 Tian-ye Zhang1,2Zi-qiong Wang1Hai-chao Hu2Zhi-qing Chen2Peng Liu2Shi-qi Gao2Fan Zhang2Long He2Peng Jin2Miao-ze Xu2Jian-ping Chen1,2*
Tian-ye Zhang1,2Zi-qiong Wang1Hai-chao Hu2Zhi-qing Chen2Peng Liu2Shi-qi Gao2Fan Zhang2Long He2Peng Jin2Miao-ze Xu2Jian-ping Chen1,2* Jian Yang2*
Jian Yang2*- 1College of Plant Protection, Nanjing Agricultural University, Nanjing, China
- 2State Key Laboratory for Quality and Safety of Agro-Products, Institute of Plant Virology, Ningbo University, Ningbo, China
N6-methyladenosine (m6A) methylation is the most prevalent internal modification of post-transcriptional modifications in mRNA, tRNA, miRNA, and long non-coding RNA in eukaryotes. m6A methylation has been proven to be involved in plant resistance to pathogens. However, there are no reports on wheat (Triticum aestivum) m6A transcriptome-wide map and its potential biological function in wheat resistance to wheat yellow mosaic virus (WYMV). To the best of our knowledge, this study is the first to determine the transcriptome-wide m6A profile of two wheat varieties with different resistances to WYMV. By analyzing m6A-sequencing (m6A-seq) data, we identified 25,752 common m6A peaks and 30,582 common m6A genes in two groups [WYMV-infected resistant wheat variety (WRV) and WYMV-infected sensitive wheat variety (WSV)], and all these peaks were mainly enriched in 3′ untranslated regions and stop codons of coding sequences. Gene Ontology analysis of m6A-seq and RNA-sequencing data revealed that genes that showed significant changes in both m6A and mRNA levels were associated with plant defense responses. Kyoto Encyclopedia of Genes and Genomes analysis revealed that these selected genes were enriched in the plant–pathogen interaction pathway. We further verified these changes in m6A and mRNA levels through gene-specific m6A real-time quantitative PCR (RT-qPCR) and normal RT-qPCR. This study highlights the role of m6A methylation in wheat resistance to WYMV, providing a solid basis for the potential functional role of m6A RNA methylation in wheat resistance to infection by RNA viruses.
Introduction
RNA molecules are crucial in all living organisms, playing significant roles in passing genetic information and regulating several biological processes (Fu et al., 2014). The structure of RNA transcripts may be adjusted through complex chemical modifications to perform specific molecular functions. To date, more than 100 types of RNA modifications have been identified. Among these post-transcriptional modifications, the most common RNA modifications are N6-methyladenosine (m6A), N5-methylcytosine, and N1-mythylcytosine (Cantara et al., 2011; Wei et al., 2017). However, m6A, which was first detected in the 1970s, is the most prevalent internal mRNA modification in eukaryotes, including mammals, plants, and yeast, as well as viruses (Wei et al., 1975). The m6A modification is induced by some m6A methyltransferases, called m6A “writers,” such as methyltransferase-like 3 and 14 (METTL3 and METTL14), as well as Wilms tumor 1-associated protein (WTAP), in mammalian cells (Fu et al., 2014; Liu et al., 2014; Ping et al., 2014). Furthermore, the m6A modification can be dynamically regulated by m6A demethylases, called m6A erasers, including fat mass and obesity-associated protein (FTO) and ALKB homolog 5 (ALKBH5), to maintain the m6A modification in a dynamic balance (Jia et al., 2011; Zheng et al., 2013; Liu et al., 2014). Another series of m6A-binding proteins (readers), such as YTHDF2 and YTHDF3, which belong to the YT521-B homology domain family, can bind specifically to m6A-modified cellular RNAs to carry out the biological function of methylation (Zhang et al., 2010; Dominissini et al., 2012; Xu et al., 2014, 2015). In general, the m6A modification can efficiently control gene expression, plant development, and physiological processes.
Through continuous research on the m6A modification, the most substantially enriched motif—RRACH (R = A/G, H = A/C/U)—has been authenticated through transcriptome-wide mapping of m6A in m6A peaks of all eukaryotes analyzed to date, consistent with early biochemical studies of eukaryote mRNA (Wei et al., 1976; Dimock and Stoltzfus, 1977; Schibler et al., 1977; Canaani et al., 1979; Nichols and Welder, 1981). Recently, another plant-specific consensus motif URUAY (R = A/G, Y = C/U) was identified using the m6A reader, ECT2 protein (Wei et al., 2018). The regulatory machinery of the m6A modification is post-transcriptionally assembled by the conserved methylation-related protease at the conserved consensus sequence, RRACH or URUAY. In plants, m6A is generally enriched in stop codons, start codons, and 3′-untranslated regions (UTRs), especially at the 3′-end of coding sequences (CDS) and the front end of 3′-UTRs (Dominissini et al., 2013; Schwartz et al., 2013, 2014). An increasing number of studies have shown that m6A can affect or regulate RNA expression and transcription in living cells under stress. For example, translation initiation for mammalian stress-responsive genes were promoted through direct binding of 5′-UTR m6A to eukaryotic initiation factor 3 (eIF3) and recruiting the 43S ribosomal complex in a cap-independent manner under the heat stress (Meyer et al., 2015). The intensity of 30% of m6A peaks is altered by ultraviolet light, heat shock, or interferon-gamma, thereby affecting gene expression and splicing (Dominissini et al., 2012). In METTL3 mutants, the translation of mRNAs with m6A modifications in the 5′-UTR is significantly reduced, and thus 5′-UTR m6A affects translation efficiency in cells (Meyer and Jaffrey, 2017). Increasing evidence indicates that m6A is also involved in regulating responses to abiotic stresses. In tobacco mosaic virus (TMV)-infected Nicotiana tabacum plants, the m6A level decreased, whereas ALKBH5-dependent m6A demethylation was promoted (Li et al., 2018). Indeed, alfalfa mosaic virus (AMV) infection induces the erasure of m6A in the viral genome and promotes systemic infection (Martínez-Pérez et al., 2017). With the development of high-throughput assays, profiling the m6A modification pattern on a transcriptome-wide scale has become possible. Currently, m6A-sequencing (m6A-seq) has been further developed, and transcriptome-wide mapping of m6A is now possible through m6A-RNA immunoprecipitation, followed by high-throughput sequencing (Dominissini et al., 2012; Meyer et al., 2012).
Wheat RNA viruses, especially wheat yellow mosaic virus (WYMV), cause a severe reduction in production in the major wheat-growing areas in China (Han et al., 2000; Liu et al., 2005). WYMV is one of the main pathogens of wheat soil-borne mosaic disease and belongs to the genus Bymovirus (Potyviridae) (Zhang et al., 2019). The WYMV genome contains two positive single RNA strands, RNA1 (7.5 kb) and RNA2 (3.6 kb) (Zhang et al., 2019). Wheat infected by WYMV show mosaic or yellow-striped leaves and plant stunting, and grain yield is reduced by 20–70% in some severely affected fields (Chen et al., 2014). In RNA viruses, including hepatitis C virus (HCV), Zika virus, dengue virus, and West Nile virus, the m6A modification has been identified in their genome (Gokhale et al., 2016; Courtney et al., 2017). Nevertheless, there have been very few studies on transcriptome-wide m6A methylome profiling of RNA viruses, especially WYMV, in different varieties of wheat. To the best of our knowledge, the present study is the first to perform transcriptome-wide m6A profiling in a WYMV-infected resistant wheat variety (yannong 999) (hereinafter referred to as WRV) and a WYMV-infected sensitive wheat variety(yannong 24) (hereinafter referred to as WSV), and found a significant variation in the m6A modification patterns between the resistant and susceptible varieties. Furthermore, different m6A RNA modifications in the two varieties demonstrated regulation of gene expression and pathogen-plant interaction-related pathways. Our study provides a basis for a comprehensive understanding of the roles of m6A modification in the molecular mechanisms underlying the interaction between wheat host and RNA viruses, especially WYMV.
Materials and Methods
Plant Materials
To obtain sufficient total RNA for IP of m6A-containing mRNA, ∼30 reviving stage plants each of yannong 999 and yannong 24 wheat infected or un-infected with WYMV were collected from a diseased nursery in Yantai city, Shandong province, China, respectively. WYMV-infected yannong 24 plants with typical yellow mosaic symptoms on the leaves, show stunted spring growth and reduced tillering. WYMV-infected yannong 999 plants show normal phenotypes without yellow mosaic symptoms (Supplementary Figure 3). Each wheat plants groups were equally divided into three mixed samples as three biological repeats respectively for RNA extraction and m6A IP sequence. These samples Stored at −80°C for RNA extraction and IP-qPCR and m6A IP sequence.
