Mitochondrial ATP Synthase is a Target of Oxidative Stress in Neurodegenerative Diseases
 Brad Ebanks
Brad Ebanks Lisa Chakrabarti
Lisa Chakrabarti- 1School of Veterinary Medicine and Science, University of Nottingham, Nottingham, United Kingdom
- 2MRC Versus Arthritis Centre for Musculoskeletal Ageing Research, Nottingham, United Kingdom
The mitochondrial ATP synthase is responsible for the production of cellular ATP, and it does so by harnessing the membrane potential of the mitochondria that is produced by the sequential oxidation of select cellular metabolites. Since the structural features of ATP synthase were first resolved nearly three decades ago, significant progress has been made in understanding its role in health and disease. Mitochondrial dysfunction is common to neurodegeneration, with elevated oxidative stress a hallmark of this dysfunction. The patterns of this oxidative stress, including molecular targets and the form of oxidative modification, can vary widely. In this mini review we discuss the oxidative modifications of ATP synthase that have been observed in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. Oxidative modifications of ATP synthase in Alzheimer’s disease are well-documented, and there is a growing body of knowledge on the subject in Parkinson’s disease. The consideration of ATP synthase as a pharmacological target in a variety of diseases underlines the importance of understanding these modifications, both as a potential target, and also as inhibitors of any pharmacological intervention.
Introduction
Neurodegenerative Diseases
Neurodegeneration is the process of progressive atrophy and loss of functional neurons in the central nervous system, and in particular the brain. The neurodegenerative process is not uniform, distinctions between which brain regions undergo atrophy, and by what molecular mechanisms the atrophy can be characterised, lead to the different neurodegenerative diseases that we now recognise (Damier et al., 1999; Sabuncu, 2011; Dugger and Dickson, 2017).
The most prevalent neurodegenerative disease is Alzheimer’s disease (AD), a form of dementia that is lived with by over 20 million people around the world, and in 2016 was the fifth largest cause of death at 2.4 million people (Nichols, 2019). As global population demographics shift towards older ages, the prevalence of the disease will increase, creating a huge health and social care as well as an economic burden (Castro et al., 2010; Marasco, 2020). The pathophysiology of AD is best characterised by three main characteristics: the aggregation of the amyloid beta (Aβ) protein into plaques (Selkoe and Hardy, 2016; Makin, 2018), the formation of neurofibrillary tangles by the tau protein (Braak and Braak, 1991; Gao et al., 2018), and the dysfunction of mitochondria (Moreira et al., 2006; Swerdlow, 2018), the cell’s bioenergetic and signalling hubs.
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease seen today. Characterised by classical motor symptoms of resting tremor and bradykinesia, much of this is due to the degeneration of the dopaminergic neurons of the substantia nigra (Hirsch et al., 1988). There are many genetic risk factors for PD, such as mutations to the SNCA gene that lead to the formation of α-synuclein aggregates known as Lewy bodies (Schulz-Schaeffer, 2010; Mahul-Mellier et al., 2020), as well as mutations to the PINK1 and parkin genes that lead to failed mitophagy (the managed intracellular degradation of dysfunctional mitochondria), neuronal defects and even apoptosis (Goldberg et al., 2003; Palacino et al., 2004; Wood-Kaczmar et al., 2008).
Other less frequent forms of neurodegenerative disease include amyotrophic lateral sclerosis (ALS), ataxia, and Huntington’s disease. Huntington’s disease (HD) is caused by an excessive number of CAG repeats in the Htt gene, resulting in PolyQ repeats near the amino terminus. Accumulation of the mutant Huntingtin protein, as well as protein fragments, leads to the formation of toxic inclusion bodies that impact cell physiology, in particular proteostatic function (Bates et al., 2015). Mitochondrial dysfunction in the form of impaired complex II activity and decreased membrane potential have also been widely discussed in HD (Damiano et al., 2010). Atrophy of the medium spiny GABAergic neurons of the striatum is the classical neurodegenerative process observed in the disease (Rikani, 2014), and the major clinical presentations include involuntary movements and bradykinesia (Roos, 2010). With higher prevalence in people of European heritage, it is shown that around one in 7,300 from this demographic are affected (Bates et al., 2015).
Redox Stress in Ageing and Neurodegenerative Disease
One common feature of neurodegenerative disease is the observation of high levels of oxidative stress within the cells of affected tissues (Lin and Beal, 2006; Smith and Cass, 2007; Sorolla, 2008; Wang, 2014; Liguori, 2018). This is in a large part due to the dysfunction of mitochondria that seems ubiquitous in these conditions. Mitochondria are the primary consumers of oxygen, they use molecular oxygen as the terminal electron acceptor at complex IV (cytochrome c oxidase) of the electron transport chain, reducing it to water (Kadenbach, 2021). This flow of electrons, provided by oxidised metabolites, facilitates the pumping of protons across the inner mitochondrial membrane and into the inter membrane space, generating a proton gradient. The activity of complexes I–IV of the electron transport chain is coupled to the activity of the enzyme ATP synthase, which harnesses the flow of protons from the inter membrane space back into the mitochondrial matrix to drive its rotary function, which ultimately provides the energy for the enzyme’s ATP synthesising catalytic activity (Gnaiger, 2020).
