 Miguel Tábuas-Pereira1,2,3*
Miguel Tábuas-Pereira1,2,3* Isabel Santana1,2,3,4
Isabel Santana1,2,3,4 Elizabeth Gibbons5
Elizabeth Gibbons5 Kimberly Paquette5
Kimberly Paquette5 Maria Rosário Almeida4
Maria Rosário Almeida4 Inês Baldeiras1,2,3,4
Inês Baldeiras1,2,3,4 Jose Bras5,6
Jose Bras5,6 Rita Guerreiro5,6
Rita Guerreiro5,6- 1Neurology Department, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
- 2Faculty of Medicine, University of Coimbra, Coimbra, Portugal
- 3Department of Innovative Biomedicine and Biotechnology (CIBB), University of Coimbra, Coimbra, Portugal
- 4Department of Neuroscience and Cell Biology (CNC), University of Coimbra, Coimbra, Portugal
- 5Department of Neurodegenerative Science, Van Andel Institute, Grand Rapids, MI, United States
- 6Division of Psychiatry and Behavioral Medicine, Michigan State University College of Human Medicine, Grand Rapids, MI, United States
Introduction: Frontotemporal dementia (FTD) is considered to be part of a continuum with amyotrophic lateral sclerosis (ALS). Many genes are associated with both ALS and FTD. Yet, many genes associated with ALS have not been shown to cause FTD. We aimed to study a Portuguese cohort of FTD patients, searching for variants in genes associated with both FTD and/or ALS.
Methods: We included 57 thoroughly characterized index FTD patients from our memory clinic, who were not carriers of pathogenic variants in GRN, MAPT or C9orf72. We performed exome sequencing and 1) prioritized potential FTD and ALS causing variants by using Exomiser to annotate and filter results; and 2) looked specifically at rare variability in genes associated with FTD (excluding GRN, MAPT and C9ORF72) and/or ALS.
Results: We identified 13 rare missense variants in 10 patients (three patients had two variants) in the following genes: FUS, OPTN, CCNF, DCTN1, TREM2, ERBB4, ANG, CHRNA4, CHRNB4 and SETX. We found an additional frameshift variant on GLT8D1 in one patient. One variant (ERBB4 p.Arg1112His) gathered enough evidence to be classified as likely pathogenic by the ACMG criteria.
Discussion: We report, for the first time, an expanded study of genes known to cause FTD-ALS, in the Portuguese population. Potentially pathogenic variants in ERBB4, FUS, SETX, ANG, CHRNA4 and CHRNB4 were identified in FTD patients. These findings provide additional evidence for the potential role of rare variability in ALS-associated genes in FTD, expanding the genetic spectrum between the two diseases.
Introduction
Frontotemporal dementia (FTD) is a clinically, genetically, and pathologically heterogeneous group of disorders that lead to progressive cognitive impairment due to degeneration of the frontal and temporal lobes as a common hallmark. FTD is the second most common cause of early-onset degenerative dementia (1) and about a third of its cases are found to be genetic (2). There are different genes shown to cause this disease, with most of the cases being associated with changes in three genes: progranulin (GRN) (3, 4), microtubule associated protein tau (MAPT) (5) and chromosome 9 open reading frame 72 (C9ORF72) (6, 7). However, the genetic landscape is rapidly expanding, with more and more genes being associated with FTD.
Over time, FTD has evolved to be considered to be part of a continuum with amyotrophic lateral sclerosis (ALS) (8). Much of the support for this association comes from genetic data, as they share many genes as the underlying cause of disease (9), such as TARDBP, FUS, VCP, TBK1, CHCHD10, SQSTM1, UBQLN2, CCNF, CHMP2B, OPTN, DCTN1, TUBA4A, hnRNP2B1 and hnRNPA1 (10) and most importantly, the C9ORF72 repeat expansion (11). Yet, many of the genes that have been associated with ALS have not been shown to cause FTD (11).
In this study, we analyzed a thoroughly characterized Portuguese cohort of 57 patients with FTD. The previous screening of FTD-causative genes (MAPT, GRN and C9ORF72) allowed us to include only cases without mutations in these genes. Here we hypothesize that, given the known genetic and clinical spectrum between FTD and ALS, genes previously only related to ALS or to FTD/ALS may carry rare variants with a role in pure FTD. In this study we search for likely pathogenic variants in genes causative of both FTD (beyond MAPT, GRN and C9ORF72), and/or ALS, through exome sequencing.
