 Carlos Cuesta-Mateos
Carlos Cuesta-Mateos Jennifer R. Brown
Jennifer R. Brown Fernando Terrón2,3
Fernando Terrón2,3 Cecilia Muñoz-Calleja
Cecilia Muñoz-Calleja- 1Immunology Department, Hospital Universitario de La Princesa, Instituto de Investigación Sanitaria- Instituto de La Princesa (IIS-IP), Madrid, Spain
- 2IMMED S.L., Immunological and Medicinal Products, Madrid, Spain
- 3Catapult Therapeutics BV, Lelystad, Netherlands
- 4Chronic Lymphocytic Leukemia (CLL) Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 5School of Medicine, Universidad Autónoma de Madrid, Madrid, Spain
The lymph node (LN) is an essential tissue for achieving effective immune responses but it is also critical in the pathogenesis of chronic lymphocytic leukemia (CLL). Within the multitude of signaling pathways aberrantly regulated in CLL the homeostatic axis composed by the chemokine receptor CCR7 and its ligands is the main driver for directing immune cells to home into the LN. In this literature review, we address the roles of CCR7 in the pathophysiology of CLL, and how this chemokine receptor is of critical importance to develop more rational and effective therapies for this malignancy.
Introduction
Lymph nodes (LN) function as a major immunological hub, essential for immune homeostasis and generation of effective immune responses (1). Yet LNs are also a fundamental tissue in the development, progression and treatment failure of several mature lymphomas/leukemias, especially for chronic lymphocytic leukemia (CLL) (2–6). In recent years growing evidence suggests that cell trafficking orchestrated by the chemokine receptor CCR7 plays a critical role in the pathophysiology of CLL. LN stromal cells secrete CCR7 ligands generating powerful chemotactic gradients that attract CLL cells into the microenvironment, where a diversity of cells, soluble factors, and matrix proteins facilitate survival and proliferative cues, thus promoting disease progression and preventing spontaneous or drug-induced apoptosis of leukemic cells. In this literature review we provide in depth insight into how CCR7-mediated functions contribute to CLL pathogenesis, and how this chemokine receptor may be a critical potential therapeutic target in CLL.
CLL
With an age-adjusted incidence of 4.3/100000 inhabitants in the United States, CLL is the most common type of leukemia in Western countries. More than 20000 newly diagnosed cases and ~4500 deaths per year are currently estimated (7). CLL is characterized by the clonal proliferation and accumulation of mature, typically CD5-positive B cells within the peripheral blood (PB), bone marrow (BM), LNs, and spleen. Despite a remarkable phenotypic and cytological homogeneity, CLL is characterized by extremely variable clinical course related to different prognostic factors including the mutational status of the immunoglobulin heavy-chain variable region (IGHV) (8–10), expression of very late antigen 4 (VLA-4), CD38 and zeta-associated-protein 70 (ZAP-70) markers (11, 12), and specific cytogenetic alterations including the most common and early event of deletion of the long arm of chromosome 13 [del(13q14)] and other alterations that occur later in the course of disease and predict worse outcome such as del(11q), and del(17p) (13–15). In recent years, whole-genome sequencing has uncovered novel recurrent somatic gene mutations that occur in CLL cells in parallel to the above-mentioned structural genomic aberrations. Of these, mutations affecting the genes NOTCH1, p53, ataxia-telangiectasia-mutated (ATM), and splicing factor 3b subunit 1 (SF3B1) seem to be more common, with a long tail of less common but nonetheless recurrent driver mutations (16–19).
Until very recently, chemoimmunotherapy, the combination of monoclonal antibodies (mAb) against CD20 with chemotherapy, was the most effective therapeutic approach in CLL. In particular, standard therapy with the combination of FCR (fludarabine, cyclophosphamide, rituximab) was shown to prolong both progression-free survival and overall survival (OS) in CLL (20) and to result in long-term remission in patients with mutated IGHV. Response in patients with TP53 aberrant disease was poor however, and patients with unmutated IGHV generally showed continuous relapse even after initial deep response, including undetectable minimal residual disease (MRD) responses. The development of Bruton´s tyrosine kinase (BTK) inhibitors in particular as well as more recently the B cell lymphoma 2 protein (BCL-2) inhibitor venetoclax has led to more effective therapy particularly for higher risk disease (21, 22). Phosphatidylinositol 3 kinase (PI3K) inhibitors also have significant activity but have been hampered by toxicity. Despite the efficacy of these drugs, continuous therapy is required with the B cell receptor (BCR) pathway inhibitors leading to toxicity and cost, as well as increasing relapse over time. The venetoclax regimens have been developed to be time-limited, and follow-up is still too short to know the durability in different disease groups. It is clear that patients who do not achieve undetectable MRD with a venetoclax regimen have steady relapse and constitute a group with unmet need. All higher risk patient groups, particularly those with p53 aberrant disease, complex karyotype and even unmutated IGHV, all have higher risk of relapse and still have significant unmet medical need for additional treatment strategies (21–23).
A hallmark of the pathophysiology of CLL is that blood circulating leukemia cells are mainly in a G0/G1 cell cycle–arrested phase, whereas CLL cells within LN are proliferating and hence promote disease progression (2, 4). In this scenario, CLL is seen as a dynamic neoplasm comprising leukemic cells that multiply and die at measurable rates (24). However, and at variance with other hematologic malignancies, CLL proliferation rates are relatively low and cell accumulation is the result of an abnormally prolonged survival rather than uncontrolled proliferation (25). Indeed, intrinsic defects in the apoptotic machinery such as overexpression of BCL-2 and myeloid-cell leukemia 1 (MCL-1) anti-apoptotic members, or impaired expression of pro-apoptotic members (Bax and Bak), and extrinsic factors consisting mainly of stromal cell–derived cytokines and chemokines (e.g. CXCL12), provide survival cues during which tumor cells transit through lymphoid tissues and tilt the balance toward prolonged lifespan of CLL B cells (6, 26).
CCR7 and Its Ligands
The homeostatic chemokine receptor CCR7 was identified in the 1990s as the first lymphocyte specific G-protein coupled receptor (GPCR) (27–29). Also known as Epstein–Barr virus-induced gene 1 (EBI1), Burkitt’s lymphoma receptor 2 (BLR2), or CD197, this 378 amino acid protein is encoded by a gene located on human chromosome 17q12-21.2 (28). CCR7 is expressed by various immune cells including double negative (DN) and single positive (SP) thymocytes, naïve, central memory and regulatory T cells (TN, TCM, TREG), naïve B cells, CD56+CD16- regulatory natural killer (NK) cells, and (semi-)mature dendritic cells (DCs) (30–32). In addition, CCR7 expression has been found in different non-immune cells, most notably in various malignancies (32). Generally, mentioned T cells subsets and mature B cells constitutively express CCR7 whereas NK cells and DCs acquire CCR7 expression upon encountering a pathogen (30). In both homeostasis and cancer, CCR7 but not other receptors, specifically drives cell homing into LN and other secondary lymphoid organs (SLO) (33–35). This GPCR orchestrates: cell trafficking, firm arrest to endothelium, extravasation, positioning within SLO, activation, and egress upon binding two cognate ligands, the chemokines CCL19 (aka ELC or MIP-3β) and CCL21 (aka SLC or 6CK), constitutively expressed by stroma cells in SLOs and present on lymphatic vessels, high-endothelial venules (HEVs), and T zones. In addition, CCL21 is produced by lymphatic endothelial cells (30, 31, 36). Both chemokines share only 32% sequence homology and are structurally and functionally distinct (37). Indeed, both molecules differ in length with CCL21 encoding a 37 aa long C-terminal tail extension, that is lacking in CCL19, and which is rich in positively charged (basic) residues. This tail, which can be proteolytically cleaved, confers high affinity for negatively charged molecules of the extracellular matrix (ECM), including glycosaminoglycans (GAGs), therefore the lack of such C-terminal basic extension in CCL19, and in CCL21 tail-less form impairs its ability to form haptotactic gradients (36, 38–40).
The LN in CLL
Upon immune activation, reactive LN acquire a characteristic structure (1, 41). Three main cellular compartments are easily distinguishable: the cortex, the paracortex and the medulla. The cortical area contains lymphoid follicles composed mostly of B cells (B zone). In reactive LN, the primary follicles evolve to secondary follicles, made up of a germinal center (GC) and surrounding mantle zone. In paracortical areas (or T zone), T cells predominate and are mixed with interdigitating DCs, plasma cells and “tingible body” macrophages (TBM). Finally, the medulla consists of the medullary cords, which contain lymphocytes, plasma cells, and macrophages, and the medullary sinuses, contiguous with the efferent lymphatics, and contain lymph, macrophages, plasma cells, and mast cells.
In homeostasis, reactive LN is an important site for B cell-Ag interaction, initiating and supporting multiple processes including heavy immunoglobulin chain class switching, somatic hypermutation and proliferation (2). These processes take place on the GC, where B cells interact with helper follicular T cells (THF) and follicular dendritic cells (FDC), that serve to increase the variety of Ag binding sites available for clonal selection (2). In this normal tissue, CCR7 is expressed in B cell and T cell zones with a marked positivity on centrocytes and centroblasts in secondary follicle GCs (42). Additional aspects related to reactive tissue structure and CCR7 expression are disclosed in Figure 1.
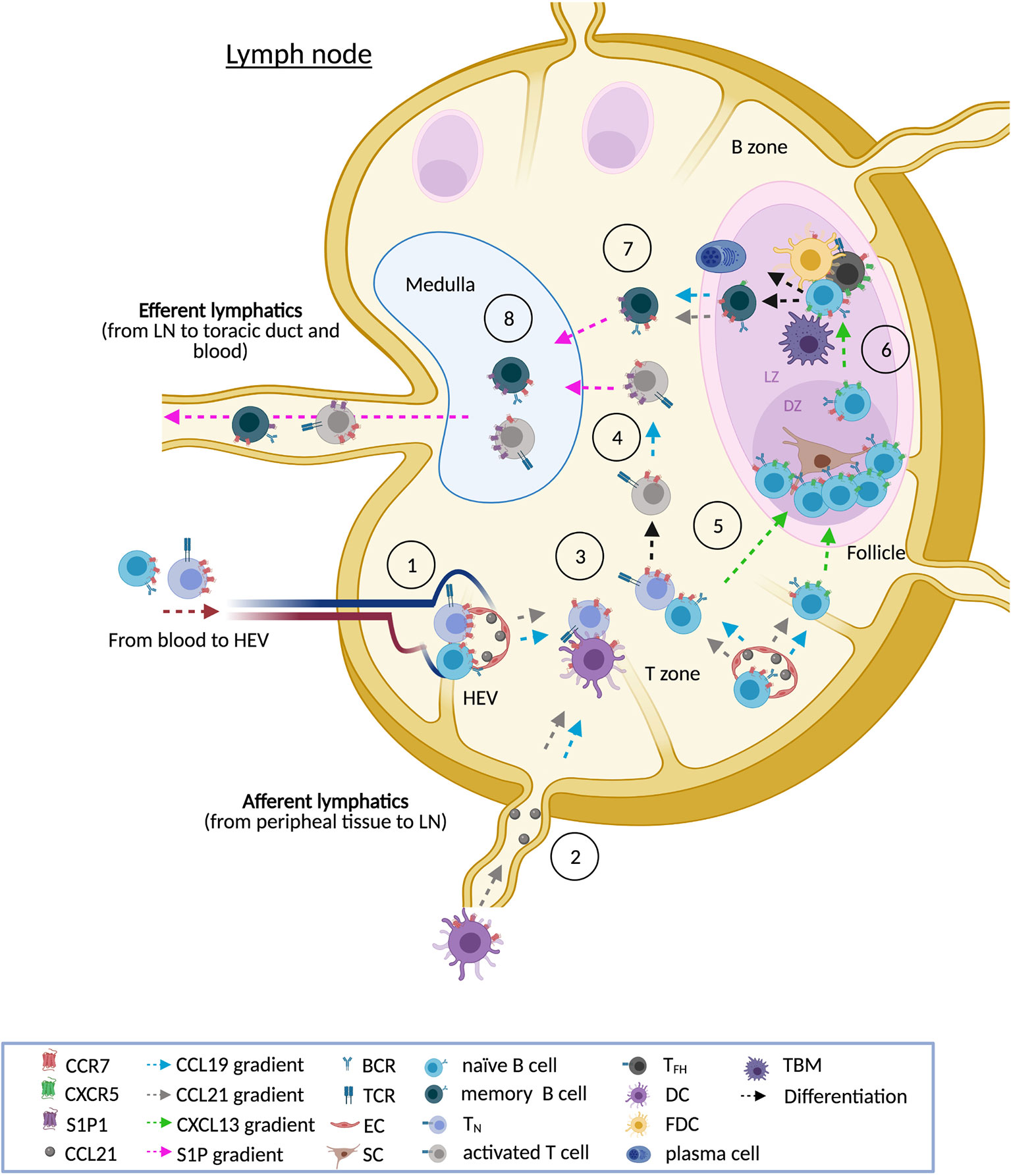
Figure 1 CCR7 and the reactive LN. In homeostasis, normal LN show three main cellular compartments: the cortex (B zone), the paracortex (T zone) and the medulla. Upon antigen stimulation, the primary follicles evolve to secondary follicles, made up of a germinal center (GC) and surrounding mantle zone. In reactive LN, CCR7 is necessary for the entry of naïve B cells, naïve T cells (TN), regulatory T cells (TREG, not shown), central memory T cells (TCM, not shown), and dendritic cells (DCs). CCR7 guides lymphocyte homing through high endothelial venules (HEVs) in the paracortex [1] whereas DCs preferentially use afferent lymphatics [2]. CCR7 also drives interstitial migration of these immune subsets in the T zone facilitating, for instance, the interaction of TN with antigen presenting cells such as B cells and DCs [3]. Upon activation, T cells are directed to the medulla following CCL19 gradients. CCL19 signaling also induces CCR7 internalization and the up-regulation of the egressing receptor S1P1. The balance between CCR7 and S1P1 is needed for the movement of activated T cells from the T zone to the medulla [4]. Similarly, a fine-tuned balance between CCR7 and CXCR5 allows the migration of activated B cells through the T zone and the follicle. In a first step, CCR7 is down-modulated while a concomitant up-regulation of CXCR5 allows activated B cells to enter into the follicle [5]. In reactive follicles, fully developed GC are polarized into two regions clearly differentiated: the dark zone (DZ) and the light zone (LZ). Although GC B cells re-express CCR7, migration of GC B cells between both regions relies on the CXCR5-CXCL13 axis [6]. In the DZ, GC B cells (centroblasts) interact with stromal cells, proliferate (clonal expansion) and undergo somatic hypermutation on the immunoglobulin genes. In the LZ, hypermutated resting GC B cells (centrocytes) interact with a dense network of CXCL13hi follicular dendritic cells (FDCs) and CXCR5hi follicular helper T cells (TFH). FDCs display antigen and secrete cytokines and chemokines (CXCL13) that attract B cells and TFH to the GC. TFH are specialized CD4+ PD-1+ T cells that express BCL-6 and secrete cytokines that promote B cell proliferation and differentiation. TFH deliver survival signals to GC B cells through a number of different pathways, including CD40-CD40L, PD1-PDL1, and IL-21. The pro-survival signals from TFH counteract pro-apoptotic signals from the FAS-FASL pathway. Crosstalk of centrocytes with FDCs and THF allows the class-switch recombination and the selection of B cells. Centrocytes with the appropriate antigen affinity are selected to become memory B cells or antibody secreting plasma cells. The centrocytes that are not selected undergo apoptosis and are removed by tingible-body macrophages (TBM). Expression of CCR7 allows memory B cells to exit from follicles back to the T zone and, from there, to the medulla [7]. S1P1-expressing T cells and B cells move towards the efferent vessels following S1P gradients [8]. Notation: this scheme shows the main cell types in a reactive LN and in the GC, however in these complex tissues participate additional subtypes not listed here that can be further reviewed elsewhere (1, 30, 31, 41).