RNA Extraction and Fragmentation
Wheat leaves were collected, frozen, and stored at −80°C until use. Total RNA was extracted from plants using Trizol reagent (Invitrogen). NanoDrop ND-1000 (NanoDrop, Wilmington, DE, United States) was used to quantify the amount and purity of RNA in each sample. RNA integrity was investigated using a Bioanalyzer 2100 (Agilent, CA, United States) with RIN number >7.0, and further confirmed through electrophoresis with denaturing agarose gel. Then, Dynabeads Oligo (dT) 25–61005 (Thermo Fisher Scientific, CA, United States) was used to purify the poly (A) RNA from 150 μg total RNA. Finally, the poly(A) RNA was randomly fragmented into small pieces using a Magnesium RNA Fragmentation Module (cat. e6150, United States).
m6A IP and Library Construction
The total RNA was fragmented and then incubated with m6A-specific antibody (No. 202003, Synaptic Systems, Germany) in IP buffer (50 mM Tris–HCl, 750 mM NaCl, and 0.5% Igepal CA-630) for 2 h at 4°C. Then, the IP RNA was reverse-transcribed to cDNA, and U-labeled second-stranded DNAs were synthesized using cDNA with E. coli DNA polymerase I, RNase H (NEB, United States), and dUTP Solution (Thermo Fisher Scientific, United States). Then, the strands were prepared with A-base and ligated to the indexed adapters containing a T-base overhang for ligating the adapter to the A-tailed fragmented DNA. AMPureXP beads were used to screen fragments of the right size. The U-labeled second-stranded DNAs treated with heat-labile UDG enzyme (NEB, United States) were next amplified by PCR to generate a cDNA library with an average insert size of 300 ± 50 bp. Finally, 2 × 150 bp paired-end sequencing (PE150) was performed on an Illumina NovaseqTM 6000 (LC-BioTechnology Co., Ltd., Hangzhou, China), following the manufacturer’s recommended protocol.
Data Analysis
The raw data were processed by the online FASTP software1 to remove reads with adaptor contamination, low quality bases, and bases with undefined default parameters. HISAT2, an online software2 to map reads to the reference genome of Triticum aestivum (Version: IWGSC v1.0). Mapped reads of IP and input data were used for analysis using the R package exomePeak document3 then m6A intensity was visualized using Integrative Genomics Viewer (IGV) software4. MEME and HOMER56 were used for motif analysis. Called peaks were annotated by intersection with gene architecture using the R package ChIP seeker. The expression levels of all mRNAs from the input libraries were determined using StringTie7 by calculating FPKM [total exon fragment/mapped reads (millions) × exon length (kB)]. Differentially expressed genes were screened out using the criteria: fold change >1.5 or fold change <0.5, and p-value < 0.05 by the R package edgeR8.
Gene-Specific m6A qPCR and Normal qPCR
Total RNA was extracted and fragmented into 300-nucleotide-long fragments and incubated with anti-m6A antibody-coupled beads, while a portion of fragmented RNA was used as an input control. After ethanol precipitation, the input RNA and immunoprecipitated RNA were eluted from the beads. Both input control and m6A-IP samples were subjected to real-time quantitative PCR (RT-qPCR) with gene-specific primers. RT-PCR analysis was performed using an ABI Q5 Sequence Detection System (Applied Biosystems, CA, United States) with AceQ qPCR SYBR Green Master Mix (Vazyme, Nanjing, Jiangsu, China). At least, three biological replicates, with three technical replicates, were used for each assay. The T. aestivum cell division cycle (TaCDC) gene (Accession Number: XM_020313450) was used as the internal reference gene for analysis to calculate the fold changes in gene expression. The fold changes were calculated using the 2–Δ Δ C(t) method (Livak and Schmittgen, 2002). The calculation of specific mRNA fragment m6A levels was performed as previously described (Miao et al., 2020). In brief, relative enrichment of each fragment was calculated by first normalizing both the number of target cDNA fragment and the internal control, after which the value for the immunoprecipitated sample and the input was also normalized. And the input was used as the internal control for analysis to calculate the fold changes using the 2–ΔΔC(t). Statistical analyses were performed done using the Student’s t-test. Asterisks indicate a significant difference when compared to the control. ∗p < 0.05; ∗∗p < 0.01.
Results
Transcriptome-Wide Detection of the m6A Modification in WYMV-Infected Resistant Wheat Variety and WYMV-Infected Sensitive Wheat Variety
Leaf tissues from the WRV and the WSV were collected. Firstly, the accumulation of WYMV was detected in the total RNA extracted from WRV and WSV leaves by qRT-PCR assay using CP-specific primers. The results assay showed that the accumulation of WYMV was significantly increased in WSV than that in WRV (Supplementary Figure 1). And then these samples were used for transcriptome-wide m6A-seq and RNA-sequencing (RNA-seq) using the Illumina NovaseqTM 6000. A total of 129–139 million reads were acquired from the m6A-seq dataset, and 124–136 million reads were obtained from the RNA-seq dataset. After statistical analysis and quality control, there were still 100–135 million and 123–133 million valid reads in the m6A IP-seq and RNA-seq datasets, respectively. Indeed, the reads with a sequencing error rate <0.1% were more than 92% (Q30 > 92%), indicating that our data were clean (Supplementary Table 1). According to the wheat (T. aestivum) (Version: IWGSC v1.0) reference genome, more than 90% of the reads in the m6A IP-seq dataset were mapped. Moreover, almost 70% of the valid reads were uniquely mapped reads, similar to the results of a previous study on rice (Supplementary Table 2; Li et al., 2014).
The total m6A peaks (actually identified as m6A modification sites) of the two groups were identified by comparing the mapped reads of IP and input [two sequencing libraries, named m6A-seq library (IP) and RNA-seq library (Input)] using the R package, exomePeak. There were 45,067 m6A peaks in the WRV group and 37,718 m6A peaks in the WSV group. Moreover, the m6A peaks in the WRV and WSV groups represented transcripts of 35,993 and 35,649 genes, respectively (Figure 1A). Through the Venn diagram method, we found 25,752 common peaks representing 30,582 common genes that were m6A-modified in both groups (Figure 1A). Furthermore, there were 5411 genes and 5067 genes that underwent m6A modification in the WRV and WSV groups, respectively (Figure 1B). The distribution of m6A peaks in the whole transcriptome for these two groups was investigated according to gene annotations in the reference genome. The result showed that the vast majority of m6A peaks were centrally located near 3′-UTRs and stop codons of CDS (Figure 1C). The unique m6A peak distribution in both WRV and WSV groups are shown in Supplementary Figure 2. HOMER, a motif analysis software, was used to identify reliable motifs in the peak region in each group of samples. Two high consensus motifs, UGUAY and GAACU, were identified in the m6A peaks (Figure 1D), consistent with previous studies (Fu et al., 2014; Miao et al., 2020). Indeed, we analyzed the numbers of m6A-modified sites per m6A-modified gene and found that more than 99% of the genes (A: 37569/41127, B:33487/35534) had only one or two m6A-modified sites. However, genes with two m6A-modified sites in the WRV group (7.8%) were more than those in the WSV group (5.3%), and the WRV group unique m6A genes had more methylated sites than those of the WSV group (Figure 1E). In summary, these data indicated significant differences in the m6A modification patterns between the WRV and WSV groups.