Methods
Subjects
In this study, we included 57 FTD index patients from the memory clinic, in Centro Hospitalar e Universitário de Coimbra, Portugal, a tertiary reference hospital from the central region of Portugal. The diagnosis was performed according to the most widely accepted criteria (12, 13), and supported by extensive characterization. Our clinical protocol included (1) a complete and systematic clinical and neuropsychological evaluation; (2) structural neuroimaging [computed tomography and/or magnetic resonance imaging]; (3) lumbar puncture to determine Alzheimer's Disease (AD) cerebrospinal fluid biomarkers and/or amyloid PET, to strengthen the diagnosis, by excluding AD.
Patients are from a convenience sample recruited between 2012 and 2016. We included 57 patients, 31 of which were female (53.4%). Mean age of onset was 54.5 (34 to 73) years. All patients were from Portuguese ancestry. Of the whole cohort, twenty patients (34.4%) had positive family history (a first-degree relative with a neurodegenerative condition).
The study was approved by the ethics committee of Coimbra University Hospital and biological samples were obtained following written informed from the patients and/or the patients legal representatives.
Exome Sequencing
Exome sequencing was performed for all patients on a HiSeq2500 with 75-100 bp pair-end reads after library preparation with the SureSelect Exome Capture Kit v4 (Agilent). Exome data processing was done following GenomeAnalysisTK best practices. Alignment was done with the Burrows-Wheeler Aligner (bwa-mem) v0.7.12 against the hg19 genome assembly. Duplicates were identified with samblaster v0.1.21 and bases were recalibrated with GenomeAnalysisTK v3.8-1 (14). Variant Quality Score Recalibration (15), and annotation with snpEff v4.2 and dbNSFP v2.9 were applied to all variants (16, 17). Variants were filtered according to their quality metrics as described by Patel and collaborators (18).
We explored variants in genes reported to cause FTD (10): FUS, TBK1, TARDBP, SQSTM1, CHMP2B, VCP, CHCHD10, UBQLN2, CCNF, OPTN, DCTN1, and TUBA4A. We also explored other genes previously linked to FTD: TREM2, TYROBP, hnRNPA1, hnRNPA2B1 and TMEM106B. We further analyzed variants in genes reported to cause ALS (19), namely: SOD1, ANG, MATR3, FIG4, VAPB, PFN1, SETX, ERBB4, ANXA11, KIF5A, PRPH, GLT8D1, TIA1, DAO, ELP3, TAF15, EWSR1, SS18L1, NEK1, C21orf2, NEFH, CHRNA3, CHRNA4, CHRNB4, PON1, PON2, and PON3. Genes known to cause ALS in an autosomal recessive pattern and genes associated with copy number variations were not examined. All patients were previously screened for C9ORF72 expansions, GRN and MAPT mutations, and were found to be negative. Variants identified in CYLD in this cohort have been reported elsewhere (20).
Only rare variants were included in the study. Variants with a minor allele frequency (MAF) > 0.001 in gnomAD overall sample, or in any of the detailed subpopulations were excluded. In silico pathogenicity prediction was assessed using SIFT (Scale-Invariant Feature Transform) (21), Polyphen-2 (22), PROVEAN (Protein Variation Effect Analyzer) (23), Mutation Taster (24) and CADD (Combined Annotation Dependent Depletion) (25). Scores and cut-offs suggested by the software's authors were considered. In CADD, a score higher than 15 was considered as deleterious. The significant findings were comprehensively assessed by considering MAF, predicted pathogenicity, disease association, and family history.
Exomiser Analysis
An adapted version of the Exomiser software (v12.1.0) was used to identify variants that could potentially cause the disease in the individual FTD cases by prioritizing variants related to FTD (HP:0002145) and ALS (HP: 0007354), using an autosomal dominant inheritance model.
The results from the individual runs of Exomiser analyses were filtered by excluding variants without alternative alleles, not passing the Exomiser filter (inheritance), synonymous, with low coverage, and high frequency. Variants that had heterozygous calls in more than three controls and more than 100 cases were filtered out. Variants that were not present in the available databases were kept and were sorted by the Exomiser Gene Combined score in a descending order. A list was compiled with the three top ranked candidate variants separately for FTD and ALS.
InterVar ACMG Criteria
An adapted version of the InterVar software (v2.2.1) was used to classify variants based on ACMG guidelines (26). Out of the 28 ACMG criteria, InterVar can automatically apply 18 criteria. The variants in Table 1 were analyzed using the InterVar classification followed by a manual analysis to determine if any of the ten manual criteria were present.