In CLL, the LN frequently shows a pattern of architectural effacement by homogeneous diffuse sheets of small CLL lymphocytes, often obliterating the normal nodal tissue, and exhibits small, if any, residual GCs. In many cases, neoplastic cells with increased mitotic activity form scattered foci that resemble germinal centers called pseudofollicles or proliferation centers (PCs) which are enriched with prolymphocytes and paraimmunoblasts (4, 43–46). In CLL patients, the PCs are much more evident in LN than in the spleen and the BM; they occur in 88% of LN biopsies. Moreover, PCs are hallmark features in the LN of patients with CLL as they are not observed in other B-cell tumors (4).
Unlike reactive GCs, the leukemic cells of PCs lack a GC B-cell phenotype, but may contain increased CD40L+CD4+ TH cells and in some cases a fine network of DCs, suggesting that PCs likely do not arise secondary to colonization of GCs by tumor cells (44). In contrast, the PCs tend to have less well-defined borders without mantle zones, lack polarity, and do not contain tingible body macrophages, allowing for easy discrimination from GC (44).
CCR7 Over-Expression in CLL
A large series of studies have reported that the CLL cells of almost all patients express high surface levels of CCR7, where a distinct, intense positive peak consisting of > 96% of positive cells is usually observed by flow cytometry (14, 42, 47–66). This expression of CCR7 in CLL is abnormally high when compared to the corresponding normal CD5+ B-cell population or pan-B cells. The CCR7 expression is consistently observed in both resting and proliferative compartments; in different groups of CLL patients regardless of the stage at diagnosis; unfavorable flow cytometry (CD38, ZAP-70, VLA-4) or genetic (IGHV mutated/un-mutated status, p53 mutated/deleted, ATM mutated/deleted, trisomy 12, NOTCH1 mutations) prognostic markers; previous treatments or the presence of relapsed or refractory (r/r) disease to different standard-of-care (SOC) therapies (Table 1). Similarly, in CLL nodes, a strong CCR7 positivity is seen in small lymphocytes and paraimmunoblasts of PCs (42), and this expression did not differ between matched LN and BM biopsy samples, hence, CCR7 levels remains high on CLL cells following their accumulation in SLOs and BM.
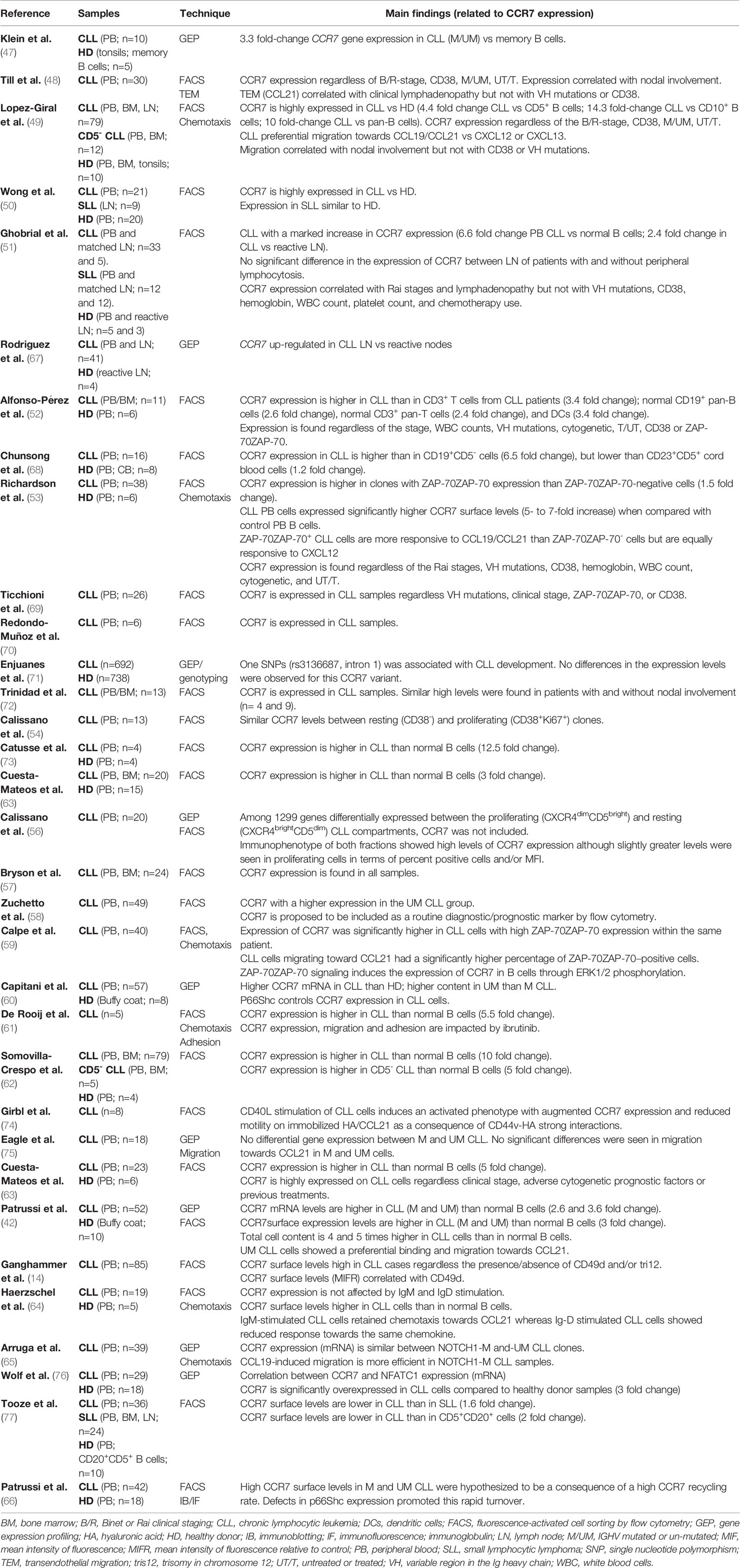
Table 1 Publications reporting CCR7 expression in CLL cells.
In several reports, CCR7 expression and/or functionality has been associated with higher LN involvement and staging but no report directly associated this receptor, or its migratory response, with CLL patient survival (14, 42, 48, 49, 51, 53, 63, 78). Nonetheless, in other blood cancers such as diffuse large B-cell lymphoma (DLBCL) or T cell prolymphocytic leukemia (T-PLL) a clear correlation was found between CCR7 expression levels at diagnosis and OS (79, 80). Moreover, indirect evidence suggests a role of CCR7 in shortening lifespan in CLL. High levels of IκBα, a known repressor of NF-κB-mediated transcription of CCR7, correlated to extended OS in CLL (67). Similarly, NOTCH1 mutations inducing a higher phosphorylation of the signal transducer and activator of transcription 3 (STAT-3) factor, and subsequent higher expression of CCR7, showed clinical characteristics of aggressive disease in a retrospective analysis on a cohort of 113 NOTCH1-mutated CLL patients (65). Moreover, serum levels for CCL19 are greater in CLL patients than in age-matched healthy subjects and those. Related to CCR7 ligands, serum levels for CCL19 were comparable between CLL patients and age-matched healthy subjects (81). However, within the CLL cohort, higher CCL19 levels independently associated with shorter survival. Moreover, when CCL19 was clustered along with other cytokines such as CCL3, CCL4, CXCL9, CXCL10, CXCL11, IFNγ, IL-5, and IL-12, this group of soluble factors was also found as an independent prognostic indicator of aggressive disease (time-to-first-treatment) and OS when compared to other clusters of cytokines (81). Therefore, it is likely that this group of cytokines provides a cytokine milieu that favors survival and growth to CLL cells.
Mechanisms Underlying CCR7 Expression in CLL
In CLL, the key factors involved in the over-expression of CCR7 still remain to be uncovered. Nonetheless, recent evidence supports the hypothesis that both the inner genetic background of leukemic cells and/or environment factors facilitate CCR7 over-expression. To our knowledge, no potential CCR7 gene mutations affecting transcription have been reported in CLL but one single-nucleotide polymorphism (SNP) in this gene was strongly associated with CLL risk (71, 82). Enjuanes et al. (81) found that the minor allele in the SNP rs3136687 (intron 1) resulted in a protective effect for the risk of CLL, although no CCR7 expression differences were observed for such allelic variants. This lack of association between CCR7 genetic variants and CCR7 over-expression suggests the presence of alternative genotypes affecting other proteins that ultimately determine different signaling pathways controlling CCR7 gene transcription and/or surface protein expression. In this regard, patient-associated DNA hypomethylation of the transcription factor NFATC1 (nuclear factor of activated T cells 1), a down-stream effector of the BCR, selectively facilitates expression of CCR7 (but not of other chemokines receptors such as CXCR4) and CCR7-induced migratory responses of CLL cells (76, 83). Other transcription factors known to regulate expression of CCR7 are NF-κB and activator protein 1 (AP-1); the latter is known to interact with NFAT protein (67, 84–86). NF-κB proteins are present in the cytoplasm bound to IκBα proteins, which are inhibitory molecules that sequester NF-κB dimers in the cytoplasm. Once BCR gets activated in CLL, IκBα is phosphorylated, ubiquitinated, and degraded in the proteasome. This facilitates NF-κB translocation to the nucleus, making the transcription of its target genes possible, including CCR7 (67). In addition, BCR signaling in primary CLL cells through ZAP70 up-regulated CCR7 via an extracellular signal-regulated kinase (ERK)-1/2-dependent mechanism (59) although similar results were not seen in ZAP-70-overexpressing OSU-CLL cell line (87) and a recent study showed no regulation of CCR7 protein levels in CLL cells after IgM and IgD stimulation while CXCR4 and CXCR5 were down-regulated (64). Whatever the reason underlying those different outcomes are, these studies clearly show that BCR-activating environment factors might orchestrate the up-regulation of CCR7 in CLL as they do in normal B cells where CCR7 expression, following engagement of the BCR, is augmented through BCR-BTK signaling (88). This up-regulation facilitates T-B cells interaction by guiding B cells from the follicle to the border between the B and T cell zones in LN thanks to pre-established CCR7 ligand chemotactic pathways (89, 90). In this areas, B-T cell cross-talk is regulated by CD40-CD40L(CD154) receptors, another environment interaction known to up-regulate CCR7 expression in CLL, B-cell precursor acute lymphoblastic leukemia (BCP-ALL), or myeloid leukemia-derived DCs (74, 91–93). Accordingly, in CLL the activation of the CCR7 transcription factor NF-κB takes place after induction by CD40-CD40L ligation (94).
Another receptor implicated in sensing environment factors which has been shown to regulate CCR7 in T cell malignancies is the NOTCH1 transmembrane protein (95). In CLL, Arruga et al. associated NOTCH1 mutations (which are found in ~10% of patients at diagnosis, ~20% of r/r patients, and ~30% in Richter’s syndrome) with STAT3-mediated CCR7 over-expression (65). This transcription factor is activated by mitogen-activated protein kinases (MAPKs) and directly regulates CCR7 gene expression (96). In wild-type NOTCH1 CLL cells, NOTCH intracellular domain (NICD) controls promoter methylation of the dual specificity protein phosphatase 22 (DUSP22) tumor suppressor gene that encodes a phosphatase that inactivates MAPKs, including c-jun N-term kinase (JNK) and p38, and dephosphorylates STAT3 (65). In a first step, NICD binds to the recombination signal binding protein RBPJk, which is bound to histone deacetylase 1 (HDAC1) in a heterodimeric repressor complex. Then, free HDAC1 binds and stabilize DNA methyltransferase 3A (DNMT3A), promoting DNMT3A activity and consequently the methylation of DUSP22 promoter. Therefore, NOTCH1 mutations leading to constitutive activation of the NICD down-regulate DUSP22 levels, increasing MAPK and STAT3 activation which result in increased CCR7 levels. This mechanism was confirmed in a cohort of 113 CLL patients (65). Those patients with a NOTCH1-mutated clone showed significant hypermethylation of DUSP22 with lower mRNA and protein levels of DUSP22, higher phosphorylation of STAT3 and expression of CCR7 and active chemotaxis to CCL19. Accordingly, patients with molecular or clinical characteristics of aggressive disease displayed significantly lower DUSP22 levels. Remarkably, another STAT family member, STAT-4 which is profoundly reduced in CLL cells (97) was implicated in in vivo down-regulation of CCR7 in TH cells (98).
The scavenger receptor CD5 is another receptor implicated in the over-expression of CCR7 (99). Since this hallmark CLL phenotype marker is up-regulated in CLL cells, it is thought that increased CD5 signaling is another cause of differential CCR7 surface levels between normal and CLL B cells. Other studies suggest that CCR7 gene is a reactive oxygen species (ROS)-responsive gene in B cells. Under normal conditions this gene is negatively controlled by the ROS-elevating activity of p66Shc, a cytoplasmatic pro-apoptotic protein, member of the Shc family of protein adaptors, and normally expressed in healthy B cells (42, 60, 66, 100). In CLL, the abnormal CCR7 surface levels were shown to be a consequence of the presence of ROS and a concomitant defect of p66Shc, which is also the cause of a rapid recycling of cell membrane CCR7 thus helping to maintain abnormal elevated membrane levels (42, 60, 66, 101). Curiously, the implication of a pro-apoptotic p66Shc protein in controlling CCR7 is not the only event of the apoptotic machinery involved in the regulation of this receptor. For example, some anti-apoptotic proteins such as Bcl-2 have been correlated with increased CCR7 expression in other tumor diseases and in non-tumor CD8+ T cells (102, 103). In these cases, a positive loop CCR7-Bcl2 promotes expression of the anti-apoptotic protein while Bcl2-signalling favors CCR7 production. Whether similar positive loops can be found in CLL is still unknown. However we do know that in CLL, activation of NF-κB after CD40-CD154 ligation, or activation of NFATC1 transcription factor, the downstream effector of BCR, results in both expression of CCR7 and Bcl2, therefore both events seem related to one shared former event (67, 76, 104, 105). Finally, it is worth noting that in solid cancers, such as breast cancer, a differential CCR7 expression between different histologic subtypes is determined by tumor microenvironment factors (e.g. hormonal, inflammatory, and growth stimulating) which may disable migration of CCR7-expressing tumor cells towards CCL21 and, consequently, the presence of LN migrations (106). Therefore, we cannot exclude finding similar requirements within the LN microenvironment for different CLL clones featured by differential combinations of adverse prognostic factors.
CCR7-Induced Chemotaxis in CLL
It is known that levels of chemokine receptors do not always correlate with a higher migrative capacity (107). Instead, in CLL, the CCR7 surface over-expression does correlate with enhanced migration in response to CCR7 ligands, with this chemotactic response being more effective in CLL cells than in normal B cells (42, 48, 49, 55). Migration in CLL is likely mediated through a variety of downstream effectors, such as Rho, ERK and PI3K kinases (55, 59). These are assisted by several clonal factors that potentiate migratory responses favoring clinical lymphadenopathy and poor outcome, including the presence of certain adverse prognostic factors such as CD38, ZAP70, CD49d, trisomy 12, NOTCH1 mutations, or un-mutated IGHV (42, 48, 49, 51, 53, 58–61, 64, 65, 75, 87, 108–111).
