 Marisa F. Nicolás1†
Marisa F. Nicolás1† Pablo Ivan Pereira Ramos2†
Pablo Ivan Pereira Ramos2† Fabíola Marques de Carvalho1†
Fabíola Marques de Carvalho1† Dhian R. A. Camargo3
Dhian R. A. Camargo3 Carlene de Fátima Morais Alves3
Carlene de Fátima Morais Alves3 Guilherme Loss de Morais1
Guilherme Loss de Morais1 Luiz G. P. Almeida1
Luiz G. P. Almeida1 Rangel C. Souza1
Rangel C. Souza1 Luciane P. Ciapina1
Luciane P. Ciapina1 Ana C. P. Vicente4
Ana C. P. Vicente4 Roney S. Coimbra5
Roney S. Coimbra5 Ana T. Ribeiro de Vasconcelos1*
Ana T. Ribeiro de Vasconcelos1*- 1Laboratório Nacional de Computação Científica, Petrópolis, Brazil
- 2Instituto Gonçalo Moniz, Fundação Oswaldo Cruz, Salvador, Brazil
- 3Fundação Ezequiel Dias, Belo Horizonte, Brazil
- 4Laboratório de Genética Molecular de Microrganismos, Instituto Oswaldo Cruz, Fundação Oswaldo Cruz, Rio de Janeiro, Brazil
- 5Neurogenômica, Fiocruz Institute Renê Rachou, Belo Horizonte, Brazil
The aim of this study was to unravel the genetic determinants responsible for multidrug (including carbapenems) resistance and virulence in a clinical isolate of Klebsiella quasipneumoniae subsp. similipneumoniae by whole-genome sequencing and comparative analyses. Eighty-three clinical isolates initially identified as carbapenem-resistant K. pneumoniae were collected from nosocomial infections in southeast Brazil. After RAPD screening, the KPC-142 isolate, showing the most divergent DNA pattern, was selected for complete genome sequencing in an Illumina HiSeq 2500 instrument. Reads were assembled into scaffolds, gaps between scaffolds were resolved by in silico gap filling and extensive bioinformatics analyses were performed, using multiple comparative analysis tools and databases. Genome sequencing allowed to correct the classification of the KPC-142 isolate as K. quasipneumoniae subsp. similipneumoniae. To the best of our knowledge this is the first complete genome reported to date of a clinical isolate of this subspecies harboring both class A beta-lactamases KPC-2 and OKP-B-6 from South America. KPC-142 has one 5.2 Mbp chromosome (57.8% G+C) and two plasmids: 190 Kbp pKQPS142a (50.7% G+C) and 11 Kbp pKQPS142b (57.3% G+C). The 3 Kbp region in pKQPS142b containing the blaKPC−2 was found highly similar to that of pKp13d of K. pneumoniae Kp13 isolated in Southern Brazil in 2009, suggesting the horizontal transfer of this resistance gene between different species of Klebsiella. KPC-142 additionally harbors an integrative conjugative element ICEPm1 that could be involved in the mobilization of pKQPS142b and determinants of resistance to other classes of antimicrobials, including aminoglycoside and silver. We present the completely assembled genome sequence of a clinical isolate of K. quasipneumoniae subsp. similipneumoniae, a KPC-2 and OKP-B-6 beta-lactamases producer and discuss the most relevant genomic features of this important resistant pathogen in comparison to several strains belonging to K. quasipneumoniae subsp. similipneumoniae (phylogroup II-B), K. quasipneumoniae subsp. quasipneumoniae (phylogroup II-A), K. pneumoniae (phylogroup I), and K. variicola (phylogroup III). Our study contributes to the description of the characteristics of a novel K. quasipneumoniae subsp. similipneumoniae strain circulating in South America that currently represent a serious potential risk for nosocomial settings.
Introduction
The prevalence of isolation of carbapenem-resistant Klebsiella pneumoniae strains in nosocomial infections is increasing, posing a serious therapeutic problem given the limited number of effective antimicrobial agents. Recently in the USA, isolates belonging to the related K. pneumoniae species, namely K. variicola, and K. quasipneumoniae, were isolated and shown to harbor capsular synthesis K type locus (KL) KL19 and KL1, respectively, and both carried the Klebsiella pneumoniae carbapenemase (KPC) gene (Long et al., 2017). Also, contrary to what was previously thought, K. quasipneumoniae and K. variicola strains can be as virulent as extended-spectrum beta-lactamases (ESBL)-producing K. pneumoniae strains, causing invasive infections, and mortality at rates statistically similar to those of K. pneumoniae strains (Long et al., 2017), which confers resistance to carbapenem antibiotics. Even more recently, a hypervirulent Klebsiella quasipneumoniae subsp. similipneumoniae isolated from a patient with chronic liver disease in India that belongs to novel sequence type ST2320 and possesses the K1 capsular serotype has been reported (Shankar et al., 2017). These findings accentuate the concern for the potential spread of multidrug resistance and increased virulence capacity among various Klebsiella species.
Because of overlapping biochemical profiles, phenotypic tests are unable to differentiate between K. pneumoniae, K. quasipneumoniae, or K. variicola (Alves et al., 2006; Bowers et al., 2016), and this limitation may lead to underreporting infections caused by the latter species. Notably, the capsular serotypes and MLST types among K. quasipneumoniae and K. variicola strains recovered from human infections are diverse and novel K-types and MLST types are being found in isolates of these two species (Brisse et al., 2013; Garza-Ramos et al., 2015; Long et al., 2017). Core chromosomal beta-lactamases have been proposed as molecular markers to differentiate Klebsiella species, i.e., K. pneumoniae (SHV restricted), K. quasipneumoniae (OKP restricted), and K. variicola (LEN restricted) (Haeggman et al., 2004; Fonseca et al., 2017). However, this method has some complications, since there are some SHV beta-lactamase genes encoded on plasmids and K. variicola isolates carrying chromosomal OKP-B instead of LEN (Long et al., 2017). Nevertheless, when genomic sequences are available, the average nucleotide identity (ANI) is a robust proxy for genomic relatedness between strains, and has been used to differentiate Klebsiella species with BLASTn (cut-off 96% identity) (Brisse et al., 2014). Thus, a better understanding of the genomics aspects of K. quasipneumoniae and K. variicola strains will help toward the development of improved diagnostics.
In 2011 we collected an isolate from a nosocomial infection in Southeast Brazil, initially identified as a KPC-producing K. pneumoniae (KPC-142) by clinical routine methods. In this report, by determining its complete genome sequence, we show that this is an isolate of KPC-producing K. quasipneumoniae subsp. similipneumoniae (KPC-Kqps) instead of KPC-producing K. pneumoniae (KPC-Kp). Genomic analyses showed that KPC-142 harbors a cps cluster related to KL16 type, an integrative conjugative element termed ICEPm1 and a novel combination of known MLST alleles. Besides blaKPC−2 it carries additional antibiotic resistance genes, including blaOKP−B−6, aph(3')-VIa for an aminoglycoside-modifying enzyme, silver resistance genes, and possess a significant amount of virulence factors. By a genomic comparison between KPC-142 and strains belonging to other Klebsiella phylogroups we highlight the most relevant genomic features, including the N-acetyl-neuraminic acid catabolism pathway that appears only in K. quasipneumoniae subsp. similipneumoniae strains and which is known to play a role in the bacterial pathogenesis.
Material and Methods
Bacterial Strains Used in This Study
From 83 KPC isolates carrying the blaKPC gene collected from nosocomial infections in Minas Gerais state (Southeast Brazil) in 2011 we selected the isolate named KPC-142, which displayed the most divergent DNA pattern by RAPD cluster analysis (Figure S1). This isolate was initially assigned as K. pneumoniae KPC-142 and susceptibility tests were performed according to CLSI guidelines 2017, using Vitek 2 system and AST-N239 cards (bioMérieux, Inc., Durham, NC) according to the manufacturer's instructions and using Gram-negative identification (GN ID) cards (Bobenchik et al., 2017).
The bacterial isolate was grown overnight on nutrient agar and the DNA was extracted using the method originally described by Coimbra et al. (1999). The isolate KPC-142 is maintained in the certified strains collection of Ezequiel Dias Foundation (FUNED), Belo Horizonte, Brazil.
For the comparative genomic analyses and determination of average nucleotide identity (ANI), we used a bacterial strain panel representative of each Klebsiella phylogroup (Holt et al., 2015) that included: (i) nine K. quasipneumoniae subsp. similipneumoniae (phylogroup II-B) (HKUOPA4, HKUOPJ4, HKUOPL4, KPC-142, ATCC 700603, 07A044 T, MGH 44, 193_KOXY, and 385_ECLO), (ii) nine K. quasipneumoniae subsp. quasipneumoniae (phylogroup II-A) (UCICRE14, 01A030 T, FI HV 2014, MGH96, 18A069, ARLG-2711, PO1285, AK_SD_007, and 21_GR_13), nine K. pneumoniae (phylogroup I) (DSM 30104 T, Kp13, 1084, CAV1193, HS11286, KCTC 2242, KP617, MGH 78578, and NTUH-K2044), and nine K. variicola (phylogroup III) (BZ19, GJ1, UCICRE10, MGH 40, MGH 20, UCI 18, BIDMC 61, BIDMC88, and MGH 80), totaling 36 bacterial genomes. An additional 31 isolates were included for phylogenetic studies (see section MLST classification and phylogeny).
Genome Sequencing, Assembly, Annotation, and Bioinformatics Analyses
Genomic DNA was sequenced on an Illumina HiSeq 2500 sequencer using Nextera XT paired-end run with a 500-bp insert library at the High-Throughput Sequencing Platform of the Oswaldo Cruz Foundation (Fiocruz, Rio de Janeiro, Brazil). The assembly of reads into scaffolds, based on 3,804,017 reads, was accomplished using a combination of Newbler v 2.6 (Roche Inc.) and SPAdes 3.10.0 (Bankevich et al., 2012) programs. 99.99% of the bases on the assembled genome had an average Phred quality of >40, calculated by Newbler. Gaps intra- and inter-scaffolds were resolved using a three-phase approach: (1) by extracting consensus sequences that did not form gaps in one of the assemblies; (2) by performing local assemblies of reads falling in gap termini; and (3) using the GapFiller tool (Boetzer and Pirovano, 2012). Further details of each step are presented in the Figure S2.
