 Qijie Zhao1,2,3†
Qijie Zhao1,2,3† Yu Jiang1,2†Shixin Xiang1,2Parham Jabbarzadeh Kaboli1,2
Yu Jiang1,2†Shixin Xiang1,2Parham Jabbarzadeh Kaboli1,2 Jing Shen1,2
Jing Shen1,2 Yueshui Zhao1,2
Yueshui Zhao1,2 Xu Wu1,2Fukuan Du1,2Mingxing Li1,2Chi Hin Cho1,2Jing Li4Qinglian Wen5Tao Liu6*
Xu Wu1,2Fukuan Du1,2Mingxing Li1,2Chi Hin Cho1,2Jing Li4Qinglian Wen5Tao Liu6* Tao Yi7*
Tao Yi7* Zhangang Xiao1,8*
Zhangang Xiao1,8*- 1Laboratory of Molecular Pharmacology, Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, China
- 2South Sichuan Institute of Translational Medicine, Luzhou, China
- 3Department of Pathophysiology, College of Basic Medical Science, Southwest Medical University, Luzhou, China
- 4Department of Oncology and Hematology, Hospital (T.C.M.) Affiliated to Southwest Medical University, Luzhou, China
- 5Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, China
- 6Department of Oncology Rehabilitation, Shenzhen Luohu People’s Hospital, Shenzhen, China
- 7School of Chinese Medicine, Hong Kong Baptist University, Hong Kong, Hong Kong
- 8Department of Pharmacy, The Affiliated Hospital of Southwest Medical University, Luzhou, China
This review provides insight into the role of engineered T-cell receptors (TCRs) in immunotherapy. Novel approaches have been developed to boost anticancer immune system, including targeting new antigens, manufacturing new engineered or modified TCRs, and creating a safety switch for endo-suicide genes. In order to re-activate T cells against tumors, immune-mobilizing monoclonal TCRs against cancer (ImmTAC) have been developed as a novel class of manufactured molecules which are bispecific and recognize both cancer and T cells. The TCRs target special antigens such as NY-ESO-1, AHNAKS2580F or ERBB2H473Y to boost the efficacy of anticancer immunotherapy. The safety of genetically modified T cells is very important. Therefore, this review discusses pros and cons of different approaches, such as ImmTAC, Herpes simplex virus thymidine kinase (HSV-TK), and inducible caspase-9 in cancer immunotherapy. Clinical trials related to TCR-T cell therapy and monoclonal antibodies designed for overcoming immunosuppression, and recent advances made in understanding how TCRs are additionally examined. New approaches that can better detect antigens and drive an effective T cell response are discussed as well.
Introduction
In 2018, GLOBOCAN reported 18.1 million new cancer cases and 9.6 million related deaths worldwide (1). Recent studies have examined the role of neo-antigens in promoting immunotherapy pointing to their promising potential to treat cancer and increase overall survival rates of patients (2). Antibodies which inhibit T-cell inhibitory receptor, programmed death 1 (PD-1), and target its ligand on tumor surface, PD-L1, in order to decrease T cell tolerance in the tumor microenvironment (TME), have recently been approved by US Food and Drug Administration (FDA) (3). Suppression of PD-1 and PD-L1 has achieved promising antitumor effects in metastatic bladder, pancreatic, ovarian, breast, gastric, and renal-cell cancers (4–9). However, more potent and targeted approaches are crucial to combat certain cancers resistant to conventional therapies (10).
The one of most powerful therapeutic strategy in immunotherapy is adoptive cell transfer (ACT) (Figure 1). Chimeric antigen receptors (CAR) and engineered T cell receptors (TCRs) are recent therapeutic manufactured receptors of T cells which are used in adoptive T-cell immunotherapy (11, 12). The TCR-engineered T cells express tumor antigen-specific receptors with α and β chains which are produced from high-quality and high-avidity antigen-specific T-cell clones. They are utilized to develop antigen-specific immunotherapy (13). Recent clinical trials assessing the effectiveness of ACT therapy, TCR modified T (TCR-T) cells, and immune checkpoint inhibitors showed good outcomes. The high affinity and cellular avidity of TCR-T cells determine specific binding of several cellular proteins, which mean TCRs play a key role in activating T cellular avidity. In contrast to low affinity of TCRs which reduce the efficiency of TCR-based therapy, the high-affinity TCRs (Affinity ≥ 2.5nM) are specific and sensitive for targeting cell-surface human leukocyte antigens (HLAs) (14). The TCR molecules belong to a superfamily of immunoglobulin and they consist of two covalently-bound polymorphic subunits, each of which is antigen-specific, and they are related to at least four different types of signal transduction chains.
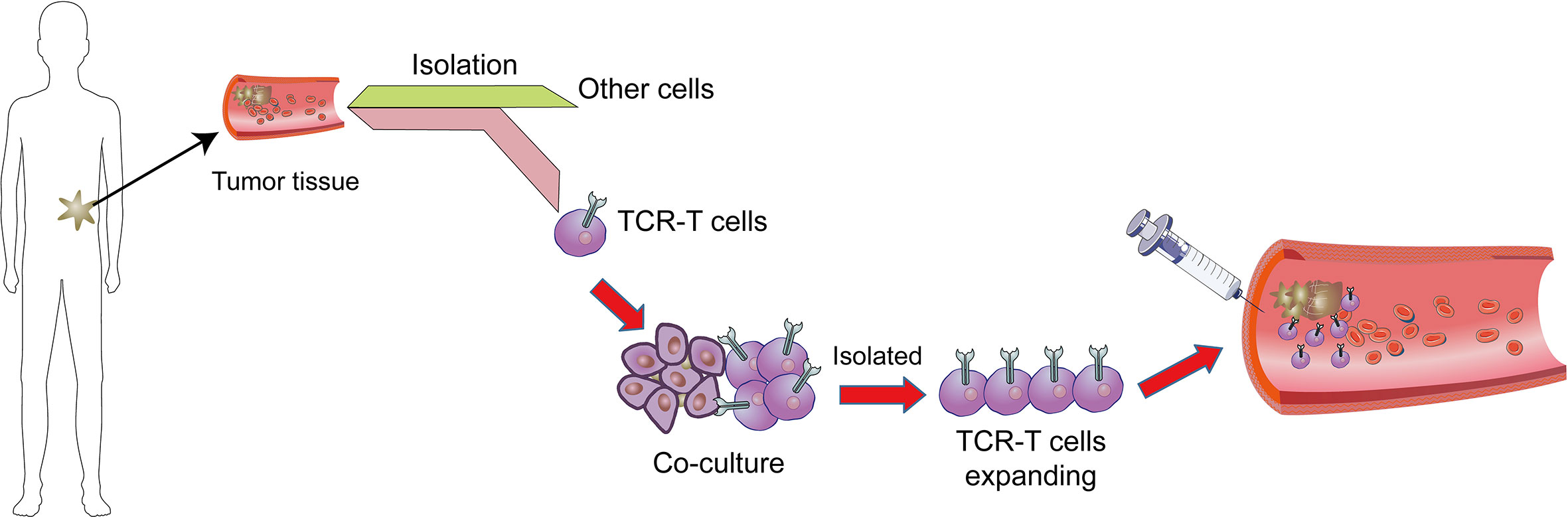
Figure 1 Adoptive T cell therapy. In adoptive cell therapy (ACT), cells are collected from cancer tissues/T cells which are then isolated from other cells, genetically manipulated by engineered TCR or CAR, co-cultured and proliferated, and eventually sent back into circulation.
In order to activate T lymphocyte, there has to be an interaction between TCR and major histocompatibility complex (MHC) (Figure 2). The strength of interaction between TCRs and pMHC (peptide-MHC) determines the fate of immature thymocytes, which is very important for the survival of naive T cells. Thus, TCR-T immunotherapy technique activates the host’s immune system through efficient interaction with MHC, especially class II molecules; the latter are specifically recognized by TCR-T cells and CAR-T cells (15). The TCR-T cells penetrate tumors but CAR-T cells are mainly distributed on the tumor periphery to access surface antigens. This makes TCR-T cells more efficient in cancer treatment (16).