It is worth noting that CLL cells also express high levels of CXCR4 and CXCR5 (49, 112, 113). Despite this, leukemic CLL cells have a preferential in vitro migration towards CCR7 ligands. This situation contrasts with chemotaxis of normal B cells where CXCR4 and CXCR5 ligands are more effective than CCL19 or CCL21 (49). In this regard, a recent study by S. McHeik et al. demonstrated that expression of CCR7 in B cells selectively inactivated CXCR4, impairing migration towards its ligand CXCL12, and facilitating emigration from BM to PB (114). Mechanistically, up-regulation of CCR7 favors the formation of CXCR4-CCR7 heterodimers, thus acting as a selective endogenous allosteric modulator of CXCR4 that impairs its ability to activate certain G-protein complexes. These results may explain our observation in CLL migration (49) since up-regulation of CCR7 in CLL cells could favor the formation of CXCR4-CCR7 heterodimers thus reducing migration towards CXCL12. Nonetheless, we still have no data corroborating the presence of such regulatory heterodimers in CLL.
Finally, CCR7 not only directs migration of leukemic cells in CLL but, in addition, it is needed for migration of non-tumor subsets such as T cells. T cells from CLL patients and healthy donors have comparable expression of CCR7 and CXCR4 surface levels (52, 115), although CLL-associated T cells are less responsive to CXCL12, CCL21 and CCL19, except in T cells from patients with ZAP-70-expressing leukemic clones where migratory responses where similar to normal T cells (115).
CCR7 and CLL Homing Into the LN
Emigration of normal T and B lymphocytes from the blood to LN is a well-defined multi-step process in normal lymphocyte trafficking, termed “homing”. Tethering and rolling of lymphocytes on HEVs mediated by selectins, chemokine receptor–induced integrin activation, integrin-dependent firm arrest on HEVs, and trans-endothelial migration are processes strongly dependent on CCR7 (31). Similarly, since the seminal work by Till et al. reported CCR7 as the main receptor involved in CLL cells entry into the LNs (48), there is much evidence to support that CCR7 takes over these processes which are exploited by CLL cells to enter LN (14, 61, 70, 72, 87, 116). In line, prominent HEVs with high amounts of CCL21 and CCL19 are found in CLL nodes (48, 74, 117).
For effective CLL cell homing, chemokine-mediated activation of the integrins lymphocyte function–associated antigen (LFA-1) and VLA-4 are key events needed for the initiation of shear-resistant arrest of B cells to endothelium (48, 87, 116, 118). Normally, CLL cells express low levels of these integrins, and thus few CLL clones enter the LN but some CLL clones with trisomy 12 or ZAP70 are associated with high levels of LFA-1 and VLA-4, allowing their entry into lymphoid organs. Accordingly, patients with trisomy 12 or ZAP-70 expression may show exacerbated lymphadenopathy (14, 58, 87, 111, 119, 120) but the molecular mechanisms underlying this clinical association have been largely unknown until recent works by Legler´s laboratory. The group identified a critical role for ZAP-70 in CCL21/CCR7-mediated, Src-dependent clustering and inside-out activation of the integrins VLA-4 and LFA-1 that facilitates CLL cell arrest on endothelial cells. ZAP-70 expression, however, seemed dispensable for other processes, such as migration velocity, chemokine-mediated crawling and diapedesis (87). Since ZAP-70 associates with IGHV mutational status (11, 12, 121, 122), it is reasonable to link CCR7/ZAP-70-induced homing to the un-mutated clones which have enhanced CCR7-triggered migration when compared to mutated ones (42, 64, 75). Mechanistically, CCR7 activation autophosphorylates Src kinase which in turn phosphorylates CCR7 at tyrosine 155 within the cytoplasmatic DRY motif of CCR7, providing a binding site for the Src homology 2 (SH2) domain containing signaling molecules (123). Then both SH2 domains of ZAP-70 directly interact with phosphorylated CCR7 and ZAP-70 promotes LFA-1 clustering which is essential for cell adhesion to intracellular adhesion molecule 1 (ICAM-1) on endothelial cells under physiologic flow conditions. Since the impact of ZAP70 is quantitative and not qualitative, Legler and colleagues propose that, because more cells arrest on CCL21-presenting endothelium, ZAP-70 expression enhances the chance of individual CLL cells to extravasate and thereby contributes to the accumulation of CLL cells in lymphoid tissues.
Despite the studies that strongly support the CCR7 axis as a main player in LN homing of CLL cells, some other chemokine receptors might also contribute to the characteristic disseminated lymphadenopathy in CLL, including CXCR3, CXCR4, or CXCR5 (48–50, 112, 124–126). Accordingly, normal B cells can exploit signaling mediated by CCR7, CXCR4, and CXCR5 to induce integrin-mediated arrest on HEVs and homing (127–130). Even though, only CCR7 seems relevant for LN homing as demonstrated in several in vivo models where CCR7 over-expression or genetic and pharmacology targeting impacted in the entry of CCR7-expressing cells into the LN (33–35, 62, 131, 132). In addition, CLL nodes have a disrupted architecture featured by the lack of follicles, which are the main source of CXCR5 cognate ligand, the chemokine CXCL13 (133, 134), and the chemokine CXCL12, the ligand of CXCR4, was not found in HEVs from CLL nodes (48). Taken together, these data suggest that CXCR4 and CXCR5 are not as relevant as CCR7 in CLL cell homing to LN. In agreement herewith, CLL cells transmigration through endothelium is more effective upon stimulation with CCL19 than with CXCL12 or CXCL13 (72). Under shear flow conditions, immortalized or primary CLL cells with trisomy 12 (associated to high levels of LFA-1 and VLA-4) significantly arrested to endothelial cells or VCAM/ICAM-1 coatings in the presence of CCL21, but not of CXCL12, since only the CCR7 ligand was capable to induce inside-out VLA-4 conformational changes as demonstrated in real-time kinetic assays (14, 87). Notably, similar results were obtained with CLL cells lacking trisomy 12. In another study, it was proposed that CCR7 might guide preferentially the homing of antigen-stimulated CLL cells since IgM activation selectively reduced migration of CLL cells towards CXCL12, but not CCL21 (64).
These arguments, substantiating the prevailing role of CCR7 in LN homing, rather than excluding the importance of CXCR5 or CXCR4 in the pathophysiology of CLL indicate that these receptors might act as synergistic, accessory molecules for CCR7 which may alter the global outcome in the process of LN homing. For example, in primary T cells and in one human Burkitt’s lymphoma cell line the exposure to CXCL12 potentiated in vitro transendothelial migration towards CCL19/CCL21, and also increased CCR7-dependent recruitment of T cells into LN in vivo (135, 136). On the other hand, CXCR4 might promote LN homing in an indirect manner, for example, favoring relocation of CLL from BM to the LN. In this respect, up-regulation of CCR7 in normal B cells selectively inactivates CXCR4 whereas mature B cells from CCR7-/- mice display higher responsiveness to CXCL12 and improved retention in the BM (114). Accordingly, CXCR4 is the main receptor driving homing of CLL cells to BM where stromal cells provide protection from spontaneous or drug-induced apoptosis (51, 113, 137). Nevertheless, in certain CLL clones, such in trisomy-12-positive cells, CXCR4 expression is decreased and BM homing barely reliant on CXCL12-induced signals, despite a fully functional CXCR4 receptor in chemotaxis assays (14). Since these cells retained the ability to activate VLA-4 and to arrest on VCAM-1 in CCL21-stimulated CLL cells, one can propose a biased extravasation into LN tissues with pathophysiological consequences.
CCR7 and LN Microenvironments in CLL
The survival of CLL cells is not a cell-autonomous, genetically determined process (138–141). Instead, CLL cells strictly depend on a permissive microenvironment that supports their survival and proliferation and consequently drives disease progression (2–4). CLL cells not only take advantage of CCR7 to home into the LN but also use this receptor for a correct positioning within the LN tissue as a result of the chemotactic routes created by stromal cells, which secrete CCR7 ligands (48, 74). In this regard, Höpken et al. demonstrated that genetic deletion of CCR7 in the syngeneic Eµ-Myc mouse lymphoma model was sufficient to exclude CCR7-deficient lymphoma cells from the T cell zone whereas wild-type lymphoma cells lodged to the stroma in close proximity to CD40L-expressing CD4+ T cells, DCs, and gp38+ fibroblastic reticular cells (FRCs), these latter providers of CCR7 ligands and the anti-apoptotic Indian hedgehog protein (Ihh) (34, 142). In turn, the lymphoma cells themselves secreted lymphotoxin through which they stimulated lymphotoxin-β-receptor–expressing FRCs. This crosstalk was necessary for the creation and preservation of protective niches in LN and spleen as demonstrated in vivo in adoptive transfer experiments since lack of CCR7 delayed disease onset and tumor burden (34, 142). Therefore, CCR7 enables distribution to different microenvironments where malignant cells may interact with supportive stroma (adhesion molecules and cells) and intra-tumor vasculature, crosstalk with CD40L-expressing cells, DCs, macrophages, and other tumor cells, recognize cognate antigens which triggers BCR signaling, and are exposed to soluble trophic factors: e.g. IL-7, IL-8, indoleamine 2,3-dioxygenase, CXCL12, CXCL13 (2–6, 34, 113, 140, 143–151). As a result, chemokines such as CCL3, CCL4, CCL19, CCL21, CCL22, CXCL12, CXCL13, are produced and a cycle of recruitment and proliferation begins in both CLL and accessory cells (147, 151–154). For example, recruited CCR7-expressing DCs in the LN may help further entry of tumor and other accessory cells directly through the production of CCL19 (131), or indirectly by promoting the secretion of vascular endothelial growth factor (VEGF) and CCL21 by FRCs (131, 155–157). The final outcome is a DC-induced vascular remodeling and generation of chemotactic cues for additional leukemic and accessory cells. Nonetheless, it should be kept in mind that increasing the presence of CCR7-expressing immune cells does not always associate with poor prognosis. The expression of CCL21 and endothelium proteins such as peripheral node addressin in LN-like vasculature found within solid tumors helps with anti-tumor T cell infiltration and positive prognosis in murine models (158). Unfortunately, similar studies showing a correlation between prognosis and the presence of CCR7-expressing infiltrating T-lymphocytes are still lacking in CLL. Finally, soluble CCL19/CCL21 also promote CCR7-induced survival of CLL cells through mechanisms not found in normal B cells. For example, CCL19 and CXCL13 cooperative signaling contributes to resistance to TNF-α-mediated apoptosis through up-regulation of paternally expressed gene 10 (PEG10) which stabilizes caspase-3 and caspase-8 (68, 78). Additional roles for CCR7-induced CLL cell survival have been described through MAP-kinases and PI3K-AKT signaling pathways upon binding of both cognate ligands (55, 69).
The knowledge about the mechanisms underlying the CCR7-guided interstitial CLL cell migration supports the idea that stop/go signals mediated by CCR7 in CLL are unique to this condition rather than resembling the well-known behavior of normal CCR7-expressing lymphocytes. In this regard, since CCR7 expression remains high within the LN and PCs (42) it is likely that CCR7 guides CLL cells to different niches assisted by other molecules. For example, interaction of wnt5 (A and B) or cordon-blue protein-like 1 (COBLL1) proteins with the receptor tyrosine kinase-like orphan receptor 1 (ROR1), a transmembrane receptor upregulated in CLL, activated pathways controlling cell polarity and migration. Specifically, ROR-1 activation increased basal migration and attenuated motility and chemotaxis toward CCL19 (159, 160) suggesting a role of these axes in fine-tuning CLL movement along CCL19 gradients and shutting migration off once the right niche is found. Interestingly, in non-tumor cells, COBLL1 expression was also found to be higher in GC B cells than naïve and memory subsets, where a marked reduction in CCR7 expression is needed to facilitate CXCR5-guided access to the GC (129, 161, 162).
The CC chemokine receptor-like 2 (CCRL2, also known as CRAM) is also aberrantly over-expressed in CLL B cells and it was proposed as a bystander molecule regulating CCR7-induced migration along CCL19 gradients (73), likely towards niches where the cognate antigen is found as suggested by the fact that CCRL2 is down-regulated upon IgD- or IgM-induced stimulation of BCR in CLL cells (64). However, the scavenging properties of CCRL2 in CLL are a matter of debate. In endothelial cells, recent evidence discards the binding of CCRL2 to CCL19 (among many other chemokines), the subsequent receptor internalization, and the ability to scavenge CCL19 (163, 164) precluding, therefore, its formerly proposed role as a shaper of CCL19 gradients. Instead, CCRL2 binds the non-chemokine chemotactic factor chemerin and presents it to cells expressing the chemokine-like receptor 1 (CMKLR1), the functional chemerin receptor (165). Since CMKLR1 is expressed in plasmacytoid DCs and endothelial cells it is tempting to speculate that the biological function of the axis CCRL2-chemerin-CMLKR1 in CLL has to do with facilitating interactions with these accessory cell types although future studies addressing this hypothesis are required to fully elucidate the mechanism by which CCRL2 regulates CLL cell functions.
Another peculiarity in CCR7-directed interstitial migration of CLL cells can be found in the important role attributed to the Cdc42-interacting protein 4 (CIP4), which is specifically overexpressed in CLL when compared with normal B cells or other subtypes of B-cell malignancies (166). For CCL19-driven directional cell steering and chemotaxis, CLL cells depend on CIP4 to modulate the Wiskott-Aldrich syndrome protein (WASP), p21-activated kinase 1 (PAK1), and p38 MAPK pathways to control the assembly of highly structured actin-rich protrusions, including lamellipodia. Remarkably all these factors (wnt-5A, wnt-5B, COBLL1, and CIP-4) are overexpressed in un-mutated IGHV CLL and are associated with poor outcome and nodal involvement. Once again, it seems reasonable that un-mutated or mutated IGHV clones interpret gradients of CCR7 ligands differently with the contribution of BCR downstream signaling proteins, such as ZAP-70 which has already been marked as an enhancer of BCR signaling and CCR7-mediated adhesion (61, 87). This differential behavior between mutated and un-mutated IGHV clones is not new. It is long known that un-mutated CLL shows a sustained interaction of competent BCR with low-affinity self-Ags, resulting in a higher proliferative rate, and only these clones usually respond to IgM stimulation (167–169). Similarly, IgM activation reduced migration of CLL cells towards CXCL12, but not CCL21, whereas IgD activation predominantly impacted on CCL21 but not CXCL12-mediated chemotaxis (64). This indicates a preferential role of CCR7 for migration of antigen-stimulated CLL cells within the lymphoid microenvironment.
Stromal proteins are also relevant players assisting CCR7 in the positioning of CLL cells close to CD40L-expressing cells, mainly CD4+ TH cells. According to the model proposed by Hartmann et al. (74), unstimulated CLL cells use the nodal reticular network, simultaneously presenting hyaluronan (HA) and CCL21, as a guiding structure for their interstitial migration. The HA triggers a robust, random CCL21-induced motility of resting CLL cells until they encounter autologous T cells. If CLL cells get activated via CD40–CD40L signaling, then N-linked glycosylations of CD44 take place (particularly associated with the variant isoform CD44v6) and this glycoprotein strongly binds to HA causing CLL cells to stop migrating and instead tightly adhere to HA-bearing stromal cells. This strong CD44–HA–dependent adhesion facilitates cell division by retaining CLL cells in close proximity to CD4+ T cells in PCs, providing survival and proliferation cues: for example, T cell–derived interleukins (e.g. IL-4) (2–4). Interestingly, Hartmann and colleagues have demonstrated that this restriction to CCL21-induced motility is only based on physical blocking interactions since activated cells retain their migratory response towards CCL21 (74). Therefore, this mechanism to modulate CLL migration seems to override the continuous high, functional expression of CCR7 in the leukemic cells. Moreover, since CLL clones with unmutated IGHV present overexpression of CD44 (75), this proposed mechanism contributes to the enhanced retention of these clones within the LN by mediating adhesion to HA following CD40-CD40L engagement.
