Update on the implication of potassium channels in autism: K+ channelautism spectrum disorder
 Luca Guglielmi1*
Luca Guglielmi1*  Ilenio Servettini1
Ilenio Servettini1  Martino Caramia2
Martino Caramia2  Luigi Catacuzzeno2
Luigi Catacuzzeno2  Fabio Franciolini2
Fabio Franciolini2  Maria Cristina D’Adamo1
Maria Cristina D’Adamo1  Mauro Pessia1*
Mauro Pessia1*
- 1Section of Physiology and Biochemistry, Department of Experimental Medicine, University of Perugia School of Medicine, Perugia, Italy
- 2Department of Chemistry, Biology and Biotechnology, University of Perugia, Perugia, Italy
Autism spectrum disorders (ASDs) are characterized by impaired ability to properly implement environmental stimuli that are essential to achieve a state of social and cultural exchange. Indeed, the main features of ASD are impairments of interpersonal relationships, verbal and non-verbal communication and restricted and repetitive behaviors. These aspects are often accompanied by several comorbidities such as motor delay, praxis impairment, gait abnormalities, insomnia, and above all epilepsy. Genetic analyses of autistic individuals uncovered deleterious mutations in several K+ channel types strengthening the notion that their intrinsic dysfunction may play a central etiologic role in ASD. However, indirect implication of K+ channels in ASD has been also reported. For instance, loss of fragile X mental retardation protein (FMRP) results in K+ channels deregulation, network dysfunction and ASD-like cognitive and behavioral symptoms. This review provides an update on direct and indirect implications of K+ channels in ASDs. Owing to a mounting body of evidence associating a channelopathy pathogenesis to autism and showing that nearly 500 ion channel proteins are encoded by the human genome, we propose to classify ASDs - whose susceptibility is significantly enhanced by ion channels defects, either in a monogenic or multigenic condition - in a new category named “channelAutism Spectrum Disorder” (channelASD; cASD) and introduce a new taxonomy (e.g., Kvx.y-channelASD and likewise Navx.y-channelASD, Cavx.y-channelASD; etc.). This review also highlights some degree of clinical and genetic overlap between K+ channelASDs and K+ channelepsies, whereby such correlation suggests that a subcategory characterized by a channelASD-channelepsy phenotype may be distinguished. Ultimately, this overview aims to further understand the different clinical subgroups and help parse out the distinct biological basis of autism that are essential to establish patient-tailored treatments.
Introduction
Nearly 70 million people worldwide suffer from ASD. 700,000 are from USA and the estimated cost to society of caring for these children has been $11.5 billion in 2011 (Lavelle et al., 2014). ASD is a group of heterogeneous neurodevelopmental disorders previously dealt with as single pathologies: autistic disorder, pervasive developmental disorder-not otherwise specified (PDD-NOS), Asperger syndrome, childhood disintegrative disorder also known as Heller syndrome, and Rett syndrome. Specific impairments before age three, in each of the following three core domains fit the diagnostic criteria for ASD: social (difficulty in interacting with people, in reading facial expressions and making eye contact), language (difficulty in using or understanding language, tending to focus attention and conversation on a limited number of topics, frequently repeat phrases, and have very limited speech ability), and restricted/repetitive behavior (excessive attention to routines and difficulty in adjusting to new surroundings or changes in routine). Given the complexity of the disorder and the variety and severity of the symptoms, ASD is thought to be caused by multiple factors interacting in a complex way. Genetic defects, both inherited or occurring spontaneously, undoubtedly play a role in increasing the susceptibility to the disorder by affecting brain development and proper neural networking. Neurological comorbidities are often observed in ASD patients and they are associated with more clinical severity. The most noted is epilepsy, comorbid in 30% of ASD, about 50 times higher than in the general population (Tuchman and Rapin, 2002; Mouridsen et al., 2011). Cognitive and behavioral deficits occurs in up to 46% of children with epilepsy (Clarke et al., 2005; Matsuo et al., 2010) suggesting that, at least in these cases, the excitability imbalance could be a common denominator. Another comorbidity observed in nearly 90% of genetic syndromes associated with ASDs is motor delay. Delays occur in both gross and fine motor domains, while deficits are documented in praxis, motor planning, gait, coordination, and postural control (Rinehart and McGinley, 2010; Van Waelvelde et al., 2010). Several lines of evidence suggest that motor delay is caused by aberrant neural circuitry mostly in the cerebellum and fronto-striatal regions (Rinehart et al., 2006; Wegiel et al., 2013).
The relative risk for a child to develop the disease is increased ~25-fold in families in which a sibling is affected (Jorde et al., 1991). Unfortunately, the genetic etiology of autism is characterized by high locus heterogeneity, where de novo and rare inherited CNVs and SNV mutations in conjunction contribute to the overall genetic risk to develop the disease. To date, we know that defined mutations, genetic syndromes and de novo CNVs account for about 30% of ASD cases (Schaaf and Zoghbi, 2011). Linkage analyses of ASD patients have allowed the identification of loss- or gain-of-function mutations and chromosomal alterations in genes coding for several channel types, including K+ channels. These proteins are highly heterogenic and widely expressed in the CNS where they set the resting membrane potential of neurons and glia, shape action potentials and regulate firing, neurotransmitter release and wiring. At network level, K+ channels are involved in post-synaptic EPSP-spike coupling, activity dependent short- and long-term synaptic plasticity and information processing. Thus, these channels provide neurons, glia and CNS networks with distinct electrical identity and their dysfunction is a major contributing factor in excitability disequilibrium and network impairment. Animal models, in which these genes are deleted or mutated, often show epilepsy (Eijkelkamp et al., 2012; D’Adamo et al., 2013, 2014), motor impairment, learning and behavioral phenotypes that resemble somehow ASD. The insights, provided by investigations with these animal models, are valuable to understand neural networks abnormalities potentially underlying ASD. The plethora of ion channel types expressed by neurons and the numerous variations discovered in their relevant genes often generate confusion in the classification of the associated diseases. To further understand the clinical subgroups of autism, help parse out distinct biological basis of autism and identify tailored treatments, we propose to name the ion channels defects contributing significantly to either monogenic or multigenic ASDs “channelASD” and suggest a new taxonomy (e.g., Kvx.y-channelASD and likewise Navx.y-channelASD; Cavx.y-channelASD; etc.), according to a previously proposed nomenclature for ion channels dysfunctions underlying epilepsy (D’Adamo et al., 2013). Interestingly, this review will also show that there is a high degree of clinical overlap between channelASD and channelepsy. This strong correlation between a distinct subgroup of patients prompt us to also define a new phenotype: “channelASD-channelepsy,” according with our previously and presently introduced nomenclature.
Voltage-Gated K+ Channels
The first voltage-gated K+ channel (Kv) was cloned from the Shaker mutant of Drosophila melanogaster (Tempel et al., 1987). Since then, a number of other genes encoding for Kv channels have been identified from many different species. Based on sequence relatedness, Kv channels have been classified in subfamilies from Kv1 to Kv12 (Chandy and Gutman, 1993). The α subunit contains six transmembrane segments with the N- and C-termini residing inside the cell. The full crystal structure, provided for a Kv channel, confirmed that this channel is composed of four homologous pore-forming α subunits. These channels may exist either as homomers, whenever four identical α-subunits are assembled or as heteromers, whenever different types of α-subunits heteropolymerize to form channels with distinct functional and pharmacological properties (Pessia, 2004).
Kv4.2 – ChannelASD
KCND2 gene encodes for the α subunit of the voltage-gated K+ channel Kv4.2 that generates an “A-type” current (IA). When assembled in homotetramers, Kv4.2 channels activate at sub-threshold membrane potentials, inactivate rapidly, and quickly recover from inactivation (Zhu et al., 1999; Birnbaum et al., 2004). The availability of this channel type, in response to membrane potential fluctuation, is regulated by its peculiar inactivation kinetics occurring from the open state by a fast N-type mechanism or (much more frequently) from pre-open closed states, when the voltage-sensor domains activate but the pore fails to open (Bahring et al., 2001; Gebauer et al., 2004; Jerng et al., 2004; Barghaan and Bahring, 2009; Bahring and Covarrubias, 2011). In vivo, other proteins further tune-up Kv4.2 inactivation kinetics: the K+ channel interacting protein (KChIP) and the dipeptidyl-peptidase-like protein (DPP) that respectively inhibits and accentuates N-type open state inactivation (Wang et al., 2002; Jerng et al., 2005, 2009; Covarrubias et al., 2008). This channel type is mainly expressed in the hippocampal CA1 pyramidal neurons where it regulates the threshold for action potential initiation and repolarization, frequency-dependent AP broadening, and back-propagation of action potentials (Chen et al., 2006; Kim et al., 2007; Nerbonne et al., 2008). These notions were further corroborated by the evidence that Kv4.2 channel deletion lowers and increases the threshold for LTP and LTD induction, respectively (Chen et al., 2006; Zhao et al., 2011). Conversely, enhancement of Kv4.2 expression prevents LTP induction (Jung et al., 2008). The presence of Kv4.2 channels in hippocampus appears fundamental, mostly at early developmental stage when neuronal activity drives synaptic maturation and network refinement. At hippocampal synapses, the gradual reduction in GluN2B/GluN2A subunit ratio, during post-natal development, is correlated with AMPA expression and synaptic maturation. Ablation of Kv4.2 in mice abolished this phenomenon and resulted in a higher number of silent synapses in the adulthood (Kim and Hoffman, 2012). Noteworthy, both NMDA subunit composition and synaptic inactivity are rescued by reentering the channel (Kim and Hoffman, 2012).
Given the importance of Kv4.2 in brain development and functioning, defects of this channel have been unsurprisingly correlated with a broad spectrum of neurological disorders. Gene deletion in mice leads to increased susceptibility to convulsant stimuli (Barnwell et al., 2009) and truncating mutation of Kv4.2 in humans leads to temporal lobe epilepsy (Singh et al., 2006). Moreover, rare variants in KCND2 have been identified in individuals with autism, namely: a submicroscopic de novo deletion (Okamoto et al., 2011), three translocation breakpoints (at 7q22.1, 7q31.2 and 7q31.3) potentially deleterious (Scherer et al., 2003), and three substitution variants, (N544S; F538S; R539L) reported in three independent cases of autism (Mikhailov et al., 2008). Recently, exome sequencing of two identical twins affected by autism, with frequent and uncontrollable seizures, revealed a de novo heterozygous variant in KCND2 gene that resulted in the substitution V404M (Lee et al., 2014). Expression and functional characterization of Kv4.2 V404M channels in Xenopus laevis oocytes showed significantly slower inactivation than wild-type, leading to grater channel availability in response to depolarization. These functional alterations persisted when the mutated subunit was co-expressed with the wild-type (in 1:1 ratio to mimic the heterozygous nature of the variation), suggesting a dominant effect. The V404M impact on channel kinetics was attributed to impaired closed-state inactivation, because the effect was still evident in the presence of the auxiliary subunits KChIP3a or DPP10a (Lee et al., 2014).
