 Patrick J. Paasila
Patrick J. Paasila Jason A. Aramideh
Jason A. Aramideh Greg T. Sutherland
Greg T. Sutherland Manuel B. Graeber
Manuel B. Graeber- 1Charles Perkins Centre, School of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Camperdown, NSW, Australia
- 2School of Medicine, Western Sydney University, Campbelltown, NSW, Australia
- 3Brain and Mind Centre, Faculty of Medicine and Health, The University of Sydney, Camperdown, NSW, Australia
Alzheimer’s disease (AD) is characterised by synaptic dysfunction accompanied by the microscopically visible accumulation of pathological protein deposits and cellular dystrophy involving both neurons and glia. Late-stage AD shows pronounced loss of synapses and neurons across several differentially affected brain regions. Recent studies of advanced AD using post-mortem brain samples have demonstrated the direct involvement of microglia in synaptic changes. Variants of the Apolipoprotein E and Triggering Receptors Expressed on Myeloid Cells gene represent important determinants of microglial activity but also of lipid metabolism in cells of the central nervous system. Here we review evidence that may help to explain how abnormal lipid metabolism, microglial activation, and synaptic pathophysiology are inter-related in AD.
Introduction
Alzheimer’s disease (AD) accounts for 60–80% of total dementia diagnoses (Brookmeyer et al., 2011; Alzheimer’s Association, 2021). Extracellular β amyloid (Aβ) plaques and intraneuronal neurofibrillary tangles (NFTs) are the major pathological characteristics of AD. The gold-standard for a definitive diagnosis of AD is post-mortem neuropathology (Hyman et al., 2012; Montine et al., 2012). However fluid analytes—including most recently the development of the serum phosphorylated tau (threonine 217) blood test (Barthélemy et al., 2020)—and imaging biomarkers allow for a reasonably confident diagnosis to be made in vivo (Jack et al., 2010; Leuzy et al., 2018).
Synapses are the sites at which neuronal communication takes place by chemical or electrical means (Alcamí and Pereda, 2019). Synaptic plasticity refers to the brain’s ability to modify neural circuitry structurally and functionally at the synaptic level to facilitate learning, memory, cognition, and the regulation of emotions and behaviours (Kennedy, 2016)—though there remains significant debate in the field as to the sole importance of synapses in these processes, particularly memory (Trettenbrein, 2016). Long-term potentiation (LTP) and long-term depression (LTD) represent well researched mechanisms of synaptic plasticity which entail long lasting and activity dependent changes in synaptic efficacy as shown by in vitro and in vivo electrophysiological recordings (Abraham et al., 2019). Normal ageing is associated with a loss of synapses (Masliah et al., 1993a). This is markedly accelerated in AD which has been described as a disease of synaptic failure (Terry, 2000; Selkoe, 2002); albeit with the caveat that synaptic changes are not specific to AD (Scheff et al., 2014).
Microglia are the central nervous system’s (CNS’s) resident phagocytes of mesodermal origin (predominantly from blood islands of the embryological yolk sac) and as such share many characteristics with macrophages of myeloid lineage (Ginhoux and Prinz, 2015). Microglia perform a number of key functions in the CNS: (1) synaptogenesis (Miyamoto et al., 2016), synaptic pruning during development (Paolicelli et al., 2011), and complement-mediated elimination (Streit et al., 1988; Stevens et al., 2007; Schafer et al., 2012) in the adult brain that may represent a potential biological mechanism of forgetting (Wang et al., 2021); (2) vasculogenesis (Ronaldson and Davis, 2020); (3) constant surveillance of their local microenvironment (Nimmerjahn et al., 2005) by means of their sensome (Hickman et al., 2013), including continual monitoring of neuronal activity (Wake et al., 2013)—notably by exerting protective effects through somatic purinergic junctions (Cserép et al., 2020); (4) regulation of myelin homeostasis (Healy et al., 2016); (5) modification of synaptic plasticity through cytokine signalling and brain-derived neurotrophic factor (BDNF) (Wu et al., 2015); (6) innate immune functions (Kofler and Wiley, 2011), including convergence on sites of injury (Davalos et al., 2005), detection of danger-associated molecular patterns (alarmins and pathogen-associated molecular patterns) (Bianchi, 2007), phagocytosis (Sierra et al., 2013), macropinocytosis (Li et al., 2013), antigen presentation (Schetters et al., 2017), secretion of anti- and pro-inflammatory cytokines (Da Pozzo et al., 2019), and regulation of neuronal apoptosis in the setting of traumatic injury (Wang et al., 2020); and (7) coordination with astrocytes in aspects of each the aforementioned functions (Matejuk and Ransohoff, 2020).
Glia have long been implicated in AD since Alzheimer himself commented on their involvement in his original case report (Alzheimer, 1907)—“The glia have formed plentiful fibres; in addition, many glial cells exhibit large fat sacks.” A more detailed follow-up of microglia in AD began toward the end of the last century (Eikelenboom and Stam, 1982; Dickson et al., 1988; Eikelenboom et al., 1989; Itagaki et al., 1989; McGeer et al., 1989; Perlmutter et al., 1990; Sasaki et al., 1997). Since then, microglial involvement has been demonstrated in many neurodegenerative (Hickman et al., 2018) and psychiatric diseases (Tay et al., 2017), including AD, frontotemporal dementia, Parkinson’s disease, Huntington’s disease, motor neurone disease, prion diseases, chronic traumatic encephalopathy, bipolar disorder, major depressive disorder, and schizophrenia. More recently in AD, microglia have been shown to internalise greater quantities of synaptic material during the symptomatic phase of the disease. This has been demonstrated using confocal (Tzioras et al., 2019) and single-molecule localisation microscopy (Paasila et al., 2021) of archival cortical tissue samples.
Lipids are enriched in the CNS and figure prominently in AD pathophysiology (Di Paolo and Kim, 2011). Bozek et al. (2015) found that 75% of molecules from a panel of some 5713 lipids were either enriched or depleted in human brain compared to other mammals. Lipids participate in cell structure, cellular signalling, energy balance, and inflammatory signalling (Cermenati et al., 2015). For instance, synaptogenesis is promoted by glia-derived cholesterol (Mauch et al., 2001)—which also serves as the critical precursor compound of neuroactive steroids capable of altering gene expression and ultimately neuron survival (Melcangi et al., 2008). The importance of lipids to brain health is highlighted by genetic connections between neurological disease and cholesterol metabolism (Björkhem et al., 2010), and particularly in AD (Kunkle et al., 2019). Most classes of lipids are involved in the pathogenesis of AD, including cholesterols, sterols, glycerolipids, glycerophospholipids, and sphingolipids (Chew et al., 2020). The APOE ε4 allele represents the most direct genetic link between lipid metabolism and AD (Corder et al., 1993). APOE is highly expressed in the liver and brain where astrocytes are thought to be a main source (Grehan et al., 2001). APOE is involved in the transportation and metabolism of lipids, functioning as a ligand for low-density and very low-density lipoprotein (LDL and VLDL, respectively) receptors that trigger receptor-mediated endocytosis of lipoprotein particles (Bu, 2009). APOE appears to exert its effect on AD risk by influencing the time of disease onset as well as the total load of Aβ pathology (Kim et al., 2009a).
Proteinopathy in Alzheimer’s Disease
β Amyloid
β Amyloid (Aβ) is derived from the large type I transmembrane protein APP (Kang et al., 1987; Dyrks et al., 1988) found at both pre- and post-synapses and which is involved in dendritic spine plasticity (Montagna et al., 2017). APP undergoes a constant cycle of trafficking through the endomembrane system: it is first routed from the endoplasmic reticulum to the plasma membrane, Golgi apparatus, or trans-Golgi network (Haass et al., 2012). Nascent APP is post-translationally modified by glycosylation, phosphorylation, and sulphation. The small proportion of APP that reaches the plasma membrane is endocytosed within minutes and recycled or degraded in lysosomes. Our recently published results also demonstrate a mechanism of “short-circuited” APP recycling governed by lactoferrin which results in increased Aβ production (Tsatsanis et al., 2021). APP may be proteolytically cleaved by γ-secretase and either α- or β-secretase. β-secretase initiates the amyloidogenic pathway responsible for the generation of Aβ peptides. β-secretase cleaves APP in the extracellular domain to form the secreted APP ectodomain (APPsβ) and the membrane-bound APP carboxyl-terminal fragment (βCTF). βCTF is subsequently cleaved by γ-secretase within the plasma membrane (termed regulated intramembrane proteolysis; Lichtenthaler et al., 2011) or the endosomal-lysosomal system. Cleavage by γ-secretase can occur at several sites; labelled the ε-, ζ-, and γ-site, respectively. The γ-cleavage site varies in its position and is therefore responsible for the production of Aβ peptides of different lengths, from Aβ37 to Aβ43 (or longer). This is of relevance to AD as Aβ39/40 predominate in cerebral amyloid angiopathy (CAA) (Prelli et al., 1988; Suzuki et al., 1994) and Aβ42/43 in parenchymal deposits (Iwatsubo et al., 1994). The latter species are thought by some to constitute the neurotoxic oligomers responsible for initiating AD (Haass and Selkoe, 2007; Saito et al., 2011; Masters and Selkoe, 2012; Selkoe and Hardy, 2016).
Aggregates of Aβ filaments are observed as plaques in post-mortem AD brains (Walker, 2020). Morphological subtypes of Aβ plaques include diffuse (also, “primitive” or “immature”), fibrillar, dense-cored (also “classical” or “mature”), and burned-out (also, “core-only”) plaques. Other Aβ plaque types include subpial bands (Thal et al., 2000), cotton-wool plaques which feature prominently in PSEN1 (also PS1; encoding presenilin 1) familial AD (Tabira et al., 2002), lake-like patches in the presubiculum (Wisniewski et al., 1998), and the recently described coarse-grained plaques in early onset AD (Boon et al., 2020). Neuritic plaques are a minority subset of Aβ plaques that are most often associated with phosphotau-positive dystrophic neurites. Aβ plaques occur in most elderly but are not universal (Braak et al., 2011; Jicha et al., 2012). Further, Aβ plaques are not sufficient to cause AD, however there is a strong association between their formation and the eventual development of AD as demonstrated by rare mutations (including APP, PSEN1, and PSEN2) in familial early- and late-onset AD (Cruchaga et al., 2012; Sassi et al., 2014; Lanoiselée et al., 2017).
Microtubule Associated Protein Tau
A hyperphosphorylated form of the microtubule associated protein tau (MAPT) (“tau” for short) is the subunit of NFTs, neuropil threads, and tau positive dystrophic neurites (Mandelkow and Mandelkow, 2012)—collectively referred to as neurofibrillary degeneration (NFD) (Iqbal et al., 2010b). Tau is a member of the type 2 microtubule associated protein (MAP) family. It is a highly soluble protein which is unfolded in its native state (Schweers et al., 1994; Mukrasch et al., 2009). It is expressed as six alternatively spliced isoforms with 0, 1, or 2 amino (N-) terminal inserts of 29 residues each (0N, 1N, or 2N; derived from exons 2 and 3) and 3 or 4 carboxyl (C-) terminal repeats of 31–32 residues (3R or 4R; derived from exon 10) (Goedert et al., 1989). The full-length protein can be divided into an N-terminal “projection domain,” which is directed away from the microtubule, followed by a C-terminal “assembly domain.” The assembly domain is further subdivided into a proline-rich region, the microtubule-binding repeats, and a C-terminal tail (Mandelkow and Mandelkow, 2012). Tau contains 85 potential serine, threonine, and tyrosine phosphorylation sites, most of which reside in the proline-rich region and the C-terminal tail (Noble et al., 2013). Tau exhibits many structural conformations, biochemical modifications, and the ability to interact with several different protein types, including chaperones, cytoskeletal proteins, kinases, motors, and phosphatases (Mandelkow and Mandelkow, 2012). However, unlike APP, mutations in MAPT cause frontotemporal dementia but not AD.
Tau is susceptible to a range of disease-related processes, including acetylation, glycation, glycosylation, methylation, nitration, oxidation, ubiquitination, truncation, and missorting to the somatodendritic compartment (Ittner and Ittner, 2018). Aggregations of phosphotau occur first as granular oligomers which then aggregate further to form fibrils (Maeda et al., 2007). Soluble oligomers form early in the pathogenesis of AD and may represent the major neurotoxic substrate (Lasagna-Reeves et al., 2012; Ward et al., 2012) and which are thought by some to initiate the AD pathophysiological process rather than Aβ (Arnsten et al., 2021). Conversely, intraneuronal, insoluble fibrillar tau inclusions may represent end-stage lesions, appearing to be inert markers of earlier pathological changes to soluble tau species (Iqbal et al., 2010a). Importantly, the density and anatomical spread of NFTs remain criteria for diagnostic and staging purposes (Montine et al., 2012).
Synapses in Alzheimer’s Disease
The overall picture is that of a widespread loss of synapses and synaptic proteins in AD (Terry and Davies, 1980; Davies et al., 1987; Masliah et al., 1989). During the symptomatic end-stage of AD the pronounced loss of synapses correlates with the severity of symptoms (DeKosky and Scheff, 1990; Terry et al., 1991; Masliah et al., 1993b; Dickson et al., 1995; Sze et al., 1997). A series of studies demonstrated this using electron microscopy (EM) (Scheff and Price, 2003) on the hippocampal dentate gyrus (Scheff et al., 1996, 2006; Scheff and Price, 1998), cingulate cortex (Scheff and Price, 2001), entorhinal cortex (Scheff et al., 1993), frontal cortex (Scheff et al., 1990), temporal cortex (Scheff and Price, 1993), and precuneus (PreC) (Scheff et al., 2013). Immunohistochemistry studies have also revealed a reduced density of synapses in the frontal cortex (Masliah et al., 1992), entorhinal cortex, and hippocampus (Masliah et al., 1994; Wakabayashi et al., 1994). The latter region also showed reduced post-synaptic drebrin (Counts et al., 2012) and PSD95 as detected by immunoblot in early AD (Sultana et al., 2010). Widespread loss of drebrin, which is involved in synaptic morphology and plasticity (Sekino et al., 2017), has been noted in the superior frontal, superior temporal, visual, inferior parietal, and anterior cingulate cortices whereas pre-synaptic synaptophysin was reduced only in the superior temporal and inferior parietal cortices while synaptotagmin levels were unchanged in all these areas (Counts et al., 2006). Furthermore, a reduction in the density of synapses is also apparent in mild cognitive impairment of the amnestic type in at least the Cornu Ammonis 1 (CA1) subregion of the hippocampus (Scheff et al., 2007) and the inferior temporal cortex (ITC) (Scheff et al., 2011).
