Non-alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease: Two Intertwined Entities
 Francisco Idalsoaga
Francisco Idalsoaga Anand V. Kulkarni
Anand V. Kulkarni Omar Y. Mousa3,4
Omar Y. Mousa3,4  Marco Arrese
Marco Arrese Juan Pablo Arab
Juan Pablo Arab- 1Departamento de Gastroenterología, Escuela de Medicina, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Department of Hepatology, Asian Institute of Gastroenterology, Hyderabad, India
- 3Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN, United States
- 4Division of Gastroenterology and Hepatology, Mayo Clinic Health System, Mankato, MN, United States
- 5Centro de Envejecimiento y Regeneración (CARE), Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide, with a prevalence of 25–30%. Since its first description in 1980, NAFLD has been conceived as a different entity from alcohol-related fatty liver disease (ALD), despite that, both diseases have an overlap in the pathophysiology, share genetic–epigenetic factors, and frequently coexist. Both entities are characterized by a broad spectrum of histological features ranging from isolated steatosis to steatohepatitis and cirrhosis. Distinction between NAFLD and ALD is based on the amount of consumed alcohol, which has been arbitrarily established. In this context, a proposal of positive criteria for NAFLD diagnosis not considering exclusion of alcohol consumption as a prerequisite criterion for diagnosis had emerged, recognizing the possibility of a dual etiology of fatty liver in some individuals. The impact of moderate alcohol use on the severity of NAFLD is ill-defined. Some studies suggest protective effects in moderate doses, but current evidence shows that there is no safe threshold for alcohol consumption for NAFLD. In fact, given the synergistic effect between alcohol consumption, obesity, and metabolic dysfunction, it is likely that alcohol use serves as a significant risk factor for the progression of liver disease in NAFLD and metabolic syndrome. This also affects the incidence of hepatocellular carcinoma. In this review, we summarize the overlapping pathophysiology of NAFLD and ALD, the current data on alcohol consumption in patients with NAFLD, and the effects of metabolic dysfunction and overweight in ALD.
Introduction
Non-alcoholic fatty liver disease (NAFLD) and alcohol-related liver disease (ALD) are the most frequent causes of chronic liver disease worldwide (1, 2). Over the past decade, both entities have been increasing in the U.S. and worldwide, contributing to the rising burden of cirrhosis and hepatocellular carcinoma (HCC) and surpassing the figures of viral hepatitis infection as chief etiologies of these conditions (3). These temporal trend shifts in the contributions of NAFLD and ALD to the total burden of liver disease are likely related to diverse factors. Among them are the changing epidemiology of viral hepatitis in the last decade, the increasing rates of obesity and type 2 diabetes (T2DM) and the changing patterns of alcohol consumption in the general population. NAFLD and ALD have a number of commonalities and may eventually coexist in the same individual. In this context, it seems timely to review some basic and clinical concepts on these two intertwined conditions.
NAFLD is closely related to obesity and overweight as well as to the presence of metabolic dysfunction, and although the occurrence of steatosis in this setting was recognized in the early 1950s (4), only in 1980 was it pointed out as a possible cause of cirrhosis in a landmark case series study by Ludwig et al. (5). At the present time, NAFLD is defined by steatosis associated with a spectrum of hepatic histopathologic changes including the presence of inflammatory infiltrates and various degrees of fibrosis and cirrhosis (1). These features develop in the absence of known factors that cause fat accumulation such as alcohol consumption (defined as <30 g/day in men and <20 g/day in women), viral liver disease, and hereditary disorders. NAFLD is usually found in patients with comorbidities, such as metabolic syndrome (MetS), obesity, insulin resistance (IR), T2DM, and dyslipidemia (6). It is estimated that between 7 and 30% of patients with NAFLD may develop an inflammatory subtype termed non-alcoholic steatohepatitis (NASH), which is hallmarked by the presence of cell ballooning and lobular inflammation (7). NASH seems to be a more aggressive form of the disease that progresses more commonly to advanced fibrosis and cirrhosis (8). Patients with NAFLD, particularly those with NASH, have an increased mortality due to liver disease, and it is likely that cardiovascular mortality could also be increased (9).
NAFLD has increased significantly worldwide over the last decades, in line with the obesity epidemic and sedentary lifestyles (8, 10–12). Currently, the global prevalence of NAFLD is around 25%, with important differences between the Middle East (32%), South America (31%), United States (24.1%), and Africa (14%) (12–15). Additionally, the prevalence varies in association with metabolic diseases. NAFLD can be detected with ever greater prevalence in ~90% of obese patients and 65% of overweight patients (13) and in up to 70% of T2DM patients (16).
ALD affects 2–2.5% of the general population and exhibits a greater prevalence in areas with higher alcohol consumption (17). In Western countries, up to 50% of the patients with end-stage liver disease have alcohol as a major etiologic factor (18). According to the World Health Organization in 2018, more than 3 million deaths every year—representing around 5% of global deaths—are attributable to alcohol consumption (19). In the United States in 2006, alcohol-related deaths (excluding accidents) accounted for 22,073 deaths, with 13,000 of those specifically attributed to ALD (20). ALD is caused by heavy chronic alcohol consumption. Heavy or hazardous drinking is defined as consumption of more than 3 standard drinks per day in men, and more than 2 drinks per day in women, or binge drinking (defined as more than 5 standard drinks in men and more than 4 in women over a 2-h period) (21), implying a greater risk of developing health problems associated with alcohol (22–24). Clinical manifestations range from no symptoms to severe acute alcoholic hepatitis (AH) with or without cirrhosis (17).
The relationship between NAFLD and ALD is complex due to overlapping clinical features and lack of positive criteria for NAFLD (25). Of note, the interaction between NAFLD and alcohol consumption has been controversial over the last few years (26). Initially, some studies suggested a protective effect of moderate doses of alcohol (27). However, recent evidence indicates that there is no safe threshold for alcohol consumption in NAFLD patients (28). Moreover, alcohol use is a significant risk factor for the progression of liver disease in these individuals, eventually impacting a mortality in those patients with NAFLD and MetS (29). On the other hand, in ALD, MetS and obesity may increase liver disease progression and the incidence and mortality of HCC (30).
In this review, we aim to summarize current data on the overlapping pathophysiology of NAFLD and ALD as well as the available information on alcohol consumption in patients with NAFLD and the effects of MetS and overweight in ALD. We underscore the need for a change in NAFLD nomenclature in order to account for the dual etiology of liver disease, which is present in a likely significant proportion of patients with concurrent alcohol consumption and metabolic disturbances. Proper consideration of these concepts should impact clinical management.
How Much Alcohol Is Bad in NAFLD?
Definition of NAFLD, and the distinction from ALD, is based on the amount of alcohol consumed, which has been established arbitrarily, and without definitive evidence. The threshold of alcohol consumption that rules out NAFLD usually is 20 g (2 units per day) in women, 30 g (3 units per day) in men, based on guidelines of scientific associations recommendations (1, 6). Alcohol consumption is reported in up to two-thirds of patients with NAFLD in the United States (31). The effect of alcohol consumption on the prognosis of NASH has been a subject of controversy for many years, with some studies suggesting a protective effect and others suggesting an increased risk of liver disease progression and HCC (32–35) (Table 1). Initial evidence for the protective effect of moderate alcohol intake in NASH dates back to 2001. Dixon et al. suggested that moderate alcohol consumption reduces the risk of NAFLD in the severely obese, probably by reducing IR (OR, 0.35; 95% CI, 0.12–1.00). Steatosis was diagnosed by laparoscopic biopsies during bariatric surgery and NASH was present only in 25% (26/105) of the cases (36). In 2007, Suzuki et al. performed a cross-sectional study in men without chronic liver diseases to determine the association between alcohol consumption (none, light, moderate, and excessive) and elevated serum aminotransferase levels. They concluded that excessive alcohol consumption was associated with increased aminotransferase levels, while light and moderate alcohol intake may protect against the development of elevated aminotransferases (37). However, aminotransferases are poor screening tools for NAFLD. The third National Health and Nutrition Examination Survey (NHANES) compared patients with no alcohol intake (n = 7,211) vs. patients who drank wine exclusively (up to 10 g/day) (n = 945). In this study, the low-dose wine consumption (but not beer or liquor) was associated with a decreased risk of elevated aminotransferase levels (OR 0.62; 95% CI 0.41–0.92) (56). In 2009, Gunji et al. developed a cross-sectional study in the Japanese population, including a large series of asymptomatic male subjects. Alcohol intake was defined through a questionnaire, and steatosis status was assessed by aminotransferases or ultrasonography (US) (38). They reported an inverse association between alcohol consumption and steatosis, with a protective effect of light and moderate alcohol intake. Later, the same group obtained similar results defining steatosis through a CT scan (independent of MetS or physical activity) (39). Another cross-sectional study in males on regular health check-ups was conducted by Hiramine et al. They utilized a questionnaire to determine the alcohol intake and classified the subjects according to alcohol consumption as none, light, moderate, and heavy drinkers (0, <20, 20–59, and ≥60 g/day, respectively). Steatosis was defined by US. They also concluded that alcohol consumption plays a protective role against fatty liver in men. It is interesting that the analysis of the drinking patterns revealed that the prevalence of fatty liver was inversely associated with the frequency of alcohol consumption (≥21 days/month) (OR 0.62, CI 0.53–0.71), but not with the volume of alcohol consumed (40). Moriya et al. reported a significant inverse correlation between drinking frequency and the prevalence of fatty liver (p < 0.001) in the Japanese population. These authors described that drinking <20 g on 1–3 days/week was associated with a lower prevalence of fatty liver assessed by US (adjusted odds ratio, 0.47; 95% confidence interval, 0.23–0.96). This study included men and women, obtaining the same results for both (men: OR 0.59; 95% CI 0.52–0.68; women: OR 0.60; 95% CI 0.45–0.80) (41). These results were consistent with the study by Hamaguchi et al., who defined the prevalence of steatosis by CT scan (42).
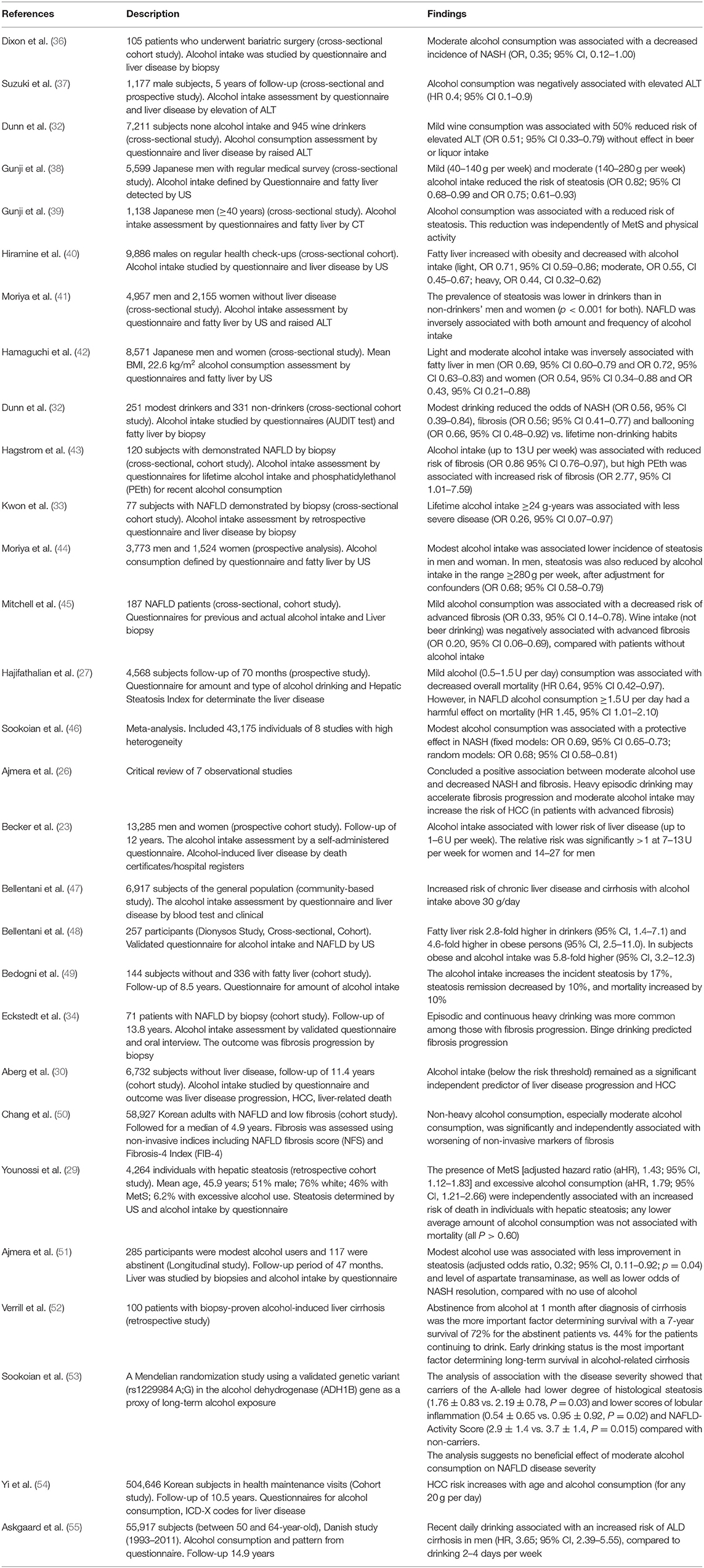
Table 1. Studies assessing the effect of moderate alcohol intake on NAFLD.