Genome annotation was performed using the System for Automated Bacterial Integrated Annotation (SABIA) (Almeida et al., 2004). The steps performed by this platform consist of an initial gene prediction process using Glimmer (Delcher et al., 1999) and GeneMark (Besemer et al., 2001) including a start codon correction routine based in the multiple alignment of similar proteins identified by BLASTP against NCBI proteins database (www.ncbi.nlm.nih.gov/protein). CDSs identified after this process were annotated applying an automated annotation pipeline, where each open reading frame (ORF) is submitted to similarity searches using both nucleotide and amino acid sequences by Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1990) against KEGG (www.genome.jp/kegg), NCBI-nr (www.ncbi.nlm.nih.gov/protein) and UniProtKB/Swiss-Prot (www.uniprot.org) databases, in addition to the prediction of protein domains and important catalytic sites using InterPro database (Finn et al., 2017), and the results are made available on the screen for the assessment of expert users. In this process, only CDSs with predicted lengths over 50 aa and at least one BLASTP hit against any of the four databases mentioned above were considered. The transfer RNAs (tRNAs) were detected by tRNAscan-SE (Lowe and Eddy, 1997) and the annotation of ribosomal RNA genes was carried out by RNAmmer (Lagesen et al., 2007).
Further bioinformatics analyses included BLAST searches against AtlasT4SS database (Souza et al., 2012)/Resfinder database (Zankari et al., 2012)/ARDB-Antibiotic Resistance Genes Database (Liu and Pop, 2009)/Comprehensive Antibiotic Resistance Database (Jia et al., 2017) and Virulence Factors database (VFDB) (Chen et al., 2005), which were performed for identification of type 4 secretion systems (T4SS), acquired antimicrobial resistance genes and bacterial virulence factors, respectively. Typing of plasmids was carried out by in silico detection using PlasmidFinder database (Carattoli et al., 2014).
Average nucleotide identity (ANI) was calculated using BLASTn (ANIb) in JSpeciesWS (Richter et al., 2016). Reference type strains of K. quasipneumoniae belonging to subspecies K. quasipneumoniae subsp. quasipneumoniae 01A030 (GenBank accession no. GCA_000751755) and K. quasipneumoniae subsp. similipneumoniae 07A044 (GenBank accession no. GCA_000613225) and type strain K. pneumoniae subsp. pneumoniae DSM 30104 (GenBank accession no. GCA_000281755), in addition to strain K. variicola GJ1 (GenBank accession no. GCA_001989495.1), were used to perform all-versus-all comparisons with a bacterial strain panel including KPC-142. An ANI threshold of ≥96% or greater was considered to delineate species boundaries as it correlates well to DNA-DNA hybridization studies (Goris et al., 2007; Federhen et al., 2016).
Comparative genomic and gene cluster analyses were performed using the Gview tools (Petkau et al., 2010) and Proteinortho (Lechner et al., 2011), respectively. The comparisons involved the bacterial strain panel. In Proteinortho, co-orthologous clustering was calculated using BLASTp, with an identity/coverage cutoff of >90% and E-values < 10−5. BLASTn Atlas was generated with Gview, applying as parameters identity >70% and E-values < 10−5 (Petkau et al., 2010).
The blaKPC−2-harboring 10,951 bp plasmid of KPC-142 (pKQPS142b) was compared with plasmids of K. pneumoniae strain Kp13 (pKp13d), K. pneumoniae strain A60136 (p60136), and E. coli (IncQ pRSF1010) by BLASTn. The genomic context of the highly similar genes to pKQPS142b were obtained using the Gview program (Petkau et al., 2010), with manual editing.
Kaptive tool (Wyres et al., 2016) was used for classification of capsular loci (cps cluster) and wzi and wzc allele typing was performed by querying the predicted nucleotides sequences of both genes against the Institut Pasteur MLST database [bigsdb.pasteur.fr/klebsiella/].
MLST Classification and Phylogeny
The allele sequences for seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) and sequence types (STs) were assigned by using the MLST (http://bigsdb.pasteur.fr/klebsiella/klebsiella.html) and NCBI (www.ncbi.nlm.nih.gov) databases.
In order to investigate the phylogenetic placement of KPC-142, the seven housekeeping genes were concatenated for each of the 36 genomes used in a bacterial strain panel, in addition to K. quasipneumoniae subsp. quasipneumoniae, K. quasipneumoniae subsp. similipneumoniae, and K. pneumoniae strains, which sequence typing were closer to that identified to KPC-142. K. variicola was used as outgroup. The sequences were aligned in MAFFT using the accurate LINSI strategy (Katoh and Standley, 2013). Poorly aligned regions were removed in the final alignment by Gblocks program (Castresana, 2000), allowing positions with a gap in less than 50% of the sequences. Twenty-four percent of the 12,219 original base positions were retained in the final alignment.
Maximum likelihood tree for the matrix consisting of the seven concatenated loci was constructed with PhyML (Guindon et al., 2009), comprising 67 nucleotide sequences. jModelTest v. 2.1.10 (Darriba et al., 2012) was used to select the best-fit model of nucleotide substitution according to the corrected Akaike information criterion measure. The evolutionary history was inferred using GTR+I+G as nucleotide substitution model, which was chosen as best-scoring in the earlier step, with 1,000 replicates of a nonparametric bootstrap as clade support. Tree editing and annotation were performed using interactive Tree of Life (iTOL) (Letunic and Bork, 2016). A phylogenetic network using the same aligned sequences as input was inferred using the NeighborNet algorithm and untransformed distances (uncorrected-p) in SplitsTree4 v. 4.14.6 (Huson and Bryant, 2006). The pairwise homoplasy index (phi) (Bruen et al., 2006) was calculated within SplitsTree4 to evaluate if the recombination events were statistically significant.
Nucleotide Sequence Accession Number
The Bioproject accession number for KPC-142 is PRJNA383559 and the complete sequences of the chromosome and plasmids pKQPS142a and pKQPS142b have been deposited in GenBank/NCBI under accession numbers CP023478, CP023479, and CP023480, respectively.
Results and Discussion
Main Features of the Chromosome and Plasmids Sequences of Isolate KPC-142
We have sequenced to closure the genome of K. quasipneumoniae subsp. similipneumoniae KPC-142. This isolate was chosen for sequencing following a RAPD analysis of a panel of multiple blaKPC−2-producing Klebsiella clinical isolates (Figure S1), in which KPC-142 presented a very divergent DNA pattern. The KPC-142 genome consists of one 5,217,996 bp chromosome with 57.84% G+C content (4,856 protein-coding genes) and two plasmids, namely pKQPS142a with 189,707 bp and 50.71% G+C content (199 protein-coding genes) and pKQPS142b with 10,951 bp and 57.32% G+C content (12 protein-coding genes). Also, KPC-142 contains eight copies of 16S rRNAs and 23S rRNA and nine copies of 5S rRNA.
A panel of 35 representative Klebsiella isolates belonging to K. quasipneumoniae, K. pneumoniae and K. variicola was chosen to comparatively investigate the genome of isolate Kqps142. The genomic characteristics of all 36 isolates (including Kqps142) are shown in Table 1, and the selected strains include the type species of the three Klebsiella species compared (and the two K. quasipneumoniae subspecies), and cover a wide range of sequence types (STs; including those closer to Kqps142), and variable composition of resistance determinants.
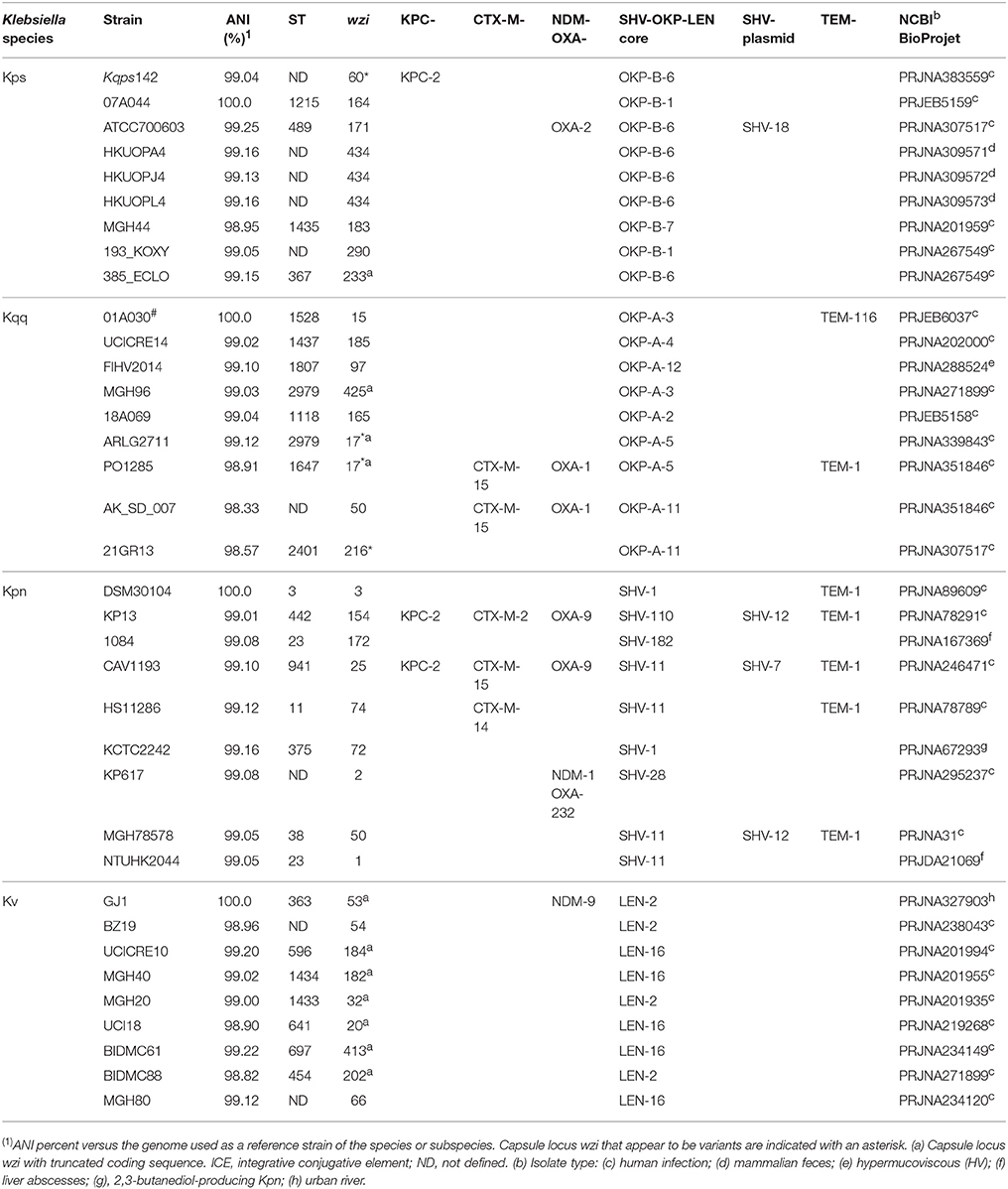
Table 1. Major genomic characteristics of the panel of K. quasipneumoniae subsp. similipneumoniae (Kqs), K. quasipneumoniae subsp. quasipneumoniae (Kqq), K. pneumoniae (Kpn), and K. variicola (Kv) strains analyzed in this study.
