 Chongjing Xia
Chongjing Xia Meinan Wang1
Meinan Wang1 Xianming Chen
Xianming Chen- 1Department of Plant Pathology, Washington State University, Pullman, WA, United States
- 2School of Biological Sciences, Washington State University, Pullman, WA, United States
- 3Wheat Health, Genetics, and Quality Research Unit, Agricultural Research Service, U.S. Department of Agriculture, Pullman, WA, United States
Stripe (yellow) rust, caused by Puccinia striiformis f. sp. tritici (Pst), is one of the most destructive diseases of wheat worldwide. Planting resistant cultivars is an effective way to control this disease, but race-specific resistance can be overcome quickly due to the rapid evolving Pst population. Studying the pathogenicity mechanisms is critical for understanding how Pst virulence changes and how to develop wheat cultivars with durable resistance to stripe rust. We re-sequenced 7 Pst isolates and included additional 7 previously sequenced isolates to represent balanced virulence/avirulence profiles for several avirulence loci in seretome analyses. We observed an uneven distribution of heterozygosity among the isolates. Secretome comparison of Pst with other rust fungi identified a large portion of species-specific secreted proteins, suggesting that they may have specific roles when interacting with the wheat host. Thirty-two effectors of Pst were identified from its secretome. We identified candidates for Avr genes corresponding to six Yr genes by correlating polymorphisms for effector genes to the virulence/avirulence profiles of the 14 Pst isolates. The putative AvYr76 was present in the avirulent isolates, but absent in the virulent isolates, suggesting that deleting the coding region of the candidate avirulence gene has produced races virulent to resistance gene Yr76. We conclude that incorporating avirulence/virulence phenotypes into correlation analysis with variations in genomic structure and secretome, particularly presence/absence polymorphisms of effectors, is an efficient way to identify candidate Avr genes in Pst. The candidate effector genes provide a rich resource for further studies to determine the evolutionary history of Pst populations and the co-evolutionary arms race between Pst and wheat. The Avr candidates identified in this study will lead to cloning avirulence genes in Pst, which will enable us to understand molecular mechanisms underlying Pst-wheat interactions, to determine the effectiveness of resistance genes and further to develop durable resistance to stripe rust.
Introduction
Wheat stripe (yellow) rust, caused by the obligate biotrophic fungus Puccinia striiformis Westend. f. sp. tritici Erikss. (Pst), is a significant threat to wheat production in the world (Chen, 2005; Wellings, 2011). Planting resistant cultivars is widely accepted as the most effective, economic, easy-to-use, and environmental-friendly way to control this disease (Chen, 2013). However, breeding resistant cultivars can be complicated by several biological characteristics of the pathogen, e.g., long-distance aerial dispersal of urediniospores and ever-changing virulent races (Brown and Hovmøller, 2002; Line, 2002; Chen, 2005; Liu et al., 2017). The rapid, long-distance aerial dispersal or unintentional introduction by travelers can lead to new invasions and reestablishments of the disease in previously disease-free or seasonally pathogen-absent regions (Wellings, 2011; Sharma-Poudyal et al., 2014). The rapid evolution of Pst results in “breakdown” of race-specific resistance after resistant cultivars are grown for only few years (Line and Qayoum, 1992; Bayles et al., 2000; de Vallavieille-Pope et al., 2012). In the United States, more than 200 different races have been identified since the 1960s with an average of 6 new races identified every year (Wan and Chen, 2014; Wan et al., 2016; Liu et al., 2017). The change in races has accelerated as more races, including new races have been detected since the year 2000. The new races, under favorable weather conditions, could shorten the life time of wheat cultivars with only race-specific resistance and cause destructive epidemics (Line and Qayoum, 1992; Chen, 2005; Chen et al., 2010; Hovmøller et al., 2016; Wan et al., 2016). Thus, understanding Pst evolution is critical and valuable not only for predicting and monitoring the population changes but also for developing cultivars with durable resistance for sustainable control of the disease. Therefore, the pathogenicity and molecular mechanisms that Pst uses to infect and interact with wheat need to be elucidated.
Although stripe rust is economically important, molecular mechanisms of Pst-wheat interactions are not fully understood. Based on the discoveries in other pathosystems, effectors, mainly protein molecules secreted from pathogens, are important components to modulate plant immunity and facilitate infection (Toruño et al., 2016). These effectors are located in different subcellular locations within plants, e.g., in the host cell and in the apoplastic space between adjacent cells, indicating their diverse functions ranging from modulating plant cellular metabolic pathways and signaling cascades, RNA silencing, anti-microbial inhibition and interfering with recognition machinery (Sharpee and Dean, 2016). To our knowledge, only a limited number of studies have been conducted to identify secreted protein (SP) genes and possible effectors in the Pst fungus (Yin et al., 2009, 2015; Huang et al., 2011; Saunders et al., 2012; Cantu et al., 2013; Cheng et al., 2015; Liu et al., 2016; Kiran et al., 2017). Among plant pathogen effectors, there is a special group, called avirulence (Avr) effectors which trigger multifaceted defense responses upon recognition by cognate plant resistance (R) proteins and therefore determine resistance specificity. The interactions between most Avr and corresponding R genes determine the gene-for-gene relationship in which “for each R gene that conditions resistance in the host there is a corresponding Avr gene that conditions pathogenicity in the parasite” (Flor, 1971). In fact, the first cloned effectors from rust and other fungi were all Avr effectors since it was easier to detect the avirulence function, e.g., the hypersensitive responses (Petre et al., 2014; de Wit, 2016). However, no Avr effectors have been identified and characterized in Pst.
The availability of Pst genomes and the advanced next generation sequencing technologies enable us to predict effector candidates from whole genome screening based on some conserved features of cloned fungi and fungi-like plant pathogen effectors, even though the cloned effectors generally show low sequence similarities among species (Sperschneider et al., 2015a). Such conserved features include secreted, small and cysteine-rich proteins which are representative for first cloned fungal Avr effector Avr9, effectors from Cladosporium fulvum and for most other cloned effectors (van Kan et al., 1991; Sperschneider et al., 2015c). Particularly in the flax rust fungus Melampsora lini (Mli), four Avr genes (AvrL567, AvrM, AvrP4, and AvrP123) were identified from 21 haustorially expressed secreted proteins (HESPs) and transient expression of these genes in flax caused resistance gene-mediated cell death, thus confirming their avirulence activities (Catanzariti et al., 2006). Haustoria are of special interest because it is a specialized infection structure through which the fungus obtains nutrients from the host and produces secreted effectors to interact with the host (Garnica et al., 2013, 2014). In the Pst fungus, Garnica et al. (2013) identified 437 HESPs as putative effectors by transcriptome sequencing. This result indicates that the mentioned conserved features are not one-size-fits-all criteria, and other rules could be added to the effector candidates mining pipeline. For example, the conserved structures of functionally characterized motifs also provide new opportunities for clustering and identifying plant pathogen effectors. More features, such as presence of long intergenic regions between effector genes, internal repeats and no PFAM domains except with known pathogenicity have been used in a comprehensive hierarchical clustering method to screen rust effectors (Saunders et al., 2012). Moreover, besides the criteria mentioned above, Cantu et al. (2013) incorporated the polymorphism analysis in haustoria-expressed genes into secretome characterization and identified five candidate effectors from 2,999 SPs for two closely related Pst isolates. However, no effectors were identified for particular avirulence genes.
In the present study, we characterized the Pst secretome and identified candidate effectors using meta-analysis of genome and transcriptome data using an advanced next generation sequencing technology and the latest pathogen-host interaction (PHI) database, e.g., PHI-base 4.0 to annotate Pst candidate effectors. We further identified candidate Avr effectors in Pst isolates corresponding to wheat resistance genes by correlating polymorphic variations with the virulence phenotypes of 14 Pst isolates. Previous studies of genetic inheritance of pathogenicity in flax-rust and stem rust-wheat pathosystems, and preliminary studies of Pst-wheat pathosystem suggested that the gene-for-gene model may also apply to Pst-wheat interactions (Flor, 1971; Tian et al., 2016; Yuan et al., 2017). Therefore, given that more than 70 permanently named and more than 60 provisionally named yellow rust (stripe rust) resistance (Yr) genes have been genetically characterized (Chen, 2013; McIntosh et al., 2013), we speculated that in the Pst genome there are hundreds of Avr genes encoding Avr effectors that are interacting specifically with corresponding R gene products following the gene-for-gene model. To screen for candidate effectors of Avr genes in Pst, we took advantages of the 18 Yr single-gene lines that are used to differentiate Pst races (Wan and Chen, 2014). By recording the incompatible or compatible reactions between each wheat line inoculated with the 14 selected Pst isolates, we were able to determine Avr (for avirulence) or avr (for virulence) in each Pst isolate corresponding to the 18 Yr genes. The virulence phenotype data were used to test the correlation with polymorphic effector genes for identifying candidates of the Yr genes. Our specific objectives were to (1) characterize the Pst secretome to identify candidate effectors by applying diverse screening criteria; (2) identify candidate Avr genes by correlation analysis of the virulence phenotypes and polymorphic effector genes; and (3) understand the virulence evolution of the Pst fungus.
Materials and Methods
Selecting and Virulence Phenotyping Pst Isolates
We selected 14 Pst isolates including seven previously sequenced isolates (Cantu et al., 2011, 2013; Cuomo et al., 2017). The other seven isolates were selected based on previous virulence data (Wan and Chen, 2014; Wan et al., 2016) to have relative balance of avirulent and virulent isolates for a maximum number of virulence loci. To confirm the virulence profiles, the 14 isolates were tested on the 18 wheat Yr single-gene differential lines, following the procedure described by Wan and Chen (2014). On each of the inoculated Yr single-gene line, the infection type (IT) of the isolate was recorded based on a 0–9 scale. Isolates with ITs 0–6 were considered as avirulent to the Yr single-gene line, while with isolates ITs 7–9 as virulent. To reduce phenotypic variation within avirulent and virulent classes, isolates with ITs 0–2 for avirulent reactions and with ITs 8–9 for virulent reactions were selected and thus intermediate ITs (3–7) were mostly avoided. Generally, the selected 14 Pst isolates represented a balanced virulent-avirulent profile to the majority of the Yr genes (Table 1).
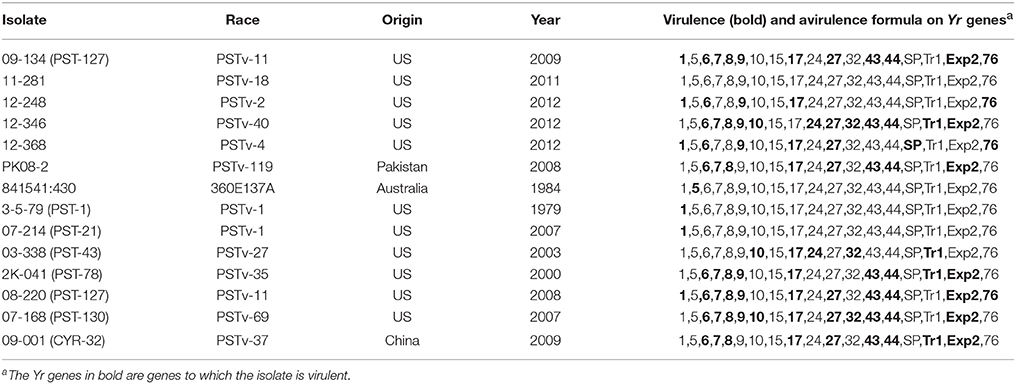
Table 1. Puccinia striiformis f. sp. tritici isolates used in this study and their virulence/avirulence formulae.