At least 4 cross-sectional studies suggested a protective role of alcohol consumption after defining steatosis by liver biopsy (32, 43, 45, 57). In the NIH NASH Clinical Research Network, modest alcohol consumption was associated with less steatohepatitis, hepatocellular ballooning, and fibrosis (32). In another study, alcohol intake up to 13 U/week was associated with lower fibrosis stage in NAFLD (OR 0.86 per U/week, 95% CI 0.76–0.97). Nevertheless, an elevated phosphatidylethanol (a biomarker for recent alcohol consumption) was associated with higher stages of fibrosis (43). Finally, Mitchell et al. had similar results, but particularly with wine consumption (not with beer) and non-binge pattern (45). The association of modest alcohol intake with survival in NAFLD has also been evaluated. This analysis was made using the NHANES data (1988–2010). NAFLD was diagnosed by hepatic steatosis index (HSI) in 4,568 subjects. Modest alcohol consumption was associated with a significant decrease in all-cause mortality (after a median follow-up of 70 months, and adjustment for race, physical activity, education level, T2DM, and fiber and polyunsaturated fatty acid intake) [hazard ratio (HR) 0.64, 95% CI 0.42–0.97], whereas drinking ≥1.5 drinks per day was to be associated with an increased in mortality (HR 1.45, 95% CI 1.01–2.10) (27). Currently, only one meta-analysis has been published (46), which included 43,175 individuals. It concluded that modest alcohol consumption was associated with a protective effect in NASH (fixed models: OR 0.69, 95% CI 0.65–0.73; random models: OR 0.68; 95% CI 0.58–0.81) based on 8 heterogeneous studies. The critical review by Ajmera et al. (26) of 7 observational studies concluded a positive association between moderate alcohol use and decreased NASH and fibrosis; however, heavy episodic drinking may accelerate fibrosis progression and moderate alcohol intake may increase the risk of HCC (in patients with advanced fibrosis). However, the studies had significant methodological limitations, including incomplete adjustment for confounding factors.
Despite that some studies suggest a beneficial effect of moderate alcohol consumption on the occurrence and progression of NAFLD, more recent evidence suggests that there is no safe limit for alcohol consumption and that the alcohol intake is associated with a higher risk of liver disease progression, including HCC (23, 30, 34, 36, 47–49, 54, 55, 58–63). The association between alcohol intake and liver damage has been reported widely since 1957 (64). Many studies showed that alcohol consumption is associated with increased prevalence and progression of NASH. In the Dionysos Study, 144 subjects without steatosis and 336 with steatosis were followed up for 8.5 years. The most relevant risk factor for steatosis incidence and remission, as well as a predictor of mortality in these patients with fatty liver, was alcohol intake (20 g/day). The incidence of fatty liver increased by 17%, steatosis remission decreased by 10%, and mortality increased by 10% in the fatty liver cohort (49). Follow-up liver biopsy in 71 patients with NAFLD showed that fibrosis progression was associated with episodic drinking (at least once per month) and higher weekly alcohol consumption. Also, the heavy episodic drinking (p < 0.001) and IR (p < 0.01) were independently associated with significant fibrosis progression. The study concluded that moderate alcohol consumption was associated with fibrosis progression in NAFLD, and the authors advised to refrain from heavy episodic drinking in patients with NAFLD (34). Recent studies showed that there is no safe limit for alcohol consumption and suggested that even light alcohol consumption is not safe in NAFLD. In 2018, a systematic analysis from the Global Burden of Diseases, Injuries, and Risk Factors Study, which included 28 million individuals and 649,000 cases with outcomes, also suggested a detrimental effect of moderate alcohol intake (65). A Finnish cohort study of 6,732 individuals without baseline liver disease, after a follow-up of 11.4 years, demonstrated that non-risky alcohol intake (<3 units per day in men and 2 units per day in women) was associated with a significant increase in the risk of liver disease progression (30). A large-scale cohort was performed in 58,927 young and middle-aged Korean individuals with NAFLD (with low baseline fibrosis scores), who were followed for a median of 4.9 years. The progression from low to intermediate or high probability of advanced fibrosis was assessed using non-invasive index including NAFLD fibrosis score (NFS) and Fibrosis-4 Index (FIB-4). They demonstrated that light (1.0–9.9 g/d) or moderate (10.0–29.9 g/d for men and 10.0–19.9 g/d for women) alcohol consumption compared with none (0 g/d) was significantly and independently associated with worsening of fibrosis. The effect was higher with moderate alcohol consumption (50). The authors also suggested that a safe limit of alcohol use may not exist. In a study that analyzed NHANES III including 4,264 adults with hepatic steatosis diagnosed by US examination, the overall mortality was significantly higher among subjects with excessive alcohol intake (32.2%) vs. subjects with non-excessive alcohol consumption (22.2%) after 5 and 20 years of follow-up (p = 0.003). The association of excessive alcohol use with mortality was significant in individuals who have MetS (aHR, 2.46; 95% CI, 1.40–4.32) but not without it (p = 0.74) (29). The impact of alcohol consumption is associated not only with incidence of steatosis, fibrosis progression, and mortality but also with less improvement in steatosis in patients with NASH and increased liver malignancies. In a longitudinal analysis of liver biopsies from patients with NAFLD, the low and modest alcohol use was associated with less improvement in steatosis, higher levels of AST, and less NASH resolution, compared with no alcohol intake (51). Another study suggested that total abstinence even prevents disease progression and is the more important factor determining survival in patients with established cirrhosis (52). Additionally, data from a Mendelian randomization study using a validated genetic variant (rs1229984 A:G) in the alcohol dehydrogenase (ADH1B) gene as a proxy of long-term alcohol exposure was used, thereby minimizing measurement bias and confounding factors. They found that carriers of the A-allele consumed significantly lower amounts of alcohol compared with non-carriers. Additionally, A-allele carriers had a lower degree of histological steatosis, lobular inflammation, and NAFLD-Activity Score (NAS) compared with non-carriers. They suggested that there is no beneficial effect of moderate alcohol consumption on NAFLD disease severity (53).
The association between alcohol intake and HCC development has been evaluated. Mild or moderate alcohol consumption may be a cofactor for the development of HCC in NAFLD. In a study that included 504,646 Korean patients (age 40–80 years) on routine health checkups, HCC incidence was associated with hepatitis B and C infection, and each 20 g/day of alcohol intake increased the risk of HCC by 6, 8, 16, and 30%, respectively, in individuals aged <50, 50–59, 60–69, and 70–80 years (54). An analysis of the participants in the Health 2000 and FINNRISK (1992–2012) databases was performed, which linked national registers for hospital admissions, malignancies, and death regarding liver, cardiovascular, and malignant disease, as well as all-cause mortality. They concluded that alcohol consumption is associated with a dose-dependent risk of advanced liver disease and neoplasia but a dose-dependent decrease in cardiovascular outcomes (21% risk reduction with ≤1 unit per day intake limited to non-smokers) (58). Alcohol intake is also associated with extrahepatic cancers, particularly breast, oral, pharyngeal, and colorectal cancer (66–69).
In summary, although some studies suggest a beneficial effect of light and moderate alcohol consumption on the occurrence and progression of NAFLD, most of them are cross-sectional studies, limiting their interpretation. These studies defined the association observed on the grounds of present alcohol intake history; nevertheless, NAFLD and ALD are processes that require long-term exposure and the damage could be driven by previous alcohol history. This is limited by the recall bias. Some studies have significant methodological limitations (including incomplete adjustment for confounding factors, as metabolic history) and several potential biases (especially in retrospective analyses), limiting their validity. On the other hand, the evidence supporting no benefit or even a detrimental effect of alcohol intake is based on solid longitudinal studies. The association between alcohol intake with fibrosis progression and cancer in these studies seems appealing and less biased than observed in cross-sectional analyses.
How Obesity and Metabolic Syndrome Affect ALD?
In recent years, the association between alcohol consumption and NAFLD progression has been clearly established. Emerging evidence has demonstrated that obesity and MetS increase the progression of ALD and HCC incidence and mortality. Indeed, a synergism between alcohol and obesity has been suggested. A cross-sectional study (48) including 257 participants of the Dionysos Study used a validated food questionnaire and ultrasound assessment of NAFLD. The prevalence of steatosis was increased in heavy drinkers (46.4% [95% CI, 34–59%]) and obese (75.8% [CI, 63–85%]) compared with controls (16.4% [CI, 8–25%]). Those heavy drinkers who are obese had an even higher prevalence of NAFLD, 94.5% (CI, 85–99%), which suggest an additive effect. Obesity doubles the risk of steatosis in heavy drinkers (48). This synergistic effect was also observed in 2 of the long-term Midspan prospective cohort studies (9,559 men) in Scotland. The body mass index (BMI) and alcohol consumption were strongly associated with liver disease mortality in analyses adjusted by other confounders (p = 0.001 and p < 0.0001, respectively). After a median follow-up of 29 years, consumers of 15 U/week or more exhibited higher rates of liver disease irrespective of BMI. In mild users (1–14 U/week), an excess of liver disease was only observed in subjects with obesity, with a synergistic effect between alcohol and BMI (synergy index, 2.89; [95% CI, 1.29–6.47]) (60). In the analysis of NHANES III, previously commented in this review, the presence of MetS [adjusted hazard ratio (aHR), 1.43; [95% CI, 1.12–1.83]] and excessive alcohol consumption (HR, 1.79; [95% CI, 1.21–2.66]) were independently associated with an increased mortality in subjects with steatosis. Additionally, alcohol intake and the presence of MetS had a synergistic effect (29). It is necessary to consider the alcohol intake as an aggravating element of overweight and obesity. A mild alcohol intake can contribute with 100–300 kcal/day, directly to weight gain and obesity, irrespective of the type of alcohol consumed (69, 70).
The association between BMI and HCC (incidence and mortality) has been demonstrated through two prospective population-based studies in Taiwan. The first of them was a prospective study that included 2,260 Taiwanese men positive for HBV infection, followed up for 14 years. HCC was diagnosed by imaging or histopathology (Cancer Registry). In this study, alcohol intake (any amount) had synergistic effects with the risk of incident HCC in analyses adjusted for age (HR, 3.41; 95% CI, 1.25–9.27; p < 0.025) and multiple variables (HR, 3.40; 95% CI, 1.24–9.34; p < 0.025). Also, the risk of HCC increased in overweight (HR, 2.4; 95% CI, 1.3–4.4), obese (HR, 2.0; 95% CI, 1.1–3.7), and extremely obese (HR, 2.9; 95% CI, 1.0–8.0) alcohol users (p for trend = 0.046) (62). Later, Loomba et al. conducted a prospective, population-based study of 23,712 Taiwanese, followed for 11.6 years for the incidence of HCC, and the study concluded with similar results. Alcohol consumption and obesity (BMI ≥30) showed a synergistic association with the risk of incident HCC in both unadjusted analyses (HR = 7.19, 95% CI: 3.69, 14.00; p < 0.01) and multivariable-adjusted analyses [age, sex, smoking, serum alanine aminotransferase (ALT), serum hepatitis B surface antigen, anti-hepatitis C virus antibody, and T2DM] (HR = 3.82, 95% CI: 1.94, 7.52; P < 0.01). Finally, the study concluded that obesity and alcohol have a synergic effect increasing the risk of incident HCC (63).