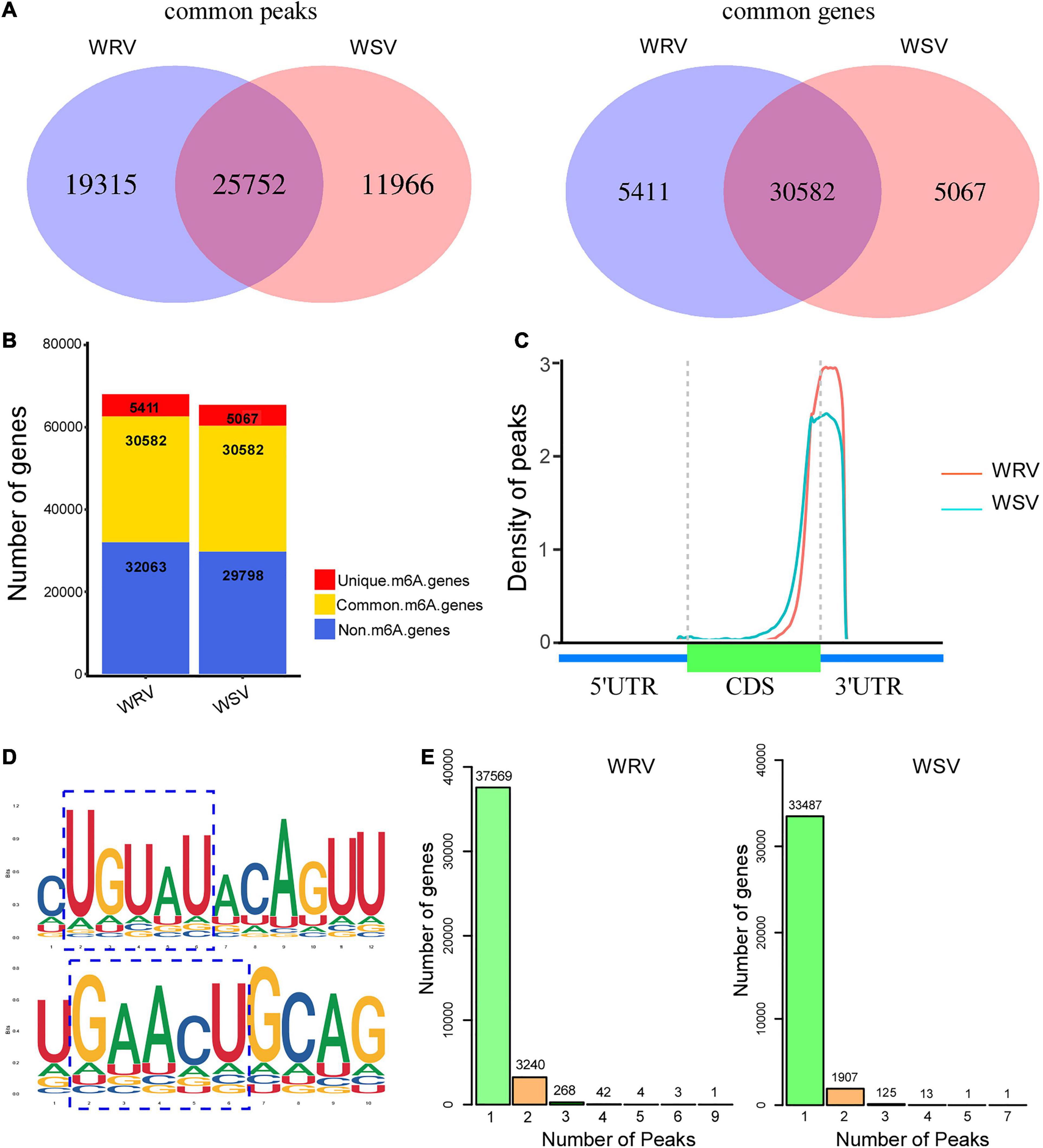
Figure 1. Overview of m6A methylome in two varieties of wheat and analysis of m6A peaks. (A) Left: numbers of WRV unique peaks and WSV unique and common peaks. Right: numbers of these peaks represent genes of the two groups. (B) Numbers of m6A-modified genes identified in m6A-seq data. (C) Density of m6A peaks along the wheat genome, which was divided into three non-overlapping regions, including 5′-UTR, CDS, and 3′-UTR. (D) Two top m6A motifs were enriched in all identified m6A peaks, RRACH and UGUAY (R is A/G, H is A/C/U, and Y is C/U). The blue dotted line box represented the conserved m6A motifs. (E) Numbers of m6A methylated genes with different numbers of m6A peaks in the two groups. WRV, WYMV-infected resistant wheat variety; WSV, WYMV-infected sensitive wheat variety.
Differentially Methylated Genes Are Enriched in Plant–Pathogen Interaction-Related Pathways
To determine the differentially methylated peaks between the WRV and WSV groups, the m6A-seq dataset of the WRV group was compared to that of the WSV group, and then 3261 differentially methylated sites were screened out, including 394 hyper-methylated m6A sites and 2867 hypo-methylated m6A sites (fold change >1.5, p < 0.05) (Figure 2A). Furthermore, we randomly selected two differentially methylated sites from the hyper-methylated and hypo-methylated site database. The genes corresponding to these two differentially methylated sites (hyper-methylated: TraesCS2B02G345000, hypo-methylated: TraesCS1D02G381400) showed altered methylation intensity, using the IGV software (Figure 2B).
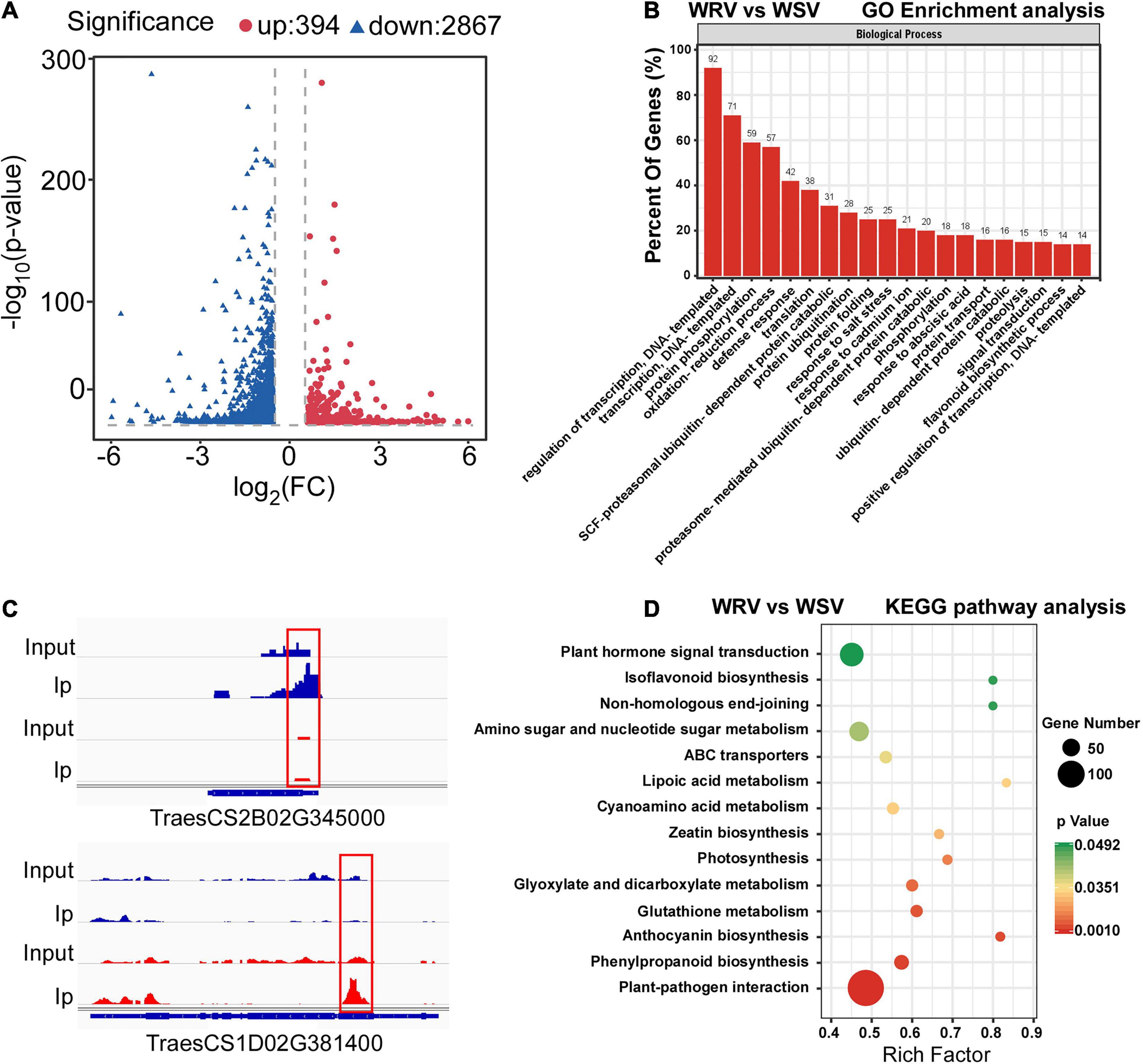
Figure 2. Global m6A modification changes in two WYMV-infected varieties and functional analysis of genes with m6A. (A) Identification of 394 hyper-methylated and 2867 hypo-methylated m6A peaks in the WRV group compared with the WSV group that showed a significant increase or decrease in m6A methylation (fold change >1.5, p < 0.05). (B) Examples of hyper-methylated and hypo-methylated genes with differential m6A peaks. The blue peaks represent the IP and Input reads from the WRV group, and the red peaks represent the IP and Input reads from the WSV group. The red boxes represent m6A peaks. (C) Gene Ontology analysis of biological processes of differentially methylated genes (DMGs). The vertical axis represents the proportion of DMGs enriched in each pathway. (D) KEGG pathway enrichment analysis of the DMGs. The horizontal axis represents the rich factor of each pathway.