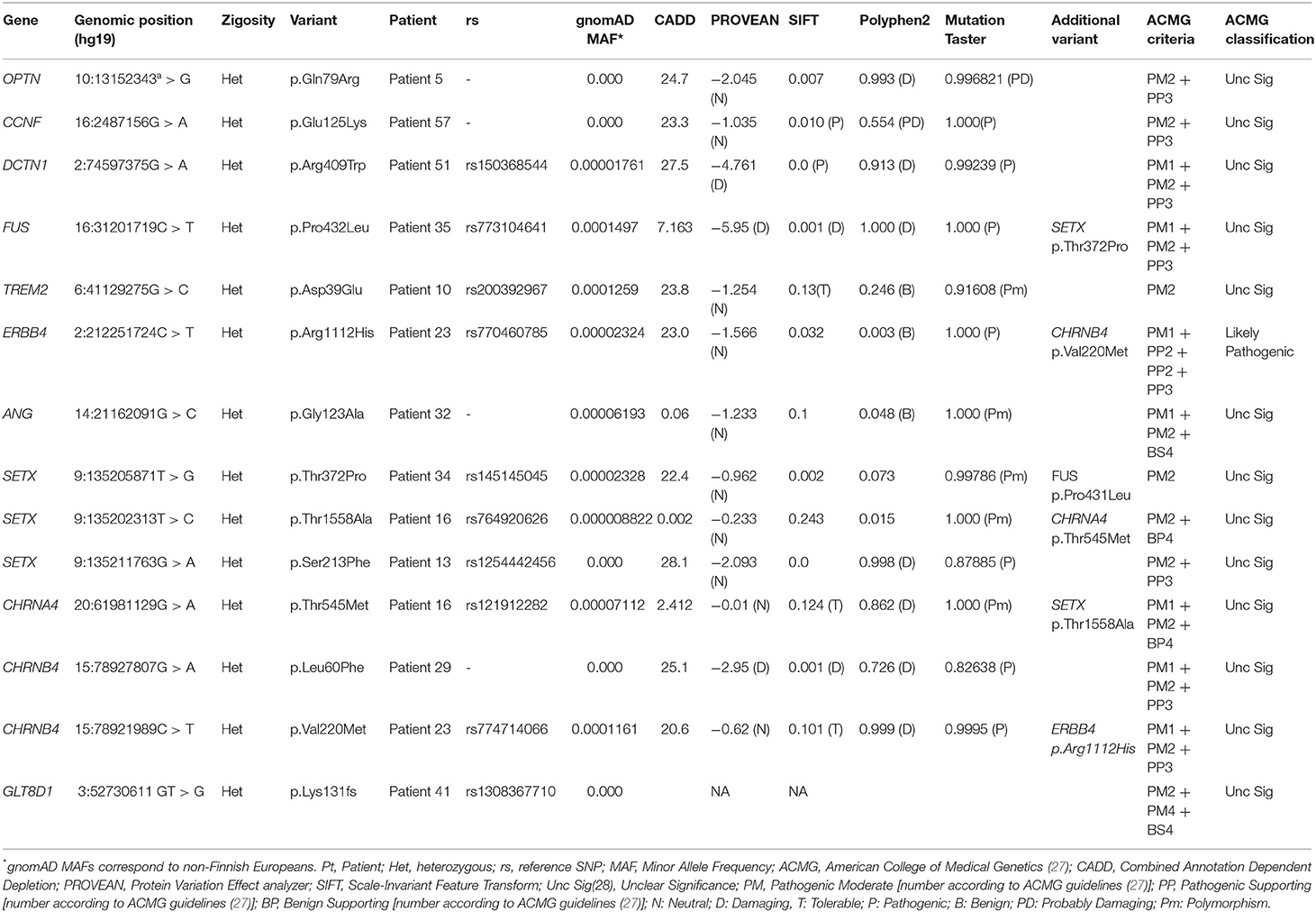
Table 1. Rare variants identified in genes previously associated with Frontotemporal and/or Amyotrophic Lateral Sclerosis in the Portuguese cohort of Frontotemporal patients.
Results
We identified 14 rare variants (13 missense and one frameshift variant) in 11 patients (three patients had two variants), which are reported in Table 1.
We found rare missense variants in FUS, OPTN, DCTN1, ERBB4, ANG, TREM2, CCNF, CHRNA4, CHRNB4 and SETX and a frameshift variant in GLT8D1 (p.Lys131fs*7), in one patient.