Whole Genome Sequencing, Alignment, and Genomic Variation Calling
Among the 14 isolates used in this study, 7 were sequenced previously using FLX chemistry with a Roche 454 and Illumina-adapted Fosmids by 454 Illumina (Cuomo et al., 2017); and Illumina (Cantu et al., 2011, 2013) sequencing approaches. Raw reads were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) or the Broad Institute Puccinia website (http://www.broadinstitute.org/) (Table 2). The remaining 7 isolates were sequenced in the present study using an Ion Proton platform. Genomic DNA was extracted from urediniospores using OmniPrep™ kit (G-Biosciences, St. Louis, MO). DNA libraries with 200-base-read for the Ion Proton™ system were prepared following the instruction of Ion Xpress™ Plus gDNA Fragment Library Preparation (Thermo Fisher Scientific Inc. 2014). The fragment size for each DNA library was checked using the Agilent®2100 Bioanalyzer® (Agilent Technologies, UK) instrument. Then the adaptors and barcodes were ligated to the sheared DNA. Fragment size selection of the library was conducted using E-Gel® SizeSelect™ agarose gel electrophoresis. The library was amplified following the Ion Xpress™ Plus gDNA Fragment Library Preparation protocol. Whole-genome sequencing was carried out using an Ion Proton™ system at the USDA-ARS Western Regional Small Grain Genotyping Laboratory at Pullman, WA. The chip for the Ion Proton™ system for this study was Ion PI™ Chip v2 which is able to ideally generate at least 10 Gb bases in one chip. On average, the isolates from Illuminia and Ion Proteon sequencing were sequenced at 26X and 32X coverage, respectively. The Ion Proton sequences of seven Pst isolates (09-134, 11-281, 12-248, 12-346, 12-368, PK08-2, and 841541:430) have been deposited in NCBI SRA under accession numbers SRR5050793, SRR5050794, SRR5050796, SRR5050790, SRR5050791, SRR5050792, and SRR5050795, respectively.
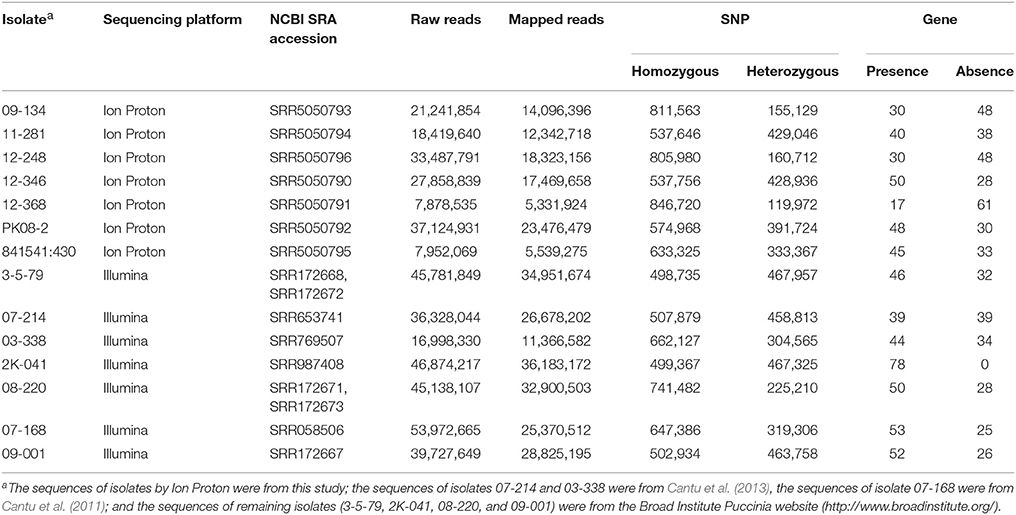
Table 2. Summary of mapping for variation calling from the genomic sequences of Puccinia striiformis f. sp. tritici isolates.
The adapter and barcode trimmed reads in the FASTQ format were corrected using Coral 1.4 (Salmela and Schröder, 2011) with sequencing technology set as −454 and other parameters as default. Quality assessment of corrected reads was conducted using the FASTQC program (Andrews, 2010). The reads that were shorter than 40 bp were discarded and all reads were cut to 246 bp by removing the 3′ low quality bases using Trimmomatic 0.33 (Bolger et al., 2014). The Illumina raw reads from NCBI SRA were also trimmed in such way. The high-quality trimmed reads in FASTQ format were used for genomic variants calling. The previously sequenced and assembled PST-78 genome (Cuomo et al., 2017) was used as a reference and a previously proposed framework by DePristo et al. (2011) for variation discovery was followed. The reference genome was indexed first and the high-quality trimmed reads from each isolate were mapped to the reference genome using bwa-mem algorithm with default setting in Burrows-Wheeler Alignment (BWA) Tool version 0.7.12 (Li and Durbin, 2010). The SAM formatted alignment information was converted to the BAM format after the duplicates were removed using SAMtools version 1.2 (Li, 2011). The BAM files were cleaned, validated and sorted using the Picard tools (version 1.129) (http://picard.sourceforge.net/), and then were indexed using SAMtools. To reduce the number of potentially mismatching insertion and deletion (Indels) in the alignment, the local realignment was conducted following two steps using Genome Analysis Toolkit (GATK) version 3.3 (van der Auwera et al., 2013). Firstly, the interval targets for local realignment were defined using GATK's RealignerTargetCreator with the default parameters. Secondly, GATK's IndelRealigner was used to perform Indel realignment of reads around defined targets from the previous step. The option -nWayout, with other default parameters, was used in this step to generate a BAM file for each isolate for further analyses. The BAM files were submitted to QualiMap v2.1 to evaluate the mapping quality (Okonechnikov et al., 2016). These analysis-ready BAM files were utilized in the following analyses. The first utilization was to call variants using GATK's HaplotypeCaller. We used VCFtools 0.1.12b to manipulate the variants in VCF format (Danecek et al., 2011). For correlation analysis, SNPs and Indels from only SP genes were used and filtered with the following criteria: –mac 3, –max-alleles 2 and –max-missing 1. The second use of the analysis-ready BAM files was to identify supercontigs with absence/presence polymorphism in each isolate. This analysis was achieved from the QualiMap results, and supercontigs were considered as absent in an isolate when no reads were mapped. The same procedure was applied to identify absence/presence polymorphic SPs. The analysis-ready BAM files were also used to generate secretome sequences for each isolate. To do so, a consensus genome sequence was generated for each isolate using the SAMtools mpileup function with -Q 10 and other default parameters. Then SP genes were extracted using the Bedtools getfasta function. These secretomes were used in the subsequent analyses to identify identical SPs among different Pst isolates.
Principle Component Analysis
Whole-genome SNPs and InDels from section Whole Genome Sequencing, Alignment, and Genomic Variation Calling were separated into three regions, SP genes, non-SP genes, and non-coding regions. Principle component analysis (PCA) was conducted for each region using the “prcomp” function, and the results were visualized using the “scatterplot3d” package in R v3.2.3 (Ligges and Maechler, 2003; R Core Team, 2014).
Correlation Analysis
The polymorphisms obtained from previous section were used to identify potential correlation relationships with the virulence phenotypes corresponding to individual Yr genes. To facilitate correlation analysis, the SNP and Indel data in the VCF file were compressed and recoded using the BLINK program (http://zzlab.net/blink/index.html; parameters: –compress –vcf, and –recode –numeric, respectively) and the numeric format data were generated. The gene absence/presence polymorphisms were also coded as numeric. For phenotype data preparation, the non-polymorphic as well as low frequent traits were excluded. For example, all isolates used in this study were avirulent to Yr15, and only one isolate was virulent to Yr5 and YrSP. Thus, these genes were excluded for correlation analysis. Also excluded were redundant traits. For example, all of the isolates virulent on Yr7 were also virulent on Yr8, Yr43, Yr44, and YrExp2. By excluding these Yr genes, eight virulence traits were used for correlation analysis. The correlation analysis was conducted using the cor function in R version 3.2.3. The formulae used were:
where Y and X were the phenotype and genotype matrix in numeric, respectively, n was the number of isolates, and p was the significance value. In this study, the Bonferroni corrected p significance, calculated as α/m in which m was the number of polymorphisms, was used as the threshold.
Comparison of the Genomes and Secretomes of the Wheat Rust Fungi
We started with three predicted proteomes of Pst, P. graminis f. sp. tritici (Pgt), and P. triticina (Pt). These three predicted proteomes were obtained from the Puccinia Group Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org/). Then we followed a well-studied classical routing pipeline to predict secretomes (Saunders et al., 2012; Cantu et al., 2013). For each predicted proteome, the SignalP version 4.1, TargetP version 1.1, and TMHMM version 2.0 algorithms were used. SignalP was used to screen the presence and location of signal peptide cleavage sites, setting the options for eukaryotic organisms with other default parameters. The input files were protein sequences in the FASTA format. TargetP was used to confirm the presence of secretory pathway signal peptides and to remove the protein sequences with chloroplast transit peptides and mitochondrial targeting peptides. Protein sequences with transmembrane helices predicted using TMHMM were excluded from final protein data sets. The filtered protein sequences for each rust fungus were defined as the final secretome for subsequent analyses. We were aware that some SPs might be exported to the extracellular space through a non-classical mechanism in fungi, and therefore, the predicted secretomes using this classical method in this study might not represent the entire secretome repertoires in the rust fungi.
The complete sets of proteomes were used to make comparisons among the three wheat rust fungi. Realizing the difficulties arising from the presence of paralogs, program Proteinortho version 5.11 was used to detect orthologs and co-orthologs (Lechner et al., 2011). The blastp function was performed using NCBI BLAST version 2.2.24+, which was installed as a part of the Proteinortho package. All analyses with Proteinortho were conducted with the default values except the following: E-value for blast was set as 1e-03, and the minimum coverage of best blast alignments in percent was set as 20. Also, selfblast and singles were applied to directly detect paralogs and to report unique sequences in each proteome, respectively. It should be noted that the E-value of 1e-03 was set arbitrarily, fewer species-specific proteins would be obtained if E-value was set as 1e-05 or even lower. The same analyses were conducted using the secretomes of the three wheat rust fungi.
The phylogenomic relationships were estimated from six selected rust fungi including Pst, Pgt, Pt, Melampsora larici-populina (Mlp) (the leaf rust fungus of popular, NCBI GenBank assembly accession GCA_000204055.1), M. lini (Mli) (the flax rust fungus) (Nemri et al., 2014) and Septobasidium spp. (Sep) (http://genome.jgi.doe.gov/Sepsp1/Sepsp1.home.html). Orthologs and paralogs among these six proteomes were detected using program Proteinortho version 5.11 (E-value as 1e-05, identity as 50%, and minimum coverage as 100%). The orthologous groups from the Proteinortho output were kept only when all six species were covered so that the protein absence/presence was not included for phylogenomic analyses. For each orthologous group only a single protein sequence was selected from each proteome and used for alignment. For each group, the six orthogous proteins were kept in one multi-FASTA file and used as input for multiple protein alignments using the CLUSTAL Omega algorithm (Sievers et al., 2011) with default parameters. Subsequently, the 63 alignment files in NEXUS format were concatenated using FASconCAT v1.0 (Kück and Meusemann, 2010). Phylogenomic inference was conducted using Bayesian-based method in program MrBayes v3.2.6 (Ronquist et al., 2012). A partitioned analysis was set up for MrBayes so that different models could be estimated and applied for each protein. Our data set was divided into 63 partitions as each protein alignment was considered as one partition. Evolutionary models were selected based on ProtTest 3.4 (Darriba et al., 2011). The best-fit model was incorporated into MrBayes as parameters and priors for each protein sequence. Then the parameters were unlinked so that each partition has its own set of parameters. We allowed all our partitions to evolve under different rates by setting overall rate to be different across partitions using commands prset applyto = (all) and ratepr = variable. Other parameters including model parameters, priors and the Markov Chain Monte Carlo sampling parameters were set as defaults. The performance was stopped when average standard deviation of split frequencies was lower than 0.01 and the performance was confirmed by checking the stationarity of the plot of the generation vs. the log probability of the data between two separated runs. Totally, four chains of 200,000 generations were run.