Kv4.2 – ChannelASD linked to Fragile X syndrome
Kv4.2 channel expression may also participate in establishing the conditions for the development of ASDs, given that Kv4.2 mRNA can bind to the fragile X mental retardation protein (FMRP), which is associated to fragile X syndrome (FXS), the most common monogenic cause of autism and inherited intellectual retardation (Garber et al., 2008) often accompanied by seizures and poor motor coordination (Garber et al., 2008). FXS is present in ~5% of ASDs cases, and is the most likely cause of autism in those individuals. FXS is caused by an expansion of CGG triplets (>200) in the 5′ untranslated region of the fragile X mental retardation gene 1 (FMR1), located on chromosome X (Krawczun et al., 1985; Fu et al., 1991), which results in the failure to produce the FMRP, required for normal neural development (Pieretti et al., 1991; Verheij et al., 1993; Santoro et al., 2012). This protein is predominantly post-synaptic, where is associated with ribosomal complexes and represses or promotes the translation of specific mRNAs (Siomi et al., 1993; Todd and Malter, 2002; Zalfa et al., 2003; Weiler et al., 2004; Muddashetty et al., 2007; Bechara et al., 2009). The function and localization of this protein make it a key regulator of synaptic plasticity, especially for all those activity dependent processes that require the synthesis of new proteins. In particular, FRMP appears to play a critical role in the maintenance of LTD and LTP in the hippocampus (Weiler et al., 1997; Huber et al., 2000, 2002; Hou et al., 2006; Lauterborn et al., 2007; Michalon et al., 2012). In hippocampal dendrites, FMRP is associated with Kv4.2 mRNA and prevents its translation leading to reduced channel expression at post-synaptic membrane (Lee et al., 2011). Consistent with this scenario, hippocampal neurons from Fmr1-null mice show higher levels of Kv4.2 protein than WT and reduced LTP, when induced by threshold stimuli (Lee et al., 2011; Table 2). The application of heteropodatoxin (HpTx2), a spider toxin that specifically blocks Kv4.2 channels, restores LTP in hippocampal slices derived from Fmr1-null mice (Lee et al., 2011). It is therefore likely that deregulated expression of Kv4.2, resulting from FMRP-dependent abnormalities, may contribute significantly to FXS pathogenesis and generate the conditions for ASDs development.
Kv7.3 – ChannelASD
The K+ channel subunit Kv7.3 is encoded by the KCNQ3 gene and is expressed in the hippocampus and cortex where it assembles predominantly with the homologous Kv7.2 to form “M” channels (Wang et al., 1998). Generation of functional M channels by heteropolymerization of Kv7.2(KCNQ2) with Kv7.4(KCNQ4) or Kv7.5(KCNQ5) subunits has also been described (Kubisch et al., 1999; Schroeder et al., 2000). This channel type was identified by Brown and Adams (1980) as the principal mediator of muscarine-induced depolarization. They are located in electrically critical regions of neuronal membrane, generate a typical sub-threshold K+ conductance and regulate the excitability of many types of neurons. In hippocampal pyramidal cells, the channel is found in the peri-somatic regions where it modulates EPSPs integration and set the threshold for EPSP-spike coupling (Shah et al., 2008). The expression of M channels is finely tuned during the development of early forms of neuronal synchrony. Indeed, in immature CA3 pyramidal cells, the “giant depolarizing potentials” (GDPs) exist thanks to the excitatory nature of GABA in synergy with M channel down-regulation. The progressive disappearance of GDPs, following the first postnatal week, is matched by enhancement of M current density (Safiulina et al., 2008) and development of more organized forms of activity, such as theta and gamma rhythms (Buzsáki and Draguhn, 2004).
KCNQ3 and KCNQ2 gene mutations segregate with various forms of Kv7.3/Kv7.2-channelepsies (Maljevic et al., 2008; D’Adamo et al., 2013) such as benign familial neonatal convulsion (BFNC) and rolandic epilepsy. Importantly, >20% of patients with rolandic epilepsy have cognitive deficits and >10% display abnormal behavioral (ADHD, anxiety, depression, and pervasive developmental disorder; Tovia et al., 2011). Furthermore, 40% of patients with BFNC show delayed psychomotor development or intellectual disability (Steinlein et al., 2007). Several KCNQ3 mutations, associated with BFNC, reduce heteromeric M current amplitudes (Singh et al., 2003). Mice carrying the heterozygous G311V mutation in the channel’s pore show a lower threshold for epileptogenesis. While, homozygous mice exhibit spontaneous tonic–clonic seizures associated with reduced M current amplitudes and increased deactivation kinetics in hippocampal CA1 pyramidal cells (Singh et al., 2008). Interestingly, a de novo interstitial deletion in the long arm of chromosome 8, encompassing KCNQ3, has been identified in two children with a broad spectrum of congenital abnormalities, psychomotor delay and convulsions (Verheij et al., 2009). Another study from Gilling et al. (2013) identified the de novo reciprocal translocation t(3;8)(q21, q24) truncating KCNQ3 gene and the missense mutation P574S in distinct autistic patients. The de novo translocation was identified in a Danish boy affected by idiopathic periodic trembling since the age of 2 days that persisted for the first 5 weeks of life. In one case, the P574S variant, which substitutes a phylogenetically conserved residue in the C-terminal region of Kv7.3, was inherited from the mother who suffered from major depression. Surprisingly, none of the patients harboring this variant had histories of seizures. Functionally, the P574S variant significantly reduces K+ current amplitude in oocytes when co-expressed with Kv7.5 subunit but not with Kv7.2 or Kv7.4 subunits (Gilling et al., 2013; Table 1). Given the importance of M current in the development of neuronal identity and regulation of excitability, its impairment in immature neurons can depolarize membrane potential increasing cell excitability, facilitating the onset of seizures and delaying the definition of complex neuronal rhythms that possibly result in autistic phenotype.
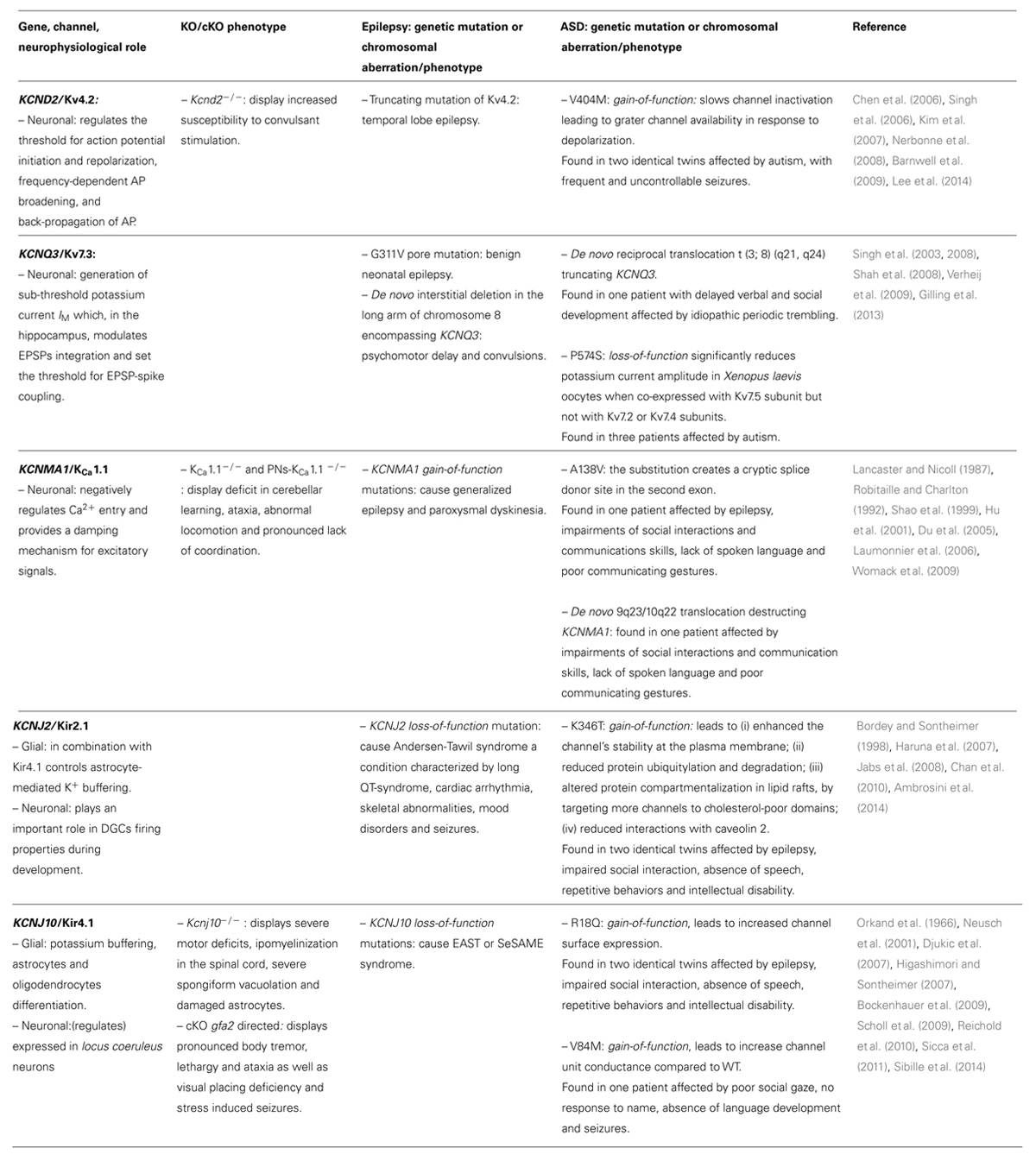
TABLE 1. Direct involvement of K+ channels in ASD.
