 Tariq Webber
Tariq Webber Katharina Ronacher
Katharina Ronacher Marli Conradie-Smit
Marli Conradie-Smit Léanie Kleynhans
Léanie Kleynhans- 1DSI-NRF Centre of Excellence for Biomedical Tuberculosis Research, South African Medical Research Council Centre for Tuberculosis Research, Division of Molecular Biology and Human Genetics, Department of Biomedical Sciences, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
- 2Translational Research Institute, Mater Research Institute – The University of Queensland, Brisbane, QLD, Australia
- 3Division of Endocrinology, Department of Medicine, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
The role of the endocrine system on the immune response, especially in the lung, remains poorly understood. Hormones play a crucial role in the development, homeostasis, metabolism, and response to the environment of cells and tissues. Major infectious and metabolic diseases, such as tuberculosis and diabetes, continue to converge, necessitating the development of a clearer understanding of the immune and endocrine interactions that occur in the lung. Research in bacterial respiratory infections is at a critical point, where the limitations in identifying and developing antibiotics is becoming more profound. Hormone receptors on alveolar and immune cells may provide a plethora of targets for host-directed therapy. This review discusses the interactions between the immune and endocrine systems in the lung. We describe hormone receptors currently identified in the lungs, focusing on the effect hormones have on the pulmonary immune response. Altered endocrine responses in the lung affect the balance between pro- and anti-inflammatory immune responses and play a role in the response to infection in the lung. While some hormones, such as leptin, resistin and lipocalin-2 promote pro-inflammatory responses and immune cell infiltration, others including adiponectin and ghrelin reduce inflammation and promote anti-inflammatory cell responses. Furthermore, type 2 diabetes as a major endocrine disease presents with altered immune responses leading to susceptibility to lung infections, such as tuberculosis. A better understanding of these interactions will expand our knowledge of the mechanisms at play in susceptibility to infectious diseases and may reveal opportunities for the development of host-directed therapies.
Introduction
There is a growing interest in the relationship between the immune and endocrine systems (1). Bidirectional communication between the immune and endocrine systems facilitates optimal host responses during infection and homeostasis. This communication is possible since endocrine organs express cytokine receptors and cells of the immune system express hormone receptors (2). Metabolic, nutritional, psychosocial, and genetic factors influence the immune response via endocrine signaling. During infection, cytokines produced by immune cells directly or indirectly alter hormonal responses, developing a feedback response between the endocrine and immune systems. Cytokines induce alterations in hormone production, by directly affecting endocrine organ function, such as IL-1, IL-6, and TNF-α. These cytokines increase the activity of the hypothalamus-pituitary-adrenal (HPA) and hypothalamus-pituitary-thyroidal (HPT) axes, and reduce that of the hypothalamus-pituitary-gonadal (HPG) axis (3–6), or indirectly by promoting cellular destruction of endocrine cells, as in the pancreatic dysfunction of type 2 diabetes (T2D) (7).
Hormones exert a profound effect on the functions of the immune system, exemplified by the anti-inflammatory functions of glucocorticoids (GCs) and androgens (8). While acute stress may be beneficial for the immune system during fight or flight, chronic stress is harmful. Chronic inflammation signal cortisol release through inflammatory signaling as well as the HPA axis and can drive HPA axis dysfunction if left unchecked. Stress-induced activation of the HPA axis results in the secretion of corticotropin releasing hormone (CRH) from the hypothalamus, which stimulates the production of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. ACTH stimulates the adrenal glands to produce cortisol, a potent anti-inflammatory hormone. To limit long-term exposure of tissues and cells to the immunosuppressive actions of cortisol, a negative feedback loop allows cortisol to regulate its own secretion. Long-term exposure to elevated levels of cortisol may lead to adaptations in HPA axis function, resulting in an initial increase in cortisol production (hypercortisolism) followed by a decrease in the production of the hormone (hypocortisolism) or cortisol resistance (9). Thus, chronic inflammatory responses and a dysregulated HPA axis result in altered cytokine signals and immune cell function (10).
Type 2 diabetes is a metabolic disease that presents with numerous alterations in hormone levels, including increased insulin and cortisol levels, and reduced free triiodothyronine (fT3) and testosterone levels (11–14) (Table 1). Common comorbidities of T2D include depression, asthma, and chronic obstructive pulmonary disease, which are co-prevalent and may indicate a relationship between stress-related hormonal alterations and lung function, as well as hypothyroidism (34). Altered endocrine responses in T2D are, in part, related to dysregulation of the HPA, HPT, and HPG axes, having downstream effects on immune functions within various organs (35–37).
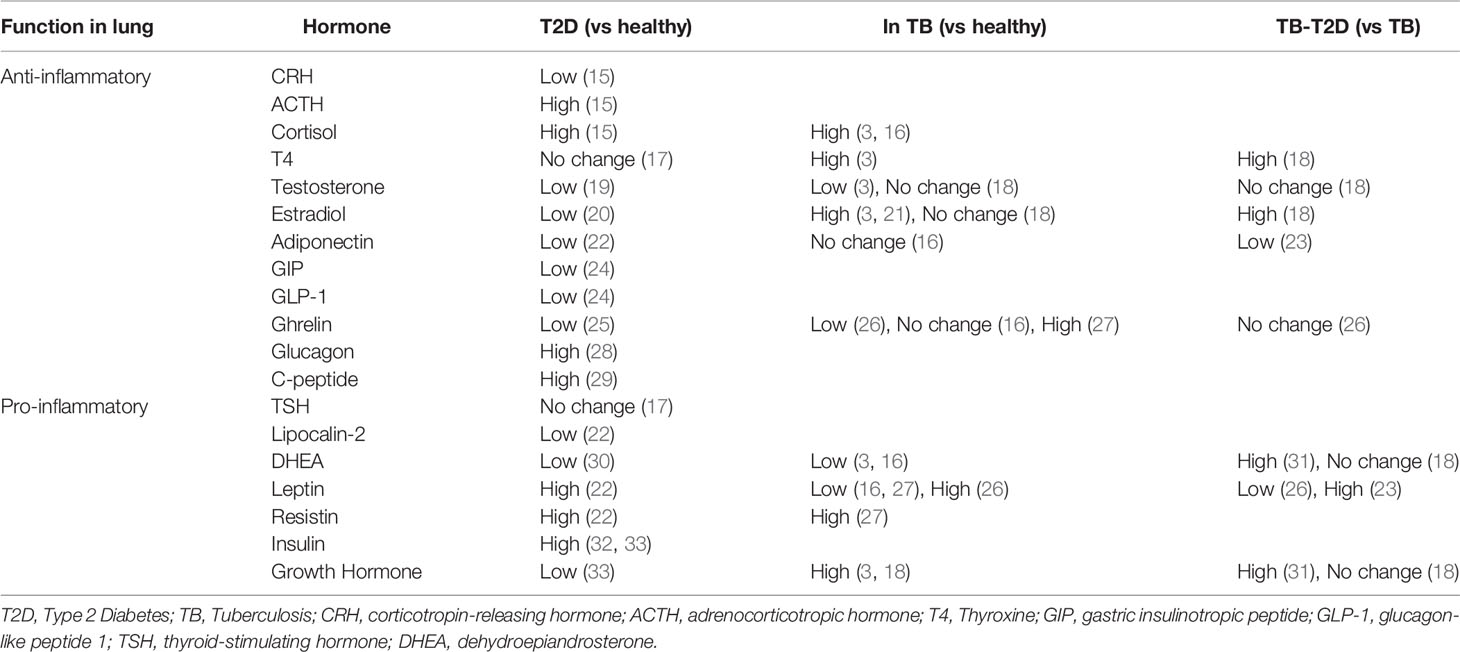
Table 1 Inflammatory Properties of hormones and their relative levels measured in the periphery.
Although the lungs are a major site of acquired infections, the effects of hormonal dysregulation on the immune response within the lung are still largely unknown. This review discusses hormones with immune modulatory potential in the lung and their influence on the immune response therein (summarized in Figure 1). Implications for susceptibility to tuberculosis (TB) are highlighted, drawing on examples of endocrine dysregulation in the form of T2D.
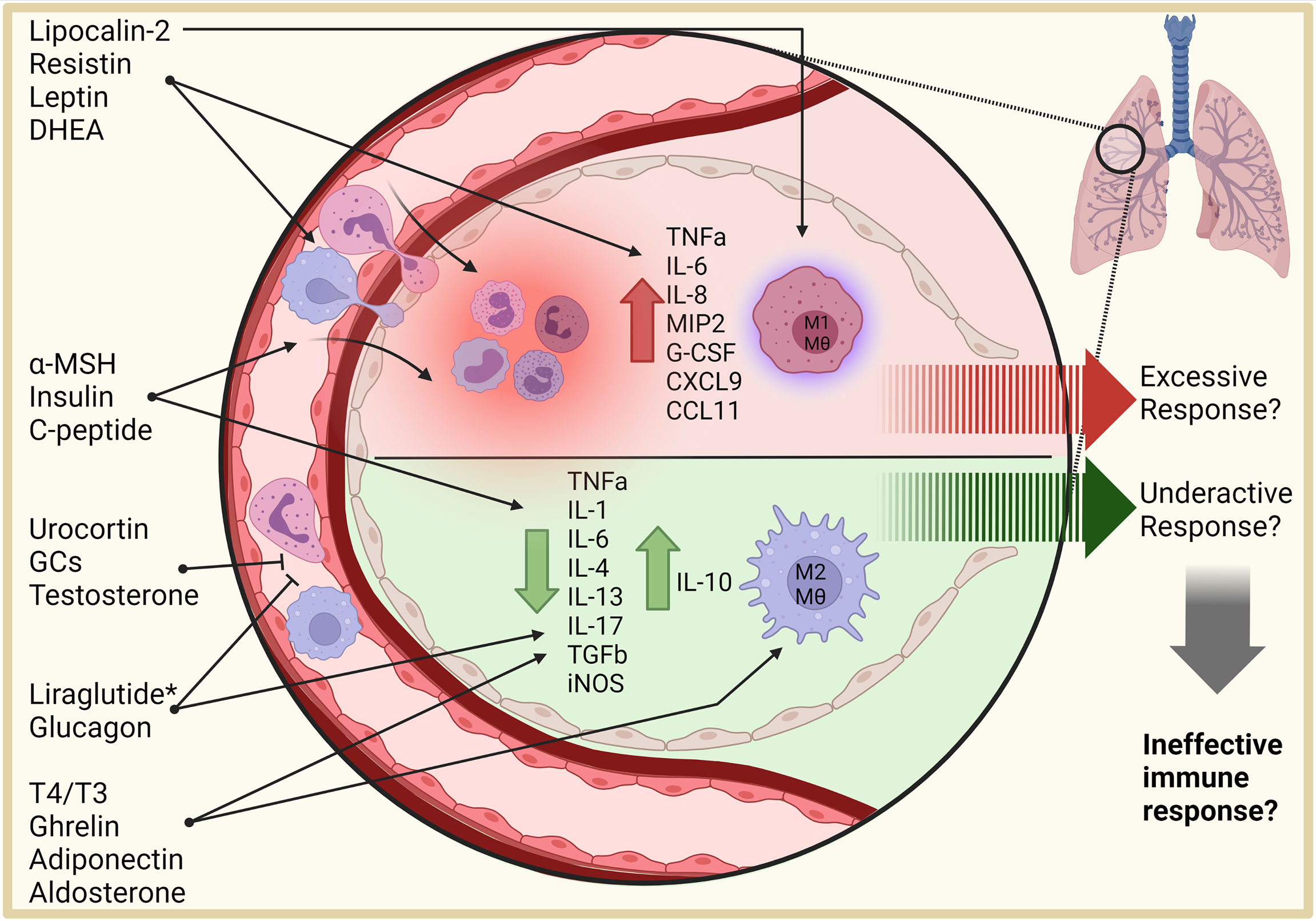
Figure 1 Effects of hormones on the lung environment. Hormones influence inflammatory responses, necessitating a clear understanding of their role in infectious disease. Pro-inflammatory adipokines, such as resistin, leptin, lipocalin-2, and DHEA collectively stimulate migration of immune cells into the lung and promote a pro-inflammatory signaling environment, with lipocalin-2 stimulating macrophage activation and M1 polarization. Anti-inflammatory hormones, including glucocorticoids, testosterone, glucagon, thyroxine, ghrelin, adiponectin, and aldosterone, reduce inflammatory signaling, immune cell recruitment, and polarize macrophages to anti-inflammatory phenotypes. Whether these combined effects are beneficial or detrimental to infectious disease response in the lung require further investigation. Additionally, agonists and antagonists (*) provide a future opportunity to regulate these processes. DHEA, dehydroepiandrosterone; α-MSH, alpha-melanocyte-stimulating hormone; T4, Thyroxine; T3, triiodothyronine; Mθ, macrophage. This figure was created with BioRender.com.
Central Endocrine (Hypothalamic and Pituitary) Hormones
Corticotropin Releasing Hormone (CRH)
CRH is primarily produced by the hypothalamus, but can also be expressed at peripheral sites where it acts as an autocrine or paracrine inflammatory modulator (38, 39). CRH binds with a high affinity to CRH receptor 1 (CRHR1) and CRH-like peptides with a high affinity to CRHR2. CRHR2 is the major receptor expressed in human lungs, however compared to other tissues (heart, stomach, liver and adrenal), the lung and skeletal muscle express the highest levels of CRHR1 (40). In mice, the CRHR1/R2 ratio changes in response to stress in different immune cell types in the lung (41). CRHR1 mRNA increases and CRHR2 decreases in dendritic cells, whereas CRHR2 increases in macrophages in response to stress. Furthermore, CRHR2 is found to be preferentially expressed in neutrophils in the lungs of mice (42).
Differential expression of CRHR1 and CRHR2 in phagocytic cells in the lungs could affect innate cell function, potentially leading to altered adaptive immune responses. Limited information is available on how targeting CRH and CRHR activity in the respiratory tract impacts bacterial disease outcome. In a murine respiratory Streptococcus pneumoniae infection model, Burnley and Jones (2017) demonstrated that intranasal CRH administration reduces the pulmonary inflammatory response, and the subsequent recruitment of neutrophils and monocytes to the lung, improving survival in these animals (43). Moffat et al. (2006) demonstrated that, urocortin, a structurally related peptide to CRH, causes bronchorelaxation and limits lipopolysaccharide (LPS)-induced pulmonary inflammation (specifically neutrophil recruitment) to a greater extent than CRH (44). During S. pneumoniae infection in the lungs of CD-1 mice, an outbred strain of albino mice, prior CRHR2 antagonist (astressin 2B) treatment reduces pulmonary bacterial growth and prevents sepsis, while CRHR1 antagonism (antalarmin) provided no benefit (39). Stimulation of CRHR in the lung, therefore, provides a promising approach for reducing excessive inflammation in the lungs and should be studied further.