Pst Secretome Characterization and Annotation
In this study, diverse characters were applied to the Pst secretome which was predicted from the previous section, and a general analysis pipeline was shown in Figure 1. These characters included (a) typical fungal effector features, e.g., secreted, protein length, number and percent of cysteine residues; (b) haustorially specific expression; (c) effector prediction by program EffectorP; (d) polymorphisms among Pst isolates; and (e) the correlation relationships with Yr genes.
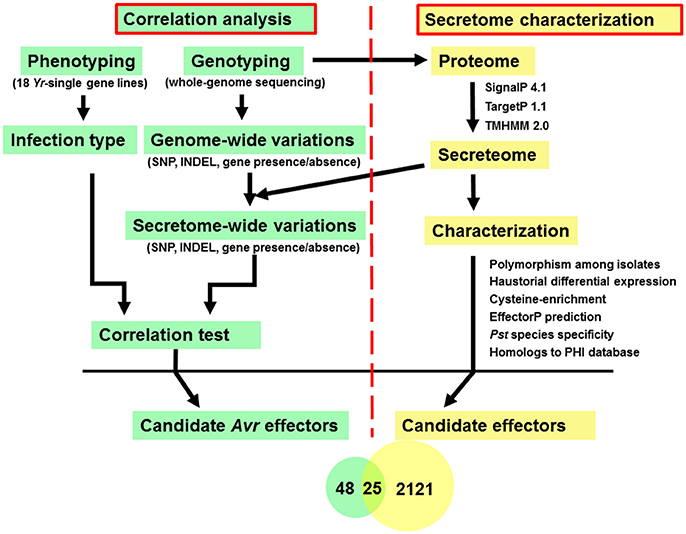
Figure 1. Analysis pipeline for identification of candidate Avr effectors in wheat stripe rust fungus Puccinia striiformis f. sp. tritici. Left: Correlation analysis between phenotypes, which were recorded as infection type by testing each of the Pst isolates on 18 Yr single-gene lines, and genotypes, which were conducted by whole-genome sequencing, to identify candidate Avr effectors corresponding to each avirulence gene. Right: Secretome characterization of a reference genome, PST-78. Secreted proteins were predicted using a classic routine pipeline from the well-annotated PST-78 proteome (http://www.broadinstitute.org/). Then each secreted protein was characterized using multiple criteria including whether it is (a) polymorphic among isolates; (b) haustorial differentially expressed; (c) cysteine-rich; (d) predicted to be effector using program EffectorP; (e) Pst species specific; and (f) homologous to known effectors by searching the PHI database.
To determine how many and which SPs were differentially expressed in haustoria, two sets of previously published HESPs were retrieved from Cantu et al. (2013) and Garnica et al. (2013). DE Seq p-value of 0.05 was used as a cut-off to set HSPs from Cantu et al. (2013). These HESPs were blasted against the PST-78 secretome using BLASTP with E-value set as 1e-10 and similarity as 90. Then we tested whether the SPs were identical or polymorphic among the sequenced isolates. To reduce the computational load, only five Pst isolates were used to identify identical protein sequences, and the incomplete proteins were excluded from the proteomes. These five Pst isolates, PK08-2, 2K041, 3-5-79, 12-248, and 09-001, were selected based on their mapping depth to avoid false polymorphisms caused by sequencing. The proteome was translated from the consensus genome sequence obtained from previous section for each isolate with the PST-78 gff3 annotation file using the gffread function in Cufflinks v2.2.1 (Trapnell et al., 2010). Each of these 6 proteomes was blasted against the remaining 5 proteomes using NCBI BLAST version 2.2.24+ with E-value set as 1e-10. The BLSAT results were filtered manually. The proteins were considered identical only when (1) the whole length of the protein was covered in the BLAST alignment, (2) no mismatches and (3) no gaps in the alignment. As expected, some of the proteins were only identical among several of these 6 isolates. In this case, only proteins that were identical among all 6 isolates were considered as highly conserved proteins in Pst. To predict effectors, the PST-78 predicted secretome was submitted to program EffectorP with default parameters (Sperschneider et al., 2015b).
The PST-78 predicted secretome was annotated using Blast2GO v3.3 (Conesa et al., 2005). LocTree3 (Goldberg et al., 2014), and NucPred (Brameier et al., 2007) were used to predict subcellular localizations. NLStradamus (Nguyen et al., 2009) was used to predict Nuclear Localization Signals. PFAM domains were annotated using HMMSCAN searches against the PFAM database in HmmerWeb v2.3.3 (Finn et al., 2015). To determine the putative functions of SPs from Pst, similar to known fungal effectors, BlastP was used to search against PHI-base 4.0 (Urban et al., 2015). The secretome was scanned to search the effector motifs [L/I]XAR, [R/K]CXXCX12H, RXLR, [Y/F/W]XC, YXSL[R/K], and G[I/F/Y][A/L/S/T]R between amino acids 10–110 using FIMO version 4.11.2, with a p-value of 1e-04 (Grant et al., 2011).
Results
Defining the Puccinia Secretome
We defined and compared the secretomes and effector repertoires of three wheat rust fungi following a classical pipeline in which SPs were predicted based on the presence of N-terminal signal peptides and the absence of transmembrane helices (Figure 1). The PST-78 secretome was predicted from the previously assembled and annotated proteome (20,482 proteins) available at the Puccinia genome database of the Broad Institute (http://www.broadinstitute.org/). The predicted PST-78 secretome contains 2,146 SPs, representing 10.48% of all proteins. The Pst secretome was similar to that of other wheat rust fungi Pgt and Pt in the percentage of SPs out of the total number of proteins. In contrast, the Pgt secretome comprised of 1,857 SPs (11.62%) over the total of 15,979 proteins, while the Pt secretome comprised of 1,425 SPs (9.08%) over the total of 15,685 proteins (Figure 2A). The proportions of SPs fall in the higher part of the range of the general proportion range (4–14%) of fungi (Lowe and Howlett, 2012).
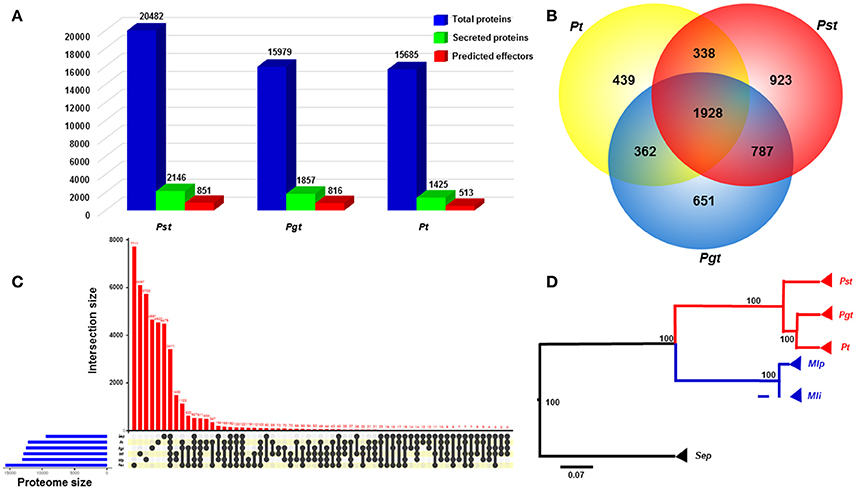
Figure 2. Comparative genomics analyses between Puccinia striiformis f. sp. tritici (Pst) and other rust fungi. (A) Comparison of the numbers of total proteins, secreted proteins and predicted effectors among three wheat rust fungi, Pst, Pgt (P. graminis f. sp. tritici), and Pt (P. triticina), indicated a large portion of secreted proteins in the three wheat rust fungi. The predicted effectors were defined as effectors predicted from a machine learning-based program EffectorP. (B) Venn diagram comparing the secretome with paralogs among the three rust fungi. A large proportion of species-specific secreted proteins was suggested in each of the three rust fungi. (C) Comparison of complete proteomes between Pst and five other Basidiomycete rust fungi. The comparison was visualized using package UpSetR in R v3.2.3. (D) A rooted phylogenetic tree showing evolutionary relationships of Pst with other five rust fungi. The phylogram suggested the early divergence of Pst from the most recent common ancestor of wheat rust fungi. The phylogenic relationship was estimated based on 63 orthologous protein sequences using MrBayes v3.2.6. Bootstrap values were shown. Scale bar indicted an evolutionary distance of 0.07 amino acid substation per position in the sequence.
Comparative Genomics Showing Pst-Specific SP Effectors
We conducted a comparison analysis to estimate the specificity of SPs among three wheat rusts. In this analysis, only SPs from Pst, Pgt, and Pt were used, resulting in a total of 5,428 proteins analyzed. Among these proteins, 1,928 proteins (in 444 orthologous families) were conserved among the three rust fungi (Figure 2B). All three rust fungi have a large number of species-specific SPs (SSSPs), with Pst having the highest proportion of 43.01% (923; in 747 orthologous families), followed by Pgt having 35.05% (651; in 523 orthologous families) and Pt having 30.80% (439; in 384 orthologous families) (Figure 2B and Supplementary Table 1). The numbers of SSSP for the three Puccinia species are comparable as the number for Pt is very close to the number of 420 candidate secreted effector proteins (CSEPs) specific for Pt (Cuomo et al., 2017). When we applied a broader comparison including the proteins of additional Pucciniomycetes proteins mentioned in the methods, the proportion of SSSPs were reduced to 34.16% (733) in Pst, 27.08% (503) in Pgt, and 22.38% (319) in Pt. The decrease in the number of SSSPs might be explained by: (i) some of them had non-secreted orthologs in other rust fungi, or (ii) some are specific among wheat rust fungi, but have orthologs in other phylogenetically distant rust fungi. Nevertheless, the reduced numbers of SSSPs are still big, and the SPs might have specific roles in each species.
We also conducted comparative genomic analysis between Pst and a set of Pucciniomycetes fungi. Based on orthologous analysis, a large proportion of phytopathogenic rust fungi proteins were found to be species-specific (Figure 2C; Lex et al., 2014). For example, Pst has 9,108 (44.46%) unique proteins grouped in 7,711 orthologous families (Supplementary Table 2). In contrast, the numbers of species-specific proteins were lower in Pt (5,358 or 34.16% in 4,537 orthologous families) and Pgt (5,394 or 33.75% in 4651 orthologous families) (Figure 2C, Supplementary Tables 2). The higher number of unique proteins in Pst than those in Pt and Pgt may reflect the relatively distant phylogenetically relationship between Pst and the other two wheat rust fungi, as shown in Figure 2D, which was inferred from 63 conserved protein sequences among six Pucciniomycetes fungi. The earlier divergence of Pst from Pgt and Pt was consistent with previous estimation from phylogenetic analysis of 1,208 single-copy orthologs (Cuomo et al., 2017).
Genome Sequencing, Variations and Genetic Diversity of Pst Isolates
In addition to the 7 previously sequenced isolates, we selected 7 Pst isolates for Ion Proton sequencing with the aim to generate a balanced virulence-avirulence profile for correlation study. A summary of the whole genome sequencing data for the 7 isolates (09-134, 11-281, 12-248, 12-346, 12-368, PK08-2, and 841541:430) produced by Ion Proton is shown in Table 2. Also shown in Table 2 are the sequence summary data for the previously sequenced 7 isolates (3-5-79, 07-214, 03-338, 2K-041, 08-220, 07-108, and 09-001) by Illumina.
