 Zachary Lamplugh
Zachary Lamplugh Yi Fan
Yi Fan- Department of Radiation Oncology, University of Pennsylvania, Philadelphia, PA, United States
Immunotherapy holds great promise for treating cancer. Nonetheless, T cell-based immunotherapy of solid tumors has remained challenging, largely due to the lack of universal tumor-specific antigens and an immunosuppressive tumor microenvironment (TME) that inhibits lymphocyte infiltration and activation. Aberrant vascularity characterizes malignant solid tumors, which fuels the formation of an immune-hostile microenvironment and induces tumor resistance to immunotherapy, emerging as a crucial target for adjuvant treatment in cancer immunotherapy. In this review, we discuss the molecular and cellular basis of vascular microenvironment-mediated tumor evasion of immune responses and resistance to immunotherapy, with a focus on vessel abnormality, dysfunctional adhesion, immunosuppressive niche, and microenvironmental stress in tumor vasculature. We provide an overview of opportunities and challenges related to these mechanisms. We also propose genetic programming of tumor endothelial cells as an alternative approach to recondition the vascular microenvironment and to overcome tumor resistance to immunotherapy.
Introduction
Tumor vasculature has presented a complex problem to achieving therapeutic success across all cancer treatment modalities. Solid tumors exhibit aberrant vasculature composed of tumor endothelial cells (ECs) that present a physical barrier to treatment as well as promote aggressive tumor phenotypes that are prone to become aggressive and metastatic. The abnormal blood circulation leads to insufficient perfusion and drug delivery and creates pockets of hypoxia, which enhances tumor heterogeneity and promotes resistance to cancer therapies such as chemotherapy, radiation, and immunotherapy. Growing evidence shows that tumor ECs drive immunosuppression through selective immune cell recruitment, metabolic competition and epigenetic remodeling, suggesting the vascular microenvironment as an emerging target for solid tumor immunotherapy. Of various blood vessel-targeting treatments, anti-angiogenic therapy that primarily blocks vascular endothelial growth factor (VEGF) pathway has been widely pursued to inhibit tumor vascularization to starve cancer of oxygen and nutrients or to normalize tumor vasculature to overcome hypoxia-mediated treatment resistance. Combination of anti-angiogenic or other next-generation vasculature-targeting therapies with immunotherapy may, therefore, offer exciting opportunities for treating solid tumors. In this review we look to highlight the challenges implicated in immunotherapy for solid tumors, the landscape of the vascular microenvironment, and examine how these unique obstacles presented by tumor ECs impact current treatment options.
T Cell-Based Immunotherapy of Solid Tumors
Current Approaches
The promise of immunotherapy as an approach to solid tumors offers a potentially significant upgrade over traditional treatments such as chemotherapy, radiation, and molecular targeted therapy. While the therapeutic windows of chemotherapy and radiation are limited to when the drug is being applied or is present inside the body, immunotherapy has the capacity to remain active for an extended period once the immune system has been modified to target cancer cells. Immune checkpoint blockade and chimeric antigen receptor T cell (CAR-T) therapy remain the two dominant approaches for current T cell-based immunotherapy. Immune checkpoint blockade sought to remove the “brakes” or inhibitory checkpoints that prevented our immune systems from reaching their full potential as a natural defense against cancer. Monoclonal antibodies targeting immune cell expressed cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed-death 1 (PD-1) along with TME expressed programmed death-ligand 1 (PD-L1) have led to approved treatment options for solid tumors such as melanoma, non-small cell lung cancer (NSCLC), and urothelial carcinoma (1). Although approved treatments exist, many factors play a role in whether immune checkpoint blockade will be successful such as age, sex, gene expression, mutation burden, epigenetic and metabolic alternation in tumor cells, and stage of disease leaving a lot to be desired in effectiveness of the treatment (2). The notion of exploiting antigen fragment recognition of T cells by introducing genetic material that encodes for antibody recognition was first proposed by Gross et al. in 1989 (3). After development of several generations of CAR constructs, the US Food and Drug Administration (FDA) has recently approved drugs developed using this method such as Novartis’s Kymriah and Kite Pharma’s Yescarta that target tumor-associated antigens (TAAs) in hematopoietic malignancies (4–6). As of now there are no FDA approved CAR-T cell treatments available for solid tumors as there are many challenges unique to solid tumors that make CAR-T cells challenging to implement.
Therapeutic Challenges
The first major challenge that exist for implementing CAR-T cell therapy for solid tumors is the absence of universal tumor-specific antigens (TSAs) or TAAs in the cancer of interest. Ideally the antigen should be expressed selectively on tumor cells or at least highly expressed on tumor cells while being minimally expressed on healthy tissues. The majority of current CAR-T cell target antigens exist on healthy tissues leading to “on-target/off-tumor” toxicity (7, 8). A second important characteristic of the antigen is that it should be expressed on all cancerous cells. If any cells within the tumor do not express the target antigen, then the chances of a treatment having a meaningful impact decrease significantly. Even if the target antigen is present on all of the tumor cells, loss of antigenicity can still occur through immune escape mechanisms such as loss of antigen presentation or mutational gains within the antigen itself (9). In addition to antigen presentation, T-cell infiltration is vital for therapeutic success. It is known that solid tumors have the capacity to limit T-cell infiltration through secretion of immunosuppressive cytokines, disruption of T-cell homing, and promoting abnormal vasculature (10). T-cells must also combat with the progressive loss of T cell effector functions known as T cell exhaustion. This is due primarily to the constant presence of antigen and inflammation, which leads to an upregulation of immunosuppressive receptors such as CTLA-4 and PD-1 (11). Immune checkpoint blockade can help alleviate immunosuppression that occurs during exhaustion, but the underlying mechanisms for exhaustion are still not fully understood making it a challenge to reinvigorate T-cells from this state.
Tumor Microenvironment and Immunotherapy
Reprogramming the TME is seen as a viable option to overcome the challenges that exist for immunotherapy in solid tumors. Normalizing tumor vasculature has the potential to reverse immune evasion and increase the infiltration of immune cells. Normalizing vasculature can be done using antiangiogenic agents (12–14). It has also been hypothesized that this could be achieved through the alternative approach of vascular detransformation, a method that would look to reverse endothelial-mesenchymal transition (Endo-MT) that occurs within the TME (15). These transformed endothelial cells have been shown in glioma to acquire a mesenchymal phenotype that leads to vessel sprouting and outgrowth making them a novel target (16, 17). Additionally, myeloid cells, specifically tumor-associated macrophages (TAMs), are also a prime target to reprogram the TME due to their ability promote angiogenesis, metastasis, and immunosuppression across various cancer types (12, 18). TAMs can be targeted using selective inhibitors of colony-stimulating factor 1 receptor (CSF1R) and PI3K kinase, neutralizing antibodies of CSF-1, Toll-like receptor (TLR) agonists, and DNA binding agents that selectively induce cell cycle arrest in monocytes and macrophages (18–21). Another group of immunosuppressive cells within the TME, known as regulatory T-cells (Treg), have been shown to be recruited by chemokines produced by tumor cells, cancer-associated fibroblasts, and an immunosuppressive subset of TAMs (22, 23). Tregs are normally required to balance T-cell effector function, however they are recruited to suppress the antitumor immune response and promote tumor progression within the TME (12, 24). Outright Treg ablation is not seen as a viable option to improve immunotherapy as the absence of Tregs would increase the occurrence of adverse events due to overactive T cell effector function (25). The reprograming of Tregs could be achieved through metabolic manipulation via protein kinase B (AKT) and through downregulation of transcription factors such as Helios, NF-kB, Eos, Bach2, and Nr4a (26). It has been found that once Tregs have been reprogrammed they will begin to exhibit a decrease in Foxp3 expression, gain immune-stimulating function, and develop a T helper cell phenotype (26).
Vascular Microenvironment and Tumor Immunity
Mechanisms Underlying Vascular Regulation of Tumor Immunity
Aberrant vascularity characterizes malignant solid tumors, fueling the formation of an immune-hostile TME and inducing tumor resistance to immunotherapy, which involves multifactorial mechanisms: tumor vasculature becomes structurally and topologically abnormal, hampering the infiltration and therapeutic delivery of T cells into the tumors; expression of adhesion proteins including ICAM-1 and VCAM-1 are down-regulated in tumor-associated ECs, impeding T cell adhesion to and diapedesis through the tumor vessel wall; tumor ECs form a vascular niche and produce multiple growth factors and cytokines that induce immunosuppressive phenotypes in TAMs and inhibit survival and activation in T cells; tumor capillaries with functional abnormalities generate a vascular microenvironment with locoregional stresses including heterogeneous hypoxia and nutrient deprivation, reducing T cell activity and inducing T cell exhaustion. All of these mechanisms facilitate pro-tumor immunity and collectively lead to tumor evasion of immune responses and resistance to immunotherapies (Figure 1).