A subsequent meta-analysis of post-mortem human brain studies confirmed a reduction of synaptic density and loss of synaptic proteins in AD (de Wilde et al., 2016). This analysis also demonstrated that pre-synaptic molecules are more affected than post-synaptic molecules—particularly in the hippocampus compared to cortical areas. Molecules of interest are listed below and a summary of their differential regulation in AD across different brain regions is provided in Table 1. Pre-synaptic markers included those involved in calcium sensing and buffering (e.g., parvalbumin; synaptotagmin—which contrasts with the immunoblot study mentioned above; and synaptophysin); cytoskeletal structures (septin 5 and 7); endocytosis (AP180, dynamin I); small GTPases (rab-3a, -5, and -7); SNAREs (complexins, SNAP25, synaptobrevin, syntaxins, VAMPs); vesicular tethering (synapsin I); and vesicles (SV2, synaptophysin, VGLUTs). Post-synaptic markers included cytoskeletal proteins (drebrin, IRSp53, MAP2, GKAP, synaptopodin); a growth/plasticity marker (GAP43); a metal ion transporter (ZnT-1); protein dephosphorylation (spinophilin); neurotransmitter receptors (GABA, muscarinic, NMDA); and molecular signalling components (CaMKII, PSD95). Common pre-/post-synaptic molecules included those involved in adhesion (catenin β, N-cadherin, NCAM); calcium homeostasis (calbindin, calretinin); cytoskeletal proteins (actin); protein phosphatase (calcineurin); receptors (TrkA); and REDOX signalling (thioredoxin). Overall, there was a loss of pre-, post-, and common-synaptic markers across several brain regions in AD. Interestingly, markers of cytoskeletal organisation were significantly elevated in hippocampal pre-synaptic areas but were decreased in the frontal, cingulate, entorhinal, and temporal cortices. The latter areas also showed increased expression of post-synaptic neurotransmitter receptors. Notably, APP was not examined in these studies despite it’s presence at both pre- and post-synapses (Montagna et al., 2017).
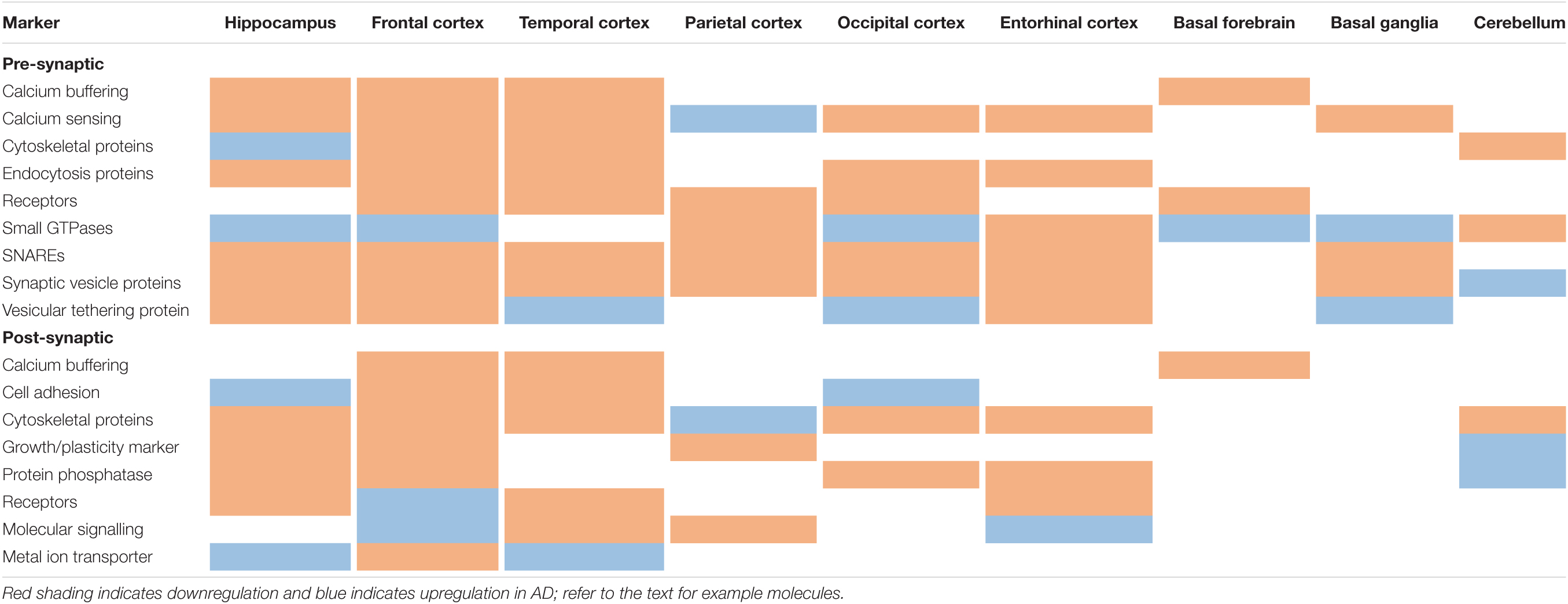
Table 1. Synaptic markers across differentially affected brain regions in AD.
Early stages of AD are characterised by an apparently compensatory inverse relationship between the increased size of pre-synaptic terminals and the loss of synapses across many cortical and limbic structures (Scheff et al., 1990; Masliah et al., 1994; Scheff and Price, 2003, 2006; Overk and Masliah, 2014). Further, deimpregnation of Golgi-stained neurons followed by Congo red staining has shown that tangle-bearing neurons in the CA1 of the hippocampus exhibit a more extensive dendritic tree than tangle-free neurons (Gertz et al., 1991). It appears that brain areas which normally show an elevated level of synaptic plasticity (for instance the mesial temporal lobe) are associated with increased expression and phosphorylation of tau and are therefore also most prone to the formation of NFTs. Tau dysfunction in these temporal and limbic areas may be further propagated by local neuronal death which would result in increased neuroplasticity demand in adjacent neurons and projection targets which would themselves subsequently be subject to tau dysfunction and the build-up of phosphotau (Mesulam, 1999).
Expression Microarrays Have Demonstrated the Differential Regulation of Synaptic Genes Across Different Stages of Alzheimer’s Disease
Expression microarray data has demonstrated the downregulation of many synaptic genes in late-stage AD (Berchtold et al., 2013; George et al., 2017; Williams et al., 2021). Berchtold et al. (2013) showed decreased expression of genes involved in synaptic vesicle trafficking and release in the hippocampus. These included SNAREs such as synaptobrevin 2 and SNAP25, and SNARE-related genes, including, synapsin I and II, synaptotagmin 1 and 5, dynamin 1, synaptopodin, rab-3a, syntaxin 6, bassoon, and piccolo, amongst others. Interestingly, several gene transcripts were increased in the hippocampus and entorhinal cortex in AD, including, SNAP23, synaptopodin 2, and synaptobrevin 1. Other pathways that were downregulated in the hippocampus included neuromodulatory peptides (e.g., BDNF, corticotropin releasing hormone, somatostatin, cortistatin, histamine, and tachykinin), voltage-gated ion channels (e.g., Ca2+, K+, and Na+), and an extensive number of transporters, receptors, and enzymes that are required for the synthesis of different neurotransmitters, including glutamate, GABA, acetylcholine, dopamine, glycine, noradrenaline, and serotonin. An earlier investigation using laser capture microdissection of tangle bearing neurons in the CA1 showed upregulation of genes regulating early (rab5) and late endosomes (rab7) which paralleled the downregulation of genes encoding neurotrophin receptors (TrkB; TrkC) over disease progression (Ginsberg et al., 2010).
In contrast, there appears to be an upregulation of genes related to synaptic plasticity before the formation of AD-type neuropathology in the medial frontal cortex of asymptomatic Braak stage II and III brains (Bossers et al., 2010). The same set of genes were only downregulated in the same region following the appearance of Aβ plaques and NFTs in more advanced Braak stages (IV–VI). Among other proteins, Braak stage II was associated with increased expression of several voltage-gated K+ channels—regulators of the action potential; GABA receptor subunits; neurotransmitter exocytosis (e.g., SNAREs such as SNAP25, complexin I, synaptotagmins, VAMP7); clathrin heavy chain 1 and protein kinase C—involved in activity-dependent bulk endocytosis; corticotropin releasing hormone; cell adhesion molecule with homology to L1CAM and doublecortin-like kinase 1—proteins involved in neuronal migration and axonal outgrowth; p21-activated kinase 1—involved in activity dependent synaptogenesis; glycine receptor β—involved in the regulation of synaptic connectivity; and stathmin-like 2—a regulator of microtubules during axonal extension. The authors suggested that these expression patterns are indicative of increased synaptic activity and plasticity over the course of Braak stages I–II. Further, they argue that these synaptic alterations are a compensatory response to increased intracellular Aβ and APP fragments containing the Aβ peptide—demonstrated by 4G8 immunoreactivity—which have inhibitory effects on synaptic plasticity in a mouse model and in vitro (He et al., 2019). Similarly, synaptoneurosomes derived from early AD cases and individuals with mild cognitive impairment show increased expression of neuroplasticity and synaptic transmission genes such as synaptic vesicle glycoprotein 2A (SV2A), growth associated protein 43 (GAP43), lipid phosphate phosphatase-related protein type 4 (LPPR4), glutamate receptor ionotropic AMPA 2 (GRIA2), cholinergic receptor muscarinic 3 (CHRM3), and 5-hydroxytryptamine receptor 2A (HTR2A) (Williams et al., 2009). Notably, somatostatin was downregulated in the study by Bossers et al. (2010) and also in our recently published RNA sequencing (RNAseq) analysis of the PreC and primary visual cortex (PVC) (Guennewig et al., 2021). The PVC and PreC also showed increased syntaxin binding protein 2 (STXBP2)—involved in intracellular vesicle trafficking. The PreC represented a relatively moderately affected area in AD compared to the PVC and showed decreased synaptotagmin 2 (SYT2) and SNAP25, suggesting that these genes are downregulated relatively soon after their upregulation in the earlier stages of AD.
Recent Advances in the Understanding of Transcriptomic Signatures of Synaptic Dynamics Highlight Their Dysregulation in Alzheimer’s Disease
Single-cell RNAseq performed in the entorhinal and prefrontal cortices have shown downregulation of excitatory transmission genes (e.g., SNAP25; RIMS1) in AD (Grubman et al., 2019; Mathys et al., 2019). The latter study found enrichment of regulators of myelination, inflammation, neuronal survival, and global stress response (especially in late-stage disease). However, early stages of AD were associated with the downregulation of both excitatory and inhibitory neuronal genes, which contrasts with findings from the gene microarrays described above. More recently, another study which examined tissues from frontal, temporal, and entorhinal cortices concluded that AD may be subclassed according to either the upregulation or downregulation of genes involved in excitatory synaptic transmission (Neff et al., 2021). This study also corroborated findings from previous RNAseq studies that AD is characterised by dysregulation of genes involved in immune activity, mitochondrial organisation, and myelination in addition to synaptic dysfunction. A single-nucleus RNAseq study also identified impairment of angiogenesis in addition to immune response, myelination, and synaptic signalling in the prefrontal cortex of AD cases (Braak stage ≥ 4) (Lau et al., 2020a). A meta-analysis of transcriptomic data from bulk human AD and mouse tissue showed that the gene signature of synaptic dysfunction in human disease broadly overlapped with the signature seen in mice with a low AD-type pathological burden, which supports the concept of synaptic and neuronal dysfunction as an early event in AD (Wan et al., 2020). Finally, a pre-print of one study which used fluorescence-activated cell sorting of NFT-bearing and NFT-free soma from Braak stage VI AD prefrontal cortex showed NFT-bearing neurons were associated with an upregulation of genes involved in synaptic transmission, including a core set of 63 genes seen across different neuronal subtypes that were enriched for synaptic vesicle cycling (e.g., SNAP25; SYT1 encoding synaptotagmin 1) and transsynaptic signalling (e.g., NTRK2 encoding the BDNF receptor TrkB) (Otero-Garcia et al., 2020). This result appears consistent with the suggestion that neurons with a high neuroplastic demand are most prone to tangle formation as discussed above.
Experimental Animal Models of Alzheimer’s Disease Have Demonstrated Reduced Synaptic Plasticity
LTP and LTD—though to a lesser extent—have been intensively investigated (and reviewed) in experimental animal models of AD (Mango et al., 2019). Electrophysiological recordings have shown reduced LTP in certain APP mouse strains (Palop et al., 2007; Balducci et al., 2011; D’Amelio et al., 2011; Tozzi et al., 2015). Similarly, APP/PS1 mice show reduced LTP (Trinchese et al., 2008; Calella et al., 2010) and LTD (Chang et al., 2006; Song et al., 2014; Yang et al., 2016). A triple transgenic model (3×Tg-AD mice expressing human APP, PS1, and MAPT mutations) displayed impaired LTP which correlated with intraneuronal Aβ before the formation of Aβ plaques and NFTs (Oddo et al., 2003). Loss of synaptic spines in primary neuronal cultures derived from APP/PS1 mice was shown to cause the loss of coordinated neuronal activity which eventually translated to impaired transmission efficiency and the breakdown of global neuronal network transmission (Kashyap et al., 2019). Conversely, other experimental systems have demonstrated that injury or denervation induced neuroplasticity leads to the upregulation of APP (Banati et al., 1993; Wallace et al., 1993; Beeson et al., 1994; Chauvet et al., 1997), the processing of which occurs at synaptic spines (Kamenetz et al., 2003; Priller et al., 2006). It is therefore conceivable that neuroplasticity changes associated with tau pathology act upstream of neuron activity-dependent generation of Aβ and its attendant neuronal network dysfunction—a scenario which is consistent with the synaptic gene microarray data outlined above and which may be especially relevant to sporadic AD (Arnsten et al., 2021). Thus pharmacological rescue of synaptic function represents an area open to pharmacological intervention (Prieto et al., 2017; Jackson et al., 2019).
The ultrastructure and quantitative biochemistry of dendritic spines were recently described in greater detail (Helm et al., 2021). In their investigation the authors combined mass spectrometry, EM, and super-resolution microscopy to annotate 47,000 spines for 110 synaptic proteins in cultured, predominantly glutamatergic, hippocampal neurons. The combination of these data allowed the authors to construct 3D models of the average stubby and mushroom spines. Both types of spines showed similar protein copy numbers and spatial organisation. It would be interesting to determine how the protein, and even lipid, content of spines might change in relation to the timing of Aβ and tau pathology across the AD continuum using similar techniques. For instance, the release of calcium from the endoplasmic reticulum can be up to 10 times greater in AD transgenic mice compared to wild type controls which can impact synaptic structure and function (Goussakov et al., 2010, 2011; Chakroborty et al., 2019).