However, variations in this collective process leads to the production of oxygen based free radicals that are thought to be important signalling molecules (Zhang, 2016; Bárcena et al., 2018), but that can be harmful in excess (Perry et al., 1998; Redza-Dutordoir and Averill-Bates, 2016; Nissanka and Moraes, 2018). The endogenous production of free radicals largely occurs due to the leak or slip of electrons from complexes I and III, before their arrival at the catalytic site of complex IV, producing reactive oxygen species such as superoxide (O2.) (Chen et al., 2003; Balaban et al., 2005; Hirst et al., 2008).
Forms of Protein Oxidation
The oxygen based free radicals such as superoxide and the hydroxyl radical, if not detoxified by antioxidants such as superoxide dismutase and catalase, go on to produce a variety of downstream radical species. In turn these species can modify the structure and function of proteins within the cell, and ultimately alter cellular physiology. While the radicals are capable of interacting with proteins directly (Stadtman and Levine, 2006), they are also known to lead to the modification of proteins through lipid peroxidation (Ayala et al., 2014), carbonylation (Fedorova et al., 2014), and also nitration (Ischiropoulos, 2009). These covalent modifications lead to often irreversible modifications to the proteins structure and therefore function, which often has detrimental outcomes. In order to determine the presence of these modifications, highly sensitive and specific assays can indicate the presence of a type of a modification within a sample (Shacter, 2000), while methods of liquid chromatography coupled with mass spectrometry can determine the presence of that modification to a specific protein (Hawkins and Davies, 2019).
ATP Synthase Structure and Function
The ATP synthase enzyme itself is a large, two component enzyme that provides many targets for oxidative species to covalently modify. The insoluble FO component is contained within the inner mitochondrial membrane and channels the protons from the intermembrane space to the mitochondrial matrix via its c-ring (composed of multiple subunit c peptides and subunit a) (Kühlbrandt and Davies, 2016). The movement of protons through the FO component drives its rotary function, with the protruding central stalk (composed of the γ, δ, and ε subunits) stimulating conformational changes in the α and β subunits of the aqueous F1 component, which undertake the catalytic process of ATP synthesis from ADP and Pi (Abrahams et al., 1994; Noji et al., 1997; Stock et al., 1999; Von Ballmoos et al., 2009).
The aqueous component of ATP synthase is a target for the oxidative species produced by mitochondria, due to the proximity to the sites of radical production and the surface exposure of the aqueous F1 component. In one model of ageing the tryptophan-503 amino acid of the α subunit was identified as frequently oxidised in the P. anserina model of ageing (Rexroth, 2012). Reversible thiol oxidation of the α-subunit in the oocytes of X. laevis has also been observed to impact the function of the enzyme’s catalytic activity (Cobley et al., 2020). Other studies have also presented ATP synthase as a target of carbonylation and 4-hydroxy-2-nonenal (HNE) modification (Prokai et al., 2007; Jeong, 2008; Guo, 2011).
Alzheimer’s Disease
Oxidative stress is widely observed in AD and is a defining characteristic of the mitochondrial hypothesis (Swerdlow, 2018; Butterfield and Halliwell, 2019). Within this hypothesis, it is described how oxidative stress is generated by intramitochondrial Aβ mediated dysfunction of the electron transport chain (Casley, 2002; Manczak, 2006). The original mitochondrial cascade hypothesis of AD describes how a gradual accumulation of oxidative damage to mitochondrial DNA, RNA, lipids, and proteins through dysfunctional electron transport chain activity contributes to disease pathology (Swerdlow and Khan, 2004). An updated consideration of this hypothesis considers how mitochondrial dysfunction can lead to oxidative stress, independent of intramitochondrial Aβ (Swerdlow, 2018). Aβ is also recognised as a source of neuroinflammation and oxidative stress in AD through its extracellular deposition in the brain that leads to the activation of microglia, the CNS macrophage (Colton et al., 2000; Koenigsknecht-Talboo and Landreth, 2005; Joshi, 2014; Zhong, 2018). Activated microglia contribute to the oxidative stress environment through the activity of the membrane-bound NADPH oxidase, and its release of the superoxide radical (Wilkinson and Landreth, 2006).
With mitochondria being a primary site of endogenous free-radical production and oxidative stress, the modification of ATP synthase by oxidative species is not unexpected. One of the first studies to identify the oxidation of ATP synthase in an AD context used C. elegans that overexpressed an aggregating form of GFP (as a proxy for protein aggregates observed in AD), and ATP synthase subunit α was identified as an oxidised protein (Boyd-Kimball, 2006). Since this preliminary observation, more detailed analyses of ATP synthase oxidation in AD have been produced.