Ca2+-Activated K+ Channels
The calcium-activated K+ (KCa) channels are highly conserved across species, and widely expressed in the human brain. The phylogenetic tree of KCa channels shows that they are made of two well distinct groups (Wei et al., 2005), the large conductance (BK; KCa1.1), and the small/intermediate-conductance (SK/IK; KCa2.1, KCa2.2, KCa2.3, KCa3.1) KCa channels. With regard to gating mechanism, the Ca2+ sensitivity of SK/IKchannels is provided by tightly bound calmodulin (Xia et al., 1998; Fanger et al., 1999), in contrast to the direct binding of Ca2+ at specific internal sites on the channel protein of KCa1.1 channels (Lee and Cui, 2010). Moreover, unlike the SK/IK channels, KCa1.1 channels are also activated by voltage. In brain neurons, KCa channels are widely distributed in axons and presynaptic terminals (Knaus et al., 1996; Blank et al., 2004), often located close to voltage-gated Ca2+ channels Cav (Marrion and Tavalin, 1998). Ca2+ influx that results from neuronal excitation activates KCa channels, whose outward K+ flux contributes to terminate the action potential, establish the afterhyperpolarization (AHP) and close Cav channels. This negative feed-back control has been generally assumed to make KCa channels critical players in opposing repetitive firing and hyperexcitability typical of epileptic disorders. To date mutations only in the KCa1.1 channel type have been clearly associated to both autism and epilepsy.
KCa1.1 – ChannelASD
The KCNMA1 gene encodes for the α subunit of KCa1.1 channel which possesses a tetrameric structure with four optional auxiliary β subunits (Sausbier et al., 2004). Channel activation requires both an increase in intracellular Ca2+ concentration and membrane depolarization (Storm, 1987; Robitaille et al., 1993). Thanks to its Ca2+- and voltage-dependence it negatively regulates Ca2+ entry and provides a damping mechanism for excitatory signals in many neuronal types. KCa1.1 channels are expressed in the hippocampus, cortex and cerebellum where they contribute to both action potential repolarization and brief AHP (Lancaster and Nicoll, 1987; Robitaille and Charlton, 1992; Shao et al., 1999; Hu et al., 2001; Womack et al., 2009). Experimentally, the contribution of presynaptic IKCa1.1 is not easily detectable under basal conditions (Storm, 1987). It can be unmasked by blocking voltage-gated K+ channels with 4-aminopyridine that broadens the presynaptic action potential. Under these conditions, blocking KCa1.1 channels in CA1 with iberiotoxin (IbTX) causes further broadening of the presynaptic compound action potential, enhancement of synaptic transmission and reduced paired-pulse facilitation ratio (Hu et al., 2001). In cerebellar Purkinje neurons (PNs), the contribution of KCa1.1 channel to the AHP phase is developmentally regulated with a greater contribution in immature neurons than in adult PNs (Womack et al., 2009). In vivo, deletion of the Kcnma1 gene profoundly alters cerebellar function: KCa1.1-/- mice show an abnormal conditioned eye blink response, abnormal locomotion and pronounced lack of coordination (Sausbier et al., 2004). These changes are, in part, the direct consequence of a profound disinhibition of deep cerebellar nuclei (DCN). Indeed, while all the wild-type PNs discharge spontaneous APs at baseline, 50% of KCa1.1-/- PNs become quiescent. Since these silent neurons have a depolarized membrane potential, a depolarization block mediated by inactivated voltage-gated Na+ channels has been invoked as the underlying molecular mechanism (Sausbier et al., 2004). Thus, KCa1.1 channels ablation dramatically lowers the overall discharge activity of PNs. The close correlation between the loss of the KCa1.1 channel in PNs and the cerebellar deficit was further confirmed by the genesis of PN-KCa1.1-/- mice, where KCa1.1 expression was specifically abolished in PNs (Chen et al., 2010). These animals recapitulate the motor deficits observed in KCa1.1-/- mice, although to a lesser extent and show, in vivo, a reduction of both simple spike (SS) and complex spike (CS) activity in PNs. Recall that SS is generated by afferent parallel fibers (PF) while CS is induced by climbing fibers (CF). In these mice, the olivo-cerebellar circuit is likely deregulated since reductions in PNs inhibitory inputs result in an enhancement of DCNs activity which, in turn, exert an excessive inhibition on downstream inferior olive (IO). Interestingly, the application of muscimol (a GABAA receptor agonist) onto DCN partially restores CS activity (Chen et al., 2010).
In humans, the KCNMA1 gain-of-function mutation D434G has been found in patients suffering from KCa1.1-channelepsy that is characterized by generalized epilepsy and paroxysmal dyskinesia (Du et al., 2005; D’Adamo et al., 2013). Expression studies indicated that the D434G mutant channel results in markedly greater macroscopic currents and single-channel open probability due to ~5-fold increase in Ca2+ sensitivity (Du et al., 2005). The enhancement of KCa1.1 channel activity may lead to increased excitability by inducing rapid repolarization of action potentials, thus allowing neurons to fire at faster rates (Du et al., 2005). The possible role of KCa1.1 channels in ASD phenotype has been highlighted by Laumonnier et al. (2006) who have identified both the substitution A138V and the de novo translocation t(9;10) (q23; q22) in the KCNMA1 gene of two unrelated autistic patients (Table 1). The autistic patient carrying the substitution displayed epilepsy, impairments of reciprocal social interactions and communications skills, lack of spoken language and poor communicating gestures. The A138V substitution is located within the second exon of KCNMA1 and changes a residue highly conserved in evolution from C. elegans to mammals. Sequence analysis showed that the substitution creates a cryptic splice donor site that likely impairs channel expression. The de novo translocation was found in a 6 year old boy, displaying typical symptoms of autism. The translocation separates the promoter of KCNMA1 from the rest of the gene, leading to a non-functional allele and haploinsufficiency. In fact, the KCNMA1 transcript levels (determined from patient’s lymphoblastoid cells) were halved. In addition, patch-clamp recordings from these cells revealed depolarized Vm, increased input resistance and halved IbTX-sensitive current (Laumonnier et al., 2006). Physiological KCa1.1 channel dosage is crucial for network functionality. Indeed, either loss or gain of KCa1.1 channel activity are both capable to unbalance cell excitability, markedly (Sausbier et al., 2004; Du et al., 2005; Chen et al., 2010). KCa1.1 loss-of-function mutations likely alter pyramidal neurons excitability and result in impairment of neural networks in hippocampus, an area implicated in cognition, mood disorders, and ASD. However, these mutations may also affect cerebellar PNs excitability, development, learning and memory processes, suggesting that KCa1.1 channels dysfunction may impact these crucial neurophysiological processes occurring within the cerebellum and result in the psychomotor development and cognition impairments of ASD (see below).
KCa1.1 – and Slack – ChannelASD linked to Fragile X syndrome
Recently, KCa1.1 channels have been implicated in ASD on a different ground, since their activity is regulated by FMRP, whose mutation produces FXS. We have mentioned previously that FMRP is mainly located postsynaptically, however, important pre-synaptic functions have been recently unmasked. FMRP is present in developing growth cones of axons (Antar et al., 2006), and it has been proposed to play pre-synaptic roles in neural wiring establishment (Hanson and Madison, 2007; Christie et al., 2009). Importantly, in pre-synaptic terminals FMRP regulates KCa1.1 channel Ca2+ sensitivity via interactions with the channel’s regulatory β4 subunit (Deng et al., 2013). Loss of FMRP, as obtained in Fmr1-null mice, reduces KCa1.1 Ca2+ sensitivity leading to smaller current, AP broadening and increased short-term plasticity (STP) in hippocampal CA3 pyramidal neurons (Deng et al., 2011, 2013; Table 2). The application of IbTX, a selective inhibitor of KCa1.1 channels, reproduced in WT mice the effects observed in Fmr1-null mice (Deng et al., 2013). In addition, Fmr1-null mice displayed abnormal eye-blink conditioning, atypically elongated PNs spines and enhanced LTD at PF-PNs synapses, leading to reduced PNs response to PF stimuli. The enhancement in LTD induction found in Fmr1-null mice was reproduced in animals where the Fmr1 gene was ablated specifically in PNs. This more direct evidence supports the notion that intrinsic PNs defects account for altered synaptic plasticity in the cerebellum of animals lacking FMRP and the crucial role that this brain structure may play in ASDs (Koekkoek et al., 2005).
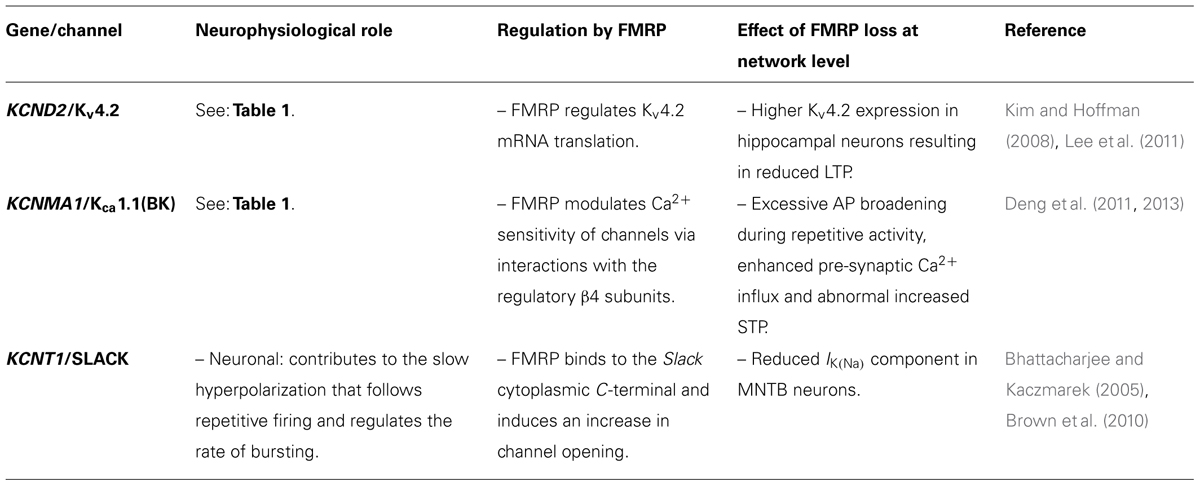
TABLE 2. Indirect involvement of K+ channels in ASD.