Microglia in Alzheimer’s Disease
Microglia constitute a dynamic population of cells in the CNS (Gertig and Hanisch, 2014). They exhibit significant functional plasticity that is also reflected by morphological diversity. It was through the study of morphology that these intrinsic cells were first conclusively implicated in diseases of the CNS (Streit et al., 1988, 2014; Kreutzberg, 1996). Microglia can be distinguished from other cells of the CNS using EM without antibody-based techniques (Savage et al., 2018). Dark microglia are the most recently described type using EM (Bisht et al., 2016). These microglia display signs of oxidative stress, including an electron dense cyto- and nucleoplasm with remodelling of chromatin. They are reactive cells seen in chronic stress, ageing, and in CXCR1 knock-out and APP/PS1 mice. The morphology of microglia is influenced by many factors, including diet and obesity (Cope et al., 2018); consumption of alcohol (Marshall et al., 2020) and associated hepatic encephalopathy (Dennis et al., 2014); drug consumption (Burkovetskaya et al., 2020); infection and traumatic injury (Giordano et al., 2021); stress (Kreisel et al., 2014); ageing and sex (Brawek et al., 2021); sleep–wake cycles (Nakanishi et al., 2021); sleep deprivation (Wadhwa et al., 2017); and autoimmune and systemic diseases (Aw et al., 2020). Interestingly, microglia may also contribute to sex differences observed in neurodegenerative diseases (Stephen et al., 2019; Chowen and Garcia-Segura, 2021). Most recently in the field of AD, microglia are suggested to contribute to the propagation of Aβ by acting as carriers in brain tissue of young human APP-expressing 5×FAD mice (d’Errico et al., 2021).
Genomic and Transcriptomic Signatures of Microglia in Alzheimer’s Disease and Models of the Disease
The understanding of the molecular signatures of microglia has been expanded considerably over the last decade (Prinz et al., 2019) and includes the development of an atlas of transcriptomic changes in microglia across age, brain regions, and disease pathologies (de Paiva Lopes et al., 2020). RNAseq in mouse models of AD has been used to identify neuroprotective “disease-associated microglia” (DAM) as described by Keren-Shaul et al. (2017) and the “microglial neurodegenerative phenotype” (MGnD) (Krasemann et al., 2017)—both of which are characterised by TREM2 activation and associated with increased APOE expression. Single-cell RNAseq has also uncovered (mouse) disease stage-specific microglial signatures, including two profiles characterised by either type I or II interferon response genes (Mathys et al., 2017). Another RNAseq study in mice showed that immunological imprinting of microglia by either training (a single intraperitoneal injection of lipopolysaccharides) or tolerance (multiple injections) exacerbated or attenuated cerebral Aβ pathology, respectively (Wendeln et al., 2018). It is important that caution is exercised when interpreting results from mouse studies given the key differences between human and mouse single-cell microglial transcriptomes (Chen and Colonna, 2021). It appears that the binding of Aβ to pattern recognition receptors, including the receptor for advanced glycation end-products, nucleotide-binding oligomerisation domain-like, scavenger, formyl peptide, and toll-like receptors—among other mechanisms—is sufficient to cause neurotoxic activation of microglia (Salminen et al., 2009). A number of mechanisms by which neurotoxic microglia exert their effects are proposed including the release of cytokines such as interleukins (ILs), interferons, and tumour necrosis factor (TNF) which are elevated in the cerebrospinal fluid (CSF) of AD patients before the onset of symptoms (Tarkowski et al., 2003) and which have been shown to suppress LTP in rats (Griffin et al., 2006); reduced secretion of trophic factors such as transforming growth factor β (TGFβ) and BDNF (Parkhurst et al., 2013; Heneka et al., 2015); inhibition of mitochondrial respiration through increased nitric oxide (Meda et al., 1995; Poderoso et al., 2019); and phagoptosis of phosphatidylserine presenting neurons (Neniskyte et al., 2011). Furthermore, Baik et al. (2019) reported that exposure to monomeric, oligomeric, or fibrillar Aβ induced microglial activation by shifting metabolism toward aerobic glycolysis via the (mTOR)-hypoxia-inducible factor-1α (HIF-1α) pathway which resulted in the increased production of cytokines such as IL1β and was associated with mitochondrial impairment in microglia. Inhibition of glycolysis in microglia exposed to Aβ by treatment with 2-deoxy-D-glucose (2DG) resulted in reduced IL1β and TNF-α and preserved mitochrondrial function compared to microglia exposed to Aβ only.
Several genome wide association studies (GWAS) have been performed for late-onset AD (Bertram et al., 2008; Harold et al., 2009; Lambert et al., 2009; Hollingworth et al., 2011; Naj et al., 2011), including with concurrent meta-analyses (Lambert et al., 2013; Marioni et al., 2018; Jansen et al., 2019; Kunkle et al., 2019). Many of the common allelic variants which exert only a small effect size on the overall risk of developing AD are those expressed by myeloid cells and microglia in particular (Jones et al., 2015; Pimenova et al., 2018; Bertram and Tanzi, 2019; Andrews et al., 2020; Podleśny-Drabiniok et al., 2020). Other gene categories implicated include those involved in the processing of APP, endosomal-lysosomal vesicle cycling, and lipid and cholesterol metabolism and transport. Gene network and proteomic analyses also suggest that microglia have a role in AD. Gene-regulatory networks have demonstrated the differential regulation of microglial genes governed by DAP12 (encoded by TYROBP) and TREM2 signalling pathways (Zhang et al., 2013; Audrain et al., 2021). A study of protein networks in end-stage cortical AD tissue showed enrichment of microglial and astroglial markers suggesting that both are important effectors of cognitive impairment late in the disease course (Seyfried et al., 2017).
TREM2 rare variants represent the strongest risk factor for developing sporadic late-onset AD after the much more common APOE variants. The R47H variant more than doubles the risk of AD (Guerreiro et al., 2013; Jonsson et al., 2013). This polymorphism causes the expression of a truncated protein and a loss of function which results in reduced clearance of Aβ (Yeh et al., 2016). Another related set of genes implicated by GWAS is the MS4A family which includes risk polymorphisms in the MS4A4A and MS4A6A subtypes—the latter being significantly upregulated in both the PreC and PVC in our recent RNAseq report (Guennewig et al., 2021). These proteins are key regulators of cellular activation and levels of soluble TREM2 (Eon Kuek et al., 2016; Deming et al., 2019). The loss of TREM2 facilitates the accumulation of tau pathology but only in the presence of Aβ (Hardy and Salih, 2021; Haass, 2021; Lee et al., 2021). Again, the roles of microglia at different stages deserve attention. For instance, one in silico transcriptomic analysis supports the role of TREM2/TYROBP signalling during early stages of disease whereas later stages were associated with soluble TREM2 and nuclear factor κβ (NFκβ) (Ji et al., 2021).
TREM2 and CSF1R are emerging targets for disease-modifying therapeutics in AD and other neurodegenerative diseases (Piccioni et al., 2021). CSF1R is a transmembrane protein expressed by myeloid cells. Its ligands include CSF1 and IL34 (Elmore et al., 2014) and its activation supports the development of myeloid cells in mice (Lei et al., 2020). The exposure of 5×FAD mice (expressing three APP and two PSEN1 human mutations) to CSF1R inhibitors show reduced proliferation of microglia which are characterised by a shift to an anti-inflammatory profile and are associated with reduced neuronal loss (Spangenberg et al., 2016). Later work also showed CSF1R inhibitors prevented the formation of parenchymal Aβ plaques, but not CAA (Spangenberg et al., 2019). TREM2 is a transmembrane immunoglobulin expressed by microglia (Ulland and Colonna, 2018) and as mentioned is implicated in the pathogenesis of AD (Gratuze et al., 2018). Notably, there is controversy surrounding the expression of TREM2 by human microglia as the recruitment of peripheral myeloid cells may have been underappreciated to date (Fahrenhold et al., 2018). Notwithstanding this caveat, it appears that an effective microglial response to Aβ is TREM2-dependent and limits the extent of phosphotau pathology (Lee et al., 2021). The loss of function of TREM2 (as seen with the R47H mutation) results in reduced binding affinity to APOE compared to wild-type protein, resulting in reduced clustering of microglia at Aβ plaques and increased Aβ load (Krasemann et al., 2017). Interestingly, TREM2(+/–) deficiency is associated with increased Aβ plaques and phosphotau-positive dystrophic neurites compared to complete knock-out of TREM2(–/–) in AD mice, demonstrating a complex relationship between microgliosis and plaque-associated neurofibrillary pathology (Delizannis et al., 2021). More broadly, the loss of TREM2 function is also associated with reduced microglial survival (McQuade et al., 2020) and impaired lipid metabolism and thus represents a druggable target of particular interest (Deczkowska et al., 2020; Lewcock et al., 2020; Hardy and Salih, 2021; Haass, 2021).
Similarly, a loss of APOE function is associated with an impaired microglial response to AD pathology (Pimenova et al., 2017). It appears the interaction between APOE and TREM2 is required for an adequate microglial response to Aβ pathology (Shi and Holtzman, 2018; Nguyen et al., 2020). Single-cell RNAseq of human microglia demonstrated the loss of a subset of microglia which highly express APOE and TREM2 in the context of AD (Olah et al., 2020). Whilst APOE deficiency in APP/PS1 mice was associated with reduced density of Aβ plaques, remaining plaques showed reduced compaction, a loss of microglial clustering around plaques, worsened NFD, and a significant downregulation of immune-related genes (Ulrich et al., 2018) and others such as Itgax and Cst7—genes which are highly expressed in DAM (Keren-Shaul et al., 2017). Unexpectedly, the complete knock-out of microglia-specific APOE in 5×FAD mice did not alter plaque load, number of microglia, or clustering of microglia but was associated with increased average plaque size (Henningfield et al., 2022)—notably in this study, astroglial APOE is still present. The APOE ε4 allele contributes to the disruption of glial homeostatic functions (Fernandez et al., 2019). APOE4 carriers show an increased number of activated microglia compared to APOE3 controls (Egensperger et al., 1998) and the expression of human APOE4 increases reactive microglia with dystrophic processes around Aβ plaques which are larger in size compared to human APOE3-expressing mice (Rodriguez et al., 2014). It is likely that other microglial pathways, such as the activation of the inflammasome with increased downstream caspase-1 and IL1β activity (Ising et al., 2019) or the hypersecretion of extracellular vesicles (Clayton et al., 2021), also contribute to the accumulation of tau pathology. The cross talk and role of other glial cells over the course of AD will also have to be closely investigated for a more complete understanding of the disease and may open new avenues of therapies. For instance, astrocytic IL3 has been shown to promote the neuroprotective activation of microglia (McAlpine et al., 2021).
Distinct microglial transcriptional profiles are associated with Aβ or tau pathology (Gerrits et al., 2021; Lemprière, 2021). Gerrits et al. (2021) described 13 transcriptional subclusters of microglia in post-mortem AD tissues from the PVC and occipitotemporal cortex (fusiform gyrus). There were two subclusters labelled “AD1” and “AD2” which were of particular interest. AD1 microglia were most prominent in cases with only Aβ pathology in which the microglia correlated with Aβ load and localised to Aβ plaques. This correlation was absent in AD cases exhibiting both Aβ and tau pathology. AD2 microglia occurred in AD cases with both Aβ and tau pathology in which these microglia correlated with phosphotau. AD1 microglia showed enrichment of markers of phagocytosis and activation, including ITGAX, LPL, GPNMB, MYO1E, and SPP1. AD2 microglia showed enrichment of homeostatic genes (e.g., CX3CR1 and P2RY12) and a number of neuron-related genes such as GRID2, ADGRB3, and DPP10. Among the eleven other subclusters was one enriched for genes of proliferation (e.g., TOP2A and MKI67), another for markers of cellular stress (e.g., early response genes—FOS and JUNB; heat-shock genes—HSPA1A and HSPA1B), and homeostatic subclusters which were inversely correlated with the load of Aβ or tau pathology. The correlation of microglial genes with Aβ pathology was noted recently by us (Tsatsanis et al., 2021) and others beforehand (Matarin et al., 2015). The latter genome-wide expression analysis also demonstrated correlations between genes of synaptic plasticity to tau pathology. The close association between Aβ and activated microglia prior to tau deposition has now been noted in post-mortem neuropathological studies (Sheffield et al., 2000; Eikelenboom et al., 2010; Paasila et al., 2020), tau transgenic mice (P301S) (Yoshiyama et al., 2007), in vitro (Hopp et al., 2018), and in PET imaging studies (Zou et al., 2020; Pascoal et al., 2021).
Post-mortem Immunohistopathology Highlights the Degeneration of Microglia in Advanced Alzheimer’s Disease
A characterisation of the morphological subtypes of microglia in AD and their relationship to Aβ and NFD across differentially affected regions of the AD brain was performed in our lab (Paasila et al., 2019). In this study, a major feature of cortical AD tissue was the dramatic reduction in the number of healthy ramified Iba1-positive microglia in the ITC—a severely affected area of the AD brain in terms of neuronal loss and cortical atrophy. The observed reduction in Iba1 and its fragmented distribution can be assumed to have serious implications for the motility (Franco-Bocanegra et al., 2019), membrane ruffling, and phagocytic capacity of the microglial cell population given its role as an actin crosslinking protein essential for actin bundling (Bartles, 2000; Ohsawa et al., 2000, 2004; Sasaki et al., 2001) and its enrichment in vitro during phagocytosis of full-length tau oligomers (Das et al., 2020). Interestingly, the ITC showed a significantly increased density of activated microglia in preclinical AD cases only (cognitive controls with Alzheimer-type pathological changes at post-mortem—CAc). Activated microglia with reduced branching also increased with age as seen in an earlier (Davies et al., 2017) and recent post-mortem investigation (Casaletto et al., 2021). Further, the density of clusters of activated microglia was significantly higher in mildly affected areas of the AD brain such as the primary motor cortex (PMC) and PVC. Findings in the PMC also demonstrated the clustering of activated microglia around Aβ plaques ahead of the formation of dystrophic neurites (Paasila et al., 2020). However, the total percentage of microglia associated with a cluster was <2% and only a minority of Aβ plaques were associated with a cluster—the proportion of which decreased in a stepwise fashion from mildly to severely affected regions. Lastly, the internalisation of synaptophysin-positive pre-synapses was found to be significantly elevated in the superior frontal cortex of AD cases compared to both CAc and controls without AD-type pathology using super-resolution microscopy (Paasila et al., 2021). Synaptophysin is one of the most severely affected synaptic markers in AD (Reddy et al., 2005). It would be of interest to determine if the microglia observed in our latter study were targeting viable neurons or those already marked for removal or undergoing apoptosis.
The concept of protective microglial activation early in the disease time course followed by degeneration associated with a loss of homeostatic function during the end-stage of disease is increasingly appreciated (Navarro et al., 2018; Bennett and Liddelow, 2019; Schwabe et al., 2020; Streit et al., 2020; Chatila and Bradshaw, 2021). A previous investigation has highlighted the stark contrast between APP-based animal models which show strong activation of microglia and post-mortem human hippocampus which contrastingly showed a weak microglial response (Sanchez-Mejias et al., 2016)—similarly described in cortical tissue by us (Paasila et al., 2019). Sanchez-Mejias et al. (2016) also presented results from an in vitro investigation showing that the soluble fraction of phosphotau is responsible for driving the degeneration observed in microglia. The loss of ramified cells in AD observed by us has been validated in a larger cohort by others—albeit without the expected loss of branching complexity of residual cells (Franco-Bocanegra et al., 2021)—and has similarly been associated with AD-type neuropathology in the brains of people with Down’s syndrome (Martini et al., 2020). Dystrophic microglia were also associated with NFD in our investigations (Paasila et al., 2020) and in others’ (Streit et al., 2009). However, in our study brain pH showed a stronger correlation with dystrophic microglia than disease status (Paasila et al., 2019), highlighting the importance of agonal factors as a cause of phenotypic changes in microglia. More broadly, this would have important implications for –omics studies using post-mortem human tissue if the prominence of the immune signalling pathways is a residual of brain pH (Monoranu et al., 2009; Durrenberger et al., 2010).