The reads of each isolate were aligned to the PST-78 reference genome to call SNPs and Indels. During filtering, SNPs and Indels were kept only when minor alleles appeared at least three times (–mac 3 in VCFtools, see Methods). Totally, 48,059 high quality variants including 41,764 SNPs and 6,295 Indels from SP genes were kept for subsequent analyses. To measure genetic diversity, we calculated the rate of He/(Ho+He), in which He and Ho represented heterozygous sites and homozygous sites, respectively. Generally, different isolates showed different heterozygosity rates (Figure 3A). We noticed that the isolates with low heterozygosity had a wider virulence spectrum compared with highly heterozygous isolates, which is consistent with the observations from our previous population study (Xia et al., 2016a).
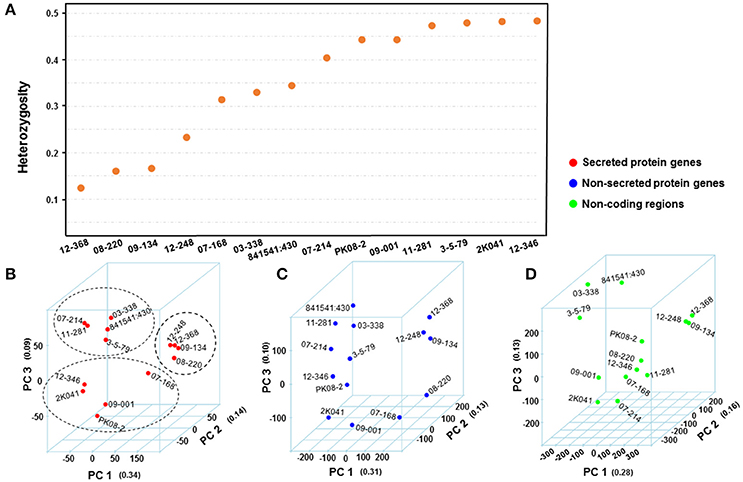
Figure 3. Comparison of Puccinia striiformis f. sp. tritici (Pst) genetic diversity from different genomic regions. (A) Distribution of mean heterozygosity over secreted protein (SP)-coding regions for each isolate. The heterozygosity was calculated first as He/(He+Ho), where He and Ho are the number heterozygous and homozygous SNP and Indel sites, respectively. (B–D) 3-dimentional plots of principle component analyses (PCA) using genomic variations from SP, non-SP, and non-coding gene regions of 14 Pst isolates. The number in quotes along each principle component (PC) is the proportion of variance explained by the PC. The values along each axis are eigenvalues calculated for isolates by PCA.
We performed principal component analysis (PCA) to evaluate the differences between secreted and non-secreted protein gene variants and variants in non-coding regions in dissecting the correlational structure. As shown in Figures 3B–D, the SNPs and Indels in SP gene regions had a potential to reveal hidden population structures, while the variants in non-SP gene and non-coding regions did not. For instance, the four isolates (08-220, 09-134, 12-368, and 12-248) with low heterozygosity and broad virulence profiles were clustered together in Figure 3B, and the five less virulent and highly heterozygous isolates (03-338, 07-214, 11-281, 3-5-79, and 841541:430) were also clustered together, whereas such patterns were not seen in Figures 3C–D. Considering that the revealed structure in Figure 3B associated with the virulence profile in Table 1, we would hypothesize that SP genes may contribute more to the virulence variability.
Prediction of Pst Effectors
To further characterize the Pst secretome and predict effectors, the 2,146 predicted SPs were evaluated using five criteria. First, 6 Pst secretomes were compared to determine the SPs that were polymorphic among these different isolates. By doing this, we assumed that the virulence-related proteins should be polymorphic since these isolates had different virulence-avirulence profiles. In total, 1,787 (83.27%) out of the 2,146 SPs were polymorphic (Figure 4). Secondly, we identified 923 SPs as Pst-specific among the three wheat rust causing species. Details of this group are illustrated in the next section. Thirdly, we used a cutoff of at least 3% of cysteine in the SP sequences as a criterion and identified 611 SPs. Fourthly, we retrieved a total of 2,154 haustorial differentially expressed (HDE) proteins from previous studies and identified 625 of Pst SPs to be homologous to these HDE proteins. Lastly, we used a machine learning-based program, EffectorP, to predict Pst putative effector proteins from SPs. A total of 851 (39.7%) of 2,146 Pst SPs were predicted to be effectors. For comparison, we also conducted the same analysis for Pgt and Pt, and predicted 816 (43.9%) of 1,857 SPs as effectors in Pgt and 513 (36.0%) of 1,425 SPs as effectors in Pt. It should be noted that most of the SPs that passed one criteria also satisfied at least one of other criteria, the group comprising of polymorphic SPs was the only exception. For example, over 96% (891 out of 923) of Pst-specific SPs had at least one of the other criterion (Figure 4). Taking all of the five criteria together, 32 Pst SPs were identified to be highly possible candidate effectors.
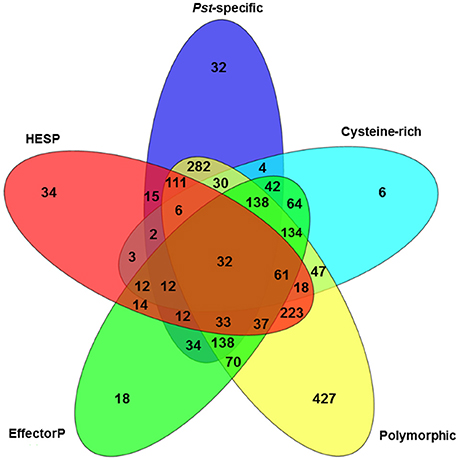
Figure 4. Characterization of the Puccinia striiformis f. sp. tritici (Pst) secretome using five criteria: cysteine-rich, polymorphic among Pst isolates, EffectorP, haustorially expressed secreted protein (HESP) and Pst-specific.
SP Genes Significantly Correlated with Avirulence Genes in Pst
Since previous genome-based effectors mining studies have not emphasized avirulence effectors (Saunders et al., 2012; Cantu et al., 2013), we conducted a correlation analysis of effector polymorphisms and avirulence phenotypes. The avirulence function of an effector can be inferred, based on the gene-for-gene concept, from the incompatible interaction between the pathogen and host. In the Pst-wheat pathosystem, the presence of an Avr gene in the isolates is indicated when they are avirulent to the corresponding Yr gene. Based on this rationale, we conducted a correlation analysis to identify Avr candidates corresponding to 18 Yr genes. Toward this goal, we selected 14 Pst isolates, confirmed their virulence/avirulence profiles for each Yr gene, and conducted the correlation analysis. We identified 73 SP or effector genes that were significantly correlated with the avirulenc/virulence patterns to 5 Yr genes, Yr6, Yr8, Yr9, Yr17, and YrTr1 (Figures 5A–E). Features of these effectors were shown in Table 3, and details were available in Supplementary Table 3.
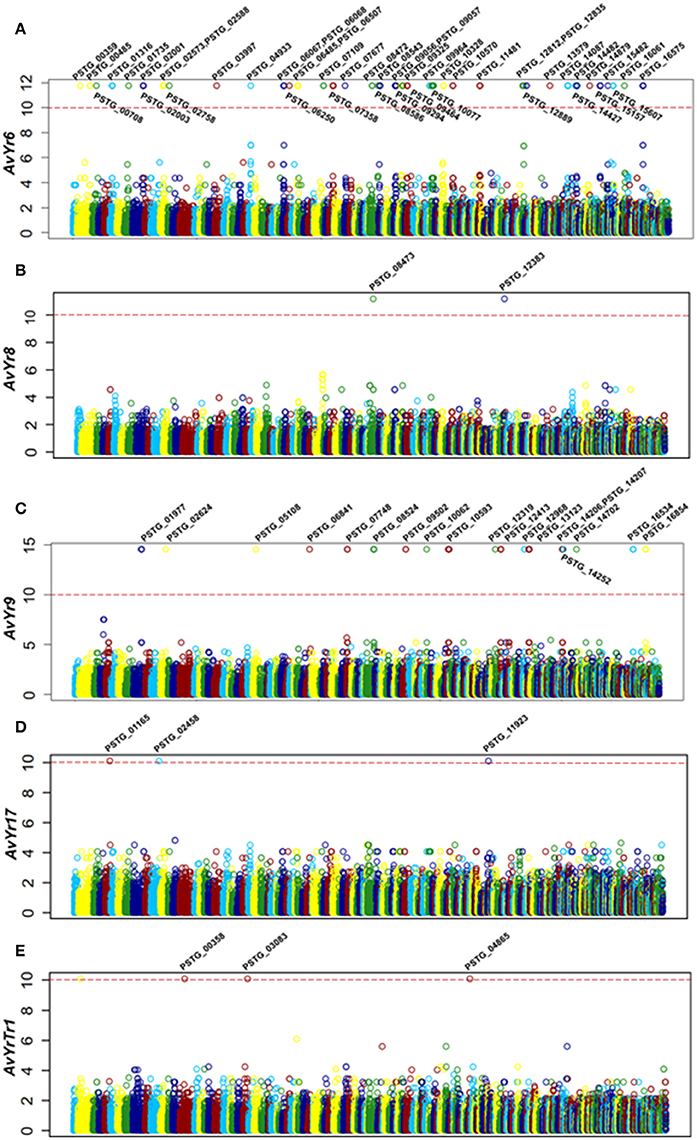
Figure 5. Correlation analysis identified secreted protein genes associated with Avr genes. (A–E) Manhattan plots showing the correlation analysis results, with each plot representing candidate Avr genes corresponding to a Yr gene. X-axis represents the genomic position of SNPs or Indels at the supercontigs of the PST-78 reference genome, and Y-axis is -log10 transformed significance P values. SNPs and Indels are represented as empty circles. The dashed red lines are Bonferroni-corrected threshold. The gene name in the significant correlation is also shown.
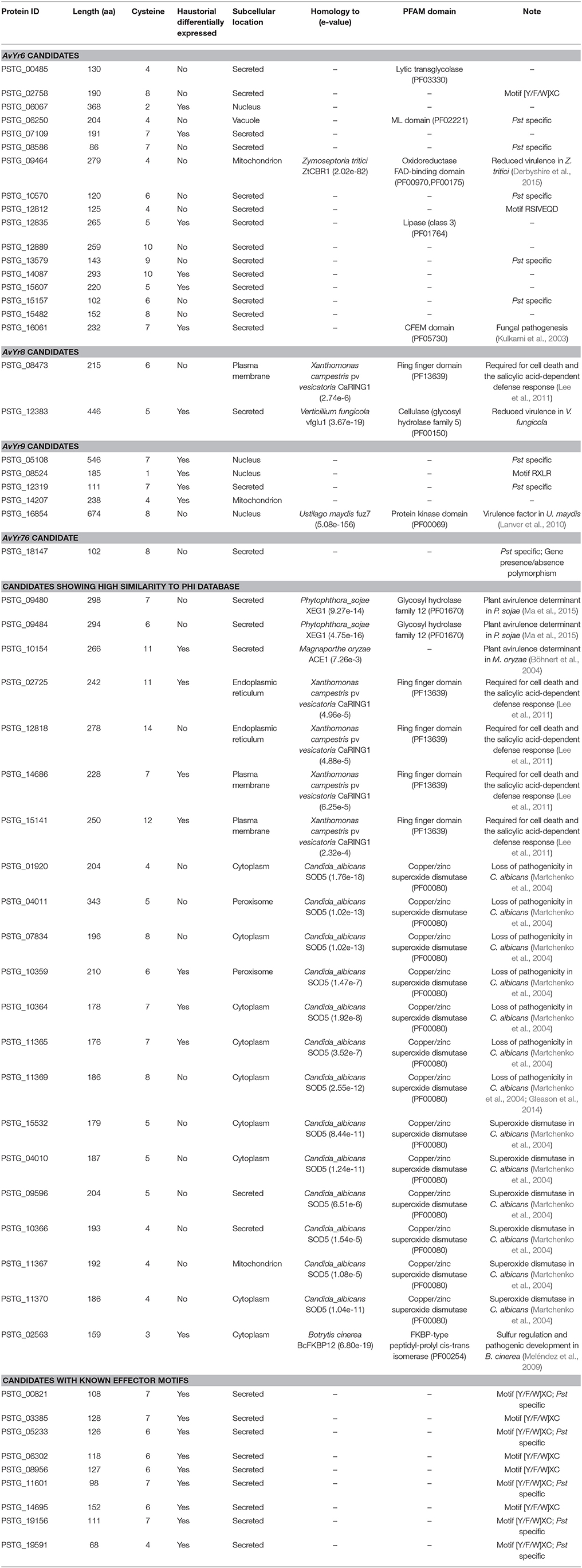
Table 3. Candidate effectors for avirulence genes in Puccinia striiformis f. sp. tritici.