Notably, FMRP can also bind to Na+-activated K+ channel Slack, and thus regulate its activity (Brown et al., 2010). This channel type is mainly expressed in the cortex, hippocampus, olfactory bulb and lateral MNTB neurons of the brainstem (Brown et al., 2008) where it contributes to the slow hyperpolarization that follows repetitive firing and regulates the rate of bursting (Bhattacharjee and Kaczmarek, 2005). At the single channel level, Slack generates a very high conductance (~180 pS) with frequent transitions to a relatively long-lived sub-state with a conductance about one-third that of the fully open state. The binding of FMRP to the cytoplasmic C-terminal of Slack induces a fast and dramatic effect on these channel gating properties, namely, the transition to the sub-states is almost completely abolished, and there is a marked increase in channel opening (Brown et al., 2010). In Fmr1-KO mice, whole-cell recording of lateral MNTB neurons shows a significantly reduced IK(Na) component compared to WT, consistent with a role for FMRP in regulating the activity of native Slack channels (Brown et al., 2010). Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is caused by mutations in the CHRNA4, CHRNA2, CHRNB2, or KCNT1(Slack) genes (Steinlein et al., 1995; De Fusco et al., 2000; Aridon et al., 2006; Heron et al., 2012). Interestingly, intellectual disability only occurs in those patients who carry mutations in Slack channels (Heron et al., 2012), further suggesting a role for this channel type in both epilepsy and cognitive disorders (reviewed in Kim and Kaczmarek, 2014).
Inwardly Rectifying K+ Channels
Inwardly rectifying K+ (Kir) channels take their name from the greater conductance at potentials negative to EK, while at more positive values the outward flow of K+ ions is variably inhibited by cytoplasmic polyamines and Mg2+, by means of affinity dependent blockade. Indeed, the different sensitivity of Kir channels to polyamines and Mg2+ dictates the degree of rectification from weak to strong (Matsuda et al., 1987; Lopatin et al., 1994; Lu and Mac Kinnon, 1994; Stanfield et al., 1994). Cells that possess large Kir conductance display a resting membrane potential (Vm) close to EK. Beside setting the Vm values, Kir channels of excitable cells play key roles in the regulation of action potential duration and excitability. Several Kir clones have been identified and classified into seven major subfamilies, Kir1.x-Kir7.x (Bond et al., 1994; Hibino et al., 2010). Structurally, a Kir subunit possesses two transmembrane domains separated by a pore-forming region, serving as the “ion-selectivity filter,” with both the N- and C-termini residing inside the cell. Four subunits are assembled as both homotetramers and heterotetramers (Hibino et al., 2010).
Kir2.1 – ChannelASD
KCNJ2 encodes for Kir2.1 channels that possess a conductance of ~30 pS and generate macroscopic currents with strong rectification properties. High levels of KCNJ2 transcript are found in the brain, heart and skeletal muscle. In the brain, Kir2.1 is predominantly expressed in the hippocampus, caudate, putamen, nucleus accumbens, and to lower levels in habenula and amygdala (Karschin et al., 1996), where it contributes to control neuronal excitability. In particular, the amplitude of Kir2.1 currents is small in young dentate granule neurons (DGCs) and increases ~3-fold in mature DGCs to optimize their excitability. Thus, Kir2.1 channels play an important role in DGCs firing properties during development (Mongiat et al., 2009). Kir2.1 is also expressed in astrocytes and, in combination with Kir4.1, controls astrocyte-mediated K+ buffering (see below; Bordey and Sontheimer, 1998; Jabs et al., 2008; Chever et al., 2010). Loss-of-function mutations in the KCNJ2 gene are responsible for the rare Andersen-Tawil syndrome (OMIM 170390) a condition characterized by long QT-syndrome, cardiac arrhythmia, skeletal abnormalities, periodic paralysis, mood disorders, and seizures (Haruna et al., 2007; Chan et al., 2010). Conversely, Kir2.1 gain-of-function mutations segregate with SQT3 syndrome (OMIM 609622), another cardiac disorder characterized by QT shortening, ventricular tachyarrhythmias and atrial fibrillation. Indeed, in the heart Kir2.1 is a component of the inward-rectifier current IK1, which provides substantial repolarizing current during the terminal repolarization phase of the cardiac action potential and is the primary conductance controlling the diastolic membrane potential (Hutter and Noble, 1960; Sanguinetti and Tristani-Firouzi, 2000).
Recently, we reported on the identification of a new K346T heterozygous mutation in the KCNJ2 of monozygotic twins displaying autism and epilepsy in cis with the previously detected R18Q variant in KCNJ10 (Sicca et al., 2011; Ambrosini et al., 2014; Table 1). The twins also showed an electrocardiogram (ECG) with a markedly short repolarization time and conspicuously narrow and peaked T waves (QTc interval, 331 ms). K346T expression resulted in larger homozygous and heterozygous K+ currents due to increased surface expression of the channel in oocytes, HEK293 and glial-like cells. Functionally, several deleterious defects were also described for this novel KCNJ2 variant which: (i) enhanced the channel’s stability at the plasma membrane; (ii) reduced protein ubiquitylation and degradation; (iii) altered protein compartmentalization in lipid rafts, by targeting more channels to cholesterol-poor domains; (iv) reduced interactions with caveolin 2. All these molecular mechanisms contributed to causing Kir2.1 gain-of-function. Notably, the mutation promotes the surface expression of the channels particularly at end-feet, filopodia-like structures and cell–cell contacts. These structures are essential for astrocyte-mediated K+ siphoning through Kir2.1, Kir4.1, and Kir5.1 channels, all of which could be influenced by the K346T mutation. Given that Kir2 channels also contribute to regulating neuronal excitability, cell differentiation, synaptic plasticity and wiring, their dysfunction may impact these crucial neurophysiological processes. Therefore, this study proposed that genetically induced Kir2.1 defects, beside causing SQT3 syndrome, may possibly result in functional impairment of neural networks where this channel type resides and contributes to ASDs pathogenesis (Ambrosini et al., 2014).
Kir4.1 – ChannelASD
The KCNJ10 gene encodes for Kir4.1 channels (Bond et al., 1994), which forms homomeric channels or polymerize with Kir5.1 (KCNJ16) to form heterotetramers (Pessia et al., 1996) highly sensitive to pH (Tucker et al., 2000; Pessia et al., 2001; Casamassima et al., 2003; D’Adamo et al., 2011b). Kir4.1 is mainly expressed in locus coeruleus neurons, oligodendrocytes and astrocytes surrounding synapses and blood vessels from cortex, thalamus, hippocampus, and brainstem (Takumi et al., 1995; Higashi et al., 2001; D’Adamo et al., 2011b). In these cells, Kir4.1 channels generate high K+ permeability driving the membrane potential near EK values. During excitatory synaptic activity, astrocytes surrounding synapses slowly depolarize because of K+ influx across their membranes (Orkand et al., 1966). This prolonged inward K+ current is mediated mainly by Kir4.1 and is synchronized with synaptic and spiking activity (Sibille et al., 2014). Thanks to the functional coupling between neurons and astrocytes the excess of extracellular K+ ions, resulting from intense neuronal firing, is taken up by astrocytes by flowing through Kir4.1 channels. Then, K+ ions are transferred toward sites where their concentration is kept at low levels by the gap-junction cell-syncytium (Kuffler and Nicholls, 1966). By this mechanism, known as “K+ siphoning,” the concentration of extracellular K+ in CNS is strictly ruled about 2–3 mM, avoiding neuronal after-discharges and depolarization block. In the mouse hippocampus, Kir4.1 is significantly up-regulated between P3 and P12 (Seifert et al., 2009) and its expression is closely related to cell differentiation. In fact, Kir4.1 channels are absent in immature proliferating cells of the glia, and its progressive expression is linked to astrocytes hyperpolarization, differentiation and exit from cell cycle (from G2/M to G0/G1; Higashimori and Sontheimer, 2007). Genetic ablation of Kir4.1 in rodents results in severe motor deficits, hypomyelination in the spinal cord, severe spongiform vacuolation, astrocytes with immature morphology and premature death before P24 (Neusch et al., 2001). Conditional ablation of Kir4.1, exclusively in astrocytes, resulted in a mouse phenotype characterized by pronounced body tremor, lethargy, ataxia as well as visual placing deficiency and stress-induced seizures (Djukic et al., 2007). In humans, KCNJ10 loss-of-function mutations segregate with the EAST or SeSAME syndrome, a rare autosomic recessive disorder characterized by epilepsy, ataxia, sensorineural deafness, and tubulopathy. The patients develop tonic–clonic seizures in infancy followed by motor delay, speech and mental retardation (Bockenhauer et al., 2009; Scholl et al., 2009; Reichold et al., 2010). In line with the pathophysiological relationship between the Kir4.1 channel impairment and both epilepsy and developmental disorders, we investigated the frequency of KCNJ10 mutations in several children with cryptogenic epilepsy and autism spectrum traits. We found two inherited heterozygous KCNJ10 mutations at residues highly conserved in mammalian and vertebrate orthologs: R18Q in two identical twins and V84M in another child. The expression of mutant channels in Xenopous laevis oocytes resulted in current/voltage relationships with greater amplitudes than WT for both mutations, suggesting a gain-of-function effect. Patch-clamp single-channel recordings revealed that the V84M mutation increased the unit conductance ~1.5 fold compared to WT. The R18Q mutation altered neither the unit conductance nor the single channel parameters, yet increased the channel surface expression. Clinically, the two identical twins showed impaired social interaction, sleep difficulties, hypotonia and both exhibited epileptic spasms occurring within the same 24 h period. Other symptoms, typical of ASD, included clumsiness, absence of speech, severe disorder of social interaction, stereotypies, repetitive behaviors, symptoms of anxiety, depression, obsessive compulsive disorder and intellectual disability (IQ: 58). The child harboring the V84M mutation showed normal psychomotor development until 12 months of age, when ASD symptoms such as poor social gaze, no response to name, absence of language development, and withdrawal behaviors became evident. At the age of six, he experienced complex partial seizures. EEG recordings showed synchronous and asynchronous paroxysmal abnormalities over frontal regions in both hemispheres, tending to spread (Sicca et al., 2011; Table 1). Co-occurrence of epilepsy and ASD in patients harboring KCNJ10 gain-of-function mutations suggests that dysfunction in the astrocytic-dependent K+ buffering may be a common mechanism contributing to seizures as well as the core behavioral features of ASD. Kir4.1 gain-of-function mutations may lead to faster and larger influx of K+ into astrocytes, during intense neuronal activity, resulting in membrane depolarization and higher intracellular Ca2+ elevations in these cells. Ca2+ elevations in astrocytes are associated with gliotransmitters’ release, such as glutamate and D-serine that trigger discharges in neurons and promote local neuronal synchrony and epileptic activity (Steinhäuser et al., 2015). Abnormal synaptic function is the common basis for ASDs. Inasmuch as the activity of many thousands of synapses is controlled by a single astrocyte and this cell type makes up 90% of all human brain cells, defective astrocyte-dependent CNS development and regulation of K+ homeostasis in the brain represent original mechanistic hypotheses linking the allelic variations we have identified in KCNJ2 and KCNJ10 and ASD.