CCR7 and Immune Tolerance in the LN
As further reviewed elsewhere (2–6), several studies suggest that CLL cells are not passive players within the tumor microenvironment (TME). Instead they actively modify it. We have already addressed their role in producing chemokines and cytokines that grant the access and/or promote proliferation of accessory gp38+ FRCs, DCs or CD40L-expressing T cells. In addition, CLL cells have the capacity to induce specific changes in myeloid and plasmacytoid DCs (170, 171), and T cells (172, 173) that can alter T cell immunological recognition and function resulting in an impaired immune response to the leukemia. Another mechanism that may contribute to creating a tolerant microenvironment for CLL is the presence of regulatory cells such as TREG and the myeloid-derived suppressor cells (MDSCs). Both cell types have elevated frequencies in patients with CLL which associate with active disease (174–182) and, remarkably, both subsets express CCR7. Notably, this receptor is needed to enter the LN and other TME where CCR7 ligands are produced and where suppressive cells expand defeating antitumor immunity through heterogeneous mechanisms that include direct contact or the production of soluble factors such as IL-10 and TGF-β (183, 184). Indeed, in CLL the proportion of CCR7+ naïve natural TREG is increased compared to healthy controls and within the overall TREG population. Despite this population being characterized by augmented co-expression CD39, a molecule closely associated with suppressive activity in TREG, their suppressive activity (tested in 5 CLL patients) was not superior to other TREG subsets (178). This result is not something unexpected since CCR7 expression is not needed for suppressive activity in TREG (183) but for homing into LN where they respond to antigen and get armed performing their immunosuppressive activity. Without this step they still home to the peripheral tissues but cannot regulate (185–187). Nevertheless, further work on CCR7+ TREG activity in CLL is needed in more samples to shed light on this aspect since patients could benefit from a therapeutic intervention consisting in the depletion of these suppressive cells. In other conditions, this therapeutic venue has been demonstrated to be effective in pre-clinical models of aggressive tumors (188, 189) and in clinical trials with mogamulizumab, an anti-CCR4 therapeutic mAb which selectively decreased the fraction of CCR4+ TREG and associated with better clinical outcome in T cell malignancies and melanoma (190, 191).
CCR7 Prolongs Residency of CLL Cells in LN Protective Niches
Besides its role in homing into SLOs, interstitial positioning, and survival, it has long been known that CCR7 is also crucial in the egress of lymphocytes from LN to PB. As shown in CCR7-deficient mice, absence of CCR7 forces a rapid egress from the LN, quicker than wild-type cells, whereas over-expression of this receptor retarded this process (192). Therefore, CCR7 expression and induced signaling pathways prolong the stay of lymphocytes in LN. Once lymphocytes find their cognate antigen, get activated, and undergo several divisions, CCR7 expression is progressively shut off (30, 192). CCL19-triggered signaling promotes down-modulation of CCR7 and a balanced up-regulation in the transcription of the counteracting sphingosine-1-phosphate receptor (S1P1) plays a crucial role in regulating the egress of T- and B cells from LN toward S1P1 rich circulatory fluids (blood and lymph), overcoming the retention signals provided by CCR7 (193).
In CLL, the work of Baldari et al. suggests that over-expression of CCR7, together with the concomitant low expression of egress S1P1, results in an altered balance that contributes to a prolonged residency of CLL cells in protective niches and subsequent lymphadenopathy (42, 60, 66, 100, 101). According to these authors, high CCR7 recycling rates in CLL cells seem to be one of the main contributors to receptor over-expression, with un-mutated CLL cells showing the highest turn-over and the more defective S1P1 expression. Mechanistically, deficiency of p66shc contributes to this process in two different ways. First, lack of p66Shc protein is the cause of CCR7 up-regulation and S1P1 down-modulation in CLL cells since the genes encoding both receptors are controlled in opposite directions by the ROS-elevating (pro-oxidant) activity of p66Shc (60, 100). Second, p66Shc inhibits the Ca2+-dependent activation of the phosphatase type 2B (PP2B, aka calcineurin) which dephosphorylates serine residues of CCR7, a step needed to release CCR7 from β-arrestin in early Rab5+ endosomes allowing transit to Rab11+ recycling endosomes (42, 66). The cause of p66shc deficiency in CLL remains unknown, however p66Shc expression is regulated by STAT-4, which is profoundly reduced in CLL cells (97). Together these findings are highly relevant as they uncover a novel regulatory mechanism for CCR7 in B cells where the receptor was thought not to be completely needed to balance stay/egress from lymphoid tissues (192). p66Shc deficiency does not impact S1P1 expression in T cells (60). Moreover, p66Shc deficiency seems highly specific to leukemic CLL B cells and more specifically in un-mutated IGHV CLL clones. However, it is important to keep in mind that other factors may contribute to the unbalanced CCR7/S1P1 signaling in CLL cells. For example, un-mutated IGHV cells show low levels of S1P1 signaling proteins such as dynamin-2 and G-protein αi (75). In addition, un-mutated clones overexpress CD44. As already discussed, CD44 contributes to the retention of CLL cells in LN by mediating adhesion to HA following CD40-CD40L engagement, thus contributing, in and additional manner, to prolong residency of CLL cells within the LN (74, 75, 91).
CCR7 as a Novel Therapeutic Target in CLL
The evidence presented here suggests that novel tools targeting CCR7 are appealing to displace cells from LN microenvironment, hitting, therefore, the “Achilles’ heel” of CLL. As such, the present authors and other teams demonstrated that CCR7 genetic deletion or pharmacological inhibition with anti-CCR7 monoclonal antibodies (mAb) reduced CCR7-triggered migration and homing into LN in both in vitro and in vivo pre-clinical models (34, 62) (52, 55, 63). Similarly, driving leukemic cells out of LN to induce “death by neglect” or forcing their apoptosis are the mechanism of action (MOA), respectively, of ibrutinib and venetoclax, the current standards-of-care (SOC) in CLL (21, 22, 194).
Recently, it was reported that allosteric antagonism by small molecules under clinical investigation interfered with CCL19-driven signaling (195). Thus this work confirmed a long-standing interest in active synthetic drugs to avoid entry of cancer cells into LN. Our laboratories’ work focused on antibodies which may provide several advantages over small molecules: selectivity, affinity, increased serum half-life and tumoricidal capacities (196, 197). Moreover, small molecules are unable to trigger host anti-tumor responses whereas therapeutic antibodies have provided such clinical benefits to cancer patients during the last decades (198, 199). We hypothesized that a higher efficacy at reducing LN tumor burden would be achieved by a neutralizing anti-CCR7 monoclonal antibody (mAb) able to immobilize cancer cells and to elicit cell killing (52, 62, 63). However, raising blocking antibodies against CCR7 and other CKR has been considered a challenging task due to the necessity of targeting specific epitopes involved in ligand binding, and to a high sequence homology between human and mouse CCR7 that impairs immunogenicity (197). In agreement herewith, few anti-CKRs are under study in pre-clinical or early clinical phases, and only one (mogamulizumab) has been approved for clinical use (197, 200–202). Fortunately, the advent of novel technologies including sophisticated purification techniques, synthesis of conformational peptides, genetic immunization techniques, production of CKR-containing liposomes or lipoparticles, or over-expression of receptors in viral particles (197, 202) has overcome cited limitations and the first therapeutic anti-CCR7 antibodies have been developed. Recently, Novartis has announced the beginning of a phase I trial with JBH492 an antibody targeting CCR7 (NCT042140704). Despite not being clear if this molecule blocks the target, the fact that it is linked to a cytotoxic molecule (antibody-drug-conjugate, ADC) indicates that killing tumor cells by means of the release of cytotoxic payloads seems its main MOA. In addition, Catapult Therapeutics disclosed first pre-clinical results of a novel humanized IgG1 antagonist (CAP-100) specifically developed for cancer therapy which will be evaluated in a first-in-human clinical trial (NCT04704323) in 2021 (203, 204). This antibody is featured by a unique dual MOA which relies on an effective combination of strong blocking and killing activities. This antibody neutralizes receptor-ligand interactions thus inhibiting CCR7-induced cell functions, such as the access of cancer cells to niches where CCR7 ligands are produced (e.g. LN or spleen). This way, tumor cells are forced to accumulate in the bloodstream where they become more accessible to indirect cell killing mediated by effector immune cells (ADCC, antibody-dependent cell-mediated cytotoxicity), to spontaneous apoptosis, or to other treatments. Targeting CCR7 is a plausible strategy not only as a monotherapy but may also contribute in a rational combination to enhance current standard-of-care treatments. For example, the use of neutralizing anti-CCR7 antibodies along with ibrutinib would synergistically target CCR7-induced integrin-mediated adhesion to lymphoid stroma (42, 61, 66), thus enhancing drug-promoted CLL cell mobilization from protective lymphoid niches into circulation. Additionally, the antibody would block recirculation and loops of LN homing. Ibrutinib would also interfere with CXCR4- and CXCR5-mediated signaling and with the production of chemokines (CXCL12, CXCL13, CCL19) by myeloid stroma cells (61, 151). Then, spontaneous death in cells deprived of critical microenvironment growth and survival signals will be supplemented by antibody-induced cell killing. Similarly, anti-CCR7 mAbs combined with PI3Kδ inhibitors such as Idelalisib (205), a selective inhibitor of the p110 delta isoform, might contribute to inhibition of integrin-mediated arrest of CLL cells on endothelial cells. In other combinations, anti-CCR7 antibodies would mainly interfere with homing into LN and with the following interstitial migration within this tissue, whereas adjuvant therapies, such as the Bcl-2 inhibitor venetoclax, would collaborate in inducing cell death. Interestingly, venetoclax is markedly less toxic towards CLL cells when co-cultured with activated T lymphocytes (206) therefore, anti-CCR7 therapies could reverse this situation by impairing the support of CLL cells by accessory T cells. Finally, it is worth mentioning that un-mutated CLL clones are preferentially retained in LN, where they are exposed to proliferative stimuli, suggesting that anti-CCR7 strategies may be particularly effective in un-mutated-CLL. In agreement with this prediction, clinical studies have shown that the overall response to ibrutinib or idelalisib is significantly higher in patients with un-mutated CLL compared with those with mutated clones (61, 205, 207–209).
Safety of Anti-CCR7 Therapies in CLL
Today, the main concerns linked to the potential of CCR7 as a therapeutic target have to do with the fact that CCR7 is a critical molecule for inducing adaptative immune responses as well as for the generation of TREG cells that control the development of self-reactive cells and subsequent auto-immunity. Although most of the knowledge on the involvement of CCR7 in these processes is derived from CCR7-deficient mice (33) and paucity of lymph-node T cells (plt) mice, a naturally occurring strain that carries an autosomal recessive deletion that contains the genes encoding CCL19 and CCL21-Ser (210, 211), the expression patterns of CCR7 in mice and humans are similar thus corroborating their use for the in vivo investigations of this CKR. From these models we learnt that lack of CCR7 signaling did not compromise life or life-span, and animals were not immuno-deficient in a strict sense. On the contrary, these phenotypes associated with retarded but effective T cell and B cell responses (33, 129, 212), especially in cases where a replicating antigen (viruses and some bacterial infections) was present (213–216). We also learnt that lack of CCR7 reduced LN homing of TREG and their right positioning in the nodal environment hampering, therefore, central and peripheral tolerance (183, 217–220). In the long-term, CCR7-defficient mice were prone to develop generalized multi-organ autoimmunity (mainly in mucosa) although had a normal life span and did not suffer from clinically aggressive diseases. Finally, it is important to remark that also in CCR7-deficient mice autoimmunity is a multi-factorial process related to mice strain (genetic predisposition) and environmental factors (219–221) and that many phenotypes in CCR7-deficient animals are a consequence of immunity development within abnormal SLO microenvironments (222). Therefore, it is coherent to think that the immune responses of a patient treated with therapies targeting CCR7 will not fully mirror the outcomes of CCR7-deficient animals. In this sense, pre-clinical studies have shown selective effects of anti-CCR7 mAbs for CLL cells while sparing healthy counterparts, even at saturating concentrations of antibody (52, 63, 223, 224). In these studies only TN and TCM cells were impacted (though not entirely removed) while other CCR7-expressing cells such as DC or B cells were surprisingly not affected. We and others believe that this effect is likely due to a lower target density in non-tumor cells or to a lower affinity of these antibodies for CCR7 expressed in these cell types. These results lead to expect a low-to-mild associated immunosuppression in patients receiving therapeutic doses of an anti-CCR7 mAb. For example, it is likely that anti-CCR7 therapy could impair new immunization processes dependent on TN cells, however it should not affect memory effector responses against infections (224). Similarly, CCR7-negative TEFF and TEM rather than CCR7-expressing naïve or TCM are necessary to effective anti-tumor responses (225). Targeting CCR7 could also affect B cell homing during antigen-dependent and independent B cell differentiation; however, CCR7-deficient mice show splenic B cell responses upon bacterial challenge (129). This regard, normal B cells are less dependent on CCR7 than CLL cells for arrest on HEVs and homing (127, 133, 192) while B cell BM precursors and plasma cells lack CCR7 (49), thus suggesting that CCR7 therapy would not suppress B cell lymphopoiesis nor immunoglobulin secretory function (33, 129).
As previously indicated, TREG is another cell type that might be affected by anti-CCR7 therapies. We have already seen how this subset is significantly increased in CLL patients and correlates with poorest clinical outcomes (174–178). However, whether targeting CCR7-expressing TREG would be beneficial or deleterious for CLL patients is a controversial issue. By one hand, interfering TREG functions is an undesired side-effect associated to the development of autoimmunity in CLL patients treated with the PI3Kδ inhibitor idelalisib (226). On the other hand, these frequent unwanted events linked to idelalisib were not as prevalent in mAb-based therapies with the discontinued anti-CD52 antibody alemtuzumab (which depletes pan-T cell populations, including TREG) (227) or with the anti-CCR4 antibody mogamulizumab (which removes CCR4+ T cell subsets, including TREG) (228). Finally, it is worth to indicate that anti-CCR7 therapy in pre-clinical syngeneic mouse models of cancer, autoimmunity, GVHD, or inflammation did not uncover un-wanted treatment-associated side effects (184, 223, 229) and CAP-100 toxicology studies in NHP did not reveal overt toxicities or autoimmune disease indicating tolerability of this novel therapy. In the coming months, first data in patients receiving a chronic administration of an anti-CCR7 mAb will shed light on these safety issues.
Conclusions
The LN is the main hub for CLL where leukemic cells find proliferation and survival cues. Among the multiple altered signaling pathways found in CLL, the axis CCR7-CCL19/CCL21 is especially relevant (Figure 2). As revised here, CCR7 over-expression is a feature of CLL that has historically been reported but, until recently, we did not begin to understand the exact mechanisms underlying this process. Interesting novel findings suggest that altered receptor recycling pathways are involved in this up-regulation. Nonetheless, these abnormal processes at the protein levels could be just the tip of the ice-berg and additional studies addressing another mechanisms, such as genetic or epigenetic dysregulation, would be of great interest. The canonical view of CCR7 as a LN homing receptor of CLL cells has been up-dated thanks to studies revealing CCR7 as a key mediator of interstitial migration towards PCs and other locations where CLL cells cross-talk with accessory cells and have access to trophic factors which promote tumor growth and progression, including CCR7’s own ligands. The extensive knowledge arisen on the pathogenic roles of CCR7 in CLL cells contrasts with the scant studies focused on receptor-mediated functions in accessory cells during the different phases of niche colonization, preservation and progression. Fortunately, the advent of novel BCR inhibitors has indirectly provided first clues for a better understanding of the mechanisms through which CCR7 creates and preserves protective and tolerogenic milieus, prolongs CLL cell residency in these niches (contributing, therefore, to lymphadenopathy), and provides escape from therapeutic agents. Gathered together, this current extensive collection of evidence confirms CCR7 as key molecule in CLL and suggests, therefore, that tools targeting CCR7 are appealing as novel therapies in CLL. Today, two novel anti-CCR7 mAbs are facing clinical evaluation. Although both compounds have shown high activity in pre-clinical models, first-in-human studies need to solve several questions, beyond efficacy, that are linked to this kind of therapy such as the associated risk for impairing adaptative immunity and/or developing auto-immunity. If positive results are obtained in this clinical trials, we will likely witness to an exciting new age not only in CLL therapy but also in other diseases where CCR7 mediates deleterious roles.