For Yr6, we identified 151 SNPs and 17 Indels located in 46 SP genes at the significance threshold of P < 1 × 10−10 in the phenotype and genotype correlation analysis. Most of the 46 genes had more than one SNP, and these SNPs occurred in intron, exon, or both regions (Supplementary Table 3). However, in 5 of the genes (PSTG_00359, PSTG_09293, PSTG_09294, PSTG_14087, and PSTG_14482), the variations occurred only in the promoter regions, indicating that they may have regulatory functions instead of changing the protein structures. Compared to the 46 SP genes in Yr6, fewer significant SPs were identified for other Yr genes, 2 for Yr8, 19 for Yr9, 3 for Yr17, and 3 for YrTr1 (Figures 5A–E). Of the 46 SP genes in Yr6, 17 satisfied at least one of the five criteria used to identify effectors (Table 3). Similarly, 2 genes in Yr8, 5 in Yr9 and 1 in Yr76 were considered as avirulence candidate effectors. In addition, 21 other SP genes had significant homologies with genes in the PHI database and other 9 genes had known effector motifs.
In addition to the candidate effectors for the five avirulence genes, we identified a candidate, PSTG_18147, for AvYr76. Different from the SNP and/or Indel polymorphisms for in the above mentioned candidate effectors, the gene showed the presence/absence polymorphism. This Pst-specific gene was present in all 10 avirulent isolates but absent in all 4 virulent isolates (Figure 6). We also detected presence/absence polymorphisms for other SP genes among the isolates at the supercontig level when mapping raw reads to the PST-78 reference genome. To assess differences in genome structure, we found that some supercontigs were present in some isolates but missing in others. The number of lost supercontigs ranged from 894 for CYR32 to 2,740 for isolate 841541:430 (Supplementary Table 4). Most of presence/absence polymorphic supercontigs were relatively short (<14 Kb), and are likely unassembled chromosomal fragments rather than dispensable chromosomes often found in other fungi.
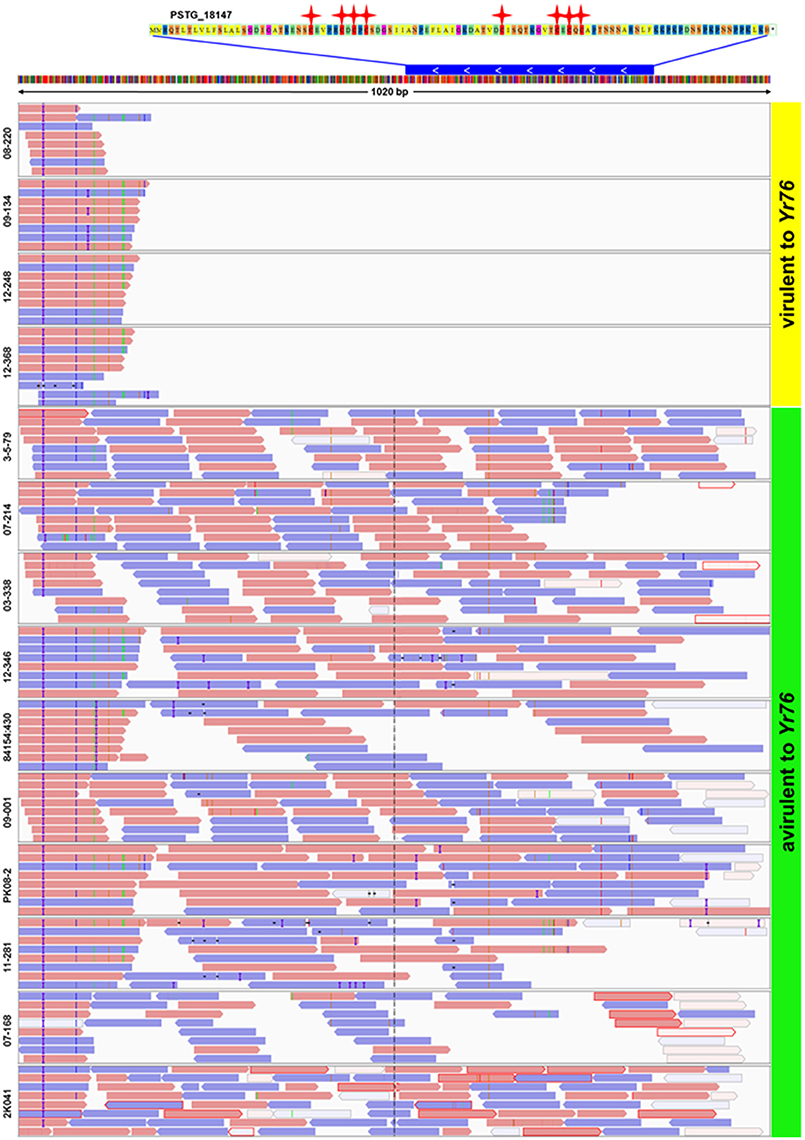
Figure 6. Presence of secreted protein gene PSTG_18147 in Yr76-avirulent and absence in Yr76-virulent isolates of Puccinia striiformis f. sp. tritici. Upper: The sequence of PSTG_18147. Red stars represent Cysteine amino acids. Blue bar represents the gene of PSTG_18147 and its position in Supercontig_1.1907. The white arrow shows the gene orientation. Lower: Results of mapping raw reads of 14 isolates to the PST-78 reference genome. Orange and blue bars show the reads and the orientation of mapping. Blank areas in the first four isolates indicate no reads are mapped to gene PSTG_18147.
Functional Features and Subcellular Localizations of Pst SPs
To investigate the potential functions, Pst SPs were searched for PFAM domains (Finn et al., 2015) and further classified into five major enzyme classes, including CAZymes (Carbohydrate-Active enZymes), lipases, oxidoreductases, peptidases and peroxidases (Figure 7A). Most SPs, 1,701 out of 2,146 (79.3%), had unknown functions, as expected, with only 445 (20.7%) being annotated. Of the annotated SPs, peptidases were the largest group with 164 (7.6%) SPs, followed by CAZymes (129; 6.0%). These two enzyme classes, together with lipases (40 SPs), suggest important roles in nutrient acquisition during the Pst colonization in the host. We predicted the protein sub-cellular localization for each of 2,146 Pst SPs. The extracellular group was the largest with 1,197 (55.8) SPs. Nucleus ranked the second, harboring 502 (23.4%) SPs, and followed by the cytoplasm group (172; 8.0%) (Figure 7B). In addition, 209 proteins with nuclear localization signals were predicted to be nuclear proteins that were transported by their import machinery of the cell (Supplementary Table 3).
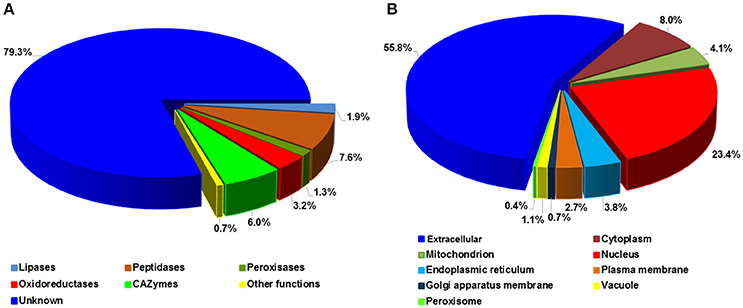
Figure 7. Functional annotation of Puccinia striiformis f. sp. tritici (Pst) secreted proteins. (A) Percentage distribution of the Pst secreted proteins with distinct enzymatic functions. (B) Percentage distribution of predicted subcellular locations of Pst secreted proteins.
Comprising of 70 (3.2%) secreted proteins, we considered oxidoreductases as an important enzyme class because their potential functions to protect the fungus against host-produced reactive oxygen species (ROS). In the further analysis, we identified 13 Pst SPs from the oxidoreductases to have copper/zinc superoxide dismutase (SOD) PF00080 domains, and 8 of them were homologous to the SOD5 gene in Candida albicans (e-value < 1e-6) (Table 3). Most of them were cysteine-rich and three were haustorial differentially expressed. The genetic relationships among the eight Pst SODs and the C. albicans SOD5 are shown in Figure 8A; their structures containing a signal peptide, a copper/zinc superoxide dismutase domain and other amino acids were illustrated in Figure 8B; and their alignment with the SOD5 residues and indication of copper and zinc ligands are given in Figure 8C.
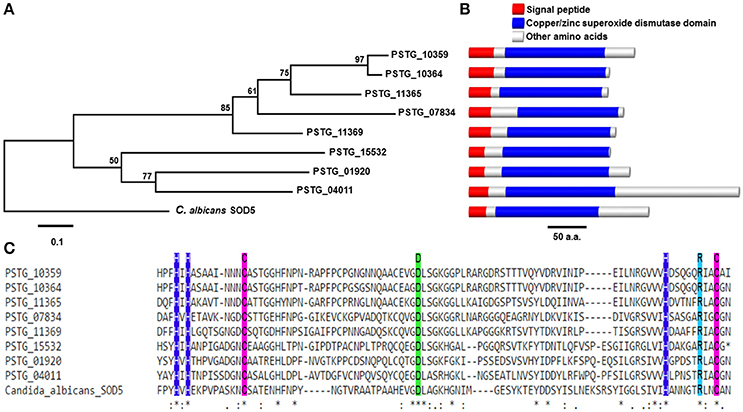
Figure 8. Phylogenetic and structural analysis of a class of SOD5-like secreted proteins in Puccinia striiformis f. sp. tritici (Pst). (A) Phylogenetic tree showing evolutionary relationships of eight SOD5-like secreted proteins in Pst. The phylogeny was estimated using the neighbor-joining algorithm, with bootstrap values indicated. The scale bar indicates an evolutionary distance of 0.1 amino acid substitution per position in the sequence. (B) The genomic structural of SOD5 and SOD5-like secreted proteins in Pst. The length of bars is scaled to the length of proteins. (C) Alignment of SOD5 residues F72-N164 against eight SOD5-like secreted proteins in Pst. Copper and zinc ligands are represented by blue and green box, respectively. The purple box represents cysteine.