For several of the analyzed genes we did not identify any relevant variants: TBK1, TARDBP, VCP, TUBA4A, UBQLN2, SQSTM1, CHMP2B, CHCHD10, hnRNPA1, hnRNPA2B1, TYROBP and TMEM106B. This was also true for the ALS genes SOD1, PFN1, VAPB, MATR3, ELP3, TAF15, EWSR1, FIG4, SIGMAR1, NEFH, DAO, KIF5A, TIA1, C21orf2, NEK1, SS18L1, ANXA11, CHRNA3, PRPH, PON1, PON2, and PON3.
The top three results from the Exomiser analysis for each FTD case are presented in Supplementary Table 1. None of the variants identified were a clear cause of disease in the respective cases. Some of the top results were identified in the same genes studied here, including MATR3, TREM2, SQSTM1, and DCTN1.
Discussion
Frontotemporal dementia is thought to be part of a spectrum with ALS (8). Although only 10% of ALS cases are considered to be familial (28), the heritability of ALS sums up to 60% (27). Currently, over 25 genes have been associated with ALS (29), but only a fraction of these has also been implicated in FTD. FTD has a strong genetic component but in many familial cases a genetic cause remains to be identified. One may speculate that some of those cases can be attributed to genes which have, so far, been described in ALS cases only.
We studied a sample of 57 FTD Portuguese patients, by performing exome sequencing and (1) prioritizing potential FTD and ALS causing variants by using Exomiser to annotate and filter results; and (2) looking specifically at rare variability in genes associated with FTD (excluding GRN, MAPT and C9ORF72) and/or ALS.
No clearly pathogenic variants were identified in the top results from Exomiser, but some of the prioritized genes overlapped with the genes studied here and known to have a role in FTD and/or ALS. Noteworthy, there was one GRN variant (p.Gly70Ser in patient 5), which has been found in controls (30) and one APP variant (p.Ala500Thr in patient 18), which has also been found in controls (31).
We identified rare variants in FUS, OPTN, CCNF and DCTN1, adding support to the role of these genes in FTD. Regarding genes previously only associated with ALS, we identified rare variants in ERBB4, ANG, CHRNA4, CHRNB4, SETX and GLT8D1, some of them predicted to be pathogenic in silico. These findings suggest that these ALS genes may also have a role in the pathogenesis of FTD. The absence of significant results in the FTD genes studied here may reflect the strong role of GRN, MAPT and C9ORF72 in the disease. Cases with variants predicted to be pathogenic in these genes were excluded from the cohort to increase the probability of finding new genetic factors associated with the disease.
When comparing the phenotypic features between patients harboring rare genetic variants in the ALS genes with those without such variants, we did not see any specific phenotypic differences. However, given the small size of our cohort and the fact that we are considering different genes, such a pattern would be difficult to notice.
Genes Previously Associated With FTD
We identified a novel OPTN variant (p.Gln79Arg), predicted to be pathogenic in silico, in a male patient with progressive primary aphasia, starting at 62 years old (patient 5). This variant is absent from gnomAD, has a high CADD score (24.7), and leads to a change in the NEMO-like domain of the protein, thought to interact with TBK1 (32). So far most variants reported in ALS are located at the end of this domain (p.Ala93Pro, p.Lys95Asn, p.Arg96Leu), but there are some variants, namely p.Glu50Lys, which have been shown to be pathogenic, causing normal tension glaucoma (32).
Another variant absent from gnomAD was identified in CCNF (p.Glu125Lys) in a female patient diagnosed with bvFTD at age 48 (patient 57). CCNF encodes the Cyclin F protein that has been shown to cause abnormal ubiquination and accumulation of ubiquinated proteins, including TDP-43 when mutated (33). This variant is predicted to be pathogenic by SIFT, Polyphen 2 and Mutation Taster and is located between the F-box and the cyclin D regions of the protein (33). Other variants have been described in this region, such as p.Lys97Arg, p.Thr181Ile and p.Ser195Arg (33). These variants have been shown to cause the mislocalization of Cyclin F to the cytoplasm and increased association of Cyclin F with VCP, resulting in activation of VCP ATPase, which plays a positive role in TDP-43 aggregation (34).
For both variants, in OPTN and CCNF the in silico analyses performed pointed to potential deleterious effects. In addition, the absence of these variants from gnomAD supports a potential pathogenic role in disease. Still, these variants had uncertain significance when using the ACMG guidelines and further evidence would need to be found to support their pathogenicity.
Other rare variants were identified in DCTN1, FUS and TREM2, but contrary to the ones described above, these are all reported in gnomAD.