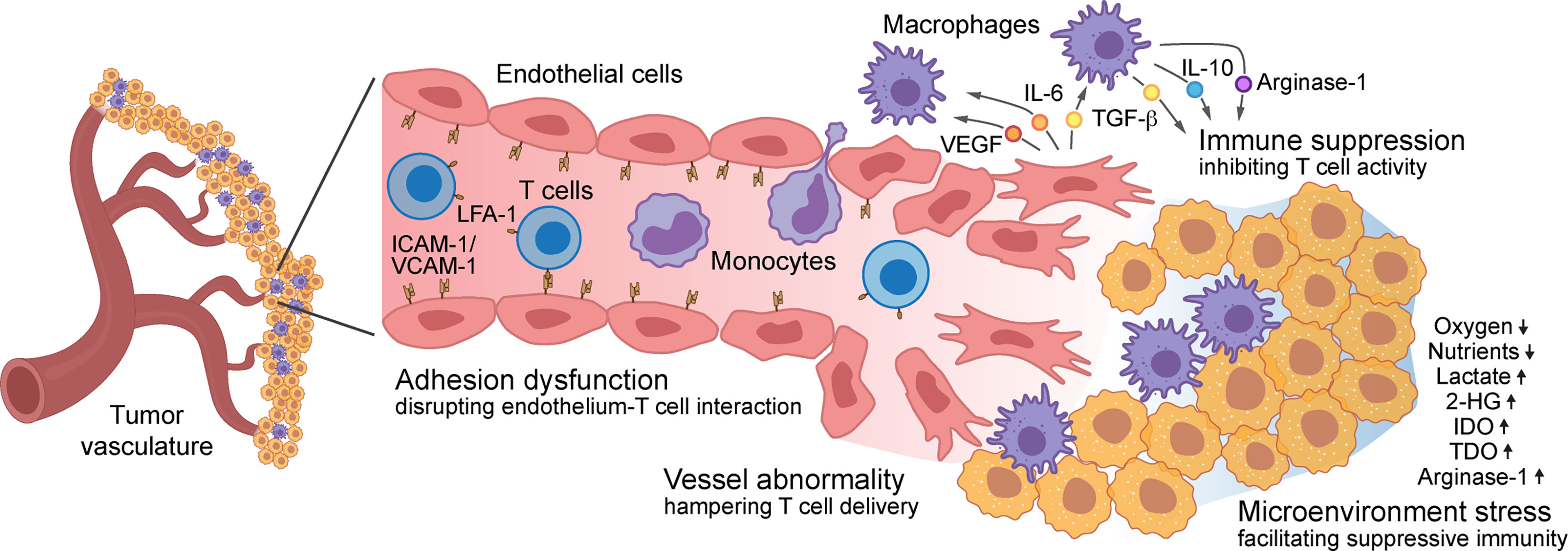
Figure 1 Role of vascular microenvironment in regulation of tumor immunity – Vascular microenvironment regulates T cell-based tumor immunity via multifactorial mechanisms: structurally and topologically abnormal vasculature hampers T cell infiltration; downregulated expression of adhesion molecules, such as ICAM-1 and VCAM-1 in tumor-associated endothelial cells (ECs), impedes T cell adhesion to and diapedesis through the tumor vessel wall; tumor ECs produce multiple cytokines that induce immunosuppressive phenotypes in macrophages and inhibit T cell survival and activation; the vascular microenvironment generates locoregional stresses including hypoxia, nutrient deprivation, and excessive immunosuppressive metabolites and enzyme, reducing T cell activity and inducing T cell exhaustion.
Vessel Abnormality
ECs have specialized roles in regulating many essential functions in the body that differs from tissue to tissue (27). However, a constant remains where endothelial cells throughout the body mediate the flow of blood, regulate the supply of nutrients, oxygen, and other solutes, and manage the migration of immune cells into and out of tissues. Vasculature begins to change when in the presence of a tumor, where these tumor ECs exhibit a defective endothelial monolayer, large intracellular openings and holes and abnormal sprouts (28, 29). The changes that occur within the vasculature could be initially driven by chronic tumor-expressed vascular endothelial growth factor (VEGF) stimulation (30), followed by EC plasticity-mediated permanent phenotypic changes (15). In addition, pericytes are vital cells that play an important role in vessel maintenance and remodeling; however, in tumor vasculature they are either absent or exhibit abnormal function leaving vessels to become leaky. The leakiness of the tumor vasculature results in irregular blood flow and ultimately dysfunctional trafficking of lymphocytes (31). These structural changes lead to vessel abnormalities and inadequate perfusion resulting in insufficient delivery of therapeutic agents and lymphocytes.
Microenvironmental Stress
Structural and topological vessel aberrancy causes regions of hypoxia and nutrient depletion within the tumors (29). Due to cancer’s intensification of metabolic demands, there is less blood oxygenation which means the vascular density does not need to be lower than normal tissue for hypoxia to occur (32). Hypoxia inducible factor 1-alpha (HIF1α), the master regulator of hypoxia, further drives upregulation of VEGF which has been shown to enhance tumor survivability through activation of the MAPK/ERK pathway (33). Additionally, as cancer consumes glucose within the TME, it secretes lactate creating an acidic environment that aids in the stabilization of HIF-1α mRNA (34). The lactate produced by the cancer cells further drives angiogenesis in tumor ECs via the lactate importer monocarboxylate transporter 1 (MCT1) which activates NF-κB and HIF-1α (34). Further crosstalk between tumor cells and ECs can influence the ECs to adopt a prothrombotic, proinflammatory, and cell-adhesive state known as EC dysfunctional activation (35). These “activated” ECs can promote aggressive tumor cell phenotypes that are drug resistant and have an increased risk of metastasizing increasing the difficulty of implementing a wide range of treatments (36, 37), and also facilitate pro-tumor immunity.
Immunosuppressive Niche
Tumor ECs produce a wide range of angiocrine factors that can promote angiogenesis, tumorigenesis, chemoresistance, and immune suppression (38–45). One such angiocrine, interleukin-6 (IL-6), has been shown to promote alternative TAM polarization via induction of HIF-2α in glioblastoma (45), leading to TAM secretion of immunosuppressive molecules, such as TGF-β, IL-10, and Argainse-1, that inhibit T cell activation. Tumor ECs also recruit TAMs through the production of CCL2, M-CSF, and VEGF which upon entry into the TME promote angiogenesis, immune suppression, and metastasis (28, 46–48). This mechanism demonstrates how tumor ECs can promote tumor progression through immunosuppression, thus further highlighting the importance of cross talk between different cells within the TME.
Adhesion Dysfunction
Adding on to the difficulties of treating solid tumors is the interaction or lack thereof, that takes place between tumor ECs and lymphocytes. Not only can tumor ECs activate or inactivate the immune cells within the TME, but they may selectively recruit immune cells to promote tumorigenesis and immune evasion. Interactions between leukocytes and ECs are mediated by expression of adhesion molecules on the cell surface of ECs, which mainly include intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), E-selectin, P-selectin, platelet–endothelial-cell adhesion molecule-1 (PECAM-1, CD31), and CD99 (49, 50). Infiltration of immune cells into the tumor is a multistep event subjected to temporospatial regulation of lymphocyte adhesion to, rolling at, and transmigration across the tumor ECs. Exposure of ECs to proinflammatory stimulus with LPS, TNF-α, or IL-1β upregulates ICAM-1 and VCAM-1 expression, as a natural process for lymphocyte recruitment and infiltration (50, 51); however, dysfunctional expression of adhesion molecules, such as ICAM-1, reduces T cell adhesion to tumor endothelium, likely due to microenvironmental cues that reprogrammed ECs, making it harder to mount an anti-tumor immune response (47, 52, 53). Adhesion molecules linked to leukocyte binding are suppressed by angiogenic factors leading tumor ECs to adopt a state known as endothelial anergy where there is a lack of an immune response to the presence of proinflammatory stimuli such as IL-1, TNF-α, and IFNγ (47, 53–55). Additionally, tumor cells upregulate the ligand enothelin-1 in ovarian cancer, which in turn binds to endothelin B receptor on tumor ECs, resulting in the inhibition of ICAM1 expression thus preventing lymphocyte infiltration (56). Alongside this unresponsive state, tumor ECs themselves have shown to downregulate adhesion molecules and chemokines. The tumor vascular microenvironment creates problems for all solid tumor treatment options, and it’s important to understand the underlying mechanisms in order to remodel the TME to increase the efficacy of therapeutics.
Immune Cell Infiltration
It is well documented that the presence of preexisting tumor-infiltrating lymphocytes (TILs), which include T cells, B cells, and natural killer (NK) cells, usually indicates a positive response to treatment (57, 58). Tumors can combat TILs by disrupting lymphocyte homing and infiltration. Trafficking of lymphocytes is regulated through chemokines and adhesion molecules. Chemokines mediate chemotaxis through gradients and initiate the active form of ligands expressed on the surface of lymphocytes allowing them to transmigrate into the tissue. Tumors have been shown to hinder lymphocyte chemotaxis by suppressing expression of chemokines that promote T-cell infiltration such as CCL4 and CCL27 in melanoma (10, 59). Lymphocyte homing and infiltration can also be altered by posttranslational nitrosylation of CCL2 to nullify its ability to recruit tumor specific cytotoxic T lymphocytes (CTLs) making it hard to implement immunotherapy using antigen specific T cells (60). Aberrant vascularization presents a significant hindrance to lymphocyte infiltration, which inhibits T cell adhesion to tumor vasculature and hampers T cell delivery into the tumor. VEGF, a pro-angiogenic factor driving tumor angiogenesis and aberrant vascularity, likewise has also been shown to downregulate the expression of ligands required for T cell extravasation (10, 31).