We calculated pairwise average nucleotide identity (ANI) of KPC-142 chromosome sequence including type strains K. pneumoniae subsp. pneumoniae DSM 30104, K. quasipneumoniae subsp. quasipneumoniae 01A030, K. quasipneumoniae subsp. similipneumoniae 07A044, K. variicola GJ1 and the remaining strains of the bacterial panel described in the material and methods (Table S1).
The results pointed that, although KPC-142 strain was originally reported as K. pneumoniae before sequencing, it did not fit genomic species boundaries threshold set at 96%, and displayed ANI with the K. pneumoniae DSM 30104 type strain of 93.7% (Table S1). Rather, comparison to the type strains of K. quasipneumoniae subsp. quasipneumoniae (ANI of 96.52%) and K. quasipneumoniae subsp. similipneumoniae (ANI of 99.04%) revealed their relatedness (Table S1). These results are in line with those previously reported by Brisse et al. (2014), further supporting the classification of KPC-142 as subspecies Klebsiella quasipneumoniae subsp. similipneumoniae (hereafter referred as Kqps142). Misidentification of Klebsiella species by conventional clinical microbiology laboratory techniques was evidenced recently by Long et al. (2017), who demonstrated that almost 30% of tentatively identified K. pneumoniae strains in their study were in fact K. quasipneumoniae. Furthermore, through this ANI analysis, it can be inferred that the K. pneumoniae strains are slightly more similar to the K. variicola strains (ANI of 94.38%, median among the compared Kv strains) than to the K. quasipneumoniae (Kqq + Kqs) strains (ANI of 93.60%, median among the compared Kqq and Kqs strains) (Table S1). This observation correlates with the findings of Holt et al. (2015), where the K. pneumoniae and K. variicola phylogroups are closer to each other than K. pneumoniae and K. quasipneumoniae phylogroups (Holt et al., 2015).
Regarding the plasmid pKQPS142a with ~190 Kbp, searches using the PlasmidFinder database classified it as a conjugative IncFIB(K) plasmid, more similar to pKPN3 plasmid (50% coverage query, 98% maximum nucleotide identity) that has very narrow host range, limited to a predominantly human-associated sub-clade of Klebsiella (Kaplan et al., 2015). While pKPN3 plasmid is conjugative, pKQPS142a appears to have lost the genes for T4SS, and has an average of 50.71% G+C content, which is lower than the K. quasipneumoniae median of 57% (reported for 103 assemblies currently available in the NCBI/Genome database (www.ncbi.nlm.nih.gov/genome). This might be due to the considerable rearrangements with several mobile genetic elements throughout the plasmid sequence. Interestingly, pKQPS142a has multiple regions with high identity (70% query coverage, 98% maximum nucleotide identity) to plasmid pKPN-332 of K. pneumoniae KPNIH39, which was recovered from nosocomial infection during long-term patient colonization (Conlan et al., 2016) (Figure 1). Several key features are in common between pKQPS142a and pKPN-332, such as the RepFIB-like origin of replication (repA/repC), the ter loci (terZABCDEF and terX) that confer resistance to tellurium as well as resistance to bacteriophage and colicins (Taylor et al., 2002), a gene encoding for creatinase (creatine amidinohydrolase), an important medical enzyme that has been used for clinical diagnosis of renal function (Liu et al., 2015), the copper-resistance operon (pcoABCDRS) that encode an efflux system to both remove the toxic metal in excess and cytoplasmic copper management (Brown et al., 1995), fibrinolysin (plasminogen activator, pla gene) and the silver resistance genes (sil) (Figure 1). The predicted amino acid sequence of fibrinolysin (pla gene KPC142_05211) was highly similar (BLASTp >80%) to the plasminogen activator protease of Yersinia pestis CO92, which has a proteolytic activity responsible for the invasive character of plague and is considered an important virulence factor in this species (Lähteenmäki et al., 2001). In Kqps142 the genetic context of pla gene is characterized by two upstream transposases (KPC142_1834 and KPC142_1835) homologous to IS3 family of Yersinia enterocolitica (NC_008800), which could be involved in recombination events leading to spread of this Yersinia virulence factor among Klebsiella species.
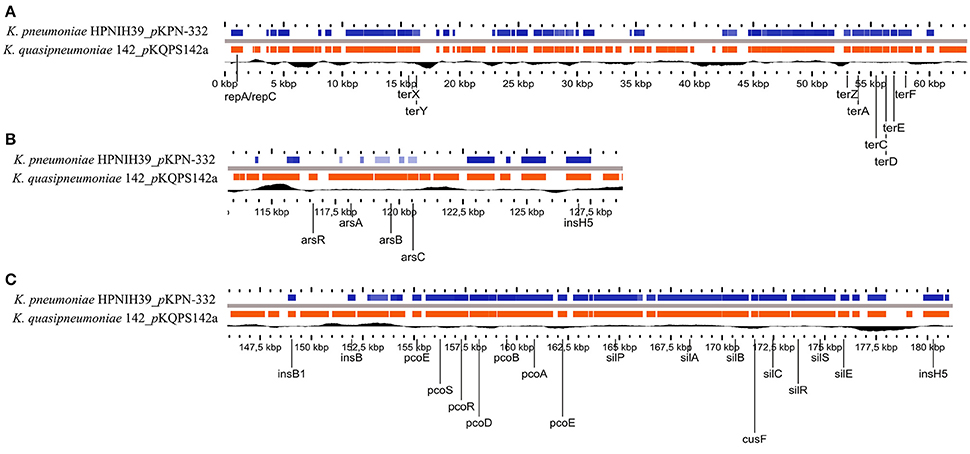
Figure 1. Genomic comparison between the plasmid pKQPS142a of Kqps142 and plasmid pKPN-332 of K. pneumoniae KPNIH39 performed by Gview program, using BlastN (>70% identity and E-values < 10−5). Black line represents GC content. (A) region of ter loci; (B) arsenical operon, and (C) copper-resistance operon and silver resistance genes.
The sil genes represent a major concern, since silver is commonly used in several types of medical devices, e.g., wound dressings, implants, catheter, and endotracheal tubes. While Klebsiella spp. are not primary wound pathogens, they have been linked with ventilator-associated pneumonia (Torres et al., 2009). In a recent Swedish tertiary hospital study with restricted consumption of silver-based products, the presence of sil operon genes was observed at high frequency, exclusively for members of the Enterobacteriaceae family and most common among Enterobacter cloacae and K. oxytoca isolates (Sütterlin et al., 2017). Furthermore, in the case of K. pneumoniae the silver resistance could be selected in a single step (i.e., by a single point mutation) from organisms harboring a repressed sil operon (Randall et al., 2015). Collectively these studies and the results showed here for a nosocomial isolate of K. quasipneumoniae subsp. similipneumoniae emphasize the advisement for the controlled use of silver-based products as well as the monitoring of silver resistance in hospital environments.
On the other hand, pKQPS142a could be considered as a multidrug plasmid, since it also carries the arsenical resistance operon (ars) that encode a specific detoxification pathway for arsenic extrusion (Achour-Rokbani et al., 2010) and a sugE gene encoding for quaternary ammonium compound efflux pump that at high-level expression leads to resistance to a subset of toxic quaternary ammonium compounds (Chung and Saier, 2002) (Figure 1). Also, we highlight another virulence factor encoded on pKQPS142a, i.e., the citrate-dependent iron(III) transport system FecABCDE, FecR, and FecI, where citrate is usually considered an external siderophore (Hussein et al., 1981).
Of special interest in Kqps142 is the plasmid pKQPS142b carrying blaKPC−2. We identified that this small plasmid has homology with the broad host-range IncQ1 plasmid pRSF1010 of E. coli (Scholz et al., 1989) in the regions involved in plasmid replication and mobilization, i.e., mob and rep genes (Figure 2). Also, pKQPS142b shows an extensive region of high identity (99% identity, from mobA, mobB, repression protein F, repA, repB to aph(3')-VIa) with the IncQ1-like p60136 plasmid of K. pneumoniae strain A60136. The p60136 carries a novel β-lactamase, namely Brazilian Klebsiella carbapenemase-1 (BKC-1) and was isolated in São Paulo, Brazil in 2009 (Nicoletti et al., 2015). While p60136 carries BKC-1, pKPC142b harbors blaKPC−2 (KPC142_06049), flanked by an upstream ISKpn6 transposase from IS1182 family and a downstream transposon resolvase Tn3. This plasmid region has around 3 Kbp and showed high similarity to the homologous region of pKp13d of K. pneumoniae Kp13 isolated in Southern Brazil in 2009, whose complete genome has been previously reported by our group (Ramos et al., 2014) (Figure 2).
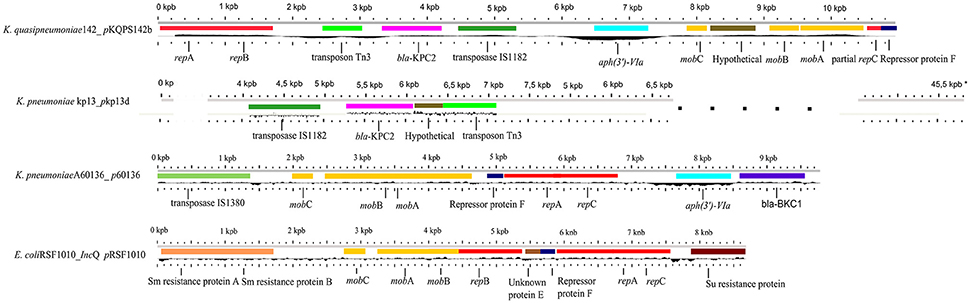
Figure 2. Comparison among the plasmids pKQPS142b of Kqps142, K. pneumoniae strain Kp13 (pKp13d), K. pneumoniae strain A60136 (p60136), and E. coli (IncQ pRSF1010). Homologous genes to plasmid pKPC142b carrying blaKPC−2 were obtained by BLASTn, and represented by the same color in other species, each color defined to a specific gene. Black line represents the GC content. Features of each plasmid were obtained using the GView program, with modifications.*The pKp13d is significantly larger than other compared elements. The genome length scales were adapted to emphasize only the region with similarity to pKQPS142b.