Lipid peroxidation is a common hallmark of the oxidative stress that is observed in AD. The complex chemical interaction between ROS and polyunsaturated fatty acids leads to the production of reactive aldehydes, such as HNE, which can covalently modify proteins, modulating their structure and function. The first observation of HNE modified ATP synthase in the context of AD was from both the hippocampus and inferior parietal lobule (IPL) of mild cognitive impairment (MCI) patient brains, where ATP synthase subunit α was identified as excessively HNE modified (Reed, 2008). ATP synthase enzymatic activity was also reduced in both tissue types.
The Butterfield research group reported that the ATP synthase subunit α was again HNE modified in the IPL of early AD patients, considered to be the intermediate between MCI and AD (Reed et al., 2009a). ATP synthase was identified as HNE modified when the IPL of AD patients were studied, and this was again coupled with the observation that ATP synthase catalytic activity was reduced (Perluigi, 2009). A study of the entorhinal cortex from patients at Braak stages I/II observed the HNE oxidation of the ATP synthase subunit α, and the activity of the ATP synthase enzyme from the AD patient samples was found to be significantly decreased, while there was no observed decrease in activity of the major electron transport chain enzyme Complex I (Terni et al., 2010). At the earliest stages of AD, HNE modification of ATP synthase and subsequent downregulation of its activity has been observed. However, enzyme activity assays have suggested that this decrease in ATP synthase activity is not concordant with a decrease in Complex I activity, which suggests that a decrease in mitochondrial ATP output in early AD might be specifically dependent upon inhibited ATP synthase activity.
The frontal cortex of AD patient brains at Braak stages V/VI was assessed for a variety of oxidative protein modifications, and a four-fold increase in Nε-(malondialdehyde)-lysine (MDAL) modification of mitochondrial ATP synthase subunit β (Pamplona, 2005). However, when the entorhinal cortices of AD patients at Braak stages V/VI were assessed for variations in protein expression and for the presence of protein carbonylation, it was reported that while ATP synthase subunit β had upregulated protein expression, there were no significant differences in the carbonylation of the protein when compared with age-matched controls (Korolainen, 2006). When considered alongside the previously discussed studies that reported HNE modification of ATP synthase subunit α, this suggests that the oxidative modifications are specific to ATP synthase subunit α, and that changes in the oxidation state of the protein could be modified throughout disease progression.
Linked to the oxidative stress that characterises AD is the production of highly reactive peroxynitrite from nitrate and superoxide radicals (Radi, 2018). ATP synthase subunit α was identified as excessively nitrated in the hippocampus of AD patients (Sultana, 2006). When the IPL of early AD patients were measured, H+-transporting ATPase had excessive levels of nitration which was also coupled with a significantly reduced ATP synthase catalytic activity (Reed et al., 2009b).
A study of the APP23 transgenic mouse model of AD in the pre-symptomatic stage considered the carbonylation of proteins that were extracted from cortex tissue (Hartl, 2012). The carbonylated forms of ATP synthase subunit α, ATP synthase subunit β, ATP synthase subunit b, ATP synthase subunit O, and the ADP/ATP translocase 1 were all more abundant in the APP23 transgenic mouse model than in the control. Primary cortical neurons of the APP23 mice were cultured and were found to have a significantly decreased ADP/ATP ratio than controls, pointing to a disrupted energy metabolism that appears at the very earliest stages of the disease. The observed oxidative modifications of ATP synthase in AD are summarised in Figure 1, adapted from Spikes et al. (2020).
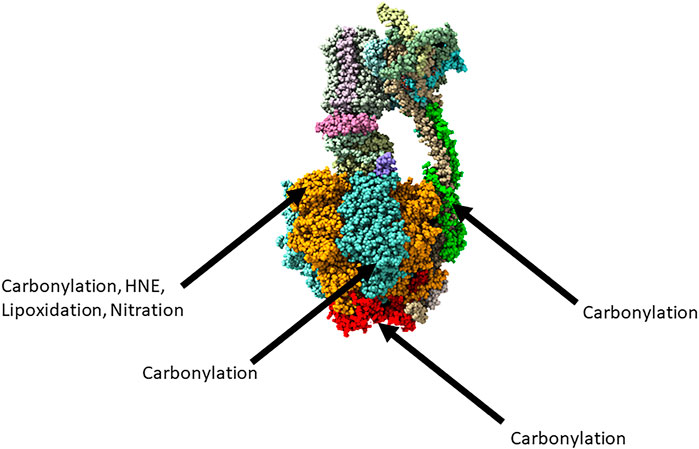
FIGURE 1. The reported oxidative modifications of ATP synthase in Alzheimer’s disease. Adapted from https://www.rcsb.org/structure/6ZPO. Subunit α represented in orange, subunit β represented in blue, subunit O represented in red, subunit D represented in green.
Huntington’s Disease
While oxidative stress is a common feature of both PD and HD, there are fewer observations of ATP synthase being a target of oxidative modifications in these diseases. However, a study of the oxidised proteins from the striatum of HD patients reported that ATP synthase subunit α is significantly carbonylated in comparison with age-matched controls (Sorolla, 2010). Enriched mitochondrial fractions were taken from the patient striatum samples and assessed ATP synthase catalytic activity, which was also significantly lower in comparison with controls.