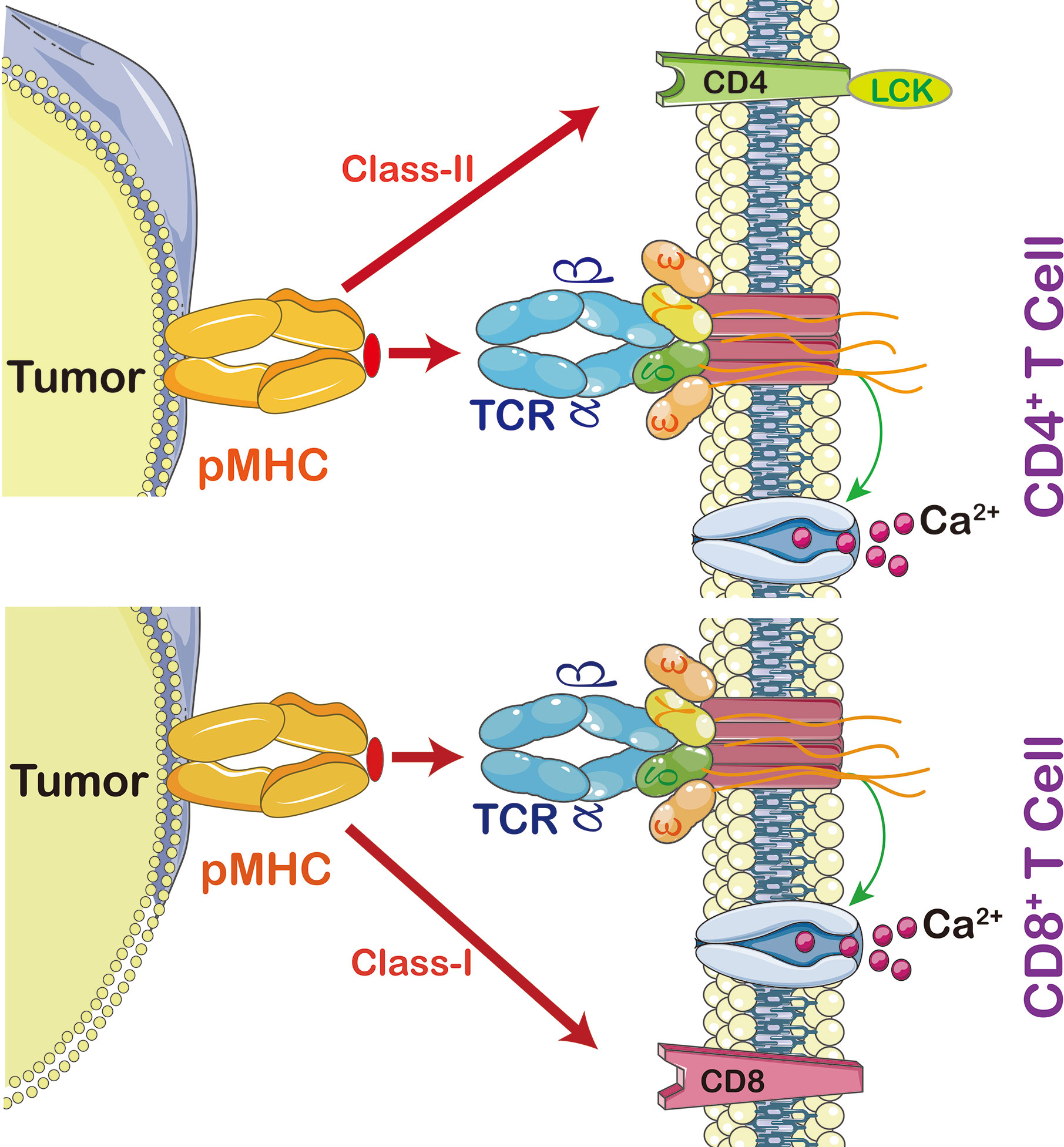
Figure 2 The interaction between peptide MHC and T cells. Cytotoxic T cells (CD8+) recognize MHC-I, CD3 coupled TCRs which activate T cell signaling cascade, including calcium-dependent and protein kinase C signaling pathways (PKC). Calcium is required for activation of nuclear factor of activated T cells (NFAT) leading to expression and secretion of Interleukin (IL)-2 and activation of PKC leading to tuning and regulating the production and secretion of ILs, including IL-2, T cell migration, T cell proliferation, and autoimmunity and graft rejection. CD4+ T cells are able to switch off the immune system by promoting regulatory T cells (Treg). In order to activate T cell proliferation, TCR also triggers p38MAPK and PI3K/Akt pathways. Once CD4 or CD8 activates TCR, it then activates Lymphocyte-Specific Protein-Tyrosine Kinase (Lck) which triggers phosphorylation of CD3 ζ chains.
This review provides insights into TCR-T cells with a special focus on its role in clinical anticancer immunotherapy. It also discusses advantages and disadvantages of different approaches in anticancer immunotherapy, including immune mobilizing monoclonal T-cell receptors against cancer (ImmTAC), inducible caspase-9, and Herpes simplex virus-thymidine kinase (HSV-TK) systems. Clinical trials on TCR-T cell therapy are discussed as well. Further, various strategies in anticancer immunotherapy with a special focus on TCR-T cell therapy are examined, highlighting efficacy and safety of each method in diagnostic and targeted cancer immunotherapy.
Comparison of CAR and TCR Therapies
In ACT, TCRs and CARs therapies, modified T cells have been successfully used as a paradigm-shifting clinical immunotherapy to treat solid tumors (17) (Figure 3). CAR T cells were engineered to transfer arbitrary specificity onto an immune effector cell, like T cell, which specifically eliminates antigen-bearing tumor cells (17). The CAR has scFv derived from antibody, CD3ζ and transmembrane domain (so-called first-generation CARs) (18). In this way, engineered CAR is able to recognize specific tumor associated-antigens (19). Therefore, the CAR has the ability to bind unprocessed tumor surface antigens without MHC processing (20) while TCRs engage with both tumor intracellular and surface antigenic peptides embedded in MHC (Table 1) (21). Generally, CAR scFv domain is used to engage with cell surface antigens (22). The scFv fragments could guide the constitutive activation and proliferation of T cells in an antigen-dependent mechanism; however, some scFv may guide T cells into an antigen-independent mechanism which lead to an unsuccessful treatment (23).
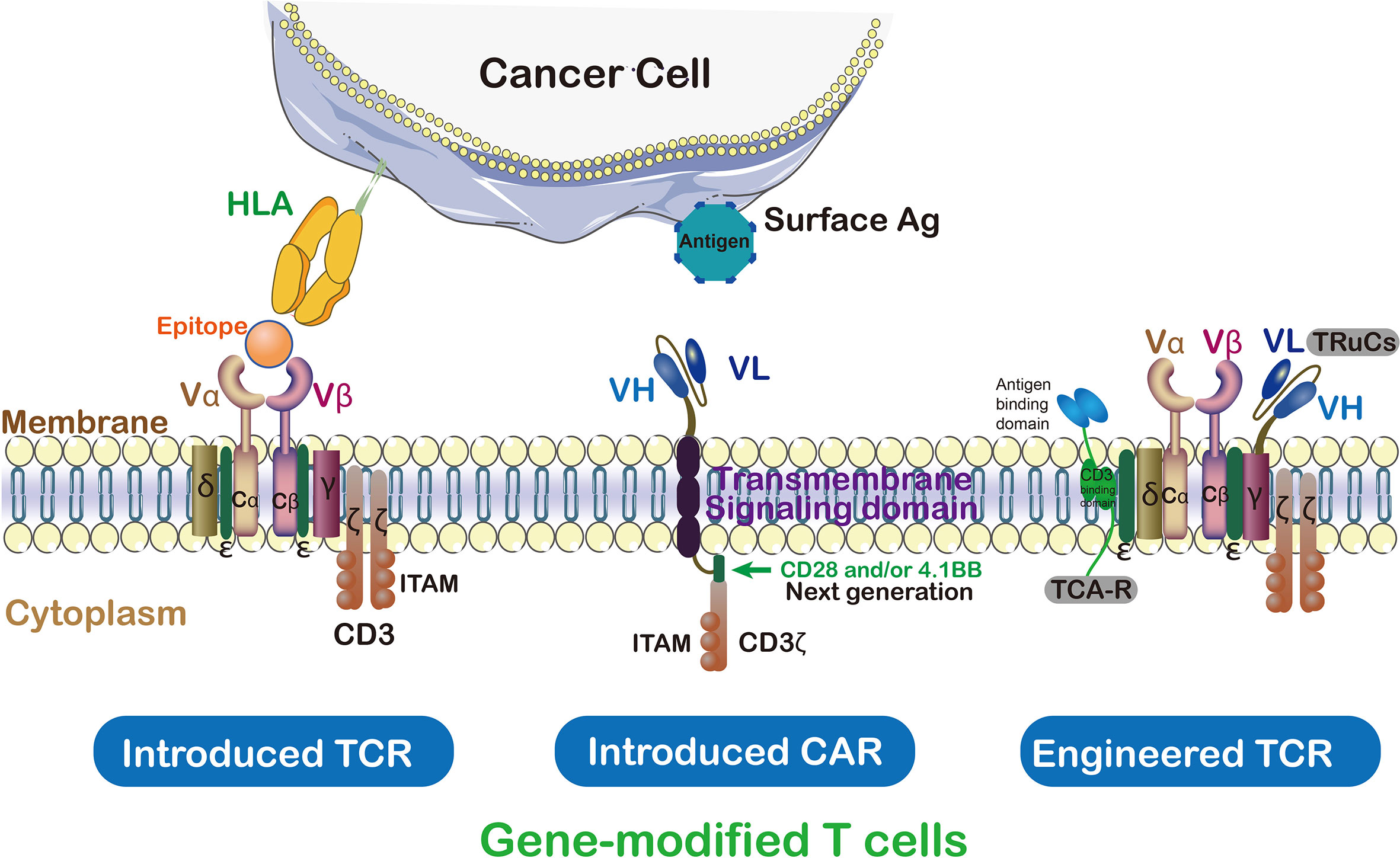
Figure 3 TCRs vs CAR and genetically modified T cells in immunotherapy. Cancer targeting receptors, TCR and CAR, are introduced to the activated T cells to empower them against special type of cancer. ζ subunit of CD3 on the surface of T cell is essential for triggering signaling cascade of T cells. In contrast to TCR which needs to be activated by endogenous CD3, CARs are hybrid receptors manufactured from a single-chain variable fragment (scFv) attached to ζ subunit of CD3. Both introduced and endogenous TCRs recognize HLA peptides on cancer cells. The CARs, instead, do not recognize HLA directly which trigger T cell signaling cascade in a TCR-independent manner.