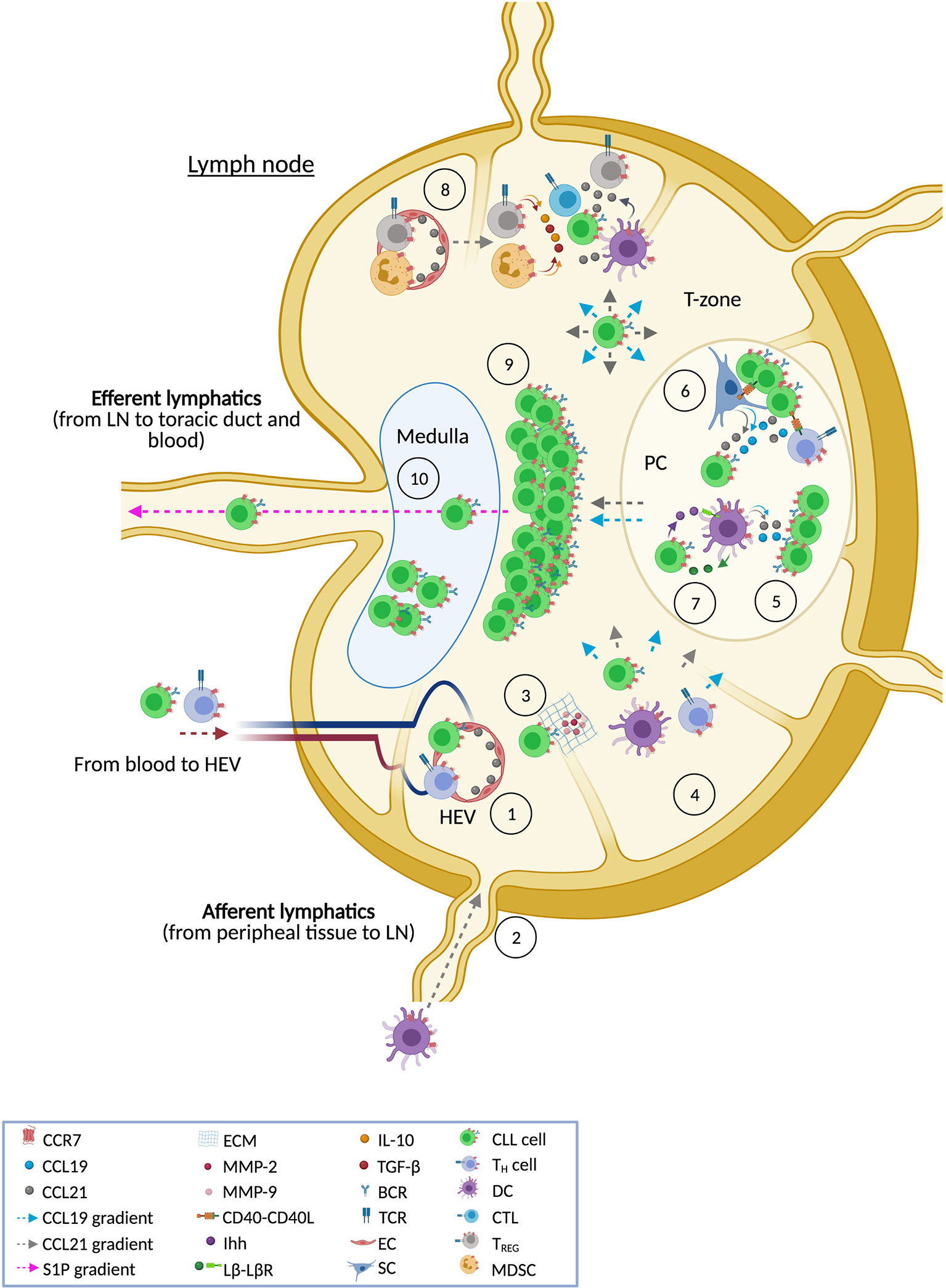
Figure 2 CCR7 and the lymph node in CLL. The figure shows the different ways in which CCR7 contributes to CLL pathobiology in the LN tissue. This receptor directs leukemic and accessory cells into the LN following CCL21 gradients that allow cells to across high endothelial venules (HEV) [1]. It is also likely that CCR7 might promote entry through a different gate, the afferent lymphatic vessels [2], although this last situation has not been reported yet in CLL cells. Both entry points are also used by accessory cells such as T cells and dendritic cells (DCs). When CLL cells get access through HEV, binding of CCL21 and subsequent CCR7 signaling promotes a more invasive phenotype, featured by enhanced production of matrix metalloproteases (MMP-2 and MMP-9) that degrade the extracellular matrix (ECM) [3]. This process facilitates trans-endothelial migration and the following interstitial migration within the LN tissue following CCL19 and CCL21 gradients favoring the right positioning of CLL cells within niches where accessory cells, stroma components, or soluble factors (e.g. cytokines and chemokines) are available [4]. Accessory and stromal cells are the main producers of CCR7 ligands thus facilitating the creation of chemotactic routes towards these niches. Similarly, some CCR7-expressing accessory cells can be directed by CCR7 ligands to these environments. Once CLL cells are driven to protective niches, such as proliferation centers (PC), tumor cells have access to CCL19 and CCL21 (which are produced by stromal cells and DCs) which rescue CLL cells from spontaneous or drug-induced apoptosis [5]. CLL cells also have access to BCR signaling and CD40-CD40L signaling [6] which regulate both CCR7 expression and chemotaxis in CLL cells further contributing to interstitial movement within the LN tissue. In the protective niches, CCR7 signaling in CLL cells is also involved in the secretion of trophic factors needed by accessory cells thus creating a positive feedback loop to preserve these tumor niches. For example, CLL cells themselves might preserve PC by means of secretion of lymphotoxin β (Lβ) which binds to Lβ-receptor in stromal cells and induces their differentiation into pro-tumor cells [7] which secrete Indian hedgehog protein (Ihh) triggering survival in malignant cells. Similarly, CLL cells can modulate activity of anti-tumor immunity through the recruitment of pro-tumor regulatory cells such as TREG and myeloid-derived suppressor cells (MDSC); both subtypes characterized by expression of CCR7 which orchestrates their homing into the LN [8]. These suppressor cells inhibit anti-tumor effector cells (CTLs, NK cells, B cells, etc) through cell-cell interactions or well by creating a tolerant milieu enriched in IL-10 and TGFβ. As a result of all these described activities [5–8], CCR7 directly or indirectly promotes tumor growth in the T cell zone of the LN [9], contributing to the typical obliterated enlarged structure in CLL nodes. Moreover, CCR7 up-regulation in CLL cells (as a consequence of an aberrantly rapid recycling rate of the receptor) leads to an impaired up-regulation of S1P1, the receptor guiding the egress of immune cells trough S1P gradients towards the efferent lymphatic vessels. Therefore, CCR7 signaling retains CLL cell within the LN, increasing the residence time in protective niches thus contributing in an additional way to bulky disease in the LN [10].
Author Contributions
CC-M, FT, JB, and CM-C contributed equally to the conception, design and writing of this manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
CC-M is an employee of Catapult Therapeutics and of Immunological and Medical Products (IMMED S.L.), and a shareholder in this last company. FT declares that he is CEO and a shareholder in the same companies. CM-C is a consultant for IMMED S.L., who has a granted patent for the use of therapeutic antibodies targeting CCR7 in cancer and has received research funds from IMMED.S.L. and Catapult Therapeutics. She also holds shares in IMMED S.L. JB has served as a consultant for Abbvie, Acerta, Astra-Zeneca, Beigene, Catapult, Dynamo Therapeutics, Eli Lilly, Genentech/Roche, Gilead, Juno/Celgene/Bristol Myers Squibb, Kite, Loxo, MEI Pharma, Nextcea, Novartis, Octapharma, Pfizer, Pharmacyclics, Rigel, Sunesis, TG Therapeutics, Verastem; received honoraria from Janssen, received research funding from Gilead, Loxo, Sun, TG Therapeutics and Verastem; and served on data safety monitoring committees for Invectys.
Acknowledgments
The authors would like to thank Lawrence Baron and Wim Mol for language editing and proofreading of the manuscript. In addition, we would like to thank BioRender.com for figure editing.
References
1. Riedel A, Shorthouse D, Haas L, Hall BA, Shields J. Tumor-induced stromal reprogramming drives lymph node transformation. Nat Immunol (2016) 17(9):1118–27. doi: 10.1038/ni.3492
2. Hayden RE, Pratt G, Roberts C, Drayson MT, Bunce CM. Treatment of chronic lymphocytic leukemia requires targeting of the protective lymph node environment with novel therapeutic approaches. Leuk Lymphoma (2012) 53(4):537–49. doi: 10.3109/10428194.2011.610014
3. Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Primers (2017) 3:16096. doi: 10.1038/nrdp.2016.96
4. Ponzoni M, Doglioni C, Caligaris-Cappio F. Chronic lymphocytic leukemia: the pathologist’s view of lymph node microenvironment. Semin Diagn Pathol (2011) 28(2):161–6. doi: 10.1053/j.semdp.2011.02.014
5. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood (2011) 117(2):563–74. doi: 10.1182/blood-2010-05-284984
6. Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematol Am Soc Hematol Educ Program (2011) 2011:96–103. doi: 10.1182/asheducation-2011.1.96
7. Hallek M. Signaling the end of chronic lymphocytic leukemia: new frontline treatment strategies. Blood (2013) 122(23):3723–34. doi: 10.1182/blood-2013-05-498287
8. Hashimoto S, Dono M, Wakai M, Allen SL, Lichtman SM, Schulman P, et al. Somatic diversification and selection of immunoglobulin heavy and light chain variable region genes in IgG+ CD5+ chronic lymphocytic leukemia B cells. J Exp Med (1995) 181(4):1507–17. doi: 10.1084/jem.181.4.1507
9. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood (1999) 94(6):1848–54. doi: 10.1182/blood.V94.6.1848.418k05_1848_1854
10. Krober A, Seiler T, Benner A, Bullinger L, Bruckle E, Lichter P, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood (2002) 100(4):1410–6. doi: 10.1182/blood.V100.4.1410.h81602001410_1410_1416
11. Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med (2003) 348(18):1764–75. doi: 10.1056/NEJMoa023143
12. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. N Engl J Med (2004) 351(9):893–901. doi: 10.1056/NEJMoa040857
13. Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med (2000) 343(26):1910–6. doi: 10.1056/NEJM200012283432602
14. Ganghammer S, Hutterer E, Hinterseer E, Brachtl G, Asslaber D, Krenn PW, et al. CXCL12-induced VLA-4 activation is impaired in trisomy 12 chronic lymphocytic leukemia cells: a role for CCL21. Oncotarget (2015) 6(14):12048–60. doi: 10.18632/oncotarget.3660
15. Grever MR, Lucas DM, Dewald GW, Neuberg DS, Reed JC, Kitada S, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol (2007) 25(7):799–804. doi: 10.1200/JCO.2006.08.3089
16. Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature (2011) 475(7354):101–5. doi: 10.1038/nature10113
17. Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet (2011) 44(1):47–52. doi: 10.1038/ng.1032
18. Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med (2011) 365(26):2497–506. doi: 10.1056/NEJMoa1109016
19. Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood (2013) 121(8):1403–12. doi: 10.1182/blood-2012-09-458265
20. Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet (2010) 376(9747):1164–74. doi: 10.1016/S0140-6736(10)61381-5
21. Brown JR. Relapsed CLL: sequencing, combinations, and novel agents. Hematol Am Soc Hematol Educ Program (2018) 2018(1):248–55. doi: 10.1182/asheducation-2018.1.248
22. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood (2018) 131(25):2745–60. doi: 10.1182/blood-2017-09-806398
23. Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med (2014) 370(24):2286–94. doi: 10.1056/NEJMoa1400029
24. Messmer BT, Messmer D, Allen SL, Kolitz JE, Kudalkar P, Cesar D, et al. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J Clin Invest (2005) 115(3):755–64. doi: 10.1172/JCI23409
25. Caligaris-Cappio F, Bertilaccio MT, Scielzo C. How the microenvironment wires the natural history of chronic lymphocytic leukemia. Semin Cancer Biol (2014) 24:43–8. doi: 10.1016/j.semcancer.2013.06.010
26. Packham G, Stevenson FK. Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunology (2005) 114(4):441–9. doi: 10.1111/j.1365-2567.2005.02117.x
27. Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, Kieff E. Epstein-Barr virus-induced genes: first lymphocyte-specific G protein-coupled peptide receptors. J Virol (1993) 67(4):2209–20. doi: 10.1128/JVI.67.4.2209-2220.1993
28. Schweickart VL, Raport CJ, Godiska R, Byers MG, Eddy RL Jr., Shows TB, et al. Cloning of human and mouse EBI1, a lymphoid-specific G-protein-coupled receptor encoded on human chromosome 17q12-q21.2. Genomics (1994) 23(3):643–50. doi: 10.1006/geno.1994.1553
29. Burgstahler R, Kempkes B, Steube K, Lipp M. Expression of the chemokine receptor BLR2/EBI1 is specifically transactivated by Epstein-Barr virus nuclear antigen 2. Biochem Biophys Res Commun (1995) 215(2):737–43. doi: 10.1006/bbrc.1995.2525
30. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE. McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev (2013) 24(3):269–83. doi: 10.1016/j.cytogfr.2013.03.001
31. Forster R, Davalos-Misslitz AC, Rot A. CCR7 and its ligands: balancing immunity and tolerance. Nat Rev Immunol (2008) 8(5):362–71. doi: 10.1038/nri2297
32. Legler DF, Uetz-von Allmen E, Hauser MA. CCR7: roles in cancer cell dissemination, migration and metastasis formation. Int J Biochem Cell Biol (2014) 54:78–82. doi: 10.1016/j.biocel.2014.07.002
33. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999) 99(1):23–33. doi: 10.1016/S0092-8674(00)80059-8
34. Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, et al. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma-permissive niche within murine secondary lymphoid organs. Blood (2011) 118(4):1020–33. doi: 10.1182/blood-2010-11-321265
35. Wiley HE, Gonzalez EB, Maki W, Wu MT, Hwang ST. Expression of CC chemokine receptor-7 and regional lymph node metastasis of B16 murine melanoma. J Natl Cancer Inst (2001) 93(21):1638–43. doi: 10.1093/jnci/93.21.1638
36. Hauser MA, Legler DF. Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J Leukoc Biol (2016) 99(6):869–82. doi: 10.1189/jlb.2MR0815-380R
37. Willimann K, Legler DF, Loetscher M, Roos RS, Delgado MB, Clark-Lewis I, et al. The chemokine SLC is expressed in T cell areas of lymph nodes and mucosal lymphoid tissues and attracts activated T cells via CCR7. Eur J Immunol (1998) 28(6):2025–34. doi: 10.1002/(SICI)1521-4141(199806)28:06<2025::AID-IMMU2025>3.0.CO;2-C
38. de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol (2007) 2(11):735–44. doi: 10.1021/cb700159m
39. Barmore AJ, Castex SM, Gouletas BA, Griffith AJ, Metz SW, Muelder NG, et al. Transferring the C-terminus of the chemokine CCL21 to CCL19 confers enhanced heparin binding. Biochem Biophys Res Commun (2016) 477(4):602–6. doi: 10.1016/j.bbrc.2016.06.098
40. Hjorto GM, Larsen O, Steen A, Daugvilaite V, Berg C, Fares S, et al. Differential CCR7 Targeting in Dendritic Cells by Three Naturally Occurring CC-Chemokines. Front Immunol (2016) 7:568. doi: 10.3389/fimmu.2016.00568
41. Muppidi JR, Klein U. Directing traffic in the germinal center roundabout. Nat Immunol (2020) 21(6):599–601. doi: 10.1038/s41590-020-0690-9
42. Patrussi L, Capitani N, Martini V, Pizzi M, Trimarco V, Frezzato F, et al. Enhanced Chemokine Receptor Recycling and Impaired S1P1 Expression Promote Leukemic Cell Infiltration of Lymph Nodes in Chronic Lymphocytic Leukemia. Cancer Res (2015) 75(19):4153–63. doi: 10.1158/0008-5472.CAN-15-0986
43. Faguet GB. Chronic lymphocytic leukemia: an updated review. J Clin Oncol (1994) 12(9):1974–90. doi: 10.1200/JCO.1994.12.9.1974
44. Gars E, Butzmann A, Ohgami R, Balakrishna JP, O’Malley DP. The life and death of the germinal center. Ann Diagn Pathol (2020) 44:151421. doi: 10.1016/j.anndiagpath.2019.151421
45. Inamdar KV, Bueso-Ramos CE. Pathology of chronic lymphocytic leukemia: an update. Ann Diagn Pathol (2007) 11(5):363–89. doi: 10.1016/j.anndiagpath.2007.08.002
46. Harris NL, Jaffe ES, Stein H, Banks PM, Chan JKC, Cleary ML, et al. A Revised European-American Classification of Lymphoid Neoplasms: A proposal from the International Lymphoma Study Group. Blood (1994) 84(5):1361–92. doi: 10.1182/blood.V84.5.1361.bloodjournal8451361
47. Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med (2001) 194(11):1625–38. doi: 10.1084/jem.194.11.1625
48. Till KJ, Lin K, Zuzel M, Cawley JC. The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood (2002) 99(8):2977–84. doi: 10.1182/blood.V99.8.2977
49. Lopez-Giral S, Quintana NE, Cabrerizo M, Alfonso-Perez M, Sala-Valdes M, De Soria VG, et al. Chemokine receptors that mediate B cell homing to secondary lymphoid tissues are highly expressed in B cell chronic lymphocytic leukemia and non-Hodgkin lymphomas with widespread nodular dissemination. J Leukoc Biol (2004) 76(2):462–71. doi: 10.1189/jlb.1203652
50. Wong S, Fulcher D. Chemokine receptor expression in B-cell lymphoproliferative disorders. Leuk Lymphoma (2004) 45(12):2491–6. doi: 10.1080/10428190410001723449
51. Ghobrial IM, Bone ND, Stenson MJ, Novak A, Hedin KE, Kay NE, et al. Expression of the chemokine receptors CXCR4 and CCR7 and disease progression in B-cell chronic lymphocytic leukemia/ small lymphocytic lymphoma. Mayo Clin Proc (2004) 79(3):318–25. doi: 10.4065/79.3.318
52. Alfonso-Perez M, Lopez-Giral S, Quintana NE, Loscertales J, Martin-Jimenez P, Munoz C. Anti-CCR7 monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J Leukoc Biol (2006) 79(6):1157–65. doi: 10.1189/jlb.1105623
53. Richardson SJ, Matthews C, Catherwood MA, Alexander HD, Carey BS, Farrugia J, et al. ZAP-70 expression is associated with enhanced ability to respond to migratory and survival signals in B-cell chronic lymphocytic leukemia (B-CLL). Blood (2006) 107(9):3584–92. doi: 10.1182/blood-2005-04-1718
54. Calissano C, Damle RN, Hayes G, Murphy EJ, Hellerstein MK, Moreno C, et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood (2009) 114(23):4832–42. doi: 10.1182/blood-2009-05-219634
55. Cuesta-Mateos C, Lopez-Giral S, Alfonso-Perez M, de Soria VG, Loscertales J, Guasch-Vidal S, et al. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp Hematol (2010) 38(9):756–64, 64 e1-4. doi: 10.1016/j.exphem.2010.05.003
56. Calissano C, Damle RN, Marsilio S, Yan XJ, Yancopoulos S, Hayes G, et al. Intraclonal complexity in chronic lymphocytic leukemia: fractions enriched in recently born/divided and older/quiescent cells. Mol Med (2011) 17(11-12):1374–82. doi: 10.2119/molmed.2011.00360
57. Bryson MS. The role of chemokines and their receptors in non-Hodgkin’s lymphoma. Glasgow, Scotland, United Kingdom: PhD Thesis University of Glasgow (2011).