ChannelASD-Channelepsy Phenotype
Mutations in K+ channels have been widely associated with several forms of channelepsies (D’Adamo et al., 2013). The analysis of the literature performed here shows that in nearly all cases affected by K+ channelASDs, epilepsy is always comorbid. The degree of clinical and genetic overlap between K+ channelASDs and some K+ channelepsies, suggests that a subcategory named channelASD-channelepsy phenotype may be distinguished. Indeed, this scenario could be envisioned for some other channel types, playing significant roles in brain development, behavior and cognition. An effective example in this direction comes from Dravet’s syndrome (DS), a devastating neurodevelopmental disorder (Catterall, 2000; De Jonghe, 2011). DS is characterized by comorbidity of epilepsy and psychiatric disorders, and linked to Nav1.1(SCN1A) channel dysfunction. Indeed, together with early-life intractable seizures, patients develop devastating psychomotor, cognitive and behavioral deficits persisting through adulthood (Bender et al., 2012). Thus, SCN1A has been suggested as a candidate gene for ASD (Weiss et al., 2003). The implication of this gene was confirmed by whole-exome sequencing of sporadic ASD cases that revealed a missense mutation in a severely affected patient with evidence of early onset, language delay, epilepsy, and mild intellectual disability. This mutation leads to replacement of the highly conserved proline 1894 with lysine, and is predicted to be functionally deleterious (O’Roak et al., 2011). Scn1a-/+ rodents recapitulate DS (Yu et al., 2006; Kalume et al., 2007). The loss of Nav1.1 channels, which is mostly expressed in GABAergic interneurons, leads to a marked reduction in the inhibitory activity of these cells that reverberates onto prefrontal and hippocampal pyramidal neurons, which consequently fire at higher frequencies (Han et al., 2012). Remarkably, these mice show epilepsy and a spectrum of behavioral abnormalities characteristic of ASD such as hyperactivity, anxiety, altered behavioral interaction and deficits in context-dependent spatial memory (Yu et al., 2006; Han et al., 2012). Thus, it could be possible to hypothesize that distinct channel-dependent autism/epilepsy cases share a common etiopathological denominator, namely an excitatory-inhibitory imbalance generating a non-permissive substrate for the physiological development of the proper cognitive and behavioral skills, resulting in channelASD-channelepsy phenotype.
Concluding Remarks
Autism spectrum disorders are amongst the most common neuropsychiatric diseases (Blumberg et al., 2013) that show increasing prevalence over the past years and no discrimination in terms of ethnicity, family income or educational levels. However, how much of this increase is due to a broader definition of ASD and better efforts in diagnosis or an effective increased incidence of the disorders, is unclear. The number of ASDs cases that could be accounted for a channel-dependent pathogenesis and classified in the channelASD subgroup, remains to be established. Inasmuch as ~70 million people suffer from ASD and nearly 500 ion channel proteins are encoded by the human genome, the worldwide number of channelASD cases might not be negligible.
Despite the efforts made, the etiology of ASD remains largely elusive because many cases arise from a mixture of multiple environmental and genetic factors. In addition, the modalities by which the combinations of different genetic variations contribute to the overall risk to develop the disease are mostly obscure. Nevertheless, a mounting body of evidence indicates that ion channel dysfunction may well enhance autism susceptibility (Schmunk and Gargus, 2013) also when other contributing alleles are co-inherited. Direct and indirect defects in K+ channels have been implicated in ASDs pathogenesis, likely altering crucial neural network processes in several brain areas including the cerebellum, a structure that emerges as critically involved in determining the core features of ASDs. Abnormal synaptic transmission and dendritic spine pathology play crucial roles in ASDs. Notably, the activity of many thousands synapses is controlled by a single astrocyte. Thus, aberrant astrocyte-dependent synaptic functions and CNS development, induced by defective ion channels, represent an interesting causative hypotheses for ASDs (D’Adamo et al., 2011a; Sicca et al., 2011; Ambrosini et al., 2014). Undeniably, the high heterogeneity of ASDs makes the solution of the “autism puzzle” an extremely difficult task. Notwithstanding, to ensure the best possible outcomes for children affected by this devastating disease, comprehensive socio–economic policies and coordinated scientific efforts are required as a matter of urgency.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ambrosini, E., Sicca, F., Brignone, M. S., D’Adamo, M. C., Napolitano, C., Servettini, I.,et al. (2014). Genetically induced dysfunctions of Kir2.1 channels: implications for short QT3 syndrome and autism-epilepsy phenotype. Hum. Mol. Genet. 23, 4875–4886. doi: 10.1093/hmg/ddu201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Antar, L. N., Li, C., Zhang, H., Carroll, R. C., and Bassell, G. J. (2006). Local functions for FMRP in axon growth cone motility and activity-dependent regulation of filopodia and spine synapses. Mol. Cell. Neurosci. 32, 37–48. doi: 10.1016/j.mcn.2006.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Aridon, P., Marini, C., Di Resta, C., Brilli, E., De Fusco, M., Politi, F.,et al. (2006). Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am. J. Hum. Genet. 79, 342–350. doi: 10.1086/506459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bahring, R., Boland, L. M., Varghese, A., Gebauer, M., and Pongs, O. (2001). Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535, 65–81. doi: 10.1111/j.1469-7793.2001.00065.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bahring, R., and Covarrubias, M. (2011). Mechanisms of closed-state inactivation in voltage-gated ion channels. J. Physiol. 589, 461–479. doi: 10.1113/jphysiol.2010.191965
Barghaan, J., and Bahring, R. (2009). Dynamic coupling of voltage sensor and gate involved in closed-state inactivation of kv4.2 channels. J. Gen. Physiol. 133, 205–224. doi: 10.1085/jgp.200810073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barnwell, L. F., Lugo, J. N., Lee, W. L., Willis, S. E., Gertz, S. J., Hrachovy, R. A.,et al. (2009). Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia 50, 1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bechara, E. G., Didiot, M. C., Melko, M., Davidovic, L., Bensaid, M., Martin, P.,et al. (2009). A novel functionfor fragile X mental retardation protein in translational activation. PLoS Biol. 7:e16. doi: 10.1371/journal.pbio.1000016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bender, A. C., Morse, R. P., Scott, R. C., Holmes, G. L., and Lenck- Santini, P. P. (2012). SCN1A mutations in Dravet syndrome: impact of interneuron dysfunction on neural networks and cogni- tive outcome. Epilepsy Behav. 23, 177–186. doi: 10.1016/j.yebeh.2011.11.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bhattacharjee, A., and Kaczmarek, L. K. (2005). For K+ channels, Na+ is the new Ca2+. Trends Neurosci. 28, 422–428. doi: 10.1016/j.tins.2005.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Birnbaum, S. G., Varga, A. W., Yuan, L. L., Anderson, A. E., Sweatt, J. D., and Schrader, L. A. (2004). Structure and function of Kv4-family transient potassium channels. Physiol. Rev. 84, 803–833. doi: 10.1152/physrev.00039.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blank, T., Nijholt, I., Kye, M. J., and Spiess, J. (2004). Small conductance Ca2+- activated K+ chan- nels as targets of CNS drug development. Curr. Drug Targets CNS Neurol. Disord. 3, 161–167. doi: 10.2174/1568007043337472
Blumberg, S. J., Bramlett, M. D., Kogan, M. D., Schieve, L. A., Jones, J. R., and Lu, M. C. (2013). Changes in Prevalence of Parent-Reported Autism Spectrum Disorder in School-Aged U.S. Children: 2007 to 2011–2012. Hyattsville, MD: National Center for Health Statistics.
Bockenhauer, D., Feather, S., Stanescu, H. C., Bandulik, S., Zdebik, A. A., Reichold, M.,et al. (2009). Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 360, 1960–1970. doi: 10.1056/NEJMoa0810276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bond, C. T., Pessia, M., Xia, X. M., Lagrutta, A., Kavanaugh, M. P., and Adelman, J. P. (1994). Cloning and expression of a family of inward rectifier potassium channels. Receptors Channels 2, 183–191.