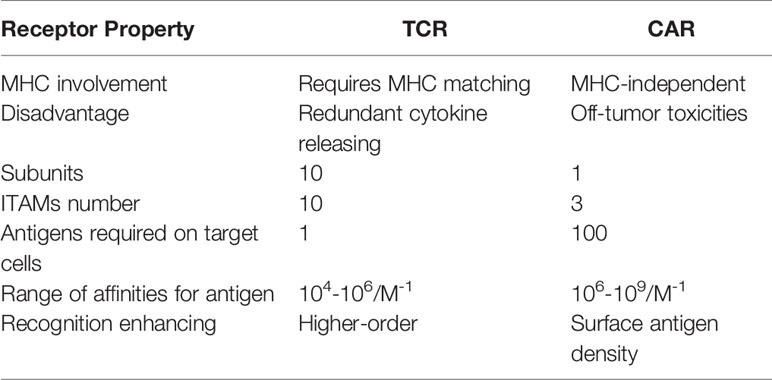
Table 1 Comparative characteristics of TCR and CAR.
Furthermore, first-generation CARs T cells showed limited expansion and relatively short persistence, which failed to evoke robust anti-tumor activity in the clinical studies (24, 25). The ‘second-generation’ CARs clinical activity can be induced by the CD3ζ domain insertion, namely co-stimulatory receptors CD28 (28ζ), 4-1BB/CD137 (BBζ) and OX40 (OX40ζ) CARs (26–28). The CD28 or CD137/4–1BB was added to CD3ζ endodomain of CAR-T cells which then promotes a more robust and durable T cells response (29, 30). Moreover, these second-generation CARs, which target CD19 antigen (CD19-specific scFv), are highly active against B cell malignancies and have promising clinical benefit (31, 32). In order to overcome the limitations of each individual costimulatory domain, the third generation CARs were proposed to simultaneously combine two co-stimulatory signaling (CD28 and 4-1BB), which presents a superior expansion and longer persistence than second-generation (33).
In contrast, TCRs are α/β heterodimers that bind to the MHC-bound antigens (Figure 3). As discussed above, CARs recognize tumor antigen which led to T cell activation with different functions compared with TCR. CAR-T cell therapy has certain disadvantages like off-tumor toxicities when targeting tumor-specific antigen (34). Compared with CARs, TCRs have several structural advantages in T cell-based therapy because they, such as more subunits in their receptor structure (ten subunits vs one subunit), greater immunoreceptor tyrosine-based activation motif (ITAMs) (ten vs three), less dependence on antigens (one vs 100), and more co-stimulate receptors (CD3, CD4, CD28, etc.) (20). The TCRs with low MHC interaction affinity range (104-106M-1) have been suggested for efficient T-cell stimulation (35). On the contrary, CARs possess a higher affinity range of (106-109M-1) and off-rates to recognize cell surface antigen (20). In order to maintain high-antigen sensitivity and to recognize pMHCs, exclusively monomeric TCR-CD3 complexes have been suggested (21). In contrast, the CAR-mediated cell sensitivity depends on higher density of cell surface antigen (36). Furthermore, T cell/antigen interaction is initiated in an immune synapse (IS) structure in which the TCR presents a ring region with peripheral LFA-1 adhesion, while CAR shows diffuse LFA-1 distribution without ring region (37). As a result, TCR-IS initiates a slower but longer duration signaling than that of CAR-IS. Meanwhile, the CAR-T cells presents faster killing function and move on to the next tumor target (serial killing), this was in stark contrast to the TCR-T cells protracted signaling and more extended killing.
Clinical trials involving with TCRs and CARs therapies have potential to suppress tumors progression. The CARs have been unable to effectively suppress malignant cells of some solid tumors and this is due to the presence of various types of antigens, their expression level, immunosuppressive environment, and construction of CARs. However, TCR-T cell transfer therapy has effectively treated some solid and hematological tumors (37).
Recombinant TCRs
The TCR is one of the body’s most complex receptors, which contain six different receptor subunits to its very broad signaling activities in T cells (38). Tumor-infiltrating lymphocytes (TILs)-TCR changes dramatically influence the tumor-dominant T cells (39). Among which, the changes in TCR will contribute to the expansion/proliferation of T cells. The TCR diversity and increased TILs were associated with anti-tumor effects, and TCR engineering in selective TILs is optimal therapy for the tumor rather than periphery (40). The TCR is composed of α and β chains that together bind to the peptide-MHC ligand, and signaling subunits of CD3 complex (ϵ, γ and δ) as well as the CD3ζ homodimer (41). All subunits except CD3ζ have extracellular immunoglobulin (Ig) domains. Based on these structures, the immune mobilizing monoclonal modified T-cell receptors (ImmTAC) were designed so that can specifically recognize the target HLA-peptide. There are some emerging techniques that utilize signaling subunits of the TCR and improve immunotherapy efficiency in HLA-independent manners, such as ImmTAC, TRuCs and TAC.
Immune Mobilizing Monoclonal T-Cell Receptors (ImmTAC)
The ImmTACs were designed using engineered, soluble, and affinity-enhanced monoclonal TCRs (mTCRs). ImmTACs are basically fusion proteins which combine an engineered TCR-based targeting system with a single chain antibody fragment (scFv) effector function. In the construction of ImmTACs, TCRs was deemed as antibodies which possess antigen recognition within the immune system. However, while antibodies only target cell surface or secreted proteins, TCRs are able to recognize peptides derived from intracellular targets presented by human leukocyte antigen (HLA) (42)(Figure 4).
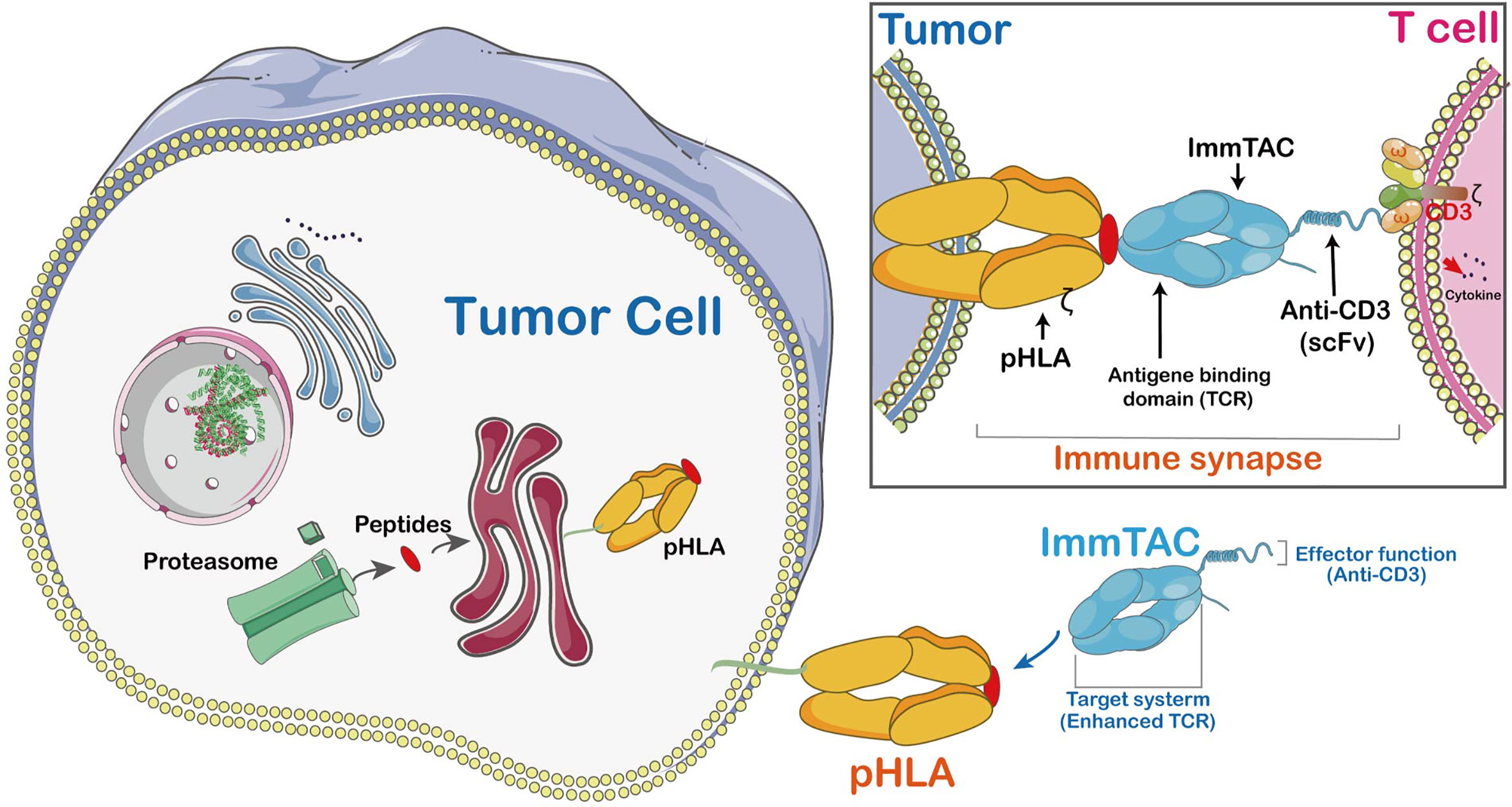
Figure 4 The mechanism of ImmTAC action. ImmTACs are designed to activate T cells against cancer and virus-infected cells. ImmTAC is specific to both MHC-peptides (pHLA) located on the surface of the tumor and CD3s located on T cells. The ImmTAC is able to directly activate CD3 and its corresponding pathways in activated T cells. In fact, the specific role of CD4 and CD8 is to stabilize the immune synapse, an important process of T cells shortened and empowered by ImmTACs.