New Names for NAFLD/ALD: A Changing Nomenclature for Fatty Liver Disorders
The use of the acronym NAFLD as an umbrella term is now recognized as a problematic issue in the field of hepatology (31). This is largely due to the significant heterogeneity of patients grouped under that denomination as well as by the absence of positive criteria for NAFLD, which makes it difficult to classify subjects with metabolic alterations drinking beyond the threshold set for NAFLD. The latter also impedes the recognition of dual etiology for liver disease in individuals with both moderate or excessive alcohol consumption and metabolic disturbances. For this reason, and in order to better characterize the disease, NAFLD nomenclature has recently been revised and a new consensus-driven acronym proposed (71). Thus, the term MAFLD, which stands for metabolic (dysfunction)-associated fatty liver disease, was suggested as a more appropriate overarching term. This revised nomenclature should allow for more precise study designs leading to decreased variability of study groups and to a better understanding of the natural history of the disease. Positive criteria to diagnose MAFLD have been also proposed by the same expert group (72), which considers the evidence of fat accumulation in the liver and presence of evidence of metabolic dysregulation. The latter is defined by the presence of at least two metabolic risk abnormalities (dyslipidemia, hypertension, abdominal obesity, prediabetes, IR, or elevated high sensitivity C-reactive protein). Since ALD also comprises a spectrum of liver lesions, some adaptations may be needed in order to acknowledge the dual etiology of patients with fatty liver disease (i.e., concomitant MAFLD and ALD). Thus, the spectrum of fatty liver disease (Figure 1) should include patients with true ALD (alcohol-associated fatty liver disease, AAFLD), patients with predominant ALD but with metabolic cofactors (ALD with MetS), those with true NAFLD with alcohol consumption near zero (MAFLD) (71), and patients with NAFLD but with alcohol consumption contributing to the disease process (i.e., MAFLD with alcohol component). A final group will be composed of those patients with both MAFLD and ALD, equally contributing (or not possible to determine which one predominates) to the disease process (both alcohol and metabolic associated fatty liver disease, BAFLD). Similarly, the term BASH has been used to describe both alcohol and metabolic associated steatohepatitis (73). Each group probably has different clinical manifestations, course, liver prognosis, and mortality. This approach recently suggested by Eslam et al. (31) proposes that patients with fatty liver and predominance of metabolic dysfunction could be stratified according to alcohol intake and patients with alcohol predominant fatty liver according to the presence of coexisting metabolic comorbidities when included in clinical studies. These distinctions may help to a more robust understanding of the natural history of these different patient populations. In Figure 2, we suggest an algorithm to be applied in patients with fatty liver, which intend to account for dual etiology and predominance in the setting of metabolic dysfunction and alcohol consumption.
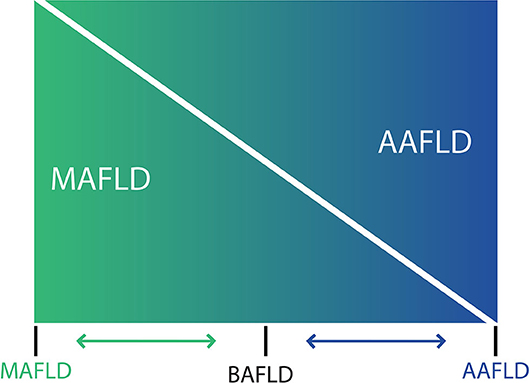
Figure 1. Spectrum of fatty liver diseases. In non-alcoholic fatty liver disease (NAFLD) and alcohol-related liver disease have been generally conceived as different entities, and despite that both conditions share an overlapping pathophysiology and frequently coexist in clinical practice, thresholds to diagnose NAFLD makes difficult to account for a dual etiology in a given patient. For that reason, a nomenclature change has been proposed [see (71) in the main text] considering that at the ends of the spectrum of fatty liver disease, there are patients with true ALD (now named alcohol-associated fatty liver disease, AAFLD) and some with true NAFLD with alcohol consumption near-zero (now named metabolic associated fatty liver disease, MAFLD) but that the vast majority of patients are between these two extremes. Thus, in clinical practice there will be patients with ALD that have metabolic cofactors (AAFLD with MetS) and patients with NAFLD that consume alcohol, which contributes to the disease process (MAFLD with alcohol component). In the middle, a large group of patients have both conditions (NAFLD and ALD) with some showing an equal contribution of alcohol and metabolic factors (proposedly named as both alcohol and metabolic associated fatty liver disease, BAFLD).
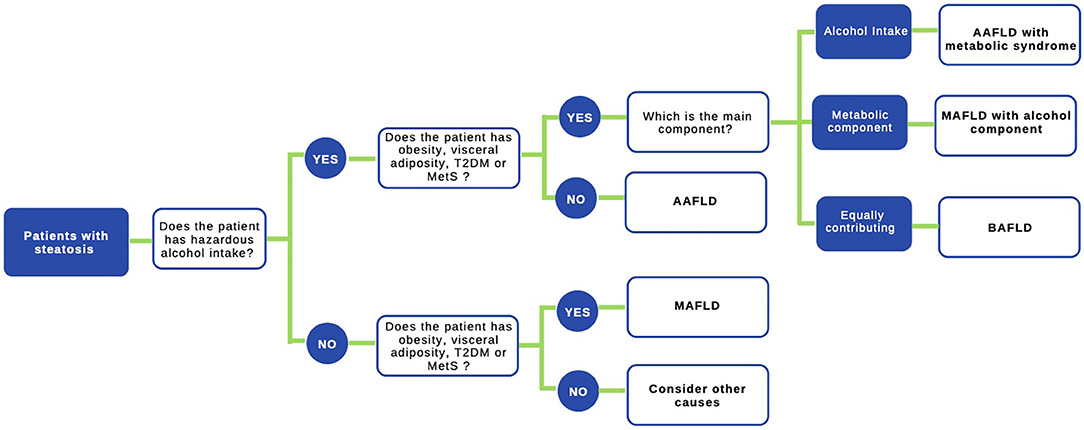
Figure 2. Proposed algorithm to approach patients with liver steatosis. A staggered algorithm to approach patients with liver steatosis is shown. This algorithm considers alcohol intake and metabolic cofactors (obesity, T2DM, and MetS) and classifies patients in AAFLD alcohol-associated fatty liver disease (true ALD) if hazardous alcohol intake is present, AAFLD with metabolic component (predominant ALD but with metabolic cofactors), MAFLD metabolic associated fatty liver disease (NAFLD with alcohol consumption near zero), MAFLD with alcohol component (NAFLD but with alcohol consumption contributing to the disease process), and finally BAFLD both alcohol and metabolic associated fatty liver disease (patients with both NAFLD and ALD equally contributing or no possible to determine which predominates). Criteria to diagnose MAFLD are suggested in (72) in the main text.
Pathogenesis of NAFLD and ALD: Overlapping Aspects and Salient Differences
Although NAFLD and ALD are two distinct biological entities, they have a number of commonalities in their pathogenetic mechanisms leading to activation of both hepatic inflammatory and fibrogenetic pathways that fuel disease progression (74, 75) (Figure 3). Disturbed lipid handling by the hepatocyte resulting in intracellular accumulation of potentially toxic bioactive lipid species is an essential phenomenon in both NAFLD and ALD followed by the occurrence of cellular stress [i.e., endoplasmic reticulum (ER) stress and mitochondrial dysfunction] and death, which in turn triggers the innate immune response and activation of hepatic stellate cells (HSCs), resulting in inflammation and excessive collagen production and deposition (76). This general sequence is highly heterogeneous in both entities and is modulated by different genetic and epigenetic factors, some of which are also common for NAFLD and ALD. This accounts for the broad phenotypic spectrum seen in both diseases. In the following paragraphs, we summarize the main mechanisms of liver injury at play in NAFLD and ALD and underscore their similarities and differences (77–80).
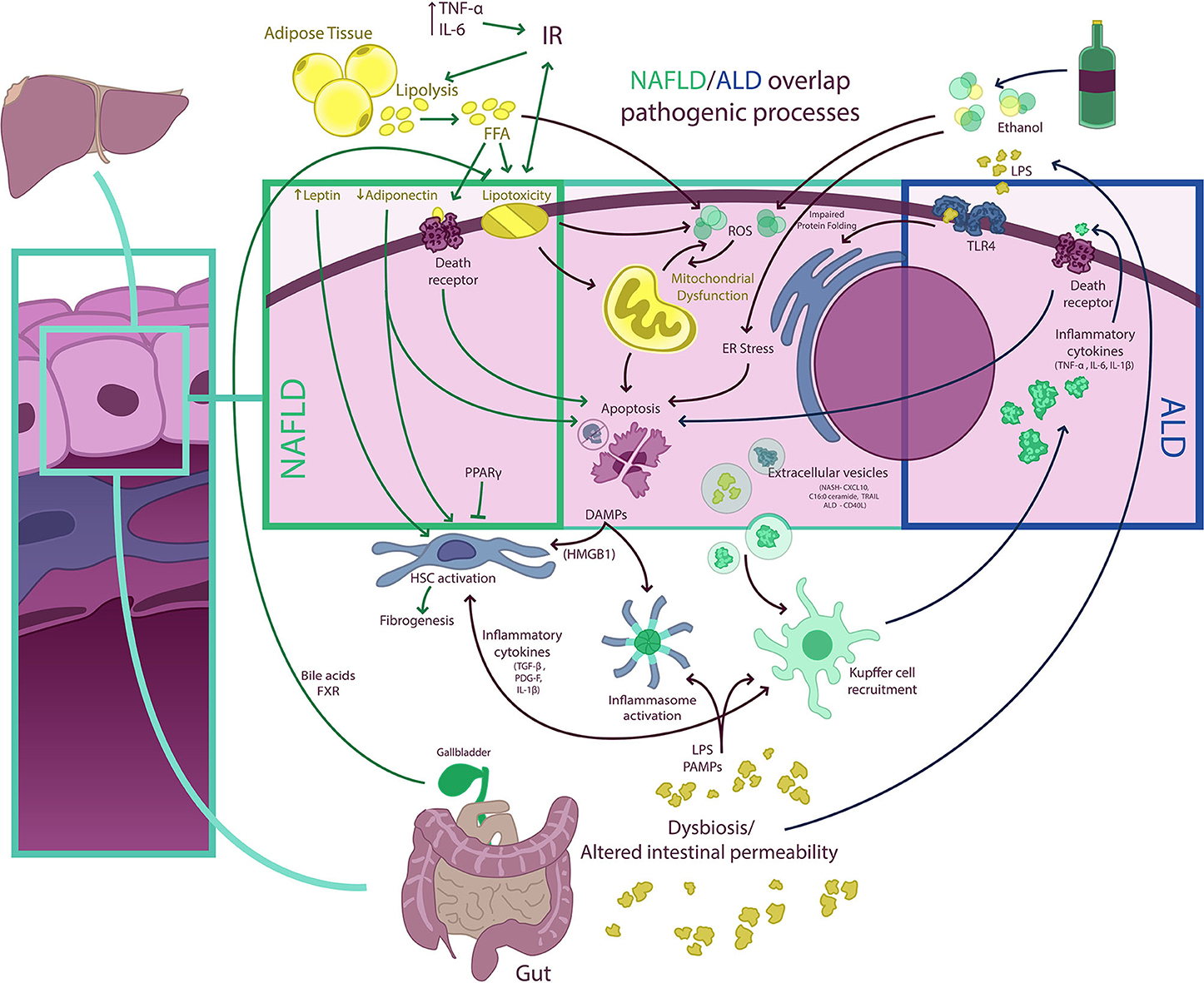
Figure 3. NAFLD/ALD overlapping pathogenic processes. Free fatty acids (FFA) and ethanol have a myriad of effects on hepatocytes determining, among other phenomena, the occurrence of mitochondrial dysfunction, endoplasmic reticulum (ER) stress (resulting in impaired protein folding), and excessive production of ROS, which result in hepatocellular injury, activation of other cell death pathways, and inflammasome activation. Damaged hepatocytes release damage-associated molecular pattern (DAMP) molecules [e.g., high-mobility group box 1 (HMGB1)] that signal to Kupffer and hepatic stellate cells (HSC), fueling inflammation and fibrogenesis. Extracellular vesicle (EV) release from hepatocytes also contributes to both Kupffer cell and HSC activation. In non-alcoholic fatty liver disease (NAFLD), insulin resistance (IR) is a central phenomenon promoting adipose tissue lipolysis and an increased FFA flux to the liver. This FFA overflow surpasses the storage capacity of hepatocytes and determines the occurrence of lipotoxicity and activation of cell death pathways. Also, adipose tissue dysfunction is associated with a proinflammatory state with elevated levels of circulating cytokines [e.g., tumor necrosis factor-α (TNF-α) and interleukin (IL)-6] and with an imbalance in circulating levels of adipose tissue-derived adipokines (i.e., a decrease in adiponectin and an increase in leptin). This may increase IR and contribute to HSC activation. Of note, some nuclear receptors such as peroxisome proliferator-activated receptors (PPARs) and farnesoid X receptor (FXR), a bile acid receptor, may have anti-inflammatory and anti-steatotic effects, which are being exploited therapeutically. Both NAFLD and ALD (alcohol-related liver disease) are associated with intestinal dysbiosis and altered gut permeability, which results in the pass of bacterial products [e.g., pathogen-associated molecular patterns (PAMPs), lipopolysaccharides (LPS), and others] into portal circulation. In the liver, LPS and PAMPS may activate different Toll-like receptors (TLRs) as well as promote inflammasome assembly determining amplification of inflammatory responses. In ALD, proinflammatory cytokines released from macrophages may also activate cell death receptors and induce apoptosis.