Immune Cell Function
The success of immunotherapies is not only dependent on the trafficking of lymphocytes, but also their function. T cells and NK cells require activation in order to produce their cytotoxic proteins, which tumors can evade through gene regulation and immune checkpoints. T cells require activation via their T cell receptor (TCR) that recognizes MHC class I molecules, conversely NK cells are inhibited by the presence of MHC class 1 molecules. MHC class I presentation is not enough to fully activate T cells; they must also receive signaling from adhesion and costimulatory molecules. It is common for tumor cells to lack these necessary components and instead express inhibitory ligands such as PD-L1 that will shut down T cell effector function (61, 62). Additionally, NK cells can be influenced within the TME to adopt a pro-angiogenic phenotype to promote tumor progression in non-small cell lung carcinomas (NSCLC) patients (63). Tumors also have the capability to downregulate MHC class I presentation, however, if the tumor cell is completely negative for MHC class I molecules then it will become susceptible to NK cells (64). In order to evade NK cells, cancer will maintain low expression of MHC class I molecules to prevent NK cells from initiating cell death, while simultaneously reducing the activation of T cells (65). In order for T cell and NK cell persistence to occur they must continually be exposed to stimulating cytokines such as IL-2 (62, 66). IL-2 has been used in clinical trials to help stimulate the anti-tumor response, but it is associated with dangerous toxicities and expansion of Treg cells (62).
The components of the TME, including hypoxia, immunosuppressive cytokines and matrix, stroma cells, and anti-inflammatory leukocytes, propel T cells to adopt a less effective state known as exhaustion. Exhausted T cells exhibit a decrease in effector function and an increase in expression of inhibitory receptors such as PD-1 and CTLA-4 (67). Chronic exposure to tumor antigen combined with the presence of inhibitory ligands is thought to be the driving forces behind T cell exhaustion (68). Additionally, expression of TGF-β by tumor cells, fibroblasts, immune cells and tumor ECs quells the expression of cytotoxic T cell genes including perforin, granzymes and cytotoxins (69). The presence of other immune cells within the TME such as TAMs and myeloid-derived suppressor cells (MDSCs) negatively impact anti-tumor immune response. Macrophages play an important role in regulating immune response by adopting two different phenotypes: the pro-inflammatory M1 phenotype and the anti-inflammatory and immunosuppressive M2 phenotype. Upon entering the TME, most TAMs are polarized towards the M2 phenotype through signaling from tumor cells, T helper 2 (TH2) cells, Treg cells, and other cells or molecules in the TME (70). These M2 TAMs promote immunosuppression through expression of TGF-β and IL-10 thus being negatively correlated with therapeutic response (71, 72). Even though TAMs primarily adopt the M2 phenotype, polarization of macrophages is all about a balance of signals with their phenotype easily being swayed by changes in signaling opening the potential for macrophage reeducation. Immunosuppressive TAMs express PD-L1, the negative regulator of T and NK cells, where blocking of PD-L1 unlocks TAMs’ potential for anti-tumor activity, suggesting that immune checkpoint blockade could be a possible avenue for macrophage reeducation (70). Not only are TAMs immunosuppressive but they also stimulate angiogenesis through the production of pro-angiogenic factors such as VEGF, epidermal growth factor (EGF), basic fibroblast growth factor 2 (FGF2), IL-8, CXCL12, and TNFα further echoing the need for anti-TAM therapies (62). Similarly, MDSCs induce tumor immunosuppression with several additional mechanisms, such as the expression of PD-L1 and CD-80 to abrogate antigen-specific immune responses, and production of reactive oxygen species (ROS) which induces posttranslational modification of TCRs rendering them unresponsive to antigen presentation (73). MDSCs have also been implicated in promoting angiogenesis through increased production of fatty acid synthase which in turn activates PPARβ/δ-dependent expression of genes including VEGF (63). Pro-inflammatory neutrophils within the TME, whose primary role is to be the first line of defense against infection, have been negatively correlated with clinical outcome (63). These cells hold significant influence over tumor ECs through the wide range of secretory factors including IL-1β, VEGF, FGF2, TGFα, hepatocyte growth factor (HGF), and angiopoietin 1 (ANG1) (63).
ECs play a significant role in immune regulation. Tumor ECs may modify the expression of adhesion molecules to recruit specific types of immune cells, leading to favored recruitment of macrophages, Treg cells, and MDSCs while inhibiting CD4+ and CD8+ T cells, NK cells, dendritic cells (DCs), and neutrophils (74). Tumor ECs lack the necessary stimulatory components needed to fully activate naïve T cells, and downregulate MHC associated genes to aid in immune evasion, as well as express inhibitory ligands such as PD-1 to further suppress T cell effector function (75). Additionally, tumor ECs can selectively induce T cell apoptosis through expression of FasL allowing them to eliminate CD8+ T cells while sparing Treg cells (76). Crosstalk between tumor ECs and MDSCs creates a positive feedback loop involving VEGF: VEGF stimulates MDSC recruitment, while recruited MDSCs promote angiogenesis and immune suppression (77). Similarly, a feedback loop exists between TAMs and ECs within the hypoxic TME where tumor ECs express common lymphatic endothelial and vascular endothelial receptor (CLEVER1), which is an adhesion molecule that selectively recruits immunosuppressive cells (78).
Immune Cell Metabolism
The TME is a battleground for metabolic resources with tumor cells, effector immune cells and immunosuppressive cells vying for limited nutrients essential for survival. Cancer cells increased metabolic demands combined with aberrant vasculature create an environment that is deficient of nutrients and hypoxic, greatly influencing what metabolic pathways cells within the TME can utilize (79). T cells switch from the mainly oxidative and fatty acid metabolism of naive and resting T cells to increased glucose uptake and glycolysis during activation. Metabolism of glucose is essential for CD4+ and CD8+ effector T cells; having received costimulatory signals these cells immediately begin to upregulate genes associated with glycolysis and the tricarboxylic acid cycle (TCA) to meet increased metabolic demands for proliferation (80). Additionally, the pentose phosphate pathway (PPP) is upregulated and provides the necessary NADPH required for fatty acid and plasma membrane synthesis (81). Nuclear factor of activated T cells (NFAT), which induces the vital stimulatory cytokine IL-2, requires ROS to be present in order for expression to occur making it strongly dependent upon the type of metabolism pathway utilized (79). Glucose deficiency in the TME causes the vital intermediate phosphoenolpyruvate, involved in NFAT expression, to become expressed thus losing IL-2 stimulation and T cell persistence (82). Mitochondrial respiration is essential for T cell effector function and consequently mitochondrial dysfunction has been identified as negatively correlated to treatment response when using CAR-T cells (83). It’s thought that a buildup of mitochondrial ROS levels causes mitochondrial dysfunction and has strongly been associated with the T cell exhaustion phenotype (79). Competition within the TME for crucial amino acids impacts the quality of the anti-tumor T cell response. Tumor cells, MDSCs, TAMs, and cancer associated fibroblasts (CAFs) actively deplete tryptophan through upregulation of indoleamine 2,3-dioxygenase (IDO), which its metabolite kynurenine has been shown to upregulate PD1 expression on CD8+ T cells (84, 85). Depletion of tryptophan is negatively correlated with patient outcome and further demonstrates the importance of metabolism to obtain a therapeutic immune response (86).
The suppressed blood circulation that exists in the vascular TME leads to locoregional accumulation of immune-hostile metabolites and enzymes including lactate, 2-hydroxyglutarate (2-HG), arginase-1, indoleamine 2,3-dioxygenase (IDO), and tryptophan 2,3-dioxygenase (TDO). For example, lactate can reach immunosuppressive concentrations of 20–30 mM in the vascular TME, as opposed to around 3 mM in normal tissues (87). Excess lactate and H+ suppress cell proliferation, survival, cytokine production and cytotoxicity in T and NK cells (88, 89). Oncometabolite 2-HG inhibits demethylases to increase histone methylation, and these epigenetic changes lead to suppressed T cell proliferation, TCR signaling, and NFAT activity (90). Arginase-1, derived from TAMs and MDSCs, depletes arginine, a critical amino acid for T cell activation (91). IDO and TDO, primarily produced by TAMs and CAFs, respectively, break down tryptophan and yield kynurenine, a suppressive metabolite that inhibits T cell functions (91–93). In addition, the aberrant vasculature of the TME contributes to promoting a hypoxic environment that inhibits effector T cell function, enhances Treg activity, and reduce NK cell cytotoxicity (94, 95). Hypoxia rewires T cell metabolism mainly via HIF-1α (96), but the precise in vivo role of HIF-1α in regulation of T cell function remains obscure (97): HIF-1α deficiency in CD8+ T cells enhances fatty acid catabolism and their anti-tumor functions (98), while HIF-1α promotes infiltration of CD8+ T cells into the tumors and enhances their effector responses to persistent antigen and promote tumor clearance (99, 100).