The PHI database was searched to predict specific functions of Pst SPs by homology to known effector proteins (Urban et al., 2015). While 22 (10.34%) had significant hits, most SPs in Pst contained no known functions (Supplementary Table 3). After we applied the general effector mining criteria, a number of candidate effectors were identified, as listed in Table 3. Among these candidates, the SOD5-like proteins, as mentioned above, formed the biggest group. Another group including five SPs (PSTG_08473, PSTG_02725, PSTG_12818, PSTG_14686, and PSTG_15141) had a ring finger domain (PF13639). One (PSTG_08473) of the five SP genes was significantly correlated with avirulence to Yr8. Another group comprised of 3 SPs like plant avirulence determinants; PSTG_09480 and PSTG_09484 were homologous to XEG1 and PSTG_10154 was homologous to ACE1. The last group had only one protein, PSTG_02563, homologous to BcFKBP12, which is a haustorial differentially expressed secreted serine protease. In addition, we identified nine SP genes that had an effector motif ([Y/F/W]XC) and haustorially expressed (Table 3). Five (PSTG_00821, PSTG_05233, PSTG_11601, PSTG_19156, and PSTG_19591) of them were Pst specific. These genes were also good candidates for Pst effectors.
Discussion
One of the prominent features of the present study was that we incorporated virulence phenotypes into a correlation analysis to identify avirulence effector candidates (Figure 1). A similar study was tried previously by analyzing SP gene polymorphisms between two UK isolates which were different mainly on two known Yr genes, and 5 candidates with single amino acid substitution were identified (Cantu et al., 2013). However, they were unable to detect other Avr candidates because of the incomplete virulence phenotypes and limited isolates. In another study, virulence phenotypes of 352 isolates from natural populations were used to associate SP gene polymorphisms associated with avirulence genes (Xia et al., 2016b), but the study was limited by the number of SP-SNPs used for genotyping. To overcome the limitations, we selected 14 Pst isolates from worldwide collections and tested them on 18 wheat lines with single Yr genes, which are currently used to differentiate Pst races (Wan and Chen, 2014; Wan et al., 2016, 2017). By doing this, we assume that there is an Avr gene present in the isolate that is avirulent to the single Yr gene. The identification of 73 Avr candidates corresponding to 5 Yr genes indicates the potential power of the correlation analysis (Figure 5).
Regarding the candidate effectors identified for the Yr genes, two points should be discussed. First, multiple candidates were identified as significantly correlated with an avirulence gene, even after they were filtered by the general effector mining criteria (Table 3). We suspect that some of the significant signals are false-positives and may not the causative variants since they are synonymous substitutions. The false-positives can be explained by the undetermined linkage disequilibrium (LD) due to the Pst clonal reproduction since the sexual recombination on alternate hosts is less important under nature conditions (Wang et al., 2015; Zhao et al., 2016). Another plausible explanation for the multiple candidates to a single Yr gene is that reaction to a single resistance gene may be controlled by multiple pathogen genes, as reported in powdery mildew-cereal interactions (Bourras et al., 2016) and as suggested by genetic studies of Pst virulence to Yr genes (Tian et al., 2016; Yuan et al., 2017). Second, the correlation analysis did not result in significant effectors for the rest Yr genes, even though they had balanced virulence-avirulence profiles, e.g., avirulence to Yr7, Yr27, Yr43, Yr44, and YrExp2. This can be caused by several reasons as discussed in Xia et al. (2016b). Here we highlighted the following four reasons. (1) The causal variations may be minor alleles which were filtered out in this study since we only kept variants having minor alleles three times or above. (2) The correlation algorithm in the present study could not take variation interactions into consideration. (3) The virulence to avirulence change may be caused by variations other than SNPs or Indels, e.g., gene presence/absence polymorphisms and copy number variations. (4) Avirulence may be controlled by non-SP genes. As our focus was on SP genes, we might miss other genes for avirulence. Therefore, future studies need to be conducted by deep sequencing more Pst isolates from both natural populations and artificially produced sexual populations with diverse virulence/avirulence profiles and using different algorithm models to improve the power of detecting avirulence genes.
The two nuclei in Pst urediniospores were suggested to be highly heterozygous in one Chinese isolate (Zheng et al., 2013) and in many US isolates (Liu et al., 2012; Cheng and Chen, 2014; Cheng et al., 2016). In the present study, we noticed different Pst isolates had tremendous variation in heterozygosity (Figure 3A). The highly homozygous isolates were represented by isolates 08-220 and 09-134 (race PST-127 based on the previous set of wheat cultivar differentials or PSTv-11 based on the current set of 18 Yr single-gene lines). The high level of homozygosity of this group was confirmed from a previous study in which 352 US isolates were genotyped with 97 SP-SNPs (Xia et al., 2016b). Isolates of this race group were distinct with other isolates based on a population study and characterized by their broadest virulence spectra and the virulence to Yr1 and Yr76 (Wan et al., 2016; Xia et al., 2016b; Xiang et al., 2016). This group was first detected in the western US in 2007 and became the most predominant race group in this region but not in other regions in 2009-2011. Since 2012, however, the frequency of this race group decreased due potentially to the fitness cost for maintaining the broad virulence (Chen et al., 2010; Wan and Chen, 2012, 2014; Wan et al., 2016). From this case, we speculate that Pst isolates with high homozygosity are less adaptive, even though they have a broad virulence spectrum. Furthermore, it seems that the heterozygosity, especially at avirulence loci, is the result of positive selection and enables Pst populations to evolve quickly (Hovmøller et al., 2008; Zheng et al., 2013; Cuomo et al., 2017; Yuan et al., 2017). Further studies on avirulence loci using artificially produced sexual populations and continual monitoring the natural populations are needed to fully test this hypothesis.
Our study indicated that all three wheat rust fungi have extensive of secretomes, ranging from 9.08 to 11.62% of the proteomes, which are also in the high part of the range of the plant pathogenic fungi (Lowe and Howlett, 2012). The results are consistent with the previous report for the three Puccinia species infecting wheat (Cuomo et al., 2017). The large secretome sizes of wheat rust fungi could be due to their complicated lifestyles: macrocyclic (the presence of all five spore stages in either natural or controlled conditions) and heteroecious (requiring of two different host species to complete the life cycle). It is presumed that different sets of SPs may be involved in the interactions between rust fungi and their cereal and alternate hosts (Krijger et al., 2014). Other rust fungi in Pucciniales also showed such relationship. For example, the heteroecious poplar leaf rust fungus Mlp has 11.2% SPs (1,829 secreted over 16,399 proteins) (Duplessis et al., 2011), whereas only 4.9% (802 of 16,271 proteins) were predicted as SPs for autoecious flax rust fungus Mli (Nemri et al., 2014).
Moreover, the SSSPs are enriched in wheat rust secretomes. This is similar to many biotrophs and symbionts that intimately interact with their host cells. The functions of most SSSPs (90%) are unknown since they do not have defined domains. However, a kingdom-wide analysis of fungal secretomes suggested that the number of SSSPs could correlate with their proteome size, lifestyle and taxonomic position (Kim et al., 2016). Generally, biotrophs, e.g., Pucciniomycotina and Ustilagomycotina species, have the highest number of SSSPs. Given the similar lifestyles and taxonomic positons of the three wheat rust fungi, we speculate that these SSSPs might be impacted by different evolutionary histories compared with conserved ones, e.g., during the coevolutionary arms-race between rust fungi and wheat hosts (Krijger et al., 2014; Kim et al., 2016). Therefore, these SSSPs coding genes may provide new insight in understanding how rust fungi interact with their primary wheat host, auxiliary grass hosts and alternate hosts (Zhao et al., 2016).
Since the draft genome of Pst was available in 2011, from our knowledge, only few genes (or their products) have been validated as fungal virulence effectors, and functionally involved in host-pathogen interactions (Cheng et al., 2015; Liu et al., 2016; Ramachandran et al., 2017). All of these validated effectors suppress host-produced ROS. In the present study, we identified 70 secreted proteins as oxidoreductases, an important enzyme class with potential functions to protect the fungus against host-produced ROS (Chi et al., 2009). By searching the PHI database, we identified 13 SPs in Pst highly homologous to the SOD5 gene in Candida albicans, which was reported to be associated with pathogenicity of this human pathogen (Martchenko et al., 2004). In C. albicans, SOD5 is an extracellular protein with an open copper site, and could accumulate outside the cell in an inactive form that subsequently utilizes host copper to transform itself into an antioxidant defense for the pathogen (Gleason et al., 2014). Therefore, these SOD5-like SPs in Pst could be a common class of virulence effectors for its defense against plant basal immunity, since suppression of ROS accumulation has been observed by cloned virulence effectors for successful Pst infection (Liu et al., 2016). Several SP genes in Pst may be related to pathogenicity or trigger hypersensitive response as they are homologous to XEG1, ACE1, or CaRING1. These proteins have been reported to be plant avirulence determinants in Phytophthora sojae (Ma et al., 2015) and Magnaporthe oryzae (Böhnert et al., 2004), or required for cell death and the salicylic acid-dependent defense response in X. campestris pv. vesicatoria (Lee et al., 2011). Nine SP genes of Pst identified in the present study had an effector motif ([Y/F/W]XC). This conserved motif is present in many fungal pathogens including Blumaria graminis (Godfrey et al., 2010) and Melampsora larici-populina (Duplessis et al., 2011). The PSTG_02563 protein identified in the present study was highly homologous to BcFKBP12, which is involved in the sulfur repression of the synthesis of a secreted serine protease in Botrytis cinerea (Meléndez et al., 2009). Particularly, this gene was haustorial differentially expressed. Some candidates for specific Avr genes in Pst are likely related to pathogenicity or virulence as they have homology to genes in other fungi with similar functions. For example, PSTG_09464, which was associated with AvYr6, is homogous to ZtCBR1, a virulence gene in Zymoseptoria tritici (Derbyshire et al., 2015). PSTG_16061, also associated to AvYr6, has a CFEM domain that has been reported for fungal pathogenesis (Kulkarni et al., 2003). PSTG_16854, a candidate SP gene for AvYr9, has a fuz7 protein kinase domain, which has been reported as a virulence factor in Ustilago maydis (Lanver et al., 2010). Therefore, these genes are excellent candidate effectors with priority for further functional validation.
The mature methods for testing the function of effector candidates include (a) delivery of effector candidates to plant cell through bacteria type III secretion system to test the ability to induce hypersensitive response or to suppress pattern triggered immunity (Yin and Hulbert, 2011), and (b) use of host-induced gene silencing to test the involvement of candidates in fungal pathogenesis (Yin et al., 2015). We speculate that these methods can also be used to test the avirulence function of some Pst candidate effectors identified in this study. Once the candidate Avr genes are confirmed and their functions are determined, the information will shed the light on the Pst-wheat interactions, and the Avr genes can be used to develop gene-specific markers for monitoring the pathogen population changes and identify effective resistance genes from germplasm of wheat and related species using an effectoromics approach as suggested by Vleeshouwers and Oliver (2014).
Author Contributions
CX, MW, DJ, and DS participated in isolates collection, genomic DNA extraction, and Ion Proton sequencing. CX, OC, and XC analyzed and interpreted the data. CX was a major contributor in writing the manuscript. XC conceived the study, designed the experiments and wrote the manuscript. All authors reviewed and approved the final manuscript.
Funding
This study was supported by the U.S. Department of Agriculture, Agricultural Research Service (Project No. 2090-22000-018-00D), Washington Grain Commission, and Idaho Wheat Commission. PPNS No. 00000, Department of Plant Pathology, College of Agricultural, Human, and Natural Resource Sciences, Agricultural Research Center, Project No. WNP00663 (Projects 13C-3061-5682; 13C-3061-4232), Washington State University, Pullman, WA 99164-6430, USA. The China Scholarship Council scholarship to CX is highly appreciated.