Parkinson’s Disease
Rotenone is a well-characterised inhibitor of the mitochondrial complex I that is used in studies of PD (Greenamyre et al., 2010). Complex I dysfunction is a major pathophysiological component of PD, and its inhibition by rotenone is utilised widely in both cell and animal disease models (KrakoJakovljevic, 2021). A study of dopaminergic PC12 cells exposed to rotenone observed that both ATP synthase subunit α and ATP synthase subunit β were carbonylated, to which the authors suggest could contribute to a reduced ATP output (Chiaradia, 2019). Mutations in the SNCA gene that encodes α-synuclein are also a feature of PD, as the aggregation of the protein leads to the formation of toxic oligomers and fibrils (Schulz-Schaeffer, 2010; Mahul-Mellier et al., 2020). Rat neuronal co-cultures treated with oligomers of α-synuclein showed a co-localisation of the oligomers with ATP synthase, which resulted in high levels of oxidised ATP synthase subunit β (Ludtmann, 2018). There is evidence of mitochondrial permeability transition pore (mPTP) opening in response to α-synuclein oligomer induced oxidative stress and compromised bioenergetics, and the role of ATP synthase as a structural component of mPTP is now increasingly better understood (Angeli, 2021).
D. melanogaster with parkin mutations are also a well-characterised disease model for PD (Greene, 2003). Studies have reported reduced Glutathione S-Transferase Omega (GSTO) in these mutants, which may result in increased susceptibility to oxidative stress due to the protective role of S-Glutathionylation against the irreversible oxidation of protein thiols (Song, 2014). When parkin mutants of D. melanogaster had GSTO1 restored, as it is downregulated in parkin mutants, the levels of S-Glutathionylation of ATP synthase subunit β increased, along with the catalytic activity of the ATP synthase enzyme (Kim et al., 2012).
Conclusion and Perspectives
The mitochondrion is a primary site of free radical production in the cell, and thus a site of high levels of oxidative stress. This mitochondrial dysfunction that leads to oxidative stress has come to characterise many neurodegenerative diseases. The protein targets of this oxidative stress are varied but distinct to given diseases, and so in order to better understand the disease process, a comprehension of the oxidised proteins is required.
Proper functioning of the ATP synthase enzyme is essential to metazoan life, and in H. sapiens, its dysfunction is widely implicated in disease (Dautant, 2018). Here we have discussed the published evidence of ATP synthase modification by processes of oxidation in different neurodegenerative diseases, with a summary of these modification presented in Table 1. A more complete understanding of ATP synthase oxidation is presented in AD, largely based upon the work of the D. Allan Butterfield lab, but there is still more to be understood about this process.
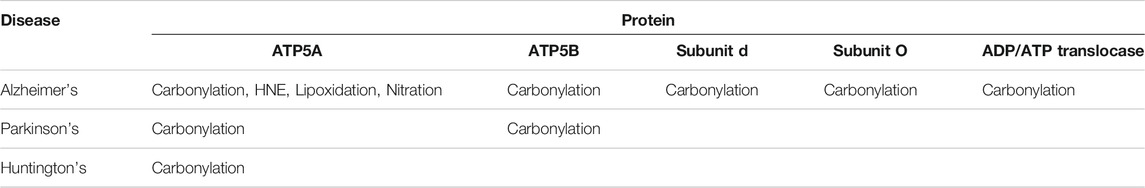
TABLE 1. The different oxidative modifications of ATP synthase reported in Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease.
Much less information is available in PD and HD but given that oxidative stress is a major feature in the pathophysiology of both diseases, effort should be directed toward understanding the oxidised proteome in both diseases, and in particular the oxidation of the ATP synthase enzyme. Other neurodegenerative conditions such not be overlooked in these investigations either, protein oxidation has long being understood to characterise ALS, but information regarding the specific proteins which are modified is lacking (Beal, 1997; Andrus et al., 1998).
Given the progression of the candidate drug J147, which targets ATP synthase, to clinical trial as a potential treatment for AD (Goldberg, 2018), as well as the deeper consideration of the enzyme as a target in anti-cancer therapies (Wang et al., 2021), a deeper understanding of the frequency, type, and location of the oxidative modifications of the mitochondrial ATP synthase enzyme should allow for the development of candidate drugs to treat these diseases in the future.
Author Contributions
BE and LC co-wrote the manuscript.
Funding
BE is supported by the Biotechnology and Biological Sciences Research Council (grant number BB/J014508/1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abrahams, J. P., Leslie, A. G. W., Lutter, R., and Walker, J. E. (1994). Structure at 2.8 Â Resolution of F1-ATPase from Bovine Heart Mitochondria. Nature 370, 621–628. doi:10.1038/370621a0
Andrus, P. K., Fleck, T. J., Gurney, M. E., and Hall, E. D. (1998). Protein Oxidative Damage in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 71, 2041–2048. doi:10.1046/j.1471-4159.1998.71052041.x
Angeli, S. (2021). The Mitochondrial Permeability Transition Pore Activates the Mitochondrial Unfolded Protein Response and Promotes Aging. Elife 10, 1–23. doi:10.7554/elife.63453
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxid. Med. Cel. Longev. 2014, 1. doi:10.1155/2014/360438
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, Oxidants, and Aging. Cell 120, 483–495. doi:10.1016/j.cell.2005.02.001
Bárcena, C., Mayoral, P., and Quirós, P. M. (2018). Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 340, 35–77.