Hepatic Fat Accumulation and Lipotoxicity
Increased lipid droplets (i.e., steatosis) inside the hepatocytes is the earliest histological finding in NAFLD and ALD (78). These lipid droplets are enriched in fully saturated triglycerides and result in the typical histological pattern of macrovesicular steatosis common to both entities (7). Excessive accumulation of triglycerides and other lipid species relates to a dysregulated hepatic lipid flux consisting in an increased hepatocellular lipid uptake, synthesis, and degradation [i.e., fatty acid oxidation (FAO)], induced by a positive caloric balance and IR in NAFLD and ethanol consumption in ALD (81). The main regulators of this process are SREBP1c (82) and PPARα and PPARδ, which are critical regulators of FAO (83). In NAFLD, excessive calorie intake increases the size and number of adipocytes and renders them insulin resistant, leading to uncontrolled lipolysis and decreased fatty acid uptake, thus promoting the release of free fatty acids (FFA) into the circulation. FFA are later uptaken by hepatocytes promoting lipid droplet formation. Of note, some data suggest that fatty acid transporters are upregulated in the setting of NAFLD (84). Also, de novo hepatic lipogenesis (DNL) seems to be upregulated in most subjects with NAFLD, which relates to inactivation of peroxisome proliferator-activated receptor-alpha (PGC1-α) and upregulation of SREBP1c (82, 85–87). Interestingly, DNL may be accompanied by a compensatory enhancement of FAO, but studies in this regard are conflicting. Human data from patients with NAFLD show that FAO may be enhanced, unchanged or decreased (84). In ALD, ethanol consumption induces a multilevel disturbance of hepatic lipid metabolism (88). One of the best-studied effects, in murine models, is the increase in SREBP1c expression (a key transcription factor in hepatic lipogenesis) (85), leading to increased expression of hepatic lipogenic genes and increased DNL. Other important enzymes controlling lipid fluxes such as acetyl-CoA carboxylase, ACC (limiting enzyme in DNL), and carnitine palmitoyltransferase, CPT (limiting enzyme for mitochondrial β-oxidation, that regulates lipid degradation), have a pivotal role in both diseases in human studies (88, 89). Ethanol increases the activity of ACC and suppresses the rate of palmitic acid oxidation, producing modifications in fatty acid metabolism and steatosis (90). Additionally, an increased ACC expression has been described in experimental murine models of NAFLD (91). Accumulation of saturated fatty acids as well as other harmful lipids such as ceramides, diacylglycerols, and lysophosphatidylcholine, among others, promote the occurrence of lipotoxicity, a phenomenon defined by the appearance of leading to cellular dysfunction and death (92). Lipotoxicity plays an important pathophysiological role in NAFLD/NASH (93) and likely drives disease progression through different mechanisms such as direct cytotoxicity, increased IR and hyperinsulinemia, cell signaling modification [through hepatic nuclear factor-α or toll-like receptors (TLR)], ER stress, upregulation of autophagic processes, and trigger of different cell death pathways (e.g., apoptosis, necrosis, pyroptosis; see below). Cell injury and death, in murine and human models, determine release of damage-associated molecular pattern (DAMP) molecules, leading to macrophage recruitment and a secondary inflammatory response (94). In ALD, although accumulation of FFA occurs similarly to NAFLD, the lipotoxic phenomenon has not been well characterized and information on the nature of intrahepatic lipid species and their cellular effects is scarce (95).
Insulin Resistance
IR plays a pivotal role in the pathogenesis of NAFLD. IR in adipose tissue is associated with an increase lipolysis in adipocytes, leading to excess FFA release into the circulation and to a higher uptake by the hepatocytes, which is a driver of steatosis. In turn, increased hepatic fat content promotes IR in hepatocytes by decreasing insulin-stimulated tyrosine phosphorylation of both insulin receptor substrate (IRS)-1 and IRS-2, which leads to increased gluconeogenesis and hepatic glucose production, which leads to hyperinsulinemia. The latter stimulates the transcription factor SREBP-1c, activating most genes involved in DNL, further increasing steatosis (96). Worsening IR is considered a potential driver of disease progression in NAFLD (97), and targeting IR is one of therapeutic strategies explored for NAFLD/NASH treatment according to evidence from in vitro and mouse models (98). With regard to the relationship between alcohol consumption and IR, data are limited. Both clinical and experimental information suggests that insulin signaling is impaired in ALD. Chronic alcohol consumption disrupts whole-body lipid metabolism, and several studies, in clinical and preclinical mouse models, suggest that alcohol may promote IR and increase the risk of T2DM (99). Indeed, it is likely that the presence of IR, which is prevalent in patients with ALD, may increase the risk of advanced liver disease through multiple mechanisms. However, current data is limited, and more studies are needed to confirm if targeting insulin signaling pathways pharmacologically may be beneficial in the setting of ALD.
Cell Death Signaling
Another important process involved in the pathogenesis of NAFLD/ALD is the activation of cell death pathways (100–102). This pathway can be activated by both intrinsic and extrinsic signaling via surface receptors belonging to the tumor necrosis factor-alpha (TNF-α) receptor family (103–105). Specifically, TRAIL (TNF-related apoptosis-inducing ligand) receptor 2 (TRAIL-R2) has been associated with hepatocyte lipoapoptosis, probably as an effect of toxic lipids (free cholesterol, ceramides, and FFA), that promote reorganization of plasma membrane domains and ligand-independent activation of TRAIL-R2 signaling leading to cell death (106–109). In addition to apoptosis, other lytic forms of cell death may be at play in NAFLD/NASH, such as necroptosis, pyroptosis, and ferroptosis, which are related to cell-membrane permeabilization (101). The TRAIL pathway has been also implicated in ALD pathogenesis. After ethanol consumption, TRAIL expression leads to hepatic steatosis and TRAIL-mediated steatosis that can be inhibited by the neutralizing TRAIL antibody (110). Also, ER stress, induced by alcohol consumption and lipotoxicity, and reactive oxygen species may activate Bcl2 initiators of apoptosis members (108) and inhibit guardian members, inducing cell deaths, through caspase activation in mouse models (111). Finally, activation of apoptosis signal-regulating kinase 1 (ASK-1), which leads to phosphorylation of p38 and JNK and activation of several stress response pathways, has been involved in apoptosis occurrence in both NASH and alcoholic hepatitis. This is of particular interest since inhibitors of this pathway are available (i.e., selonsertib). However, studies conducted to date have yielded negative results (112).
Other cell death pathways that have been studied in liver diseases is necroptosis, which is a form of programmed necrosis. Necroptosis is defined as a non-apoptotic cell-death-receptor-mediated death observed in some cell types and is dependent of a kinase cascade that involves a family of proteins known as receptor interaction protein kinases (RIPKs), in particular RIPK3 activation of mixed-lineage-kinase-domain-like (MLKL) (113). This pathway has been explored in mouse models of NAFLD and ALD with different results (113–116). Of note, RIPK3-dependent and RIPK1-independent activity activation has been described in ALD, while in NASH models studies are contradictory (116–118). Currently, new studies assessing the role of necroptosis in fatty liver are underway (119).
Autophagy, or cellular self-digestion, is a cellular pathway crucial for development, differentiation, homeostasis, and survival of cells. This process is used to eliminate potentially harmful proteins and organelles and to remove intracellular microbial pathogens. In ALD and NAFLD, a dysregulation of this process with a decreased autophagic function that can lead to liver cell death, steatohepatitis, and HCC exist. In the setting of NASH, it has been demonstrated that palmitic acid suppresses autophagy, while oleic acid may promote it. Also, mice with genetic deletion of Atg7 (a critical autophagy mediator) have shown increased hepatic fat content accumulation (120). The autophagic degradation of intracellular lipid droplets may play a role in buffering FFA toxicity and maintaining hepatic lipid homeostasis (120–123). In ALD, a normal functioning of autophagy is associated with attenuated alcohol-induced injury and less lipotoxicity (122).
Immune Response
Innate immune cells have a fundamental role in the pathogenesis of both NAFLD and ALD, sharing important characteristics but with some substantial differences (124, 125). Release of DAMPs from hepatocytes activate innate immune cells, particularly resident macrophages (i.e., Kupffer cells) (123) in in vitro studies using cultured HepG2 cells and primary mouse hepatocytes. Additionally, some specific DAMPs such as high-mobility-group protein box 1 (HMGB1) have shown to activate TLR4 in NASH and ASH, playing a pivotal role during the early progression of NAFLD (126, 127). Other DAMPs, for example, sonic hedgehog (SHH) ligands, have been also associated with progression of NAFLD and fibrosis in human studies (128).
Neutrophils are another key element in ASH and NASH. Neutrophil elastase (NE), a protease released by neutrophils, produces cellular IR, and the deletion of NE produces less tissue inflammation and is associated, in mouse models, with lower adipose tissue neutrophil and macrophage content (129). In ALD, neutrophils induce progression through the release of ROS, proteases, and proinflammatory mediators (130). Additionally, neutrophils have been associated with portal hypertension as these immune cells promote the formation of microvascular thrombosis, through neutrophil extracellular traps (NETs). Occurrence of microvascular thrombosis and fibrin may drive portal hypertension through space effects in liver sinusoids (131).
Finally, the monocyte chemoattractant protein-1 (132) is another important component in ASH and NASH. The monocyte chemotactic protein-1 (MCP-1) is an inflammatory chemokine released by hepatocytes, Kupffer cells, sinusoidal endothelial cells, and hepatic stellate cells in response to alcohol, producing chemoattraction of macrophages and maintaining mononuclear infiltration in mouse models (133–136).
Inflammasome
Inflammasomes are multiprotein complexes that are mainly expressed in hepatocytes and myeloid cells (i.e., Kupffer cells) (137) that sense pro-inflammatory signals through NOD-like receptors (NLRs) and activate caspase-1, the effector protein (138–141). Caspase-1 cleaves pro-interleukins (IL-1β, IL-18, and IL-23), which results in sterile inflammation and lytic hepatocyte cell death (i.e., pyroptosis). Activation of the NLRP3 inflammasome has been found in murine models of both NAFLD and ALD (142–144). Also in rodent models of NAFLD/NASH, mRNAs encoding the NLRP3 inflammasome complex are elevated and overexpression of NLRP3 is associated with greater degrees of liver injury (145–147). Despite the similarities, the cell types involved and the trigger signals for NLRP3 activation appear to be somewhat different between ALD and NAFLD (148). In ALD, inflammasome and IL-1 production is increased at very early stages of the disease, which seems not to occur early in NASH (147). Also, in ALD, inflammasome activation is predominantly seen in Kupffer cells, while in NASH NLRP3 is mainly activated by hepatocytes (149–151). Finally, inflammasome component deficiency, in mouse models, protects against inflammation, steatosis, and liver injury in both ALD and NASH (152–154).