Epigenetic Regulation of Immune Cells
Epigenetic regulation of immune cells, i.e., DNA methylation and histone modifications, determines their fate and activation. Understanding how these epigenetic changes impact immune cells within the TME is vital to reversal of tumor immunosuppression. As already noted, T cell exhaustion is a major problem for long term efficacy of immunotherapy. Exhausted T cells have been identified to have a unique chromatic architecture induced through epigenetic modifications (70, 101–103), implicating that epigenetic therapeutic agents could serve in adjuvant treatment for cancer immunotherapy. Likewise, PD1 blockade is a treatment option used to combat T cell exhaustion, however, this treatment does not fully restore effector T cell function suggesting the importance of the impact that epigenetic modifications have on T cell function (104). Recent studies identify HMG-box transcription factor TOX as a major regulator of genetic and epigenetic remodeling that occurs during T cell exhaustion (104–106). TOX is largely dispensable for the formation of Teffector and Tmemory cells, but it is required for the development for Texhaustion cells in chronic infection (105).
The epigenetic landscape in TAMs can be remodeled in response to acute stimulation and polarizing stimuli, which helps integrate signaling, such as NF-κB and STATs, over time and underlies reprogramming of TAMs to alter their gene expression (107). Most of the epigenetic research work has been focused on the macrophages with M1 phenotype, and the epigenetic modifiers involved in TAMs with M2 polarization remains largely unknown. Previous work shows that the histone demethylase JMJD3, induced by IL-4, promotes expression of M2-promoting transcription factor IRF4 by removing negative H3K27me3 marks at the Irf4 locus (70, 108, 109). Recent studies identify epigenetic enzymes, including DNMT1, PRMT6, and KDM6B, which regulate M2 polarization and tumor-promoting functions in TAMs (110, 111), serving as targets for disrupting TAM immunosuppressive phonotypes.
Therapeutic Effects of Radiochemotherapy on Immune Cells
Chemotherapy and radiation have been two standard treatments against solid tumors; understanding how these cytotoxic therapies impact immune cells will better strategies of using synergistic therapies. It should be noted that not all cytotoxic strategies have a positive impact on immune cells. However, cytotoxic radiochemotherapies have emerged as a potential immune stimulant due to their involvement with immunogenic cell death (ICD) (112, 113). ICD is defined by a type of cancer cell death triggered by cytotoxic therapeutics, which activates long-lasting antitumor immunity; ICD proceeds by the release of damage-associated molecular patterns (DAMPs) that allow for the processing and presentation of tumor-associated antigens, in which priming T cells can turn the immune response from a tolerogenic one to an anti-tumor immunogenic response. In addition, ICD stimulates lymphocyte trafficking and infiltration, showing promise in improving TIL numbers in breast cancer, ovarian cancer and melanoma (114–116). Interestingly, there’s been evidence that certain cytotoxic chemotherapuetics can selectively target immune suppressive cells including Treg, MDSCs and TAMs (117–119). Dosage is crucial in eliciting these selective effects, maximum tolerated dose regimens have been shown to deplete CD8+ T cells and NK cells while low dose regimens have been shown to preferentially target MDSCs and Treg cells (120–122). Better understanding of their therapeutic effects on immune cells will help design more effective immunotherapy. Similar to chemotherapy, radiation also elicits an immunogenic response via DAMPs (123). In addition, radiation can affect the tumor vasculature resulting in an upregulation of adhesion molecules, further stimulating the recruitment of T effector cells (124). Radiotherapy is one of the most widely used treatment options for solid tumors (113, 125). Recent studies show that radiation used in combination with immunotherapy, such as checkpoint blockade, elicit significant immune responses (126–128).
Reprogram Vascular Microenvironment for Immunotherapy
Anti-Angiogenic Therapy
Anti-vascular therapy was initially thought to be groundbreaking in cancer treatment (129). The concept of conventional anti-angiogenic therapy was to starve the tumor of oxygen and nutrients through eradication of its vasculature, primarily by inhibiting pro-angiogenesis factors and their downstream pathways such as VEGF (130, 131) (Figure 2). Ultimately, anti-angiogenic therapy exhibits small and transient benefits in most malignant cancers. These therapeutic difficulties and failures are due to multiple mechanisms that contributes to the tumor resistance to anti-VEGF treatment, including angiogenic pathway redundancy, compensatory activation of survival signals, and pericyte and macrophage-mediated protection (132). Furthermore, anti-angiogenic therapy-induced vascular shutdown can deteriorate tumor hypoxia, leading to more aggressive tumor phenotypes in tumor growth, invasion, and metastasis via HIF-1α; this also generates a hostile barrier for delivery of therapeutic agents and anti-tumor lymphocytes into the tumors. In addition, anti-angiogenesis has shown negative side effects including cardiac toxicity, hemorrhage, thrombosis and gastrointestinal perforation further making it hard to justify its use as a therapeutic (133–135).

Figure 2 Strategies for anti-vascular therapy – Three therapeutic strategies have been developed for vasculature-targeting anti-cancer treatments, including anti-angiogenesis, vessel normalization, and endothelial programming.
Vessel Normalization
Vessel normalization has emerged as a novel approach to combat aberrant tumor vasculature through restoration of vessel perfusion and oxygenation (136). The goal of this therapy is to structurally normalize tumor vasculature, leading to a decrease in intratumor hypoxia and an increase in the delivery of therapeutic drugs and the efficacy of radiotherapy (Figure 2). Based on the central hypothesis that vascular abnormalities are driven by imbalance of pro- and anti-angiogenic factors (29, 137), current vessel normalization therapies have focused on targeting excessive pro-angiogenic factors, such as VEGF and PlGF, using neutralizing antibodies and pharmacological inhibitors of their downstream tyrosine kinases. However, these therapies have shown transient effects on tumor oxygenation and small benefits (138–146). Ideally, these vessel-normalizing treatment can reduce intratumoral hypoxia and enhance delivery of immunotherapeutic agents or cells into the TME, providing certain opportunities for improving immunotherapy, which needs further optimization of the therapeutic dose and duration to reach maximal and persistent effects on vessel normalization.
Another therapeutic field of vessel normalization is vascular maturation, as the tumor vasculature is characterized by disrupted coverage of pericytes that stabilize vascular structure and maintain its functional integrity (137). VEGF2 blockade can recruit pericytes through activation of Ang1 and Tie2 signaling (138). The risk of hemorrhage or venous thromboembolism exists whenever targeting VEGF, which has led to research into other targets to normalize vasculature. Targeting the upregulation of PDGF-β is seen as another option as it has been reported in mouse models to increase pericyte recruitment while decreasing EC proliferation (147, 148). However, there has been instances reported where upregulation of PDGF-β led to tumor growth (17).
Endothelial Reprogramming
As an alternative process to the vascular abnormality mechanism driven by angiogenic factor-mediated vessel sprouting and outgrowth, tumor ECs undergo genetic programming to induce aberrant vascularity. Robust cell plasticity in ECs has been well characterized in embryonic development (149–153). ECs undergo endothelial mesenchymal transition (Endo-MT) to de novo generate fibroblasts, stem-like cells, and smooth muscle cells in pathological settings including cardiac, renal and liver fibrosis, ossifying myositis, vascular inflammation, and cerebral cavernous malformation (154–160). Our recent work reveals that tumor ECs retain key endothelial functions but acquire mesenchymal phenotypes including enhanced proliferative and migratory capacities via cell transformation, i.e., partial Endo-MT, therefore driving aberrant vasculature in TME (16, 161, 162); this cell plasticity-mediated mechanism provides a new insight into vascular abnormality in TME, suggesting vascular de-transformation as a new strategy for cancer therapy (15) (Figure 2). Theoretically, tumor ECs are driven by plasticity-mediated genetic reprogramming where the hope of reversing this would offer a non-transient effect on reforming vessel morphology in the TME. The strategy for EC reprogramming focuses on the key regulatory node that drives the abnormal structure features. Our recent kinome-wide genetic screening of mesenchymal-like transcriptional activation in tumor ECs identifies PAK4 as an innovative target to reprogram ECs in glioblastoma (163). Genetic ablation or pharmacological inhibition of PAK4 showed an increase in expression of adhesion molecules in tumor ECs, a decrease in vessel abnormalities with improved T-cell infiltration, rendering tumors more sensitive to CAR T immunotherapy (163).
Additional benefits for endothelial reprogramming include the potential effects on anti-tumor immunity via improving vessel perfusion to remove immunosuppressive metabolites and enzymes in the TME as well as via directly reconditioning immunosuppressive vascular niche. Our recent work reveals that transformed ECs in TMEs form an immunosuppressive vascular niche via producing IL-6 that induces M2 phenotypes in TAMs, which inhibits T cell infiltration into and activation at the TME (45, 164); endothelial reprogramming may, therefore, generate a locoregional host-friendly TME with anti-tumor immunity that allows a successful immunotherapy. These findings offer proof of concept that reprograming tumor ECs is a viable option to reverse immune suppression within the TME and improve the efficacy of T cell-based immunotherapy.