The DCTN1 variant (p.Arg409Trp), although rare and predicted to be pathogenic in silico, is located outside the cytoskeleton-associated protein-glycine-rich domain of the protein, where all the variants reported to date and considered to be pathogenic are located (35). This was found in another female patient with bvFTD, starting at the age of 48 (patient 51). This variant was also determined to be of uncertain significance by the ACMG guidelines and further evidence would be needed to support its pathogenicity.
FUS mutations in pure FTD are rare (36). Here, one female bvFTD patient with an age at onset of 53 years (patient 34) carried a missense variant (Pro432Leu), predicted to be pathogenic in silico. This variant has been previously associated with familial essential tremor (37). The proline at position 432 is highly conserved and located in the zinc finger domain. It may affect splicing as it affects the last nucleotide of exon 12. However, no other variants in this domain have been associated with ALS or FTD (36) and this was found to be of uncertain significance by the ACMG guidelines. This patient also carried a rare SETX variant (Table 1).
In TREM2, we identified an heterozygous missense variant (p.Asp39Glu), previously described and of unclear pathogenicity in FTD (38). This variant was found in a female patient with a primary progressive aphasia variant, starting at the age of 61 (patient 10).
Variants in Genes Associated Only With ALS
One of the main aims of this study was to search for rare, potentially pathogenic, variants in genes associated with ALS with no previous association with FTD. Specifically looking into these genes, we identified rare variants in ERBB4, ANG, SETX, CHRNA4, CHRNB4 and GLT8D1, the latter being a frameshift variant present in one patient.
Recently, a variant in ERBB4 has been described in one FTD-ALS patient (39), supporting the idea that genes associated with ALS may be also implicated in FTD pathogenesis. However, the assessment of cognitive impairment in that patient was complicated by dysarthria and no clear frontal impairment was documented in the study. We report a variant (p.Arg1112His, in patient 23), which is predicted to be pathogenic in silico by SIFT and Mutation Taster. This, together with the previously described ALS-FTD case, gives some support to the potential for this gene to be involved in FTD. This variant was classified as likely pathogenic by the ACMG guidelines.
Angiogenin (ANG) is a pro-angiogenic and neurotrophic factor with an important role in stress-induced injury, that acts by promoting neovascularization and neuronal survival (40). Pathogenic variants in this gene have been implicated in ALS (41). Interestingly, in an ALS-FTD cohort, the levels of angiogenin in CSF were only elevated in patients with FTD (40) and at least one patient reported in the literature seemed to have ALS-FTD (42). The variant we identified (p.Gly123Ala) is located close to the previously reported variants p.Arg121His and p.Arg122His (43, 44). The patient carrying this variant is part of a family with 3 of 4 siblings affected, and this variant was only present in two of them. It was also present in one descendant, currently not affected. This, together with the in silico data, argues against the pathogenicity of this variant, or, less likely, the presence of a phenocopy in the family.
Senataxin (SETX) is associated with juvenile ALS in an autosomal dominant inheritance pattern and with ataxia with oculomotor apraxia type 2 in a recessive inheritance pattern. Senataxin is a protein associated with R-loop regulation. An R-loop forms when an RNA transcript displaces homologous DNA sequence of one of the DNA strands of chromosomes and hybridizes to the complementary sequence of the other strand, generating a short region of RNA-DNA hybrid and single stranded DNA. R-loop dysfunction is increasingly recognized as the cause of several disorders, such as C9orf72 associated ALS (45). In our cohort, we identified three rare SETX variants. One patient carried a variant (p.Thr1558Ala, in patient 16) close to a previously reported pathogenic variant (p.Thr1863Ala) (46). Other rare variant identified in this Portuguese cohort was p.Thr372Pro (patient 34), located in the helicase domain. The third variant was p.Ser213Phe, the only one predicted to be deleterious. This variant is also not reported in gnomAD. This variant was found in a male patient with bvFTD starting at the age of 55 (patient 13).
CHRNA3, CHRNA4 and CHRNB4 have been associated with ALS (47). These genes encode neuronal nicotinic acetylcholine receptors and regulate Ca2+ influx to the neurons. Rare missense variants in the intracellular domains of these proteins have been shown to be more prevalent in sporadic ALS patients (48). They also have been shown to be associated with cognitive performance (49). In particular, CHRNB4 has been linked to the modulation of attention in attention deficit/hyperactivity disorder (50). Together, these data point to the possible role of variability in FTD risk and/or pathogenesis. We identified three rare variants in this gene cluster, with two variants located in CHRNB4 (p.Leu60Phe, in patient 29, and p.Val220Met, in patient 23) being particularly relevant, as they are very rare or absent in gnomAD and predicted to be pathogenic in silico by five of five (p.Leu60Phe) and three of five (p.Val220Met) tools (Table 1). However, all three of these variants were classified with uncertain significance by the ACMG guidelines.