Adrenocorticotropic Hormone (ACTH)
ACTH is a pituitary hormone that signals via melanocortin receptors (MCRs), of which there are five. They are expressed on a wide range of tissues, including nervous, intestinal, adipose, skeletal, and lung tissues. Among these receptors, MC1R and MC5R are expressed in lung tissues. MC1R, MC3R, and MC5R are expressed on macrophages and lymphocytes, while MC2R and MC4R are expressed on lymphocytes (45). Cleavage of the first 13 amino acids of ACTH yields alpha melanocyte-stimulating hormone (α-MSH).
ACTH and α-MSH exert broad anti-inflammatory effects in a variety of tissues by signaling through the MCRs, with the major receptor exerting these effects being MC1R (45). Additionally, numerous anti-inflammatory melanocortin-based therapies are in clinical trials for the treatment of inflammatory conditions (46). In allergic mice, intraperitoneal α-MSH reduced peribronchial airway inflammation, altering leukocyte populations in bronchoalveolar lavage (BAL) fluid, and reduced IL-4 and IL-13 levels, effects dependent on IL-10 signaling downstream of α-MSH treatment (47). In acute lung injury mouse models, α-MSH treated mice displayed reduced edema; IL-6, TNF-α, and TGF-β gene expression; and leukocyte infiltration in the lung (48, 49). Thus, MCR stimulation in the lung may provide a novel target for its anti-inflammatory properties, providing repurposing potential for current agonists used to treat obesity (setmelanotide), porphyria (afamelanotide), and reduced sexual desire in women (bremelanotide) (50).
Growth Hormone-Releasing Hormone (GHRH) and Growth Hormone (GH)
GHRH is secreted predominantly by the hypothalamus and stimulates the secretion of GH by the pituitary gland and regulates hepatic insulin-like growth factor-1 (IGF-1) production through the GH/hepatic IGF-1 axis (51). Other tissues, including the lungs also produce GHRH locally and express the receptor of GHRH (GHRHR) as well as the GHRHR splice variant 1 (SV1) (52). In normal mouse lung tissue, type 2 cells, club cells and fibroblasts, lymphocytes, and dendritic cells express GHRH (52). GHRH is involved in lung homeostasis, inflammation and fibrosis (51). Antagonists for GHRHR have anti-inflammatory, pro-apoptotic, anti-oxidative and anti-fibrotic effects in the lung. The GHRH antagonist MIA-602, frequently used in bleomycin-induced lung fibrosis models, suppresses ERK1/2 and JAK2/STAT3 pathway activation and increases P53 and pAMK levels (53). At mRNA level MIA-602 downregulates pathways involved in adaptive immune responses including T cell activation and differentiation, T cell signaling and cytokine production (54). In a sarcoidosis granuloma model, GHRHR inhibition results in reduced IL-12 and IL-17A production (54, 55). Thus, MIA-602 demonstrates the contribution of GHRHR to adaptive immunity and T cell function, and that GHRHR is a potential candidate for managing these responses when they are excessive, such as in granulomatous diseases.
The ability of GHRHR signaling to modulate lung immune responses was further demonstrated in a study assessing the vaccine-induced immune responses of GHRH knock-out mice against S. pneumoniae. Knock-out mice were significantly more susceptible to infection after vaccination than wild-type mice, with vast infiltration of neutrophils and monocytes, and reduced B cells and T cells in the lung post infection (56). These knock-out mice were unable to mount a pneumococcal vaccine-induced response, a deficiency restored by GH treatment, highlighting the importance of GH signaling in the lung immune response. The pro-inflammatory characteristics of GH have been demonstrated by exacerbating LPS-induced inflammation and cecal ligation and puncture (CLP)-induced sepsis by increasing neutrophil NF-κB activity, neutrophil presence, microvascular injury, and neutrophil CD11b expression in the lungs of rats (57, 58). In contrast, others have found GH treatment to prevent acute lung injury by reducing ICAM-1 expression and NF-κB activity in lungs (59, 60). The two studies displaying the pro-inflammatory effects of GH administered the GH subcutaneously to male Wistar rats, while the latter studies observing anti-inflammatory effects administered the GH intramuscularly in Sprague-Dawley rats and pathogen-free Kun Ming mice, respectively. Considering endocrine differences between Wistar and Sprague-Dawley rats (61), hormone-induced responses may differ and validation of observations across model organisms is key for avoiding model specific interpretations.
Adrenal Hormones
Glucocorticoids (GCs) and Mineralocorticoids
Cortisol is a known immune modulator and synthetic GCs have been used clinically to control inflammatory conditions, such as asthma, for decades. Cortisol is produced from cholesterol that is converted to pregnenolone before the steroidogenic pathway diverges toward the formation of this major class of steroid hormones. It signals via the glucocorticoid receptor (GR) and mineralocorticoid receptor (MR), which are ubiquitously expressed, including in the lung (62).
High doses of GCs reduce the number of macrophages in the respiratory tract and impair the functional activity of resident lung macrophages (63). GCs are produced in T-cells and macrophages of the lung upon stimulation with inflammatory mediators, including TNF-α, LPS, and anti-CD3, acting as an immunoregulatory mechanism to limit uncontrolled inflammatory responses in the lung. This GC stimulatory effect was promoted by intranasal leptin pretreatment in mice with LPS-induced acute lung injury (64, 65). More recently Marin-Luevano et al. (2021) demonstrated that cortisol promotes the intracellular growth of Mycobacterium tuberculosis in A549 type 2 pneumocytes and THP-1–derived macrophages, but not airway bronchial epithelial cells (66), highlighting the immunosuppressive effects of GCs in the lung. Long-term (>1 year) treatment with inhaled corticosteroids is implicated in increased risk of developing pneumonia (risk ratio of 1.41) and is associated with the risk of mycobacterial diseases, such as TB (67).
Androgens
Dehydroepiandrosterone (DHEA) is produced from cholesterol, predominantly in the zona reticularis of the adrenal glands upon ACTH stimulation, and in the gonads upon luteinizing hormone and follicle-stimulating hormone stimulation. Receptors for DHEA include the androgen receptor (AR), estrogen receptor (ER), peroxisome proliferator activated receptor-alpha (PPARα), pregnane X receptor, and neurotransmitter receptors, such as the N-methyl-D-aspartate (NMDA) receptor (68, 69).
DHEA treatment is associated with reduced pulmonary hypertension by upregulating soluble guanylate cyclase and by inhibiting src/STAT3 pathway activation, a key pathway in cytokine-induced cell activation and proliferation (70, 71). Generally DHEA counteracts the effect of cortisol, and more specifically in alveolar macrophages, restores the expression of the receptor for activated C kinase (RACK-1) and LPS-induced TNF-α and IL-8 production that is reduced due to aging in rats (72). When added to alveolar macrophages from non-smoking asbestos workers in vitro, DHEA reduces the release of superoxide anion (73), which is consistent with its role in limiting Th2 responses (74). Intratracheal administration of 16α-bromoepiandrosterone (BEA; a DHEA-related synthetic sterol) reduces the bacterial load and inflammation in the lungs of diabetic mice who are infected with M. tuberculosis (75). BEA decreases the expression of the enzyme 11-β-hydroxysteroid dehydrogenase type 1, which catalyzes the conversion of inactive cortisone into active cortisol in humans and inactive dehydrocorticosterone into active corticosterone in mice. At the same time, it increases the expression of 11-β-hydroxysteroid dehydrogenase type 2, which again converts active cortisol/corticosterone into inactive cortisone/dehydrocorticosterone. This could increase IFN-γ and TNF-α response resulting in lower bacterial loads in the lungs of these animals. The ability of DHEA and BEA to improve immune and metabolic (as shown in the TB-T2D mouse model) responses suggests that they could potentially be used as therapeutic agents, which will benefit the host’s immune, endocrine and metabolic functions.
Thyroid Hormones: Thyroxine (T4) and Triiodothyronine (T3)
The lungs express lower levels of the thyroid hormone receptors THRA1, THRA2, and THRB1 mRNA than other tissues, however the relative protein expression of THRA2 and THRB1 in lung tissue is higher than in other human tissues (76). Furthermore, THRA expression in the lung is highest in alveolar type 1 epithelial cells, while their expression is similar in lung T cells, granulocytes, type 2 alveolar cells, and macrophages. THRB on the other hand is expressed predominantly in type 2 alveolar cells (77). In rats, thyroid hormone receptors were also found in lung type 2 alveolar cells, and promote GC responses and adenylate cyclase activity in the lung (78–80). Additionally, T3 activates the MAPK/ERK1/2 pathway in rat alveolar epithelial cells and increases their sodium-potassium-ATPase pump content and activity (81).
More recently, Ning et al. (2018) described the ability of T4 to reduce the inflammatory response and senescence in an oxidized low-density lipoprotein-induced foamy macrophage model (82). In a type 2 deiodinase-knockout mouse model, active thyroid hormones are not produced in the thyroid gland. Type 2 deiodinase-knockout mice have a greater susceptibility to ventilator-induced lung injury, with increased Cxcl1, Tnf, Il1b, and Cxcl2 gene expression. Treatment with T3 ameliorated the higher cytokine and chemokine expression in knockout mice (83). Although it remains unclear whether these effects were mediated by the THR, this highlights the anti-inflammatory effects of T3 and potentiates a relevance for T3 signaling in the lung. Studies assessing the influence of thyroid hormones on lung function during infection would provide valuable information against infections.
Gonadal Hormones
Testosterone
Differences between male and female responses in disease are common, highlighting a potential role for sex hormones in disease. Testosterone acts via the AR, which is expressed in human lungs primarily in endothelial cells and club cells, fibroblasts, and type 2 alveolar cells, with lower expression in macrophages, T cells, and granulocytes (84). Testosterone exposure in mouse lungs upregulates genes involved in iron binding and oxygen transport while downregulating genes involved in DNA repair and recombination (85). In an asthma mouse model, ovalbumin sensitized male mice displayed lower susceptibility to inflammation due to lower Th2 cytokine production and lung lymphocyte levels compared to female mice (86). Becerra-Diaz et al. (2018) investigated the immune modulatory functions of AR signaling in the lungs, and found that dihydrotestosterone reconstitution in castrated mice reduced lung inflammation and enhanced M2 polarization of alveolar macrophages via IL-4 stimulation (87). Similarly, gonadectomized male mice infected with influenza A virus and treated orally with testosterone have reduced pulmonary monocyte and virus-specific CD8+ T cell infiltration resulting in improved disease outcomes (88). Testosterone decreases pro-inflammatory cytokine release from monocytes and macrophages, and increases the accumulation of cholesterol esters in human monocyte-derived macrophages (89). Additionally, type 2 innate lymphoid cells express AR, the signaling of which inhibits their maturation and IL-33–mediated lung inflammation.
Estrogen
Estrogens comprise three major forms, estrone, estradiol, and estriol. Their proportions fluctuate depending on the processes occurring in the body, with estradiol predominating most of the time. During pregnancy, enzymes in the placenta transform 16α-hydroxy-dehydroepiandrosterone sulfate (16a-OH-DHEAS) to estriol. Estrone is produced mostly during menopause. Signaling of these hormones is mediated by the α and β forms of the ER. Both ER forms are expressed in human and mouse lungs, and human lung cell lines; although in humans, ER-α presence varies among individuals (90). ER-α predominates in lung fibroblasts, with low expression in endothelial cells, granulocytes and macrophages, while ER-β predominates in human lung alveolar type 1 cells, ciliated cells, and granulocytes (84). Histological analysis in CD-1® IGS mouse and porcine lung show no ERα expression (91, 92), while RT-PCR in BALB/c mice shows expression of both receptors in the lung, with lower ERα expression (93). Kan et al. (2008) investigated the effect of 17β-estradiol (E2) on trauma-hemorrhage–induced lung injury in Sprague-Dawley rats. E2 administration induced higher endothelial nitric oxide synthase (eNOS) expression and phosphorylation, protein kinase G-1 activation, and VASP expression, resulting in reduced lung injury (94). These anti-inflammatory properties were also demonstrated in seawater induced acute lung injury in rats, which was reduced by E2 treatment (95). Despite ERβ predominance, ER-α is reported to be responsible for the anti-inflammatory effects of ER stimulation (96).
Hormones of the Gastro-Intestinal Tract
Gastric Inhibitory Polypeptide (GIP) and Glucagon-Like Peptide-1 (GLP-1)
The GIP receptor (GIPR) is present in airway ciliated and club cells, alveolar pneumocytes and lung macrophages (77). In atherosclerosis mouse models, GIP has been shown to prevent monocyte and macrophage activation (97, 98). Studies into the immune regulatory roles of GIP are scarce and may reveal additional functions of this hormone on the lung immune response.
The GLP-1 receptor (GLP-1R) is expressed in the lungs of mice and humans at a higher level than other tissues (99, 100). Numerous studies, as reviewed by Lee and Jun (2016), describe the anti-inflammatory effects of GLP-1 and the potential of GLP-1–based therapies (101). In a BALB/c mouse model of ovalbumin-induced asthma, protein kinase A (PKA) phosphorylation was inhibited and NF-κB p65 upregulated in the lung. GLP-1 agonist treatment in these mice led to PKA activation and downstream inhibition of NF-κB, mediating anti-inflammatory responses in the lung by reducing the infiltration of inflammatory cells, tissue pathology, Th2-associated cytokines in BAL fluid, and E-selectin expression (102). Gou et al. (2014) showed that bleomycin-induced pulmonary fibrosis in mice presents with an increase in macrophages and lymphocytes, as well as TGF-β1 concentrations in the BAL fluid, and VCAM-1 expression and NF-κB activation in the lung. GLP-1 agonist liraglutide, when administered intraperitoneally, reduces these characteristics of bleomycin-induced lung inflammation and fibrosis (103). Although subcutaneous liraglutide improved lung function in a chronic obstructive pulmonary disease mouse model, this effect was not accompanied by a reduction in lung inflammation. Additionally, liraglutide did not alter inflammatory cytokine responses in another study by the same group (100, 104). These differences in observations suggest that intraperitoneal administration may be required to produce the anti-inflammatory effects of GLP-1R in the lung. Other possible immunomodulatory mechanisms of GLP-1 may involve alteration of T cell function by reducing CD28 and CD86 expression, and reducing tissue factor and PAI-1 production (105). While GIPR function in the lung immune response requires further investigation, GLP-1R may reduce inflammation by mediating immune cell recruitment and activation in the lung, although it is not crucial for a balance between pro- and anti-inflammatory responses.