Bordey, A., and Sontheimer, H. (1998). Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 32, 286–303. doi: 10.1016/S0920-1211(98)00059-X
Brown, D. A., and Adams, P. R. (1980). Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283, 673–676. doi: 10.1038/283673a0
Brown, M. R., Kronengold, J., Gazula, V. R., Chen, Y., Strumbos, J. G., Sigworth, F. J.,et al. (2010). Fragile X mental retardation protein controls gating of the sodium-activated potassium channel slack. Nat. Neurosci. 13, 819–821. doi: 10.1038/nn.2563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brown, M. R., Kronengold, J., Gazula, V. R., Spilianakis, C. G., Flavell, R. A., Von Hehn, C. A.,et al. (2008). Amino-termini isoforms of the slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J. Physiol. 586, 5161–5179. doi: 10.1113/jphysiol.2008.160861
Buzsáki, G., and Draguhn, A. (2004). Neuronal oscillations in cortical networks. Science 304, 1926–1929. doi: 10.1126/science.1099745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Casamassima, M., D’Adamo, M. C., Pessia, M., and Tucker, S. J. (2003). Identification of a heteromeric interaction which influences the rectification gating and pH-sensitivity of Kir41/ Kir51 potassium channels. J. Biol. Chem. 278, 43533–43540. doi: 10.1074/jbc.M306596200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi: 10.1016/S0896-6273(00)81133-2
Chan, H. F., Chen, M. L., Su, J. J., Ko, L. C., Lin, C. H., and Wu, R. M. (2010). A novel neuropsychiatric phenotype of KCNJ2 mutation in one Taiwanese family with Andersen-Tawil syndrome. J. Hum. Genet. 55, 186–188. doi: 10.1038/jhg.2010.2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandy, K. G., and Gutman, G. A. (1993). Nomenclature form a mammalian potassium channel genes. Trends Pharmacol. Sci. 14, 434. doi: 10.1016/0165-6147(93)90181-I
Chen, X., Kovalchuk, Y., Adelsberger, H., Henning, H. A., Sausbier, M., Wietzorrek, G.,et al. (2010). Disruption of the olivo-cerebellar circuit by Purkinje neuron-specific ablation of BK channels. Proc. Natl. Acad. Sci. U.S.A. 107, 12323–12328. doi: 10.1073/pnas.1001745107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, X., Yuan, L. L., Zhao, C., Birnbaum, S. G., Frick, A., Jung, W. E.,et al. (2006). Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiationin hippocampal CA1 pyramidal neurons. J. Neurosci. 26, 12143–12151. doi: 10.1523/JNEUROSCI.2667-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chever, O., Djukic, B., McCarthy, K. D., and Amzica, F. (2010). Implication of Kir4.1 channel in excess potassium clearance: an in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 30, 15769–15777. doi: 10.1523/JNEUROSCI.2078-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Christie, S. B., Akins, M. R., Schwob, J. E., and Fallon, J. R. (2009). The FXG: a presynaptic fragile X granule expressed in a subset of developing brain circuits. J. Neurosci. 29, 1514–1524. doi: 10.1523/JNEUROSCI.3937-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clarke, D. F., Roberts, W., Daraksan, M., Dupuis, A., McCabe, J., Wood, H.,et al. (2005). The prevalence of autistic spectrum disorder in children surveyed in a tertiary care epilepsy clinic. Epilepsia 46, 1970–1977. doi: 10.1111/j.1528-1167.2005.00343.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Covarrubias, M., Bhattacharji, A., De Santiago-Castillo, J. A., Dougherty, K., Kaulin, Y. A., Na-Phuket, T. R.,et al. (2008). The neuronal Kv4 channel complex. Neurochem. Res. 33, 1558–1567. doi: 10.1007/s11064-008-9650-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D’Adamo, M. C., Catacuzzeno., L., Di Giovanni, G., Franciolini, F., and Pessia, M. (2013). K+ channelepsy: progress in the neurobiology of potassium channels and epilepsy. Front. Cell Neurosci. 7:134. doi: 10.3389/fncel.2013.00134
D’Adamo, M. C., Di Giovanni, G., and Pessia, M. (2014). “Animal models of episodic ataxia Type 1 (EA1),” in Movement Disorders: Genetics and Models, 2nd Edn, ed. M. S. LeDoux (New York, NY: Academic Press Inc./Elsevier Science Publishing Co. Inc.).
D’Adamo, M. C., Moro, F., Imbrici, P., Martino, D., Roscini, M., Santorelli, F. M.,et al. (2011a). The emerging role of the inwardly rectifying K+ channels in autism spectrum disorders and epilepsy. Malta Med. J. 23, 10–14.
D’Adamo, M. C., Shang, L., Imbrici, P., Brown, S. D. M., Pessia, M., and Tucker, S. (2011b). Genetic inactivation of Kcnj16 identifies Kir 5.1 as an important determinant of neuronal PCO2/pH sensitivity. J. Biol. Chem. 286, 192–198. doi: 10.1074/jbc.M110.189290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Fusco, M., Becchetti, A., Patrignani, A., Annesi, G., Gambardella, A., Quattrone, A.,et al. (2000). The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat. Genet. 26, 275–276. doi: 10.1038/81566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Jonghe, P. (2011). Molecular genetics of Dravet syndrome. Dev. Med. Child Neurol. 53(Suppl. 2), 7–10. doi: 10.1111/j.1469-8749.2011.03965.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, P. Y., Rotman, Z., Blundon, J. A., Cho, Y., Cui, J., Cavalli, V.,et al. (2013). FMRP regulates neurotransmitter release and synaptic information transmission by modulating action potential duration via BK channels. Neuron 77, 696–711. doi: 10.1016/j.neuron.2012.12.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deng, P. Y., Sojka, D., and Klyachko, V. A. (2011). Abnormal presynaptic short term plasticity and information processing in a mouse model of fragile X syndrome. J. Neurosci. 31, 10971–10982. doi: 10.1523/JNEUROSCI.2021-11.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Djukic, B., Casper, K. B., Philpot, B. D., Chin, L. S., and McCarthy, K. D. (2007). Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake and enhanced short-term synaptic potentiation. J. Neurosci. 27, 11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Du, W., Bautista, J. F., Yang, H., Diez-Sampedro, A., You, S.-A., Wang, L.,et al. (2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 37, 733–738. doi: 10.1038/ng1585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eijkelkamp, N., Linley, J. E., Baker, M. D., Minett, M. S., Cregg, R., Werdehausen, R.,et al. (2012). Neurological perspectives on voltage-gated sodium channels. Brain 135, 2585–2612. doi: 10.1093/brain/aws 225
Fanger, C. M., Ghanshani, S., Logsdon, N. J., Rauer, H., Kalman, K., Zhou, J.,et al. (1999). Calmodulin mediates calcium-dependent activation of the intermediate con ductance KCa channel, IKCa1. J. Biol. Chem. 274, 5746–5754. doi: 10.1074/jbc.274.9.5746
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fu, Y. H., Kuhl, D. P., Pizzuti, A., Pieretti, M., Sutcliffe, J. S., Richards, S.,et al. (1991). Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 67, 1047–1058. doi: 10.1016/0092-8674(91)90283-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garber, K. B., Visootsak, J., and Warren, S. T. (2008). Fragile X syndrome. Eur. J. Hum. Genet. 16, 666–672. doi: 10.1038/ejhg.2008.61
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gebauer, M., Isbrandt, D., Sauter, K., Callsen, B., Nolting, A., Pongs, O.,et al. (2004). N-type inactivation features of Kv4.2 channel gating. Biophys. J. 86, 210–223. doi: 10.1016/S0006-3495(04)74097-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gilling, M., Rasmussen, H. B., Calloe, K., Sequeira, A. F., Baretto, M., Oliveira, G.,et al. (2013). Dysfunction of the heteromeric KV7.3/ KV7.5 potassium channel is associated with autism spectrum disorders. Front. Genet. 4:54. doi: 10.3389/fgene.2013.00054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Han, S., Tai, C., Westenbroek, R. E., Yu, F. H., Cheah, C. S., Potter, G. B.,et al. (2012). Autistic-like behaviour in Scn1a+/- mice and rescue by enhanced GABA-mediated neurotransmission. Nature 489, 385–390. doi: 10.1038/nature11356
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanson, J. E., and Madison, D. V. (2007). Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J. Neurosci. 27, 4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haruna, Y., Kobori, A., Makiyama, T., Yoshida, H., Akao, M., Doi, T.,et al. (2007). Genotype-phenotype correlations of KCNJ2 mutations in Japanese patients with Andersen-Tawil syndrome. Hum. Mutat. 28, 208. doi: 10.1002/humu.9483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Heron, S. E., Smith, K. R., Bahlo, M., Nobili, L., Kahana, E., Licchetta, L.,et al. (2012). Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 44, 1188–1190. doi: 10.1038/ng.2440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels, their structure, function, and physiological roles. Physiol. Rev. 90, 291–366. doi: 10.1152/physrev.00021.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Higashi, K., Fujita, A., Inanobe, A., Tanemoto, M., Doi, K., Kubo, T.,et al. (2001). An inwardly rectifying K+ channel Kir4.1 expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Cell Physiol. 281, C922–C931.
Higashimori, H., and Sontheimer, H. (2007). Role of Kir4.1 channels in growth control of glia. Glia 55, 1668–1679. doi: 10.1002/glia.20574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hou, L., Antion, M. D., Hu, D., Spencer, C. M., Paylor, R., and Klann, E. (2006). Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. doi: 10.1016/j.neuron.2006.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hu, H., Shao, L. R., Chavoshy, S., Gu, N., Trieb, M., Behrens, R.,et al. (2001). Presynaptic Ca2+-activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J. Neurosci. 21, 9585–9597.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750. doi: 10.1073/pnas.122205699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Huber, K. M., Kayser, M. S., and Bear, M. F. (2000). Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. doi: 10.1126/science.288.5469.1254
Hutter, O. F., and Noble, D. (1960). Rectifying properties of heart muscle. Nature 188, 495. doi: 10.1038/188495a0
Jabs, R., Seifert, G., and Steinhäuser, C. (2008). Astrocytic function and its alteration in the epileptic brain. Epilepsia 49, 3–12. doi: 10.1111/j.1528-1167.2008.01488.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerng, H. H., Dougherty, K., Covarrubias, M., and Pfaffinger, P. J. (2009). A novel N-terminal motif of dipeptidyl peptidase-like proteins produces rapid inactivation of KV4.2 channels by a pore-blocking mechanism. Channels (Austin) 3, 448–461. doi: 10.4161/chan.3.6.10216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerng, H. H., Kunjilwar, K., and Pfaffinger, P. J. (2005). Multiprotein assembly of Kv4.2, KChIP3 and DPP10 produces ternary channel complexes with ISA-like properties. J. Physiol. 568, 767–788. doi: 10.1113/jphysiol.2005.087858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerng, H. H., Pfaffinger, P. J., and Covarrubias, M. (2004). Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell. Neurosci. 27, 343–369. doi: 10.1016/j.mcn.2004.06.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jorde, L. B., Hasstedt, S. J., Ritvo, E. R., Mason-Brothers, A., Freeman, B. J., Pingree, C.,et al. (1991). Complex segregation analysis of autism. Am. J. Hum. Genet. 49, 932–938.
Jung, S. C., Kim, J., and Hoffman, D. A. (2008). Rapid, bidirectional remodeling of synaptic NMDA receptor subunit composition by A-type K+ channel activity in hippocampal CA1 pyramidal neurons. Neuron 60, 657–671. doi: 10.1016/j.neuron.2008.08.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kalume, F., Yu, F. H., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2007). Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. 27, 11065–74. doi: 10.1523/JNEUROSCI.2162-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karschin, C., Dissmann, E., Stuhmer, W., and Karschin, A. (1996). IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J. Neurosci. 16, 3559–35570.