To investigate the potential biological processes in which the m6A-modified genes are involved, Gene Ontology (GO) enrichment analysis of these differentially methylated genes (DMGs) was performed. The results showed that the DMGs were mainly enriched in DNA transcription, protein post-translational modification (PTM), stress response including DNA-templated transcription, protein phosphorylation, ubiquitin-dependent protein catabolic process and defense response, respectively (Figure 2C). To further predict the functions of these m6A-modified genes, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed, revealing that the majority of the DMGs were enriched in plant hormone signal transduction and plant–pathogen interaction pathways (Figure 2D). Collectively, these results demonstrated that the m6A modification may be involved in a variety of biological pathways, such as PTM and defense response, while being closely related to plant host and pathogen interaction.
Differentially Expressed Gene Analysis by RNA-Seq
To explore the potential relationship between m6A modification and gene expression, differentially expressed gene analysis was performed using the input sequencing data. Through hierarchical clustering of the RNA-seq data, we found obvious differential global mRNA expression patterns between the WRV and WSV groups (Figure 3A). We then screened the RNA-seq database and identified 8179 upregulated genes and 3577 downregulated genes in the WRV samples compared to the WSV samples (fold change >1.5, p < 0.05) (Figures 3B,C). Subsequently, these selected differentially expressed genes were used for GO enrichment and KEGG pathway analyses. The results showed that more than 70% of the differentially expressed genes were enriched in protein PTMs, such as protein phosphorylation and ubiquitination processes, while 54% were enriched in defense responses, including defense against fungi (Figure 3D). To further investigate which pathways the upregulated or downregulated genes participate in, these two types of genes were used for KEGG pathway analysis. The results revealed that the upregulated and downregulated genes were both enriched in the plant–pathogen interaction pathway, consistent with the pathogenic mechanism of the virus and the immune mechanism of the plant (Figures 3E,F; Mandadi and Scholthof, 2013; Wang, 2015). Therefore, we speculated that these differentially expressed genes may be affected by virus infection and may be involved in plant immunity.
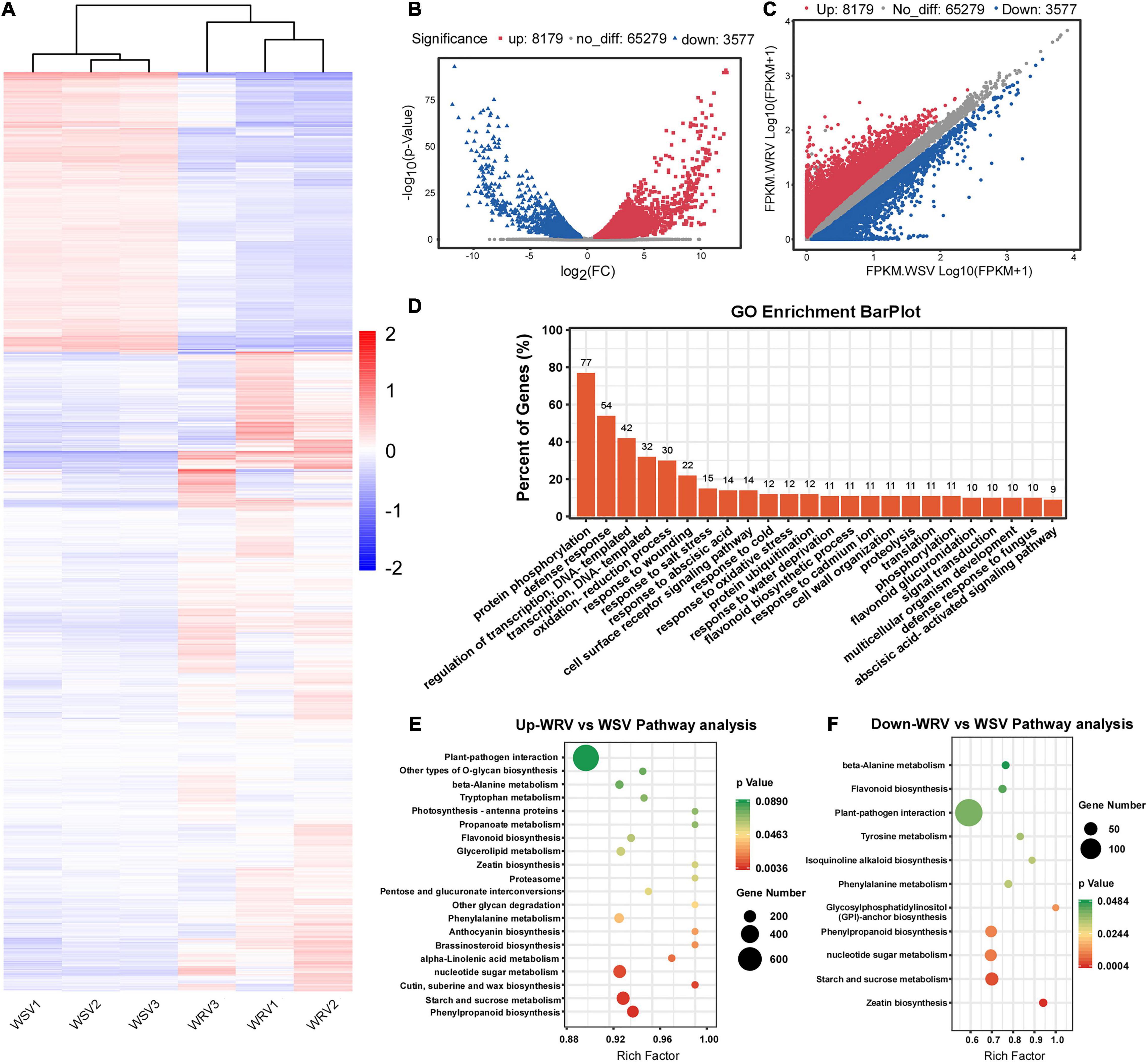
Figure 3. Overview of differentially expressed genes in the WRV group compared with the WSV group by RNA-seq and functional analysis of differentially expressed genes. (A) Heat map of RNA-seq data of WRV and WSV samples. Rows, mRNAs; columns, WRV samples and WSV samples. Red, white, and blue represent upregulation, unchanged expression, and downregulation of mRNA levels, respectively. (B) Volcano analysis of differentially expressed genes. (C) Scatter plot of differentially expressed genes. FPKM, fragments per kilobase of exon model per million mapped reads. (D) Gene Ontology analysis of the biological processes of differentially expressed genes. The vertical axis represents the proportion of differentially expressed genes enriched in each pathway. (E,F) KEGG pathway enrichment analysis of the upregulated and downregulated genes, respectively. The horizontal axis represents the rich factor of each pathway.
Association Analysis Between m6A-Seq and RNA-Seq
Previous studies have reported that m6A RNA modifications are related to gene expression (Luo et al., 2014; Lockhart, 2018). Therefore, we performed a cross-analysis of the m6A-seq and RNA-seq data (fold change >1.5, p < 0.05), and the results were displayed using a four-quadrant diagram, with the following four parts: hypo-up and hypo-down, and hyper-up and hyper-down, representing hypo/hyper m6A modifications causing gene expression up/down regulation, respectively. There were 729 differentially genes, among which 347 genes belonged to the hypo-up, 104 genes to the hyper-up, 228 genes to the hypo-down, and 50 genes to the hyper-down quadrants (Figure 4A). Furthermore, these selected genes were used for GO enrichment and KEGG pathway analyses (Figures 4B,C). Interestingly, 93% of the selected genes were related to plant defense response, while many of them were enriched in plant–pathogen interaction pathways, and some were involved in both plant–pathogen interaction and plant hormone signal transduction pathways. Next, the genes enriched in the two pathways were chosen for the four-quadrant diagram, and the majority of them had hypo m6A modifications. Among these hypo m6A-modified genes, 25 were upregulated at the mRNA level and 14 were downregulated (Figure 4D). In addition, 52 genes associated with these two pathways showed a significant difference in both m6A RNA methylation levels and mRNA expression levels (Table 1). The relationship between m6A RNA methylation level and mRNA expression level is presented more intuitively in Figure 4E, including the corresponding gene numbers. To further explore the interactions between these selected genes, we carried out protein interaction network analysis using the OmicStudio tool (Figure 4F). Many proteins were predicted to interact with SGT1 (TraesCS3D02G227500) protein, which has been reported to be associated with plant resistance to pathogens (Wang et al., 2015). Therefore, these genes and related pathways may have great significance in the protein–protein interaction network and molecular events in the viral infection process.