Lipids in Alzheimer’s Disease
Genome Wide Association Studies Have Identified Lipid Metabolism as a Major Risk Category in Alzheimer’s Disease
The plasma membrane and the internal cellular endomembranes are mainly composed of lipids. There are many factors which affect lipid metabolism, including, age, sex, genetics, diet, and physical activity (Chew et al., 2020). Lipids are transported throughout the body as lipoproteins, molecules which have a hydrophobic centre of cholesterol, esters, and triglycerides surrounded by amphipathic phospholipids with the addition of apolipoproteins. Lipids influence the trafficking and proteolytic cleavage of key proteins in AD and their propensity to self-aggregate (Di Paolo and Kim, 2011). As discussed above, AD GWAS results can be divided into anomalies in neuroimmune function, endocytosis, and lipid (and cholesterol) metabolism (Pimenova et al., 2018; Vogrinc et al., 2021). The genetics underlying lipid metabolism are of particular interest as the dissociation of phospholipids may be the defining feature of neurodegeneration in AD (Hardy, 2017). The major drivers of the enrichment of lipid metabolic pathways in GWAS (excluding APOE) include ABCA7, a key regulator of cellular cholesterol, and apolipoprotein genes APOM and APOA5 (Kunkle et al., 2019). Additional genes identified from GWAS that are involved in lipid metabolism include other apolipoproteins (APOA1, APOA4, APOC1, APOC2, APOC3, APOC4, and APOJ); an intracellular cholesterol transporter (NPC1) and membrane-bound cholesterol pumps (ABCA1, ABCA2, and ABCG4); phospholipid transporters (ATP8A1, ATP8A2, ATP8B4, PCTP, and PLCG2); intracellular lipid receptors (OSBPL7 and OSBPL9); a high-density lipoprotein receptor (SCARB1); hepatic lipase (HTGL) and endothelial lipase (LIPG); MAL-like protein (MALL)—a component of cholesterol rafts; sterol O-acyltransferase-1 (SOAT1)—involved in the synthesis of fatty acids and cholesterol esters; and a fatty acid transporter (SLC27A4) (Jones et al., 2010).
Alzheimer’s Disease Is Characterised by Broad Dysregulation of Lipid Compounds in Blood Plasma and Across Different Regions of the Brain
Alzheimer’s disease is characterised by significant dysregulation of fatty acid (Snowden et al., 2017) and lipid metabolism (Wood, 2012; Touboul and Gaudin, 2014; Wilkins and Trushina, 2017; Kao et al., 2020). For example, a study by Sáiz-Vazquez et al. (2020) showed AD is associated with high serum LDL-cholesterol. Metabolomic analyses of two brain autopsy series (Baltimore Longitudinal Study of Aging and the Religious Orders Study) showed that whilst cholesterol is unchanged in AD brains, cholesterol precursors are reduced and decomposition metabolites (including non-enzymatically generated cytotoxic oxidised forms—oxysterols) are increased (Varma et al., 2021). However, at least one other study showed elevated cholesterol levels in cortical samples of AD brains (Lazar et al., 2013). Glycerolipids such as monoacylglycerol and diacylglycerol are increased in the frontal cortex and blood plasma early in the pathogenesis of AD (Chan et al., 2012; Wood et al., 2015). Conversely, triglycerides—the predominant glycerolipid in the body—are reduced in plasma of mild cognitive impairment and AD (Bernath et al., 2020). Other plasma lipidomic studies have also demonstrated reduced levels of most cholesteryl esters (Liu et al., 2021)—particularly long chain species (Proitsi et al., 2015)—in mild cognitive impairment and AD. Glycerophospholipids are the major constituent of the cell membrane (van Meer et al., 2008). Glycerophospholipids such as phosphatidylcholine, phosphatidylethanolamine, ethanolamine plasmalogen, and cardiolipin are significantly decreased in the AD cortex, with certain species inversely correlated to AD severity (Nitsch et al., 1992; Ginsberg et al., 1995; Pettegrew et al., 2001). Blood serum also shows significant reduction of glycerophospholipid content (Mapstone et al., 2014; Whiley et al., 2014). Lastly, sphingolipids including sphingomyelins (Varma et al., 2018) and ceramides (Filippov et al., 2012) are increased in AD brains, but reduced in CSF (Fonteh et al., 2015). Other sphingolipids including sphingosine 1-phosphate (He et al., 2010), sulphatides (early in the disease) (Cheng et al., 2013), and gangliosides (Ariga, 2017) are reduced in AD brains. Notably, plaque-associated lipids have been successfully visualised (Kiskis et al., 2015). Lipid deposits co-localised with Thioflavin-S-positive Aβ plaques and showed either lamellar structure or occurred as coalescing macro-aggregates up to ∼25 μm in size. Conversely, diffuse plaques did not show the same association with lipid structures. Aβ oligomers have been shown to bind to the cell membrane to cause rupture or pore formation capable of eliciting apoptosis associated with mitochondrial death (Deshpande et al., 2006).
Experimental Models of Alzheimer’s Disease Suggest That Abnormal Lipid Metabolism, Neuropathological Tissue Changes and Cognitive Impairment Are Linked
There are several experimental systems which have been used to elucidate the mechanisms by which lipid and related pathway alterations cause neurodegeneration. APP/PS1 mice have demonstrated a connection between lipid derangements and cognitive impairment (Zhang et al., 2020). Serum triglycerides readily cross the blood-brain-barrier and contribute to the impairment of memory and learning (Banks et al., 2018) possibly by the disruption of the NMDA component of hippocampal LTP (Farr et al., 2008). Cholesteryl esters—the storage product of excess cholesterol—have been identified as an upstream regulator of tau pathology using induced-pluripotent stem cell (iPSC) lines carrying APP mutations (van der Kant et al., 2019). In this study the reduction of cholesteryl esters reduced phosphotau by increasing proteasome activity. Other studies have examined the role of lipid receptors. For instance, the deletion of low-density lipoprotein-related protein 1 (LRP1) in adult mouse forebrain neurons lead to global defects in brain lipid metabolism, including decreased cholesterol, sulphatides, ceramides, and glycerolipids which culminate in synaptic loss (Liu et al., 2010). In line with this, a recent report demonstrated that the overexpression of low-density lipoprotein receptor (LDLR) markedly reduced APOE and attenuated NFD and neurodegeneration in P301S mice (Mendiola et al., 2021; Shi et al., 2021)—similarly demonstrated in an earlier study using APP/PS1 mice (Kim et al., 2009b). In addition, changes to lipid content in PLB4 mice expressing human β-secretase 1 (hBACE1) have shown brain region-specific vulnerabilities to lipid dysregulation such as in the hippocampus and hypothalamus, suggesting that changes to lipid content may leave certain brain regions more susceptible than others (Dey et al., 2020).
The Interrelationships Between Synapses, Microglia, and Lipids in Alzheimer’s Disease
Brain cholesterol dysmetabolism is related to synaptic dysfunction in a number of neurological diseases (Petrov et al., 2016), including AD (Petrov et al., 2017). Microglia have an important role in lipid metabolism (Loving and Bruce, 2020). Microglia are capable of sensing excess saturated fats and modulating hypothalamic control of energy metabolism (Folick et al., 2021). They are also sensitive to fatty acids derived from the body’s microbiota through which they may modulate Aβ deposition (Colombo et al., 2021). Valdearcos et al. (2014) reported that microglia in the murine mediobasal hypothalamus undergo inflammatory activation in response to excess dietary saturated fatty acids. More closely related to AD, the Aβ-induced reduction in sphingosine kinase, an important regulator of lipid secretion from neurons, is associated with defective microglial phagocytosis and is also associated with increased expression of the cytokines (e.g., TNFα, IL1β, IL6, and inducible nitric oxide synthase) and reduced IL4, TGFβ, and arginase-1 (Lee et al., 2018). They are also essential for remyelination following injury via TREM2-dependent lipid droplet biogenesis—a process which incidentally involves the esterification of cholesterol (Gouna et al., 2021) and which may be relevant given the potential of age-related myelin degeneration as a driver of Aβ pathology (Depp et al., 2021). It also appears that phagocytic activity governed by TREM2 transcriptionally regulates cholesterol transport and metabolism, whereby TREM2-deficient microglia capably phagocytose myelin debris but fail to clear excess cholesterol resulting in the accumulation of cholesteryl esters (Nugent et al., 2020).
Indeed, “lipid-droplet-accumulating microglia” (LDAM) accrue with age in humans and mice and exhibit defective phagocytic capability, overproduce reactive oxygen species, and secrete excess signalling cytokines such as TNFα, IL1β, and IL6 (Marschallinger et al., 2020; cf., Figure 1). The accumulation of lipid droplets have also been noted in APP/PS1 chimeric mice xenografted with wild-type or R47H-TREM2 mutant iPSCs (Claes et al., 2021). In the latter study, the R47H mutation resulted in reduced clustering of microglia at plaques but critically did not impair the formation of lipid droplets within individual cells. Microglia also appear active in the regulation of cholesterol-modulated phosphotau pathology through activation and phagocytic pathways (Nanjundaiah et al., 2021). Excess cholesterol and other lipids are detected by microglial TREM2 (amongst other receptors) when complexed with apolipoproteins (such as APOE or APOJ) which in turn activates the PI3K/AKT/mTOR signalling cascade (Peng et al., 2010) which is linked to the inhibition of GSK3β (Hermida et al., 2017)—an important mediator of intraneuronal tau hyperphosphorylation (Hooper et al., 2008). The remote attenuation of neuronal kinases potentially occurs through reduced expression of the aforementioned TNFα, IL1β, and IL6 as described in TREM2-overexpressing P301S transgenic mice (Jiang et al., 2016)—refer to Figure 2 for an outline of this process. Further, autophagy-lysosomal dysfunction appears sufficient to cause lipid droplet formation in microglia, increased IL1β and TNFs, and is associated with enhanced intraneuronal tau pathology and substantial synaptic degeneration (Xu et al., 2021).
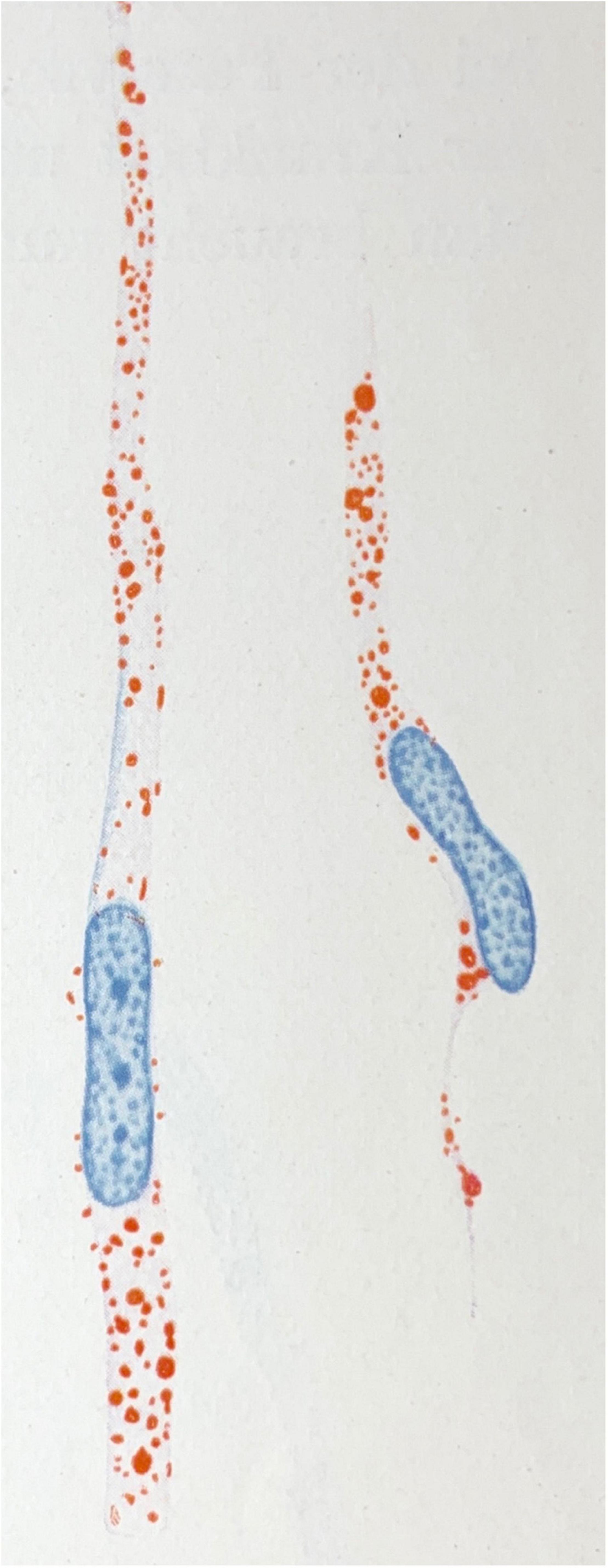
Figure 1. Microglial rod cells rich in lipoid material (scarlet red staining; Sudan IV). Drawing taken from Spielmeyer (1922).

Figure 2. Under physiological conditions TREM2 facilitates the phagocytosis of APOE-lipoprotein-bound Aβ. Lipid molecules are processed by the intracellular phagolysosomal system to yield free cholesterol which may be stored internally as cholesteryl esters or secreted by ATP-binding cassette (ABC) transporters. Aβ may be degraded in autophagic vacuoles, via the ubiquitin-proteasomal system, or by any of a number of Aβ-degrading proteases including the metalloendopeptidase and matrix metalloproteinase families of catabolic enzymes. Furthermore, DAP12 activation following TREM2 binding results in the activation of the PI3K/AKT/mTOR signalling cascade which reduces the secretion of signalling molecules such as IL1β, IL6, and TNFα which in turn is associated with reduced activation of intraneuronal GSK3β—an important protein kinase responsible for tau phosphorylation and which is also involved in the activation of γ-secretase through its interaction with presenilin 1. The R47H loss of function mutation in TREM2 and the APOE ε4 allele (both indicated in red) represent two factors which impair Aβ and lipid processing by microglia. Abnormal TREM2/DAP12 signalling may result in reduced PI3K/AKT/mTOR signalling and disinhibition of IL1β, IL6, and TNFα secretions, leading to increased intraneuronal phosphotau and the exacerbation of Aβ accumulation (indicated by the red arrows). Created with BioRender.com.