Kim, E., and Hoffman, D. A. (2012). Dynamic regulation of synaptic maturation state by voltage-gated A-type K+ channels in CA1 hippocampal pyramidal neurons. J. Neurosci. 32, 14427–14432. doi: 10.1523/JNEUROSCI.2373-12.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, G. E., and Kaczmarek, L. K. (2014). Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell Neurosci. 8:209. doi: 10.3389/fncel.2014.00209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J., and Hoffman, D. A. (2008). Potassium channels: newly found players in synaptic plasticity. Neuroscientist 14, 276–286. doi: 10.1177/1073858408315041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J., Jung, S. C., Clemens, A. M., Petralia, R. S., and Hoffman, D. A. (2007). Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 54, 933–947. doi: 10.1016/j.neuron.2007.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knaus, H. G., Schwarzer, C., Koch, R. O., Eberhart, A., Kaczorowski, G. J., Glossmann, H.,et al. (1996). Distribution of high-conductance Ca2+-activated K+ channels in rat brain: targeting to axons and nerve terminals. J. Neurosci. 16, 955–963.
Koekkoek, S. K., Yamaguchi, K., Milojkovic, B. A., Dortland, B. R., Ruigrok, T. J., Maex, R.,et al. (2005). Deletion of FMR1 in Purkinje cells enhances parallel fiber LTD, enlarges spines, and attenuates cerebellar eyelid conditioning in Fragile X syndrome. Neuron 47, 339–352. doi: 10.1016/j.neuron.2005.07.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krawczun, M. S., Jenkins, E. C., and Brown, W. T. (1985). Analysis of the fragile-X chromosome: localization and detection of the fragile site in high resolution preparations. Hum. Genet. 69, 209–211. doi: 10.1007/BF00293026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kubisch, C., Schroeder, B. C., Friedrich, T., Lutjohann, B., El-Amraoui, A., Marlin, S.,et al. (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446. doi: 10.1016/S0092-8674(00)80556-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kuffler, S. W., and Nicholls, J. G. (1966). The physiology of neuroglial cells. Ergeb. Physiol. 57, 1–90. doi: 10.1007/BF02259903
Lancaster, B., and Nicoll, R. A. (1987). Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. 389, 187–203. doi: 10.1113/jphysiol.1987.sp016653
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Laumonnier, F., Roger, S., Guérin, P., Molinari, F., M’rad, R., Cahard, D.,et al. (2006). Association of a functional deficit of the BKCa channel, a synaptic regulator of neuronal excitability, with autism and mental retardation. Am. J. Psychiatry 163, 1622–1629. doi: 10.1176/appi.ajp.163.9.1622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lauterborn, J. C., Rex, C. S., Kramár, E., Chen, L. Y., Pandyarajan, V., Lynch, G.,et al. (2007). Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci. 27, 10685–10694. doi: 10.1523/JNEUROSCI.2624-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lavelle, T. A., Weinstein, M. C., Newhouse, J. P., Munir, K., Kuhlthau, K. A., and Prosser, L. A. (2014). Economic burden of childhood autism spectrum disorders. Pediatrics 133:e520. doi: 10.1542/peds.2013-0763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H. Y., Ge, W. P., Huang, W., He, Y., Wang, G. X., Rowson-Baldwin, A.,et al. (2011). Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. doi: 10.1016/j.neuron.2011.09.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, H., Lin, M. C., Kornblum, H. I., Papazian, D. M., and Nelson, S. F. (2014). Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum. Mol. Genet. 23, 3481–3489. doi: 10.1093/hmg/ddu056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, U. S., and Cui, J. (2010). BK channel activation: structural and functional insights. Trends Neurosci. 33, 415–423. doi: 10.1016/j.tins.2010.06.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lopatin, A. N., Makhina, E. N., and Nichols, C. G. (1994). Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature 372, 366–369. doi: 10.1038/372366a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lu, Z., and Mac Kinnon, R. (1994). Electrostatic tuning of Mg2+ affinity in an inward-rectifier K+ channel. Nature 371, 243–246. doi: 10.1038/371243a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maljevic, S., Wuttke, T. V., and Lerche, H. (2008). Nervous system KV7 disorders: breakdown of a subthreshold brake. J. Physiol. 586, 1791–801. doi: 10.1113/jphysiol.2008.150656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Marrion, N. V., and Tavalin, S. J. (1998). Selective activation of Ca2+- activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905. doi: 10.1038/27674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuda, H., Saigusa, A., and Irisawa, H. (1987). Ohmic conductance through the inwardly rectifying K+ channel and blocking by internal Mg2+. Nature 325, 156–159. doi: 10.1038/325156a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuo, M., Maeda, T., Sasaki, K., Ishii, K., and Hamasaki, Y. (2010). Frequent association of autism spectrum disorder in patients with childhood onset epilepsy. Brain Dev. 32, 759–763. doi: 10.1016/j.braindev.2010.05.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Michalon, A., Sidorov, M., Ballard, T. M., Ozmen, L., Spooren, W., and Wettstein, J. G. (2012). Chronic pharmacological mGlu5 inhibition corrects Fragile X in adult mice. Neuron 74, 49–56. doi: 10.1016/j.neuron.2012.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mikhailov, A., Choufani, S., Skaug, J., Kolozsvari, D., Marshall, C., Scherer, S. W.,et al. (2008). Chromosomal translocation t(5;7)(q14;q31) and missense mutations implicate the voltage-gated potassium channel Kv4.2 gene, KCND2, on 7q31 in autism. Paper Presented at the Annual Meeting of the American Society of Human Genetics, Philadelphia, PA.
Mongiat, L. A., Espòsito, M. S., Lombardi, G., and Schinder, A. F. (2009). Reliable activation of immature neurons in the adult hippocampus. PLoS ONE 4:e5320. doi: 10.1371/journal.pone.0005320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mouridsen, S. E., Rich, B., and Isager, T. (2011). A longitudinal study of epilepsy and other central nervous system diseases in individuals with and without a history of infantile autism. Brain Dev. 33, 361–366. doi: 10.1016/j.braindev.2010.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muddashetty, R., Kelić, S., Gross, C., Xu, M., and Bassell, G. (2007). Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J. Neurosci. 27, 5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nerbonne, J. M., Gerber, B. R., Norris, A., and Burkhalter, A. (2008). Electrical remodelling maintains firing properties in cortical pyramidal neurons lacking KCND2-encoded A-type K+ currents. J. Physiol. 586, 1565–1579. doi: 10.1113/jphysiol.2007.146597
Neusch, C., Rozengurt, N., Jacobs, R. E., Lester, H. A., and Kofuji, P. (2001). Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J. Neurosci. 21, 5429–5438.
Okamoto, N., Hatsukawa, Y., Shimojima, K., and Yamamoto, T. (2011). Submicroscopic deletion in 7q31 encompassing CADPS2 and TSPAN12 in a child with autism spectrum disorder and PHPV. Am. J. Med. Genet. A. 155A, 1568–1573. doi: 10.1002/ajmg.a.34028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Orkand, R. K., Nicholls, J. G., and Kuffler, S. W. (1966). Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 29, 788–806.
O’Roak, B. J., Deriziotis, P., Lee, C., Vives, L., Schwartz, J. J., Girirajan, S.,et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43, 585–589. doi: 10.1038/ng.835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pessia, M. (2004). “Ion channels and electrical activity,” in Molecular Biology of the Neuron, 2nd Edn, eds W. R. Davies and B. J. Morris (Oxford: Oxford University Press), 103–137. doi: 10.1093/acprof:oso/9780198509981.003.0005
Pessia, M., Imbrici, P., D’Adamo, M. C., Salvatore, L., and Tucker, S. J. (2001). Differential pH-sensitivity of Kir4.1and Kir4.2 and modulation by heteropolymerisation with Kir5.1. J. Physiol. 532, 359–367. doi: 10.1111/j.1469-7793.2001.0359f.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pessia, M., Tucker, S. J., Lee, K., Bond, C. T., and Adelman, J. P. (1996). Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J. 15, 2980–2987.
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T.,et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-I
Reichold, M., Zdebik, A. A., Lieberer, E., Rapedius, M., Schmidt, K., Bandulik, S.,et al. (2010). KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. U.S.A. 107, 14490–14495. doi: 10.1073/pnas.1003072107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rinehart, N., and McGinley, J. (2010). Is motor dysfunction core to autism spectrum disorder? Dev. Med. Child Neurol. 52, 697. doi: 10.1111/j.1469-8749.2010.03631.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rinehart, N. J., Tonge, B. J., Iansek, R., McGinley, J., Brereton, A. V., Enticott, P. G.,et al. (2006). Gait function in newly diagnosed children with autism: cerebellar and basal ganglia related motor disorder. Dev. Med. Child Neurol. 48, 819–824. doi: 10.1017/S0012162206001769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Robitaille, R., and Charlton, M. P. (1992). Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J. Neurosci. 12, 297–305.
Robitaille, R., Garcia, M. L., Kaczorowski, G. J., and Charlton, M. P. (1993). Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11, 645–655. doi: 10.1016/0896-6273(93)90076-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Safiulina, V. F., Zacchi, P., Taglialatela, M., Yaari, Y., and Cherubini, E. (2008). Low expression of Kv7/M channels facilitates intrinsic and network bursting in the developing rat hippocampus. J. Physiol. 586, 5437–5453. doi: 10.1113/jphysiol.2008.156257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sanguinetti, M. C., and Tristani-Firouzi, M. (2000). “Delayed and inward rectifier potassium channels,” in Cardiac Electrophysiology From Cell to Bedside, 3rd Edn, eds D. P. Zipes and J. Jalife (Philadelphia, PA: Elsevier/W.B. Saunders), 79–85.