Bates, G. P., Dorsey, R., Gusella, J. F., Hayden, M. R., Kay, C., Leavitt, B. R., et al. (2015). Huntington Disease. Nat. Rev. Dis. Primers 1, 15005–15021. doi:10.1038/nrdp.2015.5
Beal, M. F. (1997). Increased 3-nitrotyrosine in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. Ann. Neurol. 42, 644–654. doi:10.1002/ana.410420416
Boyd-Kimball, D. (2006). Proteomic Identification of Proteins Specifically Oxidized in Caenorhabditis elegans Expressing Human Aβ(1-42): Implications for Alzheimer’s Disease. Neurobiol. Aging 27, 1239–1249. doi:10.1016/j.neurobiolaging.2005.07.001
Braak, H., and Braak, E. (1991). Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 82, 239–259. doi:10.1007/bf00308809
Butterfield, D. A., and Halliwell, B. (2019). Oxidative Stress, Dysfunctional Glucose Metabolism and Alzheimer Disease. Nat. Rev. Neurosci. 20, 148–160. doi:10.1038/s41583-019-0132-6
Casley, C. S. (2002). β-Amyloid Fragment 25-35 Causes Mitochondrial Dysfunction in Primary Cortical Neurons. Neurobiol. Dis. 10, 258–267. doi:10.1006/nbdi.2002.0516
Castro, D. M., Dillon, C., Machnicki, G., and Allegri, R. F. (2010). The Economic Cost of Alzheimer's Disease: Family or Public-Health burden? Dement. Neuropsychol. 4, 262–267. doi:10.1590/s1980-57642010dn40400003
Chen, Q., Vazquez, E. J., Moghaddas, S., Hoppel, C. L., and Lesnefsky, E. J. (2003). Production of Reactive Oxygen Species by Mitochondria: Central Role of Complex III. J. Biol. Chem. 278, 36027–36031. doi:10.1074/jbc.m304854200
Chiaradia, E. (2019). Protein Carbonylation in Dopaminergic Cells Exposed to Rotenone. Toxicol. Lett. 309, 20–32. doi:10.1016/j.toxlet.2019.04.002
Cobley, J., Noble, A., Bessell, R., Guille, M., and Husi, H. (2020). Reversible Thiol Oxidation Inhibits the Mitochondrial ATP Synthase in Xenopus Laevis Oocytes. Antioxidants 9, 1. doi:10.3390/antiox9030215
Colton, C. A., Chernyshev, O. N., Gilbert, D. L., and Vitek, M. P. (2000). Microglial Contribution to Oxidative Stress in Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 899, 292–307. doi:10.1111/j.1749-6632.2000.tb06195.x
Damiano, M., Galvan, L., Déglon, N., and Brouillet, E. (2010). Mitochondria in Huntington's Disease. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1802, 52–61. doi:10.1016/j.bbadis.2009.07.012
Damier, P., Hirsch, E. C., Agid, Y., and Graybiel, A. M. (1999). The Substantia Nigra of the Human Brain. Brain 122, 1437–1448. doi:10.1093/brain/122.8.1437
Dautant, A. (2018). ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 9, 329. doi:10.3389/fphys.2018.00329
Dugger, B. N., and Dickson, D. W. (2017). Pathology of Neurodegenerative Diseases. Cold Spring Harb Perspect. Biol. 9, a028035. doi:10.1101/cshperspect.a028035
Fedorova, M., Bollineni, R. C., and Hoffmann, R. (2014). Protein Carbonylation as a Major Hallmark of Oxidative Damage: Update of Analytical Strategies. Mass. Spectrom. Rev. 33, 79–97. doi:10.1002/mas.21381
Gao, Y.-L., Wang, N., Sun, F.-R., Cao, X.-P., Zhang, W., and Yu, J.-T. (2018). Tau in Neurodegenerative Disease. Ann. Transl. Med. 6, 175. doi:10.21037/atm.2018.04.23
Gnaiger, E. (2020). Mitochondrial Pathways and Respiratory Control: An Introduction to OXPHOS Analysis. Innsbruck, Austria: Bioenergetic Communications, 5.
