.2018.03.042
115. Svensson M, Andersson E, Manouchehrian O, Yang Y, Deierborg T. Voluntary running does not reduce neuroinflammation or improve non-cognitive behavior in the 5xFAD mouse model of Alzheimer’s disease. Sci Rep (2020) 10(1):1–10. doi: 10.1038/s41598-020-58309-8
116. Tapia-Rojas C, Aranguiz F, Varela-Nallar L, Inestrosa NC. Voluntary Running Attenuates Memory Loss, Decreases Neuropathological Changes and Induces Neurogenesis in a Mouse Model of A lzheimer’s Disease. Brain Pathol (2016) 26(1):62–74. doi: 10.1111/bpa.12255
117. Xu ZQ, Zhang LQ, Wang Q, Marshall C, Xiao N, Gao JY, et al. Aerobic Exercise Combined with Antioxidative Treatment does not Counteract Moderate-or Mid-Stage A lzheimer-Like Pathophysiology of APP/PS 1 Mice. CNS Neurosci Ther (2013) 19(10):795–803. doi: 10.1111/cns.12139
118. Zhang J, Guo Y, Wang Y, Song L, Zhang R, Du Y. Long-term treadmill exercise attenuates Aβ burdens and astrocyte activation in APP/PS1 mouse model of Alzheimer’s disease. Neurosci Lett (2018) 666:70–7. doi: 10.1016/j.neulet.2017.12.025
119. Gibbons T. Enhancing learning and memory in the aged: Interactions between dietary supplementation and exercise. Urbana: University of Illinois (2014). http://hdl.handle.net/2142/49842Ñ20.
120. Speisman RB. Identifying inflammatory biomarkers of cognitive aging through relationships between measures of inflammation, neurogenesis and cognition in aged rats. Florida: University of Florida (2013). https://ufdc.ufl.edu/UFE0045275/00001
121. Caslin HL, Taruselli MT, Haque T, Pondicherry N, Baldwin EA, Barnstein BO, et al. Inhibiting Glycolysis and ATP Production Attenuates IL-33-Mediated Mast Cell Function and Peritonitis. Front Immunol (2018) 9:3026. doi: 10.3389/fimmu.2018.03026
122. Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia (2017) 31(11):2326–35. doi: 10.1038/leu.2017.81
123. Kelly B, Tannahill GM, Murphy MP, O’Neill LA. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1beta (IL-1beta) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J Biol Chem (2015) 290(33):20348–59. doi: 10.1074/jbc.M115.662114
124. Ou Z, Kong X, Sun X, He X, Zhang L, Gong Z, et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun (2018) 69:351–63. doi: 10.1016/j.bbi.2017.12.009
125. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal (2010) 3(122):ra38. doi: 10.1126/scisignal.2000500
126. Pedicone C, Fernandes S, Dungan OM, Dormann SM, Viernes DR, Adhikari AA, et al. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J Cell Sci (2020) 133(5):1–12. doi: 10.1242/jcs.238030
127. Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol (2009) 35(3):306–28. doi: 10.1111/j.1365-2990.2008.01006.x
128. Gulyás B, Makkai B, Nagy K, Vas Á, Kása P, Andersson J, et al. In vitro evidence for competitive tspo binding of the imaging biomarker candidates vinpocetine and two iodinated daa1106 analogues in post mortem autoradiography experiments on whole hemisphere human brain slices. Curr Radiopharm (2009) 2(1):42–8. doi: 10.2174/1874471010902010042
129. Gong J, Szego ÉM, Leonov A, Benito E, Becker S, Fischer A, et al. Translocator protein ligand protects against neurodegeneration in the MPTP mouse model of Parkinsonism. J Neurosci (2019) 39(19):3752–69. doi: 10.1523/JNEUROSCI.2070-18.2019
130. Leva G, Klein C, Benyounes J, Hallé F, Bihel F, Collongues N, et al. The translocator protein ligand XBD173 improves clinical symptoms and neuropathological markers in the SJL/J mouse model of multiple sclerosis. Biochim Biophys Acta (BBA)-Mol Basis Dis (2017) 1863(12):3016–27. doi: 10.1016/j.bbadis.2017.09.007
131. Barron AM, Garcia-Segura LM, Caruso D, Jayaraman A, Lee JW, Melcangi RC, et al. Ligand for translocator protein reverses pathology in a mouse model of Alzheimer’s disease. J Neurosci (2013) 33(20):8891–7. doi: 10.1523/JNEUROSCI.1350-13.2013
132. Yao R, Pan R, Shang C, Li X, Cheng J, Xu J, et al. Translocator Protein 18 kDa (TSPO) Deficiency Inhibits Microglial Activation and Impairs Mitochondrial Function. Front Pharmacol (2020) 11:986. doi: 10.3389/fphar.2020.00986
133. Tu LN, Zhao AH, Hussein M, Stocco DM, Selvaraj V. Translocator Protein (TSPO) Affects Mitochondrial Fatty Acid Oxidation in Steroidogenic Cells. Endocrinology (2016) 157(3):1110–21. doi: 10.1210/en.2015-1795
134. Zeineh N, Denora N, Laquintana V, Franco M, Weizman A, Gavish M. Efficaciousness of Low Affinity Compared to High Affinity TSPO Ligands in the Inhibition of Hypoxic Mitochondrial Cellular Damage Induced by Cobalt Chloride in Human Lung H1299 Cells. Biomedicines (2020) 8(5):1–18. doi: 10.3390/biomedicines8050106
Keywords: beta amyloid (Aβ), neurodegeneration, metabolism, tau, microglia, mitochondria
Citation: Fairley LH, Wong JH and Barron AM (2021) Mitochondrial Regulation of Microglial Immunometabolism in Alzheimer’s Disease. Front. Immunol. 12:624538. doi: 10.3389/fimmu.2021.624538
Received: 31 October 2020; Accepted: 21 January 2021;
Published: 25 February 2021.
Edited by:
Edecio Cunha-Neto, University of São Paulo, BrazilReviewed by:
Ravikanth Velagapudi, Duke University, United StatesMaryna Skok, Palladin Institute of Biochemistry (NAS Ukraine), Ukraine
Copyright © 2021 Fairley, Wong and Barron. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna M. Barron, barron@ntu.edu.sg