Several studies have identified distinct transcriptomic profiles in murine DAM for pattern recognition, lipid metabolism, and lysosomal pathways. Keren-Shaul et al. (2017) demonstrated an increased presence of DAM characterised by intracellular Aβ particles and elevated Lpl, Cst7, and Cd9—molecules involved in lipid uptake and phagocytosis—in a 5×FAD model of AD. Ofengeim et al. (2017) reported increased surface-bound enzyme cholesterol 25 hydroxylase (Ch25h) and Cst7 levels in microglia from APP/PS1 mice which were associated with reduced phagocytic capacity and lysosomal pathway impairment. Recently, Lau et al. (2020b) reported that IL33 injection in an APP/PS1 mouse model resulted in Aβ-plaque-associated microglia acquiring the DAM transcriptomic profile with increased levels of Apoe, Axl, Cst7, Lpl, and Trem2 and which was associated with increased clearance of Aβ. Together these findings demonstrate a close relationship between microglial lipid metabolism and phagocytosis.
The role of the complement pathway in microglia-mediated clearance of synapses in certain CNS regions is well established (Stevens et al., 2007). Graeber et al. (1988) first reported a striking upregulation of CR3 complement receptor expression in activated microglia under conditions of synaptic plasticity, suggesting the non-immunological involvement of the complement pathway in neuronal repair following axotomy. More recently, Lim and Ruthazer (2021) observed microglial trogocytosis of axons that was enhanced by neuronal expression of complement receptors in vivo in the Xenopus laevis retinotectal circuit. However, Weinhard et al. (2018) did not observe changes to trogocytosis following knockout of CR3 in mice, which suggested to the authors no role for complement pathway in trogocytosis. Mutations in the sushi repeat protein X-linked 2 (SRPX2) protein expressed by neurons have been identified in controlling complement pathway-mediated synapse elimination by microglia. Cong et al. (2020) reported that SRPX2 was able to block C1q activation and thereby inhibit the classical complement-mediated elimination of synapses. They also reported that SRPX2(–/Y) knockout mice have increased C3 deposition and microglial synapse engulfment in the dorsal lateral geniculate nucleus (dLGN). Avila-Martin et al. (2017) further reported that albumin-hydroxyoleic acid complex (A-HOA) promoted sensorimotor function recovery in rats with spinal cord injury (SCI) by upregulating several genes including SRPX2, suggesting that SRPX2 may play a role in reestablishing vascularisation and recovering synapse loss associated with SCI. In another study, Cong et al. (2021) showed that the deletion of C1q resulted in a persistent decrease in microglia-mediated synapse elimination and engulfment in the visual cortex. In our RNAseq study, C4A (encoding C4) and C5AR1 expression was increased in the PreC, with the latter also increased in the PVC (Guennewig et al., 2021). Interestingly CFHR5 (involved in the regulation of the alternative complement pathway) expression was substantially reduced in the PVC.
A number of recent studies highlight the impact of lipid metabolism on complement signalling pathways. For instance, complement factor C3 and C4 levels have been associated with cardiometabolic risk factors such as obesity and insulin resistance (Arias de la Rosa et al., 2020), fat distribution (Fu et al., 2020), and metabolic syndrome and diabetes (Copenhaver et al., 2020) in humans. In animal studies, findings have also demonstrated that peptide antagonists of complement receptors C3aR and C5aR inhibited diet-induced obesity, adipose inflammation, and metabolic dysfunction in rats and ameliorated inflammatory responses in murine macrophages (Lim et al., 2013)—a topic reviewed by Barbu et al. (2015). In the brain, Madore et al. (2020) reported that low omega-3 fatty acid intake altered the expression of complement cascade proteins both in microglia and at the synapse and exacerbated spine phagocytosis. In AD, clearance of Aβ has been shown to be significantly impacted by the presence of lipids as well as by lipid metabolism which may indirectly inhibit complement-mediated microglial clearance of Aβ plaques. Time-lapse atomic force microscopy has demonstrated that the presence of cholesterol in the cell lipid bilayer significantly enhances Aβ42 aggregation (Banerjee et al., 2021). Lipid membranes containing cholesterol promote Aβ42 aggregation via a heterogeneous nucleation pathway (Habchi et al., 2018). Finally, TREM2 may exert toxic effects later in the disease through a failure of the PI3K/AKT/mTOR pathway and increased IL1β, IL6, TNFα, and complement proteins despite showing protective effects earlier in the disease through the clearance of Aβ. Linnartz-Gerlach et al. (2019) demonstrated that aged TREM2 knock-out mice had lower transcription of C1qa, C1qb, C1qc, C3, and C4b. Thus the reactivation of complement-mediated synaptic pruning is a distinct possibility in AD (Stephan et al., 2012; Heppner et al., 2015; Brucato and Benjamin, 2020; Gomez-Arboledas et al., 2021), a scenario that has been also observed in human mutant APP mice (J20) (Hong et al., 2016). Refer to Figure 3 which presents a basic mechanism linking LDAM and aberrant synaptic elimination by microglia which may be mediated by increased complement deposition. On a positive note, it has been shown that age-related microglial activation can be reduced by increased physical activity given a recent retrospective study which found associations between late life physical activity, reduced microglial activation, reduced synaptic loss, and better cognitive outcomes (Casaletto et al., 2021). Although microglia have been a focus here, the role of other glial cells bear careful consideration. For instance, oligodendrocytes may be an important source of complement deposition in AD (Hosokawa et al., 2003) and astrocytes may exert neurotoxic effects following exposure to saturated lipids (Guttenplan et al., 2021) or microglial IL1α, TNF, and C1q (Liddelow et al., 2017). Indeed, single-nucleus RNAseq has demonstrated diverse astrocytic signatures in AD characterised by the enrichment of inflammatory, proteostatic genes, and metal ion homeostatic genes (Smith et al., 2021). Notably, the involvement of metal ions in AD have recently been reviewed by Lei et al. (2021).
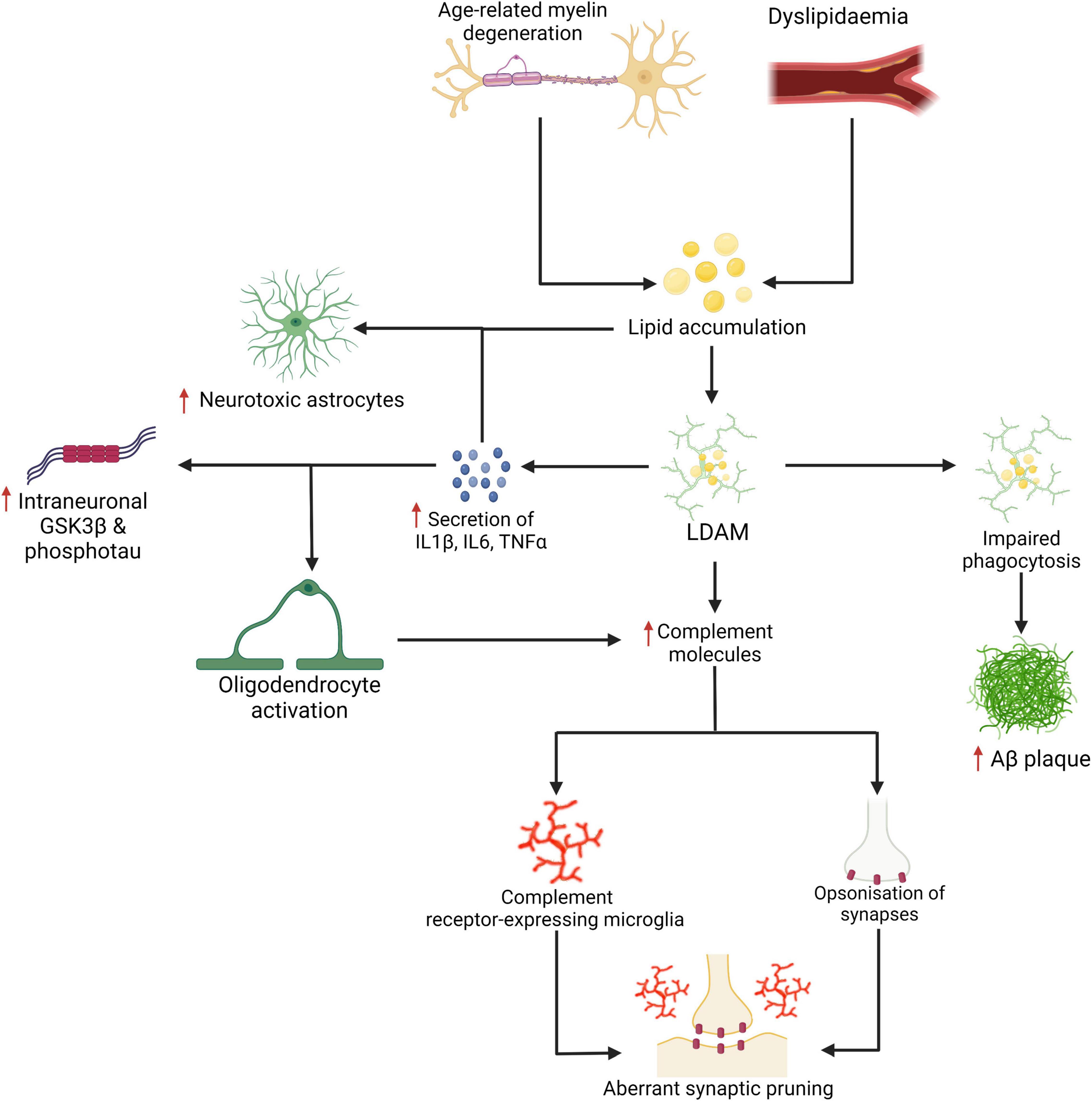
Figure 3. Age-related myelin degeneration and dyslipidemia represent two risk factors for the accumulation of saturated fats and other lipids in the brain. These lipids may be internalised by microglia and stored as cholesteryl esters. Lipid-droplet-accumulating microglia (LDAM) accumulate with age and are associated with increased secretion of IL1β, IL6, TNFα, complement molecules, and show impaired maturation of phagosomes and reduced phagocytic capacity. As demonstrated in Figure 2, the increased secretion of certain cytokines and impaired phagocytosis are linked with more severe neuropathological changes. The signalling cytokines are also implicated in the activation of oligodendrocytes (which may be a source of excess complement deposition) and the neurotoxic activation of astrocytes (for which saturated fats are a sufficient driver). Increased expression and deposition of complement proteins may represent a key event in the targeting of synapses by microglia in AD. Created with BioRender.com.
Conclusion
A purely biochemical, cell-autonomous view of AD—in which neurotoxic species of Aβ and phosphotau are solely responsible for neurodegeneration—has been described as “untenable” (De Strooper and Karran, 2016). Our own work suggests that the activation of microglia early in the disease time course is neuroprotective. The basic science on microglial biology is accelerating but remains a knowledge gap in our understanding of the pathogenesis of AD. For instance, it appears that at least some of the lipid associations seen in GWAS manifest through the dysfunction of microglia rather than simply the disruption of cellular membranes or neuronal function. The activities of microglia in AD represents one avenue for therapeutic intervention (Takata et al., 2021). However, given the complexity of the disease it is not unreasonable to expect that personalised treatment (Gauthier et al., 2018) or a combination of therapies targeting several lines of pathological processes—analogous to the therapeutic strategies used in cancer, HIV, tuberculosis, and cardiovascular disease—will be necessary for its successful management (Cummings et al., 2019; Salloway et al., 2020; Ju and Tam, 2021). To add complexity, further consideration for mixed pathology will also be essential in the management of patients in whom the dementing syndrome is caused by the cumulative effect of different disease states (Kapasi et al., 2017; Thomas et al., 2020) as AD neuropathology infrequently occurs in isolation (Schneider et al., 2007; James et al., 2012; Rahimi and Kovacs, 2014; Brenowitz et al., 2017; Boyle et al., 2018; De Reuck et al., 2018; McAleese et al., 2021). Lastly, the accumulation of lipids in amyloid plaques as well as in glial cells including microglia (Figure 4) deserves further study—age-related myelin degeneration and synaptic terminal membranes could be two relevant sources.
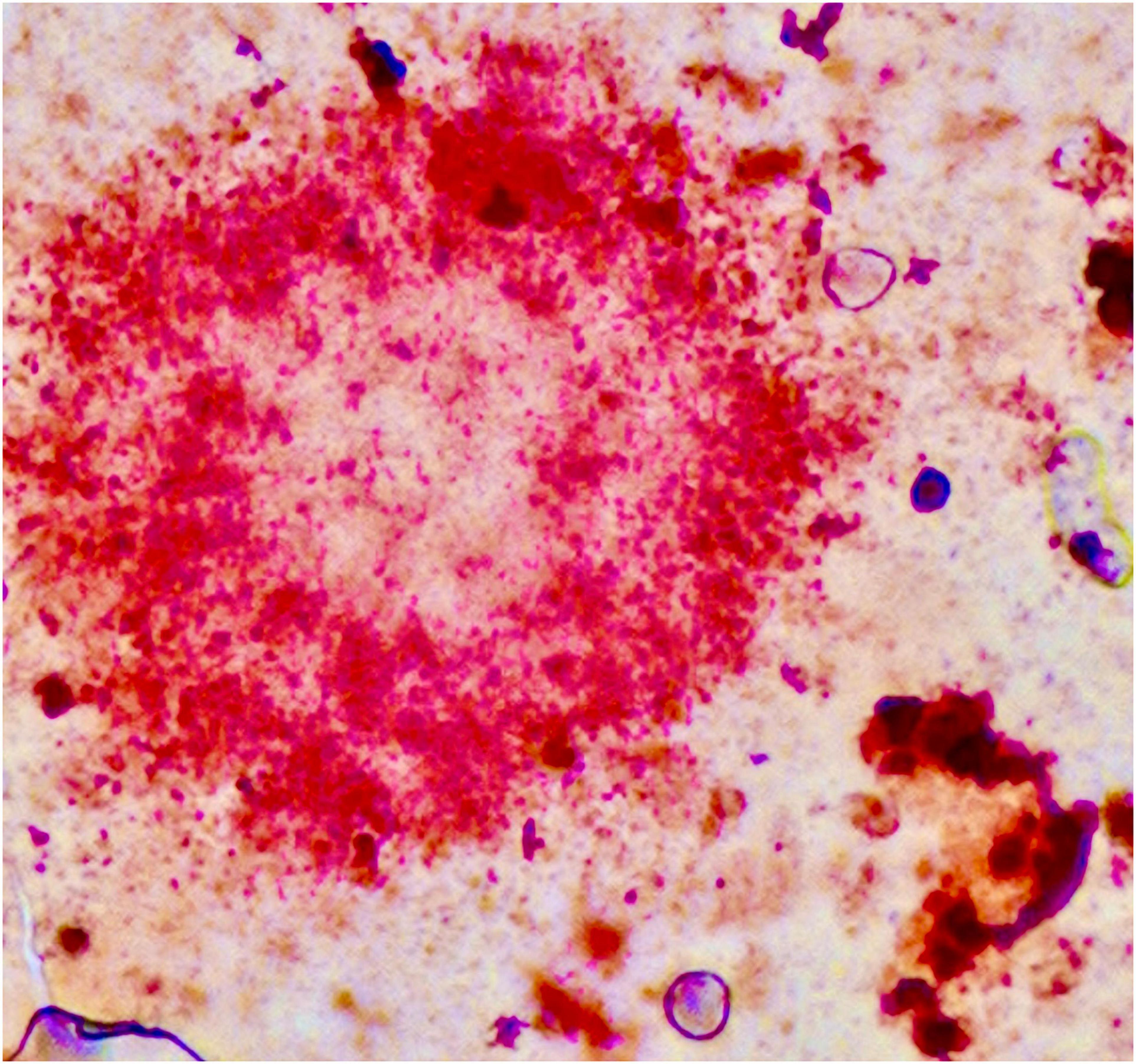
Figure 4. Amyloid plaque stained using the Herxheimer technique (Sudan IV). Significant amounts of lipids are found inside the plaque and in neighbouring glial cells. One cell probably representing a microglial cell is shown in the lower right. Formalin-fixed brain tissue, frozen section. Photograph taken by the authors (MBG): 20× oil primary magnification. Tissue section from Alois Alzheimer’s laboratory (Alzheimer, 1911; Graeber et al., 1997).