Goldberg, J. (2018). The Mitochondrial ATP Synthase Is a Shared Drug Target for Aging and Dementia. Aging Cell 17, e12715. doi:10.1111/acel.12715
Goldberg, M. S., Fleming, S. M., Palacino, J. J., Cepeda, C., Lam, H. A., Bhatnagar, A., et al. (2003). Parkin-deficient Mice Exhibit Nigrostriatal Deficits but Not Loss of Dopaminergic Neurons. J. Biol. Chem. 278, 43628–43635. doi:10.1074/jbc.m308947200
Greenamyre, J. T., Cannon, J. R., Drolet, R., and Mastroberardino, P. G. (2010). Lessons from the Rotenone Model of Parkinson’s Disease. Trends Pharmacol. Sci. 31, 141–142. doi:10.1016/j.tips.2009.12.006
Greene, J. C. (2003). Mitochondrial Pathology and Apoptotic Muscle Degeneration in Drosophila Parkin Mutants. Proc. Natl. Acad. Sci. 100, 4078–4083. doi:10.1073/pnas.0737556100
Guo, J. (2011). Protein Targets for Carbonylation by 4-Hydroxy-2-Nonenal in Rat Liver Mitochondria. J. Proteomics 74, 2370–2379. doi:10.1016/j.jprot.2011.07.009
Hartl, D. (2012). Presymptomatic Alterations in Energy Metabolism and Oxidative Stress in the APP23 Mouse Model of Alzheimer Disease. J. Proteome Res. 11, 3295–3304. doi:10.1021/pr300021e
Hawkins, C. L., and Davies, M. J. (2019). Detection, Identification, and Quantification of Oxidative Protein Modifications. J. Biol. Chem. 294, 19683–19708. doi:10.1074/jbc.rev119.006217
Hirsch, E., Graybiel, A. M., and Agid, Y. A. (1988). Melanized Dopaminergic Neurons Are Differentially Susceptible to Degeneration in Parkinson's Disease. Nature 334, 345–348. doi:10.1038/334345a0
Hirst, J., King, M. S., and Pryde, K. R. (2008). The Production of Reactive Oxygen Species by Complex I. Biochem. Soc. Trans. 36, 976–980. doi:10.1042/bst0360976
Ischiropoulos, H. (2009). Protein Tyrosine Nitration-An Update. Arch. Biochem. Biophys. 484, 117–121. doi:10.1016/j.abb.2008.10.034
Jeong, H. J. (2008). Mitochondrial ATP Synthase Is a Target for TNBS-Induced Protein Carbonylation in XS-106 Dendritic Cells. Proteomics 8, 2384–2393. doi:10.1002/pmic.200700962
Joshi, P. (2014). Microglia Convert Aggregated Amyloid-β into Neurotoxic Forms through the Shedding of Microvesicles. Cell Death Differ 21, 582–593. doi:10.1038/cdd.2013.180
Kadenbach, B. (2021). Complex IV – the Regulatory center of Mitochondrial Oxidative Phosphorylation. Mitochondrion 58, 296–302. doi:10.1016/j.mito.2020.10.004
Kim, K., Kim, S. H., Kim, J., Kim, H., and Yim, J. (2012). Glutathione S-Transferase omega 1 Activity Is Sufficient to Suppress Neurodegeneration in a Drosophila Model of Parkinson Disease. J. Biol. Chem. 287, 6628–6641. doi:10.1074/jbc.m111.291179
Koenigsknecht-Talboo, J., and Landreth, G. E. (2005). Microglial Phagocytosis Induced by Fibrillar β-amyloid and IgGs Are Differentially Regulated by Proinflammatory Cytokines. J. Neurosci. 25, 8240–8249. doi:10.1523/jneurosci.1808-05.2005
Korolainen, M. A. (2006). Oxidative Modification of Proteins in the Frontal Cortex of Alzheimer’s Disease Brain. Neurobiol. Aging 27, 42–53. doi:10.1016/j.neurobiolaging.2004.11.010
Krako Jakovljevic, N. (2021). Mitochondrial Homeostasis in Cellular Models of Parkinson’s Disease. Bioenerg. Commun. 2021, 1–31.
Kühlbrandt, W., and Davies, K. M. (2016). Rotary ATPases: A New Twist to an Ancient Machine. Trends Biochem. Sci. 41, 106–116.
Liguori, I. (2018). Oxidative Stress, Aging, and Diseases. Clin. Interv. Aging 13, 757–772. doi:10.2147/cia.s158513
Lin, M. T., and Beal, M. F. (2006). Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 443, 787–795. doi:10.1038/nature05292
Ludtmann, M. H. R. (2018). α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 9, 1. doi:10.1038/s41467-018-04422-2
Mahul-Mellier, A.-L., Burtscher, J., Maharjan, N., Weerens, L., Croisier, M., Kuttler, F., et al. (2020). The Process of Lewy Body Formation, rather Than Simply α-synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 117, 4971–4982. doi:10.1073/pnas.1913904117
Makin, S. (2018). The Amyloid Hypothesis on Trial. Nature 559, S4–S7. doi:10.1038/d41586-018-05719-4
Manczak, M. (2006). Mitochondria Are a Direct Site of Aβ Accumulation in Alzheimer’s Disease Neurons: Implications for Free Radical Generation and Oxidative Damage in Disease Progression. Hum. Mol. Genet. 15, 1437–1449. doi:10.1093/hmg/ddl066
Marasco, R. A. (2020). Economic burden of Alzheimer Disease and Managed Care Considerations. Am. J. Manag. Care 26, S177–S183.