Extracellular Vesicles and MicroRNAs
In response to injury, damaged cells release extracellular vesicles (EVs) which are membrane-surrounded structures (66) released by almost all types of cells. EVs can contain a wide variety of cargoes [e.g., proteins, lipids, and nucleic acids [coding and non-coding RNA] and mitochondrial DNA] that mediate intercellular communication. In the setting of liver damage, hepatocytes increase EV release, which may act on different target cells leading to pivotal pathobiological processes, such as activation of macrophages, endothelial cells, and HSCs, thus promoting proinflammatory, angiogenic, and fibrotic responses (155–158). Observations made in mouse models suggest that these EV-mediated processes are relevant events in the pathogenesis of both NAFLD and ALD (159–161). Moreover, EVs are promising candidates to serve as disease biomarkers. Also, their therapeutic use as a liver-specific delivery method of different compounds is being studied (155). For an in-depth discussion of current knowledge about the role of EVs in NAFLD and ALD, the reader is referred to a recent review (155).
Changes in microRNA (miR) expression are involved in pathogenesis ALD and NAFLD. In NASH, it has been shown that an upregulation of miR-34a and a downregulation of let7d (miR precursor) decrease FAO, promoting fat synthesis in murine models (83, 162, 163), while miR-122 has been linked with steatosis and fibrosis (164). On the other hand, miR-132 may trigger fibrogenesis secondary to ethanol intake, and miR-155 is associated with ethanol-induced inflammation, probably mediated by TNF-α (165). In mouse models of NASH, miR-155 is also induced without a defined role (166, 167).
Microbiota
Another interesting factor in the pathogenesis of ALD and NAFLD is the effect of the intestinal microbiota. The number of microorganisms inhabiting the gastrointestinal tract has been estimated to exceed 1014, with extremely diverse features (168, 169). Dysbiosis has been described in both ALD and NAFLD; however, the exact role in the NAFLD and ALD disease processes remains unclear (170). The intestinal microbiota composition is associated with the stage of fibrosis and also on the ethanol consumption pattern, being different between chronic, binge, and “social” drinkers (132, 171, 172). Chronic alcohol consumption disrupts tight-junction proteins and increases intestinal permeability, resulting in increased translocation of endotoxins (lipopolysaccharides) and bacterial DNA into the portal circulation, which increases even more by the overgrowth of gram-negative bacteria. This process activates Kupffer cells through activation of TLRs (TLR4 and TLR9) (173, 174), which may also contribute to steatosis and hepatic fibrosis via stimulation of TLR9-dependent profibrotic pathways in mouse models (152, 175). The peptidoglycan and flagellin, other bacteria-derived toxins, also have an impact on TLR signaling producing proinflammatory cytokines (176). Changes in microbiota have been described in NAFLD (mainly decreased Bacteroidetes and increased Prevotella and Porphyromonas species) (177). This dysbiosis may be an important factor in causing NASH, in mouse models and human, through different mechanisms like deregulating energy homeostasis, modulation of choline and bile acid metabolism, and generation of bacteria-derived toxins, such as lipopolysaccharide (LPS), and increased hepatic TNF-a expression (through TLR4 and TLR9-dependent profibrotic pathways) in hepatic Kupffer cells (178–181). Therefore, microbiota plays an important role in ALD and NAFLD, but with some differences. The TLR4 can activate two distinct pathways: one pathway is MyD88-dependent (producing activation of NF-κB and proinflammatory cytokines), and the other pathway is MyD88-independent (inducing type I IFNs and NF-κB) (151). MyD88-dependent signaling seems to have a relevant role in NAFLD, but not in ALD (in vitro and in vivo in murine models) (152, 182, 183). Furthermore, a role may have the adipocytokines that can inhibit MyD88-dependent pathways in macrophages (184).
Bile Acids and Nuclear Receptors
Nuclear receptors (NR) are ligand-activated transcription factors that have a key role in regulating lipid homeostasis and inflammation in the NAFLD/NASH process. The NRs act as receptors for fatty acids, cholesterol, oxysterols, and xenobiotics and regulate the cell metabolism, cell differentiation, and cellular homeostasis. The principal NR studied in NAFLD/NASH are liver X receptor (LXR), pregnane X receptor (PXR), peroxisome proliferator-activated receptor-gamma (PPARγ), Farnesoid X receptor (FXR), and HNF4α, hepatocyte nuclear factor 4α (185–189). PPARα has been studied in NAFLD and ALD. PPARα induces FAO in the mitochondria, thus decreasing steatosis. PPARγ ligands can inhibit inflammatory responses by decreasing IL-6, TNF-α, and IL-1β secretion and iNOS production in macrophages and Kupffer cells (187, 188, 190). Polymorphism in the PPARγ gene is associated with the susceptibility to NAFLD. LXR and RXRα also play a role in ALD due to their actions in lipid homeostasis and inflammation, with a particular role of RXRα in alcohol detoxification. HNF4α is constitutively active through the binding of integral fatty acids. HNF4α has an important role in the maintenance of hepatocyte differentiation and the regulation of bile acid and lipid homeostasis genes, mainly in mouse models (191–193).
Bile acids (BA) are not only detergents that stimulate hepatic bile flow and biliary excretion and aid in the digestion and absorption of fats from the intestinal lumen but also relevant signaling molecules that act on hepatic and extrahepatic tissues to regulate lipid and carbohydrate metabolic pathways (77). FXR is highly expressed in the liver, small intestinal mucosa, and kidneys, with effects on glucose and lipid metabolism; acts as a sensor for BA; and regulates the BA synthesis, protecting hepatocytes from the toxic effect of BA and reducing the triglyceride levels (194). FXR has anti-inflammatory and anti-steatotic effects, promoting FAO through upregulation of PPARα and repressing lipogenesis (by the modulation of SREBP-1c expression). Activation of FXR in the ileal enterocytes after active intestinal BA uptake also has important metabolic implications via FXR-stimulated local production of FGF15 (FGF19 in humans) (195). In hepatocytes, FGF15/19 is a major regulator of BA synthesis, through FGF receptor 4 (FGFR4) (196), and also decreases hepatic lipogenesis and indirectly stimulates mitochondrial FAO, in mouse models (196, 197). FXR also has a beneficial role in glucose metabolism, and it is important in vascular remodeling (198–200). Experimental models with FXR-null mice fed a high-cholesterol/high-fat diet develop massive steatosis (201) and exhibit decreased insulin sensitivity. Conversely, treatment with the selective, non-steroidal FXR agonist GW4064 improved IR and glucose homeostasis in obese ob/ob and diabetic db/db mice (202). Many other specific BA-activated receptors, including members of the nuclear receptor superfamily (FXR, NR1H4), a vitamin D receptor (NR1I1), PXR (NR1I2), members of the G protein–coupled receptor superfamily (TGR5 and sphingosine 1 receptor 2), and transporters such as ileal apical sodium-dependent bile acid transporter (ASBT), have a role in insulin sensitivity and NAFLD pathogenesis and are a target for novel therapies.
Conclusions
NAFLD and ALD share a number of features and often coexist. Alcohol consumption is often a confounding factor in patients with NAFLD due to inaccurate reporting of the magnitude of alcohol intake and the ill-defined impact of alcohol consumption, even within the arbitrary thresholds considered to diagnose NAFLD, on liver disease progression in these patients. Although initially some studies suggested protective effects in moderate doses, current evidence shows that there is no safe threshold for alcohol consumption in the setting of NAFLD. On the other hand, the presence of MetS and obesity increases the progression of ALD as well the incidence of HCC and mortality. Considering the high prevalence of obesity and MetS and the changing patterns of alcohol consumption worldwide, which may impact the incidence of advanced liver disease, it is necessary to better define both diseases, acknowledge the presence of a dual etiology of liver disease in a group of patients, and develop a multidisciplinary approach focused on preventive measures.
Author Contributions
FI, AK, and OM contributed to the review concept and drafting of the manuscript. MA and JA contributed to the review concept, critical revision of the manuscript for important intellectual content, supervision, and final version approval. All authors contributed to the article and approved the submitted version.
Funding
This work was funded, in part, by grants from the Fondo Nacional De Ciencia y Tecnología de Chile (FONDECYT #1191145 to MA and #1200227 to JA) and the Comisión Nacional de Investigación, Ciencia y Tecnología (CONICYT, AFB170005, CARE, Chile, UC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Valentina Riquelme, Faculty of Arts, Pontificia Universidad Católica, for her contribution in making the illustrations of this manuscript.
Abbreviations
NAFLD, non-alcoholic fatty liver disease; ALD, alcohol-related liver disease; FFA, free fatty acids; IR, insulin resistance; ER, endoplasmic reticulum; ROS, reactive oxygen species; EV, extracellular vesicles; DAMPs, damage-associated molecular patterns; HCC, hepatocellular carcinoma; HSC, hepatic stellate cell; IL-6, interleukin 6; LPS, lipopolysaccharide; MetS, metabolic syndrome; PDGF, platelet-derived growth factor; TLR, toll-like receptors; TLR4, Toll-like receptor 4; TNFα, tumor necrosis factor α; TGF-β, transforming growth factor-beta; T2DM, type 2 diabetes mellitus; DNL, de novo hepatic lipogenesis; FAO, fatty acid oxidation; ACC, acetyl-CoA carboxylase; CPT, carnitine palmitoyltransferase; IRS, insulin receptor substrate; TRAIL-R2, TRAIL receptor 2; HMGB1, high-mobility-group protein box; SHH, sonic hedgehog; NE, neutrophil elastase; NETs, neutrophil extracellular traps; NLRs, NOD-like receptors; miRs, microRNAs; NR, nuclear receptors; LXR, liver X receptor; PXR, pregnane X receptor; PPARγ, peroxisome proliferator-activated receptor-gamma; FXR, farnesoid X receptor; FGFR4, FGF receptor 4; ASBT, apical sodium-dependent bile acid transporter; BMI, body mass index; ALT, serum alanine aminotransferase; MAFLD, metabolic (dysfunction) associated fatty liver disease; AAFLD, alcohol-associated fatty liver disease.
References
1. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. (2012) 55:2005–23. doi: 10.1002/hep.25762
2. Younossi Z, Henry L. Contribution of alcoholic and nonalcoholic fatty liver disease to the burden of liver-related morbidity and mortality. Gastroenterology. (2016) 150:1778–85. doi: 10.1053/j.gastro.2016.03.005
3. Paik JM, Golabi P, Biswas R, Alqahtani S, Venkatesan C, Younossi ZM. Nonalcoholic fatty liver disease and alcoholic liver disease are major drivers of liver mortality in the United States. Hepatol Commun. (2020) 4:890–903. doi: 10.1002/hep4.1510
4. Zelman S. The liver in obesity. AMA Arch Intern Med. (1952) 90:141–56. doi: 10.1001/archinte.1952.00240080007002
5. Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. (1980) 55:434–8.