58. Zucchetto A, Cattarossi I, Nanni P, Zaina E, Prato G, Gilestro M, et al. Cluster analysis of immunophenotypic data: the example of chronic lymphocytic leukemia. Immunol Lett (2011) 134(2):137–44. doi: 10.1016/j.imlet.2010.09.017
59. Calpe E, Codony C, Baptista MJ, Abrisqueta P, Carpio C, Purroy N, et al. ZAP-70 enhances migration of malignant B lymphocytes toward CCL21 by inducing CCR7 expression via IgM-ERK1/2 activation. Blood (2011) 118(16):4401–10. doi: 10.1182/blood-2011-01-333682
60. Capitani N, Patrussi L, Trentin L, Lucherini OM, Cannizzaro E, Migliaccio E, et al. S1P1 expression is controlled by the pro-oxidant activity of p66Shc and is impaired in B-CLL patients with unfavorable prognosis. Blood (2012) 120(22):4391–9. doi: 10.1182/blood-2012-04-425959
61. de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood (2012) 119(11):2590–4. doi: 10.1182/blood-2011-11-390989
62. Somovilla-Crespo B, Alfonso-Perez M, Cuesta-Mateos C, Carballo-de Dios C, Beltran AE, Terron F, et al. Anti-CCR7 therapy exerts a potent anti-tumor activity in a xenograft model of human mantle cell lymphoma. J Hematol Oncol (2013) 6:89. doi: 10.1182/blood-2011-11-390989
63. Cuesta-Mateos C, Loscertales J, Kreutzman A, Colom-Fernandez B, Portero-Sainz I, Perez-Villar JJ, et al. Preclinical activity of anti-CCR7 immunotherapy in patients with high-risk chronic lymphocytic leukemia. Cancer Immunol Immunother (2015) 64(6):665–76. doi: 10.1007/s00262-015-1670-z
64. Haerzschel A, Catusse J, Hutterer E, Paunovic M, Zirlik K, Eibel H, et al. BCR and chemokine responses upon anti-IgM and anti-IgD stimulation in chronic lymphocytic leukaemia. Ann Hematol (2016) 95(12):1979–88. doi: 10.1007/s00277-016-2788-6
65. Arruga F, Gizdic B, Bologna C, Cignetto S, Buonincontri R, Serra S, et al. Mutations in NOTCH1 PEST domain orchestrate CCL19-driven homing of chronic lymphocytic leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia (2017) 31(9):1882–93. doi: 10.1038/leu.2016.383
66. Patrussi L, Capitani N, Cattaneo F, Manganaro N, Gamberucci A, Frezzato F, et al. p66Shc deficiency enhances CXCR4 and CCR7 recycling in CLL B cells by facilitating their dephosphorylation-dependent release from beta-arrestin at early endosomes. Oncogene (2018) 37(11):1534–50. doi: 10.1038/s41388-017-0066-2
67. Rodriguez A, Martinez N, Camacho FI, Ruiz-Ballesteros E, Algara P, Garcia JF, et al. Variability in the degree of expression of phosphorylated IkappaBalpha in chronic lymphocytic leukemia cases with nodal involvement. Clin Cancer Res (2004) 10(20):6796–806. doi: 10.1158/1078-0432.CCR-04-0753
68. Chunsong H, Yuling H, Li W, Jie X, Gang Z, Qiuping Z, et al. CXC chemokine ligand 13 and CC chemokine ligand 19 cooperatively render resistance to apoptosis in B cell lineage acute and chronic lymphocytic leukemia CD23+CD5+ B cells. J Immunol (2006) 177(10):6713–22. doi: 10.4049/jimmunol.177.10.6713
69. Ticchioni M, Essafi M, Jeandel PY, Davi F, Cassuto JP, Deckert M, et al. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene (2007) 26(50):7081–91. doi: 10.1038/sj.onc.1210519
70. Redondo-Munoz J, Jose Terol M, Garcia-Marco JA, Garcia-Pardo A. Matrix metalloproteinase-9 is up-regulated by CCL21/CCR7 interaction via extracellular signal-regulated kinase-1/2 signaling and is involved in CCL21-driven B-cell chronic lymphocytic leukemia cell invasion and migration. Blood (2008) 111(1):383–6. doi: 10.1182/blood-2007-08-107300
71. Enjuanes A, Benavente Y, Bosch F, Martin-Guerrero I, Colomer D, Perez-Alvarez S, et al. Genetic variants in apoptosis and immunoregulation-related genes are associated with risk of chronic lymphocytic leukemia. Cancer Res (2008) 68(24):10178–86. doi: 10.1158/0008-5472.CAN-08-2221
72. Trinidad EM, Ballesteros M, Zuloaga J, Zapata A, Alonso-Colmenar LM. An impaired transendothelial migration potential of chronic lymphocytic leukemia (CLL) cells can be linked to ephrin-A4 expression. Blood (2009) 114(24):5081–90. doi: 10.1182/blood-2009-03-210617
73. Catusse J, Leick M, Groch M, Clark DJ, Buchner MV, Zirlik K, et al. Role of the atypical chemoattractant receptor CRAM in regulating CCL19 induced CCR7 responses in B-cell chronic lymphocytic leukemia. Mol Cancer (2010) 9:297. doi: 10.1186/1476-4598-9-297
74. Girbl T, Hinterseer E, Grossinger EM, Asslaber D, Oberascher K, Weiss L, et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res (2013) 73(2):561–70. doi: 10.1158/0008-5472.CAN-12-2749
75. Eagle GL, Zhuang J, Jenkins RE, Till KJ, Jithesh PV, Lin K, et al. Total proteome analysis identifies migration defects as a major pathogenetic factor in immunoglobulin heavy chain variable region (IGHV)-unmutated chronic lymphocytic leukemia. Mol Cell Proteomics (2015) 14(4):933–45. doi: 10.1074/mcp.M114.044479
76. Wolf C, Garding A, Filarsky K, Bahlo J, Robrecht S, Becker N, et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int J Cancer (2017) 2017(18):31057. doi: 10.1002/ijc.31057
77. Tooze JA, Hamzic E, Willis F, Pettengell R. Differences between chronic lymphocytic leukaemia and small lymphocytic lymphoma cells by proteomic profiling and SNP microarray analysis. Cancer Genet (2017) 218-219:20–38. doi: 10.1016/j.cancergen.2017.09.002
78. Hu C, Xiong J, Zhang L, Huang B, Zhang Q, Li Q, et al. PEG10 activation by co-stimulation of CXCR5 and CCR7 essentially contributes to resistance to apoptosis in CD19+CD34+ B cells from patients with B cell lineage acute and chronic lymphocytic leukemia. Cell Mol Immunol (2004) 1(4):280–94.