Nicoletti et al. demonstrated that p60136 is a non-transferable plasmid by conjugation from E.coli transformant cells, but it can be mobilized at a high frequency by helper conjugative plasmids (Nicoletti et al., 2015). However, we found no evidence of T4SS genes in the sequence of the largest plasmid pKQPS142a that would support plasmid-mediated conjugation functions provided in trans.
On the other hand, we found an integrative conjugative element ICEPm1 that is conserved in Proteus mirabilis, Providencia stuartii, and Morganella morganii (Flannery et al., 2009) and it was exclusive when compared to a panel of 35 strains representative of each Klebsiella phylogroup (Table 1 and see R7 in Figure 4). In Kqps142, the ICEPm1 is a 92,220 bp region that extends from a phage integrase encoding gene (KPC142_03753) to a chromosome partitioning-related encoding gene (KPC142_05483), which contains a core segment showing homology to a T4SS for DNA transfer (KPC142_03739 to KPC142_03716). Interestingly, and in a fashion similar to plasmids, ICEs have the ability to transfer other mobile elements in trans (Meyer, 2009). Another ICE, namely R391/SXT family of ICEs of Vibrio cholerae O139 has been demonstrated to mobilize the IncQ plasmid RSF1010 in an oriT-independent manner. This and further studies suggested that other mechanisms must account for this unexpected oriT-independent mobilization of RSF1010, which are still under investigation (Hochhut et al., 2000; Poulin-Laprade et al., 2015). Also, ICEPm1 and STX are compatible, since both ICEs were found in a single bacterium of P. mirabilis HI4320 (Flannery et al., 2011). These findings raise the question whether the ICEPm1 is capable of transferring the pKQPS142b (IncQ plasmid) in trans, further aiding in the dissemination of blaKPC−2.
Additionally, in pKQPS142b the inverted right repeat (IRR) of transposon Tn3 was identified between ISKpn6 and blaKPC−2, but as previously reported in pKp13d of K. pneumoniae Kp13 no inverted left repeat (IRL) was detected. This can be regarded as evidence of horizontal spread of blaKPC−2 gene between K. pneumoniae and K. quasipneumoniae by recombination events involving ISKpn6 and the flanking Tn3-family sequences and facilitated by the broad host-range mobilizable IncQ1 plasmid. In addition, the blaKPC−2-carrying IncQ pKQPS142b plasmid found here is highly relevant in view of few reports of plasmids associated to the spread of such relevant resistance gene (Carattoli, 2011; Nicoletti et al., 2015; Mollenkopf et al., 2017).
Analysis of Multilocus Sequence and Capsule Typing
Multilocus sequence typing (MLST) analysis revealed a novel combination of known K. pneumoniae MLST alleles (gapA−18, infB−22, mdh−56, pgi−61, phoE−74, rpoB−38, tonB−99), with ST2736 being the closest sequence type, differing only in pgi (allele 16) and phoE (allele 11) loci, with ST1032, ST1361, ST1413, ST1703, ST2119, and ST2137 differing in three alleles.
Maximum likelihood phylogenetic analysis performed using the concatenated MLST genes showed the existence of three main groups. The first, Clade I, comprising all K. variicola sequences; Clade II grouped only K. pneumoniae strains, while Clade III was composed by strains from both K. quasipneumoniae subspecies, as well as some K. pneumoniae isolates (Figure 3A). The phylogenetic placement of Kqps142 was closer to Clade III strains, which included all K. quasipneumoniae strains, reinforcing its classification as K. quasipneumoniae. Kqps142 grouped with K. quasipneumoniae subsp. quasipneumoniae AJ055, an ST2119 isolate differing by three MLST alleles. Also in Clade III were strains with STs differing by few alleles compared to Kqps142, such as K. pneumoniae ERKP033 (ST1032), K. pneumoniae BK44389 (ST1703), and K. quasipneumoniae subsp. quasipneumoniae K38An (ST2137). In order to better elucidate the complex relationships between these strains, a split decomposition analysis was performed, which yielded a network-like structure suggestive of high levels of recombination in the studied population (Figure 3B and Figure S3). The pairwise homoplasy index (phi) was significant (p-value = 0.0), indicating strong statistical support for the recombination events in the split network. In this network, the three main clades identified in the maximum likelihood tree were recapitulated, however, the relationships within Clade III could be better delineated, as this branch was separated into Clade III-A (grouping both subspecies of K. quasipneumoniae and some K. pneumoniae strains) and Clade III-B (containing only K. quasipneumoniae subsp. quasipneumoniae strains) (Figure 3B). These latter strains could be regarded as closer to the type strain of this subspecies (strain 01A030), while those in Clade III-A, including Kqps142, probably underwent more recent recombination events leading to their clustering in the split network.
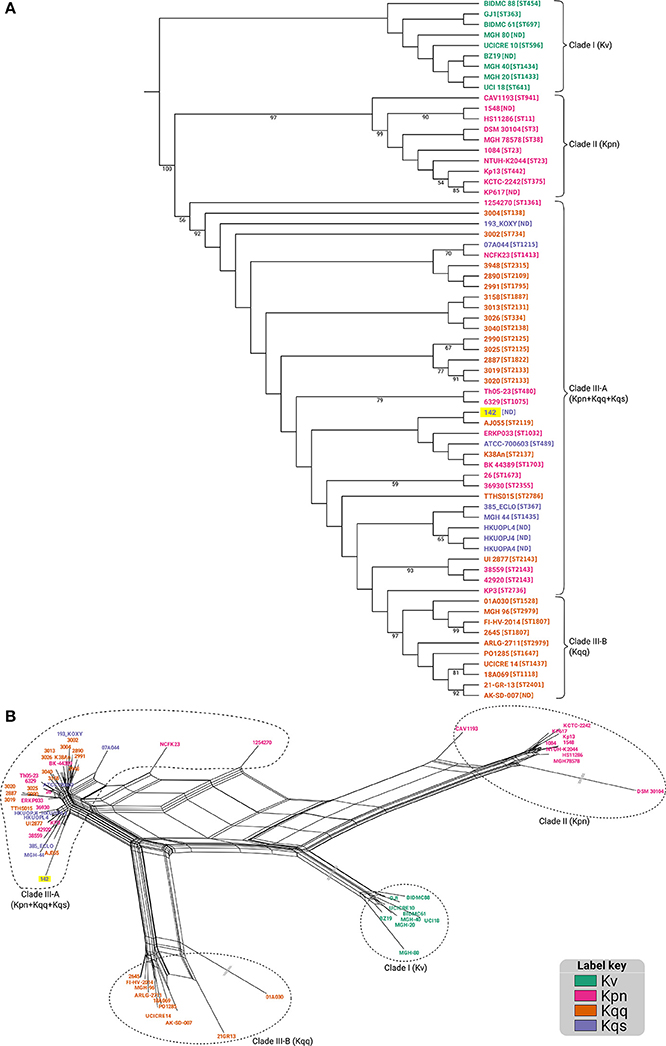
Figure 3. (A) Maximum likelihood tree for the matrix consisting of the concatenated allele sequences for seven housekeeping genes (gapA, infB, mdh, pgi, phoE, rpoB, and tonB) loci, constructed with PhyML, using the GTR+I+G model of nucleotide substitution. 1,000 bootstrap replicates were calculated to assess robustness. 1,000 bootstrap replicates were calculated to assess robustness, and support values >50% are shown. (B) Split decomposition analysis using the seven MLST alleles reveals a network-like structure, suggestive of recombination events. “||” represent long branches that were truncated to fit (see Figure S3 for the complete network). In both panels, KPC-142 is labeled on a yellow background and in the (panel A) the ST number of each isolate/strain is in brackets.
In silico capsule typing was performed using a standard nomenclature to refer to the capsular locus type (K-locus are designated as KL) (Wyres et al., 2016) and comparisons against the K. pneumoniae BIGSdb database (http://bigsdb.web.pasteur.fr). This identified that Kqps142 belongs to KL16 type, containing 22 genes with high similarity to the best match reference locus (97.2% identity and 100% coverage) with manB, manC, and wzc allele type 17. The capsular region also carries a novel wzi, a variant that differ to the most closely related allele 60 in two bases: 444T444G (base depth of coverage 63 without variants) 447C447T (base depth of coverage 63 without variants), raising the possibility that if this wzi allele is indeed novel, it could associate to the K16 capsular locus.
Antimicrobial Susceptibility Tests Associated with Resistance Factors
The antimicrobial susceptibilities of Kqps142 are summarized in Table 2. The isolate presented resistance to all tested beta-lactam antibiotics (ampicillin, ampicillin-sulbactam, cefepime, cefoxitin, ceftazidime, ceftriaxone, cefuroxime, ertapenem, imipenem, meropenem, and piperacilin-tazobactam). Susceptibilities were identified for amikacin (aminoglycoside), gentamicin (aminoglycoside), ciprofloxacin (fluoroquinolone), tigecycline, and colistin (polymyxin E). Correlation between phenotypic antibiotic susceptibility and the resistome, particularly for beta-lactam resistance, showed that besides KPC-2 (gene locus KPC142_06049, found in pKQPS142b), Kqps142 also harbors the chromosomal gene encoding for OKP-B-6 (KPC142_02249). While pKQPS142b carries aph(3')-Via (synonymous to aphA6), the MICs of the aminoglycoside antibiotics tested in this strain were low (amikacin, MIC 8 mg/L and gentamicin, MIC ≤ 1 mg/L). Similarly, Yoon et al. also observed an unexpected amikacin susceptibility of the aphA6-carrying Acinetobacter guillouiae strains, and the authors correlated this phenotype with low level of gene expression (Yoon et al., 2014), which remains to be tested in the isolate KPC-142.

Table 2. Antimicrobial susceptibility profile of KPC-Kqps142.
Chromosomal Gene Content Comparisons among the Analyzed Klebsiella Strains
We compared the chromosomal gene content between Kqps142 and strains belonging to K. quasipneumoniae subsp. similipneumoniae (phylogroup II-B), K. quasipneumoniae subsp. quasipneumoniae (phylogroup II-A), K. pneumoniae (phylogroup I), and K. variicola (phylogroup III), supporting a panel of 36 strains representative of each Klebsiella phylogroup (Holt et al., 2015).