ImmTACs have the potential to suppress tumor growth as they exert their activity through T cell redirection. Generally, ImmTACs comprising tumor-associated antigen-specific monoclonal TCR which strongly boost the affinity to pMHC (43). They effectively redirect these cells to eliminate cancer cells (44). The ImmTAC binds to the cancer cell through specific targeting of HLA-peptide complexes on their cell surface. The picomolar affinity of TCR to pMHC results in the coating of target cells by ImmTACs, and facilitates T-cell mediated effector function via interaction between scFv antibody fragments and CD3. Moreover, ImmTAC also activates CD8+ T cells in a dose-dependent manner with a low picomolar range of EC50 values (45). It has been shown that an ImmTAC, IMCgp100 effectively redirects and activates effector and memory CD8+ and CD4+ cells (46). The ImmTAC exhibits a polyfunctional response through secretion of several types of cytokines, such as tumor necrosis factor-α (TNF-α), interferon-γ, IL-6 pro-inflammatory cytokines, macrophage inflammatory protein-1α-β (MIP1α-β) and IFN-γ-inducible protein-10. The TNF-α and IFN-γ are highly effective inflammatory agents which promote apoptosis of tumor cell and stimulate inflammation of endothelial cells to enhance immune cell adhesion and extravasation in TME (47). MIP1α-β proteins involved in tumor proliferation or associated inflammation are potent chemo-attractants for monocytes. The MIP1α-β proteins are only secreted by CTLs and are mainly modulated by CD4+ T lymphocytes. An earlier study has shown IMCgp100-redirected T cells have a polyfunctional phenotype, a powerful anti-cancer response (46).
Furthermore, selecting appropriate target antigens is crucial for manufacturing ImmTACs. Certain dysregulated non-mutated proteins are presented as a tumor-associated antigen. Two decades ago, it was necessary to distinguish whether these targets are useful for immunotherapy by comparing their expression levels between tumor and normal tissues. Mass spectrometry has been used to identify low-abundance peptides because it has the sensitivity and ability to identify post-translational modifications for tumor antigens. While, failure to detect a pHLA complex using mass spectrometry does not mean it is absent. A pHLA target has the ability to induce an antigen-specific T cell response. The MHC-multimer technology can be used to profile T cell responses from immuno-stimulated patients. It also facilitates identification of suitable antigens (46). In order to compare ImmTAC targets, antigen expression, which is correlated with ImmTAC-mediated response, can be analyzed in vitro and in vivo studies (48). Of note, unexpected off-target reactivity of TCR-engineered T cells has been led to fatal cardiac toxicity, which was observed to recognize both epitope of MAGE-A3 and unrelated muscle protein titin in cardiac tissue in two series of clinical events (49). Hence, natural presentation of off-target peptide must be first confirmed using mass spectroscopy, and then followed by in vitro assessment of ImmTAC recognition. Since the therapeutic window maybe affected by ImmTAC off-target interactions, their affinity should be estimated (44). Overall, ImmTACs have been shown to enhance TCR-T cell anti-tumor response, but its safety needs further scrutiny.
T Cell Receptor Fusion Constructs (TRuCs)
More recently, reprogrammed TCR-T cells with a new target specificity and the potential for HLA-independent cells was developed. T cell receptor fusion constructs (TRuCs), antibody-based binding domain fused to T cell receptor (TCR) subunit, which was designed for effective recognition of tumor surface antigens (29, 50). The TRuCs, consisted of specific ligand antibody fused to the extracellular N-termini of five TCR subunits (TCRα, TCRβ, CD3ϵ, CD3γ and CD3δ), provide the engineered T cell with new target specificity and HLA-independent target cell elimination ability which can be activated by corresponding target cells (51). Upon lentiviral transduction, the TRuCs will be integrated into native TCR complex on the T cells surface. Therefore, activation and effector function of T cells are retained. This method on TCR engineering showed better anti-tumor effect compared to the second-generation CAR-T cells. Moreover, the TRuCs dominate full signaling machinery of the TCR complex, while CARs only utilized limited signaling of isolated CD3ζ cytoplasmic tail (50).
T Cell Antigen Coupler (TAC)
The T cell antigen coupler (TAC) is another platform that co-opts the endogenous TCR with MHC-independent manner to induce more efficient anti-tumor responses and reduces toxicity (52). The TAC chimeric proteins coupled the TCR to recognize antigen via CD3 domain binding, resulting in a TCR/CD3 complex formation and achieving more T cell responses (52). In addition, the activity of TAC receptor was critically dependent upon the choice of CD3-binding domain; hence, the appropriate scFvs improve the combination of phenotypic and functional characteristics. For example, the scFv derived from OKT3 (muromonab-CD3), one of the most commonly used agonistic anti-CD3 antibodies has lower cytokine production and cytotoxicity compared to UCHT1 (53). Thus, the delicate difference in integrating with the CD3 complex may contribute to substantially different functional outcomes. Compared to the second-generation CARs, the TAC engineered T cells did not merely favor the greater infiltration of solid tumors after adoptive injected but also reduced T cells expansion in healthy tissues that express antigens and off-tumor toxicities.
Others
Recently, several novel ACT technologies have been proposed. The Natural killer cells (NKs) were emerged as a promising source of CAR-based therapies (CAR-NKs), which is safety and availability (54). The NKs initiate innate immune responses against infections and malignancies with natural cytotoxicity (55). The CAR modified NKs shown specific and potent cytotoxic activity against target cells (56), and the CAR-NKs treatment significantly reduced the tumor growth (57, 58). Some preclinical data indicated that CAR-NKs may be advantageous over CAR-T cells in T-lymphoid malignancies, as the shared expression of targetable antigens on both malignant and normal T lymphocytes (e.g. CD5 and CD28) (54, 59). Moreover, recent studies have demonstrated that stem cells can differentiate into functional immune cells and/or crosstalk with immune cells to modulate the tumor immune environment (60). The pluripotent stem cells differentiated Cytotoxic T cells and NKs shown several advantages compared to primary immune cells, such as improved anti‐tumor activity and produced in essentially unlimited numbers (61). Meanwhile, induced pluripotent stem cells (iPSC) as a novel cancer vaccine achieved a promising preventive and therapeutic effects to various types of cancers (62). Kooreman et, al. indicated that autologous iPSC-based vaccine can elicit anti-tumor responses in breast cancer, mesothelioma, and melanoma models and reduce the local or distant relapse after primary tumor resection (63). Among which, in melanoma, iPSC vaccine is associated with fewer Th17 cells and promoted antigen-specific anti-tumor T cells response. Furthermore, the iPSC plus CpG vaccine can evoked a strong cancer-immunity by upregulating mature APCs, effector T cells and cytotoxic T cells, as well as decreasing the amount of regulatory T cells (Tregs) (62).
TCRs Side-Effects
The ACT with genetically engineered T cells has shown high sensitivity but with some severe adverse events in some clinical studies (11). Optimal TCR affinity in engineered T cells is vital and accordingly, receptor avidity is able to determine the safety/efficacy of T cell therapy (64). For example, in melanoma and neuroblastoma, some preclinical studies indicated the advanced intensity and durable antitumor effect of T cells. In a recent multi-target cell-based immunotherapy, patients were treated with CAR-Ts/TCRs against certain tumorigenic antigens, such as interventional studies of 10 and 4 tumor specific CAR-T/TCR in combination with cyclophosphamide or fludarabine (NCT03638206; NCT03941626). Meanwhile, the treatment-related adverse events were monitored based on Common Terminology Criteria for Adverse Events (CTCAE) v4.03 within 30 days after the last infusion. Not surprising, increased TCR expression and high frequency of TCR modified T cells within the graft improved TCR-modified T cells anti-tumor potency. In addition, combing the TCR therapy with irradiation therapy further improves efficiency of TCR therapy (65) (Table 2).