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This study was received funding from the Australian Research Council (ARC grant, DP150104472).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abraham, W. C., Jones, O. D., and Glanzman, D. L. (2019). Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn. 4:9. doi: 10.1038/s41539-019-0048-y
Alcamí, P., and Pereda, A. E. (2019). Beyond plasticity: the dynamic impact of electrical synapses on neural circuits. Nat. Rev. Neurosci. 20, 253–271. doi: 10.1038/s41583-019-0133-5
Alzheimer, A. (1907). Über eine eigenartige erkrankung der hirnrinde. Allg. Zeitschr. Psychiatr. 64, 146–148.
Alzheimer, A. (1911). Über eigenartige krankheitsfälle des späteren alters. Zeitschr. Gesamte Neurol. Psychiatr. 4, 356–385. doi: 10.1007/bf02866241
Alzheimer’s Association (2021). 2021 Alzheimer’s disease facts and figures Alzheimers Dement. 17, 327–406. doi: 10.1002/alz.12068
Andrews, S. J., Fulton-Howard, B., and Goate, A. (2020). Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol. 19, 326–335. doi: 10.1016/s1474-4422(19)30435-1
Arias de la Rosa, I., Font, P., Escudero-Contreras, A., López-Montilla, M. D., Pérez-Sánchez, C., Ábalos-Aguilera, M. C., et al. (2020). Complement component 3 as biomarker of disease activity and cardiometabolic risk factor in rheumatoid arthritis and spondyloarthritis. Ther. Adv. Chronic. Dis. 11:2040622320965067. doi: 10.1177/2040622320965067
Ariga, T. (2017). The pathogenic role of ganglioside metabolism in Alzheimer’s disease-cholinergic neuron-specific gangliosides and neurogenesis. Mol. Neurobiol. 54, 623–638. doi: 10.1007/s12035-015-9641-0
Arnsten, A. F. T., Datta, D., Tredici, K. D., and Braak, H. (2021). Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 17, 115–124. doi: 10.1002/alz.12192
Audrain, M., Haure-Mirande, J. V., Mleczko, J., Wang, M., Griffin, J. K., St George-Hyslop, P. H., et al. (2021). Reactive or transgenic increase in microglial TYROBP reveals a TREM2-independent TYROBP-APOE link in wild-type and Alzheimer’s-related mice. Alzheimers Dement. 17, 149–163. doi: 10.1002/alz.12256
Avila-Martin, G., Mata-Roig, M., Galán-Arriero, I., Taylor, J. S., Busquets, X., and Escribá, P. V. (2017). Treatment with albumin-hydroxyoleic acid complex restores sensorimotor function in rats with spinal cord injury: efficacy and gene expression regulation. PLoS One 12:e0189151. doi: 10.1371/journal.pone.0189151
Aw, E., Zhang, Y., and Carroll, M. (2020). Microglial responses to peripheral type 1 interferon. J. Neuroinflammation 17:340. doi: 10.1186/s12974-020-02003-z
Baik, S. H., Kang, S., Lee, W., Choi, H., Chung, S., Kim, J. I., et al. (2019). A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 30, 493–507.e496. doi: 10.1016/j.cmet.2019.06.005
Balducci, C., Mehdawy, B., Mare, L., Giuliani, A., Lorenzini, L., Sivilia, S., et al. (2011). The γ-secretase modulator CHF5074 restores memory and hippocampal synaptic plasticity in plaque-free Tg2576 mice. J. Alzheimers Dis. 24, 799–816. doi: 10.3233/jad-2011-101839
Banati, R. B., Gehrmann, J., Czech, C., Mönning, U., Jones, L. L., König, G., et al. (1993). Early and rapid de novo synthesis of Alzheimer β A4-amyloid precursor protein (APP) in activated microglia. Glia 9, 199–210. doi: 10.1002/glia.440090305
Banerjee, S., Hashemi, M., Zagorski, K., and Lyubchenko, Y. L. (2021). Cholesterol in membranes facilitates aggregation of amyloid β protein at physiologically relevant concentrations. ACS Chem. Neurosci. 12, 506–516. doi: 10.1021/acschemneuro.0c00688
Banks, W. A., Farr, S. A., Salameh, T. S., Niehoff, M. L., Rhea, E. M., Morley, J. E., et al. (2018). Triglycerides cross the blood-brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. (Lond.) 42, 391–397. doi: 10.1038/ijo.2017.231
Barbu, A., Hamad, O. A., Lind, L., Ekdahl, K. N., and Nilsson, B. (2015). The role of complement factor C3 in lipid metabolism. Mol. Immunol. 67, 101–107. doi: 10.1016/j.molimm.2015.02.027
Barthélemy, N. R., Horie, K., Sato, C., and Bateman, R. J. (2020). Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 217:e20200861. doi: 10.1084/jem.20200861
Bartles, J. R. (2000). Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 12, 72–78. doi: 10.1016/s0955-0674(99)00059-9
Beeson, J. G., Shelton, E. R., Chan, H. W., and Gage, F. H. (1994). Age and damage induced changes in amyloid protein precursor immunohistochemistry in the rat brain. J. Comp. Neurol. 342, 69–77. doi: 10.1002/cne.903420108
Bennett, F. C., and Liddelow, S. A. (2019). Microglia metabolic breakdown drives Alzheimer’s pathology. Cell Metab. 30, 405–406. doi: 10.1016/j.cmet.2019.08.017
Berchtold, N. C., Coleman, P. D., Cribbs, D. H., Rogers, J., Gillen, D. L., and Cotman, C. W. (2013). Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 34, 1653–1661. doi: 10.1016/j.neurobiolaging.2012.11.024
Bernath, M. M., Bhattacharyya, S., Nho, K., Barupal, D. K., Fiehn, O., Baillie, R., et al. (2020). Serum triglycerides in Alzheimer disease: relation to neuroimaging and CSF biomarkers. Neurology 94, e2088–e2098. doi: 10.1212/wnl.0000000000009436
Bertram, L., and Tanzi, R. E. (2019). Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 15, 191–192. doi: 10.1038/s41582-019-0158-4
Bertram, L., Lange, C., Mullin, K., Parkinson, M., Hsiao, M., Hogan, M. F., et al. (2008). Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 83, 623–632. doi: 10.1016/j.ajhg.2008.10.008
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5. doi: 10.1189/jlb.0306164
Bisht, K., Sharma, K. P., Lecours, C., Sánchez, M. G., El Hajj, H., Milior, G., et al. (2016). Dark microglia: A new phenotype predominantly associated with pathological states. Glia 64, 826–839. doi: 10.1002/glia.22966
Björkhem, I., Leoni, V., and Meaney, S. (2010). Genetic connections between neurological disorders and cholesterol metabolism. J. Lipid Res. 51, 2489–2503. doi: 10.1194/jlr.R006338
Boon, B. D. C., Bulk, M., Jonker, A. J., Morrema, T. H. J., van den Berg, E., Popovic, M., et al. (2020). The coarse-grained plaque: a divergent Aβ plaque-type in early-onset Alzheimer’s disease. Acta Neuropathol. 140, 811–830. doi: 10.1007/s00401-020-02198-8
Bossers, K., Wirz, K. T., Meerhoff, G. F., Essing, A. H., van Dongen, J. W., Houba, P., et al. (2010). Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 133(Pt 12), 3699–3723. doi: 10.1093/brain/awq258
Boyle, P. A., Yu, L., Wilson, R. S., Leurgans, S. E., Schneider, J. A., and Bennett, D. A. (2018). Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 83, 74–83. doi: 10.1002/ana.25123
Bozek, K., Wei, Y., Yan, Z., Liu, X., Xiong, J., Sugimoto, M., et al. (2015). Organization and evolution of brain lipidome revealed by large-scale analysis of human, chimpanzee, macaque, and mouse tissues. Neuron 85, 695–702. doi: 10.1016/j.neuron.2015.01.003
Braak, H., Thal, D. R., Ghebremedhin, E., and Del Tredici, K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. doi: 10.1097/NEN.0b013e318232a379
Brawek, B., Skok, M., and Garaschuk, O. (2021). Changing functional signatures of microglia along the axis of brain aging. Int. J. Mol. Sci. 22:1091. doi: 10.3390/ijms22031091
Brenowitz, W. D., Hubbard, R. A., Keene, C. D., Hawes, S. E., Longstreth, W. T. Jr., Woltjer, R. L., et al. (2017). Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement. 13, 654–662. doi: 10.1016/j.jalz.2016.09.015
Brookmeyer, R., Evans, D. A., Hebert, L., Langa, K. M., Heeringa, S. G., Plassman, B. L., et al. (2011). National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement. 7, 61–73. doi: 10.1016/j.jalz.2010.11.007
Brucato, F. H., and Benjamin, D. E. (2020). Synaptic pruning in Alzheimer’s disease: role of the complement system. Glob. J. Med. Res. 20. doi: 10.34257/gjmrfvol20is6pg1
Bu, G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344. doi: 10.1038/nrn2620
Burkovetskaya, M. E., Small, R., Guo, L., Buch, S., and Guo, M. L. (2020). Cocaine self-administration differentially activates microglia in the mouse brain. Neurosci. Lett. 728:134951. doi: 10.1016/j.neulet.2020.134951
Calella, A. M., Farinelli, M., Nuvolone, M., Mirante, O., Moos, R., Falsig, J., et al. (2010). Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med. 2, 306–314. doi: 10.1002/emmm.201000082
Casaletto, K. B., Lindbergh, C. A., VandeBunte, A., Neuhaus, J., Schneider, J. A., Buchman, A. S., et al. (2021). Microglial correlates of late life physical activity: Relationship with synaptic and cognitive aging in older adults. J. Neurosci. doi: 10.1523/jneurosci.1483-21.2021
Cermenati, G., Mitro, N., Audano, M., Melcangi, R. C., Crestani, M., De Fabiani, E., et al. (2015). Lipids in the nervous system: from biochemistry and molecular biology to patho-physiology. Biochim. Biophys. Acta 1851, 51–60. doi: 10.1016/j.bbalip.2014.08.011
Chakroborty, S., Hill, E. S., Christian, D. T., Helfrich, R., Riley, S., Schneider, C., et al. (2019). Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol. Neurodegener. 14:7. doi: 10.1186/s13024-019-0307-7
Chan, R. B., Oliveira, T. G., Cortes, E. P., Honig, L. S., Duff, K. E., Small, S. A., et al. (2012). Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 287, 2678–2688. doi: 10.1074/jbc.M111.274142
Chang, E. H., Savage, M. J., Flood, D. G., Thomas, J. M., Levy, R. B., Mahadomrongkul, V., et al. (2006). AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad Sci. U.S.A. 103, 3410–3415. doi: 10.1073/pnas.0507313103
Chatila, Z. K., and Bradshaw, E. M. (2021). Alzheimer’s disease genetics: a dampened microglial response? Neuroscientist doi: 10.1177/10738584211024531
Chauvet, N., Apert, C., Dumoulin, A., Epelbaum, J., and Alonso, G. (1997). Mab22C11 antibody to amyloid precursor protein recognizes a protein associated with specific astroglial cells of the rat central nervous system characterized by their capacity to support axonal outgrowth. J. Comp. Neurol. 377, 550–564.