Moreira, P. I., Cardoso, S. M., Santos, M. S., and Oliveira, C. R. (2006). The Key Role of Mitochondria in Alzheimer's Disease. Jad 9, 101–110. doi:10.3233/jad-2006-9202
Nichols, E. (2019). Global, Regional, and National burden of Alzheimer's Disease and Other Dementias, 1990-2016: a Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 88–106. doi:10.1016/S1474-4422(18)30403-4
Nissanka, N., and Moraes, C. T. (2018). Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett. 592, 728–742. doi:10.1002/1873-3468.12956
Noji, H., Yasuda, R., Yoshida, M., and Kinosita, K. (1997). Direct Observation of the Rotation of F1-ATPase. Nature 386, 299–302. doi:10.1038/386299a0
Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., et al. (2004). Mitochondrial Dysfunction and Oxidative Damage in Parkin-Deficient Mice. J. Biol. Chem. 279, 18614–18622. doi:10.1074/jbc.m401135200
Pamplona, R. (2005). Proteins in Human Brain Cortex Are Modified by Oxidation, Glycoxidation, and Lipoxidation: Effects of Alzheimer Disease and Identification of Lipoxidation Targets. J. Biol. Chem. 280, 21522–21530. doi:10.1074/jbc.m502255200
Perluigi, M. (2009). Redox Proteomics Identification of 4-hydroxynonenalmodified Brain Proteins in Alzheimer’s Disease: Role of Lipid Peroxidation in Alzheimer’s Disease Pathogenesis. Proteomics - Clin. Appl. 3, 682–693. doi:10.1002/prca.200800161
Perry, G., Castellani, R. J., Hirai, K., and Smith, M. A. (1998). Reactive Oxygen Species Mediate Cellular Damage in Alzheimer Disease. J. Alzheimer’s Dis. 1, 45–55. doi:10.3233/jad-1998-1103
Prokai, L., Yan, L. J., Vera-Serrano, J. L., Stevens, S. M., and Forster, M. J. (2007). Mass Spectrometry-Based Survey of Age-Associated Protein Carbonylation in Rat Brain Mitochondria. J. Mass. Spectrom. 42, 1583–1589. doi:10.1002/jms.1345
Radi, R. (2018). Oxygen Radicals, Nitric Oxide, and Peroxynitrite: Redox Pathways in Molecular Medicine. Proc. Natl. Acad. Sci. U. S. A. 115, 5839–5848. doi:10.1073/pnas.1804932115
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta - Mol. Cell Res. 1863, 2977–2992. doi:10.1016/j.bbamcr.2016.09.012
Reed, T. (2008). Redox Proteomic Identification of 4-Hydroxy-2-Nonenal-Modified Brain Proteins in Amnestic Mild Cognitive Impairment: Insight into the Role of Lipid Peroxidation in the Progression and Pathogenesis of Alzheimer’s Disease. Neurobiol. Dis. 30, 107–120. doi:10.1016/j.nbd.2007.12.007
Reed, T. T., Pierce, W. M., Markesbery, W. R., and Butterfield, D. A. (2009). Proteomic Identification of HNE-Bound Proteins in Early Alzheimer Disease: Insights into the Role of Lipid Peroxidation in the Progression of AD. Brain Res. 1274, 66–76. doi:10.1016/j.brainres.2009.04.009
Reed, T. T., Pierce, W. M., Turner, D. M., Markesbery, W. R., and Allan Butterfield, D. (2009). Proteomic Identification of Nitrated Brain Proteins in Early Alzheimer’s Disease Inferior Parietal Lobule. J. Cel. Mol. Med. 13, 2019–2029. doi:10.1111/j.1582-4934.2008.00478.x
Rexroth, S. (2012). Reactive Oxygen Species Target Specific Tryptophan Site in the Mitochondrial ATP Synthase. Biochim. Biophys. Acta - Bioenerg. 1817, 381–387. doi:10.1016/j.bbabio.2011.11.006
Rikani, A. A. (2014). The Mechanism of Degeneration of Striatal Neuronal Subtypes in Huntington Disease. Ann. Neurosci. 21, 112–114. doi:10.5214/ans.0972.7531.210308
Roos, R. A. (2010). Huntington’s Disease: a Clinical Review. Orphanet J. Rare Dis. 5, 40. doi:10.1186/1750-1172-5-40
Sabuncu, M. R. (2011). The Dynamics of Cortical and Hippocampal Atrophy in Alzheimer Disease. Arch. Neurol. 68, 1040–1048. doi:10.1001/archneurol.2011.167
Schulz-Schaeffer, W. J. (2010). The Synaptic Pathology of α-synuclein Aggregation in Dementia with Lewy Bodies, Parkinson's Disease and Parkinson's Disease Dementia. Acta Neuropathol. 120, 131–143. doi:10.1007/s00401-010-0711-0
Selkoe, D. J., and Hardy, J. (2016). The Amyloid Hypothesis of Alzheimer's Disease at 25 Years. EMBO Mol. Med. 8, 595–608. doi:10.15252/emmm.201606210