Ghrelin
Since its discovery in 1999, numerous actions of ghrelin have been described ranging from metabolic homeostasis, circadian rhythms, learning and memory to immune modulation. Ghrelin binds to the growth-hormone secretagogue receptor 1a (GHSR-1a), inducing GH release from anterior pituitary cells in a process distinct from that of GHRH (106). In Hartley guinea-pigs, Wistar rats and male C57BL6/J mice, the expression of GHSR-1a mRNA was very low in the lung (107, 108), with the lung also having the highest ghrelin expression in Wistar rats (109). In two studies assessing human lungs, mRNA and protein expression of ghrelin and biologically inactive GHSR-1b was detectable, while those of GHSR-1a were not (110–112).
Despite this, ghrelin treatment is associated with alterations in inflammatory responses in lung tissues, including a reduction in inflammatory cytokine release, NF-κB pathway activation, neutrophil infiltration, and improved survival in rats with CLP-induced lung injury (113). Additionally, these anti-inflammatory effects may be related to the acylation status of ghrelin, since only acyl ghrelin and not des-acyl ghrelin binds to GHSR-1a (114). Interestingly, it has been proposed that GHSR-1a is not required for the anti-inflammatory actions of ghrelin, suggesting another as yet unknown signaling mechanism mediating these effects (115). LPS-induced apoptosis of alveolar macrophages is attenuated by ghrelin, with GHSR-1a–mediated JNK inhibition and Wnt/B-catenin activation, promoting alveolar macrophage survival and their anti-inflammatory responses. In GHRS-1a knockdown mediated by siRNA, these effects were abrogated, highlighting the need for GHRS-1a signaling in these anti-inflammatory and anti-apoptotic responses (116). In a CLP-induced Sprague Dawley rat sepsis model, ghrelin treatment improved survival and reduced peritoneal bacterial load and pulmonary TNF-α and IL-6. Signaling via the GSHR-1a receptor was responsible for improving mortality. Additionally, the CLP-treated group without ghrelin administration had significantly lower ghrelin content in the lung (117). A more recent study also noted that in the lung with elastase-induced emphysema, ghrelin polarized alveolar macrophages to the M2 phenotype, decreased keratinocyte-derived chemokine (a mouse IL-8 analogue), TGF-β, and TNF-α, increased IL-10 levels, and promoted lung tissue recovery in C57BL/6 mice (118). Exogenous administration of ghrelin reduced pulmonary hypertension in a monocrotaline-induced mouse model (119). Ghrelin may play a key role in altering the immune response, possibly linking gastro-intestinal tract function and metabolism to immunity in the lung. Further, studying the effects of ghrelin receptor agonists on the lung may provide an important avenue for host-directed therapy during lung infection and inflammatory conditions.
Pancreatic Hormones
C-Peptide
C-peptide is a 31 amino acid peptide that links the insulin A and B subunits and is cleaved from proinsulin to form insulin. Despite evidence of C-peptide–induced intracellular signaling activity in KATOII cells (a human gastric tumor cell line), a specific receptor for C-peptide has been elusive and a proposed receptor, GPR146, remains contentious (120, 121). However, its chemotactic properties were noted when in vitro experiments showed that C-peptide increases human monocyte chemotaxis in a concentration dependent manner (122). Several studies identified beneficial effects of C-peptide in the lungs. C-peptide treatment ameliorates the inflammatory response and lung inflammation resulting from hemorrhagic shock in male Wistar rats, reducing plasma IL-1, IL-6, MIP-1α, and CXCL1 (123). Jeon et al. (2019) found that C-peptide prevents vascular leakage by inhibiting VEGF-induced transglutaminase 2 activation in the lungs of streptozotocin-treated mice and human pulmonary microvascular endothelial cells (124). These findings were also corroborated in C57BL/6 mice treated with C-peptide after hemorrhagic shock and resuscitation, showing a reduction in inflammatory markers and pulmonary protein leakage (125). Furthermore, Vish et al. (2007) used a Swiss albino mouse model with LPS-induced endotoxic shock to investigate the effects of C-peptide treatment. LPS treatment in the lungs of mice reduced PPARγ gene expression, stimulated ERK1/2 activation, and induced lung injury. These effects were reversed by C-peptide treatment (126). While PPARγ has been shown to have anti-inflammatory effects, ERK1/2 activation is involved in developing a pro-inflammatory response. These findings indicate an anti-inflammatory nature of C-peptide signaling in the lung, though further studies in this field are required to determine insights in its pulmonary effects and influence during infection. The identification and characterization of the signaling mechanisms of C-peptide are key to understanding its effects and potential applications.
Insulin
The insulin receptor (IR) is expressed in varying degrees across diverse tissues, including the lungs. In the lungs, IR gene expression predominates in endothelial cells, club cells and ciliated cells, and alveolar type 1 and type 2 cells, with lower expression in lung immune cells (84). The two IR isoforms, IR-A and IR-B, and IGF-1R bind their ligands, insulin, proinsulin, IGF-1, and IGF-2, with differing potencies (EC50). IR-A displays the lowest EC50 values for insulin, proinsulin, and IGF-2 (127). IR-B predominates in the liver and adipose tissue, while leukocytes only express the IR-A isoform (128). Different pathways are activated by the two IR isoforms (129) indicating that an altered ratio of the isoforms can alter the outcomes of insulin signaling.
GC treatment increases IR expression, while hypothyroidism induction reduces the IR expression in fetal rabbit lung, indicating the influence of other hormones on the lung’s insulin response (130). During allergic lung inflammation in rats, insulin secretion and IR expression is increased on infiltrating inflammatory cells, predominantly monocytes and macrophages (131). In an elastase-induced emphysema Wistar rat model with alloxan-induced diabetes, diabetic rats had significantly lower leukocyte infiltration into the lungs, which was significantly increased upon subcutaneous treatment with insulin (132). While suggesting that insulin treatment improves the deficient immune cell migration in diabetes, it also may indicate pro-inflammatory effects due to leukocyte recruitment, although this study did not use a vehicle treatment control to compare with the insulin treated group.
Martins et al. (2008) isolated alveolar macrophages by BAL from male Wistar rats and stimulated them with LPS to assess the inflammatory response. In the insulin treated cells after LPS-stimulation, there was a significant reduction in the LPS-induced p38 MAPK, PKC, and Akt activation, and TNF-α secretion (133). A similar study in specific-pathogen-free C57BL/6 mice with alloxan induced diabetes found that insulin treatment of alveolar macrophages decreased LPS-induced TNF-α and IL-6 production ex vivo (134). Thus, the response of the lung tissue to insulin is still unclear, although some studies indicate anti-inflammatory changes in gene and cytokine expression in alveolar macrophages treated with insulin, others suggest that insulin treatment promotes leukocyte migration to the lung. There is however limited information available regarding the effects of insulin resistance and hyperinsulinemia on immune function in the lung, despite numerous studies on respiratory function in these cases. Understanding these lung responses to insulin is crucial in the application of insulin treatments, including inhalants and injectables, and understanding the pathogenesis of infection in the lungs of patients with diabetes mellitus.
Glucagon
Reports of glucagon receptor (GCGR) expression and function in the lung are scarce. While two studies suggest little to no expression of GCGR in the lungs of rats, another study suggests its predominant expression in mouse lungs, liver, kidney, adrenal glands, and stomach (135–137). Glucagon receptors have been identified in lymphocytic cells from mice and rats, and in lymphoid cell lines (138).
Nebulized glucagon delivery to the lung improves the forced expiratory volume by 22% in asthmatic patients with methacholine-induced bronchospasm (139). This indicates responsiveness of the lung smooth muscle to glucagon. It was later shown that glucagon induces smooth muscle relaxation in the lungs of male A/J mice by inducing cAMP response element binding protein (CREB), eNOS, and cyclooxygenase 1 (COX-1) activity, and the subsequent release of second messengers nitric oxide and prostaglandin E2 (140). In this study, glucagon treatment one hour before LPS administration limited LPS-induced lung inflammation and airway hyperreactivity, preventing increases in TNF-α levels. Unlike neutrophils, LPS-induced monocyte infiltration was not reduced by glucagon pretreatment, indicating that glucagon may not influence monocyte migration and chemotaxis. This group further assessed the anti-inflammatory properties of glucagon in ovalbumin-induced lung inflammation and airway hyperreactivity (141). Glucagon treatment prevented airway hyperreactivity and eosinophilia, and reduced IL-4, IL-5, IL-13, TNF-α, CCL11, and CCL24 levels in lung tissue.
Adipose Hormones
Adiponectin
As summarized in Ye et al. (2013), adiponectin affects metabolic functions, such as glucose metabolism, insulin sensitivity, oxidative stress, and inflammation, in numerous systems, including adipose tissue, B cells, and macrophages (142). The mRNA expression of adiponectin receptors AdipoR1 and AdipoR2 have been demonstrated by Northern blot in mouse and human lung tissues, although a considerably lower expression was observed in human lung tissue compared to other tissues (143). Constant adiponectin infusion (using osmotic pumps) in BALB/cJ mice reduces allergic airway inflammation and hyperresponsiveness induced by ovalbumin challenge, while the lack of adiponectin in knockout mice promotes greater allergic airway inflammation (144). Additionally, adiponectin knockout mice have a higher frequency of TNF-α producing alveolar macrophages, which is reduced upon adiponectin supplementation, and increased transcription of IL-1α, IL-6, IL-12β, IL-17, and TNF-α in their lungs during aspergillosis, than wild type mice (145).
In humans, several studies report an inverse association of serum adiponectin and asthma prevalence and severity (146). Adiponectin promotes an anti-inflammatory M2 macrophage phenotype, while inhibiting an M1 phenotype. This was observed when adiponectin deficient mice displayed a predominance of M1-polarized macrophages and fewer M2-polarized macrophages in the peritoneum and adipose tissue. Treatment with adiponectin reversed this observation, promoting an M2 phenotype in human monocyte-derived macrophages and adipose tissue cells, and reducing LPS-stimulated TNF-α, iNOS, and MCP-1 gene expression (147). Agonist therapy of the adiponectin receptor shows promise against inflammatory conditions such as systemic sclerosis, obesity-related disorders, and T2D. This was shown using the adiponectin agonist, AdipoRon, which ameliorated signs of diabetes in db/db and high fat diet fed mice, and ameliorated dermal fibrosis in mice, also promoting a Th2/Th17 immune response (148, 149). While adiponectin is an abundant adipokine and displays anti-inflammatory functions, more information is needed on whether its levels are altered to compensate for inflammation. Additionally, the applications of adiponectin agonist therapy against lung inflammatory responses require further investigation.
Leptin
Leptin regulates innate and adaptive functions of immune cells. Leptin upregulates phagocytosis and pro-inflammatory cytokine production via phospholipase activation, and stimulates lineage marker expression, including HLA-DR, CD11b, and CD11c in monocytes (150). Thus, leptin is an important pro-inflammatory mediator predominantly produced in adipose tissue. Leptin receptor mRNA, including OB-Ra, -Rb, and -Re subtypes, are expressed in the mouse lung (151, 152).
Leptin receptor deficient db/db mice infected with M. tuberculosis have a higher lung bacterial load, disorganized granulomas, and abnormalities in lung cytokine production, with delayed IFN-γ production as compared to wild-type mice (153). Similarly, leptin deficient ob/ob mice with pulmonary infection by Klebsiella pneumoniae, S. pneumoniae, or M. abscessus have reduced survival (154). Suzukawa et al. (2015) used bronchial cells in ex vivo cultures to assess the effects of leptin stimulation on lung cells, notably these specimens were obtained from lung with localized tumors. Leptin treatment increased CCL11, G-CSF, VEGF, and IL-6 production and upregulated ICAM-1 expression, a change that coincided with activation of NF-κB, highlighting the pro-inflammatory potential of leptin and its ability to induce migration and chemotaxis of immune cells (155). This has implications for increasing the inflammatory response and leukocyte infiltration into the lung in individuals with increased leptin levels, such as during obesity. Additionally, OB-R expression was upregulated by IFN-y and IL-1β, implying that during type 1 inflammatory responses, the lung epithelial cells are more sensitive to these pro-inflammatory effects of leptin. These effects were recapitulated in later work by the same group in a lung fibroblast cell line (156).
Thus, although leptin presence may be required for a functional immune response to infection, higher levels may promote excessive inflammation. These effects require further investigation given the importance of increased leptin during obesity and T2D, which display higher prevalence of comorbid lung diseases (157).
Lipocalin-2
Lipocalin-2, or neutrophil gelatinase associated lipocalin (NGAL), is an adipokine with broad tissue expression. Zhang et al. (2012) investigated this using tissue microarray technology across humans tissues from embryos, fetuses, neonates, and adults (158). This study demonstrated expression of NGAL and its receptor (NGALR) in nervous, renal, adrenal, pituitary, spleen, lymph node, and skin tissues, with NGALR additionally expressed in lung and pancreatic tissues in adults.
NGAL influences the immune response by polarizing macrophages to an M1 phenotype (159), and altering neutrophil chemotaxis and cytokine secretion (160). Wang et al. (2019) investigated these immune modulating capabilities of NGAL, finding defective neutrophil chemotaxis, and reduced inflammatory cytokine and chemokine production in neutrophils and macrophages in NGAL deficient mice infected with E. coli, and increased macrophage migration and phagocytosis upon in vitro NGAL treatment (161). These findings were recapitulated in pulmonary M. tuberculosis infection in mice, whereby NGAL promoted neutrophil recruitment and alveolar macrophage G-CSF and CXCL1 production, while reducing T cell recruitment and CXCL9 production. NGAL deficiency resulted in larger granuloma size in chronic M. tuberculosis infection, coinciding with elevated lung T cell and CXCL9 presence (162). Furthermore, NGAL is important in the lung mucosal immunity against K. pneumoniae infection, playing a role in bacterial clearance (163). This study showed that NGAL is expressed on human bronchial epithelial cells in response to in vitro stimulation with IL-1β, IL-17A, IL-17F, or TNF-α, and in vivo upon K. pneumoniae infection in the lungs of C57BL/6 mice in a TLR4-dependent manner via the MyD88 signaling pathway. TLR4 knockout and NGAL knockout mice had significantly higher lung bacterial loads at 12 hours post infection, which was reduced significantly by exogenous NGAL administration 4 hours prior to sacrifice. Expression of NGAL is critical to the immune response in the lung and demonstrates essential functions during infection.