Finally, we found one female patient (patient 42) with an age at onset of FTD at 66 years carrying a frameshift variant in GLT8D1, a gene recently associated with ALS (51). It encodes a glycosyltransferase enzyme of unknown function, possibly linked to ganglioside synthesis. GLT8D1 has also been linked with schizophrenia (52, 53). So far, the vast majority of variants reported to cause ALS are missense and located in exon 4 (51). One variant in exon 10 has also been described in a Chinese cohort (47). The variant identified in our cohort (which is not present in gnomAD) leads to a change in the same glycosyltransferase domain of the protein but is predicted to cause a premature stop codon. It is unclear if variants causing a truncated protein will have the same functional effect as the missense variants previously identified in ALS. However, given that in this case we were able to assess the sister of the proband that also presented with FTD-ALS and did not carry the variant, the likelihood of pathogenicity for this variant is very low.
This study has some limitations, including the small size of the cohort, the lack of pathological diagnoses and the unavailability of samples from family members to test co-segregation of variants with disease. Only one of the FTD patients had a pathological confirmation of diagnosis, although all patients in the cohort were clinically evaluated and thoroughly studied in an experienced dementia focused centre. Due to the absence of family history or the unavailability of samples from family members we did not perform segregation analyses to further study the pathogenicity of most of the reported variants. However, the frequency of the variants, together with the use of in silico prediction tools and functional annotation allowed us to weight the possible role of the identified variants.
This study described for the first time the genetic variability beyond GRN, MAPT and C9ORF72, in a clinically well characterized cohort of FTD cases from Portugal. The identification of rare variants in FUS, SETX, CHRNB4, CHRNA4 and ERBB4 in FTD patients provides additional support for a role of ALS-associated genes in pure FTD cases. This expands the spectrum between FTD and ALS on a genetic level. Replication and extension of these findings can have a significant impact on the genetic counseling of affected patients and families and on the understanding of FTD mechanisms.
Data Availability Statement
The variants identified in this study have been submitted into online repositories. The name of the repository and accession numbers can be found below: National Center for Biotechnology Information (NCBI) ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/, VCV001344510-VCV001344518, VCV000586548, VCV000648503, VCV000905893, VCV000037069, and VCV000098320.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of Coimbra University Hospital, Coimbra, Portugal. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
MT-P has drafted the manuscript and prepared the final version. MT-P, RG, and JB have designed the study. IS, MT-P, IB, and MA have contributed in the collection of samples and patients study. MT-P, EG, KP, MA, RG, and JB have contributed to the analysis of the bioinformatic data. MT-P, IS, KP, EG, RG, and JB have reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Number R01AG067426.
Author Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2022.886379/full#supplementary-material
References
1. Almeida MR, Baldeiras I, Ribeiro MH, Santiago B, Machado C, Massano J, et al. Progranulin peripheral levels as a screening tool for the identification of subjects with progranulin mutations in a Portuguese cohort. Neurodegener Dis. (2014) 13:214–23. doi: 10.1159/000352022
2. Rohrer JD, Guerreiro R, Vandrovcova J, Uphill J, Reiman D, Beck J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. (2009) 73:1451–6. doi: 10.1212/WNL.0b013e3181bf997a
3. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
4. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
5. Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. (1998) 393:702–5. doi: 10.1038/31508
6. Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. (2011) 72:257–68. doi: 10.1016/j.neuron.2011.09.010
7. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) 72:245–56. doi: 10.1016/j.neuron.2011.09.011