Shacter, E. (2000). Quantification and Significance of Protein Oxidation in Biological Samples. Drug Metab. Rev. 32, 307–326. doi:10.1081/dmr-100102336
Smith, M. P., and Cass, W. A. (2007). Oxidative Stress and Dopamine Depletion in an Intrastriatal 6-hydroxydopamine Model of Parkinson’s Disease. Neuroscience 144, 1057–1066. doi:10.1016/j.neuroscience.2006.10.004
Song, J. (2014). Glutathione Protects Brain Endothelial Cells from Hydrogen Peroxide-Induced Oxidative Stress by Increasing Nrf2 Expression. Exp. Neurobiol. 23, 93–103. doi:10.5607/en.2014.23.1.93
Sorolla, M. A. (2010). Protein Oxidation in Huntington Disease Affects Energy Production and Vitamin B6 Metabolism. Free Radic. Biol. Med. 49, 612–621. doi:10.1016/j.freeradbiomed.2010.05.016
Sorolla, M. A. (2008). Proteomic and Oxidative Stress Analysis in Human Brain Samples of Huntington Disease. Free Radic. Biol. Med. 45, 667–678. doi:10.1016/j.freeradbiomed.2008.05.014
Spikes, T. E., Montgomery, M. G., and Walker, J. E. (2020). Structure of the Dimeric ATP Synthase from Bovine Mitochondria. Proc. Natl. Acad. Sci. U. S. A. 117 (38), 23519–23526. doi:10.1073/pnas.2013998117
Stadtman, E. R., and Levine, R. L. (2006). Oxidatively Modified Proteins Proteins by Reactive Oxygen Species. Redox Proteomics Protein Modif. Cel. Dysfunct. Dis. 32 (9), 797–803. doi:10.1002/0471973122.ch1
Stock, D., Leslie, A. G. W., and Walker, J. E. (1999). Molecular Architecture of the Rotary Motor in ATP Synthase. Science (80-. ) 286, 1700–1705. doi:10.1126/science.286.5445.1700
Sultana, R. (2006). Identification of Nitrated Proteins in Alzheimer’s Disease Brain Using a Redox Proteomics Approach. Neurobiol. Dis. 22, 76–87. doi:10.1016/j.nbd.2005.10.004
Swerdlow, R. H., and Khan, S. M. (2004). A ‘mitochondrial cascade Hypothesis’ for Sporadic Alzheimer’s Disease. Med. Hypotheses 63, 8–20. doi:10.1016/j.mehy.2003.12.045
Swerdlow, R. H. (2018). Mitochondria and Mitochondrial Cascades in Alzheimer's Disease. Jad 62, 1403–1416. doi:10.3233/jad-170585
Terni, B., Boada, J., Portero-Otin, M., Pamplona, R., and Ferrer, I. (2010). Mitochondrial ATP-Synthase in the Entorhinal Cortex Is a Target of Oxidative Stress at Stages I/II of Alzheimer’s Disease Pathology. Brain Pathol. 20, 222–233. doi:10.1111/j.1750-3639.2009.00266.x
Von Ballmoos, C., Wiedenmann, A., and Dimroth, P. (2009). Essentials for ATP Synthesis by F1F0 ATP Synthases. Annu. Rev. Biochem. 78, 649–672. doi:10.1146/annurev.biochem.78.081307.104803
Wang, T., Ma, F., and Qian, H. (2021). Defueling the Cancer: ATP Synthase as an Emerging Target in Cancer Therapy. Mol. Ther. - Oncolytics 23, 82–95. doi:10.1016/j.omto.2021.08.015
Wang, X. (2014). Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease. Biochim. Biophys. Acta - Mol. Basis Dis. 1842, 1240–1247. doi:10.1016/j.bbadis.2013.10.015
Wilkinson, B. L., and Landreth, G. E. (2006). The Microglial NADPH Oxidase Complex as a Source of Oxidative Stress in Alzheimer’s Disease. J. Neuroinflammation 3, 1–12. doi:10.1186/1742-2094-3-30
Wood-Kaczmar, A., Gandhi, S., Yao, Z., Abramov, A. S. Y., Miljan, E. A., Keen, G., et al. (2008). PINK1 Is Necessary for Long Term Survival and Mitochondrial Function in Human Dopaminergic Neurons. PLoS One 3, e2455. doi:10.1371/journal.pone.0002455
Zhang, J. (2016). ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cel. Longev. 2016, 1. doi:10.1155/2016/4350965
Keywords: mitochondria, ATP synthase, oxidative stress, neurodegenarative disease, oxidative phoshorylation
Citation: Ebanks B and Chakrabarti L (2022) Mitochondrial ATP Synthase is a Target of Oxidative Stress in Neurodegenerative Diseases. Front. Mol. Biosci. 9:854321. doi: 10.3389/fmolb.2022.854321
Received: 13 January 2022; Accepted: 26 January 2022;
Published: 14 February 2022.
Edited by:
Fabrizio Gentile, University of Molise, ItalyReviewed by:
Katarzyna Kaczynska, Mossakowski Medical Research Centre (PAN), PolandCopyright © 2022 Ebanks and Chakrabarti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brad Ebanks, mbybdpo@nottingham.ac.uk