Santoro, M. R., Bray, S, M., and Warren, S. T. (2012). Molecular mechanisms of Fragile X syndrome: a twenty-year perspective. Annu. Rev. Pathol. 7, 219–245. doi: 10.1146/annurev-pathol-011811-132457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sausbier, M., Hu, H., Arntz, C., Feil, S., Kamm, S., Adelsberger, H.,et al. (2004). Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc. Natl. Acad. Sci. U.S.A. 101, 9474–9478. doi: 10.1073/pnas.0401702101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaaf, C. P., and Zoghbi, H. Y. (2011). Solving the autism puzzle a few pieces at a time. Neuron 70, 806–808. doi: 10.1016/j.neuron.2011.05.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scherer, S. W., Cheung, J., MacDonald, J. R., Osborne, L. R., Nakabayashi, K., Herbrick, J. A.,et al. (2003). Human chromosome 7: DNA sequence and biology. Science 300, 767–772. doi: 10.1126/science.1083423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmunk, G., and Gargus, J. J. (2013). Channelopathy pathogenesis in autism spectrum disorders. Front. Genet. 4:222. doi: 10.3389/fgene.2013.00222
Scholl, U. I., Choi, M., Liu, T., Ramaekers, V. T., Häusler, M. G., Grimmer, J.,et al. (2009). Seizures sensorineural deafness ataxia mental retardation and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. U.S.A. 106, 5842–5847. doi: 10.1073/pnas.0901749106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schroeder, B. C., Hechenberger, M., Weinreich, F., Kubisch, C., and Jentsch, T. J. (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095. doi: 10.1074/jbc.M003245200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seifert, G., Hüttmann, K., Binder, D. K., Hartmann, C., Wyczynski, A., Neusch, C.,et al. (2009). Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J. Neurosci. 29, 7474–7488. doi: 10.1523/JNEUROSCI.3790-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shah, M. M., Migliore, M., Valencia, I., Cooper, E. C., and Brown, D. A. (2008). Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 7869–7874. doi: 10.1073/pnas.0802805105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shao, L. R., Halvorsrud, R., Borg-Graham, L., and Storm, J. F. (1999). The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J. Physiol. 521, 135–146. doi: 10.1111/j.1469-7793.1999.00135.x
Sibille, J., Pannasch, U., and Rouach, N. (2014). Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J. Physiol. 592, 87–102. doi: 10.1113/jphysiol.2013.261735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sicca, F., Imbrici, P., D’Adamo, M. C., Moro, F., Bonatti, F., Brovedani, P.,et al. (2011). Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol. Dis. 43, 239–247. doi: 10.1016/j.nbd.2011.03.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, B., Ogiwara, I., Kaneda, M., Tokonami, N., Mazaki, E., Baba, K.,et al. (2006). A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 24, 245–253. doi: 10.1016/j.nbd.2006.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, N. A., Otto, J. F., Dahle, E. J., Pappas, C., Leslie, J. D., Vilaythong, A.,et al. (2008). Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J. Physiol. 586, 3405–3423. doi: 10.1113/jphysiol.2008.154971
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Singh, N. A., Westenskow, P., Charlier, C., Pappas, C., Leslie, J., Dillon, J.,et al. (2003). KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 126, 2726–2737. doi: 10.1093/brain/awg286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Siomi, H., Siomi, M. C., Nussbaum, R. L., and Dreyfuss, G. (1993). The protein product of thefragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298. doi: 10.1016/0092-8674(93)90420-U
Stanfield, P. R., Davies, N. W., Shelton, P. A., Khan, I. A., Brammar, W. J., Standen, N. B.,et al. (1994). The intrinsic gating of inward rectifier K+ channels expressed from the murine IRK1gene depends on voltage, K+, and Mg2+. J. Physiol. 475, 1–7. doi: 10.1113/jphysiol.1994.sp020044
Steinhäuser, C., Grunnet, M., and Carmignoto, G. (2015). Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience doi: 10.1016/j.neuroscience.2014.12.047 [Epub ahead of print].
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinlein, O. K., Conrad, C., and Weidner, B. (2007). Benign familial neonatal convulsions: always benign? Epilepsy Res. 73, 245–249. doi: 10.1016/j.eplepsyres.2006.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R.,et al. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 11, 201–203. doi: 10.1038/ng1095-201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Storm, J. F. (1987). Intracellular injection of a Ca2+ chelator inhibits spike repolarization in hippocampal neurons. Brain Res. 435, 387–392. doi: 10.1016/0006-8993(87)91631-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takumi, T., Ishii, T., Horio, Y., Morishige, K., Takahashi, N., Yamada, M.,et al. (1995). A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 270, 16339–16346. doi: 10.1074/jbc.270.27.16339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tempel, B. L., Papazian, D. M., Schwarz, T. L., Jan, Y. N., and Jan, L. Y. (1987). Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 237, 770–775. doi: 10.1126/science.2441471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Todd, P., and Malter, J. (2002). Fragile X mental retardation protein in plasticity and disease. J. Neurosci. Res. 70, 623–630. doi: 10.1002/jnr.10453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tovia, E., Goldberg-Stern, H., Ben, Z. B., Heyman, E., Watemberg, N., Fattal- Valevski, A.,et al. (2011). The prevalence of atypical presentations and comorbidities of benign childhood epilepsy with centrotemporal spikes. Epilepsia 52, 1483–1488. doi: 10.1111/j.1528-1167.2011.03136.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tuchman, R., and Rapin, I. (2002). Epilepsy in autism. Lancet Neurol. 1, 352–358. doi: 10.1016/S1474-4422(02)00160-6
Tucker, J. S., Imbrici, P., Salvatore, L., D’Adamo, M. C., and Pessia, M. (2000). pH-dependence of the inwardly rectifying potassium channel Kir5.1and localisation in renal tubular epithelia. J. Biol. Chem. 275, 16404–16407. doi: 10.1074/jbc.C000127200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Waelvelde, H., Oostra, A., Dewitte, G., Van Den Broeck, C., and Jongmans, M. J. (2010). Stability of motor problems in young children with or at risk of autism spectrum disorders, ADHD, and or developmental coordination disorder. Dev. Med. Child Neurol. 52, e174–e178. doi: 10.1111/j.1469-8749.2009.03606.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verheij, C., Bakker, C. E., de Graaff, E., Keulemans, J., Willemsen, R., Verkerk, A. J.,et al. (1993). Characterization and localization of the FMR-1 gene product associated with fragile X syndrome. Nature 363, 722–724. doi: 10.1038/363722a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Verheij, J. B., de Munnik, S. A., Dijkhuizen, T., de Leeuw, N., Olde Weghuis, D., van den Hoek, G. J.,et al. (2009). An 8.35 Mb overlapping interstitial deletion of 8q24 in two patients with coloboma, congenital heart defect, limb abnormalities, psychomotor retardation and convulsions. Eur. J. Med. Genet. 52, 353–357. doi: 10.1016/j.ejmg.2009.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S.,et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wang, S., Patel, S. P., Qu, Y., Hua, P., Strauss, H. C., and Morales, M. J. (2002). Kinetic properties of Kv4.3 and their modulation by KChIP2b. Biochem. Biophys. Res. Commun. 295, 223–229. doi: 10.1016/S0006-291X(02)00658-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wegiel, J., Kuchna, I., Nowicki, K., Imaki, H., Wegiel, J., Ma, S. Y.,et al. (2013). Contribution of olivofloccular circuitry developmental defects to atypical gaze in autism. Brain Res. 1512, 106–122. doi: 10.1016/j.brainres.2013.03.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wei, A. D., Gutman, G. A., Aldrich, R., Chandy, K. G., Grissmer, S., and Wulff, H. (2005). International Union of Pharmacology LII Nomenclature and molecular relationships of calcium- activated potassium channels. Pharmacol. Rev. 57, 463–472. doi: 10.1124/pr.57.4.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K.,et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400. doi: 10.1073/pnas.94.10.5395
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiler, I., Spangler, C., Klintsova, A., Grossman, A., Kim, S., Bertaina-Anglade, V.,et al. (2004). Fragile X mental retardation protein is necessary for neurotransmitter activated protein translation at synapses. Proc. Natl. Acad. Sci. U.S.A 101, 17504–17509. doi: 10.1073/pnas.0407533101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weiss, L. A., Escayg, A., Kearney, J. A., Trudeau, M., MacDonald, B. T., Mori, M.,et al. (2003). Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 8, 186–194. doi: 10.1038/sj.mp.40x01241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Womack, M. D., Hoang, C., and Khodakhah, K. (2009). Large conductance calcium-activated potassium channels affect both spontaneous firing and intracellular calcium concentration in cerebellar Purkinje neurons. Neuroscience 162, 989–1000. doi: 10.1016/j.neuroscience.2009.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xia, X. M., Fakler, B., Rivard, A., Wayman, G., Johnson-Pais, T., Keen, J. E.,et al. (1998). Mechanism of calcium gating in small conductance calcium-activated potassium channels. Nature 395, 503–507. doi: 10.1038/26758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A.,et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zalfa, F., Giorgi, M., Primerano, B., Moro, A., Di Penta, A., Reis, S.,et al. (2003). The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112, 317–327. doi: 10.1016/S0092-8674(03)00079-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhao, C., Wang, L., Netoff, T., and Yuan, L. L. (2011). Dendritic mechanisms controlling the threshold and timing requirement of synaptic plasticity. Hippocampus 21, 288–297 doi: 10.1002/hipo.20748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: K+ channels, Kv4.2 (KCND2), Kv7.3(KCNQ3), KCa1.1 (KCNMA1), Kir2.1 (KCNJ2), Kir4.1 (KCNJ10), FMRP (FMR1), channelASD-channelepsy phenotype
Citation: Guglielmi L, Servettini I, Caramia M, Catacuzzeno L, Franciolini F, D’Adamo MC and Pessia M (2015) Update on the implication of potassium channels in autism: K+ channelautism spectrum disorder. Front. Cell. Neurosci. 9:34. doi: 10.3389/fncel.2015.00034
Received: 30 November 2014; Accepted: 20 January 2015;
Published online: 02 March 2015.
Edited by:
Tycho M. Hoogland, Netherlands Institute for Neuroscience, NetherlandsReviewed by:
Noritaka Ichinohe, National Institute of Neuroscience – National Center of Neurology and Psychiatry, JapanMohamed Jaber, University of Poitiers, France
Copyright © 2015 Guglielmi, Servettini, Caramia, Catacuzzeno, Franciolini, D’Adamo and Pessia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luca Guglielmi and Mauro Pessia, Section of Physiology and Biochemistry, Department of Experimental Medicine, University of Perugia School of Medicine, P.le Gambuli, Edificio D-Piano 1, 06132 San Sisto, Perugia, Italy e-mail: lucamormy@gmail.com; mauro.pessia@unipg.it