Table 2 Clinical trials activated on engineered TCR-T cells.
With on-target off-tumor toxicities, avidity becomes a main obstacle to the clinical success of ACT. When an antigen-specific receptor is used, in terms of efficacy, the avidity should be high enough for proper T cell activation (66). On the other hand, low avidity TCR interaction is sufficient to activate T cells, but strong avidity is required to sustain T-cell expansion (67). In phase I/II ACT clinical trial, the low-avidity engineered T cells showed safer profile, but they had a weaker anti-tumor response (68). Therefore, optimal avidity is a key factor in safety/efficacy of ACT. Recently, the reversible Ni2+-nitrilotriacetic acid histidine tags (NTAmers) technique was developed to efficiently separate high-avidity cytotoxic T-cells (69). Through recognizing TCR–pMHC interaction of T cells, engineered T cells can be isolated into high and low-intermediate avidity subtypes. Herein, an advance technology for avidity monitoring is necessary to ensure safe treatment.
Herpes Simplex Virus Thymidine Kinase
Immunotherapy using T lymphocytes is an attractive strategy to treat many types of malignancies. However, the side-effects and off-target of T cell immunotherapy necessitates finding a safety switch mechanism which should be based on engineered T cells. Many clinical trials tried to eliminate potentially harmful cells using suicide genes (70). Thymidine kinase gene derived from herpes simplex virus I (HSV-TK) is one of the most common suicide genes. Therefore, transcriptional connection between HSV-TK and cell-division gene (CDK1) has been engineered and quantified based on mathematical models to determine the safety of this therapy (71). Cell batches in the suicide system and a homozygous HSV-TK-CDK1 boost safe-cell level (SCL) while ensuring a clinically validated safety range.
A serious complication related to hematopoietic allografts can happen when the Graft-versus-host disease (GvHD) is occurred. This, as a result of T cell transplantation, can cause tissue and organ damage. Transplantation of T cells recognizes the host’s histocompatibility in about 80% of patients (72). In order to prevent GvHD, T cells have been genetically modified with suicide gene HSV-TK using prodrug ganciclovir (GCV) (73)(Figure 5). Safety switches (suicide genes) are of particular value in long-term cell-dependent immunotherapies. They also avoid T cell off-target interactions. Herein, the suicide genes were able to control therapeutic process and can be initiated by early clinical interventions. For example, in clinical trial, HSV-TK modified T cells can be monitored by positron emission tomography (PET)/CT when they are migrating to an unexpected location (74).
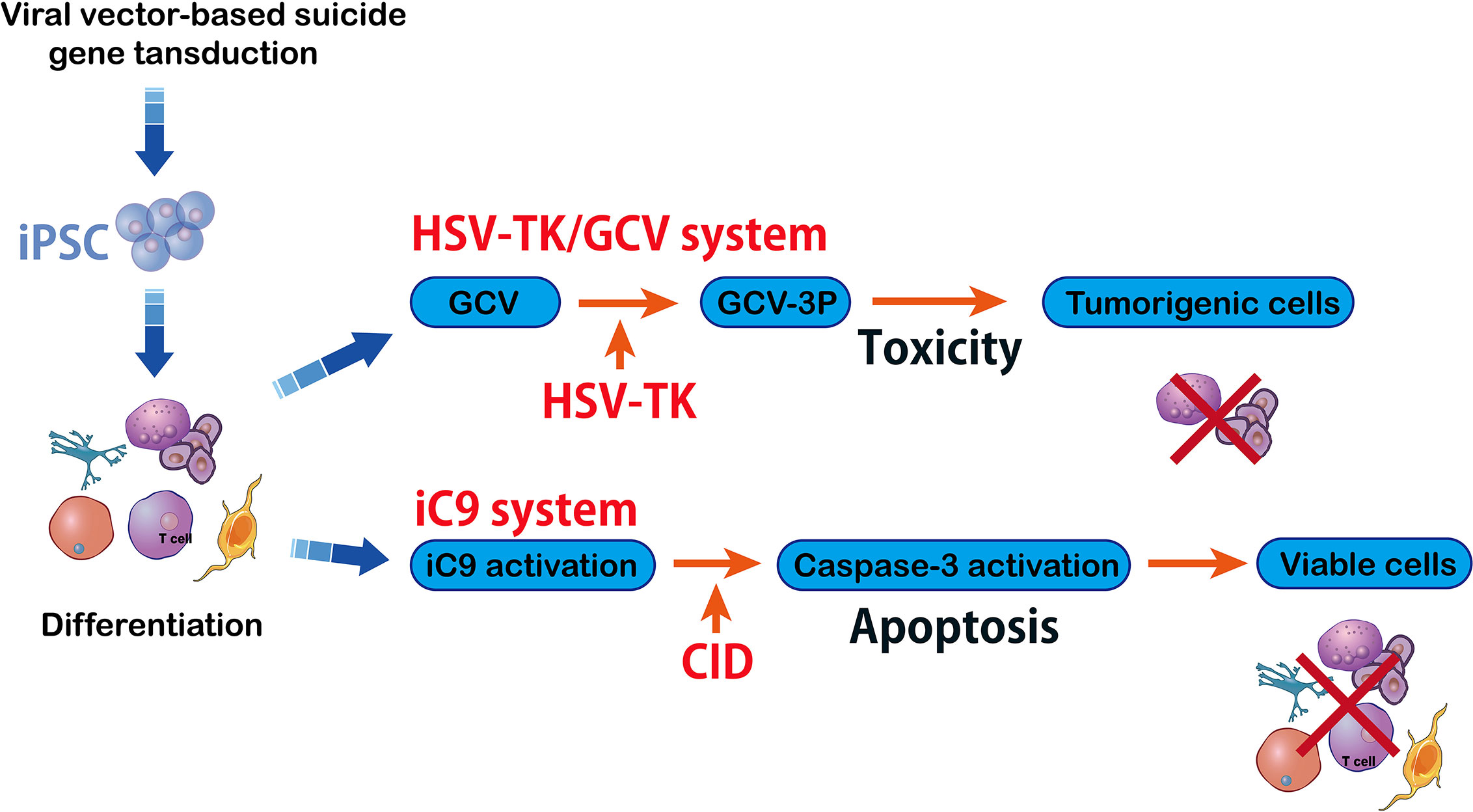
Figure 5 HSV-TK/GCV system vs inducible caspase-9 system. The herpes simplex virus–Thymidine Kinase/ganciclovir (HSV-TK/GCV) system eliminates tumorigenic cells and it is efficient and specific against inducible pluripotent stem cells (iPSC) that can kill the cells whose HSV-TK expression has been silenced. The HSV-TK/GCV system is used as a safety switch and it produces a toxic compound that kills the transduced cells. Another method in suicide gene therapy is introducing inducible caspase-9 (iC9) into iPSC. The iC9 dimerization activates iC9 which then triggers a caspase cascade leading to elimination of tumors originating from iPSC. Specific chemical inducers of dimerization (CID) induce iC9.
There are several limitations. First, previous studies observed a small number of patients with HSV-TK engineered T cells had lost sensitivity to GCV. Second, the pro-drug GCV not only activates HSV-TK which precludes its administration, but it also acts as an anti-viral drug for other indications like cytomegalovirus (CMV) infections (75). Finally, HSV-TK engineered T cells have potential immunogenic activity. Autologous immune response to activate the suicide gene may result in transduced T-cell elimination, and therefore, reduces their therapeutic efficiency (76). Non-immunogenic suicide genes with low toxicity, stable expression, and high eliminating strength are urgently required in newly engineered transduced T cells.
Inducible Caspase-9
Although HSV-TK shows safety in cell-based immunotherapy, the phosphorylated nucleoside analogs into DNA synthesis is required to complete elimination of tumor cells (77). Specifically, a quick elimination of the infused cells is required for cancer cellular therapies and regenerative medicine. An original inducible T-cell safety switch is brought to the donor T cell called caspase-9 (78) (Figure 5). Inducible caspase-9 is a fusion of human induced caspase-9 (iC9) which is a modified human FK-binding protein and it can be activated via a small-molecule compound AP1903 (79). This process depends on mitochondrial apoptotic pathway. After the pro-drug administration, the iC9-mediated cell clearance ratio was raised to ninety percent in half an hour (79). The iC9 suicide gene is less immunogenic, triggering reduced immune response against transgenic cells. This is to maintain a stable cell level in patients. After AP1903 treatment for one or two weeks in one study, polyclonal iC9-positive T cells were detected in peripheral blood with specific reactivity (79). Thus, the iC9 cell-suicide system is proven to be activated to guard T-cell-based immunotherapies and expand their clinical applications. The iC9-based safety switch has been shown to have better potential than the preexisting suicide genes for cellular therapy. In previous studies, iC9-T cells have been reported which deplete their alloreactive components ex vivo (80). A new study showed iC9 allodepletion could be done in vivo instead, and iC9-T cells can be eliminated within 30 minutes of AP1903 absorption (81).