Figure 4. Conjoint analysis of m6A-seq and RNA-seq data. (A) A four-image map to analyze the relationship between differential genes and differential peaks and to screen out genes with a significant change in both m6A and mRNA levels in WRV samples compared with WSV samples (fold change >1.5, p < 0.05). (B) Gene Ontology analysis of biological processes of genes with a significant change in both m6A and mRNA levels. The vertical axis represents the proportion of selected genes enriched in each pathway. (C) KEGG pathway enrichment analysis of the genes with a significant change in both m6A and mRNA levels. The horizontal axis represents the rich factor of each pathway. (D) Distribution of genes with a significant change in both m6A and mRNA levels that were enriched in the plant–pathogen interaction pathway and plant hormone signal transduction pathway (fold change >1.5, p < 0.05). (E) Heat map and bar diagram of “hyper-up,” “hyper-down,” “hypo-up,” and “hypo-down” genes represented in D. Left, m6A-seq; right, RNA-seq. The red column represents genes enriched in both pathways, and blue column represents genes enriched only in plant–pathogen interaction pathway. Red, white, and blue represent upregulation, unchanged expression, and downregulation of mRNA level, respectively. (F) Graph of the protein interaction network of abnormally m6A-modified genes enriched in the plant–pathogen interaction pathway generated with the OmicStudio tool at https://www.omicstudio.cn/tool/56. Red and blue circles represent the source and target genes, respectively. The size of the circle represents the complexity of the interaction.
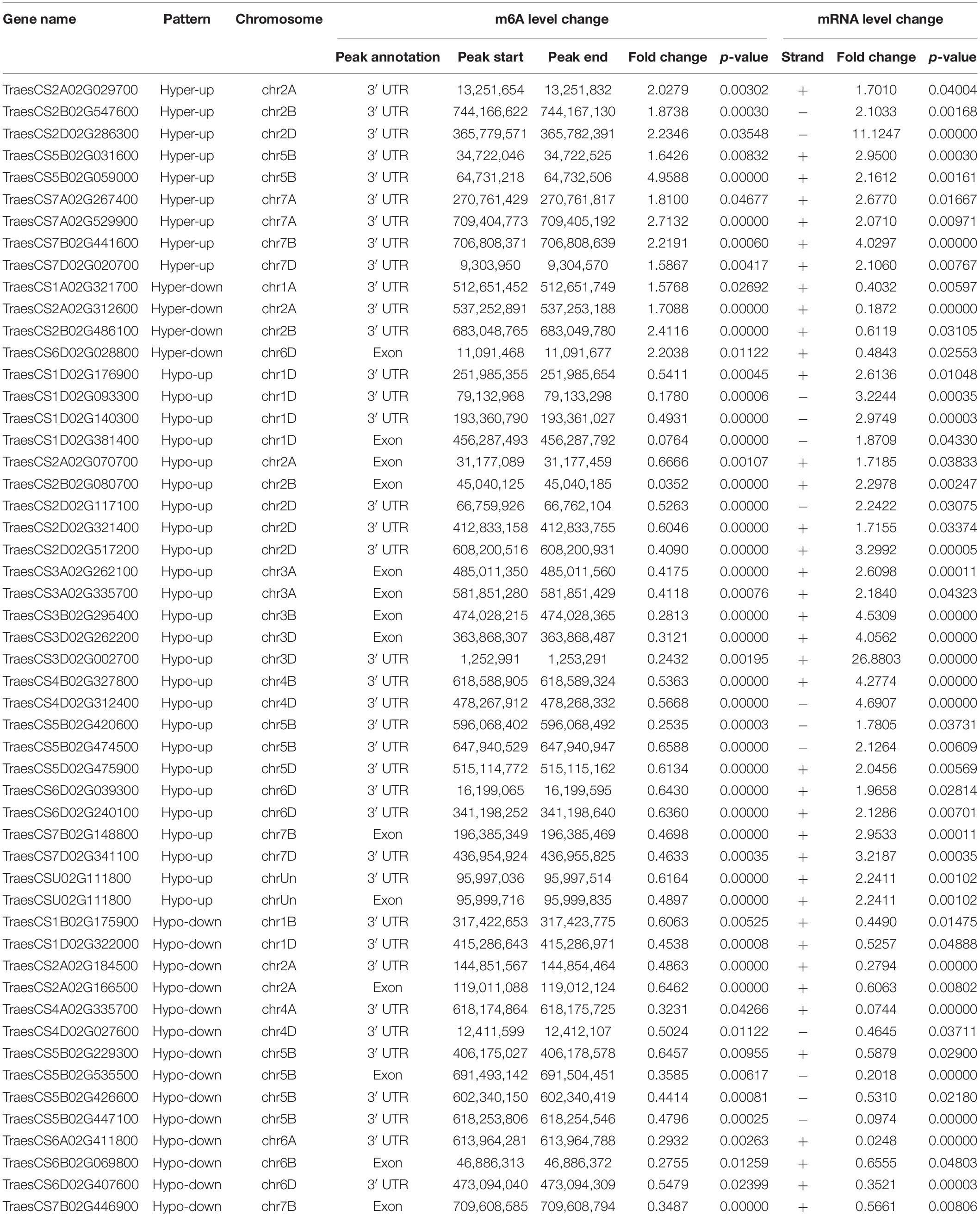
Table 1. List of 52 genes that exhibit a significant change in both m6A level and mRNA transcript abundance in WRV compared with WSV.
Gene-Specific m6A RT-qPCR Assay and Normal RT-qPCR
To further confirm the results of our m6A-seq data, eight genes (TraesCS7B02G446900, TraesCS7A02G267400, TraesCS5B02G474500, TraesCS5B02G447100, TraesCS7 D02G341100, TraesCS1D02G381400, TraesCS2A02G312600, and TraesCS1D02G176900) among the central proteins shown in the protein interaction network with complicated interaction relationship, and two gene (TraesCS1B02G175900 and TraesCS2D02G321400) related to both plant–pathogen interaction and plant hormone signal transduction pathways were randomly selected for gene-specific m6A qPCR and normal RT-qPCR. To eliminate the effect from the different genotypes of two wheat varieties to our result, we first detected the m6A levels and mRNA expression of these selected genes in two healthy wheat variety plants. Then the results displayed that most genes had no significant changes either in m6A levels or mRNA levels between two varieties (Figures 5A,B). Subsequently, we performed m6A qPCR and normal RT-qPCR assays for these genes in WRV and WSV group and the results showed that vast majority of these genes (8/10) exhibited similar m6A-level changes, consistent with the m6A-seq data and demonstrating the reliability of our m6A-seq data (Figure 5C). Furthermore, normal RT-qPCR of these genes in three pairs of WRV samples and WSV samples was performed, and the results showed the same tendency to RNA-seq data (Figure 5D). Therefore, these results confirmed that not only the m6A-levels but also the mRNA levels of most genes all had similar tendencies to our seq-data.