Proteinortho co-orthologous clustering analysis revealed a total of 1,049 core genes conserved in all 36 genomes (Table S2), whereas we found 1,597 genes that were shared by ≥ 97% of the genomes that composed our bacterial panel of strains belonging to phylogroups KpI, KpII, and KpIII. We identified a pangenome of 18,330 unique protein-coding sequences among the 36 analyzed Klebsiella genomes. These results are commensurable with those shown by Holt et al. (2015), who identified a pangenome of 29,886 unique protein-coding sequences among 328 K. pneumoniae genomes and revealed an open pangenome (Holt et al., 2015). Concerning the “exclusive” genes, a total of 9,795 genes had no orthologos among the 36 genomes (yielding a median of 272 “exclusive” genes per genome). Particularly, this comparative analysis revealed 181 “exclusive” genes in the genome of isolate Kqps142, most of which are genes of unknown function, or genes related to ICEPm1 and capsule biogenesis (wzb, wzc, wzx, and wzy) (Table S2).
Major chromosomal features, common or exclusive, among compared Klebsiella strains are depicted in Figure 4. Genome Atlas shows that Kqps142 harbors seven main regions of genomic plasticity in comparison to other 35 Klebsiella genomes, almost all of which with variations in G+C content (except R5). R3 presents the capsular polysaccharide biosynthesis genes, while R1, R2, R4, and R6 are composed by ORFs involved in phage integration. R5 includes the allantoin operon and R7 contains phage sequences and the integrative conjugative element ICEPm1.
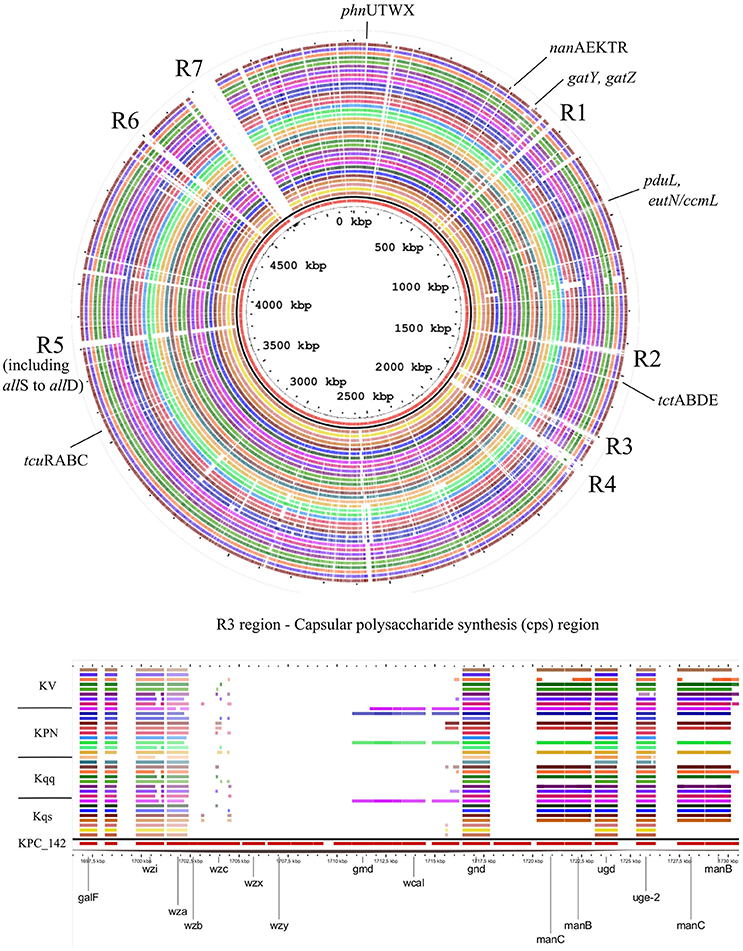
Figure 4. Genomic Atlas showing the common or exclusives chromosomal genes of Kqps142 (inner circle, in red), in comparison to: eight K. quasipneumoniae subsp. similipneumoniae strains (in order, inner to outer circles—HKUOPA4, HKUOPJ4, HKUOPL4, ATCC 700603, 07A044, MGH44, 193 KOXY, and 385 ECLO); nine K. quasipneumoniae subsp. quasipneumoniae (in order, inner to outer circles—UCICRE14, 01A030, FI_HV_2014, MGH96, 18A069, ARLG2711, PO1285, AKSD007, and 21_GR_13); nine K. pneumoniae (in order, inner to outer circles—DSM_30104, KP13, 1084, CAV1193, HS11286, KCTC-2242, KP617, MGH 78578, and NTUH-K2044); and nine K. variicola (in order, inner to outer circles–BZ19, GJ1, UCICRE10, MGH40, MGH20, UCI18, BIDMC61, BIDMC88, and MGH80). Comparison was obtained by Gview program, using BlastN (>70% identity and E-values < 10−5). Black line represents GC content. Regions containing interesting genes or features (named R1 to R7) are highlighted. R7 contains phage sequences and the integrative conjugative element ICEPm1. Genes of capsular polysaccharide synthesis are shown in details. Kqs, K. quasipneumoniae subsp. similipneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kpn, K. pneumoniae; and Kv, K. variicola.
Virulence genes identified in the 36 genomes from each Klebsiella phylogroup, including the isolate Kqps142, are shown in Figure 5. These included genes associated with the biosynthesis of siderophores, fimbriae, lipid A, capsule, or genes encoding for microcin, allantoinase, and the ferric uptake operon kfuABC, which have been reported as virulence factors of K. pneumoniae on the basis of murine models of infection (Paczosa and Mecsas, 2016). Most of these genes were detected in all Klebsiella phylogroups, since the majority of compared Klebsiella strains were isolated from human infections (Table 1). However, a few genes were identified in just one or two Klebsiella phylogroups, namely those genes encoding for the siderophore yersiniabactin (found in five K. pneumoniae and one K. variicola), capsular gmd gene (found in only one K. pneumoniae and three K. quasipneumoniae subsp. quasipneumoniae), rfbB gene (found only in six K. pneumoniae), and the N-acetyl-neuraminic acid gene cluster (found exclusively in all K. quasipneumoniae subsp. similipneumoniae) (see also Table 3).
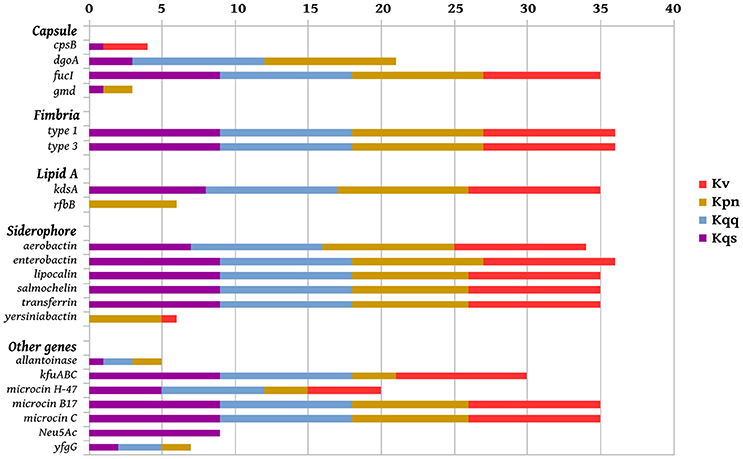
Figure 5. Virulence genes in Klebsiella strain panel, including Kqps142. Presence of the virulence genes per genome (from 0 to 9) of K. pneumoniae (Kpn), K. quasipneumoniae subsp. quasipneumoniae (Kqq), K. quasipneumoniae subsp. similipneumoniae (Kqs), and K. variicola (Kv).
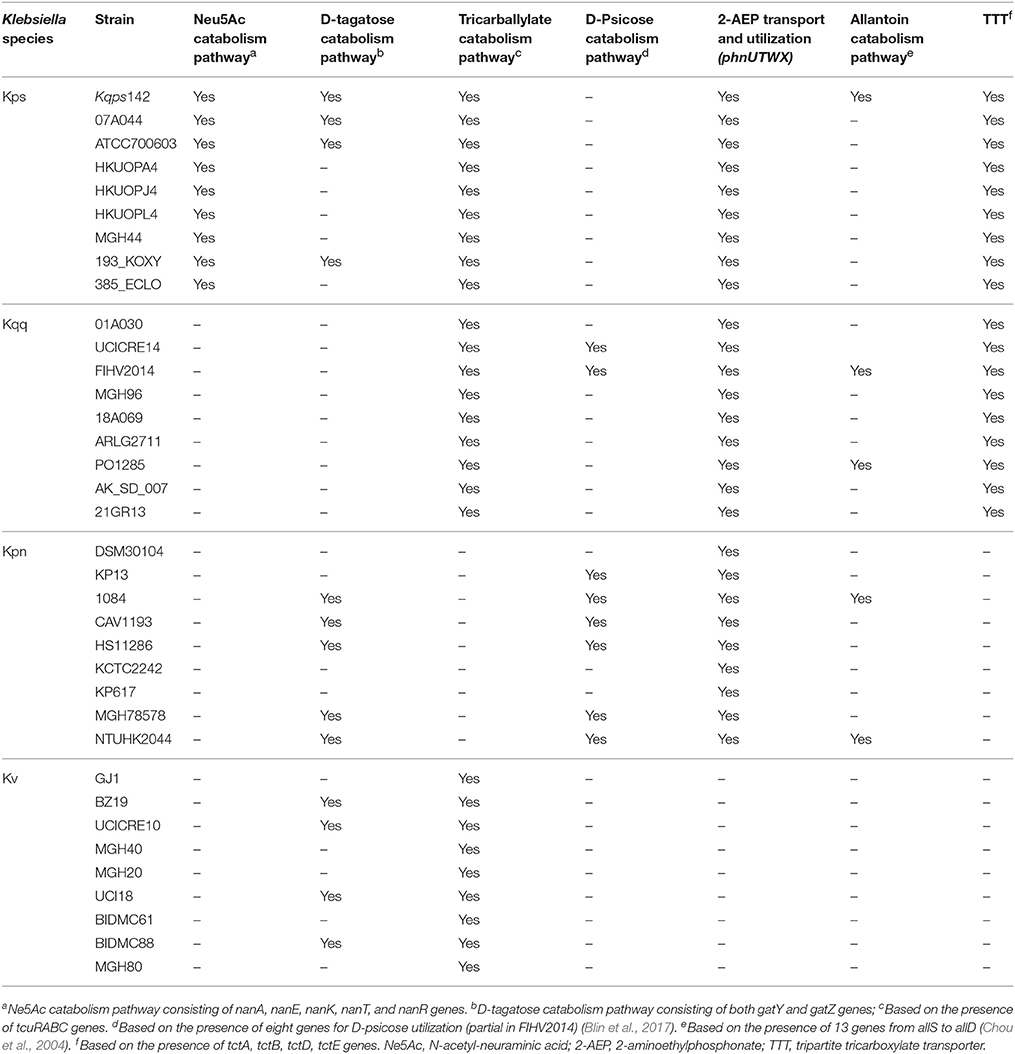
Table 3. Genomic characteristics of the K. quasipneumoniae subsp. similipneumoniae (Kqs), K. quasipneumoniae subsp. quasipneumoniae (Kqq), K. pneumoniae (Kpn), and K. variicola (Kv) strains, related to metabolism of carbon, nitrogen, and phosphorous sources and other genes.