79. Du H, Zhang L, Li G, Liu W, Tang W, Zhang H, et al. CXCR4 and CCR7 Expression in Primary Nodal Diffuse Large B-Cell Lymphoma-A Clinical and Immunohistochemical Study. Am J Med Sci (2019) 357(4):302–10. doi: 10.1016/j.amjms.2019.01.008
80. Cuesta-Mateos C, Fuentes P, Schrader A, Juárez-Sánchez R, Loscertales J, Mateu-Albero T, et al. CCR7 as a novel therapeutic target in t-cell PROLYMPHOCYTIC leukemia. Biomarker Res (2020) 8:54. doi: 10.1186/s40364-020-00234-z
81. Yan XJ, Dozmorov I, Li W, Yancopoulos S, Sison C, Centola M, et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood (2011) 118(19):5201–10. doi: 10.1182/blood-2011-03-342436
82. Chen Q, Zheng T, Lan Q, Lerro C, Zhao N, Qin Q, et al. Single-nucleotide polymorphisms in genes encoding for CC chemokines were not associated with the risk of non-Hodgkin lymphoma. Cancer Epidemiol Biomarkers Prev (2013) 22(7):1332–5. doi: 10.1158/1055-9965.EPI-13-0328
83. Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics (2012) 7(6):567–78. doi: 10.4161/epi.20237
84. Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene (2001) 20(19):2476–89. doi: 10.1038/sj.onc.1204386
85. Al Akoum C, Akl I, Rouas R, Fayyad-Kazan M, Falha L, Renno T, et al. NFAT-1, Sp-1, Sp-3, and miR-21: New regulators of chemokine C receptor 7 expression in mature human dendritic cells. Hum Immunol (2015) 76(5):307–17. doi: 10.1016/j.humimm.2015.03.010
86. Mathas S, Lietz A, Janz M, Hinz M, Jundt F, Scheidereit C, et al. Inhibition of NF-kappaB essentially contributes to arsenic-induced apoptosis. Blood (2003) 102(3):1028–34. doi: 10.1182/blood-2002-04-1154
87. Laufer JM, Lyck R, Legler DF. ZAP70 expression enhances chemokine-driven chronic lymphocytic leukemia cell migration and arrest by valency regulation of integrins. FASEB J (2018) 32(9):4824–35. doi: 10.1096/fj.201701452RR
88. Hinman RM, Bushanam JN, Nichols WA, Satterthwaite AB. B cell receptor signaling down-regulates forkhead box transcription factor class O 1 mRNA expression via phosphatidylinositol 3-kinase and Bruton’s tyrosine kinase. J Immunol (2007) 178(2):740–7. doi: 10.4049/jimmunol.178.2.740
89. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, et al. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PloS Biol (2005) 3(6):e150. doi: 10.1371/journal.pbio.0030150
90. Kehrl JH. Chemoattractant receptor signaling and the control of lymphocyte migration. Immunol Res (2006) 34(3):211–27. doi: 10.1385/IR:34:3:211
91. Luczynski W, Stasiak-Barmuta A, Piszcz J, Ilendo E, Kowalczuk O, Krawczuk-Rybak M. B-cell chronic lymphocytic leukemia-derived dendritic cells stimulate allogeneic T-cell response and express chemokines involved in T-cell migration. Neoplasma (2007) 54(6):527–35. doi: 10.1385/IR:34:3:211
92. Luczynski W, Ilendo E, Kovalchuk O, Krawczuk-Rybak M, Malinowska I, Koltan A, et al. Acute lymphoblastic leukaemia cells express CCR7 but not higher amounts of IL-10 after CD40 ligation. Scand J Clin Lab Invest (2006) 66(8):695–703. doi: 10.1080/00365510600931098
93. Kim KD, Choi SC, Noh YW, Kim JW, Paik SG, Yang Y, et al. Impaired responses of leukemic dendritic cells derived from a human myeloid cell line to LPS stimulation. Exp Mol Med (2006) 38(1):72–84. doi: 10.1038/emm.2006.9
94. Furman RR, Asgary Z, Mascarenhas JO, Liou HC, Schattner EJ. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J Immunol (2000) 164(4):2200–6. doi: 10.4049/jimmunol.164.4.2200
95. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nat (2009) 459(7249):1000–4. doi: 10.1038/nature08020
96. Jiang K, Krous LC, Knowlton N, Chen Y, Frank MB, Cadwell C, et al. Ablation of Stat3 by siRNA alters gene expression profiles in JEG-3 cells: a systems biology approach. Placenta (2009) 30(9):806–15. doi: 10.1016/j.placenta.2009.06.006
97. Cattaneo F, Patrussi L, Capitani N, Frezzato F, D’Elios MM, Trentin L, et al. Expression of the p66Shc protein adaptor is regulated by the activator of transcription STAT4 in normal and chronic lymphocytic leukemia B cells. Oncotarget (2016) 7(35):57086–98. doi: 10.18632/oncotarget.10977
98. Kim SH, Gunst KV, Sarvetnick N. STAT4/6-dependent differential regulation of chemokine receptors. Clin Immunol (2006) 118(2-3):250–7. doi: 10.1016/j.clim.2003.10.002
99. Gary-Gouy H, Sainz-Perez A, Marteau JB, Marfaing-Koka A, Delic J, Merle-Beral H, et al. Natural phosphorylation of CD5 in chronic lymphocytic leukemia B cells and analysis of CD5-regulated genes in a B cell line suggest a role for CD5 in malignant phenotype. J Immunol (2007) 179(7):4335–44. doi: 10.4049/jimmunol.179.7.4335
100. Capitani N, Lucherini OM, Sozzi E, Ferro M, Giommoni N, Finetti F, et al. Impaired expression of p66Shc, a novel regulator of B-cell survival, in chronic lymphocytic leukemia. Blood (2010) 115(18):3726–36. doi: 10.1182/blood-2009-08-239244
101. Patrussi L, Capitani N, Ulivieri C, Manganaro N, Granai M, Cattaneo F, et al. p66Shc deficiency in the Eμ-TCL1 mouse model of chronic lymphocytic leukemia enhances leukemogenesis by altering the chemokine receptor landscape. Haematologica (2019) 104(10):2040–52. doi: 10.3324/haematol.2018.209981
102. Mo M, Zhou M, Wang L, Qi L, Zhou K, Liu LF, et al. CCL21/CCR7 enhances the proliferation, migration, and invasion of human bladder cancer T24 cells. PloS One (2015) 10(3):e0119506. doi: 10.1371/journal.pone.0119506
103. Kim JW, Ferris RL, Whiteside TL. Chemokine C receptor 7 expression and protection of circulating CD8+ T lymphocytes from apoptosis. Clin Cancer Res (2005) 11(21):7901–10. doi: 10.1158/1078-0432.CCR-05-1346
104. Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP, et al. NF- kappa B2/p100 induces Bcl-2 expression. Leukemia (2003) 17(7):1349–56. doi: 10.1038/sj.leu.2402982
105. Zaninoni A, Imperiali FG, Pasquini C, Zanella A, Barcellini W. Cytokine modulation of nuclear factor-kappaB activity in B-chronic lymphocytic leukemia. Exp Hematol (2003) 31(3):185–90. doi: 10.1016/S0301-472X(02)01046-9
106. Weitzenfeld P, Kossover O, Körner C, Meshel T, Wiemann S, Seliktar D, et al. Chemokine axes in breast cancer: factors of the tumor microenvironment reshape the CCR7-driven metastatic spread of luminal-A breast tumors. J Leukoc Biol (2016) 99(6):1009–25. doi: 10.1189/jlb.3MA0815-373R
107. Scandella E, Men Y, Legler DF, Gillessen S, Prikler L, Ludewig B, et al. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood (2004) 103(5):1595–601. doi: 10.1182/blood-2003-05-1643
108. Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D’Arena G, et al. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med (2010) 16(3-4):87–91. doi: 10.2119/molmed.2009.00146
109. Hartmann TN, Grabovsky V, Wang W, Desch P, Rubenzer G, Wollner S, et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res (2009) 69(7):3121–30. doi: 10.1158/0008-5472.CAN-08-4136
110. Brachtl G, Sahakyan K, Denk U, Girbl T, Alinger B, Hofbauer SW, et al. Differential bone marrow homing capacity of VLA-4 and CD38 high expressing chronic lymphocytic leukemia cells. PloS One (2011) 6(8):e23758. doi: 10.1371/journal.pone.0023758
111. Zucchetto A, Caldana C, Benedetti D, Tissino E, Rossi FM, Hutterer E, et al. CD49d is overexpressed by trisomy 12 chronic lymphocytic leukemia cells: evidence for a methylation-dependent regulation mechanism. Blood (2013) 122(19):3317–21. doi: 10.1182/blood-2013-06-507335
112. Burkle A, Niedermeier M, Schmitt-Graff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood (2007) 110(9):3316–25. doi: 10.1182/blood-2007-05-089409
113. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood (1999) 94(11):3658–67. doi: 10.1182/blood.V94.11.3658.423k11_3658_3667
114. McHeik S, Van Eeckhout N, De Poorter C, Galés C, Parmentier M, Springael J-Y. Coexpression of CCR7 and CXCR4 During B Cell Development Controls CXCR4 Responsiveness and Bone Marrow Homing. Front Immunol (2019) 10:2970. doi: 10.3389/fimmu.2019.02970
115. Borge M, Nannini PR, Galletti JG, Morande PE, Avalos JS, Bezares RF, et al. CXCL12-induced chemotaxis is impaired in T cells from patients with ZAP-70-negative chronic lymphocytic leukemia. Haematologica (2010) 95(5):768–75. doi: 10.3324/haematol.2009.013995
116. Montresor A, Bolomini-Vittori M, Simon SI, Rigo A, Vinante F, Laudanna C. Comparative analysis of normal versus CLL B-lymphocytes reveals patient-specific variability in signaling mechanisms controlling LFA-1 activation by chemokines. Cancer Res (2009) 69(24):9281–90. doi: 10.1158/0008-5472.CAN-09-2009
117. Csanaky G. High endothelial venules and cell adhesion molecules in B-cell chronic lymphocytic leukaemia and related low grade B-cell lymphoma/leukaemia: I. High endothelial venules and lymphocyte migration. Histol Histopathol (1994) 9(3):571–5.
118. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
119. Riches JC, O’Donovan CJ, Kingdon SJ, McClanahan F, Clear AJ, Neuberg DS, et al. Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood (2014) 123(26):4101–10. doi: 10.1182/blood-2014-01-552307
120. Liso V, Capalbo S, Lapietra A, Pavone V, Guarini A, Specchia G. Evaluation of trisomy 12 by fluorescence in situ hybridization in peripheral blood, bone marrow and lymph nodes of patients with B-cell chronic lymphocytic leukemia. Haematologica (1999) 84(3):212–7.
121. Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood (2002) 100(13):4609–14. doi: 10.1182/blood-2002-06-1683
122. Wiestner A, Rosenwald A, Barry TS, Wright G, Davis E, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood (2003) 101:4944–51. doi: 10.1182/blood-2002-10-3306
123. Hauser MA, Schaeuble K, Kindinger I, Impellizzieri D, Krueger WA, Hauck CR, et al. Inflammation-Induced CCR7 Oligomers Form Scaffolds to Integrate Distinct Signaling Pathways for Efficient Cell Migration. Immunity (2016) 44(1):59–72. doi: 10.1016/j.immuni.2015.12.010
124. Jones D, Benjamin RJ, Shahsafaei A, Dorfman DM. The chemokine receptor CXCR3 is expressed in a subset of B-cell lymphomas and is a marker of B-cell chronic lymphocytic leukemia. Blood (2000) 95(2):627–32. doi: 10.1182/blood.V95.2.627
125. Barretina J, Junca J, Llano A, Gutierrez A, Flores A, Blanco J, et al. CXCR4 and SDF-1 expression in B-cell chronic lymphocytic leukemia and stage of the disease. Ann Hematol (2003) 82(8):500–5. doi: 10.1007/s00277-003-0679-0
126. Bennett F, Rawstron A, Plummer M, de Tute R, Moreton P, Jack A, et al. B-cell chronic lymphocytic leukaemia cells show specific changes in membrane protein expression during different stages of cell cycle. Br J Haematol (2007) 139(4):600–4. doi: 10.1111/j.1365-2141.2007.06790.x
127. Okada T, Ngo VN, Ekland EH, Forster R, Lipp M, Littman DR, et al. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J Exp Med (2002) 196(1):65–75. doi: 10.1084/jem.20020201
128. Muller G, Lipp M. Shaping up adaptive immunity: the impact of CCR7 and CXCR5 on lymphocyte trafficking. Microcirculation (2003) 10(3-4):325–34. doi: 10.1080/mic.10.3-4.325.334
129. Hopken UE, Achtman AH, Kruger K, Lipp M. Distinct and overlapping roles of CXCR5 and CCR7 in B-1 cell homing and early immunity against bacterial pathogens. J Leukoc Biol (2004) 76(3):709–18. doi: 10.1189/jlb.1203643
130. Velaga S, Herbrand H, Friedrichsen M, Jiong T, Dorsch M, Hoffmann MW, et al. Chemokine receptor CXCR5 supports solitary intestinal lymphoid tissue formation, B cell homing, and induction of intestinal IgA responses. J Immunol (2009) 182(5):2610–9. doi: 10.4049/jimmunol.0801141
131. Wendland M, Willenzon S, Kocks J, Davalos-Misslitz AC, Hammerschmidt SI, Schumann K, et al. Lymph node T cell homeostasis relies on steady state homing of dendritic cells. Immunity (2011) 35(6):945–57. doi: 10.1016/j.immuni.2011.10.017
132. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature (2001) 410(6824):50–6. doi: 10.1038/35065016
133. Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell (1996) 87(6):1037–47. doi: 10.1016/S0092-8674(00)81798-5
134. Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med (1998) 187(4):655–60. doi: 10.1084/jem.187.4.655
135. Bai Z, Hayasaka H, Kobayashi M, Li W, Guo Z, Jang MH, et al. CXC chemokine ligand 12 promotes CCR7-dependent naive T cell trafficking to lymph nodes and Peyer’s patches. J Immunol (2009) 182(3):1287–95. doi: 10.4049/jimmunol.182.3.1287
136. Zabel BA, Lewén S, Berahovich RD, Jaén JC, Schall TJ. The novel chemokine receptor CXCR7 regulates trans-endothelial migration of cancer cells. Mol Cancer (2011) 10:73. doi: 10.1186/1476-4598-10-73
137. Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood (2005) 106(5):1824–30. doi: 10.1182/blood-2004-12-4918
138. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med (2005) 352(8):804–15. doi: 10.1056/NEJMra041720
139. Burger JA, Gandhi V. The lymphatic tissue microenvironments in chronic lymphocytic leukemia: in vitro models and the significance of CD40-CD154 interactions. Blood (2009) 114(12):2560–1; author reply 1-2. doi: 10.1182/blood-2009-06-228981
140. Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood (1998) 91(7):2387–96. doi: 10.1182/blood.V91.7.2387.2387_2387_2396
141. Collins RJ, Verschuer LA, Harmon BV, Prentice RL, Pope JH, Kerr JF. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br J Haematol (1989) 71(3):343–50. doi: 10.1111/j.1365-2141.1989.tb04290.x
142. Hopken UE, Rehm A. Homeostatic chemokines guide lymphoma cells to tumor growth-promoting niches within secondary lymphoid organs. J Mol Med (Berl) (2012) 90(11):1237–45. doi: 10.1007/s00109-012-0906-z
143. Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer (2010) 10(1):37–50. doi: 10.1038/nrc2764
144. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood (2009) 114(16):3367–75. doi: 10.1182/blood-2009-06-225326
145. Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood (2010) 115(22):4497–506. doi: 10.1182/blood-2009-07-233692
146. Maby-El Hajjami H, Ame-Thomas P, Pangault C, Tribut O, DeVos J, Jean R, et al. Functional alteration of the lymphoma stromal cell niche by the cytokine context: role of indoleamine-2,3 dioxygenase. Cancer Res (2009) 69(7):3228–37. doi: 10.1158/0008-5472.CAN-08-3000
147. Burger JA, Quiroga MP, Hartmann E, Burkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood (2009) 113(13):3050–8. doi: 10.1182/blood-2008-07-170415
148. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood (2000) 96(8):2655–63. doi: 10.1182/blood.V96.8.2655.h8002655_2655_2663
149. Burger JA, Zvaifler NJ, Tsukada N, Firestein GS, Kipps TJ. Fibroblast-like synoviocytes support B-cell pseudoemperipolesis via a stromal cell-derived factor-1- and CD106 (VCAM-1)-dependent mechanism. J Clin Invest (2001) 107(3):305–15. doi: 10.1172/JCI11092
150. Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood (2002) 100(5):1795–801. doi: 10.1182/blood.V100.5.1795.h81702001795_1795_1801
151. Ping L, Ding N, Shi Y, Feng L, Li J, Liu Y, et al. The Bruton’s tyrosine kinase inhibitor ibrutinib exerts immunomodulatory effects through regulation of tumor-infiltrating macrophages. Oncotarget (2017) 8(24):39218–29. doi: 10.18632/oncotarget.16836
152. Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood (2010) 115(13):2578–85. doi: 10.1182/blood-2009-08-236471
153. Burger JA, Kipps TJ. CXCR4: a key receptor in the crosstalk between tumor cells and their microenvironment. Blood (2006) 107(5):1761–7. doi: 10.1182/blood-2005-08-3182
154. Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia B cells are endowedwith the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol (2002) 32(5):1403–13. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y
155. Webster B, Ekland EH, Agle LM, Chyou S, Ruggieri R, Lu TT. Regulation of lymph node vascular growth by dendritic cells. J Exp Med (2006) 203(8):1903–13. doi: 10.1084/jem.20052272