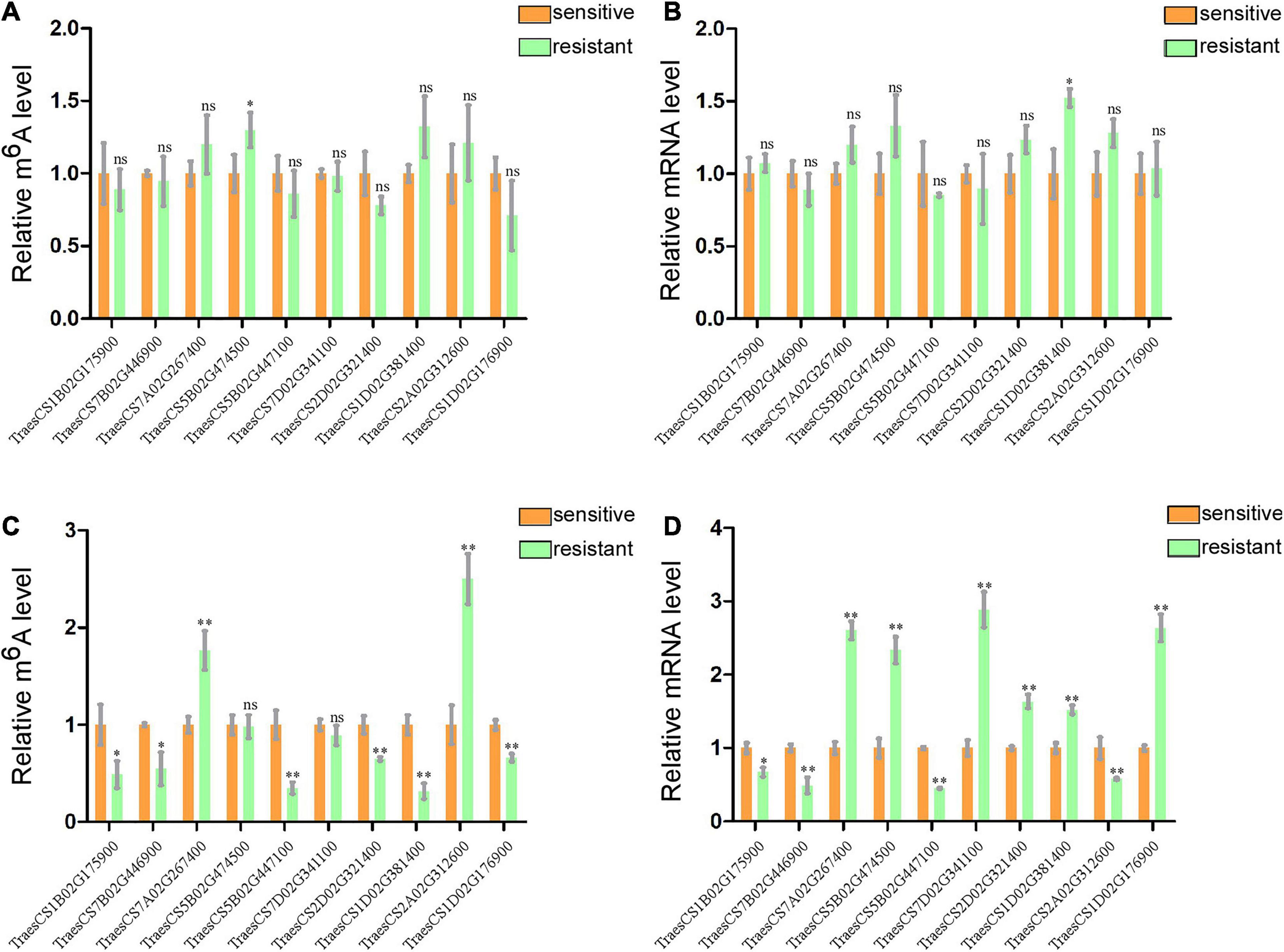
Figure 5. Gene-specific m6A/normal qPCR assays. (A) m6A IP qPCR validation of m6A methylation level of 10 randomly selected genes from Table 1 in two healthy wheat variety plants. (B) Relative mRNA levels of the 10 genes were detected via real-time qPCR in two healthy group plants. (C) m6A methylation level of 10 randomly selected genes in WRV and WSV group respectively. (D) Relative mRNA expression level of 10 genes in WRV and WSV group plants. The levels of the 10 genes in the sensitive variety group were normalized to 1. Statistical analyses were performed using the Student’s t-test. Asterisks indicate a significant difference when compared to the control. *p < 0.05; **p < 0.01.
Finally, we screened out many genes associated with plant resistance to pathogens through a series of bioinformatics analyses performed on the m6A-seq data and RNA-seq data. m6A qPCR also verified that eight of these genes indeed exhibited m6A methylation. In general, these results indicated that the differential m6A modifications between the two varieties may disturb these host-pathogen interaction pathways by regulating the expression of related genes, thereby affecting the virus infection process.
Discussion
N6-methyladenosine methylation is the most prevalent post-transcriptional modification, which is extensively present in mRNA, tRNA, miRNA, and long non-coding RNA, and plays important roles in plant responses to various biotic and abiotic stresses (Cantara et al., 2011; Wei et al., 2017; Arribas-Hernández and Brodersen, 2020). Previous studies have shown that RNA viruses, especially mammalian RNA viruses, infecting host plants are N6-methylated to affect viral replication and infection (Tan and Gao, 2018; Tan et al., 2018; Hao et al., 2019). Nevertheless, it remains unclear whether plant RNA viruses can be N6-methylated after infection and whether such methylation can induce variations in transcriptome-wide m6A modification patterns to influence virus infection. In this study, we, for the first time, illustrated different global m6A modification patterns in two wheat varieties, with different resistances to WYMV and provided new insights into m6A modification regulation during WYMV infection.
To date, a large number of m6A transcriptome-wide analyses in plants have been carried out, with the development of m6A-seq. It has been reported that the m6A modification is not randomly distributed in RNA transcripts, but is mainly distributed in 3′-UTRs and stop codons, especially near the end of the 3′-terminal of CDS and the front of 3′-UTRs in plants (Batista, 2017; Yang et al., 2018). In the present study, more than 90% of the m6A peaks were found in 3′-UTRs and the stop codons of CDS (Figure 1C). We found that two high consensus motifs, UGUAY and GAACU, were significantly enriched in the m6A peaks identified in wheat (Figure 1D), similar to the observation in other plant species, such as rice and maize, in some previous studies (Li et al., 2014; Du et al., 2020). These results further demonstrated the conserved features of m6A methylation in plants.
The m6A modification has been proven to be a significant RNA modification, playing a critical role in various steps of mRNA function, including mRNA stability, degradation, and expression (Luo et al., 2014; Visvanathan and Somasundaram, 2018). In addition, there is also evidence that the m6A modification is involved in the regulation of biotic stress response and that different stresses can cause a transcriptome-wide redistribution of m6A (Yue et al., 2019). For example, AMV infection increases m6A levels in Arabidopsis (Martínez-Pérez et al., 2017). In the present study, conjoint analysis of m6A-seq and mRNA-seq data identified 729 genes in WRV that showed differences in both m6A levels and mRNA expression levels (Figure 4A). Half of these genes were negatively correlated with m6A modification, and the other half showed a positive correlation, which may be due to the different m6A modification regions in transcripts. Luo et al. (2014) found that m6A modifications in 3′-UTR and 5′-UTR regions are positively correlated with gene expression, while m6A modifications in other regions result in lower gene expression in Arabidopsis. Moreover, these genes are involved in many biological processes, such as protein phosphorylation, defense response, and the abscisic acid (ABA)-activated signaling pathway (Figures 4B,C). According to a previous study, phosphorylation and ABA are both closely related to plant-virus interactions (Alazem and Lin, 2017). For example, the Barley stripe mosaic virus γb protein can suppress RNA silencing and host cell death response via PKA-mediated phosphorylation to promote infection (Zhang et al., 2018). Therefore, we speculated that the genes involved in these pathways may perform important functions in the wheat-WYMV interaction-related pathways and are worthy of further investigation. Furthermore, the gene-specific m6A RT-qPCR verified the different m6A methylation modifications in TraesCS7B02G446900, TraesCS7A02G267400, TraesCS1B02G175900, TraesCS5B02G474500, TraesCS5B02G 447100, TraesCS7D02G341100, TraesCS1D02G381400, TraesCS2A02G312600, TraesCS1D02G176900, and TraesCS 2D02G321400 between the WRV and WSV groups, which may be due to the abnormal expression of key m6A enzymes. We found that one gene (TraesCS4D02G261802) encoding a writer protein TaFIP37-1 and another gene (TraesCS4D02G261812) encoding an eraser protein TaALKBH29B both exhibited abnormal expression according to our RNA-seq data (Supplementary Table 3). We detected the mRNA expression of these two genes in two healthy wheat variety plants and WYMV-infected two wheat variety plants, the results showed no significant changes in health group but there are obvious changes in WYMV infected group (Supplementary Figure 4). Therefore, our results inferred that TaFIP37-1 and TaALKBH29B may be involved in the different m6A modifications in the WRV and WSV groups, but further experiments are needed to confirm this result.