Table 3 shows the gene content related to the most relevant metabolic pathways among 36 strains representative of each Klebsiella phylogroup, including the isolate Kqps142. Regarding the protein-encoding genes shared only by K. quasipneumoniae subsp. similipneumoniae strains are of special interest the N-acetyl-neuraminic acid gene cluster encompassing nanTEAR (KPC142_01465 to KPC142_01468) and nanK (KPC142_01464), which are shared by the nine analyzed strains of K. quasipneumoniae subsp. similipneumoniae (Table 3, Figure 4 and Table S2). Blin et al. (2017) showed that the ability to use N-acetyl-neuraminic acid (Neu5Ac) could be a phenotypic marker for K. quasipneumoniae subsp. similipneumoniae (Blin et al., 2017). Neu5Ac is especially abundant in the epithelial mucus of eukaryotic host and bacteria can scavenge this canonical sialic acid, for either catabolism and/or sialylation of their cell surface, which serves as a key determinant of pathogenesis (Haines-Menges et al., 2015) (see also Figure S4).
The metabolism of allantoin is used by bacteria as a source of carbon and nitrogen and is considered an important determinant of virulence in K. pneumoniae (Paczosa and Mecsas, 2016), since the operon genes from allS to allD are upregulated in hypervirulent (HV) K. pneumoniae strains compared to classical strains (Liu et al., 1986; Chou et al., 2004). The presence of allantoin operon genes among the 36 analyzed Klebsiella strains was very rare, limited only to Kqps142, K. quasipneumoniae subsp. quasipneumoniae strains FIHV2014 and PO1285 and K. pneumoniae strains 1084 and NTUH K2044 (Table 3 and Figure 4). Indeed, strain NTUH K2044 causes liver abscess and meningitis (Wu et al., 2009), strain FIHV2014 was described as a hypermucoviscosity-positive clinical isolate (Arena et al., 2015) and although 1084 was described as a hypermucoviscosity-negative clinical isolate it was capable of inducing liver abscesses in a murine model (Lin et al., 2011, 2012). On the other hand, while strains PO1285 (NCBI BioProject PRJNA351846) and Kqps142 are clinical isolates from human infections, their virulence in vivo remains to be investigated.
The genes involved in the metabolism of D-tagatose were not found among nine K. quasipneumoniae subsp. quasipneumoniae genomes and were rare among Kqq, Kqs, and Kpn strains (Table 3 and Figure 4). These genes encode for tagatose-bisphosphate aldolase subunit GatY (EC number 4.1.2.40) (KPC142_01383) and the subunit GatZ (KPC142_01387) (Table S2) that functions as a chaperone-like for the proper and stable folding of GatY (Brinkkötter et al., 2002). Fermentation and substrate assimilation tests were previously performed showing that K. quasipneumoniae subsp. similipneumoniae strains produce acid from D-tagatose and can grow on this sugar (Brisse et al., 2014; Blin et al., 2017).
The tcuRABC genes that encode functions needed for the utilization of tricarballylate as a carbon and energy source were absent only in the nine analyzed K. pneumoniae strains (Table 3 and Figure 4). These genes were described in the Salmonella enterica serovar Typhimurium LT2 (Lewis et al., 2004) and the utilization of tricarballylic acid was experimentally demonstrated as very rare in K. pneumoniae (Blin et al., 2017). A putative operon involved in the ability to use D-psicose was more frequent in the K. pneumoniae strains and it was also identified in two K. quasipneumoniae subp. quasipneumoniae genomes (UCICRE14 and FIHV2014, Table 3), which are in agreement with reported phenotypic findings (Blin et al., 2017).
An operon (phnUTWX) involved in the metabolism of 2-aminoethylphosphonate (2-AEP) as phosphorus source was only absent in the nine analyzed K. variicola strains (Table 3 and Figure 4). In Enterobacter aerogenes and Salmonella enterica subsp. enterica serovar Typhimurium, 2-AEP catabolism might be controlled as part of the Pho regulon and these bacteria can use 2-AEP as an alternative source of phosphate only under conditions of P limitation (Jiang et al., 1995), and in Pseudomonas putida str. NG2 and Pseudomonas aeruginosa str. PAO1 2-AEP can be mineralized as the sole C, N and P source (Ternan and Quinn, 1998; McGrath et al., 2013).
In bacteria, the tripartite tricarboxylate transporter (TTT) TctCAB is involved in the citrate uptake, which then can be utilized as a carbon and energy source by bacterial cell. The system was well-studied in Bordetella pertussis (Antoine et al., 2003), Salmonella typhimurium (Widenhorn et al., 1989), and Corynebacterium glutamicum, a nonpathogenic species that is of interest due to its biotechnological importance as a producer of L-glutamate and L-lysine (Brocker et al., 2009). Here, we found the tctCAB operon genes were restricted to strains belonging to K. quasipneumoniae species (Table 3 and Figure 4).
Also, protein-encoding genes involved in two pathways, i.e., 1,2- propanediol (1,2-PD) and ethanolamine (Eut) utilization were found in almost all compared strains (except for K. pneumoniae DSM 30104, Table S2 and Figure 4). Both the coenzyme B12-dependent catabolism of 1,2-PD (PduL EC: 2.3.1.222, KPC142_03505) and B12-dependent degradation of ethanolamine (eut operon) use the bacterial microcompartment (BMC) structures encoded by pduA (KPC142_03506) and eutN/ccmL (KPC142_03510), respectively. The BMC function is to optimize biochemical pathways by confining toxic by-products or volatile metabolic intermediates (Bobik et al., 2015). The ethanolamine and 1,2-PD degradations seem to be important to enteric pathogenesis (Thiennimitr et al., 2011) and their relevance in the pathogenesis or fitness of Klebsiella species still remains to be experimentally examined.
Conclusions
While the association of K. pneumoniae as responsible for infections in nosocomial settings is well-established, there are currently few reports associating K. quasipneumoniae as culprit in this context. Here, we reported the complete, closed genome of isolate K. quasipneumoniae subsp. similipneumoniae KPC-142, composed of one chromosome and two plasmids. To the best of our knowledge, this is the first complete genome of a clinical isolate of this subspecies harboring both beta-lactamases KPC-2 and OKP-B-6 responsible for a nosocomial infection from South America. The identification of multiple virulence and resistance factors coded in this genome, along with its drug resistance spectrum, reveals that, as in K. pneumoniae, this bacterium has great potential to establish infection and overcome the action of antibiotics currently used to treat these patients. The genomic characterization of K. quasipneumoniae strains will contribute to improve diagnostics and to the understanding of the epidemiological relevance of the dissemination of this Klebsiella species.
Author Contributions
CMA, DC, and RC provided the isolate Kqps142 and performed initial bacterial identification and susceptibility tests. Randomly Amplified Polymorphic DNA (RAPD) analysis was performed by RC and DC. ACV conceived the sequencing strategy. Complete genome assembly was carried out by LA and GL. Manual annotation and bioinformatic analyses were performed by ATR, FMC, LC, LA, MN, PPR, and RS. The manuscript was prepared by MN, PPR, FMC, and ATR. All authors read and approved the final manuscript.
Funding
This work was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (process no. 23038.010041/2013-13) through grant awarded to ATR.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Kary Ann del Carmen Ocaña Gautherot and Artur T. Lopo de Queiroz for help during the phylogenetic analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00220/full#supplementary-material
Table S1. Average nucleotide identity (ANI) comparisons of Klebsiella strain panel, including Kqps142. ANI was calculated using BLASTn (ANIb) in JSpeciesWS, using K. quasipneumoniae subsp. quasipneumoniae 01A030, K. quasipneumoniae subsp. similipneumoniae 07A044, K. pneumoniae subsp. pneumoniae DSM 30104, as reference type strains, in addition to strain K. variicola GJ1. An ANI threshold of ≥96% was considered to delineate species boundaries.
Table S2 (A). Total clusters of co-orthologous coding sequences generated from comparisons among 36 Klebsiella strains. (B) Clusters of co-orthologous coding sequences of core genes conserved in all 36 genomes. (C) List of “exclusive” genes in the genome of the isolate Kqps142. Comparisons were performed through Proteinortho co-orthologous clustering using BLASTp with an identity/coverage cutoff of >90% and E-values < 10.5.
Figure S1. RAPD analysis. Initially, 83 KPC isolates carrying the blaKPC gene were collected from nosocomial infections in Minas Gerais state (Southeast Brazil) in 2011. These isolates were preliminarily clustered by Randomly Amplified Polymorphic DNA (RAPD) with primer M13 (5′-GTAAAACGACGGCCAG-3′) using previously published reaction conditions (Wong et al., 1994). Seven clusters were disclosed and the isolate named 142, which displayed the most divergent DNA pattern by RAPD cluster analysis selected for sequencing analysis (Figure S1). Fragment sizes in RAPD fingerprints were estimated using GelAnalyzer (GelAnalyzer.com) and a distance matrix was calculated with MST (Coimbra et al., 2010). Distance tree was built with NEIGHBOR program within the PHYLIP package (Felsenstein, 1989) and drawn with MEGA 6.0 (Tamura et al., 2013). A red dot highlights the isolate KPC-142 selected for whole-genome sequencing.
Figure S2. An illustration of the assembly and gap-closing strategies. Gaps intra- and inter-scaffolds were resolved using an hybrid assembly strategy with the SPAdes and Newbler programs, with local assemblies in gap regions using Newbler.
Figure S3. Split decomposition analysis using the seven MLST alleles reveals a network-like structure, suggestive of recombination events.