In summary, we overview the role of the vascular TME in tumor evasion of immune responses and resistance to immunotherapy, with a focus on vessel abnormality, dysfunctional adhesion, immunosuppressive niche, and microenvironmental stress (Figure 1). We propose that development of new therapeutic approaches in order to reprogram tumor ECs may offer exciting opportunities to recondition the TME and to overcome tumor resistance to T cell-based immunotherapy (Figure 2).
Author Contributions
ZL wrote the manuscript. YF supervised the work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by National Institutes of Health (NIH) grants T32GM008076 (to ZL) and R01NS094533, R01NS106108 and R01CA241501 (to YF). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ribas A, Wolchok JD. Cancer Immunotherapy Using Checkpoint Blockade. Science (2018) 359:1350–5. doi: 10.1126/science.aar4060
2. Navani V, Graves MC, Bowden NA, van der Westhuizen A. Immune Checkpoint Blockade in Solid Organ Tumours: Choice, Dose and Predictors of Response. Br J Clin Pharmacol (2020) 86:1736–52. doi: 10.1111/bcp.14352
3. Gross G, Waks T, Eshhar Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors With Antibody-Type Specificity. Proc Natl Acad Sci USA (1989) 86:10024–8. doi: 10.1073/pnas.86.24.10024
4. Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L. Trial Watch: Adoptively Transferred Cells for Anticancer Immunotherapy. Oncoimmunology (2017) 6:e1363139. doi: 10.1080/2162402X.2017.1363139
5. Waldman AD, Fritz JM, Lenardo MJ. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat Rev Immunol (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
6. Prasad V. Immunotherapy: Tisagenlecleucel - the First Approved CAR-T-Cell Therapy: Implications for Payers and Policy Makers. Nat Rev Clin Oncol (2018) 15:11–2. doi: 10.1038/nrclinonc.2017.156
7. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and Management in CAR T-Cell Therapy. Mol Ther Oncolytics (2016) 3:16011. doi: 10.1038/mto.2016.11
8. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T Cell Immunotherapy for Human Cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
9. Beatty GL, Gladney WL. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin Cancer Res (2015) 21:687–92. doi: 10.1158/1078-0432.CCR-14-1860
10. Melero I, Rouzaut A, Motz GT, Coukos G. T-Cell and NK-Cell Infiltration Into Solid Tumors: A Key Limiting Factor for Efficacious Cancer Immunotherapy. Cancer Discov (2014) 4:522–6. doi: 10.1158/2159-8290.CD-13-0985
11. Pauken KE, Wherry EJ. Overcoming T Cell Exhaustion in Infection and Cancer. Trends Immunol (2015) 36:265–76. doi: 10.1016/j.it.2015.02.008
12. Datta M, Coussens LM, Nishikawa H, Hodi FS, Jain RK. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am Soc Clin Oncol Educ Book (2019) 39:165–74. doi: 10.1200/EDBK_237987
13. Martin JD, Seano G, Jain RK. Normalizing Function of Tumor Vessels: Progress, Opportunities, and Challenges. Annu Rev Physiol (2019) 81:505–34. doi: 10.1146/annurev-physiol-020518-114700
14. Jain RK. Normalization of Tumor Vasculature: An Emerging Concept in Antiangiogenic Therapy. Science (2005) 307:58–62. doi: 10.1126/science.1104819
15. Fan Y. Vascular Detransformation for Cancer Therapy. Trends Cancer (2019) 5:460–3. doi: 10.1016/j.trecan.2019.05.007
16. Huang M, Liu T, Ma P, Mitteer RA Jr., Zhang Z, Kim HJ, et al. C-Met-Mediated Endothelial Plasticity Drives Aberrant Vascularization and Chemoresistance in Glioblastoma. J Clin Invest (2016) 126:1801–14. doi: 10.1172/JCI84876
17. Liu J, Liao S, Huang Y, Samuel R, Shi T, Naxerova K, et al. PDGF-D Improves Drug Delivery and Efficacy via Vascular Normalization, But Promotes Lymphatic Metastasis by Activating CXCR4 in Breast Cancer. Clin Cancer Res (2011) 17:3638–48. doi: 10.1158/1078-0432.CCR-10-2456
18. Petty AJ, Yang Y. Tumor-Associated Macrophages: Implications in Cancer Immunotherapy. Immunotherapy (2017) 9:289–302. doi: 10.2217/imt-2016-0135
19. DeNardo DG, Ruffell B. Macrophages as Regulators of Tumour Immunity and Immunotherapy. Nat Rev Immunol (2019) 19:369–82. doi: 10.1038/s41577-019-0127-6
20. Akkari L, Bowman RL, Tessier J, Klemm F, Handgraaf SM, de Groot M, et al. Dynamic Changes in Glioma Macrophage Populations After Radiotherapy Reveal CSF-1R Inhibition as a Strategy to Overcome Resistance. Sci Transl Med (2020) 12. doi: 10.1126/scitranslmed.aaw7843
21. Feng Y, Mu R, Wang Z, Xing P, Zhang J, Dong L, et al. A Toll-Like Receptor Agonist Mimicking Microbial Signal to Generate Tumor-Suppressive Macrophages. Nat Commun (2019) 10:2272. doi: 10.1038/s41467-019-10354-2
22. Togashi Y, Shitara K, Nishikawa H. Regulatory T Cells in Cancer Immunosuppression - Implications for Anticancer Therapy. Nat Rev Clin Oncol (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
23. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage Plasticity and Polarization in Tissue Repair and Remodelling. J Pathol (2013) 229:176–85. doi: 10.1002/path.4133
24. Dart A. Throwing Treg Cells Off Balance. Nat Rev Cancer (2019) 19:367. doi: 10.1038/s41568-019-0163-3
25. Shitara K, Nishikawa H. Regulatory T Cells: A Potential Target in Cancer Immunotherapy. Ann N Y Acad Sci (2018) 1417:104–15. doi: 10.1111/nyas.13625
26. Moreno Ayala MA, Li Z, DuPage M. Treg Programming and Therapeutic Reprogramming in Cancer. Immunology (2019) 157:198–209. doi: 10.1111/imm.13058
27. Kalucka J, de Rooij L, Goveia J, Rohlenova K, Dumas SJ, Meta E, et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell (2020) 180:764–79.e20. doi: 10.1016/j.cell.2020.01.015
28. Dudley AC. Tumor Endothelial Cells. Cold Spring Harb Perspect Med (2012) 2:a006536. doi: 10.1101/cshperspect.a006536
29. Carmeliet P, Jain RK. Principles and Mechanisms of Vessel Normalization for Cancer and Other Angiogenic Diseases. Nat Rev Drug Discov (2011) 10:417–27. doi: 10.1038/nrd3455
30. Nagy JA, Dvorak AM, Dvorak HF. VEGF-A and the Induction of Pathological Angiogenesis. Annu Rev Pathol (2007) 2:251–75. doi: 10.1146/annurev.pathol.2.010506.134925
31. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T Cells Into Tumors. Cancer Res (2014) 74:7168–74. doi: 10.1158/0008-5472.CAN-14-2458
32. Forster JC, Harriss-Phillips WM, Douglass MJ, Bezak E. A Review of the Development of Tumor Vasculature and Its Effects on the Tumor Microenvironment. Hypoxia (Auckl) (2017) 5:21–32. doi: 10.2147/HP.S133231
33. Baek JH, Jang JE, Kang CM, Chung HY, Kim ND, Kim KW. Hypoxia-Induced VEGF Enhances Tumor Survivability via Suppression of Serum Deprivation-Induced Apoptosis. Oncogene (2000) 19:4621–31. doi: 10.1038/sj.onc.1203814
34. De Palma M, Biziato D, Petrova TV. Microenvironmental Regulation of Tumour Angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
35. Goerge T, Barg A, Schnaeker EM, Poppelmann B, Shpacovitch V, Rattenholl A, et al. Tumor-Derived Matrix Metalloproteinase-1 Targets Endothelial Proteinase-Activated Receptor 1 Promoting Endothelial Cell Activation. Cancer Res (2006) 66:7766–74. doi: 10.1158/0008-5472.CAN-05-3897
36. Maishi N, Hida K. Tumor Endothelial Cells Accelerate Tumor Metastasis. Cancer Sci (2017) 108:1921–6. doi: 10.1111/cas.13336
37. Stacer AC, Fenner J, Cavnar SP, Xiao A, Zhao S, Chang SL, et al. Endothelial CXCR7 Regulates Breast Cancer Metastasis. Oncogene (2016) 35:1716–24. doi: 10.1038/onc.2015.236
38. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-Like Character in PDGF-Induced Glioma Cells. Cell Stem Cell (2010) 6:141–52. doi: 10.1016/j.stem.2010.01.001
39. Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, et al. A Vascular Niche and a VEGF-Nrp1 Loop Regulate the Initiation and Stemness of Skin Tumours. Nature (2011) 478:399–403. doi: 10.1038/nature10525
40. Butler JM, Kobayashi H, Rafii S. Instructive Role of the Vascular Niche in Promoting Tumour Growth and Tissue Repair by Angiocrine Factors. Nat Rev Cancer (2010) 10:138–46. doi: 10.1038/nrc2791
41. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell (2007) 11:69–82. doi: 10.1016/j.ccr.2006.11.020
42. Ghajar CM, Peinado H, Mori H, Matei IR, Evason KJ, Brazier H, et al. The Perivascular Niche Regulates Breast Tumour Dormancy. Nat Cell Biol (2013) 15:807–17. doi: 10.1038/ncb2767
43. Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, et al. Endothelial Cells Promote the Colorectal Cancer Stem Cell Phenotype Through a Soluble Form of Jagged-1. Cancer Cell (2013) 23:171–85. doi: 10.1016/j.ccr.2012.12.021
44. Cao Z, Ding BS, Guo P, Lee SB, Butler JM, Casey SC, et al. Angiocrine Factors Deployed by Tumor Vascular Niche Induce B Cell Lymphoma Invasiveness and Chemoresistance. Cancer Cell (2014) 25:350–65. doi: 10.1016/j.ccr.2014.02.005
45. Wang Q, He Z, Huang M, Liu T, Wang Y, Xu H, et al. Vascular Niche IL-6 Induces Alternative Macrophage Activation in Glioblastoma Through HIF-2alpha. Nat Commun (2018) 9:559. doi: 10.1038/s41467-018-03050-0
46. Dvorak HF. Tumors: Wounds That do Not Heal. Similarities Between Tumor Stroma Generation and Wound Healing. N Engl J Med (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
47. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial Intercellular Adhesion Molecule-1 Expression is Suppressed in Human Malignancies: The Role of Angiogenic Factors. Cancer Res (1996) 56:1111–17.
48. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-Associated Macrophages (TAM) as Major Players of the Cancer-Related Inflammation. J Leukoc Biol (2009) 86:1065–73. doi: 10.1189/jlb.0609385
49. Muller WA. Leukocyte-Endothelial-Cell Interactions in Leukocyte Transmigration and the Inflammatory Response. Trends Immunol (2003) 24:327–34. doi: 10.1016/S1471-4906(03)00117-0
50. Langer HF, Chavakis T. Leukocyte-Endothelial Interactions in Inflammation. J Cell Mol Med (2009) 13:1211–20. doi: 10.1111/j.1582-4934.2009.00811.x
51. Schlesinger M, Bendas G. Vascular Cell Adhesion Molecule-1 (VCAM-1)–an Increasing Insight Into Its Role in Tumorigenicity and Metastasis. Int J Cancer (2015) 136:2504–14. doi: 10.1002/ijc.28927
52. Yoong KF, McNab G, Hubscher SG, Adams DH. Vascular Adhesion Protein-1 and ICAM-1 Support the Adhesion of Tumor-Infiltrating Lymphocytes to Tumor Endothelium in Human Hepatocellular Carcinoma. J Immunol (1998) 160:3978–88.
53. Griffioen AW, Damen CA, Blijham GH, Groenewegen G. Tumor Angiogenesis is Accompanied by a Decreased Inflammatory Response of Tumor-Associated Endothelium. Blood (1996) 88:667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
54. Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial Vascular Cell Adhesion Molecule 1 Expression is Suppressed by Melanoma and Carcinoma. J Exp Med (1995) 181:811–6. doi: 10.1084/jem.181.2.811
55. Griffioen AW, Damen CA, Mayo KH, Barendsz-Janson AF, Martinotti S, Blijham GH, et al. Angiogenesis Inhibitors Overcome Tumor Induced Endothelial Cell Anergy. Int J Cancer (1999) 80:315–9. doi: 10.1002/(SICI)1097-0215(19990118)80:2<315::AID-IJC23>3.0.CO;2-L
56. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B Receptor Mediates the Endothelial Barrier to T Cell Homing to Tumors and Disables Immune Therapy. Nat Med (2008) 14:28–36. doi: 10.1038/nm1699
57. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T Cells, Recurrence, and Survival in Epithelial Ovarian Cancer. N Engl J Med (2003) 348:203–13. doi: 10.1056/NEJMoa020177
58. Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-Infiltrating Lymphocytes in the Immunotherapy Era. Cell Mol Immunol (2021) 18:842–59. doi: 10.1038/s41423-020-00565-9
59. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine Expression in Melanoma Metastases Associated With CD8+ T-Cell Recruitment. Cancer Res (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
60. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine Nitration Prevents Intratumoral Infiltration of Antigen-Specific T Cells. J Exp Med (2011) 208:1949–62. doi: 10.1084/jem.20101956
61. Pardoll D. Does the Immune System See Tumors as Foreign or Self? Annu Rev Immunol (2003) 21:807–39. doi: 10.1146/annurev.immunol.21.120601.141135
62. Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on Tumor Cells With PD-1 on Tumor-Specific T Cells as a Mechanism of Immune Evasion: Implications for Tumor Immunotherapy. Cancer Immunol Immunother (2005) 54:307–14. doi: 10.1007/s00262-004-0593-x
63. Albini A, Bruno A, Noonan DM, Mortara L. Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Front Immunol (2018) 9:527. doi: 10.3389/fimmu.2018.00527
64. Garrido F, Algarra I. MHC Antigens and Tumor Escape From Immune Surveillance. Adv Cancer Res (2001) 83:117–58. doi: 10.1016/S0065-230X(01)83005-0
65. Soloski MJ. Recognition of Tumor Cells by the Innate Immune System. Curr Opin Immunol (2001) 13:154–62. doi: 10.1016/S0952-7915(00)00198-9
66. Jafarzadeh L, Masoumi E, Fallah-Mehrjardi K, Mirzaei HR, Hadjati J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front Immunol (2020) 11:702. doi: 10.3389/fimmu.2020.00702
67. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring Function in Exhausted CD8 T Cells During Chronic Viral Infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
68. Jiang Y, Li Y, Zhu B. T-Cell Exhaustion in the Tumor Microenvironment. Cell Death Dis (2015) 6:e1792. doi: 10.1038/cddis.2015.162
69. Thomas DA, Massague J. TGF-Beta Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012
70. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
71. Lewis CE, Pollard JW. Distinct Role of Macrophages in Different Tumor Microenvironments. Cancer Res (2006) 66:605–12. doi: 10.1158/0008-5472.CAN-05-4005
72. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol (2002) 23:549–55. doi: 10.1016/S1471-4906(02)02302-5
73. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
74. De Sanctis F, Ugel S, Facciponte J, Facciabene A. The Dark Side of Tumor-Associated Endothelial Cells. Semin Immunol (2018) 35:35–47. doi: 10.1016/j.smim.2018.02.002
75. Nagl L, Horvath L, Pircher A, Wolf D. Tumor Endothelial Cells (TECs) as Potential Immune Directors of the Tumor Microenvironment - New Findings and Future Perspectives. Front Cell Dev Biol (2020) 8:766. doi: 10.3389/fcell.2020.00766
76. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med (2014) 20:607–15. doi: 10.1038/nm.3541
77. Horikawa N, Abiko K, Matsumura N, Hamanishi J, Baba T, Yamaguchi K, et al. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity Through the Accumulation of Myeloid-Derived Suppressor Cells. Clin Cancer Res (2017) 23:587–99. doi: 10.1158/1078-0432.CCR-16-0387
78. Karikoski M, Marttila-Ichihara F, Elima K, Rantakari P, Hollmen M, Kelkka T, et al. Clever-1/Stabilin-1 Controls Cancer Growth and Metastasis. Clin Cancer Res (2014) 20:6452–64. doi: 10.1158/1078-0432.CCR-14-1236
79. Leone RD, Powell JD. Metabolism of Immune Cells in Cancer. Nat Rev Cancer (2020) 20:516–31. doi: 10.1038/s41568-020-0273-y
80. Menk AV, Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep (2018) 22:1509–21. doi: 10.1016/j.celrep.2018.01.040
81. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol Regulatory Element-Binding Proteins Are Essential for the Metabolic Programming of Effector T Cells and Adaptive Immunity. Nat Immunol (2013) 14:489–99. doi: 10.1038/ni.2570
82. Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-Tumor T Cell Responses. Cell (2015) 162:1217–28. doi: 10.1016/j.cell.2015.08.012
83. van Bruggen JAC, Martens AWJ, Fraietta JA, Hofland T, Tonino SH, Eldering E, et al. Chronic Lymphocytic Leukemia Cells Impair Mitochondrial Fitness in CD8(+) T Cells and Impede CAR T-Cell Efficacy. Blood (2019) 134:44–58. doi: 10.1182/blood.2018885863
84. Liu M, Wang X, Wang L, Ma X, Gong Z, Zhang S, et al. Targeting the IDO1 Pathway in Cancer: From Bench to Bedside. J Hematol Oncol (2018) 11:100. doi: 10.1186/s13045-018-0644-y
85. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8(+) T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell (2018) 33:480–94.e7. doi: 10.1016/j.ccell.2018.02.005
86. Godin-Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3-Dioxygenase Expression in Human Cancers: Clinical and Immunologic Perspectives. Clin Cancer Res (2011) 17:6985–91. doi: 10.1158/1078-0432.CCR-11-1331
87. Pucino V, Cucchi D, Mauro C. Lactate Transporters as Therapeutic Targets in Cancer and Inflammatory Diseases. Expert Opin Ther Targets (2018) 22:735–43. doi: 10.1080/14728222.2018.1511706
88. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
89. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
90. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of Antitumor T Cell Immunity by the Oncometabolite (R)-2-Hydroxyglutarate. Nat Med (2018) 24:1192–203. doi: 10.1038/s41591-018-0095-6
91. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
92. Hsu YL, Hung JY, Chiang SY, Jian SF, Wu CY, Lin YS, et al. Lung Cancer-Derived Galectin-1 Contributes to Cancer Associated Fibroblast-Mediated Cancer Progression and Immune Suppression Through TDO2/kynurenine Axis. Oncotarget (2016) 7:27584–98. doi: 10.18632/oncotarget.8488
93. Zhao Q, Kuang DM, Wu Y, Xiao X, Li XF, Li TJ, et al. Activated CD69+ T Cells Foster Immune Privilege by Regulating IDO Expression in Tumor-Associated Macrophages. J Immunol (2012) 188:1117–24. doi: 10.4049/jimmunol.1100164
94. Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, et al. Hypoxia Induced Impairment of NK Cell Cytotoxicity Against Multiple Myeloma can be Overcome by IL-2 Activation of the NK Cells. PLoS One (2013) 8:e64835. doi: 10.1371/journal.pone.0064835
95. Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, et al. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol Biochem (2017) 41:1271–84. doi: 10.1159/000464429
96. Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 Regulation of mTOR and Hypoxia-Inducible Factor 1 Integrate Metabolism and Migration of CD8+ T Cells. J Exp Med (2012) 209:2441–53. doi: 10.1084/jem.20112607
97. Phan AT, Goldrath AW. Hypoxia-Inducible Factors Regulate T Cell Metabolism and Function. Mol Immunol (2015) 68:527–35. doi: 10.1016/j.molimm.2015.08.004
98. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism Within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell (2017) 32:377–91.e9. doi: 10.1016/j.ccell.2017.08.004
99. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-Inducible Factors Enhance the Effector Responses of CD8(+) T Cells to Persistent Antigen. Nat Immunol (2013) 14:1173–82. doi: 10.1038/ni.2714
100. Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, et al. An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell (2017) 32:669–83.e5. doi: 10.1016/j.ccell.2017.10.003
101. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin States Define Tumour-Specific T Cell Dysfunction and Reprogramming. Nature (2017) 545:452–6. doi: 10.1038/nature22367
102. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The Epigenetic Landscape of T Cell Exhaustion. Science (2016) 354:1165–9. doi: 10.1126/science.aae0491
103. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic Stability of Exhausted T Cells Limits Durability of Reinvigoration by PD-1 Blockade. Science (2016) 354:1160–5. doi: 10.1126/science.aaf2807
104. Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and Epigenetic Regulation of T-Cell Exhaustion. Nat Metab (2020) 2:1001–12. doi: 10.1038/s42255-020-00280-9
105. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX Transcriptionally and Epigenetically Programs CD8(+) T Cell Exhaustion. Nature (2019) 571:211–8. doi: 10.1038/s41586-019-1325-x
106. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a Critical Regulator of Tumour-Specific T Cell Differentiation. Nature (2019) 571:270–4. doi: 10.1038/s41586-019-1324-y
107. Ivashkiv LB. Epigenetic Regulation of Macrophage Polarization and Function. Trends Immunol (2013) 34:216–23. doi: 10.1016/j.it.2012.11.001
108. Satoh T, Takeuchi O, Vandenbon A, Yasuda K, Tanaka Y, Kumagai Y, et al. The Jmjd3-Irf4 Axis Regulates M2 Macrophage Polarization and Host Responses Against Helminth Infection. Nat Immunol (2010) 11:936–44. doi: 10.1038/ni.1920
109. Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic Regulation of the Alternatively Activated Macrophage Phenotype. Blood (2009) 114:3244–54. doi: 10.1182/blood-2009-04-217620
110. Wang HC, Chen CW, Yang CL, Tsai IM, Hou YC, Chen CJ, et al. Tumor-Associated Macrophages Promote Epigenetic Silencing of Gelsolin Through DNA Methyltransferase 1 in Gastric Cancer Cells. Cancer Immunol Res (2017) 5:885–97. doi: 10.1158/2326-6066.CIR-16-0295
111. Xun J, Du L, Gao R, Shen L, Wang D, Kang L, et al. Cancer-Derived Exosomal miR-138-5p Modulates Polarization of Tumor-Associated Macrophages Through Inhibition of KDM6B. Theranostics (2021) 11:6847–59. doi: 10.7150/thno.51864
112. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat Rev Immunol (2017) 17:97–111. doi: 10.1038/nri.2016.107
113. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation Fosters Dose-Dependent and Chemotherapy-Induced Immunogenic Cell Death. Oncoimmunology (2014) 3:e28518. doi: 10.4161/onci.28518
114. Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, et al. Development of Tumor-Infiltrating Lymphocytes in Breast Cancer After Neoadjuvant Paclitaxel Chemotherapy. Clin Cancer Res (2001) 7:3025–30.
115. Peng J, Hamanishi J, Matsumura N, Abiko K, Murat K, Baba T, et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res (2015) 75:5034–45. doi: 10.1158/0008-5472.CAN-14-3098
116. Hong M, Puaux AL, Huang C, Loumagne L, Tow C, Mackay C, et al. Chemotherapy Induces Intratumoral Expression of Chemokines in Cutaneous Melanoma, Favoring T-Cell Infiltration and Tumor Control. Cancer Res (2011) 71:6997–7009. doi: 10.1158/0008-5472.CAN-11-1466
117. Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential Impairment of Regulatory T Cells Rather Than Effector T Cells by Paclitaxel-Based Chemotherapy. Clin Immunol (2008) 129:219–29. doi: 10.1016/j.clim.2008.07.013
118. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil Selectively Kills Tumor-Associated Myeloid-Derived Suppressor Cells Resulting in Enhanced T Cell-Dependent Antitumor Immunity. Cancer Res (2010) 70:3052–61. doi: 10.1158/0008-5472.CAN-09-3690
119. Cubas R, Moskalenko M, Cheung J, Yang M, McNamara E, Xiong H, et al. Chemotherapy Combines Effectively With Anti-PD-L1 Treatment and Can Augment Antitumor Responses. J Immunol (2018) 201:2273–86. doi: 10.4049/jimmunol.1800275
120. Chang CL, Hsu YT, Wu CC, Lai YZ, Wang C, Yang YC, et al. Dose-Dense Chemotherapy Improves Mechanisms of Antitumor Immune Response. Cancer Res (2013) 73:119–27. doi: 10.1158/0008-5472.CAN-12-2225
121. Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, et al. Metronomic Chemotherapy Prevents Therapy-Induced Stromal Activation and Induction of Tumor-Initiating Cells. J Exp Med (2016) 213:2967–88. doi: 10.1084/jem.20151665
122. Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T Regulatory Cell Function Implicated in Enhanced Immune Response by Low-Dose Cyclophosphamide. Blood (2005) 105:2862–8. doi: 10.1182/blood-2004-06-2410
123. Derer A, Deloch L, Rubner Y, Fietkau R, Frey B, Gaipl US. Radio-Immunotherapy-Induced Immunogenic Cancer Cells as Basis for Induction of Systemic Anti-Tumor Immune Responses - Pre-Clinical Evidence and Ongoing Clinical Applications. Front Immunol (2015) 6:505. doi: 10.3389/fimmu.2015.00505
124. Hallahan D, Kuchibhotla J, Wyble C. Cell Adhesion Molecules Mediate Radiation-Induced Leukocyte Adhesion to the Vascular Endothelium. Cancer Res (1996) 56:5150–5.