6. European Association for the Study of the L, European Association for the Study of D, European Association for the Study of O. EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. (2016) 64:1388–402. doi: 10.1016/j.jhep.2015.11.004
7. Bedossa P. Pathology of non-alcoholic fatty liver disease. Liver Int. (2017) 37(Suppl. 1):85–9. doi: 10.1111/liv.13301
8. Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. (2020) 158:1851–64. doi: 10.1053/j.gastro.2020.01.052
9. Targher G, Byrne CD, Tilg H. NAFLD and increased risk of cardiovascular disease: clinical associations, pathophysiological mechanisms and pharmacological implications. Gut. (2020). doi: 10.1136/gutjnl-2020-320622
10. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. (2018) 67:123–33. doi: 10.1002/hep.29466
11. Mitra S, De A, Chowdhury A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl Gastroenterol Hepatol. (2020) 5:16. doi: 10.21037/tgh.2019.09.08
12. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. (2016) 64:73–84. doi: 10.1002/hep.28431
13. Argo CK, Caldwell SH. Epidemiology and natural history of non-alcoholic steatohepatitis. Clin Liver Dis. (2009) 13:511–31. doi: 10.1016/j.cld.2009.07.005
14. Goh GB, McCullough AJ. Natural history of nonalcoholic fatty liver disease. Dig Dis Sci. (2016) 61:1226–33. doi: 10.1007/s10620-016-4095-4
15. Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. (2019) 16:377–86. doi: 10.1038/s41575-019-0144-8
16. Arab JP, Barrera F, Gallego C, Valderas JP, Uribe S, Tejos C, et al. High prevalence of undiagnosed liver cirrhosis and advanced fibrosis in type 2 diabetic patients. Ann Hepatol. (2016) 15:721–8. doi: 10.5604/16652681.1212434
17. Arab JP, Roblero JP, Altamirano J, Bessone F, Chaves Araujo R, Higuera-De la Tijera F, et al. Alcohol-related liver disease: clinical practice guidelines by the Latin American Association for the Study of the Liver (ALEH). Ann Hepatol. (2019) 18:518–35. doi: 10.1016/j.aohep.2019.04.005
18. Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therap Adv Gastroenterol. (2011) 4:63–81. doi: 10.1177/1756283X10378925
19. Poznyak V, Fleischmann A, Rekve D, Rylett M, Rehm J, Gmel G. The world health organization's global monitoring system on alcohol and health. Alcohol Res. (2013) 35:244–9.
20. Heron M, Hoyert DL, Murphy SL, Xu J, Kochanek KD, Tejada-Vera B. Deaths: final data for 2006. Natl Vital Stat Rep. (2009) 57:1–134.
21. Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol. (2018) 113:175–94. doi: 10.1038/ajg.2017.469
22. Thun MJ, Peto R, Lopez AD, Monaco JH, Henley SJ, Heath CW, et al. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N Engl J Med. (1997) 337:1705–14. doi: 10.1056/NEJM199712113372401
23. Becker U, Deis A, Sorensen TI, Gronbaek M, Borch-Johnsen K, Muller CF, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. (1996) 23:1025–9. doi: 10.1002/hep.510230513
24. Fuchs CS, Stampfer MJ, Colditz GA, Giovannucci EL, Manson JE, Kawachi I, et al. Alcohol consumption and mortality among women. N Engl J Med. (1995) 332:1245–50. doi: 10.1056/NEJM199505113321901
25. Craciun A, Lackner C, Cortez-Pinto H. Nonalcoholic fatty liver disease versus alcohol-related liver disease: is it really so different? Curr Pharm Des. (2020) 26:1093–109. doi: 10.2174/1381612826666200122152417
26. Ajmera VH, Terrault NA, Harrison SA. Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology. (2017) 65:2090–9. doi: 10.1002/hep.29055
27. Hajifathalian K, Torabi Sagvand B, McCullough AJ. Effect of alcohol consumption on survival in nonalcoholic fatty liver disease: A National Prospective Cohort Study. Hepatology. (2019) 70:511–21. doi: 10.1002/hep.30226
28. Cainelli F, Thao T, Pung C, Vento S. Alcohol? Not for non-alcoholic fatty liver disease patients. Front Med. (2020) 7:14. doi: 10.3389/fmed.2020.00014
29. Younossi ZM, Stepanova M, Ong J, Yilmaz Y, Duseja A, Eguchi Y, et al. Effects of alcohol consumption and metabolic syndrome on mortality in patients with nonalcoholic and alcohol-related fatty liver disease. Clin Gastroenterol Hepatol. (2019) 17:1625–33 e1. doi: 10.1016/j.cgh.2018.11.033
30. Aberg F, Helenius-Hietala J, Puukka P, Farkkila M, Jula A. Interaction between alcohol consumption and metabolic syndrome in predicting severe liver disease in the general population. Hepatology. (2018) 67:2141–9. doi: 10.1002/hep.29631
31. Eslam M, Sanyal AJ, George J. Toward more accurate nomenclature for fatty liver diseases. Gastroenterology. (2019) 157:590–3. doi: 10.1053/j.gastro.2019.05.064
32. Dunn W, Sanyal AJ, Brunt EM, Unalp-Arida A, Donohue M, McCullough AJ, et al. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J Hepatol. (2012) 57:384–91. doi: 10.1016/j.jhep.2012.03.024
33. Kwon HK, Greenson JK, Conjeevaram HS. Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int. (2014) 34:129–35. doi: 10.1111/liv.12230
34. Ekstedt M, Franzen LE, Holmqvist M, Bendtsen P, Mathiesen UL, Bodemar G, et al. Alcohol consumption is associated with progression of hepatic fibrosis in non-alcoholic fatty liver disease. Scand J Gastroenterol. (2009) 44:366–74. doi: 10.1080/00365520802555991
35. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. (2010) 51:1972–8. doi: 10.1002/hep.23527
36. Dixon JB, Bhathal PS, O'Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. (2001) 121:91–100. doi: 10.1053/gast.2001.25540
37. Suzuki A, Angulo P, St Sauver J, Muto A, Okada T, Lindor K. Light to moderate alcohol consumption is associated with lower frequency of hypertransaminasemia. Am J Gastroenterol. (2007) 102:1912–9. doi: 10.1111/j.1572-0241.2007.01274.x
38. Gunji T, Matsuhashi N, Sato H, Fujibayashi K, Okumura M, Sasabe N, et al. Light and moderate alcohol consumption significantly reduces the prevalence of fatty liver in the Japanese male population. Am J Gastroenterol. (2009) 104:2189–95. doi: 10.1038/ajg.2009.361
39. Gunji T, Sato H, Iijima K, Fujibayashi K, Okumura M, Sasabe N, et al. Modest alcohol consumption has an inverse association with liver fat content. Hepatogastroenterology. (2012) 59:2552–6. doi: 10.5754/hge12175
40. Hiramine Y, Imamura Y, Uto H, Koriyama C, Horiuchi M, Oketani M, et al. Alcohol drinking patterns and the risk of fatty liver in Japanese men. J Gastroenterol. (2011) 46:519–28. doi: 10.1007/s00535-010-0336-z
41. Moriya A, Iwasaki Y, Ohguchi S, Kayashima E, Mitsumune T, Taniguchi H, et al. Alcohol consumption appears to protect against non-alcoholic fatty liver disease. Aliment Pharmacol Ther. (2011) 33:378–88. doi: 10.1111/j.1365-2036.2010.04520.x
42. Hamaguchi M, Kojima T, Ohbora A, Takeda N, Fukui M, Kato T. Protective effect of alcohol consumption for fatty liver but not metabolic syndrome. World J Gastroenterol. (2012) 18:156–67. doi: 10.3748/wjg.v18.i2.156
43. Hagstrom H, Nasr P, Ekstedt M, Kechagias S, Onnerhag K, Nilsson E, et al. Low to moderate lifetime alcohol consumption is associated with less advanced stages of fibrosis in non-alcoholic fatty liver disease. Scand J Gastroenterol. (2017) 52:159–65. doi: 10.1080/00365521.2016.1239759
44. Moriya A, Iwasaki Y, Ohguchi S, Kayashima E, Mitsumune T, Taniguchi H, et al. Roles of alcohol consumption in fatty liver: a longitudinal study. J Hepatol. (2015) 62:921–7. doi: 10.1016/j.jhep.2014.11.025
45. Mitchell T, Jeffrey GP, de Boer B, MacQuillan G, Garas G, Ching H, et al. Type and pattern of alcohol consumption is associated with liver fibrosis in patients with non-alcoholic fatty liver disease. Am J Gastroenterol. (2018) 113:1484–93. doi: 10.1038/s41395-018-0133-5
46. Sookoian S, Castano GO, Pirola CJ. Modest alcohol consumption decreases the risk of non-alcoholic fatty liver disease: a meta-analysis of 43 175 individuals. Gut. (2014) 63:530–2. doi: 10.1136/gutjnl-2013-305718
47. Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. (1997) 41:845–50. doi: 10.1136/gut.41.6.845
48. Bellentani S, Saccoccio G, Masutti F, Croce LS, Brandi G, Sasso F, et al. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. (2000) 132:112–7. doi: 10.7326/0003-4819-132-2-200001180-00004
49. Bedogni G, Miglioli L, Masutti F, Castiglione A, Croce LS, Tiribelli C, et al. Incidence and natural course of fatty liver in the general population: the Dionysos study. Hepatology. (2007) 46:1387–91. doi: 10.1002/hep.21827
50. Chang Y, Cho YK, Kim Y, Sung E, Ahn J, Jung HS, et al. Nonheavy drinking and worsening of noninvasive fibrosis markers in nonalcoholic fatty liver disease: a Cohort study. Hepatology. (2019) 69:64–75. doi: 10.1002/hep.30170
51. Ajmera V, Belt P, Wilson LA, Gill RM, Loomba R, Kleiner DE, et al. Among patients with nonalcoholic fatty liver disease, modest alcohol use is associated with less improvement in histologic steatosis and steatohepatitis. Clin Gastroenterol Hepatol. (2018) 16:1511–20 e5. doi: 10.1016/j.cgh.2018.01.026
52. Verrill C, Markham H, Templeton A, Carr NJ, Sheron N. Alcohol-related cirrhosis–early abstinence is a key factor in prognosis, even in the most severe cases. Addiction. (2009) 104:768–74. doi: 10.1111/j.1360-0443.2009.02521.x
53. Sookoian S, Flichman D, Castano GO, Pirola CJ. Mendelian randomisation suggests no beneficial effect of moderate alcohol consumption on the severity of nonalcoholic fatty liver disease. Aliment Pharmacol Ther. (2016) 44:1224–34. doi: 10.1111/apt.13828
54. Yi SW, Choi JS, Yi JJ, Lee YH, Han KJ. Risk factors for hepatocellular carcinoma by age, sex, and liver disorder status: a prospective cohort study in Korea. Cancer. (2018) 124:2748–57. doi: 10.1002/cncr.31406
55. Askgaard G, Gronbaek M, Kjaer MS, Tjonneland A, Tolstrup JS. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: a prospective cohort study. J Hepatol. (2015) 62:1061–7. doi: 10.1016/j.jhep.2014.12.005
56. Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decreased prevalence of suspected nonalcoholic fatty liver disease. Hepatology. (2008) 47:1947–54. doi: 10.1002/hep.22292
57. Seitz HK, Mueller S, Hellerbrand C, Liangpunsakul S. Effect of chronic alcohol consumption on the development and progression of non-alcoholic fatty liver disease (NAFLD). Hepatobiliary Surg Nutr. (2015) 4:147–51. doi: 10.3978/j.issn.2304-3881.2014.12.01
58. Aberg F, Puukka P, Salomaa V, Mannisto S, Lundqvist A, Valsta L, et al. Risks of light and moderate alcohol use in fatty liver disease: follow-up of population cohorts. Hepatology. (2020) 71:835–48. doi: 10.1002/hep.30864
59. Bellentani S, Pozzato G, Saccoccio G, Crovatto M, Croce LS, Mazzoran L, et al. Clinical course and risk factors of hepatitis C virus related liver disease in the general population: report from the Dionysos study. Gut. (1999) 44:874–80. doi: 10.1136/gut.44.6.874
60. Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. (2010) 340:c1240. doi: 10.1136/bmj.c1240
61. Hezode C, Lonjon I, Roudot-Thoraval F, Pawlotsky JM, Zafrani ES, Dhumeaux D. Impact of moderate alcohol consumption on histological activity and fibrosis in patients with chronic hepatitis C, and specific influence of steatosis: a prospective study. Aliment Pharmacol Ther. (2003) 17:1031–7. doi: 10.1046/j.1365-2036.2003.01546.x
62. Loomba R, Yang HI, Su J, Brenner D, Iloeje U, Chen CJ. Obesity and alcohol synergize to increase the risk of incident hepatocellular carcinoma in men. Clin Gastroenterol Hepatol. (2010) 8:891–8. doi: 10.1016/j.cgh.2010.06.027
63. Loomba R, Yang HI, Su J, Brenner D, Barrett-Connor E, Iloeje U, et al. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: a prospective cohort study. Am J Epidemiol. (2013) 177:333–42. doi: 10.1093/aje/kws252