Figure S4. Schematic representation of the utilization of N-acetyl-neuraminic acid (Neu5Ac) in a hypothetical K. quasipneumoniae subsp. similipneumoniae cell. The Kqs cell has potentially the ability to scavenge host Neu5Ac and use it for catabolism and/or sialylation of its cell surface structures, since it carries the gene encoding for a sialidase (nanH), a porin (nanT), sialic acid catabolism (SAC), and cell surface sialylation (Haines-Menges et al., 2015). The genome of Kqq, Kpn, and Kv strains carries nanH encoding for a sialidase and genes encoding for cell surface sialylation, but the genes encoding for SAC are absent, so these strains can potentially use Neu5Ac only for sialylation of their cell surface structures. Kqs, K. quasipneumoniae subsp. similipneumoniae; Kqq, K. quasipneumoniae subsp. quasipneumoniae; Kpn, K. pneumonia; and Kv, K. variicola
References
Achour-Rokbani, A., Cordi, A., Poupin, P., Bauda, P., and Billard, P. (2010). Characterization of the ars gene cluster from extremely arsenic-resistant Microbacterium sp. strain A33. Appl. Environ. Microbiol. 76, 948–955. doi: 10.1128/AEM.01738-09
Almeida, L. G. P., Paixão, R., Souza, R. C., da Costa, G. C., Barrientos, F. J. A., dos Santos, M. T., et al. (2004). A System for Automated Bacterial (genome) Integrated Annotation–SABIA. Bioinformatics 20, 2832–2833. doi: 10.1093/bioinformatics/bth273
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Alves, M. S., da Silva Dias, R. S., de Castro, A. C. D., Riley, L. W., and Moreira, B. M. (2006). Identification of clinical isolates of indole-positive and indole-negative Klebsiella spp. J. Clin. Microbiol. 44, 3640–3646. doi: 10.1128/JCM.00940-06
Antoine, R., Jacob-Dubuisson, F., Drobecq, H., Willery, E., Lesjean, S., and Locht, C. (2003). Overrepresentation of a gene family encoding extracytoplasmic solute receptors in Bordetella. J. Bacteriol. 185, 1470–1474. doi: 10.1128/JB.185.4.1470-1474.2003
Arena, F., Henrici De Angelis, L., Pieralli, F., Di Pilato, V., Giani, T., Torricelli, F., et al. (2015). Draft genome sequence of the first hypermucoviscous klebsiella quasipneumoniae subsp. quasipneumoniae Isolate from a bloodstream infection. Genome Announc. 3:e00952-15. doi: 10.1128/genomeA.00952-15
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Blin, C., Passet, V., Touchon, M., Rocha, E. P. C., and Brisse, S. (2017). Metabolic diversity of the emerging pathogenic lineages of Klebsiella pneumoniae. Environ. Microbiol. 19, 1881–1898. doi: 10.1111/1462-2920.13689
Bobenchik, A. M., Deak, E., Hindler, J. A., Charlton, C. L., and Humphries, R. M. (2017). Performance of Vitek 2 for antimicrobial susceptibility testing of Acinetobacter baumannii, Pseudomonas aeruginosa, and Stenotrophomonas maltophilia with Vitek 2 (2009 FDA) and CLSI M100S 26th edition breakpoints. J. Clin. Microbiol. 55, 450–456. doi: 10.1128/JCM.01859-16
Bobik, T. A., Lehman, B. P., and Yeates, T. O. (2015). Bacterial microcompartments: widespread prokaryotic organelles for isolation and optimization of metabolic pathways. Mol. Microbiol. 98, 193–207. doi: 10.1111/mmi.13117
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13:R56. doi: 10.1186/gb-2012-13-6-r56
Bowers, J. R., Lemmer, D., Sahl, J. W., Pearson, T., Driebe, E. M., Wojack, B., et al. (2016). KlebSeq, a diagnostic tool for surveillance, detection, and monitoring of Klebsiella pneumoniae. J. Clin. Microbiol. 54, 2582–2596. doi: 10.1128/JCM.00927-16
Brinkkötter, A., Shakeri-Garakani, A., and Lengeler, J. W. (2002). Two class II D-tagatose-bisphosphate aldolases from enteric bacteria. Arch. Microbiol. 177, 410–419. doi: 10.1007/s00203-002-0406-6
Brisse, S., Passet, V., and Grimont, P. A. D. (2014). Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int. J. Syst. Evol. Microbiol. 64, 3146–3152. doi: 10.1099/ijs.0.062737-0
Brisse, S., Passet, V., Haugaard, A. B., Babosan, A., Kassis-Chikhani, N., Struve, C., et al. (2013). wzi Gene sequencing, a rapid method for determination of capsular type for Klebsiella strains. J. Clin. Microbiol.. 51, 4073–4078. doi: 10.1128/JCM.01924-13
Brocker, M., Schaffer, S., Mack, C., and Bott, M. (2009). Citrate utilization by Corynebacterium glutamicum is controlled by the CitAB two-component system through positive regulation of the citrate transport genes citH and tctCBA. J. Bacteriol. 191, 3869–3880. doi: 10.1128/JB.00113-09
Brown, N. L., Barrett, S. R., Camakaris, J., Lee, B. T. O., and Rouch, D. A. (1995). Molecular genetics and transport analysis of the copper-resistance determinant (pco) from Escherichia coli plasmid pRJ1004. Mol. Microbiol. 17, 1153–1166. doi: 10.1111/j.1365-2958.1995.mmi_17061153.x
Bruen, T. C., Philippe, H., and Bryant, D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. doi: 10.1534/genetics.105.048975
Carattoli, A. (2011). Plasmids in Gram negatives: molecular typing of resistance plasmids. Int. J. Med. Microbiol. 301, 654–658. doi: 10.1016/j.ijmm.2011.09.003
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chou, H.-C., Lee, C.-Z., Ma, L.-C., Fang, C.-T., Chang, S.-C., and Wang, J.-T. (2004). Isolation of a chromosomal region of Klebsiella pneumoniae associated with allantoin metabolism and liver infection. Infect. Immun. 72, 3783–3792. doi: 10.1128/IAI.72.7.3783-3792.2004
Chung, Y. J., and Saier, M. H. (2002). Overexpression of the Escherichia coli sugE gene confers resistance to a narrow range of quaternary ammonium compounds. J. Bacteriol. 184, 2543–2545. doi: 10.1128/JB.184.9.2543-2545.2002
Coimbra, R. S., Artiguenave, F., Jacques, L. S. R. Z., and Oliveira, G. C. (2010). MST (molecular serotyping tool): a program for computer-assisted molecular identification of Escherichia coli and Shigella O antigens. J. Clin. Microbiol. 48, 1921–1923. doi: 10.1128/JCM.00357-10
Coimbra, R. S., Grimont, F., and Grimont, P. A. (1999). Identification of Shigella serotypes by restriction of amplified O-antigen gene cluster. Res. Microbiol. 150, 543–553. doi: 10.1016/S0923-2508(99)00103-5
Conlan, S., Park, M., Deming, C., Thomas, P. J., Young, A. C., Coleman, H., et al. (2016). Plasmid Dynamics in KPC-Positive Klebsiella pneumoniae during long-term patient colonization. MBio 7:e00742-16. doi: 10.1128/mBio.00742-16
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9:772. doi: 10.1038/nmeth.2109
Delcher, A. L., Harmon, D., Kasif, S., White, O., and Salzberg, S. L. (1999). Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27, 4636–4641. doi: 10.1093/nar/27.23.4636
Federhen, S., Rossello-Mora, R., Klenk, H.-P., Tindall, B. J., Konstantinidis, K. T., Whitman, W. B., et al. (2016). Meeting report: GenBank microbial genomic taxonomy workshop (12–13 May, 2015). Stand. Genomics Sci. 11:15. doi: 10.1186/s40793-016-0134-1
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Flannery, E. L., Antczak, S. M., and Mobley, H. L. T. (2011). Self-transmissibility of the integrative and conjugative element ICEPm1 between clinical isolates requires a functional integrase, relaxase, and type IV secretion system. J. Bacteriol. 193, 4104–4112. doi: 10.1128/JB.05119-11
Flannery, E. L., Mody, L., and Mobley, H. L. T. (2009). Identification of a modular pathogenicity island that is widespread among urease-producing uropathogens and shares features with a diverse group of mobile elements. Infect. Immun. 77, 4887–4894. doi: 10.1128/IAI.00705-09
Fonseca, E. L., da Veiga Ramos, N., Andrade, B. G. N., Morais, L. L. C. S., Marin, M. F. A., and Vicente, A. C. P. (2017). A one-step multiplex PCR to identify Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae in the clinical routine. Diagn. Microbiol. Infect. Dis. 87, 315–317. doi: 10.1016/j.diagmicrobio.2017.01.005
Garza-Ramos, U., Silva-Sánchez, J., Martínez-Romero, E., Tinoco, P., Pina-Gonzales, M., Barrios, H., et al. (2015). Development of a multiplex-PCR probe system for the proper identification of Klebsiella variicola. BMC Microbiol. 15:64. doi: 10.1186/s12866-015-0396-6
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Guindon, S., Delsuc, F., Dufayard, J.-F., and Gascuel, O. (2009). Estimating maximum likelihood phylogenies with PhyML. Methods Mol. Biol. 537, 113–137. doi: 10.1007/978-1-59745-251-9_6
Haeggman, S., Löfdahl, S., Paauw, A., Verhoef, J., and Brisse, S. (2004). Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 48, 2400–2408. doi: 10.1128/AAC.48.7.2400-2408.2004
Haines-Menges, B. L., Whitaker, W. B., Lubin, J. B., and Boyd, E. F. (2015). Host sialic acids: a delicacy for the pathogen with discerning taste. Microbiol. Spectr. 3. doi: 10.1128/microbiolspec.MBP-0005-2014
Hochhut, B., Marrero, J., and Waldor, M. K. (2000). Mobilization of plasmids and chromosomal DNA mediated by the SXT element, a constin found in Vibrio cholerae O139. J. Bacteriol. 182, 2043–2047. doi: 10.1128/JB.182.7.2043-2047.2000
Holt, K. E., Wertheim, H., Zadoks, R. N., Baker, S., Whitehouse, C. A., Dance, D., et al. (2015). Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. U.S.A. 112, E3574–E3581. doi: 10.1073/pnas.1501049112
Huson, D. H., and Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267. doi: 10.1093/molbev/msj030
Hussein, S., Hantke, K., and Braun, V. (1981). Citrate-dependent iron transport system in Escherichia coli K-12. Eur. J. Biochem. 117, 431–437. doi: 10.1111/j.1432-1033.1981.tb06357.x