Studies have reported that Oryza sativa glucose−regulated protein 94 (OsGRP94), a homologous gene of TraesCS7B02G4 46900.1, is downregulated under dithiothreitol -induced endoplasmic reticulum stress, and thus OsGRP94 may participate in ER stress-induced autophagy and programmed cell death (Cui et al., 2016). Indeed, the HCV E2 protein can upregulate GRP94 expression to inhibit the apoptosis induced by HCV infection and the host immune system (Lee et al., 2008). The TraesCS1B02G175900.1 gene encodes a cysteine-rich receptor-like protein kinase, the homologous gene of which has been reported to be related to wheat resistance to stripe rust fungus (Zhou et al., 2007). TraesCS7A02G267400.1 encodes PTI1-like tyrosine protein kinase 3, and its homologous gene, PTI1-like tyrosine-protein kinase 1, which has been identified as a putative candidate resistant gene for soil-borne wheat mosaic virus (SBWMV) (Liu et al., 2020).
Conclusion
In summary, the genes investigated in this study were all closely related to plant immunity and plant resistance to pathogens. Therefore, they can be used as candidate genes to explore the strategies of wheat resistance to viral infection, and to elucidate the mechanisms by which viruses successfully infect wheat. Further functional experiments are needed to verify the regulatory role of m6A RNA modifications in the expression of these candidate genes in plants against viral infection.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: National Center for Biotechnology Information (NCBI) BioProject, https://www.ncbi.nlm.nih.gov/bioproject/, PRJNA694346.
Author Contributions
JY and JC conceived the project and designed the experiments. TZ and JY carried out the experiments with assistance from ZW, ZC, HH, PL, SG, FZ, LH, MX, and PJ. JY, TZ, and JC wrote the manuscript. All authors analyzed and discussed the results.
Funding
This work was funded by the National Key R&D Plan in China (2018YFD0200408, 2018YFD0200507, and 2017YFD-0201701), National Natural Science Foundation of China (31672017 and 31901954), Public Projects of Ningbo City (202002N3004), Natural Science Foundation of Ningbo City (2019A610415 and 2019A610410), National Key Project for Research on Transgenic Biology (2016ZX08002-001), China Agriculture Research System from the Ministry of Agriculture of the P.R. China (CARS-03), and K.C. Wong Magna Funding in Ningbo University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank LC-BIOTECHNOLOGIES (HANGZHOU) CO., LTD., for the m6A sequencing service and bioinformatics support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2021.656302/full#supplementary-material
Supplementary Figure 1 | RT-qPCR assays of WYMV CP in WRV and WSV samples.
Supplementary Figure 2 | Unique m6A peak distribution in the indicated regions in WRV and WSV samples.
Supplementary Figure 3 | The phenotype of two variety plants infected with WYMV.
Supplementary Figure 4 | RT-qPCR assay of TaFIP37-1 and TaALKBH29B.
Supplementary Table 1 | Statistics and quality control of raw data generated by sequencing.
Supplementary Table 2 | Summary of the RNA-seq and m6A-seq of two variety.
Supplementary Table 3 | List of the expressions of key m6A enzymes according to RNA-seq data.
Supplementary Table 4 | The sequences of the primers used in these experiments.
Footnotes
- ^ https://github.com/OpenGene/fastp
- ^ http://daehwankimlab.github.io/hisat2
- ^ https://bioconductor.org/packages/exomePeak
- ^ http://www.igv.org
- ^ http://meme-suite.org
- ^ http://homer.ucsd.edu/homer/motif
- ^ https://ccb.jhu.edu/software/stringtie
- ^ https://bioconductor.org/packages/edgeR
References
Alazem, M., and Lin, N. S. (2017). Antiviral Roles of Abscisic Acid in Plants. Front. Plant Sci. 8:1760. doi: 10.3389/fpls.2017.01760
Arribas-Hernández, L., and Brodersen, P. (2020). Occurrence and Functions of m6A and Other Covalent Modifications in Plant mRNA. Plant Physiol. 182, 79–96. doi: 10.1104/pp.19.01156
Batista, P. J. (2017). The RNA Modification N6-methyladenosine and Its Implications in Human Disease. Genomics Proteom. Bioinformat. 15, 154–163. doi: 10.1016/j.gpb.2017.03.002
Canaani, D., Kahana, C., Lavi, S., and Groner, Y. (1979). Identification and mapping of N6-methyladenosine containing sequences in simian virus 40 RNA. Nucleic Acids Res. 6, 2879–2899. doi: 10.1093/nar/6.8.2879
Cantara, W. A., Crain, P. F., Rozenski, J., McCloskey, J. A., Harris, K. A., Zhang, X., et al. (2011). The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res. 39, D195–D201.
Chen, M., Sun, L., Wu, H., Chen, J., Ma, Y., Zhang, X., et al. (2014). Durable field resistance to wheat yellow mosaic virus in transgenic wheat containing the antisense virus polymerase gene. Plant Biotechnol. J. 12, 447–456. doi: 10.1111/pbi.12151
Courtney, D. G., Kennedy, E. M., Dumm, R. E., Bogerd, H. P., Tsai, K., Heaton, N. S., et al. (2017). Epitranscriptomic Enhancement of Influenza A Virus Gene Expression and Replication. Cell Host Microbe 22, 377.e–386.e.
Cui, J., Chen, B., Wang, H., Han, Y., Chen, X., and Zhang, W. (2016). Glucosidase II β-subunit, a novel substrate for caspase-3-like activity in rice, plays as a molecular switch between autophagy and programmed cell death. Sci. Rep. 6:31764.
Dimock, K., and Stoltzfus, C. M. (1977). Sequence specificity of internal methylation in B77 avian sarcoma virus RNA subunits. Biochemistry 16, 471–478. doi: 10.1021/bi00622a021
Dominissini, D., Moshitch-Moshkovitz, S., Salmon-Divon, M., Amariglio, N., and Rechavi, G. (2013). Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 8, 176–189. doi: 10.1038/nprot.2012.148
Dominissini, D., Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg, S., et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485:201. doi: 10.1038/nature11112
Du, X., Fang, T., Liu, Y., Wang, M., Zang, M., Huang, L., et al. (2020). Global profiling of N6 -methyladenosine methylation in maize callus induction. Plant Genome 13:e20018.
Fu, Y., Dominissini, D., Rechavi, G., and He, C. (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15:293. doi: 10.1038/nrg3724
Gokhale, N. S., McIntyre, A. B. R., McFadden, M. J., Roder, A. E., Kennedy, E. M., Gandara, J. A., et al. (2016). N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 20, 654–665. doi: 10.1016/j.chom.2016.09.015
Han, C., Li, D., Xing, Y., Zhu, K., Tian, Z., Cai, Z., et al. (2000). Wheat yellow mosaic virus Widely Occurring in Wheat (Triticum aestivum) in China. Plant Dis. 84, 627–630. doi: 10.1094/pdis.2000.84.6.627
Hao, H., Hao, S., Chen, H., Chen, Z., Zhang, Y., Wang, J., et al. (2019). N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res. 47, 362–374. doi: 10.1093/nar/gky1007
Jia, G., Fu, Y., Zhao, X., Dai, Q., Zheng, G., Yang, Y., et al. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chemical Biol. 7, 885–887. doi: 10.1038/nchembio.687
Lee, S. H., Song, R., Lee, M. N., Kim, C. S., Lee, H., Kong, Y. Y., et al. (2008). A molecular chaperone glucose-regulated protein 94 blocks apoptosis induced by virus infection. Hepatology 47, 854–866. doi: 10.1002/hep.22107
Li, Y., Wang, X., Li, C., Hu, S., Yu, J., and Song, S. (2014). Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 11, 1180–1188. doi: 10.4161/rna.36281
Li, Z., Shi, J., Yu, L., Zhao, X., Ran, L., Hu, D., et al. (2018). N 6 –methyl-adenosine level in Nicotiana tabacum is associated with tobacco mosaic virus. Virol. J. 15:87.
Liu, J., Yue, Y., Han, D., Wang, X., Fu, Y., Zhang, L., et al. (2014). A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chemical Biol. 10, 93–95. doi: 10.1038/nchembio.1432
Liu, S., Bai, G., Lin, M., Luo, M., Zhang, D., Jin, F., et al. (2020). Identification of candidate chromosome region of Sbwm1 for Soil-borne wheat mosaic virus resistance in wheat. Sci. Rep. 10:8119.