Resistin
Resistin is an adipokine that binds to TLR4 and channel-activating protease 1 (CAP1), a serine protease. Two studies demonstrated resistin binding to TLR4, with resistin activating TLR4 in porcine alveolar macrophages and competing with LPS for TLR4 in human monocytes (164, 165). TLR4 and CAP1 are expressed on human alveolar macrophages and type 2 alveolar cells, indicating that resistin could play a role in the lung immune response during infection (77, 166). Resistin binds to decorin and ROR1 in mice with a lower affinity, though these are rarely expressed on human monocytes and macrophages (167), ROR1 is expressed on human alveolar type 1 cells and decorin is expressed on human lung fibroblasts and endothelial cells.
Using a humanized mouse model, Jiang et al. (2014) demonstrated that resistin expression exacerbated LPS-induced lung injury via neutrophil recruitment to the lung during LPS treatment and promoted their activation in the lung, with increased TNF-α and MIP-2 release, and increased neutrophil extracellular trap formation (168). Additionally, resistin-like molecules (RELMs) are homologues of resistin and have immunomodulatory effects, demonstrated in lung tissue, with pro-inflammatory and tissue remodeling capabilities (169, 170). Inflammation-mediated lung injury can occur via activation of TLR4/NF-κB pathways (171, 172). However, further studies are necessary to assess the effects of resistin signaling on lung immune function during infection.
Type 2 Diabetes Impacts Lung Immune-Endocrine Interactions: Implications for TB
Endocrine alterations play a crucial role in the development and progression of T2D. Alterations in hormone levels have been identified in the plasma of T2D patients, implicating endocrine signaling in the dysregulation of inflammatory responses in T2D patients (173). Hormones with suggested pro-inflammatory properties in the lung are increased in the blood of T2D patients, such as TSH, leptin, resistin, and insulin (Table 1). Conversely, many hormones with potential anti-inflammatory effects are reduced during T2D, such as CRH, DHEA, T4, testosterone, adiponectin, NGAL, GIP, GLP-1, and ghrelin, while ACTH, cortisol, glucagon, and C-peptide also have anti-inflammatory potential but are raised in T2D patient blood plasma. This shift toward pro-inflammatory hormone signaling in T2D may be a mechanism for the low-grade chronic inflammation in T2D patients and provides an impetus for investigating immune-endocrine alterations in the lungs of these patients. It also indicates that a complex interaction between these hormones and the immune system is at play, requiring deconvolution before useful and exploitable insights can be gained.
Under the influence of diet and GIT function, gastric hormones contribute to the control of systemic inflammatory responses. Although GIP may play a role in compensating for insulin resistance, lower GLP-1 and ghrelin may indicate a predominance of inflammatory molecules and downregulation of anti-inflammatory mechanisms. Additionally, a role for the gut microbiome in influencing the immune response via gastric hormones lays an intriguing path for further research into T2D immune responses.
Foamy macrophage formation, which provides a niche for mycobacterial survival and supply crucial lipid nutrients to the bacilli, is suppressed by GIP and GLP-1 (174). During T2D, GLP-1 and GIP are reduced and dyslipidemia typically presents with higher levels of triglycerides (175), which accumulate in foamy macrophages (176).
TB is a leading cause of death worldwide, with an incidence of 10 million and a mortality between 1.1 and 1.3 million in 2019 (177). T2D is a highly prevalent risk factor for increased disease severity, poor treatment outcomes, relapse, and death in TB patients, and is projected to rise from an estimated 9.3% in 2019 to 10.9% by 2045 (178). Pleural fluid of TB patients contains elevated levels of GH and cortisol compared to healthy controls (18, 179), while DHEA concentration is reportedly lower in the plasma of TB patients compared to that of healthy controls (3, 18).
Hormone levels change differently over the course of TB treatment between treatment failure and cured patients. These include cortisol, T4, and total amylin levels, which are higher in treatment failure compared to cured patients at month 6 of TB treatment, and decrease over the course of treatment in the cured patients only (1). These hormones may promote immunosuppression during treatment, reducing the function of immune cells to work synergistically with antibiotics to clear mycobacteria from the lung.
Santucci et al. (2011) and Fernandez et al. (2020) found lower leptin levels and higher cortisol levels in TB patients compared to controls, coinciding with higher inflammatory biomarkers, such as CRP, IL-6, and IL-1β (16, 31). Furthermore, Santucci et al. (2011) found leptin and cortisol changes to be more extreme in severe TB disease than in mild disease. OB gene knockout experiments in mice show that leptin is necessary for mycobacterial control and IFN-y response (180). Characterising the effects of hormones on lung homeostasis and immune responses to infection is paramount. Experiments including overexpression and knockout of hormones and their receptor may highlight such effects.
The immune response to infection, such as during TB, involves recognition and phagocytosis of pathogens by macrophages and dendritic cells, killing by phagolysosome fusion, and presentation on major histocompatibility complexes allowing the activation of adaptive immune responses (181–183). However, T2D alters the immune response to M. tuberculosis, having reduced pathogen recognition, phagocytosis, killing, and presentation due to reduced pattern recognition receptor and major histocompatibility complex expression on macrophages. Altered macrophage lipid trafficking results in the formation of foam cells, with reduced phagocytic function and increased apoptosis, a cholesterol rich environment develops, providing nutrients to persisting mycobacteria. Lymphocytes dictate the developing immune response and altered lymphocyte levels in T2D include higher anti-inflammatory responses (Th2 and Treg) and reduced pathogen specific memory Th17 and Th1 responses, additionally natural killer cells are increased but their responses are reduced (184–186). The influence of hormones in determining these altered immune responses requires further investigation and their alteration during T2D provides a useful model for these altered immune responses (Figure 2).
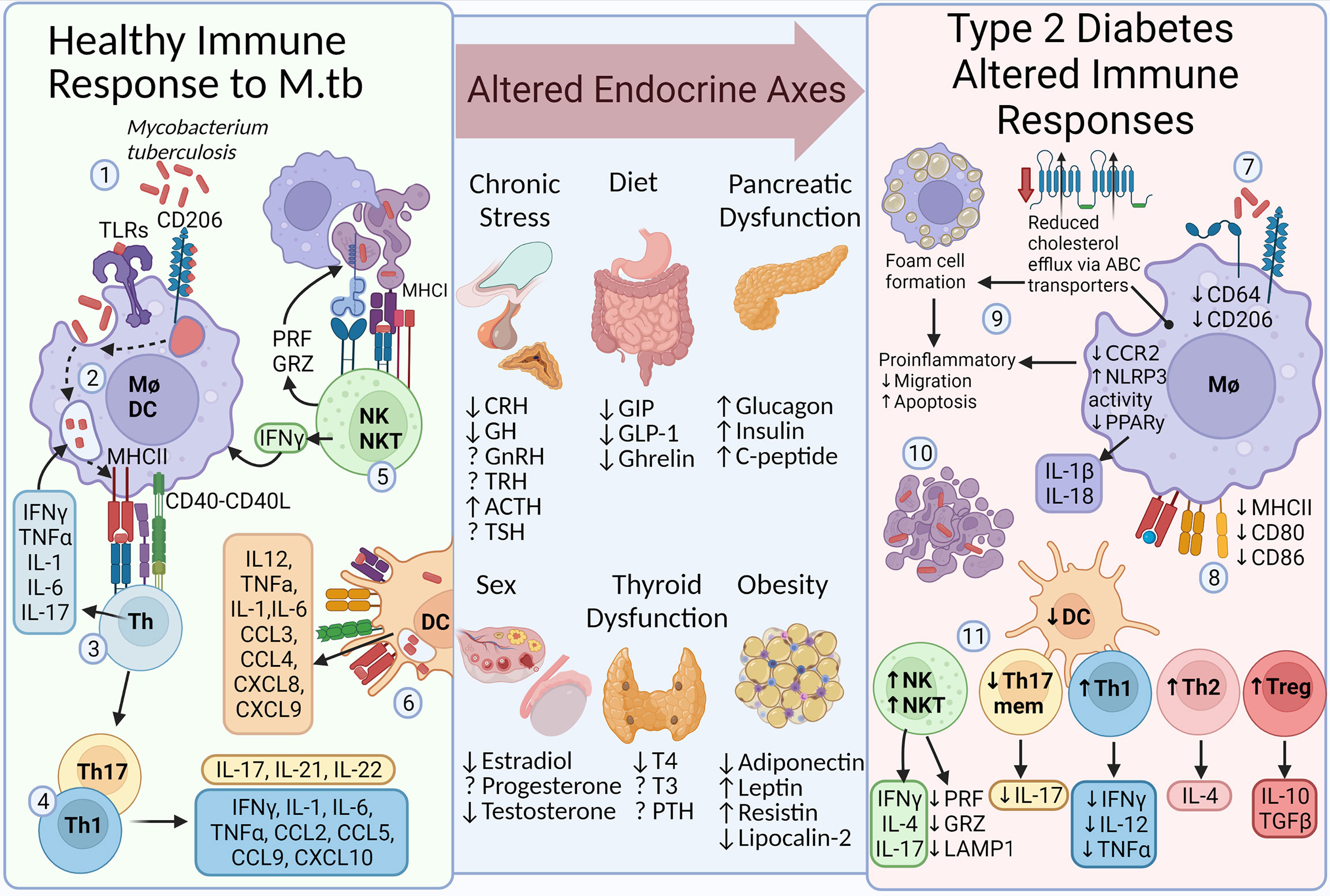
Figure 2 Altered endocrine signaling affects the appropriate immune response to infection. In an otherwise healthy immune system, the response to Mycobacterium tuberculosis includes several essential functions. 1, PAMP-PRR interaction (e.g. TLR4 and CD206) and phagocytosis. 2, Phagosome-lysosome fusion and presentation (MHCII). 3, Activation of adaptive immune responses (CD40-CD40L) enhances the innate killing response via cytokines (e.g. IFNγ). 4, Th1 and Th17 predominate, stimulating mycobacterial control and immune cell influx via cytokine and chemokine release. 5, NK cells induce apoptosis, allowing efferocytosis by macrophages, and enhance killing by IFNγ release. 6, DCs also promote killing, antigen presentation, and immune cell chemotaxis. The immune response is influenced by sex hormone production, diet-induced gastric hormones, stress-related responses from the brain stem, thyroid dysfunction, and the development of chronic metabolic diseases such as T2D and obesity. Thus, the immune response to M. tuberculosis is altered during T2D. 7, Macrophage PRR expression (e.g. CD64 and CD206), phagocytosis, and killing is diminished. 8, Expression of antigen presenting proteins is reduced. 9, Expression of cholesterol efflux transporters (ABC) is reduced, leading to cholesterol accumulation and foam cell formation. Foam cells have reduced migratory capacity and increased rate of apoptosis. 10, Reduced efferocytosis results in a cholesterol rich environment and providing a niche for mycobacterial growth. 11, Additionally, altered ratios of Th cell subsets and dendritic cells influence cytokine levels. PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; TLR, toll-like receptor; MHC, major histocompatibility complex; Mφ, macrophage; DC, Dendritic cell; Th, CD4+ helper T cell; PRF, perforin; GRZ, granzyme; Treg, regulatory T cell; ABC, ATP-binding cassette cholesterol transporter; T2D, Type 2 Diabetes; TB, Tuberculosis; CRH, corticotropin-releasing hormone; GH, growth hormone; GnRH, gonadotropin-releasing hormone; TRH, thyrotropin -releasing hormone; ACTH, adrenocorticotropic hormone; T4, Thyroxine; T3, triiodothyronine; TSH, thyroid-stimulating hormone; PTH, parathyroid hormone; GIP, gastric insulinotropic peptide; GLP-1, glucagon-like peptide 1; TSH, thyroid-stimulating hormone; DHEA, dehydroepiandrosterone; This figure was created with BioRender.com.
Clinically, T2D delays TB treatment response, while also increasing the risk of poor TB treatment outcomes, relapse, and death (187–189). In addition, more severe presentations of TB are seen in T2D patients (190). Comorbid T2D alters TB and latent TB infection adipokine levels, with the pro-inflammatory adipokine leptin levels increased, while anti-inflammatory adiponectin levels decreased compared to those of TB patients without T2D (23), Kumar et al. (2016) found this change to be independent of BMI (23, 31). However, others have found opposite results, for example, Zheng et al. (2013) found higher leptin levels in TB patients and lower levels in TB-T2D patients. This result is unexpected, since wasting during TB disease is usually accompanied by decreased adiposity and leptin levels (26). This study also found lower ghrelin levels in TB patients with and without T2D. These disparities may be due to different genetic and socio-economic backgrounds within the study populations. Clarity regarding these hormones may require larger studies in participants from diverse genetic backgrounds.
Discussion
Endocrine alterations contribute to changes in immune responses. While some hormones have been studied extensively for their immune-regulatory properties, few have been sufficiently investigated in the lung immune response to infection and compared to their effects outside the lung. Fewer still have been assessed in the lungs during T2D, a major endocrine dysregulating disease, and TB, a bacterial lung disease. While we have discussed more defined hormones in this review, others including progesterone, thyroid-stimulating hormone, parathyroid hormone, epinephrine, norepinephrine, melotonin, adipsin, require further investigation. Additionally, while some evidence suggests that receptors for hormones are not expressed in the lung tissue, such as C-peptide and ghrelin, other studies have described responses of lung tissue or immune cells to these hormones, indicating that alternative pathways of stimulation may be responsible for these effects or more sensitive methods are required for their evaluation in lung tissue. Elucidating the role of these hormones in modulating immune cell function in the lung will bridge a critical gap in our understanding of immune responses in the lungs.
Many studies thus far have investigated hormone-induced cellular responses in cell lines, peripheral blood cells, and in mouse and rat models. Cells under specific environmental conditions respond to hormonal stimulation differently. Thus, interpretation of these studies must be considered according to the cellular compartment or animal model they were performed in. While providing informative results, the relevance of these results to disease in the human lung remains to be validated with studies based on the human lung. Future studies assessing endocrine driven immune responses in the lung should incorporate human tissues or cells, like lung biopsies or resections and BALF, to build on current knowledge in this field with more relevance to human disease.