The transferred T cells can also secrete pro-inflammatory cytokines leading to life-threatening GvHD-associated cytokine release syndrome (CRS) (82). The CRS is caused by increased levels of cytokines including IL-6 and IFN-γ. Immunosuppressants, for example tocilizumab as an anti-IL-6 receptor, with or without corticosteroids, can reverse this situation (83). The results indicate iC9 activation is sufficiently potent to promote allodepletion and treat GvHD, which leads to rapid resolution of CRS. Despite the iC9 system is engineered to prevent side-effect of T cell immunotherapy, integration of any transgene is mutagenic and potentially oncogenic. Hence, it is vital to assess its potential risks and benefits.
Discussion and Concluding Remarks
Tyrosine kinases belong to a key category of oncogenic proteins including Her2, EGFR, and VEGFR which are targeted by tyrosine kinase inhibitors (TKIs), such as lapatinib, gefitinib and sunitinib (84, 85). However, treatment with TKIs showed acquired resistance to chemotherapy after a few weeks of administration (86). Hence, lower toxicity and higher efficacy in anticancer therapeutic strategy are vital. Recently, gene-modified T cells with least adverse effects have been shown as extremely effective in treating solid and liquid tumors. Therapies which activate the immune system, such as those using antibodies against immune checkpoint PD-1, were shown to have a great potential (87). In addition, antigen-targeted approaches of monoclonal antibodies, CAR-T cell therapy, and TCR-based therapy have shown varied successes against specific tumors (11, 88). Therapies that use modified TCR-T cells for preclinical and clinical investigations have tremendous potential (89, 90). In lung cancer, however, solo use of ImmTAC therapy has failed. In order to recovery ImmTAC potency and tumor regression, the anti-PD-1 monoclonal antibody showed beneficial effects (91). ImmTAC efficiently redirects and activates effector and memory T cells within the CD8+ and CD4+ repertoires. This results in cytotoxic activity against melanoma cells. Apart from its direct therapy effect, ImmTAC can redirect lymphocytes to secrete several key cytokines and chemokines in T cells, such as IFN-γ, ILs (IL-2 and IL-6) and TNF-α, which result in a sustained anti-tumor defense in ACT (46).
The TME is known to suppress TILs (92). Engineered TCRs are able to boost T-cell trafficking and activation in TME (93, 94). Recently, E7 TCR gene therapy showed potential therapeutic value against HPV cancers (89). The ACT from TCR engineered human T cells has shown encouraging results. However, these approaches need further validation (95). In ACT, GVHD maybe observable with different and unpredictable kinetics (96). It has also been shown TCR/CD3 inhibition triggers T cell apoptosis (97).
A quick elimination of infused cells is important for cancer cellular therapies as engineered T cells expressing suicide genes with a specific prodrug are rapidly eliminated by apoptosis. Caspase-9 is a key element in apoptosis and is involved in various chemotherapy stimuli (78). Incorporating NK cells with an iC9 suicide switch enhances safety of anti-leukemia approaches which allows for their safe clinical application (98). However, this method also leads to a rapid inactivation of effector T cells in the event of adverse reactions (99). Anti-cancer and suicide mechanisms can co-exist without effects on cells and hamper tumoricidal activity (100). The iC9 safety switch of transduced T-cells also proved to be reliable in Phase I trial involving patients undergoing haplo-identical stem cell transplantation (90). Furthermore, HSV-TK has been observed to be another safety switch to trigger apoptosis in the cell line of head and neck carcinoma (101). Novel and highly efficient technique called TransfeX delivers HSV-TK into cervical, oral and pharyngeal with tumoricidal effects (102). Hence, TCR-T cell engineered with HSV-TK suicide gene may also enhance oncotherapy. It is evident therefore that TCR-T-cell immunotherapy has the potential to eradicate tumors in addition to lowering risks associated with immunotherapy (103).
In conclusion, TCR-T ACT has a good potential to treat cancers. TCR tuning is vital for T cell re-activity, immune responses, and its clinical effects on foreign antigens. The engineered TCRs targeting special antigens also boost efficiency of immunotherapy. The safety of genetically modified T cells for ACT is also vital. The present article reviewed advances of TCR T cell immunotherapy and proximal new techniques which can detect antigens and drive a T cell response. However, there are some limitations which need to be addressed in future research.
Author Contributions
QJZ, YJ, JS, PJK and SXX wrote the paper, YSZ, XW, FKD and MXL edited the manuscript, CHO, JL and QLW prepared and adjusted the figures, TL, TY and ZGX designed the study, provided funding and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grants: 81503093, 81972643, and 81672444) and the Joint Funds of the Southwest Medical University & Luzhou (Grants: 2016LZXNYD-T01, 2017LZXNYD-Z05 and 2019LXXNYKD-07) and Science & Technology Department of Sichuan Province(Grants: 2018JY0079).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Yarchoan M, Johnson BA,3, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer (2017) 17:569. doi: 10.1038/nrc.2017.74
3. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
4. Fan Z, Liang Y, Yang X, Li B, Cui L, Luo L, et al. A meta-analysis of the efficacy and safety of PD-1/PD-L1 immune checkpoint inhibitors as treatments for metastatic bladder cancer. OncoTargets Ther (2019) 12:1791–801. doi: 10.2147/ott.S186271
5. Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, et al. PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett (2017) 407:57–65. doi: 10.1016/j.canlet.2017.08.006
6. Hurkmans DP, Basak EA, van Dijk T, Mercieca D, Schreurs MWJ, Wijkhuijs AJM, et al. A prospective cohort study on the pharmacokinetics of nivolumab in metastatic non-small cell lung cancer, melanoma, and renal cell cancer patients. J Immunother Cancer (2019) 7:192. doi: 10.1186/s40425-019-0669-y
7. Pawłowska A, Suszczyk D, Okła K, Barczyński B, Kotarski J, Wertel I. Immunotherapies based on PD-1/PD-L1 pathway inhibitors in ovarian cancer treatment. Clin Exp Immunol (2019) 195:334–44. doi: 10.1111/cei.13255
8. Solinas C, Aiello M, De Silva P, Gu-Trantien C, Migliori E, Willard-Gallo K. Targeting PD-1 in cancer: Biological insights with a focus on breast cancer. Crit Rev Oncol Hematol (2019) 142:35–43. doi: 10.1016/j.critrevonc.2019.07.011
9. Wang F, Wei XL, Wang FH, Xu N, Shen L, Dai GH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann Oncol Off J Eur Soc Med Oncol (2019) 30:1479–86. doi: 10.1093/annonc/mdz197
10. Kaboli PJ, Zhang L, Xiang S, Shen J, Li M, Zhao Y, et al. Molecular Markers of Regulatory T Cells in Cancer Immunotherapy with Special Focus on Acute Myeloid Leukemia (AML) - A Systematic Review. Curr Med Chem (2020) 27:4673–98. doi: 10.2174/0929867326666191004164041
11. Ikeda H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. Int Immunol (2016) 28:349–53. doi: 10.1093/intimm/dxw022
12. Scherer LD, Brenner MK, Mamonkin M. Chimeric Antigen Receptors for T-Cell Malignancies. Front Oncol (2019) 9:126:6. doi: 10.3389/fonc.2019.00126
13. Xu Y, Morales AJ, Cargill MJ, Towlerton AMH, Coffey DG, Warren EH, et al. Preclinical development of T-cell receptor-engineered T-cell therapy targeting the 5T4 tumor antigen on renal cell carcinoma. Cancer Immunol Immunother CII (2019) 68:1979–93. doi: 10.1007/s00262-019-02419-4
14. Ohta R, Demachi-Okamura A, Akatsuka Y, Fujiwara H, Kuzushima K. Improving TCR affinity on 293T cells. J Immunol Methods (2019) 466:1–8. doi: 10.1016/j.jim.2018.11.010
15. Maus MV, Plotkin J, Jakka G, Stewart-Jones G, Rivière I, Merghoub T, et al. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Mol Ther Oncolytics (2016) 3:1–9. doi: 10.1038/mto.2016.23
16. Segaliny AI, Li G, Kong L, Ren C, Chen X, Wang JK, et al. Functional TCR T cell screening using single-cell droplet microfluidics. Lab Chip (2018) 18:3733–49. doi: 10.1039/c8lc00818c
17. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Sci (New York NY) (2018) 359:1361–5. doi: 10.1126/science.aar6711
18. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T cells. Biomarker Res (2017) 5:22. doi: 10.1186/s40364-017-0102-y
19. Liu D, Zhao J, Song Y. Engineering switchable and programmable universal CARs for CAR T therapy. J Hematol Oncol (2019) 12:69. doi: 10.1186/s13045-019-0763-0
20. Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol Sci (2016) 37:220–30. doi: 10.1016/j.tips.2015.11.004
21. Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, et al. Monomeric TCRs drive T cell antigen recognition. Nat Immunol (2018) 19:487–96. doi: 10.1038/s41590-018-0092-4
22. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, Kochenderfer JN, Neelapu SS, Maus MV, et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer (2018) 6:137. doi: 10.1186/s40425-018-0460-5
23. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res (2015) 3:356–67. doi: 10.1158/2326-6066.Cir-14-0186
24. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14:1264–70. doi: 10.1038/nm.1882
25. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
26. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
27. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New Engl J Med (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
28. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. New Engl J Med (2018) 378:449–59. doi: 10.1056/NEJMoa1709919
29. Getts D, Hofmeister R, Quintás-Cardama A. Synthetic T cell receptor-based lymphocytes for cancer therapy. Adv Drug Deliv Rev (2019) 141:47–54. doi: 10.1016/j.addr.2019.04.002
30. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest (2011) 121:1822–6. doi: 10.1172/jci46110
31. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118:4817–28. doi: 10.1182/blood-2011-04-348540
32. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
33. Nonon Saa KB, Maneh N, Vonor K, Amedome K, Togo M, Dzidzinyo K, et al. [Erratum to “Manual small incision cataract surgery: Experience of a regional eye care service in Togo” [J. Fr. Ophtalmol. 41 (3) (2018) 255-61 https://doi.org/10.1016/j.jfo.2017.09.009]]. J Francais D’ophtalmol (2018) 41:898. doi: 10.1016/j.jfo.2018.09.002
34. Bedoya F, Frigault MJ, Maus MV. The Flipside of the Power of Engineered T Cells: Observed and Potential Toxicities of Genetically Modified T Cells as Therapy. Mol Ther J Am Soc Gene Ther (2017) 25:314–20. doi: 10.1016/j.ymthe.2016.11.011
35. Huang J, Brameshuber M, Zeng X, Xie J, Li QJ, Chien YH, et al. A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4(+) T cells. Immunity (2013) 39:846–57. doi: 10.1016/j.immuni.2013.08.036
36. Stone JD, Aggen DH, Schietinger A, Schreiber H, Kranz DM. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell Engagers (BiTEs). Oncoimmunology (2012) 1:863–73. doi: 10.4161/onci.20592
37. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci USA (2018) 115:E2068–e76. doi: 10.1073/pnas.1716266115
38. Schamel WW, Alarcon B, Höfer T, Minguet S. The Allostery Model of TCR Regulation. J Immunol (Baltimore Md 1950) (2017) 198:47–52. doi: 10.4049/jimmunol.1601661
39. Poschke IC, Hassel JC, Rodriguez-Ehrenfried A, Lindner KAM, Heras-Murillo I, Appel LM, et al. The Outcome of Ex Vivo TIL Expansion Is Highly Influenced by Spatial Heterogeneity of the Tumor T-Cell Repertoire and Differences in Intrinsic In Vitro Growth Capacity between T-Cell Clones. Clin Cancer Res (2020) 26:4289–301. doi: 10.1158/1078-0432.CCR-19-3845
40. Hosoi A, Takeda K, Nagaoka K, Iino T, Matsushita H, Ueha S, et al. Increased diversity with reduced “diversity evenness” of tumor infiltrating T-cells for the successful cancer immunotherapy. Sci Rep (2018) 8:1058. doi: 10.1038/s41598-018-19548-y
41. Schamel WW, Arechaga I, Risueño RM, van Santen HM, Cabezas P, Risco C, et al. Coexistence of multivalent and monovalent TCRs explains high sensitivity and wide range of response. J Exp Med (2005) 202:493–503. doi: 10.1084/jem.20042155
42. Harper J, Adams KJ, Bossi G, Wright DE, Stacey AR, Bedke N, et al. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PloS One (2018) 13:e0205491. doi: 10.1371/journal.pone.0205491
43. He Q, Jiang X, Zhou X, Weng J. Targeting cancers through TCR-peptide/MHC interactions. J Hematol Oncol (2019) 12:139. doi: 10.1186/s13045-019-0812-8
44. McCormack E, Adams KJ, Hassan NJ, Kotian A, Lissin NM, Sami M, et al. Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1- and LAGE-1-positive tumors. Cancer Immunol Immunother CII (2013) 62:773–85. doi: 10.1007/s00262-012-1384-4
45. Liddy N, Bossi G, Adams KJ, Lissina A, Mahon TM, Hassan NJ, et al. Monoclonal TCR-redirected tumor cell killing. Nat Med (2012) 18:980–7. doi: 10.1038/nm.2764
46. Boudousquie C, Bossi G, Hurst JM, Rygiel KA, Jakobsen BK, Hassan NJ. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8(+) and CD4(+) T cells. Immunology (2017) 152:425–38. doi: 10.1111/imm.12779
47. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9:271–85. doi: 10.1038/nri2526
48. Oates J, Hassan NJ, Jakobsen BK. ImmTACs for targeted cancer therapy: Why, what, how, and which. Mol Immunol (2015) 67:67–74. doi: 10.1016/j.molimm.2015.01.024
49. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
50. Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, et al. Synthetic TRuC receptors engaging the complete T cell receptor for potent anti-tumor response. Nat Commun (2019) 10:2087. doi: 10.1038/s41467-019-10097-0
51. Alcover A, Alarcón B, Di Bartolo V. Cell Biology of T Cell Receptor Expression and Regulation. Annu Rev Immunol (2018) 36:103–25. doi: 10.1146/annurev-immunol-042617-053429
52. Helsen CW, Hammill JA, Lau VWC, Mwawasi KA, Afsahi A, Bezverbnaya K, et al. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat Commun (2018) 9:3049. doi: 10.1038/s41467-018-05395-y
53. Arnett KL, Harrison SC, Wiley DC. Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc Natl Acad Sci USA (2004) 101:16268–73. doi: 10.1073/pnas.0407359101
54. Rafei H, Daher M, Rezvani K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: leveraging the power of innate immunity. Br J Haematol (2020). doi: 10.1111/bjh.17186
55. Hesker PR, Krupnick AS. The role of natural killer cells in pulmonary immunosurveillance. Front Biosci (Schol Ed) (2013) 5:575–87. doi: 10.2741/s391
56. Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly Efficient Generation of Transgenically Augmented CAR NK Cells Overexpressing CXCR4. Front Immunol (2020) 11:2028. doi: 10.3389/fimmu.2020.02028
57. Xia N, Haopeng P, Gong JU, Lu J, Chen Z, Zheng Y, et al. Robo1-specific CAR-NK Immunotherapy Enhances Efficacy of (125)I Seed Brachytherapy in an Orthotopic Mouse Model of Human Pancreatic Carcinoma. Anticancer Res (2019) 39:5919–25. doi: 10.21873/anticanres.13796
58. Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci Rep (2015) 5:11483. doi: 10.1038/srep11483
59. Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood (1993) 82:343–62. doi: 10.1182/blood.V82.2.343.bloodjournal822343
60. Wildes TJ, Flores CT, Mitchell DA. Concise Review: Modulating Cancer Immunity with Hematopoietic Stem and Progenitor Cells. Stem Cells (2019) 37:166–75. doi: 10.1002/stem.2933
61. Zhu H, Lai YS, Li Y, Blum RH, Kaufman DS. Concise Review: Human Pluripotent Stem Cells to Produce Cell-Based Cancer Immunotherapy. Stem Cells (2018) 36:134–45. doi: 10.1002/stem.2754
62. Wang L, Pegram MD, Wu JC. Induced pluripotent stem cells as a novel cancer vaccine. Expert Opin Biol Ther (2019) 19:1191–7. doi: 10.1080/14712598.2019.1650909
63. Kooreman NG, Kim Y, de Almeida PE, Termglinchan V, Diecke S, Shao NY, et al. Autologous iPSC-Based Vaccines Elicit Anti-tumor Responses In Vivo. Cell Stem Cell (2018) 22:501–13.e7. doi: 10.1016/j.stem.2018.01.016
64. D’Ippolito E, Schober K, Nauerth M, Busch DH. T cell engineering for adoptive T cell therapy: safety and receptor avidity. Cancer Immunol Immunother CII (2019) 68:1701–12. doi: 10.1007/s00262-019-02395-9