64. Summerskill WH, Wolfe SJ, Davidson CS. Response to alcohol in chronic alcoholics with liver disease; clinical, pathological, and metabolic changes. Lancet. (1957) 272:335–40. doi: 10.1016/S0140-6736(57)90402-6
65. Collaborators GBDA. Alcohol use and burden for 195 countries and territories, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2018) 392:1015–35. doi: 10.1016/S0140-6736(18)31310-2
66. Hamajima N, Hirose K, Tajima K, Rohan T, Calle EE, Heath CW, et al. Alcohol, tobacco and breast cancer–collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer. (2002) 87:1234–45. doi: 10.1038/sj.bjc.6600596
67. Liu Y, Nguyen N, Colditz GA. Links between alcohol consumption and breast cancer: a look at the evidence. Womens Health. (2015) 11:65–77. doi: 10.2217/WHE.14.62
68. Chen WY, Rosner B, Hankinson SE, Colditz GA, Willett WC. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA. (2011) 306:1884–90. doi: 10.1001/jama.2011.1590
69. Wannamethee SG, Shaper AG. Alcohol, body weight, and weight gain in middle-aged men. Am J Clin Nutr. (2003) 77:1312–7. doi: 10.1093/ajcn/77.5.1312
70. Bendsen NT, Christensen R, Bartels EM, Kok FJ, Sierksma A, Raben A, et al. Is beer consumption related to measures of abdominal and general obesity? A systematic review and meta-analysis. Nutr Rev. (2013) 71:67–87. doi: 10.1111/j.1753-4887.2012.00548.x
71. Eslam M, Sanyal AJ, George J, an international consensus p. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
72. Eslam M, Newsome PN, Anstee QM, Targher G, Gomez MR, Zelber-Sagi S, et al. A new definition for metabolic associated fatty liver disease: an international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.03.039
74. Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. (2018) 15:349–64. doi: 10.1038/s41575-018-0009-6
75. Gao B, Ahmad MF, Nagy LE, Tsukamoto H. Inflammatory pathways in alcoholic steatohepatitis. J Hepatol. (2019) 70:249–59. doi: 10.1016/j.jhep.2018.10.023
76. Lackner C, Tiniakos D. Fibrosis and alcohol-related liver disease. J Hepatol. (2019) 70:294–304. doi: 10.1016/j.jhep.2018.12.003
77. Arab JP, Arrese M, Trauner M. Recent insights into the pathogenesis of nonalcoholic fatty liver disease. Annu Rev Pathol. (2018) 13:321–50. doi: 10.1146/annurev-pathol-020117-043617
78. Greuter T, Malhi H, Gores GJ, Shah VH. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic Steatohepatitis: exploiting similarities and differences in pathogenesis. JCI Insight. (2017) 2:e95354. doi: 10.1172/jci.insight.95354
79. Neuman MG, French SW, French BA, Seitz HK, Cohen LB, Mueller S, et al. Alcoholic and non-alcoholic steatohepatitis. Exp Mol Pathol. (2014) 97:492–510. doi: 10.1016/j.yexmp.2014.09.005
80. Joshi-Barve S, Kirpich I, Cave MC, Marsano LS, McClain CJ. Alcoholic, nonalcoholic, and toxicant-associated steatohepatitis: mechanistic similarities and differences. Cell Mol Gastroenterol Hepatol. (2015) 1:356–67. doi: 10.1016/j.jcmgh.2015.05.006
81. Lakshman MR. Some novel insights into the pathogenesis of alcoholic steatosis. Alcohol. (2004) 34:45–8. doi: 10.1016/j.alcohol.2004.08.004
82. Higuchi N, Kato M, Shundo Y, Tajiri H, Tanaka M, Yamashita N, et al. Liver X receptor in cooperation with SREBP-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol Res. (2008) 38:1122–9. doi: 10.1111/j.1872-034X.2008.00382.x
83. Lakshman R, Shah R, Reyes-Gordillo K, Varatharajalu R. Synergy between NAFLD and AFLD and potential biomarkers. Clin Res Hepatol Gastroenterol. (2015) 39(Suppl. 1):S29–34. doi: 10.1016/j.clinre.2015.05.007
84. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. (2018) 75:3313–27. doi: 10.1007/s00018-018-2860-6
85. You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem. (2002) 277:29342–7. doi: 10.1074/jbc.M202411200
86. Mitsuyoshi H, Yasui K, Harano Y, Endo M, Tsuji K, Minami M, et al. Analysis of hepatic genes involved in the metabolism of fatty acids and iron in nonalcoholic fatty liver disease. Hepatol Res. (2009) 39:366–73. doi: 10.1111/j.1872-034X.2008.00464.x
87. Kohjima M, Higuchi N, Kato M, Kotoh K, Yoshimoto T, Fujino T, et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int J Mol Med. (2008) 21:507–11. doi: 10.3892/ijmm.21.4.507
88. Jeon S, Carr R. Alcohol effects on hepatic lipid metabolism. J Lipid Res. (2020) 61:470–9. doi: 10.1194/jlr.R119000547
89. Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. (2014) 146:726–35. doi: 10.1053/j.gastro.2013.11.049
90. You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. (2004) 127:1798–808. doi: 10.1053/j.gastro.2004.09.049
91. Morgan K, Uyuni A, Nandgiri G, Mao L, Castaneda L, Kathirvel E, et al. Altered expression of transcription factors and genes regulating lipogenesis in liver and adipose tissue of mice with high fat diet-induced obesity and nonalcoholic fatty liver disease. Eur J Gastroenterol Hepatol. (2008) 20:843–54. doi: 10.1097/MEG.0b013e3282f9b203
92. Bays H. Adiposopathy, “sick fat,” Ockham's razor, and resolution of the obesity paradox. Curr Atheroscler Rep. (2014) 16:409. doi: 10.1007/s11883-014-0409-1
93. Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. (2020) 4:478–92. doi: 10.1002/hep4.1479
94. Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. (2016) 57:233–45. doi: 10.1194/jlr.M063412
95. Wei X, Shi X, Zhong W, Zhao Y, Tang Y, Sun W, et al. Chronic alcohol exposure disturbs lipid homeostasis at the adipose tissue-liver axis in mice: analysis of triacylglycerols using high-resolution mass spectrometry in combination with in vivo metabolite deuterium labeling. PLoS ONE. (2013) 8:e55382. doi: 10.1371/journal.pone.0055382
96. Jou J, Choi SS, Diehl AM. Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin Liver Dis. (2008) 28:370–9. doi: 10.1055/s-0028-1091981
97. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. (2018) 24:908–22. doi: 10.1038/s41591-018-0104-9
98. Qureshi K, Neuschwander-Tetri BA. The molecular basis for current targets of NASH therapies. Expert Opin Investig Drugs. (2020) 29:151–61. doi: 10.1080/13543784.2020.1703949
99. Carr RM, Correnti J. Insulin resistance in clinical and experimental alcoholic liver disease. Ann N Y Acad Sci. (2015) 1353:1–20. doi: 10.1111/nyas.12787
100. Aizawa S, Brar G, Tsukamoto H. Cell death and liver disease. Gut Liver. (2020) 14:20–9. doi: 10.5009/gnl18486
101. Gautheron J, Gores GJ, Rodrigues CMP. Lytic cell death in metabolic liver disease. J Hepatol. (2020) 73:394–408. doi: 10.1016/j.jhep.2020.04.001
102. Wang S, Pacher P, De Lisle RC, Huang H, Ding WX. A mechanistic review of cell death in alcohol-induced liver injury. Alcohol Clin Exp Res. (2016) 40:1215–23. doi: 10.1111/acer.13078
103. Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. (2005) 10:3093–9. doi: 10.2741/1765
104. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. (2003) 125:437–43. doi: 10.1016/S0016-5085(03)00907-7
105. Casey CA, Nanji A, Cederbaum AI, Adachi M, Takahashi T. Alcoholic liver disease and apoptosis. Alcohol Clin Exp Res. (2001) 25:49S−53S. doi: 10.1111/j.1530-0277.2001.tb02373.x
106. Idrissova L, Malhi H, Werneburg NW, LeBrasseur NK, Bronk SF, Fingas C, et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J Hepatol. (2015) 62:1156–63. doi: 10.1016/j.jhep.2014.11.033
107. Affo S, Dominguez M, Lozano JJ, Sancho-Bru P, Rodrigo-Torres D, Morales-Ibanez O, et al. Transcriptome analysis identifies TNF superfamily receptors as potential therapeutic targets in alcoholic hepatitis. Gut. (2013) 62:452–60. doi: 10.1136/gutjnl-2011-301146
108. Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. (2006) 281:12093–101. doi: 10.1074/jbc.M510660200
109. Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. (2007) 56:1124–31. doi: 10.1136/gut.2006.118059
110. Mundt B, Wirth T, Zender L, Waltemathe M, Trautwein C, Manns MP, et al. Tumour necrosis factor related apoptosis inducing ligand (TRAIL) induces hepatic steatosis in viral hepatitis and after alcohol intake. Gut. (2005) 54:1590–6. doi: 10.1136/gut.2004.056929
111. Hirsova P, Gores GJ. Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. (2015) 1:17–27. doi: 10.1016/j.jcmgh.2014.11.005
112. Ntandja Wandji LC, Gnemmi V, Mathurin P, Louvet A. Combined alcoholic and non-alcoholic steatohepatitis. JHEP Rep. (2020) 2:100101. doi: 10.1016/j.jhepr.2020.100101
113. Czabotar PE, Murphy JM. A tale of two domains - a structural perspective of the pseudokinase, MLKL. FEBS J. (2015) 282:4268–78. doi: 10.1111/febs.13504
114. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. (2009) 137:1100–11. doi: 10.1016/j.cell.2009.05.021
115. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. (2005) 1:112–9. doi: 10.1038/nchembio711
116. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. (2013) 57:1773–83. doi: 10.1002/hep.26200
117. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med. (2014) 6:1062–74. doi: 10.15252/emmm.201403856
118. Hatting M, Zhao G, Schumacher F, Sellge G, Al Masaoudi M, Gabetaler N, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology. (2013) 57:2189–201. doi: 10.1002/hep.26271
119. Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. (2016) 2:16089. doi: 10.1038/cddiscovery.2016.89
120. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
121. Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci. (2016) 61:1304–13. doi: 10.1007/s10620-015-4025-x
122. Cursio R, Colosetti P, Codogno P, Cuervo AM, Shen HM. The role of autophagy in liver diseases: mechanisms and potential therapeutic targets. Biomed Res Int. (2015) 2015:480508. doi: 10.1155/2015/480508
123. Mei S, Ni HM, Manley S, Bockus A, Kassel KM, Luyendyk JP, et al. Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J Pharmacol Exp Ther. (2011) 339:487–98. doi: 10.1124/jpet.111.184341
124. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate immunity and inflammation in NAFLD/NASH. Dig Dis Sci. (2016) 61:1294–303. doi: 10.1007/s10620-016-4049-x
125. Li S, Tan HY, Wang N, Feng Y, Wang X, Feng Y. Recent insights into the role of immune cells in alcoholic liver disease. Front Immunol. (2019) 10:1328. doi: 10.3389/fimmu.2019.01328
126. Seo YS, Kwon JH, Yaqoob U, Yang L, De Assuncao TM, Simonetto DA, et al. HMGB1 recruits hepatic stellate cells and liver endothelial cells to sites of ethanol-induced parenchymal cell injury. Am J Physiol Gastrointest Liver Physiol. (2013) 305:G838–48. doi: 10.1152/ajpgi.00151.2013
127. Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology. (2011) 54:1620–30. doi: 10.1002/hep.24552
128. Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology. (2012) 55:1711–21. doi: 10.1002/hep.25559
129. Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. (2012) 18:1407–12. doi: 10.1038/nm.2885
130. Ramaiah SK, Jaeschke H. Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol. (2007) 35:757–66. doi: 10.1080/01926230701584163
131. Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M, et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology. (2019) 157:193–209 e9. doi: 10.1053/j.gastro.2019.03.013
132. Bala S, Marcos M, Gattu A, Catalano D, Szabo G. Acute binge drinking increases serum endotoxin and bacterial DNA levels in healthy individuals. PLoS ONE. (2014) 9:e96864. doi: 10.1371/journal.pone.0096864
133. Mandrekar P, Ambade A, Lim A, Szabo G, Catalano D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology. (2011) 54:2185–97. doi: 10.1002/hep.24599
134. Miura K, Yang L, van Rooijen N, Ohnishi H, Seki E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G1310–21. doi: 10.1152/ajpgi.00365.2011
135. Connolly MK, Bedrosian AS, Malhotra A, Henning JR, Ibrahim J, Vera V, et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immunogenicity. J Immunol. (2010) 185:2200–8. doi: 10.4049/jimmunol.1000332