Understanding the factors contributing to the development, progression, and treatment response of lung diseases is crucial for improving the avenues of treatment, such as host-directed therapies, especially since antibiotics in development are deemed a short-term solution needing supplementation by other means of treatment (191). Hormone receptors may provide improved adjunctive therapeutic targets, given an improvement in our understanding of their functions in the lung under disease.
Author Contributions
TW and LK conceived the idea for the review. TW, LK, and KR reviewed the current literature. TW and LK wrote the manuscript. TW, LK, MCS, and KR critically reviewed the manuscript.
Funding
This study was supported by the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (NIAID) and the South African Medical Research Council under the US-South African Program for Collaborative Biomedical Research (grant number R01AI116039). TW was funded by the National Research Foundation of South Africa. The content is solely the responsibility of the authors and does not necessarily represent the official views of any of the funding agencies.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kleynhans L, Ruzive S, Ehlers L, Thiart L, Chegou NN, Conradie M, et al. Changes in Host Immune–Endocrine Relationships During Tuberculosis Treatment in Patients With Cured and Failed Treatment Outcomes. Front Immunol (2017) 8:690. doi: 10.3389/fimmu.2017.00690
2. Csaba G. Hormones in the Immune System and Their Possible Role. A Critical Review. Acta Microbiol Immunol Hung (2014) 61:241–60. doi: 10.1556/AMicr.61.2014.3.1
3. Del Rey A, Mahuad CV, Bozza VV, Bogue C, Farroni MA, Bay ML, et al. Endocrine and Cytokine Responses in Humans With Pulmonary Tuberculosis. Brain Behav Immun (2007) 21:171–9. doi: 10.1016/j.bbi.2006.06.005
4. Besedovsky HO, del Rey A, Klusman I, Furukawa H, Monge Arditi G, Kabiersch A. Cytokines as Modulators of the Hypothalamus-Pituitary-Adrenal Axis. J Steroid Biochem Mol Biol (1991) 40:613–8. doi: 10.1016/0960-0760(91)90284-c
5. Valentino R, Savastano S, Tommaselli AP, Riccio A, Mariniello P, Pronesti G, et al. Hormonal Pattern in Women Affected by Rheumatoid Arthritis. J Endocrinol Invest (1993) 16:619–24. doi: 10.1007/BF03347683
6. Guzmán C, Hernández-Bello R, Morales-Montor J. Regulation of Steroidogenesis in Reproductive, Adrenal and Neural Tissues by Cytokines. Open Neuroendocrinol J (2010) 3:161–9. doi: 10.2174/1876528901003010161
7. Tsoli M, Boutzios G, Kaltsas G. Immune System Effects on the Endocrine System. Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, et al, editors. Endotext. South Dartmouth (MA: MDText.com, Inc (2000).
8. Bereshchenko O, Bruscoli S, Riccardi C. Glucocorticoids, Sex Hormones, and Immunity. Front Immunol (2018) 9:1332. doi: 10.3389/fimmu.2018.01332
9. Fries E, Hesse J, Hellhammer J, Hellhammer DH. A New View on Hypocortisolism. Psychoneuroendocrinology (2005) 30:1010–6. doi: 10.1016/j.psyneuen.2005.04.006
10. Dhabhar FS. Effects of Stress on Immune Function: The Good, the Bad, and the Beautiful. Immunol Res (2014) 58:193–210. doi: 10.1007/s12026-014-8517-0
11. Reynolds RM, Labad J, Strachan MWJ, Braun A, Fowkes FGR, Lee AJ, et al. Elevated Fasting Plasma Cortisol Is Associated With Ischemic Heart Disease and Its Risk Factors in People With Type 2 Diabetes: The Edinburgh Type 2 Diabetes Study. J Clin Endocrinol Metab (2010) 95:1602–8. doi: 10.1210/jc.2009-2112
12. Dias JP, Joseph JJ, Kluwe B, Zhao S, Shardell M, Seeman T, et al. The Longitudinal Association of Changes in Diurnal Cortisol Features With Fasting Glucose: MESA. Psychoneuroendocrinology (2020) 119:104698. doi: 10.1016/j.psyneuen.2020.104698
13. Islam S, Yesmine S, Khan SA, Alam NH, Islam S. A Comparative Study of Thyroid Hormone Levels in Diabetic and Non-Diabetic Patients. Southeast Asian J Trop Med Public Health (2008) 39:913–6.
14. Cheung KKT, Luk AOY, So WY, Ma RCW, Kong APS, Chow FCC, et al. Testosterone Level in Men With Type 2 Diabetes Mellitus and Related Metabolic Effects: A Review of Current Evidence. J Diabetes Investig (2015) 6:112–23. doi: 10.1111/jdi.12288
15. Hashimoto K, Nishioka T, Takao T, Numata Y. Low Plasma Corticotropin-Releasin Hormone (CRH) Levels in Patients With Non-Insulin Dependent Diabetes Mellitus (NIDDM). Endocr J (1993) 40:705–9. doi: 10.1507/endocrj.40.705
16. Santucci N, D’Attilio L, Kovalevski L, Bozza V, Besedovsky H, del Rey A, et al. A Multifaceted Analysis of Immune-Endocrine-Metabolic Alterations in Patients With Pulmonary Tuberculosis. PLoS One (2011) 6:e26363. doi: 10.1371/journal.pone.0026363
17. Cho JH, Kim HJ, Lee JH, Park IR, Moon JS, Yoon JS, et al. Poor Glycemic Control Is Associated With the Risk of Subclinical Hypothyroidism in Patients With Type 2 Diabetes Mellitus. Korean J Intern Med (2016) 31:703–11. doi: 10.3904/kjim.2015.198
18. Fernández R, Díaz A, D’Attilio L, Bongiovanni B, Santucci N, Bertola D, et al. An Adverse Immune-Endocrine Profile in Patients With Tuberculosis and Type 2 Diabetes. Tuberculosis (2016) 101:95–101. doi: 10.1016/j.tube.2016.09.001
19. Al Hayek AA, Khader YS, Jafal S, Khawaja N, Robert AA, Ajlouni K. Prevalence of Low Testosterone Levels in Men With Type 2 Diabetes Mellitus: A Cross-Sectional Study. J Fam Community Med (2013) 20:179–86. doi: 10.4103/2230-8229.122006
20. Hofman A, Darwish Murad S, van Duijn CM, Franco OH, Goedegebure A, Ikram MA, et al. The Rotterdam Study: 2014 Objectives and Design Update. Eur J Epidemiol (2013) 28:889–926. doi: 10.1007/s10654-013-9866-z
21. Erbay G, Senol G, Anar C, Meral AR, Tuzel O. Relationship Between Tuberculosis and Female Hormone Levels in Post-Menopausal. Southeast Asian J Trop Med Public Health (2016) 47:78–83.
22. De la Chesnaye E, Manuel-Apolinar L, Zarate A, Damasio L, Espino N, Revilla-Monsalve MC, et al. Lipocalin-2 Plasmatic Levels Are Reduced in Patients With Long-Term Type 2 Diabetes Mellitus. Int J Clin Exp Med (2015) 8:2853–9.
23. Pavan Kumar N, Nair D, Banurekha VV, Dolla C, Kumaran P, Sridhar R, et al. Type 2 Diabetes Mellitus Coincident With Pulmonary or Latent Tuberculosis Results in Modulation of Adipocytokines. Cytokine (2016) 79:74–81. doi: 10.1016/j.cyto.2015.12.026
24. Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L, Egan JM. GIP Contributes to Islet Trihormonal Abnormalities in Type 2 Diabetes. J Clin Endocrinol Metab (2014) 99:2477–85. doi: 10.1210/jc.2013-3994
25. Pöykkö SM, Kellokoski E, Hörkkö S, Kauma H, Kesäniemi YA, Ukkola O. Low Plasma Ghrelin Is Associated With Insulin Resistance, Hypertension, and the Prevalence of Type 2 Diabetes. Diabetes (2003) 52:2546–53. doi: 10.2337/diabetes.52.10.2546
26. Zheng Y, Ma A, Wang Q, Han X, Cai J, Schouten EG, et al. Relation of Leptin, Ghrelin and Inflammatory Cytokines With Body Mass Index in Pulmonary Tuberculosis Patients With and Without Type 2 Diabetes Mellitus. PLoS One (2013) 8:e80122. doi: 10.1371/journal.pone.0080122
27. Chang SW, Pan WS, Beltran DL, Baldelomar LO, Solano MA, Tuero I, et al. Gut Hormones, Appetite Suppression and Cachexia in Patients With Pulmonary TB. PLoS One (2013) 8:e54564. doi: 10.1371/journal.pone.0054564
28. Omar-Hmeadi M, Lund P-E, Gandasi NR, Tengholm A, Barg S. Paracrine Control of α-Cell Glucagon Exocytosis Is Compromised in Human Type-2 Diabetes. Nat Commun (2020) 11:1896. doi: 10.1038/s41467-020-15717-8
29. Handelsman Y, Bloomgarden ZT, Grunberger G, Umpierrez G, Zimmerman RS, Bailey TS, et al. American Association of Clinical Endocrinologists and American College of Endocrinology – Clinical Practice Guidelines for Developing A Diabetes Mellitus Comprehensive Care Plan – 2015 — Executive Summary. Endocr Pract (2015) 21:413–37. doi: 10.4158/EP15672.GL
30. Kumari UR, Padma K. Dehydroepiandrosterone Levels in Type 2 Diabetes. Int J Med Res Health Sci (2014) 3:411–5. doi: 10.5958/j.2319-5886.3.2.083
31. Fernández RDV, Díaz A, Bongiovanni B, Gallucci G, Bértola D, Gardeñez W, et al. Evidence for a More Disrupted Immune-Endocrine Relation and Cortisol Immunologic Influences in the Context of Tuberculosis and Type 2 Diabetes Comorbidity. Front Endocrinol (2020) 11:126. doi: 10.3389/fendo.2020.00126
32. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and Treatment of Type 2 Diabetes: Perspectives on the Past, Present and Future. Lancet (2014) 383:1068–83. doi: 10.1016/S0140-6736(13)62154-6
33. Lee ZS, Chan JC, Yeung VT, Chow CC, Lau MS, Ko GT, et al. Plasma Insulin, Growth Hormone, Cortisol, and Central Obesity Among Young Chinese Type 2 Diabetic Patients. Diabetes Care (1999) 22:1450–7. doi: 10.2337/diacare.22.9.1450
34. Nowakowska M, Zghebi SS, Ashcroft DM, Buchan I, Chew-Graham C, Holt T, et al. The Comorbidity Burden of Type 2 Diabetes Mellitus: Patterns, Clusters and Predictions From a Large English Primary Care Cohort. BMC Med (2019) 17:145. doi: 10.1186/s12916-019-1373-y
35. Champaneri S, Wand GS, Malhotra SS, Casagrande SS, Golden SH. Biological Basis of Depression in Adults With Diabetes. Curr Diabetes Rep (2010) 10:396–405. doi: 10.1007/s11892-010-0148-9
36. Costanzo PR, Knoblovits P. Male Gonadal Axis Function in Patients With Type 2 Diabetes. Horm Mol Biol Clin Investig (2016) 26:129–34. doi: 10.1515/hmbci-2016-0014
37. Biondi B, Kahaly GJ, Robertson RP. Thyroid Dysfunction and Diabetes Mellitus: Two Closely Associated Disorders. Endocr Rev (2019) 40:789–824. doi: 10.1210/er.2018-00163
38. Kalantaridou S, Makrigiannakis A, Zoumakis E, Chrousos GP. Peripheral Corticotropin-Releasing Hormone Is Produced in the Immune and Reproductive Systems: Actions, Potential Roles and Clinical Implications. Front Biosci J Virtual Libr (2007) 12:572–80. doi: 10.2741/2083
39. Webster EL, Lewis DB, Torpy DJ, Zachman EK, Rice KC, Chrousos GP. In Vivo and In Vitro Characterization of Antalarmin, a Nonpeptide Corticotropin-Releasing Hormone (CRH) Receptor Antagonist: Suppression of Pituitary ACTH Release and Peripheral Inflammation. Endocrinology (1996) 137:5747–50. doi: 10.1210/endo.137.12.8940412
40. Hiroi N, Wong ML, Licinio J, Park C, Young M, Gold PW, et al. Expression of Corticotropin Releasing Hormone Receptors Type I and Type II mRNA in Suicide Victims and Controls. Mol Psychiatry (2001) 6:540–6. doi: 10.1038/sj.mp.4000908
41. Gonzales XF, Deshmukh A, Pulse M, Johnson K, Jones HP. Stress-Induced Differences in Primary and Secondary Resistance Against Bacterial Sepsis Corresponds With Diverse Corticotropin Releasing Hormone Receptor Expression by Pulmonary CD11c+ MHC II+ and CD11c- MHC II+ APCs. Brain Behav Immun (2008) 22:552–64. doi: 10.1016/j.bbi.2007.11.005
42. Kim B-J, Kayembe K, Simecka JW, Pulse M, Jones HP. Corticotropin-Releasing Hormone Receptor-1 and 2 Activity Produces Divergent Resistance Against Stress-Induced Pulmonary Streptococcus Pneumoniae Infection. J Neuroimmunol (2011) 237:57–65. doi: 10.1016/j.jneuroim.2011.06.016
43. Burnley B P, Jones H. Corticotropin-Releasing Hormone Improves Survival in Pneumococcal Pneumonia by Reducing Pulmonary Inflammation. Physiol Rep (2017) 5:e13000. doi: 10.14814/phy2.13000
44. Moffatt JD, Lever R, Page CP. Activation of Corticotropin-Releasing Factor Receptor-2 Causes Bronchorelaxation and Inhibits Pulmonary Inflammation in Mice. FASEB J (2006) 20:1877–9. doi: 10.1096/fj.05-5315fje
45. Brzoska T, Luger TA, Maaser C, Abels C, Böhm M. α-Melanocyte-Stimulating Hormone and Related Tripeptides: Biochemistry, Antiinflammatory and Protective Effects In Vitro and In Vivo, and Future Perspectives for the Treatment of Immune-Mediated Inflammatory Diseases. Endocr Rev (2008) 29:581–602. doi: 10.1210/er.2007-0027
46. Montero-Melendez T. ACTH: The Forgotten Therapy. Semin Immunol (2015) 27:216–26. doi: 10.1016/j.smim.2015.02.003
47. Raap U, Brzoska T, Sohl S, Päth G, Emmel J, Herz U, et al. α-Melanocyte-Stimulating Hormone Inhibits Allergic Airway Inflammation. J Immunol (2003) 171:353–9. doi: 10.4049/jimmunol.171.1.353
48. Colombo G, Gatti S, Sordi A, Turcatti F, Carlin A, Rossi C, et al. Production and Effects of α-Melanocyte-Stimulating Hormone During Acute Lung Injury. Shock (2007) 27:326–33. doi: 10.1097/01.shk.0000239764.80033.7e
49. Deng J, Hu X, Yuen PST, Star RA. Alpha-Melanocyte-Stimulating Hormone Inhibits Lung Injury After Renal Ischemia/Reperfusion. Am J Respir Crit Care Med (2004) 169:749–56. doi: 10.1164/rccm.200303-372OC
50. Ericson MD, Lensing CJ, Fleming KA, Schlasner KN, Doering SR, Haskell-Luevano C. Bench-Top to Clinical Therapies: A Review of Melanocortin Ligands From 1954 to 2016. Biochim Biophys Acta BBA Mol Basis Dis (2017) 1863:2414–35. doi: 10.1016/j.bbadis.2017.03.020
51. Zhang C, Cui T, Cai R, Wangpaichitr M, Mirsaeidi M, Schally AV, et al. Growth Hormone-Releasing Hormone in Lung Physiology and Pulmonary Disease. Cells (2020) 9:2331. doi: 10.3390/cells9102331
52. Christodoulou C, Schally AV, Chatzistamou I, Kondi-Pafiti A, Lamnissou K, Kouloheri S, et al. Expression of Growth Hormone-Releasing Hormone (GHRH) and Splice Variant of GHRH Receptors in Normal Mouse Tissues. Regul Pept (2006) 136:105–8. doi: 10.1016/j.regpep.2006.05.001
53. Uddin MA, Akhter MS, Singh SS, Kubra K-T, Schally AV, Jois S, et al. GHRH Antagonists Support Lung Endothelial Barrier Function. Tissue Barriers (2019) 7:1669989. doi: 10.1080/21688370.2019.1669989
54. Zhang C, Cai R, Lazerson A, Delcroix G, Wangpaichitr M, Mirsaeidi M, et al. Growth Hormone-Releasing Hormone Receptor Antagonist Modulates Lung Inflammation and Fibrosis Due to Bleomycin. Lung (2019) 197:541–9. doi: 10.1007/s00408-019-00257-w
55. Zhang C, Tian R, Dreifus EM, Holt G, Cai R, Griswold A, et al. Activity of the GHRH Antagonist MIA602 and Its Underlying Mechanisms of Action in Sarcoidosis. Clin Transl immunol (2021) 5:e1310. doi: 10.1002/cti2.1310
56. Farhat K, Bodart G, Charlet-Renard C, Desmet CJ, Moutschen M, Beguin Y, et al. Growth Hormone (GH) Deficient Mice With GHRH Gene Ablation Are Severely Deficient in Vaccine and Immune Responses Against Streptococcus Pneumoniae. Front Immunol (2018) 9:2175. doi: 10.3389/fimmu.2018.02175
57. Liu Z, Yu Y, Jiang Y, Li J. Growth Hormone Increases Circulating Neutrophil Activation and Provokes Lung Microvascular Injury in Septic Peritonitis Rats. J Surg Res (2002) 105:195–9. doi: 10.1006/jsre.2002.6374
58. Liu Z-H, Yu Y-Q, Li W-Q, Li J-S. Growth Hormone Increases Lung Microvascular Injury in Lipopolysaccharide Peritonitis Rats: Possible Involvement of NF-kappaB Activation in Circulating Neutrophils. Acta Pharmacol Sin (2002) 23:887–92.