Chen, Y., and Colonna, M. (2021). Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 218:e20202717. doi: 10.1084/jem.20202717
Cheng, H., Wang, M., Li, J. L., Cairns, N. J., and Han, X. (2013). Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: an early event in disease pathogenesis. J. Neurochem. 127, 733–738. doi: 10.1111/jnc.12368
Chew, H., Solomon, V. A., and Fonteh, A. N. (2020). Involvement of lipids in Alzheimer’s disease pathology and potential therapies. Front. Physiol. 11:598. doi: 10.3389/fphys.2020.00598
Chowen, J. A., and Garcia-Segura, L. M. (2021). Role of glial cells in the generation of sex differences in neurodegenerative diseases and brain aging. Mech. Ageing Dev. 196:111473. doi: 10.1016/j.mad.2021.111473
Claes, C., Danhash, E. P., Hasselmann, J., Chadarevian, J. P., Shabestari, S. K., England, W. E., et al. (2021). Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol. Neurodegener. 16:50. doi: 10.1186/s13024-021-00473-0
Clayton, K., Delpech, J. C., Herron, S., Iwahara, N., Ericsson, M., Saito, T., et al. (2021). Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 16:18. doi: 10.1186/s13024-021-00440-9
Colombo, A. V., Sadler, R. K., Llovera, G., Singh, V., Roth, S., Heindl, S., et al. (2021). Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. Elife 10:e59826. doi: 10.7554/eLife.59826
Cong, Q., Soteros, B. M., Huo, A., Li, Y., Tenner, A. J., and Sia, G. M. (2021). C1q and SRPX2 regulate microglia mediated synapse elimination during early development in the visual thalamus but not the visual cortex. Glia 1–15. doi: 10.1002/glia.24114
Cong, Q., Soteros, B. M., Wollet, M., Kim, J. H., and Sia, G. M. (2020). The endogenous neuronal complement inhibitor SRPX2 protects against complement-mediated synapse elimination during development. Nat. Neurosci. 23, 1067–1078. doi: 10.1038/s41593-020-0672-0
Cope, E. C., LaMarca, E. A., Monari, P. K., Olson, L. B., Martinez, S., Zych, A. D., et al. (2018). Microglia play an active role in obesity-associated cognitive decline. J. Neurosci. 38, 8889–8904. doi: 10.1523/jneurosci.0789-18.2018
Copenhaver, M. M., Yu, C. Y., Zhou, D., and Hoffman, R. P. (2020). Relationships of complement components C3 and C4 and their genetics to cardiometabolic risk in healthy, non-Hispanic white adolescents. Pediatr. Res. 87, 88–94. doi: 10.1038/s41390-019-0534-1
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Counts, S. E., He, B., Nadeem, M., Wuu, J., Scheff, S. W., and Mufson, E. J. (2012). Hippocampal drebrin loss in mild cognitive impairment. Neurodegener. Dis. 10, 216–219. doi: 10.1159/000333122
Counts, S. E., Nadeem, M., Lad, S. P., Wuu, J., and Mufson, E. J. (2006). Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol. 65, 592–601. doi: 10.1097/00005072-200606000-00007
Cruchaga, C., Haller, G., Chakraverty, S., Mayo, K., Vallania, F. L., Mitra, R. D., et al. (2012). Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS One 7:e31039. doi: 10.1371/journal.pone.0031039
Cserép, C., Pósfai, B., Lénárt, N., Fekete, R., László, Z. I., Lele, Z., et al. (2020). Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537. doi: 10.1126/science.aax6752
Cummings, J. L., Tong, G., and Ballard, C. (2019). Treatment combinations for Alzheimer’s disease: current and future pharmacotherapy options. J. Alzheimers Dis. 67, 779–794. doi: 10.3233/jad-180766
d’Errico, P., Ziegler-Waldkirch, S., Aires, V., Hoffmann, P., Mezö, C., Erny, D., et al. (2021). Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nat. Neurosci. doi: 10.1038/s41593-021-00951-0
Da Pozzo, E., Tremolanti, C., Costa, B., Giacomelli, C., Milenkovic, V. M., Bader, S., et al. (2019). Microglial pro-inflammatory and anti-inflammatory phenotypes are modulated by translocator protein activation. Int. J. Mol. Sci. 20. doi: 10.3390/ijms20184467
D’Amelio, M., Cavallucci, V., Middei, S., Marchetti, C., Pacioni, S., Ferri, A., et al. (2011). Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat. Neurosci. 14, 69–76. doi: 10.1038/nn.2709
Das, R., Balmik, A. A., and Chinnathambi, S. (2020). Phagocytosis of full-length tau oligomers by actin-remodeling of activated microglia. J. Neuroinflammation 17:10. doi: 10.1186/s12974-019-1694-y
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davies, C. A., Mann, D. M., Sumpter, P. Q., and Yates, P. O. (1987). A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J. Neurol. Sci. 78, 151–164. doi: 10.1016/0022-510x(87)90057-8
Davies, D. S., Ma, J., Jegathees, T., and Goldsbury, C. (2017). Microglia show altered morphology and reduced arborization in human brain during aging and Alzheimer’s disease. Brain Pathol. 27, 795–808. doi: 10.1111/bpa.12456
de Paiva Lopes, K., Snijders, G. J. L., Humphrey, J., Allan, A., Sneeboer, M., Navarro, E., et al. (2020). Atlas of genetic effects in human microglia transcriptome across brain regions, aging and disease pathologies. bioRxiv [Preprint] doi: 10.1101/2020.10.27.356113
De Reuck, J., Maurage, C. A., Deramecourt, V., Pasquier, F., Cordonnier, C., Leys, D., et al. (2018). Aging and cerebrovascular lesions in pure and in mixed neurodegenerative and vascular dementia brains: a neuropathological study. Folia Neuropathol. 56, 81–87. doi: 10.5114/fn.2018.76610
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer’s disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
de Wilde, M. C., Overk, C. R., Sijben, J. W., and Masliah, E. (2016). Meta-analysis of synaptic pathology in Alzheimer’s disease reveals selective molecular vesicular machinery vulnerability. Alzheimers Dement. 12, 633–644. doi: 10.1016/j.jalz.2015.12.005
Deczkowska, A., Weiner, A., and Amit, I. (2020). The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell 181, 1207–1217. doi: 10.1016/j.cell.2020.05.003
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464. doi: 10.1002/ana.410270502
Delizannis, A. T., Nonneman, A., Tsering, W., De Bondt, A., Van den Wyngaert, I., Zhang, B., et al. (2021). Effects of microglial depletion and TREM2 deficiency on Aβ plaque burden and neuritic plaque tau pathology in 5XFAD mice. Acta Neuropathol. Commun. 9:150. doi: 10.1186/s40478-021-01251-1
Deming, Y., Filipello, F., Cignarella, F., Cantoni, C., Hsu, S., Mikesell, R., et al. (2019). The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci. Transl. Med. 11:eaau2291. doi: 10.1126/scitranslmed.aau2291
Dennis, C. V., Sheahan, P. J., Graeber, M. B., Sheedy, D. L., Kril, J. J., and Sutherland, G. T. (2014). Microglial proliferation in the brain of chronic alcoholics with hepatic encephalopathy. Metab. Brain Dis. 29, 1027–1039. doi: 10.1007/s11011-013-9469-0
Depp, C., Sun, T., Sasmita, A. O., Spieth, L., Berghoff, S., Steixner-Kumar, A., et al. (2021). Ageing-associated myelin dysfunction drives amyloid deposition in mouse models of Alzheimer’s disease. bioRxiv [Preprint] doi: 10.1101/2021.07.31.454562
Deshpande, A., Mina, E., Glabe, C., and Busciglio, J. (2006). Different conformations of amyloid β induce neurotoxicity by distinct mechanisms in human cortical neurons. J. Neurosci. 26, 6011–6018. doi: 10.1523/jneurosci.1189-06.2006
Dey, M., Gunn-Moore, F. J., Platt, B., and Smith, T. K. (2020). Brain region-specific lipid alterations in the PLB4 hBACE1 knock-in mouse model of Alzheimer’s disease. Lipids Health Dis. 19:201. doi: 10.1186/s12944-020-01367-8
Di Paolo, G., and Kim, T. W. (2011). Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat. Rev. Neurosci. 12, 284–296. doi: 10.1038/nrn3012
Dickson, D. W., Crystal, H. A., Bevona, C., Honer, W., Vincent, I., and Davies, P. (1995). Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol. Aging 16, 285–298; discussion 298–304. doi: 10.1016/0197-4580(95)00013-5
Dickson, D. W., Farlo, J., Davies, P., Crystal, H., Fuld, P., and Yen, S. H. (1988). Alzheimer’s disease: a double-labeling immunohistochemical study of senile plaques. Am. J. Pathol. 132, 86–101.
Durrenberger, P. F., Fernando, S., Kashefi, S. N., Ferrer, I., Hauw, J. J., Seilhean, D., et al. (2010). Effects of antemortem and postmortem variables on human brain mRNA quality: a BrainNet Europe study. J. Neuropathol. Exp. Neurol. 69, 70–81. doi: 10.1097/NEN.0b013e3181c7e32f
Dyrks, T., Weidemann, A., Multhaup, G., Salbaum, J. M., Lemaire, H. G., Kang, J., et al. (1988). Identification, transmembrane orientation and biogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J. 7, 949–957. doi: 10.1002/j.1460-2075.1988.tb02900.x
Egensperger, R., Kösel, S., von Eitzen, U., and Graeber, M. B. (1998). Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol. 8, 439–447. doi: 10.1111/j.1750-3639.1998.tb00166.x
Eikelenboom, P., and Stam, F. C. (1982). Immunoglobulins and complement factors in senile plaques: an immunoperoxidase study. Acta Neuropathol. 57, 239–242. doi: 10.1007/bf00685397
Eikelenboom, P., Hack, C. E., Rozemuller, J. M., and Stam, F. C. (1989). Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 56, 259–262. doi: 10.1007/bf02890024
Eikelenboom, P., van Exel, E., Hoozemans, J. J., Veerhuis, R., Rozemuller, A. J., and van Gool, W. A. (2010). Neuroinflammation – an early event in both the history and pathogenesis of Alzheimer’s disease. Neurodegener. Dis. 7, 38–41. doi: 10.1159/000283480
Elmore, M. R., Najafi, A. R., Koike, M. A., Dagher, N. N., Spangenberg, E. E., Rice, R. A., et al. (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. doi: 10.1016/j.neuron.2014.02.040
Eon Kuek, L., Leffler, M., Mackay, G. A., and Hulett, M. D. (2016). The MS4A family: counting past 1, 2 and 3. Immunol. Cell Biol. 94, 11–23. doi: 10.1038/icb.2015.48
Fahrenhold, M., Rakic, S., Classey, J., Brayne, C., Ince, P. G., Nicoll, J. A. R., et al. (2018). TREM2 expression in the human brain: a marker of monocyte recruitment? Brain Pathol. 28, 595–602. doi: 10.1111/bpa.12564
Farr, S. A., Yamada, K. A., Butterfield, D. A., Abdul, H. M., Xu, L., Miller, N. E., et al. (2008). Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology 149, 2628–2636. doi: 10.1210/en.2007-1722
Fernandez, C. G., Hamby, M. E., McReynolds, M. L., and Ray, W. J. (2019). The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci. 11:14. doi: 10.3389/fnagi.2019.00014
Filippov, V., Song, M. A., Zhang, K., Vinters, H. V., Tung, S., Kirsch, W. M., et al. (2012). Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J. Alzheimers Dis. 29, 537–547. doi: 10.3233/jad-2011-111202
Folick, A., Koliwad, S. K., and Valdearcos, M. (2021). Microglial lipid biology in the hypothalamic regulation of metabolic homeostasis. Front. Endocrinol. (Lausanne) 12:668396. doi: 10.3389/fendo.2021.668396
Fonteh, A. N., Ormseth, C., Chiang, J., Cipolla, M., Arakaki, X., and Harrington, M. G. (2015). Sphingolipid metabolism correlates with cerebrospinal fluid β amyloid levels in Alzheimer’s disease. PLoS One 10:e0125597. doi: 10.1371/journal.pone.0125597
Franco-Bocanegra, D. K., George, B., Lau, L. C., Holmes, C., Nicoll, J. A. R., and Boche, D. (2019). Microglial motility in Alzheimer’s disease and after Aβ42 immunotherapy: a human post-mortem study. Acta Neuropathol. Commun. 7:174. doi: 10.1186/s40478-019-0828-x
Franco-Bocanegra, D. K., Gourari, Y., McAuley, C., Chatelet, D. S., Johnston, D. A., Nicoll, J. A. R., et al. (2021). Microglial morphology in Alzheimer’s disease and after Aβ immunotherapy. Sci. Rep. 11:15955. doi: 10.1038/s41598-021-95535-0
Fu, S., Yao, Y., Wu, S., Deng, J., Lv, F., and Zhao, Y. (2020). Inverse association between periumbilical fat and longevity mediated by complement C3 and cardiac structure. Aging (Albany NY) 12, 23296–23305. doi: 10.18632/aging.104113
Gauthier, S., Ng, K. P., Pascoal, T. A., Zhang, H., and Rosa-Neto, P. (2018). Targeting Alzheimer’s disease at the right time and the right place: validation of a personalized approach to diagnosis and treatment. J. Alzheimers Dis. 64, S23–S31. doi: 10.3233/jad-179924
George, C., Gontier, G., Lacube, P., François, J. C., Holzenberger, M., and Aïd, S. (2017). The Alzheimer’s disease transcriptome mimics the neuroprotective signature of IGF-1 receptor-deficient neurons. Brain 140, 2012–2027. doi: 10.1093/brain/awx132
Gerrits, E., Brouwer, N., Kooistra, S. M., Woodbury, M. E., Vermeiren, Y., Lambourne, M., et al. (2021). Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696. doi: 10.1007/s00401-021-02263-w
Gertig, U., and Hanisch, U. K. (2014). Microglial diversity by responses and responders. Front. Cell Neurosci. 8:101. doi: 10.3389/fncel.2014.00101
Gertz, H. J., Krüger, H., Patt, S., and Cervos-Navarro, J. (1991). Tangle-bearing neurons show more extensive dendritic trees than tangle-free neurons in area CA1 of the hippocampus in Alzheimer’s disease. Brain Res. 548, 260–266. doi: 10.1016/0006-8993(91)91130-s
Ginhoux, F., and Prinz, M. (2015). Origin of microglia: current concepts and past controversies. Cold Spring Harb. Perspect. Biol. 7:a020537. doi: 10.1101/cshperspect.a020537
Ginsberg, L., Rafique, S., Xuereb, J. H., Rapoport, S. I., and Gershfeld, N. L. (1995). Disease and anatomic specificity of ethanolamine plasmalogen deficiency in Alzheimer’s disease brain. Brain Res. 698, 223–226. doi: 10.1016/0006-8993(95)00931-f
Ginsberg, S. D., Alldred, M. J., Counts, S. E., Cataldo, A. M., Neve, R. L., Jiang, Y., et al. (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 68, 885–893. doi: 10.1016/j.biopsych.2010.05.030
Giordano, K. R., Denman, C. R., Dubisch, P. S., Akhter, M., and Lifshitz, J. (2021). An update on the rod microglia variant in experimental and clinical brain injury and disease. Brain Commun. 3:fcaa227. doi: 10.1093/braincomms/fcaa227
Goedert, M., Spillantini, M. G., Jakes, R., Rutherford, D., and Crowther, R. A. (1989). Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 3, 519–526. doi: 10.1016/0896-6273(89)90210-9
Gomez-Arboledas, A., Acharya, M. M., and Tenner, A. J. (2021). The role of complement in synaptic pruning and neurodegeneration. Immunotargets Ther. 10, 373–386. doi: 10.2147/itt.S305420
Gouna, G., Klose, C., Bosch-Queralt, M., Liu, L., Gokce, O., Schifferer, M., et al. (2021). TREM2-dependent lipid droplet biogenesis in phagocytes is required for remyelination. J. Exp. Med. 218:e20210227. doi: 10.1084/jem.20210227
Goussakov, I., Chakroborty, S., and Stutzmann, G. E. (2011). Generation of dendritic Ca2+ oscillations as a consequence of altered ryanodine receptor function in AD neurons. Channels (Austin) 5, 9–13. doi: 10.4161/chan.5.1.14124
Goussakov, I., Miller, M. B., and Stutzmann, G. E. (2010). NMDA-mediated Ca(2+) influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer’s disease mice. J. Neurosci. 30, 12128–12137. doi: 10.1523/jneurosci.2474-10.2010
Graeber, M. B., Kösel, S., Egensperger, R., Banati, R. B., Müller, U., Bise, K., et al. (1997). Rediscovery of the case described by Alois Alzheimer in 1911: historical, histological and molecular genetic analysis. Neurogenetics 1, 73–80. doi: 10.1007/s100480050011