136. Sprenger H, Kaufmann A, Garn H, Lahme B, Gemsa D, Gressner AM. Differential expression of monocyte chemotactic protein-1 (MCP-1) in transforming rat hepatic stellate cells. J Hepatol. (1999) 30:88–94. doi: 10.1016/S0168-8278(99)80011-7
137. Szabo G, Iracheta-Vellve A. Inflammasome activation in the liver: focus on alcoholic and non-alcoholic steatohepatitis. Clin Res Hepatol Gastroenterol. (2015) 39(Suppl. 1):S18–23. doi: 10.1016/j.clinre.2015.06.012
138. Franchi L, Eigenbrod T, Nunez G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol. (2009) 183:792–6. doi: 10.4049/jimmunol.0900173
139. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
140. Stehlik C, Fiorentino L, Dorfleutner A, Bruey JM, Ariza EM, Sagara J, et al. The PAAD/PYRIN-family protein ASC is a dual regulator of a conserved step in nuclear factor kappaB activation pathways. J Exp Med. (2002) 196:1605–15. doi: 10.1084/jem.20021552
141. Stehlik C, Lee SH, Dorfleutner A, Stassinopoulos A, Sagara J, Reed JC. Apoptosis-associated speck-like protein containing a caspase recruitment domain is a regulator of procaspase-1 activation. J Immunol. (2003) 171:6154–63. doi: 10.4049/jimmunol.171.11.6154
142. Luan J, Ju D. Inflammasome: a double-edged sword in liver diseases. Front Immunol. (2018) 9:2201. doi: 10.3389/fimmu.2018.02201
143. Ganz M, Csak T, Nath B, Szabo G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J Gastroenterol. (2011) 17:4772–8. doi: 10.3748/wjg.v17.i43.4772
144. Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, et al. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J Leukoc Biol. (2013) 94:171–82. doi: 10.1189/jlb.1212659
145. Cabrera D, Wree A, Povero D, Solis N, Hernandez A, Pizarro M, et al. Andrographolide ameliorates inflammation and fibrogenesis and attenuates inflammasome activation in experimental non-alcoholic steatohepatitis. Sci Rep. (2017) 7:3491. doi: 10.1038/s41598-017-03675-z
146. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. (2014) 59:898–910. doi: 10.1002/hep.26592
147. Ganz M, Szabo G. Immune and inflammatory pathways in NASH. Hepatol Int. (2013) 7(Suppl. 2):771–81. doi: 10.1007/s12072-013-9468-6
148. Knorr J, Wree A, Tacke F, Feldstein AE. The NLRP3 inflammasome in alcoholic and nonalcoholic steatohepatitis. Semin Liver Dis. (2020). doi: 10.1055/s-0040-1708540
149. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. (2012) 122:3476–89. doi: 10.1172/JCI60777
150. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. (2011) 54:133–44. doi: 10.1002/hep.24341
151. Petrasek J, Csak T, Ganz M, Szabo G. Differences in innate immune signaling between alcoholic and non-alcoholic steatohepatitis. J Gastroenterol Hepatol. (2013) 28(Suppl. 1):93–8. doi: 10.1111/jgh.12020
152. Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, et al. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology. (2010) 139:323–34 e7. doi: 10.1053/j.gastro.2010.03.052
153. Dixon LJ, Berk M, Thapaliya S, Papouchado BG, Feldstein AE. Caspase-1-mediated regulation of fibrogenesis in diet-induced steatohepatitis. Lab Invest. (2012) 92:713–23. doi: 10.1038/labinvest.2012.45
154. Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. (2011) 55:1086–94. doi: 10.1016/j.jhep.2011.01.048
155. Hernandez A, Arab JP, Reyes D, Lapitz A, Moshage H, Banales JM, et al. Extracellular vesicles in NAFLD/ALD: from pathobiology to therapy. Cells. (2020) 9:817. doi: 10.3390/cells9040817
156. Hernandez A, Reyes D, Geng Y, Arab JP, Cabrera D, Sepulveda R, et al. Extracellular vesicles derived from fat-laden hepatocytes undergoing chemical hypoxia promote a pro-fibrotic phenotype in hepatic stellate cells. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165857. doi: 10.1016/j.bbadis.2020.165857
157. Hernandez A, Geng Y, Sepulveda R, Solis N, Torres J, Arab JP, et al. Chemical hypoxia induces pro-inflammatory signals in fat-laden hepatocytes and contributes to cellular crosstalk with Kupffer cells through extracellular vesicles. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165753. doi: 10.1016/j.bbadis.2020.165753
158. Malhi H. Emerging role of extracellular vesicles in liver diseases. Am J Physiol Gastrointest Liver Physiol. (2019) 317:G739–G49. doi: 10.1152/ajpgi.00183.2019
159. Hirsova P, Ibrahim SH, Bronk SF, Yagita H, Gores GJ. Vismodegib suppresses TRAIL-mediated liver injury in a mouse model of nonalcoholic steatohepatitis. PLoS ONE. (2013) 8:e70599. doi: 10.1371/journal.pone.0070599
160. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. (2016) 63:731–44. doi: 10.1002/hep.28252
161. Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. (2016) 64:651–60. doi: 10.1016/j.jhep.2015.11.020
162. Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol. (2013) 58:785–91. doi: 10.1016/j.jhep.2012.11.042
163. Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. (2012) 287:9817–26. doi: 10.1074/jbc.M111.333534
164. Miyaaki H, Ichikawa T, Kamo Y, Taura N, Honda T, Shibata H, et al. Significance of serum and hepatic microRNA-122 levels in patients with non-alcoholic fatty liver disease. Liver Int. (2014) 34:e302–7. doi: 10.1111/liv.12429
165. Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (TNF{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. (2011) 286:1436–44. doi: 10.1074/jbc.M110.145870
166. Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. (2013) 10:542–52. doi: 10.1038/nrgastro.2013.87
167. Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. (2013) 368:1685–94. doi: 10.1056/NEJMoa1209026
168. Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. (2009) 587(Pt 17):4153–8. doi: 10.1113/jphysiol.2009.174136
169. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. (2017) 474:1823–36. doi: 10.1042/BCJ20160510
170. Betrapally NS, Gillevet PM, Bajaj JS. Changes in the intestinal microbiome and alcoholic and nonalcoholic liver diseases: causes or effects? Gastroenterology. (2016) 150:1745–55 e3. doi: 10.1053/j.gastro.2016.02.073
171. Rivera CA, Bradford BU, Seabra V, Thurman RG. Role of endotoxin in the hypermetabolic state after acute ethanol exposure. Am J Physiol. (1998) 275:G1252–8. doi: 10.1152/ajpgi.1998.275.6.G1252
172. Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, et al. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. (2012) 302:G966–78. doi: 10.1152/ajpgi.00380.2011
173. Roh YS, Seki E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J Gastroenterol Hepatol. (2013) 28(Suppl. 1):38–42. doi: 10.1111/jgh.12019
174. Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. (2009) 136:65–80. doi: 10.1053/j.gastro.2008.10.080
175. Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, et al. Innate immunity in alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. (2011) 300:G516–25. doi: 10.1152/ajpgi.00537.2010
176. Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, et al. Differential liver sensitization to toll-like receptor pathways in mice with alcoholic fatty liver. Hepatology. (2006) 43:989–1000. doi: 10.1002/hep.21138
177. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. (2013) 57:601–9. doi: 10.1002/hep.26093
178. Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, et al. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci USA. (2011) 108(Suppl. 1):4523–30. doi: 10.1073/pnas.1006734107
179. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. (2011) 472:57–63. doi: 10.1038/nature09922
180. Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM, et al. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia. (2007) 50:2374–83. doi: 10.1007/s00125-007-0791-0
181. Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. (2012) 482:179–85. doi: 10.1038/nature10809
182. Petrasek J, Dolganiuc A, Csak T, Kurt-Jones EA, Szabo G. Type I interferons protect from Toll-like receptor 9-associated liver injury and regulate IL-1 receptor antagonist in mice. Gastroenterology. (2011) 140:697–708 e4. doi: 10.1053/j.gastro.2010.08.020
183. Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, et al. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol. (2008) 181:3049–56. doi: 10.4049/jimmunol.181.5.3049
184. Mandal P, Roychowdhury S, Park PH, Pratt BT, Roger T, Nagy LE. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. (2010) 185:4928–37. doi: 10.4049/jimmunol.1002060
185. Machado MV, Cortez-Pinto H. Nuclear receptors: how do they position in non-alcoholic fatty liver disease treatment? Liver Int. (2014) 34:1291–4. doi: 10.1111/liv.12578
186. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. (2001) 294:1866–70. doi: 10.1126/science.294.5548.1866
187. Wang H, LeCluyse EL. Role of orphan nuclear receptors in the regulation of drug-metabolising enzymes. Clin Pharmacokinet. (2003) 42:1331–57. doi: 10.2165/00003088-200342150-00003
188. Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. (2003) 3:561–72. doi: 10.2174/1566524033479537
189. Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. J Nutr. (2011) 141:603–10. doi: 10.3945/jn.110.135210
190. Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J Exp Med. (2001) 193:827–38. doi: 10.1084/jem.193.7.827
191. Dhe-Paganon S, Duda K, Iwamoto M, Chi YI, Shoelson SE. Crystal structure of the HNF4 alpha ligand binding domain in complex with endogenous fatty acid ligand. J Biol Chem. (2002) 277:37973–6. doi: 10.1074/jbc.C200420200
192. Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol. (2001) 21:1393–403. doi: 10.1128/MCB.21.4.1393-1403.2001
193. Wisely GB, Miller AB, Davis RG, Thornquest AD Jr, Johnson R, Spitzer T, et al. Hepatocyte nuclear factor 4 is a transcription factor that constitutively binds fatty acids. Structure. (2002) 10:1225–34. doi: 10.1016/S0969-2126(02)00829-8
194. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. (2009) 89:147–91. doi: 10.1152/physrev.00010.2008
195. Ferrebee CB, Dawson PA. Metabolic effects of intestinal absorption and enterohepatic cycling of bile acids. Acta Pharm Sin B. (2015) 5:129–34. doi: 10.1016/j.apsb.2015.01.001
196. Jahn D, Rau M, Hermanns HM, Geier A. Mechanisms of enterohepatic fibroblast growth factor 15/19 signaling in health and disease. Cytokine Growth Factor Rev. (2015) 26:625–35. doi: 10.1016/j.cytogfr.2015.07.016
197. Fuchs CD, Traussnigg SA, Trauner M. Nuclear receptor modulation for the treatment of nonalcoholic fatty liver disease. Semin Liver Dis. (2016) 36:69–86. doi: 10.1055/s-0036-1571296
198. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. (2013) 145:574–82 e1. doi: 10.1053/j.gastro.2013.05.042
199. Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. (2011) 53:1023–34. doi: 10.1002/hep.24148
200. Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. (2012) 17:988–97. doi: 10.1016/j.drudis.2012.05.012
201. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. (2000) 102:731–44. doi: 10.1016/S0092-8674(00)00062-3
Keywords: non-alcoholic fatty liver disease, steatosis, cirrhosis, NAFLD, NASH, alcohol, alcohol-related liver disease, ALD
Citation: Idalsoaga F, Kulkarni AV, Mousa OY, Arrese M and Arab JP (2020) Non-alcoholic Fatty Liver Disease and Alcohol-Related Liver Disease: Two Intertwined Entities. Front. Med. 7:448. doi: 10.3389/fmed.2020.00448
Received: 28 April 2020; Accepted: 06 July 2020;
Published: 20 August 2020.
Edited by:
Javier Egea, Institute of Health Research of the University Hospital of La Princesa, SpainReviewed by:
Roberto Gramignoli, Karolinska Institutet (KI), SwedenZuzana Macek Jilkova, Centre Hospitalier Universitaire de Grenoble, France
Copyright © 2020 Idalsoaga, Kulkarni, Mousa, Arrese and Arab. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Pablo Arab, jparab@uc.cl