59. Yi C, Wang SR, Zhang SY, Yu SJ, Jiang CX, Zhi MH, et al. Effects of Recombinant Human Growth Hormone on Acute Lung Injury in Endotoxemic Rats. Inflammation Res (2006) 55:491–7. doi: 10.1007/s00011-006-6011-4
60. Yi C, Cao Y, Mao SH, Liu H, Ji LL, Xu SY, et al. Recombinant Human Growth Hormone Improves Survival and Protects Against Acute Lung Injury in Murine Staphylococcus Aureus Sepsis. Inflamm Res (2009) 58:855–62. doi: 10.1007/s00011-009-0056-0
61. Kühn ER, Bellon K, Huybrechts L, Heyns W. Endocrine Differences Between the Wistar and Sprague-Dawley Laboratory Rat: Influence of Cold Adaptation. Horm Metab Res Horm Stoffwechselforschung Horm Metab (1983) 15:491–8. doi: 10.1055/s-2007-1018767
62. Suzuki T, Sasano H, Suzuki S, Hirasawa G, Takeyama J, Muramatsu Y, et al. 11Beta-Hydroxysteroid Dehydrogenase Type 2 in Human Lung: Possible Regulator of Mineralocorticoid Action. J Clin Endocrinol Metab (1998) 83:4022–5. doi: 10.1210/jcem.83.11.5227
63. Gudewicz PW, Ferguson JL, Kapin MA, Jaffe RC. The Effects of Cortisone Treatment and Burn Injury on Plasma and Lung Lavage Cortisol Concentrations and Alveolar Macrophage Activity. Adv Shock Res (1981) 5:123–32.
64. Hostettler N, Bianchi P, Gennari-Moser C, Kassahn D, Schoonjans K, Corazza N, et al. Local Glucocorticoid Production in the Mouse Lung Is Induced by Immune Cell Stimulation. Allergy (2012) 67:227–34. doi: 10.1111/j.1398-9995.2011.02749.x
65. Landgraf MA, Silva RC, Corrêa-Costa M, Hiyane MI, Carvalho MHC, Landgraf RG, et al. Leptin Downregulates LPS-Induced Lung Injury: Role of Corticosterone and Insulin. Cell Physiol Biochem (2014) 33:835–46. doi: 10.1159/000358656
66. Marin-Luevano SP, Rodriguez-Carlos A, Jacobo-Delgado Y, Valdez-Miramontes C, Enciso-Moreno JA, Rivas-Santiago B. Steroid Hormone Modulates the Production of Cathelicidin and Human β-Defensins in Lung Epithelial Cells and Macrophages Promoting Mycobacterium Tuberculosis Killing. Tuberc Edinb Scotl (2021) 128:102080. doi: 10.1016/j.tube.2021.102080
67. Miravitlles M, Auladell-Rispau A, Monteagudo M, Vázquez-Niebla JC, Mohammed J, Nuñez A, et al. Systematic Review on Long-Term Adverse Effects of Inhaled Corticosteroids in the Treatment of COPD. Eur Respir Rev (2021) 30:210075. doi: 10.1183/16000617.0075-2021
68. Lu S-F, Mo Q, Hu S, Garippa C, Simon NG. Dehydroepiandrosterone Upregulates Neural Androgen Receptor Level and Transcriptional Activity. J Neurobiol (2003) 57:163–71. doi: 10.1002/neu.10260
69. Webb SJ, Geoghegan TE, Prough RA, Miller KKM. The Biological Actions of Dehydroepiandrosterone Involves Multiple Receptors. Drug Metab Rev (2006) 38:89–116. doi: 10.1080/03602530600569877
70. Oka M, Karoor V, Homma N, Nagaoka T, Sakao E, Golembeski SM, et al. Dehydroepiandrosterone Upregulates Soluble Guanylate Cyclase and Inhibits Hypoxic Pulmonary Hypertension. Cardiovasc Res (2007) 74:377–87. doi: 10.1016/j.cardiores.2007.01.021
71. Paulin R, Meloche J, Jacob MH, Bisserier M, Courboulin A, Bonnet S. Dehydroepiandrosterone Inhibits the Src/STAT3 Constitutive Activation in Pulmonary Arterial Hypertension. Am J Physiol Heart Circ Physiol (2011) 301:H1798–1809. doi: 10.1152/ajpheart.00654.2011
72. Corsini E, Lucchi L, Meroni M, Racchi M, Solerte B, Fioravanti M, et al. In Vivo Dehydroepiandrosterone Restores Age-Associated Defects in the Protein Kinase C Signal Transduction Pathway and Related Functional Responses. J Immunol (2002) 168:1753–8. doi: 10.4049/jimmunol.168.4.1753
73. Rom WN, Harkin T. Dehydroepiandrosterone Inhibits the Spontaneous Release of Superoxide Radical by Alveolar Macrophages In Vitro in Asbestosis. Environ Res (1991) 55:145–56. doi: 10.1016/S0013-9351(05)80171-9
74. Choi IS, Cui Y, Koh Y-A, Lee H-C, Cho Y-B, Won Y-H. Effects of Dehydroepiandrosterone on Th2 Cytokine Production in Peripheral Blood Mononuclear Cells From Asthmatics. Korean J Intern Med (2008) 23:176–81. doi: 10.3904/kjim.2008.23.4.176
75. López-Torres MO, Marquina-Castillo B, Ramos-Espinosa O, Mata-Espinosa D, Barrios-Payan JA, Baay-Guzman G, et al. 16α-Bromoepiandrosterone as a New Candidate for Experimental Diabetes-Tuberculosis Co-Morbidity Treatment. Clin Exp Immunol (2021) 205:232–45. doi: 10.1111/cei.13603
76. Shahrara S, Drvota V, Sylven C. Organ Specific Expression of Thyroid Hormone Receptor mRNA and Protein in Different Human Tissues. Biol Pharm Bull (1999) 22:1027–33. doi: 10.1248/bpb.22.1027
77. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-Based Map of the Human Proteome. Science (2015) 347:1260419. doi: 10.1126/science.1260419
78. Smith DM, Hitchcock KR. Thyroid Hormone Binding to Adult Rat Alveolar Type II Cells. An Autoradiographic Study. Exp Lung Res (1983) 5:141–53. doi: 10.3109/01902148309061510
79. Morishige WK. Thyroid Hormone Influences Glucocorticoid Receptor Levels in the Neonatal Rat Lung. Endocrinology (1982) 111:1017–9. doi: 10.1210/endo-111-3-1017
80. Scarpace PJ, Abrass IB. Thyroid Hormone Regulation of Rat Heart, Lymphocyte, and Lung β-Adrenergic Receptors. Endocrinology (1981) 108:1007–11. doi: 10.1210/endo-108-3-1007
81. Lei J, Mariash CN, Bhargava M, Wattenberg EV, Ingbar DH. T3 Increases Na-K-ATPase Activity via a MAPK/ERK1/2-Dependent Pathway in Rat Adult Alveolar Epithelial Cells. Am J Physiol-Lung Cell Mol Physiol (2008) 294:L749–54. doi: 10.1152/ajplung.00335.2007
82. Ning Y, Zhang M, Du Y-H, Zhang H-N, Li L-Y, Qin Y-W, et al. [Effects of Thyroid Hormone on Macrophage Dysfunction Induced by Oxidized Low-Density Lipoprotein]. Sheng Li Xue Bao (2018) 70:141–8.