Disclaimer
Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U. S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Scot Hulbert and Tobin Peever for critical review of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02394/full#supplementary-material
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Bayles, R. A., Flath, K., Hovmøller, M. S., and de Vallavieille-Pope, C. (2000). Breakdown of the Yr17 resistance to yellow rust of wheat in northern Europe. Agronomie 20, 805–811. doi: 10.1051/agro:2000176
Böhnert, H. U., Fudal, I., Dioh, W., Tharreau, D., Notteghem, J. L., and Lebrun, M. H. (2004). A putative polyketide synthase/peptide synthetase from Magnaporthe grisea signals pathogen attack to resistant rice. Plant Cell 169, 2499–2513. doi: 10.1105/tpc.104.022715
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bourras, S., McNally, K. E., Müller, M. C., Wicker, T., and Keller, B. (2016). Avirulence genes in cereal powdery mildews: the gene-for-gene hypothesis 2.0. Front. Plant Sci. 7:214. doi: 10.3389/fpls.2016.00241
Brameier, M., Krings, A., and Maccallum, R. M. (2007). NucPred - predicting nuclear localization of proteins. Bioinformatics 23, 1159–1160. doi: 10.1093/bioinformatics/btm066
Brown, J. K., and Hovmøller, M. S. (2002). Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science 297, 537–541. doi: 10.1126/science.1072678
Cantu, D., Govindarajulu, M., Kozik, A., Wang, M., Chen, X., Kojima, K. K., et al. (2011). Next generation sequencing provides rapid access to the genome of Puccinia striiformis f. sp. tritici, the causal agent of wheat stripe rust. PLoS ONE 6:e24230. doi: 10.1371/journal.pone.0024230
Cantu, D., Segovia, V., MacLean, D., Bayles, R., Chen, X., Kamoun, S., et al. (2013). Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics 14:270. doi: 10.1186/1471-2164-14-270
Catanzariti, A. M., Dodds, P. N., Lawrence, G. J., Ayliffe, M. A., and Ellis, J. G. (2006). Haustorially expressed secreted proteins from flax rust are highly enriched for avirulence elicitors. Plant Cell 18, 243–256. doi: 10.1105/tpc.105.035980
Chen, X. M. (2005). Epidemiology and control of stripe rust [Puccinia striiformis f. sp. tritici] on wheat. Can. J. Plant Pathol. 27, 314–337. doi: 10.1080/07060660509507230
Chen, X. M. (2013). High-temperature adult-plant resistance, key for sustainable control of stripe rust. Am. J. Plant Sci. 4, 608–627. doi: 10.4236/ajps.2013.43080
Chen, X. M., Penman, L., Wan, A. M., and Cheng, P. (2010). Virulence races of Puccinia striiformis f. sp. tritici in 2006 and 2007 and development of wheat stripe rust and distributions, dynamics, and evolutionary relationships of races from 2000 to 2007 in the United States. Can. J. Plant Pathol. 32, 315–333. doi: 10.1080/07060661.2010.499271
Cheng, P., and Chen, X. M. (2014). Virulence and molecular analyses support asexual reproduction of Puccinia striiformis f. sp. tritici in the U.S. Pacific Northwest. Phytopathology 104, 1208–1220. doi: 10.1094/PHYTO-11-13-0314-R
Cheng, P., Chen, X. M., and See, D. (2016). Grass hosts harbor more diverse isolates of Puccinia striiformis than cereal crops. Phytopathology 106, 362–371. doi: 10.1094/PHYTO-07-15-0155-R
Cheng, Y., Wang, X., Yao, J., Voegele, R. T., Zhang, Y., Wang, W., et al. (2015). Characterization of protein kinase PsSRPKL, a novel pathogenicity factor in the wheat stripe rust fungus. Environ. Microbiol. 17, 2601–2617. doi: 10.1111/1462-2920.12719
Chi, M. H., Park, S. Y., Kim, S., and Lee, Y. H. (2009). A novel pathogenicity gene is required in the rice blast fungus to suppress the basal defenses of the host. PLoS Pathog. 5:e1000401. doi: 10.1371/journal.ppat.1000401
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Cuomo, C. A., Bakkeren, G., Khalil, H. B., Panwar, V., Joly, D., Linning, R., et al. (2017). Comparative analysis highlights variable genome content of wheat rusts and divergence of the mating loci. G3 (Bethesda) 7, 371–376. doi: 10.1534/g3.116.032797
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. doi: 10.1038/ng.806
Derbyshire, M. C., Michaelson, L., Parker, J., Kelly, S., Thacker, U., Powers, S., et al. (2015). Analysis of cytochrome b(5) reductase-mediated metabolism in the phytopathogenic fungus Zymoseptoria tritici reveals novel functionalities implicated in virulence. Fungal Genet. Biol. 82, 69–84. doi: 10.1016/j.fgb.2015.05.008
de Vallavieille-Pope, C., Ali, S., Leconte, M., Enjalbert, J., Delos, M., and Jacques, R. (2012). Virulence dynamics and regional structuring of Puccinia striiformis f. sp. tritici in France between 1984 and 2009. Plant Dis. 96, 131–140. doi: 10.1094/PDIS-02-11-0078
de Wit, P. J. (2016). Cladosporium fulvum effectors: weapons in the arms race with tomato. Annu. Rev. Phytopathol. 54, 1–23. doi: 10.1146/annurev-phyto-011516-040249
Duplessis, S., Cuomo, C. A., Lin, Y. C., Aerts, A., Tisserant, E., Veneault-Fourrey, C., et al. (2011). Obligate biotrophy features unraveled by the genomic analysis of rust fungi. Proc. Natl. Acad. Sci. U.S.A. 108, 9166–9171. doi: 10.1073/pnas.1019315108
Finn, R. D., Clements, J., Arndt, W., Miller, B. L., Wheeler, T. J., Schreiber, F., et al. (2015). HMMER web server: 2015 update. Nucleic Acids Res. 43, W30–W38. doi: 10.1093/nar/gkv397
Flor, H. H. (1971). Current status of the gene-for-gene concept. Annu. Rev. Phytopathol. 9, 275–296. doi: 10.1146/annurev.py.09.090171.001423
Garnica, D. P., Nemri, A., Upadhyaya, N. M., Rathjen, J. P., and Dodds, P. N. (2014). The ins and outs of rust haustoria. PLoS Pathog. 10:e1004329. doi: 10.1371/journal.ppat.1004329
Garnica, D. P., Upadhyaya, N. M., Dodds, P. N., and Rathjen, J. P. (2013). Strategies for wheat stripe rust pathogenicity identified by transcriptome sequencing. PLoS ONE 8:e67150. doi: 10.1371/journal.pone.0067150
Gleason, J. E., Galaleldeen, A., Peterson, R. L., Taylor, A. B., Holloway, S. P., Waninger-Saroni, J., et al. (2014). Candida albicans SOD5 represents the prototype of an unprecedented class of Cu-only superoxide dismutases required for pathogen defense. Proc. Natl. Acad. Sci. U.S.A. 111, 5866–5871. doi: 10.1073/pnas.1400137111
Godfrey, D., Böhlenius, H., Pedersen, C., Zhang, Z., Emmersen, J., and Thordal-Christensen, H. (2010). Powdery mildew fungal effector candidates share N-terminal Y/F/WxC-motif. BMC Genomics 11:317. doi: 10.1186/1471-2164-11-317
Goldberg, T., Hecht, M., Hamp, T., Karl, T., Yachdav, G., Nielsen, H., et al. (2014). LocTree3 prediction of localization. Nucleic Acids Res. 42, W350–W355. doi: 10.1093/nar/gku396
Grant, C. E., Bailey, T. L., and Noble, W. S. (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. doi: 10.1093/bioinformatics/btr064
Hovmøller, M. S., Walter, S., Bayles, R. A., Hubbard, A., Flath, K., Sommerfeldt, N., et al. (2016). Replacement of the European wheat yellow rust population by new races from the centre of diversity in the near-Himalayan region. Plant Pathol. 65, 402–411. doi: 10.1111/ppa.12433
Hovmøller, M. S., Yahyaoui, A. H., Milus, E. A., and Justesen, A. F. (2008). Rapid global spread of two aggressive strains of a wheat rust fungus. Mol. Ecol. 17, 3818–3826. doi: 10.1111/j.1365-294X.2008.03886.x
Huang, X., Chen, X., Coram, T., Wang, M., and Kang, Z. (2011). Gene expression profiling of Puccinia striiformis f. sp. tritici during development reveals a highly dynamic transcriptome. J. Genet. Genome 38, 357–371. doi: 10.1016/j.jgg.2011.07.004
Kim, K. T., Jeon, J., Choi, J., Cheong, K., Song, H., Choi, G., et al. (2016). Kingdom-wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front. Plant Sci. 7:186. doi: 10.3389/fpls.2016.00186
Kiran, K., Rawal, H. C., Dubey, H., Jaswal, R., Bhardwaj, S. C., Prasad, P., et al. (2017). Dissection of genomic features and variations of three pathotypes of Puccinia striiformis through whole genome sequencing. Sci. Rep. 7:42419. doi: 10.1038/srep42419
Krijger, J. J., Thon, M. R., Deising, H. B., and Wirsel, S. G. (2014). Compositions of fungal secretomes indicate a greater impact of phylogenetic history than lifestyle adaptation. BMC Genomics 15:722. doi: 10.1186/1471-2164-15-722
Kück, P., and Meusemann, K. (2010). FASconCAT: convenient handling of data matrices. Mol. Phylogen Evol. 56, 1115–1118. doi: 10.1016/j.ympev.2010.04.024
Kulkarni, R. D., Kelkar, H. S., and Dean, R. A. (2003). An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 28, 118–121. doi: 10.1016/S0968-0004(03)00025-2
Lanver, D., Mendoza-Mendoza, A., Brachmann, A., and Kahmann, R. (2010). Sho1 and Msb2-related proteins regulate appressorium development in the smut fungus Ustilago maydis. Plant Cell 22, 2085–2101. doi: 10.1105/tpc.109.073734
Lechner, M., Findeiss, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Lee, D. H., Choi, H. W., and Hwang, B. K. (2011). The pepper E3 ubiquitin ligase RING1 gene, CaRING1, is required for cell death and the salicylic acid-dependent defense response. Plant Physiol. 156, 2011–2025. doi: 10.1104/pp.111.177568
Lex, A., Gehlenborg, N., Strobelt, H., Vuillemot, R., and Pfister, H. (2014). UpSet: visualization of intersecting sets. IEEE Trans. Vis. Comput. Graphics 20, 1983–1992. doi: 10.1109/TVCG.2014.2346248
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Ligges, U., and Maechler, M. (2003). Scatterplot3d – an R package for visualizing multivariate data. J. Stat. Softw. 8, 1–20. doi: 10.18637/jss.v008.i11
Line, R. F. (2002). Stripe rust of wheat and barley in North America: a retrospective historical review. Annu. Rev. Phytopathol. 40, 75–118. doi: 10.1146/annurev.phyto.40.020102.111645
Line, R. F., and Qayoum, A. (1992). Virulence, aggressiveness, evolution, and distribution of races of Puccinia striiformis (the cause of stripe rust of wheat) in North America, 1968–1987. US Department of Agriculture, Agriculture Research Services Techn. Bull. 1992;1788, 44.