65. de Witte MA, Jorritsma A, Kaiser A, van den Boom MD, Dokter M, Bendle GM, et al. Requirements for effective antitumor responses of TCR transduced T cells. J Immunol (Baltimore Md 1950) (2008) 181:5128–36. doi: 10.4049/jimmunol.181.7.5128
66. Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin Exp Immunol (2015) 180:255–70. doi: 10.1111/cei.12570
67. Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature (2009) 458:211–4. doi: 10.1038/nature07657
68. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol Off J Am Soc Clin Oncol (2015) 33:1688–96. doi: 10.1200/jco.2014.58.0225
69. Hebeisen M, Schmidt J, Guillaume P, Baumgaertner P, Speiser DE, Luescher I, et al. Identification of Rare High-Avidity, Tumor-Reactive CD8+ T Cells by Monomeric TCR-Ligand Off-Rates Measurements on Living Cells. Cancer Res (2015) 75:1983–91. doi: 10.1158/0008-5472.Can-14-3516
70. Li W, Xiang AP. Safeguarding clinical translation of pluripotent stem cells with suicide genes. Organogenesis (2013) 9:34–9. doi: 10.4161/org.24317
71. Liang Q, Monetti C, Shutova MV, Neely EJ, Hacibekiroglu S, Yang H, et al. Linking a cell-division gene and a suicide gene to define and improve cell therapy safety. Nature (2018) 563:701–4. doi: 10.1038/s41586-018-0733-7
72. Daron C, Soubrier M, Mathieu S. Occurrence of rheumatic symptoms in celiac disease: A meta-analysis: Comment on the article “Osteoarticular manifestations of celiac disease and non-celiac gluten hypersensitivity” by Dos Santos and Lioté. Joint Bone Spine 2016, doi:10.1016/j.jbspin.2016.09.007. Joint Bone Spine (2017) 84:645–6. doi: 10.1016/j.jbspin.2017.03.006
73. Zhan H, Gilmour K, Chan L, Farzaneh F, McNicol AM, Xu JH, et al. Production and first-in-man use of T cells engineered to express a HSVTK-CD34 sort-suicide gene. PloS One (2013) 8:e77106. doi: 10.1371/journal.pone.0077106
74. Eissenberg LG, Rettig M, Dehdashti F, Piwnica-Worms D, DiPersio JF. Suicide genes: monitoring cells in patients with a safety switch. Front Pharmacol (2014) 5:241. doi: 10.3389/fphar.2014.00241
75. Vanpouille C, Bernatchez JA, Lisco A, Arakelyan A, Saba E, Götte M, et al. A common anti-cytomegalovirus drug, ganciclovir, inhibits HIV-1 replication in human tissues ex vivo. AIDS (London England) (2017) 31:1519–28. doi: 10.1097/qad.0000000000001532
76. Bonini C, Mondino A. Adoptive T-cell therapy for cancer: The era of engineered T cells. Eur J Immunol (2015) 45:2457–69. doi: 10.1002/eji.201545552
77. Ciceri F, Bonini C, Stanghellini MT, Bondanza A, Traversari C, Salomoni M, et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. Lancet Oncol (2009) 10:489–500. doi: 10.1016/s1470-2045(09)70074-9
78. Li P, Zhou L, Zhao T, Liu X, Zhang P, Liu Y, et al. Caspase-9: structure, mechanisms and clinical application. Oncotarget (2017) 8:23996–4008. doi: 10.18632/oncotarget.15098
79. Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. New Engl J Med (2011) 365:1673–83. doi: 10.1056/NEJMoa1106152
80. Zhou X, Di Stasi A, Tey SK, Krance RA, Martinez C, Leung KS, et al. Long-term outcome after haploidentical stem cell transplant and infusion of T cells expressing the inducible caspase 9 safety transgene. Blood (2014) 123:3895–905. doi: 10.1182/blood-2014-01-551671
81. Zhou X, Dotti G, Krance RA, Martinez CA, Naik S, Kamble RT, et al. Inducible caspase-9 suicide gene controls adverse effects from alloreplete T cells after haploidentical stem cell transplantation. Blood (2015) 125:4103–13. doi: 10.1182/blood-2015-02-628354
82. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther J Am Soc Gene Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
83. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
84. Jabbarzadeh Kaboli P, Leong MP, Ismail P, Ling KH. Antitumor effects of berberine against EGFR, ERK1/2, P38 and AKT in MDA-MB231 and MCF-7 breast cancer cells using molecular modelling and in vitro study. Pharmacol Rep PR (2019) 71:13–23. doi: 10.1016/j.pharep.2018.07.005
85. Zhao Y, Zhao Q, Kaboli PJ, Shen J, Li M, Wu X, et al. m1A Regulated Genes Modulate PI3K/AKT/mTOR and ErbB Pathways in Gastrointestinal Cancer. Trans Oncol (2019) 12:1323–33. doi: 10.1016/j.tranon.2019.06.007
86. Jabbarzadeh Kaboli P, Ismail P, Ling KH. Molecular modeling, dynamics simulations, and binding efficiency of berberine derivatives: A new group of RAF inhibitors for cancer treatment. PloS One (2018) 13:e0193941. doi: 10.1371/journal.pone.0193941
87. Wu Y, Gu W, Li J, Chen C, Xu ZP. Silencing PD-1 and PD-L1 with nanoparticle-delivered small interfering RNA increases cytotoxicity of tumor-infiltrating lymphocytes. Nanomed (London England) (2019) 14:955–67. doi: 10.2217/nnm-2018-0237
88. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Sci (New York NY) (2015) 348:62–8. doi: 10.1126/science.aaa4967
89. Jin BY, Campbell TE, Draper LM, Stevanović S, Weissbrich B, Yu Z, et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight (2018) 3:e99488. doi: 10.1172/jci.insight.99488
90. Zhang P, Raju J, Ullah MA, Au R, Varelias A, Gartlan KH, et al. Phase I Trial of Inducible Caspase 9 T Cells in Adult Stem Cell Transplant Demonstrates Massive Clonotypic Proliferative Potential and Long-term Persistence of Transgenic T Cells. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:1749–55. doi: 10.1158/1078-0432.Ccr-18-3069
91. Zhang H, Li Y, Liu X, Liang Z, Yan M, Liu Q, et al. ImmTAC/Anti-PD-1 antibody combination to enhance killing of cancer cells by reversing regulatory T-cell-mediated immunosuppression. Immunology (2018) 155:238–50. doi: 10.1111/imm.12954
92. Zhang J, Wang L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol Cancer Res Treat (2019) 18:1533033819831068. doi: 10.1177/1533033819831068
93. Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell (2014) 157:357–68. doi: 10.1016/j.cell.2014.02.053
94. Lou J, Rossy J, Deng Q, Pageon SV, Gaus K. New Insights into How Trafficking Regulates T Cell Receptor Signaling. Front Cell Dev Biol (2016) 4:77. doi: 10.3389/fcell.2016.00077
95. Chhabra A, Mukherji B, Batra D. Activation induced cell death (AICD) of human melanoma antigen-specific TCR engineered CD8 T cells involves JNK, Bim and p53. Expert Opin Ther Targets (2017) 21:117–29. doi: 10.1080/14728222.2017.1270941
96. Anwer F, Shaukat AA, Zahid U, Husnain M, McBride A, Persky D, et al. Donor origin CAR T cells: graft versus malignancy effect without GVHD, a systematic review. Immunotherapy (2017) 9:123–30. doi: 10.2217/imt-2016-0127
97. Zhu L, Anasetti C. Cell cycle control of apoptosis in human leukemic T cells. J Immunol (Baltimore Md 1950) (1995) 154:192–200.
98. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia (2018) 32:520–31. doi: 10.1038/leu.2017.226
99. Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer (2019) 145:1935–45. doi: 10.1002/ijc.32269
100. Rossignoli F, Grisendi G, Spano C, Golinelli G, Recchia A, Rovesti G, et al. Inducible Caspase9-mediated suicide gene for MSC-based cancer gene therapy. Cancer Gene Ther (2019) 26:11–6. doi: 10.1038/s41417-018-0034-1
101. Srivastava D, Joshi G, Somasundaram K, Mulherkar R. Mode of cell death associated with adenovirus-mediated suicide gene therapy in HNSCC tumor model. Anticancer Res (2011) 31:3851–7.
102. Düzgüneş N, Cheung J, Konopka K. Non-viral suicide gene therapy in cervical, oral and pharyngeal carcinoma cells with CMV- and EEV-plasmids. J Gene Med (2018) 20:e3054. doi: 10.1002/jgm.3054
Keywords: ImmTAC, immunosuppression, immunotherapy, T-cell receptors, suicide genes
Citation: Zhao Q, Jiang Y, Xiang S, Kaboli PJ, Shen J, Zhao Y, Wu X, Du F, Li M, Cho CH, Li J, Wen Q, Liu T, Yi T and Xiao Z (2021) Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front. Immunol. 12:658753. doi: 10.3389/fimmu.2021.658753
Received: 26 January 2021; Accepted: 02 March 2021;
Published: 30 March 2021.
Edited by:
Peng Qu, National Institutes of Health (NIH), United StatesReviewed by:
Guoyuan Zhu, Macau University of Science and Technology, MacauMenghua Wu, Jinan University, China
Copyright © 2021 Zhao, Jiang, Xiang, Kaboli, Shen, Zhao, Wu, Du, Li, Cho, Li, Wen, Liu, Yi and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Liu, tao2020@sohu.com; Tao Yi, yitao@hkbu.edu.hk; Zhangang Xiao, xzg555898@hotmail.com
†These authors have contributed equally to this work