125. Carvalho HA, Villar RC. Radiotherapy and Immune Response: The Systemic Effects of a Local Treatment. Clinics (Sao Paulo) (2018) 73:e557s. doi: 10.6061/clinics/2018/e557s
126. Weichselbaum RR, Liang H, Deng L, Fu YX. Radiotherapy and Immunotherapy: A Beneficial Liaison? Nat Rev Clin Oncol (2017) 14:365–79. doi: 10.1038/nrclinonc.2016.211
127. Demaria S, Bhardwaj N, McBride WH, Formenti SC. Combining Radiotherapy and Immunotherapy: A Revived Partnership. Int J Radiat Oncol Biol Phys (2005) 63:655–66. doi: 10.1016/j.ijrobp.2005.06.032
128. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, et al. Radiation and Dual Checkpoint Blockade Activate Non-Redundant Immune Mechanisms in Cancer. Nature (2015) 520:373–7. doi: 10.1038/nature14292
129. Folkman J. Tumor Angiogenesis: Therapeutic Implications. N Engl J Med (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
130. Folkman J. Angiogenesis: An Organizing Principle for Drug Discovery? Nat Rev Drug Discov (2007) 6:273–86. doi: 10.1038/nrd2115
131. Carmeliet P. Angiogenesis in Life, Disease and Medicine. Nature (2005) 438:932–6. doi: 10.1038/nature04478
132. Bergers G, Hanahan D. Modes of Resistance to Anti-Angiogenic Therapy. Nat Rev Cancer (2008) 8:592–603. doi: 10.1038/nrc2442
133. Chen HX, Cleck JN. Adverse Effects of Anticancer Agents That Target the VEGF Pathway. Nat Rev Clin Oncol (2009) 6:465–77. doi: 10.1038/nrclinonc.2009.94
134. Hapani S, Sher A, Chu D, Wu S. Increased Risk of Serious Hemorrhage With Bevacizumab in Cancer Patients: A Meta-Analysis. Oncology (2010) 79:27–38. doi: 10.1159/000314980
135. Nalluri SR, Chu D, Keresztes R, Zhu X, Wu S. Risk of Venous Thromboembolism With the Angiogenesis Inhibitor Bevacizumab in Cancer Patients: A Meta-Analysis. JAMA (2008) 300:2277–85. doi: 10.1001/jama.2008.656
136. Jain RK. Antiangiogenesis Strategies Revisited: From Starving Tumors to Alleviating Hypoxia. Cancer Cell (2014) 26:605–22. doi: 10.1016/j.ccell.2014.10.006
137. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the Vasculature for Treatment of Cancer and Other Diseases. Physiol Rev (2011) 91:1071–121. doi: 10.1152/physrev.00038.2010
138. Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, et al. Kinetics of Vascular Normalization by VEGFR2 Blockade Governs Brain Tumor Response to Radiation: Role of Oxygenation, Angiopoietin-1, and Matrix Metalloproteinases. Cancer Cell (2004) 6:553–63. doi: 10.1016/j.ccr.2004.10.011
139. Dings RP, Loren M, Heun H, McNiel E, Griffioen AW, Mayo KH, et al. Scheduling of Radiation With Angiogenesis Inhibitors Anginex and Avastin Improves Therapeutic Outcome via Vessel Normalization. Clin Cancer Res (2007) 13:3395–402. doi: 10.1158/1078-0432.CCR-06-2441
140. McGee MC, Hamner JB, Williams RF, Rosati SF, Sims TL, Ng CY, et al. Improved Intratumoral Oxygenation Through Vascular Normalization Increases Glioma Sensitivity to Ionizing Radiation. Int J Radiat Oncol Biol Phys (2010) 76:1537–45. doi: 10.1016/j.ijrobp.2009.12.010
141. Fischer C, Jonckx B, Mazzone M, Zacchigna S, Loges S, Pattarini L, et al. Anti-PlGF Inhibits Growth of VEGF(R)-Inhibitor-Resistant Tumors Without Affecting Healthy Vessels. Cell (2007) 131:463–75. doi: 10.1016/j.cell.2007.08.038
142. Eichhorn ME, Strieth S, Luedemann S, Kleespies A, Noth U, Passon A, et al. Contrast Enhanced MRI and Intravital Fluorescence Microscopy Indicate Improved Tumor Microcirculation in Highly Vascularized Melanomas Upon Short-Term Anti-VEGFR Treatment. Cancer Biol Ther (2008) 7:1006–13. doi: 10.4161/cbt.7.7.5997
143. Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, et al. Tumor Vascular Changes Mediated by Inhibition of Oncogenic Signaling. Cancer Res (2009) 69:6347–54. doi: 10.1158/0008-5472.CAN-09-0657
144. Cerniglia GJ, Pore N, Tsai JH, Schultz S, Mick R, Choe R, et al. Epidermal Growth Factor Receptor Inhibition Modulates the Microenvironment by Vascular Normalization to Improve Chemotherapy and Radiotherapy Efficacy. PLoS One (2009) 4:e6539. doi: 10.1371/journal.pone.0006539
145. Hansen-Algenstaedt N, Stoll BR, Padera TP, Dolmans DE, Hicklin DJ, Fukumura D, et al. Tumor Oxygenation in Hormone-Dependent Tumors During Vascular Endothelial Growth Factor Receptor-2 Blockade, Hormone Ablation, and Chemotherapy. Cancer Res (2000) 60:4556–60.
146. Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, et al. AZD2171, a Pan-VEGF Receptor Tyrosine Kinase Inhibitor, Normalizes Tumor Vasculature and Alleviates Edema in Glioblastoma Patients. Cancer Cell (2007) 11:83–95. doi: 10.1016/j.ccr.2006.11.021
147. McCarty MF, Somcio RJ, Stoeltzing O, Wey J, Fan F, Liu W, et al. Overexpression of PDGF-BB Decreases Colorectal and Pancreatic Cancer Growth by Increasing Tumor Pericyte Content. J Clin Invest (2007) 117:2114–22. doi: 10.1172/JCI31334
148. Reis M, Czupalla CJ, Ziegler N, Devraj K, Zinke J, Seidel S, et al. Endothelial Wnt/beta-Catenin Signaling Inhibits Glioma Angiogenesis and Normalizes Tumor Blood Vessels by Inducing PDGF-B Expression. J Exp Med (2012) 209:1611–27. doi: 10.1084/jem.20111580
149. Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-To-Mesenchymal and Endothelial-to-Mesenchymal Transition: From Cardiovascular Development to Disease. Circulation (2012) 125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352
150. Eilken HM, Nishikawa S, Schroeder T. Continuous Single-Cell Imaging of Blood Generation From Haemogenic Endothelium. Nature (2009) 457:896–900. doi: 10.1038/nature07760
151. Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In Vivo Imaging of Haematopoietic Cells Emerging From the Mouse Aortic Endothelium. Nature (2010) 464:116–20. doi: 10.1038/nature08764
152. Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D. Haematopoietic Stem Cells Derive Directly From Aortic Endothelium During Development. Nature (2010) 464:108–11. doi: 10.1038/nature08738
153. Lancrin C, Sroczynska P, Stephenson C, Allen T, Kouskoff V, Lacaud G. The Haemangioblast Generates Haematopoietic Cells Through a Haemogenic Endothelium Stage. Nature (2009) 457:892–5. doi: 10.1038/nature07679
154. Piera-Velazquez S, Jimenez SA. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol Rev (2019) 99:1281–324. doi: 10.1152/physrev.00021.2018
155. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, et al. Endothelial-To-Mesenchymal Transition Contributes to Cardiac Fibrosis. Nat Med (2007) 13:952–61. doi: 10.1038/nm1613
156. Li J, Qu X, Bertram JF. Endothelial-Myofibroblast Transition Contributes to the Early Development of Diabetic Renal Interstitial Fibrosis in Streptozotocin-Induced Diabetic Mice. Am J Pathol (2009) 175:1380–8. doi: 10.2353/ajpath.2009.090096
157. Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R. Fibroblasts in Kidney Fibrosis Emerge via Endothelial-to-Mesenchymal Transition. J Am Soc Nephrol (2008) 19:2282–7. doi: 10.1681/ASN.2008050513
158. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of Vascular Endothelial Cells Into Multipotent Stem-Like Cells. Nat Med (2010) 16:1400–6. doi: 10.1038/nm.2252
159. Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. FGF Regulates TGF-Beta Signaling and Endothelial-to-Mesenchymal Transition via Control of Let-7 miRNA Expression. Cell Rep (2012) 2:1684–96. doi: 10.1016/j.celrep.2012.10.021
160. Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, et al. EndMT Contributes to the Onset and Progression of Cerebral Cavernous Malformations. Nature (2013) 498:492–6. doi: 10.1038/nature12207
161. Liu T, Ma W, Xu H, Huang M, Zhang D, He Z, et al. PDGF-Mediated Mesenchymal Transformation Renders Endothelial Resistance to Anti-VEGF Treatment in Glioblastoma. Nat Commun (2018) 9:3439. doi: 10.1038/s41467-018-05982-z
162. Huang M, Zhang D, Wu JY, Xing K, Yeo E, Li C, et al. Wnt-Mediated Endothelial Transformation Into Mesenchymal Stem Cell-Like Cells Induces Chemoresistance in Glioblastoma. Sci Transl Med (2020) 12. doi: 10.1126/scitranslmed.aay7522
163. Ma W, Wang Y, Zhang R, Yang F, Zhang D, Huang M, et al. Targeting PAK4 to Reprogram the Vascular Microenvironment and Improve CAR-T Immunotherapy for Glioblastoma. Nat Cancer (2021) 2:83–97. doi: 10.1038/s43018-020-00147-8
Keywords: immunity, immunotherapy, tumor microenvironment, endothelial cells, immune suppression, T cells, infiltration, exhaustion
Citation: Lamplugh Z and Fan Y (2021) Vascular Microenvironment, Tumor Immunity and Immunotherapy. Front. Immunol. 12:811485. doi: 10.3389/fimmu.2021.811485
Received: 08 November 2021; Accepted: 29 November 2021;
Published: 20 December 2021.
Edited by:
Lijie Zhai, Northwestern Medicine, United StatesReviewed by:
Lorenzo Mortara, University of Insubria, ItalyCopyright © 2021 Lamplugh and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Fan, fanyi@upenn.edu