Liu, B., Chen, X. M., and Kang, Z. S. (2012). Gene sequencing reveals heterokaryotic variations and evolutionary mechanisms in Puccinia striiformis. Open J. Genomics 1:1. Available online at: http://rossscience.org/ojgen/articles/2075-9061-1-1.pdf
Liu, C., Pedersen, C., Schultz-Larsen, T., Aguilar, G. B., Madriz-Orde-ana, K., Hovmøller, M. S., et al. (2016). The stripe rust fungal effector PEC6 suppresses pattern-triggered immunity in a host species-independent manner and interacts with adenosine kinases. New Phytol. 213:1556. doi: 10.1111/nph.14034
Liu, T. L., Wan, A. M., Liu, D. C., and Chen, X. M. (2017). Changes of races and virulence genes of Puccinia striiformis f. sp. tritici, the wheat stripe rust pathogen, in the United States from 1968 to 2009. Plant Dis. 101, 1522–1532. doi: 10.1094/PDIS-12-16-1786-RE
Lowe, R. G., and Howlett, B. J. (2012). Indifferent, affectionate, or deceitful: lifestyles and secretomes of fungi. PLoS Pathog. 8:e1002515. doi: 10.1371/journal.ppat.1002515
Ma, Z., Song, T., Zhu, L., Ye, W., Wang, Y., Shao, Y., et al. (2015). A Phytophthora sojae glycoside hydrolase 12 protein is a major virulence factor during soybean infection and is recognized as a PAMP. Plant Cell 27, 2057–2072. doi: 10.1105/tpc.15.00390
Martchenko, M., Alarco, A. M., Harcus, D., and Whiteway, M. (2004). Superoxide dismutases in Candida albicans: transcriptional regulation and functional characterization of the hyphal-induced SOD5 gene. Mol. Biol. Cell 15, 456–467. doi: 10.1091/mbc.E03-03-0179
McIntosh, R. A., Yamazaki, Y., Dubcovsky, J., Rogers, J., Morris, C., Appels, R., et al. (2013). “Catalogue of gene symbols for wheat,” in 12th International Wheat. Genetics Symposium (Yokohama). Available online at: http://www.shigen.nig.ac.jp/wheat/komugi/genes/download.jsp
Meléndez, H. G., Billon-Grand, G., Fèvre, M., and Mey, G. (2009). Role of the Botrytis cinerea Fkbp12 ortholog in pathogenic development and in sulfur regulation. Fungal Genet. Biol. 46, 308–320. doi: 10.1016/j.fgb.2008.11.011
Nemri, A., Saunders, D. G., Anderson, C., Upadhyaya, N. M., Win, J., Lawrence, G. J., et al. (2014). The genome sequence and effector complement of the flax rust pathogen Melampsora lini. Front. Plant Sci. 5:98. doi: 10.3389/fpls.2014.00098
Nguyen Ba, A. N., Pogoutse, A., Provart, N., and Moses, A. M. (2009). NLStradamus: a simple Hidden Markov Model for nuclear localization signal prediction. BMC Bioinformatics 10:202. doi: 10.1186/1471-2105-10-202
Okonechnikov, K., Conesa, A., and García-Alcalde, F. (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. doi: 10.1093/bioinformatics/btv566
Petre, B., Joly, D. L., and Duplessis, S. (2014). Effector proteins of rust fungi. Front. Plant Sci. 5:416. doi: 10.3389/fpls.2014.00416
Ramachandran, S. R., Yin, C., Kud, J., Tanaka, K., Xiao, F., and Hulbert, S. H. (2017). Effectors from wheat rust fungi suppress multiple plant defense responses. Phytopathology 107, 75–83. doi: 10.1094/PHYTO-02-16-0083-R
R Core Team (2014). R: A Language and Environment for Statistical Computing. Avalable online at: https://www.r-project.org/
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a Large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Salmela, L., and Schröder, J. (2011). Correcting errors in short reads by multiple alignments. Bioinformatics 27, 1455–1461. doi: 10.1093/bioinformatics/btr170
Saunders, D. G., Win, J., Cano, L. M., Szabo, L. J., Kamoun, S., and Raffaele, S. (2012). Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PLoS ONE 7:e29847. doi: 10.1371/journal.pone.0029847
Sharma-Poudyal, D., Chen, X., and Rupp, R. A. (2014). Potential oversummering and overwintering regions for the wheat stripe rust pathogen in the contiguous United States. Int. J. Biometeorol. 58, 987–997. doi: 10.1007/s00484-013-0683-6
Sharpee, W. C., and Dean, R. A. (2016). Form and function of fungal and oomycete effectors. Fungal Biol. Rev. 30, 62–73. doi: 10.1016/j.fbr.2016.04.001
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Sperschneider, J., Dodds, P. N., Gardiner, D. M., Manners, J. M., Singh, K. B., and Taylor, J. M. (2015a). Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11:e1004806. doi: 10.1371/journal.ppat.1004806
Sperschneider, J., Gardiner, D. M., Dodds, P. N., Tini, F., Covarelli, L., Singh, K. B., et al. (2015b). EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 210, 743–761. doi: 10.1111/nph.13794
Sperschneider, J., Gardiner, D. M., Thatcher, L. F., Lyons, R., Singh, K. B., Manners, J. M., et al. (2015c). Genome-wide analysis in three Fusarium pathogens identifies rapidly evolving chromosomes and genes associated with pathogenicity. Genome Biol. Evol. 7, 1613–1627. doi: 10.1093/gbe/evv092
Tian, Y., Zhan, G. M., Chen, X. M., Tungruentragoon, A., Lu, X., Zhao, J., et al. (2016). Virulence and simple sequence repeat marker segregation in a Puccinia striiformis f. sp. tritici population produced by selfing a Chinese isolate on Berberis shensiana. Phytopathology 106, 185–191. doi: 10.1094/PHYTO-07-15-0162-R
Toruño, T. Y., Stergiopoulos, I., and Coaker, G. (2016). Plant-pathogen effectors: cellular probes interfering with plant defenses in spatial and temporal manners. Annu. Rev. Phytopathol. 54, 419–441. doi: 10.1146/annurev-phyto-080615-100204
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Urban, M., Pant, R., Raghunath, A., Irvine, A. G., Pedro, H., and Hammond-Kosack, K. (2015). The pathogen-host interactions database: additions and future developments. Nucleic Acids Res. 43, D645–D655. doi: 10.1093/nar/gku1165
van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., Del Angel, G., Levy-Moonshine, A., et al. (2013). From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43
van Kan, J. A., van den Ackerveken, G. F., and de Wit, P. J. (1991). Cloning and characterization of cDNA of avirulence gene avr9 of the fungal pathogen Cladosporium fulvum, causal agent of tomato leaf mold. Mol. Plant Microbe Interact. 4, 52–59. doi: 10.1094/MPMI-4-052
Vleeshouwers, V. G., and Oliver, R. P. (2014). Effectors as tools in disease resistance breeding against biotrophic, hemibiotrophic, and nectrotrophic plant pathogens. Mol. Plant Microbe Interact. 27, 196–206. doi: 10.1094/MPMI-10-13-0313-IA
Wan, A. M., and Chen, X. M. (2012). Virulence, frequency, and distribution of races of Puccinia striiformis f. sp. tritici and P. striiformis f. sp. hordei identified in the United States in 2008 and 2009. Plant Dis. 96, 67–74. doi: 10.1094/PDIS-02-11-0119
Wan, A. M., and Chen, X. M. (2014). Virulence characterization of Puccinia striiformis f. sp. tritici using a new set of Yr single-gene line differentials in the United States in 2010. Plant Dis. 98, 1534–1542. doi: 10.1094/PDIS-01-14-0071-RE
Wan, A. M., Chen, X. M., and Yuen, J. (2016). Races of Puccinia striiformis f. sp. tritici in the United States in 2011 and 2012 and comparison with races in 2010. Plant Dis. 100, 966–975. doi: 10.1094/PDIS-10-15-1122-RE
Wan, A. M., Muleta, K. T., Zegeye, H., Hundie, B., Pumphrey, M. O., and Chen, X. M. (2017). Virulence characterization of wheat stripe rust fungus Puccinia striiformis f. sp. tritici in Ethiopia and evaluation of Ethiopian wheat germplasm for resistance to races of the pathogen from Ethiopia and the United States. Plant Dis. 101, 73–80. doi: 10.1094/PDIS-03-16-0371-RE
Wang, M. N., Wan, A. M., and Chen, X. M. (2015). Barberry as alternate host is important for Puccinia graminis f. sp. tritici but not for Puccinia striiformis f. sp. tritici in the U.S. Pacific Northwest. Plant Dis. 99, 1507–1516. doi: 10.1094/PDIS-12-14-1279-RE
Wellings, C. R. (2011). Global status of stripe rust: a review of historical and current threats. Euphytica 179, 129–141. doi: 10.1007/s10681-011-0360-y
Xia, C. J., Wan, A., Wang, M., Jiwan, D. A., See, D. R., and Chen, X. (2016a). Secreted protein gene derived-single nucleotide polymorphisms (SP-SNPs) reveal population diversity and differentiation of Puccinia striiformis f. sp. tritici in the United States. Fungal Biol. 120, 729–744. doi: 10.1016/j.funbio.2016.02.007
Xia, C. J., Wang, M. N., Wan, A. M., Jiwan, D. A., See, D. R., and Chen, X. M. (2016b). Association analysis of SP-SNPs and avirulence genes in Puccinia striiformis f. sp. tritici, the wheat stripe rust pathogen. Am. J. Plant Sci. 7, 126–137. doi: 10.4236/ajps.2016.71014
Xiang, C., Feng, J. Y., Wang, M. N., Chen, X. M., See, D. R., Wan, A. M., et al. (2016). Molecular mapping of Yr76 for resistance to stripe rust in winter club wheat cultivar Tyee. Phytopathology 106, 1186–1193. doi: 10.1094/PHYTO-01-16-0045-FI
Yin, C., Downey, S. I., Klages-Mundt, N. L., Ramachandran, S., Chen, X., Szabo, L. J., et al. (2015). Identification of promising host-induced silencing targets among genes preferentially transcribed in haustoria of Puccinia. BMC Genomics 16:579. doi: 10.1186/s12864-015-1791-y
Yin, C., and Hulbert, S. (2011). Prospects for functional analysis of effectors from cereal rust fungi. Euphytica 179, 57–67. doi: 10.1007/s10681-010-0285-x
Yin, C., Chen, X., Wang, X., Han, Q., Kang, Z., and Hulbert, S. H. (2009). Generation and analysis of expression sequence tags from haustoria of the wheat stripe rust fungus Puccinia striiformis f. sp. tritici. BMC Genomics 10:626. doi: 10.1186/1471-2164-10-626
Yuan, C., Wang, M., Skinner, D. Z., See, D. R., Xia, C. J., Guo, X., et al. (2017). Inheritance of virulence, construction of a linkage map, and mapping of dominant virulence genes in Puccinia striiformis f. sp. tritici through characterization of a sexual population with genotyping-by-sequencing. Phytopathology. doi: 10.1094/PHYTO-04-17-0139-R. [Epub ahead of print].
Zhao, J., Wang, M. N., Chen, X. M., and Kang, Z. S. (2016). Role of alternate hosts in epidemiology and pathogen variation of cereal rusts. Annu. Rev. Phytopathol. 54, 207–228. doi: 10.1146/annurev-phyto-080615-095851
Keywords: avirulence genes, effectors, Puccinia striiformis f. sp. tritici, secretome, stripe rust, yellow rust
Citation: Xia C, Wang M, Cornejo OE, Jiwan DA, See DR and Chen X (2017) Secretome Characterization and Correlation Analysis Reveal Putative Pathogenicity Mechanisms and Identify Candidate Avirulence Genes in the Wheat Stripe Rust Fungus Puccinia striiformis f. sp. tritici. Front. Microbiol. 8:2394. doi: 10.3389/fmicb.2017.02394
Received: 17 September 2017; Accepted: 20 November 2017;
Published: 11 December 2017.
Edited by:
Haiwei Luo, The Chinese University of Hong Kong, Hong KongReviewed by:
Benjamin Schwessinger, Australian National University, AustraliaJonathan Michael Plett, Western Sydney University, Australia
Copyright © 2017 Xia, Wang, Cornejo, Jiwan, See and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xianming Chen, xianming.chen@ars.usda.gov