156. Chyou S, Ekland EH, Carpenter AC, Tzeng TC, Tian S, Michaud M, et al. Fibroblast-type reticular stromal cells regulate the lymph node vasculature. J Immunol (2008) 181(6):3887–96. doi: 10.4049/jimmunol.181.6.3887
157. Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity (2005) 23(5):539–50. doi: 10.1016/j.immuni.2005.10.002
158. Peske JD, Thompson ED, Gemta L, Baylis RA, Fu YX, Engelhard VH. Effector lymphocyte-induced lymph node-like vasculature enables naive T-cell entry into tumours and enhanced anti-tumour immunity. Nat Commun (2015) 6:7114. doi: 10.1038/ncomms8114
159. Janovska P, Poppova L, Plevova K, Plesingerova H, Behal M, Kaucka M, et al. Autocrine Signaling by Wnt-5a Deregulates Chemotaxis of Leukemic Cells and Predicts Clinical Outcome in Chronic Lymphocytic Leukemia. Clin Cancer Res (2016) 22(2):459–69. doi: 10.1158/1078-0432.CCR-15-0154
160. Plešingerová H, Janovská P, Mishra A, Smyčková L, Poppová L, Libra A, et al. Expression of COBLL1 encoding novel ROR1 binding partner is robust predictor of survival in chronic lymphocytic leukemia. Haematologica (2018) 103(2):313–24. doi: 10.3324/haematol.2017.178699
161. Ansel KM, Ngo VN, Hyman PL, Luther SA, Forster R, Sedgwick JD, et al. A chemokine-driven positive feedback loop organizes lymphoid follicles. Nature (2000) 406(6793):309–14. doi: 10.1038/35018581
162. Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med (2000) 192(11):1545–52. doi: 10.1084/jem.192.11.1545
163. Bachelerie F, Ben-Baruch A, Burkhardt AM, Combadiere C, Farber JM, Graham GJ, et al. International Union of Basic and Clinical Pharmacology. [corrected]. LXXXIX. Update on the extended family of chemokine receptors and introducing a new nomenclature for atypical chemokine receptors. Pharmacol Rev (2014) 66(1):1–79. doi: 10.1124/pr.113.04er14a
164. Mazzotti C, Gagliostro V, Bosisio D, Del Prete A, Tiberio L, Thelen M, et al. The Atypical Receptor CCRL2 (C-C Chemokine Receptor-Like 2) Does Not Act As a Decoy Receptor in Endothelial Cells. Front Immunol (2017) 8:1233. doi: 10.3389/fimmu.2017.01233
165. De Henau O, Degroot G-N, Imbault V, Robert V, De Poorter C, McHeik S, et al. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PloS One (2016) 11(10):e0164179. doi: 10.1371/journal.pone.0164179
166. Malet-Engra G, Viaud J, Ysebaert L, Farce M, Lafouresse F, Laurent G, et al. CIP4 controls CCL19-driven cell steering and chemotaxis in chronic lymphocytic leukemia. Cancer Res (2013) 73(11):3412–24. doi: 10.1158/0008-5472.CAN-12-3564
167. Petlickovski A, Laurenti L, Li X, Marietti S, Chiusolo P, Sica S, et al. Sustained signaling through the B-cell receptor induces Mcl-1 and promotes survival of chronic lymphocytic leukemia B cells. Blood (2005) 105(12):4820–7. doi: 10.1182/blood-2004-07-2669
168. Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood (2007) 109(10):4424–31. doi: 10.1182/blood-2006-11-056648
169. Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F, Packham G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood (2003) 101(3):1087–93. doi: 10.1182/blood-2002-06-1822
170. Mami NB, Mohty M, Aurran-Schleinitz T, Olive D, Gaugler B. Blood dendritic cells in patients with chronic lymphocytic leukaemia. Immunobiology (2008) 213(6):493–8. doi: 10.1016/j.imbio.2007.11.009
171. Lee J-J, Foon KA, Mailliard RB, Muthuswamy R, Kalinski P. Type 1-polarized dendritic cells loaded with autologous tumor are a potent immunogen against chronic lymphocytic leukemia. J Leukoc Biol (2008) 84(1):319–25. doi: 10.1189/jlb.1107737
172. Gorgun G, Holderried TA, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest (2005) 115(7):1797–805. doi: 10.1172/JCI24176
173. Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest (2008) 118(7):2427–37. doi: 10.1172/JCI35017
174. Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endl E, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood (2005) 106(6):2018–25. doi: 10.1182/blood-2005-02-0642
175. Giannopoulos K, Schmitt M, Własiuk P, Chen J, Bojarska-Junak A, Kowal M, et al. The high frequency of T regulatory cells in patients with B-cell chronic lymphocytic leukemia is diminished through treatment with thalidomide. Leukemia (2008) 22(1):222–4. doi: 10.1038/sj.leu.2404869
176. Jak M, Mous R, Remmerswaal EB, Spijker R, Jaspers A, Yague A, et al. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk Lymphoma (2009) 50(5):788–801. doi: 10.1080/10428190902803677
177. D’Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res (2011) 35(3):363–8. doi: 10.1016/j.leukres.2010.08.010
178. Biancotto A, Dagur PK, Fuchs JC, Wiestner A, Bagwell CB, McCoy JP. Phenotypic complexity of T regulatory subsets in patients with B-chronic lymphocytic leukemia. Mod Pathol (2012) 25(2):246–59. doi: 10.1038/modpathol.2011.164
179. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124(5):750–60. doi: 10.1182/blood-2013-12-546416
180. Zirlik K. MDSCs: the final frontier of the microenvironment in CLL? Blood (2014) 124(5):666–8. doi: 10.1182/blood-2014-06-578880
181. Tang C-H, Chang S, Hashimoto A, Chen Y-J, Kang CW, Mato AR, et al. Secretory IgM Exacerbates Tumor Progression by Inducing Accumulations of MDSCs in Mice. Cancer Immunol Res (2018) 6(6):696–710. doi: 10.1158/2326-6066.CIR-17-0582
182. Zahran AM, Moeen SM, Thabet AF, Rayan A, Abdel-Rahim MH, Mohamed WMY, et al. Monocytic myeloid-derived suppressor cells in chronic lymphocytic leukemia patients: a single center experience. Leuk Lymphoma (2020) 61(7):1645–52. doi: 10.1080/10428194.2020.1728747
183. Schneider MA, Meingassner JG, Lipp M, Moore HD, Rot A. CCR7 is required for the in vivo function of CD4+ CD25+ regulatory T cells. J Exp Med (2007) 204(4):735–45. doi: 10.1084/jem.20061405
184. Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (2010) 328(5979):749–52. doi: 10.1126/science.1185837
185. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13(15 Pt 1):4345–54. doi: 10.1158/1078-0432.CCR-07-0472
186. Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol (2007) 178(1):320–9. doi: 10.4049/jimmunol.178.1.320
187. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med (2014) 211(1):121–36. doi: 10.1084/jem.20131142
188. Beyer M, Schultze JL. Regulatory T cells in cancer. Blood (2006) 108(3):804–11. doi: 10.1182/blood-2006-02-002774
189. Taylor NA, Vick SC, Iglesia MD, Brickey WJ, Midkiff BR, McKinnon KP, et al. Treg depletion potentiates checkpoint inhibition in claudin-low breast cancer. J Clin Invest (2017) 127(9):3472–83. doi: 10.1172/JCI90499
190. Ni X, Jorgensen JL, Goswami M, Challagundla P, Decker WK, Kim YH, et al. Reduction of regulatory T cells by Mogamulizumab, a defucosylated anti-CC chemokine receptor 4 antibody, in patients with aggressive/refractory mycosis fungoides and Sezary syndrome. Clin Cancer Res (2015) 21(2):274–85. doi: 10.1158/1078-0432.CCR-14-0830
191. Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci USA (2013) 110(44):17945–50. doi: 10.1073/pnas.1316796110
192. Pham TH, Okada T, Matloubian M, Lo CG, Cyster JG. S1P1 receptor signaling overrides retention mediated by G alpha i-coupled receptors to promote T cell egress. Immunity (2008) 28(1):122–33. doi: 10.1016/j.immuni.2007.11.017
193. Shannon LA, McBurney TM, Wells MA, Roth ME, Calloway PA, Bill CA, et al. CCR7/CCL19 controls expression of EDG-1 in T cells. J Biol Chem (2012) 287(15):11656–64. doi: 10.1074/jbc.M111.310045
194. Jain N. Selecting Frontline Therapy for CLL in 2018. Hematol Am Soc Hematol Educ Program (2018) 2018(1):242–7. doi: 10.1182/asheducation-2018.1.242
195. Jaeger K, Bruenle S, Weinert T, Guba W, Muehle J, Miyazaki T, et al. Structural Basis for Allosteric Ligand Recognition in the Human CC Chemokine Receptor 7. Cell (2019) 178(5):1222–30.e10. doi: 10.1016/j.cell.2019.07.028
196. Hutchings CJ, Koglin M, Marshall FH. Therapeutic antibodies directed at G protein-coupled receptors. MAbs (2010) 2(6):594–606. doi: 10.4161/mabs.2.6.13420
197. Hutchings CJ, Koglin M, Olson WC, Marshall FH. Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat Rev Drug Discovery (2017) 16(9):1–24. doi: 10.1038/nrd.2017.173
198. Chiavenna SM, Jaworski JP, Vendrell A. State of the art in anti-cancer mAbs. J BioMed Sci (2017) 24(1):15. doi: 10.1186/s12929-016-0311-y
199. Jarboe J, Gupta A, Saif W. Therapeutic human monoclonal antibodies against cancer. Methods Mol Biol (2014) 1060:61–77. doi: 10.1007/978-1-62703-586-6_4
200. Yamamoto K, Utsunomiya A, Tobinai K, Tsukasaki K, Uike N, Uozumi K, et al. Phase I study of KW-0761, a defucosylated humanized anti-CCR4 antibody, in relapsed patients with adult T-cell leukemia-lymphoma and peripheral T-cell lymphoma. J Clin Oncol (2010) 28(9):1591–8. doi: 10.1200/JCO.2009.25.3575
201. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol (2015) 27(1):11–20. doi: 10.1093/intimm/dxu079
202. Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol (2015) 6:12. doi: 10.3389/fimmu.2015.00012
203. Cuesta C, Munoz-Callega C, Loscertales J, Terron F, Mol W. CAP-100: First-in-class antibody for CCR7+ hematological malignancies. J Clin Oncol (2019) 37(15_suppl):e19008–e. doi: 10.1200/JCO.2019.37.15_suppl.e19008
204. Cuesta-Mateos C, Muñoz-Calleja C, Loscertales J, Terron F, Mol W. Abstract 4849: CAP-100: first-in-class anti-CCR7 antibody for CLL. Cancer Res (2019) 79(13 Supplement):4849. doi: 10.1158/1538-7445.AM2019-4849
205. Fiorcari S, Brown WS, McIntyre BW, Estrov Z, Maffei R, O’Brien S, et al. The PI3-Kinase Delta Inhibitor Idelalisib (GS-1101) Targets Integrin-Mediated Adhesion of Chronic Lymphocytic Leukemia (CLL) Cell to Endothelial and Marrow Stromal Cells. PloS One (2013) 8(12):e83830. doi: 10.1371/journal.pone.0083830
206. Elías EE, Almejún MB, Colado A, Cordini G, Vergara-Rubio M, Podaza E, et al. Autologous T-cell activation fosters ABT-199 resistance in chronic lymphocytic leukemia: rationale for a combined therapy with SYK inhibitors and anti-CD20 monoclonal antibodies. Haematologica (2018) 103(10):e458–e61. doi: 10.3324/haematol.2018.188680
207. Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol (2012) 31(1):88–94. doi: 10.1200/JCO.2012.42.7906
208. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med (2013) 369(1):32–42. doi: 10.1056/NEJMoa1215637
209. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med (2014) 370(11):997–1007. doi: 10.1056/NEJMoa1315226
210. Vassileva G, Soto H, Zlotnik A, Nakano H, Kakiuchi T, Hedrick JA, et al. The reduced expression of 6Ckine in the plt mouse results from the deletion of one of two 6Ckine genes. J Exp Med (1999) 190(8):1183–8. doi: 10.1084/jem.190.8.1183
211. Luther SA, Tang HL, Hyman PL, Farr AG, Cyster JG. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc Natl Acad Sci U S A (2000) 97(23):12694–9. doi: 10.1073/pnas.97.23.12694
212. Moschovakis GL, Bubke A, Dittrich-Breiholz O, Braun A, Prinz I, Kremmer E, et al. Deficient CCR7 signaling promotes TH2 polarization and B-cell activation in vivo. Eur J Immunol (2012) 42(1):48–57. doi: 10.1002/eji.201141753
213. Junt T, Scandella E, Forster R, Krebs P, Krautwald S, Lipp M, et al. Impact of CCR7 on priming and distribution of antiviral effector and memory CTL. J Immunol (2004) 173(11):6684–93. doi: 10.4049/jimmunol.173.11.6684
214. Junt T, Nakano H, Dumrese T, Kakiuchi T, Odermatt B, Zinkernagel RM, et al. Antiviral Immune Responses in the Absence of Organized Lymphoid T Cell Zones in plt/plt Mice. J Immunol (2002) 168(12):6032–40. doi: 10.4049/jimmunol.168.12.6032
215. Kursar M, Hopken UE, Koch M, Kohler A, Lipp M, Kaufmann SH, et al. Differential requirements for the chemokine receptor CCR7 in T cell activation during Listeria monocytogenes infection. J Exp Med (2005) 201(9):1447–57. doi: 10.1084/jem.20041204
216. Scandella E, Fink K, Junt T, Senn BM, Lattmann E, Forster R, et al. Dendritic cell-independent B cell activation during acute virus infection: a role for early CCR7-driven B-T helper cell collaboration. J Immunol (2007) 178(3):1468–76. doi: 10.4049/jimmunol.178.3.1468
217. Hintzen G, Ohl L, del Rio ML, Rodriguez-Barbosa JI, Pabst O, Kocks JR, et al. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J Immunol (2006) 177(10):7346–54. doi: 10.4049/jimmunol.177.10.7346
218. Worbs T, Bode U, Yan S, Hoffmann MW, Hintzen G, Bernhardt G, et al. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J Exp Med (2006) 203(3):519–27. doi: 10.1084/jem.20052016
219. Davalos-Misslitz AC, Rieckenberg J, Willenzon S, Worbs T, Kremmer E, Bernhardt G, et al. Generalized multi-organ autoimmunity in CCR7-deficient mice. Eur J Immunol (2007) 37(3):613–22. doi: 10.1002/eji.200636656
220. Kurobe H, Liu C, Ueno T, Saito F, Ohigashi I, Seach N, et al. CCR7-dependent cortex-to-medulla migration of positively selected thymocytes is essential for establishing central tolerance. Immunity (2006) 24(2):165–77. doi: 10.1016/j.immuni.2005.12.011
221. Hopken UE, Wengner AM, Loddenkemper C, Stein H, Heimesaat MM, Rehm A, et al. CCR7 deficiency causes ectopic lymphoid neogenesis and disturbed mucosal tissue integrity. Blood (2007) 109(3):886–95. doi: 10.1182/blood-2006-03-013532
222. Vander Lugt B, Tubo NJ, Nizza ST, Boes M, Malissen B, Fuhlbrigge RC, et al. CCR7 plays no appreciable role in trafficking of central memory CD4 T cells to lymph nodes. J Immunol (2013) 191(6):3119–27. doi: 10.4049/jimmunol.1200938
223. Moschovakis GL, Bubke A, Friedrichsen M, Ristenpart J, Back JW, Falk CS, et al. The chemokine receptor CCR7 is a promising target for rheumatoid arthritis therapy. Cell Mol Immunol (2019) 16(10):791–9. doi: 10.1038/s41423-018-0056-5
224. Cuesta-Mateos C, Portero-Sainz I, García-Peydró M, Alcain J, Fuentes P, Juárez-Sánchez R, et al. Evaluation of therapeutic targeting of CCR7 in acute graft-versus-host disease. Bone Marrow Transplant (2020) 55(10):1935–45. doi: 10.1038/s41409-020-0830-8
225. Soudja SM, Henri S, Mello M, Chasson L, Mas A, Wehbe M, et al. Disrupted lymph node and splenic stroma in mice with induced inflammatory melanomas is associated with impaired recruitment of T and dendritic cells. PloS One (2011) 6(7):e22639. doi: 10.1371/journal.pone.0022639
226. Brown JR. Phosphatidylinositol 3 Kinase δ Inhibitors: Present and Future. Cancer J (Sudbury Mass) (2019) 25(6):394–400. doi: 10.1097/PPO.0000000000000414
227. Fiegl M, Stauder R, Steurer M, Mian M, Hopfinger G, Brychtova Y, et al. Alemtuzumab in chronic lymphocytic leukemia: final results of a large observational multicenter study in mostly pretreated patients. Ann Hematol (2014) 93(2):267–77. doi: 10.1007/s00277-013-1966-z
228. Lewis DJ, Rook AH. Mogamulizumab in the treatment of advanced mycosis fungoides and Sézary syndrome: safety and efficacy. Expert Rev Anticancer Ther (2020) 20(6):447–52. doi: 10.1080/14737140.2020.1760096
Keywords: CCR7, CLL (chronic lymphocytic leukemia), lymph node, pathophysiology, immunotherapy
Citation: Cuesta-Mateos C, Brown JR, Terrón F and Muñoz-Calleja C (2021) Of Lymph Nodes and CLL Cells: Deciphering the Role of CCR7 in the Pathogenesis of CLL and Understanding Its Potential as Therapeutic Target. Front. Immunol. 12:662866. doi: 10.3389/fimmu.2021.662866
Received: 01 February 2021; Accepted: 09 March 2021;
Published: 24 March 2021.
Edited by:
Nurit Hollander, Tel Aviv University, IsraelReviewed by:
Laura Patrussi, University of Siena, ItalyDaniel F. Legler, Biotechnology Institute Thurgau, Switzerland
Copyright © 2021 Cuesta-Mateos, Brown, Terrón and Muñoz-Calleja. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Cuesta-Mateos, carlos.cuesta@salud.madrid.org