8. Burrell JR, Halliday GM, Kril JJ, Ittner LM, Gotz J, Kiernan MC, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. (2016) 388:919–31. doi: 10.1016/S0140-6736(16)00737-6
9. Ji AL, Zhang X, Chen WW, Huang WJ. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet. (2017) 54:145–54. doi: 10.1136/jmedgenet-2016-104271
10. Guerreiro R, Gibbons E, Tabuas-Pereira M, Kun-Rodrigues C, Santo GC, Bras J. Genetic architecture of common non-Alzheimer's disease dementias. Neurobiol Dis. (2020) 18:104946. doi: 10.1016/j.nbd.2020.104946
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci. (2020) 14:42. doi: 10.3389/fnins.2020.00042
12. Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. (2011) 134:2456–77. doi: 10.1093/brain/awr179
13. Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. (2011) 76:1006–14. doi: 10.1212/WNL.0b013e31821103e6
14. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. (2010) 20:1297–303. doi: 10.1101/gr.107524.110
15. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From fastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. (2013) 43:1101–33. doi: 10.1002/0471250953.bi1110s43
16. Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. (2012) 6:80–92. doi: 10.4161/fly.19695
17. Liu X, Jian X, Boerwinkle E. dbNSFP v20: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. (2013) 34:E2393–402. doi: 10.1002/humu.22376
18. Patel ZH, Kottyan LC, Lazaro S, Williams MS, Ledbetter DH, Tromp H, et al. The struggle to find reliable results in exome sequencing data: filtering out Mendelian errors. Front Genet. (2014) 5:16. doi: 10.3389/fgene.2014.00016
19. Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA, et al. Genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. (2019) 13:1310. doi: 10.3389/fnins.2019.01310
20. Tabuas-Pereira M, Santana I, Kun-Rodrigues C, Bras J, Guerreiro R. CYLD variants in frontotemporal dementia associated with severe memory impairment in a Portuguese cohort. Brain. (2020) 143:e67. doi: 10.1093/brain/awaa183
21. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. (2009) 4:1073–81. doi: 10.1038/nprot.2009.86
22. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
23. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. (2015) 31:2745–7. doi: 10.1093/bioinformatics/btv195
24. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/nmeth.2890
25. Rentzsch P, Schubach M, Shendure J, Kircher M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. (2021) 13:31. doi: 10.1186/s13073-021-00835-9
26. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
27. Al-Chalabi A, Fang F, Hanby MF, Leigh PN, Shaw CE, Ye W, et al. An estimate of amyotrophic lateral sclerosis heritability using twin data. J Neurol Neurosurg Psychiatry. (2010) 81:1324–6. doi: 10.1136/jnnp.2010.207464
28. Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R, et al. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. (2009) 72:725–31. doi: 10.1212/01.wnl.0000343008.26874.d1
29. Nguyen HP, Van Broeckhoven C, van der Zee J. ALS Genes in the Genomic Era and their Implications for FTD. Trends in genetics: TIG. (2018) 34:404–23. doi: 10.1016/j.tig.2018.03.001
30. Yu CE, Bird TD, Bekris LM, Montine TJ, Leverenz JB, Steinbart E, et al. The spectrum of mutations in progranulin: a collaborative study screening 545 cases of neurodegeneration. Arch Neurol. (2010) 67:161–70. doi: 10.1001/archneurol.2009.328
31. Schulte EC, Fukumori A, Mollenhauer B, Hor H, Arzberger T, Perneczky R, et al. Rare variants in beta-amyloid precursor protein (APP) and Parkinson's disease. Eur J Human Genet EJHG. (2015) 23:1328–33. doi: 10.1038/ejhg.2014.300
32. Toth RP, Atkin JD. Dysfunction of optineurin in amyotrophic lateral sclerosis and glaucoma. Front Immunol. (2018) 9:1017. doi: 10.3389/fimmu.2018.01017
33. Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. (2016) 7:11253.
34. Yu Y, Nakagawa T, Morohoshi A, Nakagawa M, Ishida N, Suzuki N, et al. Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum Mol Genet. (2019) 28:3486–97. doi: 10.1093/hmg/ddz119
35. Konno T, Ross OA, Teive HAG, Slawek J, Dickson DW, Wszolek ZK. DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat Disord. (2017) 41:14–24. doi: 10.1016/j.parkreldis.2017.06.004
36. Deng H, Gao K, Jankovic J. The role of FUS gene variants in neurodegenerative diseases. Nat Rev Neurol. (2014) 10:337–48. doi: 10.1038/nrneurol.2014.78