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Jiang, W., Metcalf, W. W., Lee, K. S., and Wanner, B. L. (1995). Molecular cloning, mapping, and regulation of Pho regulon genes for phosphonate breakdown by the phosphonatase pathway of Salmonella typhimurium LT2. J. Bacteriol. 177, 6411–6421. doi: 10.1128/jb.177.22.6411-6421.1995
Kaplan, E., Sela, N., Doron-Faigenboim, A., Navon-Venezia, S., Jurkevitch, E., and Cytryn, E. (2015). Genomic and functional characterization of qnr-encoding plasmids from municipal wastewater biosolid Klebsiella pneumoniae isolates. Front. Microbiol. 6:1354. doi: 10.3389/fmicb.2015.01354
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H.-H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lähteenmäki, K., Kuusela, P., and Korhonen, T. K. (2001). Bacterial plasminogen activators and receptors. FEMS Microbiol. Rev. 25, 531–552. doi: 10.1111/j.1574-6976.2001.tb00590.x
Lechner, M., Findeiß, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (co-) orthologs in large-scale analysis. BMC Bioinformatics 12:124. doi: 10.1186/1471-2105-12-124
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Lewis, J. A., Horswill, A. R., Schwem, B. E., and Escalante-Semerena, J. C. (2004). The Tricarballylate utilization (tcuRABC) genes of Salmonella enterica serovar Typhimurium LT2. J. Bacteriol. 186, 1629–1637. doi: 10.1128/JB.186.6.1629-1637.2004
Lin, A.-C., Liao, T.-L., Lin, Y.-C., Lai, Y.-C., Lu, M.-C., and Chen, Y.-T. (2012). Complete genome sequence of Klebsiella pneumoniae 1084, a hypermucoviscosity-negative K1 clinical strain. J. Bacteriol. 194:6316. doi: 10.1128/JB.01548-12
Lin, Y.-C., Lu, M.-C., Tang, H.-L., Liu, H.-C., Chen, C.-H., Liu, K.-S., et al. (2011). Assessment of hypermucoviscosity as a virulence factor for experimental Klebsiella pneumoniae infections: comparative virulence analysis with hypermucoviscosity-negative strain. BMC Microbiol. 11:50. doi: 10.1186/1471-2180-11-50
Liu, B., and Pop, M. (2009). ARDB–Antibiotic Resistance Genes Database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, S., Dai, J., Kang, Z., Li, J., Chen, J., and Du, G. (2015). Production of novel NaN3-resistant creatine amidinohydrolase in recombinant Escherichia coli. Bioengineered 6, 248–250. doi: 10.1080/21655979.2015.1052919
Liu, Y. C., Cheng, D. L., and Lin, C. L. (1986). Klebsiella pneumoniae liver abscess associated with septic endophthalmitis. Arch. Intern. Med. 146, 1913–1916. doi: 10.1001/archinte.1986.00360220057011
Long, S. W., Linson, S. E., Ojeda Saavedra, M., Cantu, C., Davis, J. J., Brettin, T., et al. (2017). Whole-genome sequencing of human clinical Klebsiella pneumoniae isolates reveals misidentification and misunderstandings of Klebsiella pneumoniae, Klebsiella variicola, and Klebsiella quasipneumoniae. mSphere 2:e00290-17. doi: 10.1128/mSphereDirect.00290-17
Lowe, T. M., and Eddy, S. R. (1997). tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.0955
McGrath, J. W., Chin, J. P., and Quinn, J. P. (2013). Organophosphonates revealed: new insights into the microbial metabolism of ancient molecules. Nat. Rev. Microbiol. 11, 412–419. doi: 10.1038/nrmicro3011
Meyer, R. (2009). Replication and conjugative mobilization of broad host-range IncQ plasmids. Plasmid 62, 57–70. doi: 10.1016/j.plasmid.2009.05.001
Mollenkopf, D. F., Stull, J. W., Mathys, D. A., Bowman, A. S., Feicht, S. M., Grooters, S. V., et al. (2017). Carbapenemase-producing enterobacteriaceae recovered from the environment of a swine farrow-to-finish operation in the United States. Antimicrob. Agents Chemother. 61:e02348-16. doi: 10.1128/AAC.01298-16
Nicoletti, A. G., Marcondes, M. F. M., Martins, W. M. B. S., Almeida, L. G. P., Nicolás, M. F., Vasconcelos, A. T. R., et al. (2015). Characterization of BKC-1 class A carbapenemase from Klebsiella pneumoniae clinical isolates in Brazil. Antimicrob. Agents Chemother. 59, 5159–5164. doi: 10.1128/AAC.00158-15
Paczosa, M. K., and Mecsas, J. (2016). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/MMBR.00078-15
Petkau, A., Stuart-Edwards, M., Stothard, P., and Van Domselaar, G. (2010). Interactive microbial genome visualization with GView. Bioinformatics 26, 3125–3126. doi: 10.1093/bioinformatics/btq588
Poulin-Laprade, D., Carraro, N., and Burrus, V. (2015). The extended regulatory networks of SXT/R391 integrative and conjugative elements and IncA/C conjugative plasmids. Front. Microbiol. 6:837. doi: 10.3389/fmicb.2015.00837
Ramos, P. I. P., Picão, R. C., de Almeida, L. G. P., Lima, N. C. B., Girardello, R., Vivan, A. C. P., et al. (2014). Comparative analysis of the complete genome of KPC-2-producing Klebsiella pneumoniae Kp13 reveals remarkable genome plasticity and a wide repertoire of virulence and resistance mechanisms. BMC Genomics 15:54. doi: 10.1186/1471-2164-15-54
Randall, C. P., Gupta, A., Jackson, N., Busse, D., and O'Neill, A. J. (2015). Silver resistance in Gram-negative bacteria: a dissection of endogenous and exogenous mechanisms. J. Antimicrob. Chemother. 70, 1037–1046. doi: 10.1093/jac/dku523
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Scholz, P., Haring, V., Wittmann-Liebold, B., Ashman, K., Bagdasarian, M., and Scherzinger, E. (1989). Complete nucleotide sequence and gene organization of the broad-host-range plasmid RSF1010. Gene 75, 271–288. doi: 10.1016/0378-1119(89)90273-4
Shankar, C., Nabarro, L. E. B., Muthuirulandi Sethuvel, D. P., Raj, A., Devanga Ragupathi, N. K., Doss, G. P., et al. (2017). Draft genome of a hypervirulent Klebsiella quasipneumoniae subsp. similipneumoniae with novel sequence type ST2320 isolated from a chronic liver disease patient. J. Glob. Antimicrob. Resist. 9, 30–31. doi: 10.1016/j.jgar.2017.01.004
Souza, R. C., del Rosario Quispe Saji, G., Costa, M. O. C., Netto, D. S., Lima, N. C. B., Klein, C. C., et al. (2012). AtlasT4SS: a curated database for type IV secretion systems. BMC Microbiol. 12:172. doi: 10.1186/1471-2180-12-172
Sütterlin, S., Dahlö, M., Tellgren-Roth, C., Schaal, W., and Melhus, Å. (2017). High frequency of silver resistance genes in invasive isolates of Enterobacter and Klebsiella species. J. Hosp. Infect. 96, 256–261. doi: 10.1016/j.jhin.2017.04.017
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Taylor, D. E., Rooker, M., Keelan, M., Ng, L.-K., Martin, I., Perna, N. T., et al. (2002). Genomic variability of o islands encoding tellurite resistance in enterohemorrhagic Escherichia coli O157:H7 isolates. J. Bacteriol. 184, 4690–4698. doi: 10.1128/JB.184.17.4690-4698.2002
Ternan, N. G., and Quinn, J. P. (1998). Phosphate starvation-independent 2-aminoethylphosphonic acid biodegradation in a newly isolated strain of Pseudomonas putida, NG2. Syst. Appl. Microbiol. 21, 346–352. doi: 10.1016/S0723-2020(98)80043-X
Thiennimitr, P., Winter, S. E., Winter, M. G., Xavier, M. N., Tolstikov, V., Huseby, D. L., et al. (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U.SA. 108, 17480–17485. doi: 10.1073/pnas.1107857108
Torres, A., Ewig, S., Lode, H., Carlet, J., and The European HAP working group. (2009). Defining, treating and preventing hospital acquired pneumonia: European perspective. Intensive Care Med. 35, 9–29. doi: 10.1007/s00134-008-1336-9
Widenhorn, K. A., Somers, J. M., and Kay, W. W. (1989). Genetic regulation of the tricarboxylate transport operon (tctI) of Salmonella typhimurium. J. Bacteriol. 171, 4436–4441. doi: 10.1128/jb.171.8.4436-4441.1989
Wong, N. A., Linton, C. J., Jalal, H., and Millar, M. R. (1994). Randomly amplified polymorphic DNA typing: a useful tool for rapid epidemiological typing of Klebsiella pneumoniae. Epidemiol. Infect. 113, 445–454. doi: 10.1017/S095026880006845X
Wu, K.-M., Li, L.-H., Yan, J.-J., Tsao, N., Liao, T.-L., Tsai, H.-C., et al. (2009). Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J. Bacteriol. 191, 4492–4501. doi: 10.1128/JB.00315-09
Wyres, K. L., Wick, R. R., Gorrie, C., Jenney, A., Follador, R., Thomson, N. R., et al. (2016). Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genomics 2:e000102. doi: 10.1099/mgen.0.000102
Yoon, E.-J., Goussard, S., Touchon, M., Krizova, L., Cerqueira, G., Murphy, C., et al. (2014). Origin in Acinetobacter guillouiae and dissemination of the aminoglycoside-modifying enzyme Aph(3')-VI. MBio 5, e01972–e01914. doi: 10.1128/mBio.01972-14
Keywords: Klebsiella quasipneumoniae subsp similipneumoniae, KPC-2, OKP-B-6, silver resistance, nosocomial infection, complete genome sequence
Citation: Nicolás MF, Ramos PIP, Marques de Carvalho F, Camargo DRA, de Fátima Morais Alves C, Loss de Morais G, Almeida LGP, Souza RC, Ciapina LP, Vicente ACP, Coimbra RS and Ribeiro de Vasconcelos AT (2018) Comparative Genomic Analysis of a Clinical Isolate of Klebsiella quasipneumoniae subsp. similipneumoniae, a KPC-2 and OKP-B-6 Beta-Lactamases Producer Harboring Two Drug-Resistance Plasmids from Southeast Brazil. Front. Microbiol. 9:220. doi: 10.3389/fmicb.2018.00220
Received: 06 October 2017; Accepted: 30 January 2018;
Published: 16 February 2018.
Edited by:
Daniela Ceccarelli, Wageningen Bioveterinary Research (WBVR), NetherlandsReviewed by:
Elena Perrin, University of Florence, ItalyChristopher John Grim, United States Food and Drug Administration, United States
Copyright © 2018 Nicolás, Ramos, Marques de Carvalho, Camargo, de Fátima Morais Alves, Loss de Morais, Almeida, Souza, Ciapina, Vicente, Coimbra and Ribeiro de Vasconcelos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana T. Ribeiro de Vasconcelos, atrv@lncc.br
†These authors have contributed equally to this work.