Liu, W., Nie, H., Wang, S., Li, X., He, Z., Han, C., et al. (2005). Mapping a resistance gene in wheat cultivar Yangfu 9311 to yellow mosaic virus, using microsatellite markers. TAG Theoret. Appl. Genet. 111, 651–657. doi: 10.1007/s00122-005-2012-x
Livak, K. J., and Schmittgen, T. D. (2002). Analysis of Relative Gene Expression Data using Real-Time Quantitative PCR. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lockhart, J. (2018). A Tale of Three Studies: Uncovering the Crucial Roles of m6A Readers. Plant Cell 30:947. doi: 10.1105/tpc.18.00352
Luo, G. Z., MacQueen, A., Zheng, G., Duan, H., Dore, L. C., Lu, Z., et al. (2014). Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5:5630.
Mandadi, K. K., and Scholthof, K. B. (2013). Plant immune responses against viruses: how does a virus cause disease? Plant Cell 25, 1489–1505. doi: 10.1105/tpc.113.111658
Martínez-Pérez, M., Aparicio, F., López-Gresa, M. P., Bellés, J. M., Sánchez-Navarro, J. A., and Pallás, V. (2017). Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. U S A. 114, 10755–10760. doi: 10.1073/pnas.1703139114
Meyer, K. D., and Jaffrey, S. R. (2017). Rethinking m6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 33:319. doi: 10.1146/annurev-cellbio-100616-060758
Meyer, K. D., Patil, D. P., Zhou, J., Zinoviev, A., Skabkin, M. A., Elemento, O., et al. (2015). 5’ UTR m6A Promotes Cap-Independent Translation. Cell 163, 999–1010. doi: 10.1016/j.cell.2015.10.012
Meyer, K. D., Saletore, Y., Zumbo, P., Elemento, O., Mason, C. E., and Jaffrey, S. R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646. doi: 10.1016/j.cell.2012.05.003
Miao, Z., Zhang, T., Qi, Y., and Song, J. (2020). Evolution of the RNA N (6)-Methyladenosine Methylome Mediated by Genomic Duplication. Plant Physiol. 182, 345–360. doi: 10.1104/pp.19.00323
Nichols, J. L., and Welder, L. (1981). Nucleotides adjacent to N6-methyladenosine in maize poly(A)-containing RNA. Plant Sci. Lett. 21, 75–81. doi: 10.1016/0304-4211(81)90071-7
Ping, X.-L., Sun, B.-F., Wang, L., Xiao, W., Yang, X., Wang, W.-J., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. doi: 10.1038/cr.2014.3
Schibler, U., Kelley, D. E., and Perry, R. P. (1977). Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol. 115, 695–714. doi: 10.1016/0022-2836(77)90110-3
Schwartz, S., Agarwala, S. D., Mumbach, M. R., and Jovanovic, M. (2013). High-Resolution Mapping Reveals a Conserved, Widespread, Dynamic mRNA Methylation Program in Yeast Meiosis. Cell 155, 1409–1421. doi: 10.1016/j.cell.2013.10.047
Schwartz, S., Mumbach, M., Jovanovic, M., Wang, T., Maciag, K., Bushkin, G. G., et al. (2014). Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5’ Sites. Cell Rep. 8, 284–296. doi: 10.1016/j.celrep.2014.05.048
Tan, B., and Gao, S. J. (2018). RNA epitranscriptomics: Regulation of infection of RNA and DNA viruses by N(6) -methyladenosine (m6A). Rev. Med. Virol. 28:e1983. doi: 10.1002/rmv.1983
Tan, B., Liu, H., Zhang, S., da Silva, S. R., Zhang, L., Meng, J., et al. (2018). Viral and cellular N(6)-methyladenosine and N(6),2’-O-dimethyladenosine epitranscriptomes in the KSHV life cycle. Nat. Microbiol. 3, 108–120. doi: 10.1038/s41564-017-0056-8
Visvanathan, A., and Somasundaram, K. (2018). mRNA Traffic Control Reviewed: N6-Methyladenosine (m(6) A) Takes the Driver’s Seat. BioEssays News Rev. Mol. Cell. Dev. Biol. 40:1700093. doi: 10.1002/bies.201700093
Wang, A. (2015). Dissecting the molecular network of virus-plant interactions: the complex roles of host factors. Annu. Rev. Phytopathol. 53, 45–66. doi: 10.1146/annurev-phyto-080614-120001
Wang, G. F., Fan, R., Wang, X., Wang, D., and Zhang, X. (2015). TaRAR1 and TaSGT1 associate with TaHsp90 to function in bread wheat (Triticum aestivum L.) seedling growth and stripe rust resistance. Plant Mol. Biol. 87, 577–589. doi: 10.1007/s11103-015-0298-x
Wei, C. M., Gershowitz, A., and Moss, B. et al. (1976). 5’-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry 15, 397–401. doi: 10.1021/bi00647a024
Wei, C.-M., Gershowitz, A., and Moss, B. (1975). Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 4, 379–386.
Wei, L. H., Song, P., Wang, Y., Lu, Z., Tang, Q., Yu, Q., et al. (2018). The m6A Reader ECT2 Controls Trichome Morphology by Affecting mRNA Stability in Arabidopsis. Plant Cell 30, 968–985. doi: 10.1105/tpc.17.00934
Wei, W., Ji, X., Guo, X., and Ji, S. (2017). Regulatory Role of N6−methyladenosine (m6A) methylation in RNA processing and human diseases. J. Cell. Biochem. 118, 2534–2543. doi: 10.1002/jcb.25967
Xu, C., Liu, K., Ahmed, H., Loppnau, P., Schapira, M., and Min, J. (2015). Structural Basis for the Discriminative Recognition of N6-Methyladenosine RNA by the Human YT521-B Homology Domain Family of Proteins. J. Biol. Chem. 290, 24902–24913. doi: 10.1074/jbc.m115.680389
Xu, C., Wang, X., Liu, K., Roundtree, I. A., Tempel, W., and Li, Y et al. (2014). Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chemical Biol. 10, 927–929. doi: 10.1038/nchembio.1654
Yang, Y., Hsu, P. J., Chen, Y. S., and Yang, Y. G. (2018). Dynamic transcriptomic m(6)A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 28, 616–624. doi: 10.1038/s41422-018-0040-8
Yue, H., Nie, X., Yan, Z., and Weining, S. (2019). N6-methyladenosine regulatory machinery in plants: composition, function and evolution. Plant Biotechnol. J. 17, 1194–1208. doi: 10.1111/pbi.13149
Zhang, T., Liu, P., Zhong, K., Zhang, F., Xu, M., He, L., et al. (2019). Wheat Yellow Mosaic Virus NIb Interacting with Host Light Induced Protein (LIP) Facilitates Its Infection through Perturbing the Abscisic Acid Pathway in Wheat. Biology 8:80. doi: 10.3390/biology8040080
Zhang, X., Dong, K., Xu, K., Zhang, K., Jin, X., Yang, M., et al. (2018). Barley stripe mosaic virus infection requires PKA-mediated phosphorylation of γb for suppression of both RNA silencing and the host cell death response. New Phytol. 218, 1570–1585. doi: 10.1111/nph.15065
Zhang, Z., Theler, D., Kaminska, K. H., Hiller, M., de la Grange, P., Pudimat, R., et al. (2010). The YTH domain is a novel RNA binding domain. J. Biol. Chem. 285, 14701–14710.
Zheng, G., Dahl, J. A., Niu, Y., Fedorcsak, P., Huang, C.-M., Li, C. J., et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29. doi: 10.1016/j.molcel.2012.10.015
Keywords: wheat, m6A methylation, m6A-seq, RNA-seq, WYMV, plant-pathogen interacting, transcriptional regulation
Citation: Zhang T-y, Wang Z-q, Hu H-c, Chen Z-q, Liu P, Gao S-q, Zhang F, He L, Jin P, Xu M-z, Chen J-p and Yang J (2021) Transcriptome-Wide N6-Methyladenosine (m6A) Profiling of Susceptible and Resistant Wheat Varieties Reveals the Involvement of Variety-Specific m6A Modification Involved in Virus-Host Interaction Pathways. Front. Microbiol. 12:656302. doi: 10.3389/fmicb.2021.656302
Received: 21 January 2021; Accepted: 29 April 2021;
Published: 26 May 2021.
Edited by:
Dragan Perovic, Julius Kühn-Institut, GermanyReviewed by:
Jinhong Kan, Institute of Crop Sciences, Chinese Academy of Agricultural Sciences, ChinaWanxin Chen, Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Germany
Copyright © 2021 Zhang, Wang, Hu, Chen, Liu, Gao, Zhang, He, Jin, Xu, Chen and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-ping Chen, jpchen2001@126.com; Jian Yang, nather2008@163.com