37. Merner ND, Girard SL, Catoire H, Bourassa CV, Belzil VV, Riviere JB, et al. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am J Hum Genet. (2012) 91:313–9. doi: 10.1016/j.ajhg.2012.07.002
38. Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. (2014) 35:726 e11–9. doi: 10.1016/j.neurobiolaging.2013.09.009
39. Sun L, Cheng B, Zhou Y, Fan Y, Li W, Qiu Q, et al. ErbB4 mutation that decreased NRG1-ErbB4 signaling involved in the pathogenesis of amyotrophic lateral sclerosis/frontotemporal dementia. J Alzheimers Dis. (2020) 74:535–44. doi: 10.3233/JAD-191230
40. Morelli C, Tiloca C, Colombrita C, Zambon A, Soranna D, Lafronza A, et al. CSF angiogenin levels in amyotrophic lateral sclerosis-frontotemporal dementia spectrum. Amyotroph Lateral Scler Frontotemporal Degener. (2020) 21:63–9. doi: 10.1080/21678421.2019.1704016
41. Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, et al. ANG mutations segregate with familial and 'sporadic' amyotrophic lateral sclerosis. Nat Genet. (2006) 38:411–3. doi: 10.1038/ng1742
42. Fernandez-Santiago R, Hoenig S, Lichtner P, Sperfeld AD, Sharma M, Berg D, et al. Identification of novel angiogenin (ANG) gene missense variants in German patients with amyotrophic lateral sclerosis. J Neurol. (2009) 256:1337–42. doi: 10.1007/s00415-009-5124-4
43. Padhi AK, Narain P, Gomes J. Rare angiogenin and ribonuclease 4 variants associated with amyotrophic lateral sclerosis exhibit loss-of-function: a comprehensive in silico study. Metab Brain Dis. (2019) 34:1661–77. doi: 10.1007/s11011-019-00473-6
44. Paubel A, Violette J, Amy M, Praline J, Meininger V, Camu W, et al. Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch Neurol. (2008) 65:1333–6. doi: 10.1001/archneur.65.10.1333
45. Perego MGL, Taiana M, Bresolin N, Comi GP, Corti S. R-Loops in motor neuron diseases. Mol Neurobiol. (2019) 56:2579–89. doi: 10.1007/s12035-018-1246-y
46. Kenna KP, McLaughlin RL, Byrne S, Elamin M, Heverin M, Kenny EM, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. (2013) 50:776–83. doi: 10.1136/jmedgenet-2013-101795
47. Li W, Liu Z, Sun W, Yuan Y, Hu Y, Ni J, et al. Mutation analysis of GLT8D1 and ARPP21 genes in amyotrophic lateral sclerosis patients from mainland China. Neurobiol Aging. (2020) 85:156 e1–4. doi: 10.1016/j.neurobiolaging.2019.09.013
48. Sabatelli M, Eusebi F, Al-Chalabi A, Conte A, Madia F, Luigetti M, et al. Rare missense variants of neuronal nicotinic acetylcholine receptor altering receptor function are associated with sporadic amyotrophic lateral sclerosis. Hum Mol Genet. (2009) 18:3997–4006. doi: 10.1093/hmg/ddp339
49. Winterer G, Mittelstrass K, Giegling I, Lamina C, Fehr C, Brenner H, et al. Risk gene variants for nicotine dependence in the CHRNA5-CHRNA3-CHRNB4 cluster are associated with cognitive performance. Am J Med Genet B Neuropsychiatr Genet. (2010) 153B:1448–58. doi: 10.1002/ajmg.b.31126
50. Alemany S, Ribases M, Vilor-Tejedor N, Bustamante M, Sanchez-Mora C, Bosch R, et al. New suggestive genetic loci and biological pathways for attention function in adult attention-deficit/hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet. (2015) 168:459–70. doi: 10.1002/ajmg.b.32341
51. Cooper-Knock J, Moll T, Ramesh T, Castelli L, Beer A, Robins H, et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. (2019) 26:2298–306 e5. doi: 10.1016/j.celrep.2019.02.006
52. Sasayama D, Hori H, Yamamoto N, Nakamura S, Teraishi T, Tatsumi M, et al. ITIH3 polymorphism may confer susceptibility to psychiatric disorders by altering the expression levels of GLT8D1. J Psychiatr Res. (2014) 50:79–83. doi: 10.1016/j.jpsychires.2013.12.002
Keywords: frontotemporal dementia, ALS (amyotrophic lateral sclerosis), phenotype, cohort, Portuguese
Citation: Tábuas-Pereira M, Santana I, Gibbons E, Paquette K, Almeida MR, Baldeiras I, Bras J and Guerreiro R (2022) Exome Sequencing of a Portuguese Cohort of Frontotemporal Dementia Patients: Looking Into the ALS-FTD Continuum. Front. Neurol. 13:886379. doi: 10.3389/fneur.2022.886379
Received: 28 February 2022; Accepted: 31 May 2022;
Published: 07 July 2022.
Edited by:
Liyong Wu, Capital Medical University, ChinaReviewed by:
Carol Dobson-Stone, The University of Sydney, AustraliaCaroline Vance, King's College London, United Kingdom
Copyright © 2022 Tábuas-Pereira, Santana, Gibbons, Paquette, Almeida, Baldeiras, Bras and Guerreiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel Tábuas-Pereira, miguelatcp@gmail.com