83. Barca-Mayo O, Liao X-H, DiCosmo C, Dumitrescu A, Moreno-Vinasco L, Wade MS, et al. Role of Type 2 Deiodinase in Response to Acute Lung Injury (ALI) in Mice. Proc Natl Acad Sci U S A (2011) 108:E1321–9. doi: 10.1073/pnas.1109926108
84. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, et al. A Cellular Census of Human Lungs Identifies Novel Cell States in Health and in Asthma. Nat Med (2019) 25:1153–63. doi: 10.1038/s41591-019-0468-5
85. Mikkonen L, Pihlajamaa P, Sahu B, Zhang F-P, Jänne OA. Androgen Receptor and Androgen-Dependent Gene Expression in Lung. Mol Cell Endocrinol (2010) 317:14–24. doi: 10.1016/j.mce.2009.12.022
86. Melgert BN, Postma DS, Kuipers I, Geerlings M, Luinge MA, van der Strate BWA, et al. Female Mice are More Susceptible to the Development of Allergic Airway Inflammation Than Male Mice. Clin Exp Allergy J Br Soc Allergy Clin Immunol (2005) 35:1496–503. doi: 10.1111/j.1365-2222.2005.02362.x
87. Becerra-Díaz M, Strickland AB, Keselman A, Heller NM. Androgen and Androgen Receptor as Enhancers of M2 Macrophage Polarization in Allergic Lung Inflammation. J Immunol (2018) 201:2923–33. doi: 10.4049/jimmunol.1800352
88. vom Steeg LG, Dhakal S, Woldetsadik YA, Park H-S, Mulka KR, Reilly EC, et al. Androgen Receptor Signaling in the Lungs Mitigates Inflammation and Improves the Outcome of Influenza in Mice. PLoS Pathog (2020) 16:e1008506. doi: 10.1371/journal.ppat.1008506
89. Becerra-Diaz M, Song M, Heller N. Androgen and Androgen Receptors as Regulators of Monocyte and Macrophage Biology in the Healthy and Diseased Lung. Front Immunol (2020) 11:1698. doi: 10.3389/fimmu.2020.01698
90. Mollerup S, Jørgensen K, Berge G, Haugen A. Expression of Estrogen Receptors Alpha and Beta in Human Lung Tissue and Cell Lines. Lung Cancer Amst Neth (2002) 37:153–9. doi: 10.1016/s0169-5002(02)00039-9
91. Carvalho O, Gonçalves C. Expression of Oestrogen Receptors in Foetal Lung Tissue of Mice: Expression of Oestrogen Receptors. Anat Histol Embryol (2012) 41:1–6. doi: 10.1111/j.1439-0264.2011.01096.x
92. Knapczyk K, Duda M, Szafranska B, Wolsza K, Panasiewicz G, Koziorowski M, et al. Immunolocalisation of Oestrogen Receptors Alpha (Erα) and Beta (Erβ) in Porcine Embryos and Fetuses at Different Stages of Gestation. Acta Vet Hung (2008) 56:221–33. doi: 10.1556/avet.56.2008.2.10
93. Beyer C, Küppers E, Karolczak M, Trotter A. Ontogenetic Expression of Estrogen and Progesterone Receptors in the Mouse Lung. Neonatology (2003) 84:59–63. doi: 10.1159/000071445
94. Kan W-H, Hsu J-T, Schwacha MG, Choudhry MA, Bland KI, Chaudry IH. Estrogen Ameliorates Trauma-Hemorrhage–Induced Lung Injury via Endothelial Nitric Oxide Synthase-Dependent Activation of Protein Kinase G. Ann Surg (2008) 248:294–302. doi: 10.1097/SLA.0b013e318180a3db
95. Fan Q, Zhao P, Li J, Xie X, Xu M, Zhang Y, et al. 17β-Estradiol Administration Attenuates Seawater Aspiration-Induced Acute Lung Injury in Rats. Pulm Pharmacol Ther (2011) 24:673–81. doi: 10.1016/j.pupt.2011.07.002
96. Vegeto E, Cuzzocrea S, Crisafulli C, Mazzon E, Sala A, Krust A, et al. Estrogen Receptor-α as a Drug Target Candidate for Preventing Lung Inflammation. Endocrinology (2010) 151:174–84. doi: 10.1210/en.2009-0876
97. Nogi Y, Nagashima M, Terasaki M, Nohtomi K, Watanabe T, Hirano T. Glucose-Dependent Insulinotropic Polypeptide Prevents the Progression of Macrophage-Driven Atherosclerosis in Diabetic Apolipoprotein E-Null Mice. PLoS One (2012) 7:e35683. doi: 10.1371/journal.pone.0035683
98. Kahles F, Liberman A, Halim C, Rau M, Möllmann J, Mertens RW, et al. The Incretin Hormone GIP Is Upregulated in Patients With Atherosclerosis and Stabilizes Plaques in ApoE-/- Mice by Blocking Monocyte/Macrophage Activation. Mol Metab (2018) 14:150–7. doi: 10.1016/j.molmet.2018.05.014
99. Dunphy JL, Taylor RG, Fuller PJ. Tissue Distribution of Rat Glucagon Receptor and GLP-1 Receptor Gene Expression. Mol Cell Endocrinol (1998) 141:179–86. doi: 10.1016/s0303-7207(98)00096-3
100. Viby N-E, Isidor MS, Buggeskov KB, Poulsen SS, Hansen JB, Kissow H. Glucagon-Like Peptide-1 (GLP-1) Reduces Mortality and Improves Lung Function in a Model of Experimental Obstructive Lung Disease in Female Mice. Endocrinology (2013) 154:4503–11. doi: 10.1210/en.2013-1666
101. Lee Y-S, Jun H-S. Anti-Inflammatory Effects of GLP-1-Based Therapies Beyond Glucose Control. Mediators Inflamm (2016) 2016:e3094642. doi: 10.1155/2016/3094642
102. Zhu T, Wu X, Zhang W, Xiao M. Glucagon Like Peptide-1 (GLP-1) Modulates OVA-Induced Airway Inflammation and Mucus Secretion Involving a Protein Kinase A (PKA)-Dependent Nuclear Factor-κb (NF-κb) Signaling Pathway in Mice. Int J Mol Sci (2015) 16:20195–211. doi: 10.3390/ijms160920195
103. Gou S, Zhu T, Wang W, Xiao M, Wang X, Chen Z. Glucagon Like Peptide-1 Attenuates Bleomycin-Induced Pulmonary Fibrosis, Involving the Inactivation of NF-κb in Mice. Int Immunopharmacol (2014) 22:498–504. doi: 10.1016/j.intimp.2014.07.010
104. Balk-Møller E, Windeløv JA, Svendsen B, Hunt J, Ghiasi SM, Sørensen CM, et al. Glucagon-Like Peptide 1 and Atrial Natriuretic Peptide in a Female Mouse Model of Obstructive Pulmonary Disease. J Endocr Soc (2019) 4:bvz034. doi: 10.1210/jendso/bvz034
105. Hosokawa S, Haraguchi G, Maejima Y, Doi S, Isobe M. Abstract 13535: The Synergistic Effects of Incretin-Related Drugs for the Treatment of Pulmonary Arterial Hypertension. Circulation (2014) 130:A13535–5. doi: 10.1161/circ.130.suppl_2.13535
106. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide From Stomach. Nature (1999) 402:656–60. doi: 10.1038/45230
107. Kitazawa T, Nakamura T, Saeki A, Teraoka H, Hiraga T, Kaiya H. Molecular Identification of Ghrelin Receptor (GHS-R1a) and Its Functional Role in the Gastrointestinal Tract of the Guinea-Pig. Peptides (2011) 32:1876–86. doi: 10.1016/j.peptides.2011.07.026
108. Sun Y, Garcia JM, Smith RG. Ghrelin and Growth Hormone Secretagogue Receptor Expression in Mice During Aging. Endocrinology (2007) 148:1323–9. doi: 10.1210/en.2006-0782
109. Ghelardoni S, Carnicelli V, Frascarelli S, Ronca-Testoni S, Zucchi R. Ghrelin Tissue Distribution: Comparison Between Gene and Protein Expression. J Endocrinol Invest (2006) 29:115–21. doi: 10.1007/BF03344083
110. Gnanapavan S, Kola B, Bustin SA, Morris DG, McGee P, Fairclough P, et al. The Tissue Distribution of the mRNA of Ghrelin and Subtypes of Its Receptor, GHS-R, in Humans. J Clin Endocrinol Metab (2002) 87:2988–91. doi: 10.1210/jcem.87.6.8739
111. Volante M, Fulcheri E, Allìa E, Cerrato M, Pucci A, Papotti M. Ghrelin Expression in Fetal, Infant, and Adult Human Lung. J Histochem Cytochem (2002) 50:1013–21. doi: 10.1177/002215540205000803
112. Ueberberg B, Unger N, Saeger W, Mann K, Petersenn S. Expression of Ghrelin and Its Receptor in Human Tissues. Horm Metab Res (2009) 41:814–21. doi: 10.1055/s-0029-1233462
113. Peng Z, Zhu Y, Zhang Y, Wilhelmsen K, Jia C, Jin J, et al. Effects of Ghrelin on Pulmonary NOD2 mRNA Expression and NF-κb Activation When Protects Against Acute Lung Injury in Rats Challenged With Cecal Ligation and Puncture. Int Immunopharmacol (2012) 13:440–5. doi: 10.1016/j.intimp.2012.04.006
114. Harvey RE, Howard VG, Lemus MB, Jois T, Andrews ZB, Sleeman MW. The Ghrelin/GOAT System Regulates Obesity-Induced Inflammation in Male Mice. Endocrinology (2017) 158:2179–89. doi: 10.1210/en.2016-1832
115. Liu H, Luo J, Guillory B, Chen J, Zang P, Yoeli JK, et al. (In-Gi), Anderson B, Storie M, Et al. Ghrelin Ameliorates Tumor-Induced Adipose Tissue Atrophy and Inflammation via Ghrelin Receptor-Dependent and -Independent Pathways. Oncotarget (2020) 11:3286–302. doi: 10.18632/oncotarget.27705
116. Li B, Zeng M, He W, Huang X, Luo L, Zhang H, et al. Ghrelin Protects Alveolar Macrophages Against Lipopolysaccharide-Induced Apoptosis Through Growth Hormone Secretagogue Receptor 1a-Dependent C-Jun N-Terminal Kinase and Wnt/β-Catenin Signaling and Suppresses Lung Inflammation. Endocrinology (2015) 156:203–17. doi: 10.1210/en.2014-1539
117. Wu R, Dong W, Zhou M, Zhang F, Marini CP, Ravikumar TS, et al. Ghrelin Attenuates Sepsis-Induced Acute Lung Injury and Mortality in Rats. Am J Respir Crit Care Med (2007) 176:805–13. doi: 10.1164/rccm.200604-511OC
118. Rocha N de N, de Oliveira MV, Braga CL, Guimarães G, de Albuquerque Maia L, de Araújo Padilha G, et al. Ghrelin Therapy Improves Lung and Cardiovascular Function in Experimental Emphysema. Respir Res (2017) 18:185. doi: 10.1186/s12931-017-0668-9
119. Henriques-Coelho T, Correia-Pinto J, Roncon-Albuquerque R, Baptista MJ, Lourenço AP, Oliveira SM, et al. Endogenous Production of Ghrelin and Beneficial Effects of its Exogenous Administration in Monocrotaline-Induced Pulmonary Hypertension. Am J Physiol-Heart Circ Physiol (2004) 287:H2885–90. doi: 10.1152/ajpheart.01122.2003
120. Yosten GLC, Kolar GR, Redlinger LJ, Samson WK. Evidence for an Interaction Between Proinsulin C-Peptide and GPR146. J Endocrinol (2013) 218:B1–8. doi: 10.1530/JOE-13-0203
121. Lindfors L, Sundström L, Fröderberg Roth L, Meuller J, Andersson S, Kihlberg J. Is GPR146 Really the Receptor for Proinsulin C-Peptide? Bioorg Med Chem Lett (2020) 30:127208. doi: 10.1016/j.bmcl.2020.127208
122. Marx N, Walcher D, Raichle C, Aleksic M, Bach, Grüb, et al. C-Peptide Colocalizes With Macrophages in Early Arteriosclerotic Lesions of Diabetic Subjects and Induces Monocyte Chemotaxis. In Vitro Arterioscler Thromb Vasc Biol (2004) 24:540–5. doi: 10.1161/01.ATV.0000116027.81513.68
123. Chima RS, LaMontagne T, Piraino G, Hake PW, Denenberg A, Zingarelli B. C-Peptide, a Novel Inhibitor of Lung Inflammation Following Hemorrhagic Shock. Am J Physiol-Lung Cell Mol Physiol (2011) 300:L730–9. doi: 10.1152/ajplung.00308.2010
124. Jeon H-Y, Lee Y-J, Kim Y-S, Kim S-Y, Han E-T, Park WS, et al. Proinsulin C-Peptide Prevents Hyperglycemia-Induced Vascular Leakage and Metastasis of Melanoma Cells in the Lungs of Diabetic Mice. FASEB J (2019) 33:750–62. doi: 10.1096/fj.201800723R
125. Kao RLC, Xu X, Xenocostas A, Parry N, Mele T, Martin CM, et al. C-Peptide Attenuates Acute Lung Inflammation in a Murine Model of Hemorrhagic Shock and Resuscitation by Reducing Gut Injury. J Trauma Acute Care Surg (2017) 83:256–62. doi: 10.1097/TA.0000000000001539
126. Vish MG, Mangeshkar P, Piraino G, Denenberg A, Hake PW, O’Connor M, et al. Proinsulin C-Peptide Exerts Beneficial Effects in Endotoxic Shock in Mice. Crit Care Med (2007) 35:1348–55. doi: 10.1097/01.CCM.0000260245.61343.B3
127. Belfiore A, Malaguarnera R, Vella V, Lawrence MC, Sciacca L, Frasca F, et al. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr Rev (2017) 38:379–431. doi: 10.1210/er.2017-00073
128. Benecke H, Flier JS, Moller DE. Alternatively Spliced Variants of the Insulin Receptor Protein. Expression in Normal and Diabetic Human Tissues. J Clin Invest (1992) 89:2066–70. doi: 10.1172/JCI115819
129. Leibiger B, Leibiger IB, Moede T, Kemper S, Kulkarni RN, Kahn CR, et al. Selective Insulin Signaling Through A and B Insulin Receptors Regulates Transcription of Insulin and Glucokinase Genes in Pancreatic β Cells. Mol Cell (2001) 7:559–70. doi: 10.1016/S1097-2765(01)00203-9
130. Devaskar SU, Ganguli S, Devaskar UP, Sperling MA. Glucocorticoids and Hypothyroidism Modulate Development of Fetal Lung Insulin Receptors. Am J Physiol-Endocrinol Metab (1982) 242:E384–91. doi: 10.1152/ajpendo.1982.242.6.E384
131. Ma Y, He Q. [Study of the Role of Insulin and Insulin Receptor in Allergic Airway Inflammation of Rats]. Zhonghua Yi Xue Za Zhi (2005) 85:3419–24.
132. Di Petta A, Greco KV, Castro EO, Lopes FDTQS, Martins MA, Capelozzi VL, et al. Insulin Modulates Inflammatory and Repair Responses to Elastase-Induced Emphysema in Diabetic Rats. Int J Exp Pathol (2011) 92:392–9. doi: 10.1111/j.1365-2613.2011.00787.x
133. Martins J, Ferracini M, Ravanelli N, Landgraf RG, Jancar S. Insulin Inhibits LPS-Induced Signaling Pathways in Alveolar Macrophages. Cell Physiol Biochem (2008) 21:297–304. doi: 10.1159/000129388
134. Tessaro FHG, Ayala TS, Nolasco EL, Bella LM, Martins JO. Insulin Influences LPS-Induced TNF-α and IL-6 Release Through Distinct Pathways in Mouse Macrophages From Different Compartments. Cell Physiol Biochem (2017) 42:2093–104. doi: 10.1159/000479904
135. Iwanij V, Vincent AC. Characterization of the Glucagon Receptor and Its Functional Domains Using Monoclonal Antibodies. J Biol Chem (1990) 265:21302–8.
136. Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative Quantitative Analysis of Glucagon Receptor mRNA in Rat Tissues. Mol Cell Endocrinol (1994) 105:131–7. doi: 10.1016/0303-7207(94)90162-7
137. Burcelin R, Li J, Charron MJ. Cloning and Sequence Analysis of the Murine Glucagon Receptor-Encoding Gene. Gene (1995) 164:305–10. doi: 10.1016/0378-1119(95)00472-I
138. Koh WS, Lee M, Yang K-H, Kaminski NE. Expression of Functional Glucagon Receptors on Lymphoid Cells. Life Sci (1996) 58:741–51. doi: 10.1016/0024-3205(95)02352-6
139. Melanson SW, Bonfante G, Heller MB. Nebulized Glucagon in the Treatment of Bronchospasm in Asthmatic Patients. Am J Emerg Med (1998) 16:272–5. doi: 10.1016/S0735-6757(98)90100-0
140. Insuela DBR, Daleprane JB, Coelho LP, Silva AR, Silva PMRe, Martins MA, et al. Glucagon Induces Airway Smooth Muscle Relaxation by Nitric Oxide and Prostaglandin E2. J Endocrinol (2015) 225:205–17. doi: 10.1530/JOE-14-0648
141. Insuela DBR, Azevedo CT, Coutinho DS, Magalhães NS, Ferrero MR, Ferreira TPT, et al. Glucagon Reduces Airway Hyperreactivity, Inflammation, and Remodeling Induced by Ovalbumin. Sci Rep (2019) 9:6478. doi: 10.1038/s41598-019-42981-6
142. Ye R, Scherer PE. Adiponectin, Driver or Passenger on the Road to Insulin Sensitivity? Mol Metab (2013) 2:133–41. doi: 10.1016/j.molmet.2013.04.001
143. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of Adiponectin Receptors That Mediate Antidiabetic Metabolic Effects. Nature (2003) 423:762–9. doi: 10.1038/nature01705
144. Shore SA, Terry RD, Flynt L, Xu A, Hug C, Kong H. Adiponectin Attenuates Allergen-Induced Airway Inflammation and Hyperresponsiveness in Mice. J Allergy Clin Immunol (2006) 118:389–95. doi: 10.1016/j.jaci.2006.04.021
145. Amarsaikhan N, Tsoggerel A, Hug C, Templeton SP. The Metabolic Cytokine Adiponectin Inhibits Inflammatory Lung Pathology in Invasive Aspergillosis. J Immunol (2019) 203:956–63. doi: 10.4049/jimmunol.1900174
146. Garcia P, Sood A. Adiponectin in Pulmonary Disease and Critically Ill Patients. Curr Med Chem (2012) 19:5493–500. doi: 10.2174/092986712803833263
147. Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin Promotes Macrophage Polarization Toward an Anti-Inflammatory Phenotype*. J Biol Chem (2010) 285:6153–60. doi: 10.1074/jbc.M109.088708
148. Yamashita T, Lakota K, Taniguchi T, Yoshizaki A, Sato S, Hong W, et al. An Orally-Active Adiponectin Receptor Agonist Mitigates Cutaneous Fibrosis, Inflammation and Microvascular Pathology in a Murine Model of Systemic Sclerosis. Sci Rep (2018) 8:11843. doi: 10.1038/s41598-018-29901-w
149. Okada-Iwabu M, Yamauchi T, Iwabu M, Honma T, Hamagami K, Matsuda K, et al. A Small-Molecule AdipoR Agonist for Type 2 Diabetes and Short Life in Obesity. Nature (2013) 503:493–9. doi: 10.1038/nature12656
150. Pérez-Pérez A, Vilariño-García T, Fernández-Riejos P, Martín-González J, Segura-Egea JJ, Sánchez-Margalet V. Role of Leptin as a Link Between Metabolism and the Immune System. Cytokine Growth Factor Rev (2017) 35:71–84. doi: 10.1016/j.cytogfr.2017.03.001
151. Löllmann B, Grüninger S, Stricker-Krongrad A, Chiesi M. Detection and Quantification of the Leptin Receptor Splice Variants Ob-Ra, B, and, E in Different Mouse Tissues. Biochem Biophys Res Commun (1997) 238:648–52. doi: 10.1006/bbrc.1997.7205
152. Tsuchiya T, Shimizu H, Horie T, Mori M. Expression of Leptin Receptor in Lung: Leptin as a Growth Factor. Eur J Pharmacol (1999) 365:273–9. doi: 10.1016/S0014-2999(98)00884-X
153. Lemos MP, Rhee KY, McKinney JD. Expression of the Leptin Receptor Outside of Bone Marrow-Derived Cells Regulates Tuberculosis Control and Lung Macrophage MHC Expression. J Immunol (2011) 187:3776–84. doi: 10.4049/jimmunol.1003226
154. Vernooy JHJ, Ubags NDJ, Brusselle GG, Tavernier J, Suratt BT, Joos GF, et al. Leptin as Regulator of Pulmonary Immune Responses: Involvement in Respiratory Diseases. Pulm Pharmacol Ther (2013) 26:464–72. doi: 10.1016/j.pupt.2013.03.016
155. Suzukawa M, Koketsu R, Baba S, Igarashi S, Nagase H, Yamaguchi M, et al. Leptin Enhances ICAM-1 Expression, Induces Migration and Cytokine Synthesis, and Prolongs Survival of Human Airway Epithelial Cells. Am J Physiol Lung Cell Mol Physiol (2015) 309:L801–811. doi: 10.1152/ajplung.00365.2014
156. Watanabe K, Suzukawa M, Arakawa S, Kobayashi K, Igarashi S, Tashimo H, et al. Leptin Enhances Cytokine/Chemokine Production by Normal Lung Fibroblasts by Binding to Leptin Receptor. Allergol Int (2019) 68S:S3–8. doi: 10.1016/j.alit.2019.04.002
157. Zammit C, Liddicoat H, Moonsie I, Makker H. Obesity and Respiratory Diseases. Int J Gen Med (2010) 3:335–43. doi: 10.2147/IJGM.S11926
158. Zhang P-X, Zhang F-R, Xie J-J, Tao L-H, Lü Z, Xu X-E, et al. Expression of NGAL and NGALR in Human Embryonic, Fetal and Normal Adult Tissues. Mol Med Rep (2012) 6:716–22. doi: 10.3892/mmr.2012.980
159. Guo H, Jin D, Chen X. Lipocalin 2 Is a Regulator Of Macrophage Polarization and NF-κb/STAT3 Pathway Activation. Mol Endocrinol (2014) 28:1616–28. doi: 10.1210/me.2014-1092
160. Shao S, Cao T, Jin L, Li B, Fang H, Zhang J, et al. Increased Lipocalin-2 Contributes to the Pathogenesis of Psoriasis by Modulating Neutrophil Chemotaxis and Cytokine Secretion. J Invest Dermatol (2016) 136:1418–28. doi: 10.1016/j.jid.2016.03.002
161. Wang Q, Li S, Tang X, Liang L, Wang F, Du H. Lipocalin 2 Protects Against Escherichia Coli Infection by Modulating Neutrophil and Macrophage Function. Front Immunol (2019) 10:2594. doi: 10.3389/fimmu.2019.02594
162. Guglani L, Gopal R, Rangel-Moreno J, Junecko BF, Lin Y, Berger T, et al. Lipocalin 2 Regulates Inflammation During Pulmonary Mycobacterial Infections. PLoS One (2012) 7:e50052. doi: 10.1371/journal.pone.0050052
163. Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, et al. Lipocalin 2 Is Required for Pulmonary Host Defense Against Klebsiella Infection. J Immunol (2009) 182:4947–56. doi: 10.4049/jimmunol.0803282
164. Li B, Fang J, Zuo Z, Yin S, He T, Yang M, et al. Activation of the Porcine Alveolar Macrophages via Toll-Like Receptor 4/NF-κb Mediated Pathway Provides a Mechanism of Resistin Leading to Inflammation. Cytokine (2018) 110:357–66. doi: 10.1016/j.cyto.2018.04.002
165. Tarkowski A, Bjersing J, Shestakov A, Bokarewa MI. Resistin Competes With Lipopolysaccharide for Binding to Toll-Like Receptor 4. J Cell Mol Med (2010) 14:1419–31. doi: 10.1111/j.1582-4934.2009.00899.x
166. Planès C, Leyvraz C, Uchida T, Angelova MA, Vuagniaux G, Hummler E, et al. In Vitro and In Vivo Regulation of Transepithelial Lung Alveolar Sodium Transport by Serine Proteases. Am J Physiol-Lung Cell Mol Physiol (2005) 288:L1099–109. doi: 10.1152/ajplung.00332.2004
167. Zhao C-W, Gao Y-H, Song W-X, Liu B, Ding L, Dong N, et al. An Update on the Emerging Role of Resistin on the Pathogenesis of Osteoarthritis. Mediators Inflammation (2019) 2019:e1532164. doi: 10.1155/2019/1532164
168. Jiang S, Park DW, Tadie J-M, Gregoire M, Deshane J, Pittet JF, et al. Human Resistin Promotes Neutrophil Pro-Inflammatory Activation, Neutrophil Extracellular Trap Formation, and Increases Severity of Acute Lung Injury. J Immunol (2014) 192:4795–803. doi: 10.4049/jimmunol.1302764
169. Wang W-Y, Chen Y, Su X, Tang D, Ben Q-W, Yao W-Y, et al. Resistin-Like Molecule-α Causes Lung Injury in Rats With Acute Pancreatitis by Activating the PI-3k/Akt-NF-κb Pathway and Promoting Inflammatory Cytokine Release. Curr Mol Med (2016) 16:677–87. doi: 10.2174/1566524016666160802145700
170. Mishra A, Wang M, Schlotman J, Nikolaidis NM, DeBrosse CW, Karow ML, et al. Resistin-Like Molecule-Beta is an Allergen-Induced Cytokine With Inflammatory and Remodeling Activity in the Murine Lung. Am J Physiol Lung Cell Mol Physiol (2007) 293:L305–313. doi: 10.1152/ajplung.00147.2007
171. Meng L, Li L, Lu S, Li K, Su Z, Wang Y, et al. The Protective Effect of Dexmedetomidine on LPS-Induced Acute Lung Injury Through the HMGB1-Mediated TLR4/NF-κb and PI3K/Akt/mTOR Pathways. Mol Immunol (2018) 94:7–17. doi: 10.1016/j.molimm.2017.12.008
172. Ye R, Liu Z. ACE2 Exhibits Protective Effects Against LPS-Induced Acute Lung Injury in Mice by Inhibiting the LPS-TLR4 Pathway. Exp Mol Pathol (2020) 113:104350. doi: 10.1016/j.yexmp.2019.104350
173. Fuentes N, Silveyra P. Endocrine Regulation of Lung Disease and Inflammation. Exp Biol Med (2018) 243:1313–22. doi: 10.1177/1535370218816653
174. Tashiro Y, Sato K, Watanabe T, Nohtomi K, Terasaki M, Nagashima M, et al. A Glucagon-Like Peptide-1 Analog Liraglutide Suppresses Macrophage Foam Cell Formation and Atherosclerosis. Peptides (2014) 54:19–26. doi: 10.1016/j.peptides.2013.12.015
175. Mooradian AD. Dyslipidemia in Type 2 Diabetes Mellitus. Nat Clin Pract Endocrinol Metab (2009) 5:150–9. doi: 10.1038/ncpendmet1066
176. Guerrini V, Prideaux B, Blanc L, Bruiners N, Arrigucci R, Singh S, et al. Storage Lipid Studies in Tuberculosis Reveal That Foam Cell Biogenesis Is Disease-Specific. PLoS Pathog (2018) 14:e1007223. doi: 10.1371/journal.ppat.1007223
177. World Health Organization. Global Tuberculosis Report 2020. Geneva: World Health Organization (2020).
178. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results From the International Diabetes Federation Diabetes Atlas, 9th Edition. Diabetes Res Clin Pract (2019) 157:107483. doi: 10.1016/j.diabres.2019.107843
179. D’Attilio L, Díaz A, Fernández RDV, Bongiovanni B, Santucci N, Dídoli G, et al. The Neuro-Endocrine-Immune Relationship in Pulmonary and Pleural Tuberculosis: A Better Local Profile in Pleural Fluid. Int J Tuberc Lung Dis (2018) 22:321–7. doi: 10.5588/ijtld.17.0270
180. Wieland CW, Florquin S, Chan ED, Leemans JC, Weijer S, Verbon A, et al. Pulmonary Mycobacterium Tuberculosis Infection in Leptin-Deficient Ob/Ob Mice. Int Immunol (2005) 17:1399–408. doi: 10.1093/intimm/dxh317
181. Mihret A. The Role of Dendritic Cells in Mycobacterium Tuberculosis Infection. Virulence (2012) 3:654–9. doi: 10.4161/viru.22586
182. Handzel ZT. The Immune Response to Mycobacterium Tuberculosis Infection in Humans. London: IntechOpen (2013). doi: 10.5772/54986
183. de Martino M, Lodi L, Galli L, Chiappini E. Immune Response to Mycobacterium Tuberculosis: A Narrative Review. Front Pediatr (2019) 7:350. doi: 10.3389/fped.2019.00350
184. Kumar Nathella P, Babu S. Influence of Diabetes Mellitus on Immunity to Human Tuberculosis. Immunology (2017) 152:13–24. doi: 10.1111/imm.12762
185. Ayelign B, Negash M, Genetu M, Wondmagegn T, Shibabaw T. Immunological Impacts of Diabetes on the Susceptibility of Mycobacterium Tuberculosis. J Immunol Res (2019) 2019:e6196532. doi: 10.1155/2019/6196532
186. Daryabor G, Atashzar MR, Kabelitz D, Meri S, Kalantar K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front Immunol (2020) 11:1582. doi: 10.3389/fimmu.2020.01582
187. Jiménez-Corona ME, Cruz-Hervert LP, García-García L, Ferreyra-Reyes L, Delgado-Sánchez G, Bobadilla-del-Valle M, et al. Association of Diabetes and Tuberculosis: Impact on Treatment and Post-Treatment Outcomes. Thorax (2013) 68:214–20. doi: 10.1136/thoraxjnl-2012-201756
188. Alisjahbana B, Sahiratmadja E, Nelwan EJ, Purwa AM, Ahmad Y, Ottenhoff THM, et al. The Effect of Type 2 Diabetes Mellitus on the Presentation and Treatment Response of Pulmonary Tuberculosis. Clin Infect Dis (2007) 45:428–35. doi: 10.1086/519841
189. Baker MA, Harries AD, Jeon CY, Hart JE, Kapur A, Lönnroth K, et al. The Impact of Diabetes on Tuberculosis Treatment Outcomes: A Systematic Review. BMC Med (2011) 9:81. doi: 10.1186/1741-7015-9-81
190. Gil-Santana L, Almeida-Junior JL, Oliveira CAM, Hickson LS, Daltro C, Castro S, et al. Diabetes Is Associated With Worse Clinical Presentation in Tuberculosis Patients From Brazil: A Retrospective Cohort Study. PLoS One (2016) 11:e0146876. doi: 10.1371/journal.pone.0146876
Keywords: immuno-endocrine interactions, hormone, type 2 diabetes, tuberculosis, hormone receptor
Citation: Webber T, Ronacher K, Conradie-Smit M and Kleynhans L (2022) Interplay Between the Immune and Endocrine Systems in the Lung: Implications for TB Susceptibility. Front. Immunol. 13:829355. doi: 10.3389/fimmu.2022.829355
Received: 05 December 2021; Accepted: 02 February 2022;
Published: 22 February 2022.
Edited by:
Christoph Siegfried Niki Klose, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Jingbo Pang, University of Illinois at Chicago, United StatesRand T. Akasheh, American University of Madaba, Jordan
Copyright © 2022 Webber, Ronacher, Conradie-Smit and Kleynhans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Léanie Kleynhans, leaniek@sun.ac.za