Graeber, M. B., Streit, W. J., and Kreutzberg, G. W. (1988). Axotomy of the rat facial nerve leads to increased CR3 complement receptor expression by activated microglial cells. J. Neurosci. Res. 21, 18–24. doi: 10.1002/jnr.490210104
Gratuze, M., Leyns, C. E. G., and Holtzman, D. M. (2018). New insights into the role of TREM2 in Alzheimer’s disease. Mol. Neurodegener. 13:66. doi: 10.1186/s13024-018-0298-9
Grehan, S., Tse, E., and Taylor, J. M. (2001). Two distal downstream enhancers direct expression of the human apolipoprotein E gene to astrocytes in the brain. J. Neurosci. 21, 812–822. doi: 10.1523/jneurosci.21-03-00812.2001
Griffin, R., Nally, R., Nolan, Y., McCartney, Y., Linden, J., and Lynch, M. A. (2006). The age-related attenuation in long-term potentiation is associated with microglial activation. J. Neurochem. 99, 1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x
Grubman, A., Chew, G., Ouyang, J. F., Sun, G., Choo, X. Y., McLean, C., et al. (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097. doi: 10.1038/s41593-019-0539-4
Guennewig, B., Lim, J., Marshall, L., McCorkindale, A. N., Paasila, P. J., Patrick, E., et al. (2021). Defining early changes in Alzheimer’s disease from RNA sequencing of brain regions differentially affected by pathology. Sci. Rep. 11:4865. doi: 10.1038/s41598-021-83872-z
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Guttenplan, K. A., Weigel, M. K., Prakash, P., Wijewardhane, P. R., Hasel, P., Rufen-Blanchette, U., et al. (2021). Neurotoxic reactive astrocytes induce cell death via saturated lipids. Nature 599, 102–107. doi: 10.1038/s41586-021-03960-y
Haass, C. (2021). Loss of TREM2 facilitates tau accumulation, spreading, and brain atrophy, but only in the presence of amyloid pathology. Neuron 109, 1243–1245. doi: 10.1016/j.neuron.2021.03.029
Haass, C., and Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Haass, C., Kaether, C., Thinakaran, G., and Sisodia, S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2:a006270. doi: 10.1101/cshperspect.a006270
Habchi, J., Chia, S., Galvagnion, C., Michaels, T. C. T., Bellaiche, M. M. J., Ruggeri, F. S., et al. (2018). Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat. Chem. 10, 673–683. doi: 10.1038/s41557-018-0031-x
Hardy, J. (2017). Membrane damage is at the core of Alzheimer’s disease. Lancet Neurol. 16:342. doi: 10.1016/s1474-4422(17)30091-1
Hardy, J., and Salih, D. (2021). TREM2-mediated activation of microglia breaks link between amyloid and tau. Lancet Neurol. 20, 416–417. doi: 10.1016/s1474-4422(21)00133-2
Harold, D., Abraham, R., Hollingworth, P., Sims, R., Gerrish, A., Hamshere, M. L., et al. (2009). Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 41, 1088–1093. doi: 10.1038/ng.440
He, X., Huang, Y., Li, B., Gong, C. X., and Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
He, Y., Wei, M., Wu, Y., Qin, H., Li, W., Ma, X., et al. (2019). Amyloid β oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol–4,5–bisphosphate. Nat. Commun. 10:1193. doi: 10.1038/s41467-019-09114-z
Healy, L. M., Perron, G., Won, S. Y., Michell-Robinson, M. A., Rezk, A., Ludwin, S. K., et al. (2016). MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J. Immunol. 196, 3375–3384. doi: 10.4049/jimmunol.1502562
Helm, M. S., Dankovich, T. M., Mandad, S., Rammner, B., Jähne, S., Salimi, V., et al. (2021). A large-scale nanoscopy and biochemistry analysis of postsynaptic dendritic spines. Nat. Neurosci. 24, 1151–1162. doi: 10.1038/s41593-021-00874-w
Heneka, M. T., Golenbock, D. T., and Latz, E. (2015). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Henningfield, C. M., Arreola, M. A., Soni, N., Spangenberg, E. E., and Green, K. N. (2022). Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression. Glia 70, 287–302. doi: 10.1002/glia.24105
Heppner, F. L., Ransohoff, R. M., and Becher, B. (2015). Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. doi: 10.1038/nrn3880
Hermida, M. A., Dinesh Kumar, J., and Leslie, N. R. (2017). GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 65, 5–15. doi: 10.1016/j.jbior.2017.06.003
Hickman, S. E., Kingery, N. D., Ohsumi, T. K., Borowsky, M. L., Wang, L. C., Means, T. K., et al. (2013). The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905. doi: 10.1038/nn.3554
Hickman, S., Izzy, S., Sen, P., Morsett, L., and El Khoury, J. (2018). Microglia in neurodegeneration. Nat. Neurosci. 21, 1359–1369. doi: 10.1038/s41593-018-0242-x
Hollingworth, P., Harold, D., Sims, R., Gerrish, A., Lambert, J. C., Carrasquillo, M. M., et al. (2011). Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 43, 429–435. doi: 10.1038/ng.803
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hooper, C., Killick, R., and Lovestone, S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 104, 1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x
Hopp, S. C., Lin, Y., Oakley, D., Roe, A. D., DeVos, S. L., Hanlon, D., et al. (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15:269. doi: 10.1186/s12974-018-1309-z
Hosokawa, M., Klegeris, A., Maguire, J., and McGeer, P. L. (2003). Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia 42, 417–423. doi: 10.1002/glia.10234
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Iqbal, K., Wang, X., Blanchard, J., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010b). Alzheimer’s disease neurofibrillary degeneration: pivotal and multifactorial. Biochem. Soc. Trans. 38, 962–966. doi: 10.1042/bst0380962
Iqbal, K., Liu, F., Gong, C. X., and Grundke-Iqbal, I. (2010a). Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 7, 656–664. doi: 10.2174/156720510793611592
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S. V., Vieira-Saecker, A., et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Itagaki, S., McGeer, P. L., Akiyama, H., Zhu, S., and Selkoe, D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24, 173–182. doi: 10.1016/0165-5728(89)90115-x
Ittner, A., and Ittner, L. M. (2018). Dendritic tau in Alzheimer’s disease. Neuron 99, 13–27. doi: 10.1016/j.neuron.2018.06.003
Iwatsubo, T., Odaka, A., Suzuki, N., Mizusawa, H., Nukina, N., and Ihara, Y. (1994). Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ 42(43). Neuron 13, 45–53. doi: 10.1016/0896-6273(94)90458-8
Jack, C. R. Jr., Knopman, D. S., Jagust, W. J., Shaw, L. M., Aisen, P. S., Weiner, M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. doi: 10.1016/s1474-4422(09)70299-6
Jackson, J., Jambrina, E., Li, J., Marston, H., Menzies, F., Phillips, K., et al. (2019). Targeting the synapse in Alzheimer’s disease. Front. Neurosci. 13:735. doi: 10.3389/fnins.2019.00735
James, B. D., Bennett, D. A., Boyle, P. A., Leurgans, S., and Schneider, J. A. (2012). Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA 307, 1798–1800. doi: 10.1001/jama.2012.3556
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413.
Ji, Z., Liu, C., Zhao, W., Soto, C., and Zhou, X. (2021). Multi-scale modeling for systematically understanding the key roles of microglia in AD development. Comput. Biol. Med. 133, 104374. doi: 10.1016/j.compbiomed.2021.104374
Jiang, T., Zhang, Y. D., Chen, Q., Gao, Q., Zhu, X. C., Zhou, J. S., et al. (2016). TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 105, 196–206. doi: 10.1016/j.neuropharm.2016.01.028
Jicha, G. A., Abner, E. L., Schmitt, F. A., Kryscio, R. J., Riley, K. P., Cooper, G. E., et al. (2012). Preclinical AD Workgroup staging: pathological correlates and potential challenges. Neurobiol. Aging 33, 622.e621–622 e622. doi: 10.1016/j.neurobiolaging.2011.02.018
Jones, L., Holmans, P. A., Hamshere, M. L., Harold, D., Moskvina, V., Ivanov, D., et al. (2010). Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 5:e13950. doi: 10.1371/journal.pone.0013950
Jones, L., Lambert, J.-C., Wang, L.-S., Choi, S.-H., Harold, D., Vedernikov, A., et al. (2015). Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement. 11, 658–671. doi: 10.1016/j.jalz.2014.05.1757
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Ju, Y., and Tam, K. Y. (2021). Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 17, 543–549. doi: 10.4103/1673-5374.320970
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., Iwatsubo, T., et al. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/s0896-6273(03)00124-7
Kang, J., Lemaire, H. G., Unterbeck, A., Salbaum, J. M., Masters, C. L., Grzeschik, K. H., et al. (1987). The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325, 733–736. doi: 10.1038/325733a0
Kao, Y. C., Ho, P. C., Tu, Y. K., Jou, I. M., and Tsai, K. J. (2020). Lipids and Alzheimer’s disease. Int. J. Mol. Sci. 21:1505. doi: 10.3390/ijms21041505
Kapasi, A., DeCarli, C., and Schneider, J. A. (2017). Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 134, 171–186. doi: 10.1007/s00401-017-1717-7
Kashyap, G., Bapat, D., Das, D., Gowaikar, R., Amritkar, R. E., Rangarajan, G., et al. (2019). Synapse loss and progress of Alzheimer’s disease –a network model. Sci. Rep. 9:6555. doi: 10.1038/s41598-019-43076-y
Kennedy, M. B. (2016). Synaptic signaling in learning and memory. Cold Spring Harb. Perspect. Biol. 8:a016824. doi: 10.1101/cshperspect.a016824
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e1217. doi: 10.1016/j.cell.2017.05.018
Kim, J., Basak, J. M., and Holtzman, D. M. (2009a). The role of apolipoprotein E in Alzheimer’s disease. Neuron 63, 287–303. doi: 10.1016/j.neuron.2009.06.026
Kim, J., Castellano, J. M., Jiang, H., Basak, J. M., Parsadanian, M., Pham, V., et al. (2009b). Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular Aβ clearance. Neuron 64, 632–644. doi: 10.1016/j.neuron.2009.11.013
Kiskis, J., Fink, H., Nyberg, L., Thyr, J., Li, J. Y., and Enejder, A. (2015). Plaque-associated lipids in Alzheimer’s diseased brain tissue visualized by nonlinear microscopy. Sci. Rep. 5:13489. doi: 10.1038/srep13489
Kofler, J., and Wiley, C. A. (2011). Microglia: key innate immune cells of the brain. Toxicol. Pathol. 39, 103–114. doi: 10.1177/0192623310387619
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e569. doi: 10.1016/j.immuni.2017.08.008
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. doi: 10.1038/mp.2013.155
Kreutzberg, G. W. (1996). Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318. doi: 10.1016/0166-2236(96)10049-7
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Lambert, J.-C., Heath, S., Even, G., Campion, D., Sleegers, K., Hiltunen, M., et al. (2009). Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 41, 1094–1099. doi: 10.1038/ng.439
Lambert, J.-C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lanoiselée, H. M., Nicolas, G., Wallon, D., Rovelet-Lecrux, A., Lacour, M., Rousseau, S., et al. (2017). APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: a genetic screening study of familial and sporadic cases. PLoS Med. 14:e1002270. doi: 10.1371/journal.pmed.1002270
Lasagna-Reeves, C. A., Castillo-Carranza, D. L., Sengupta, U., Sarmiento, J., Troncoso, J., Jackson, G. R., et al. (2012). Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 26, 1946–1959. doi: 10.1096/fj.11-199851
Lau, S. F., Cao, H., Fu, A. K. Y., and Ip, N. Y. (2020a). Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 117, 25800–25809. doi: 10.1073/pnas.2008762117
Lau, S. F., Chen, C., Fu, W. Y., Qu, J. Y., Cheung, T. H., Fu, A. K. Y., et al. (2020b). IL-33-PU.1 transcriptome reprogramming drives functional state transition and clearance activity of microglia in Alzheimer’s disease. Cell Rep. 31:107530. doi: 10.1016/j.celrep.2020.107530
Lazar, A. N., Bich, C., Panchal, M., Desbenoit, N., Petit, V. W., Touboul, D., et al. (2013). Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathol. 125, 133–144. doi: 10.1007/s00401-012-1041-1
Lee, J. Y., Han, S. H., Park, M. H., Baek, B., Song, I. S., Choi, M. K., et al. (2018). Neuronal SphK1 acetylates COX2 and contributes to pathogenesis in a model of Alzheimer’s Disease. Nat. Commun. 9:1479. doi: 10.1038/s41467-018-03674-2
Lee, S. H., Meilandt, W. J., Xie, L., Gandham, V. D., Ngu, H., Barck, K. H., et al. (2021). Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 109, 1283–1301.e1286. doi: 10.1016/j.neuron.2021.02.010
Lei, F., Cui, N., Zhou, C., Chodosh, J., Vavvas, D. G., and Paschalis, E. I. (2020). CSF1R inhibition by a small-molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proc. Natl. Acad. Sci. U.S.A. 117, 23336–23338. doi: 10.1073/pnas.1922788117
Lei, P., Ayton, S., and Bush, A. I. (2021). The essential elements of Alzheimer’s disease. J. Biol. Chem. 296:100105. doi: 10.1074/jbc.REV120.008207
Lemprière, S. (2021). Distinct microglial profiles associated with amyloid and tau pathology in Alzheimer disease. Nat. Rev. Neurol. 17:194. doi: 10.1038/s41582-021-00482-z
Leuzy, A., Heurling, K., Ashton, N. J., Schöll, M., and Zimmer, E. R. (2018). In vivo detection of Alzheimer’s disease. Yale J. Biol. Med. 91, 291–300.
Lewcock, J. W., Schlepckow, K., Di Paolo, G., Tahirovic, S., Monroe, K. M., and Haass, C. (2020). Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 108, 801–821. doi: 10.1016/j.neuron.2020.09.029
Li, H. Q., Chen, C., Dou, Y., Wu, H. J., Liu, Y. J., Lou, H. F., et al. (2013). P2Y4 receptor-mediated pinocytosis contributes to amyloid β-induced self-uptake by microglia. Mol. Cell Biol. 33, 4282–4293. doi: 10.1128/mcb.00544-13
Lichtenthaler, S. F., Haass, C., and Steiner, H. (2011). Regulated intramembrane proteolysis—lessons from amyloid precursor protein processing. J. Neurochem. 117, 779–796. doi: 10.1111/j.1471-4159.2011.07248.x
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lim, J., Iyer, A., Suen, J. Y., Seow, V., Reid, R. C., Brown, L., et al. (2013). C5aR and C3aR antagonists each inhibit diet-induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 27, 822–831. doi: 10.1096/fj.12-220582
Lim, T. K., and Ruthazer, E. S. (2021). Microglial trogocytosis and the complement system regulate axonal pruning in vivo. Elife 10:e62167. doi: 10.7554/eLife.62167
Linnartz-Gerlach, B., Bodea, L. G., Klaus, C., Ginolhac, A., Halder, R., Sinkkonen, L., et al. (2019). TREM2 triggers microglial density and age-related neuronal loss. Glia 67, 539–550. doi: 10.1002/glia.23563
Liu, Q., Trotter, J., Zhang, J., Peters, M. M., Cheng, H., Bao, J., et al. (2010). Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 30, 17068–17078. doi: 10.1523/jneurosci.4067-10.2010
Liu, Y., Thalamuthu, A., Mather, K. A., Crawford, J., Ulanova, M., Wong, M. W. K., et al. (2021). Plasma lipidome is dysregulated in Alzheimer’s disease and is associated with disease risk genes. Transl. Psychiatry 11:344. doi: 10.1038/s41398-021-01362-2
Loving, B. A., and Bruce, K. D. (2020). Lipid and lipoprotein metabolism in microglia. Front. Physiol. 11:393. doi: 10.3389/fphys.2020.00393
Madore, C., Leyrolle, Q., Morel, L., Rossitto, M., Greenhalgh, A. D., Delpech, J. C., et al. (2020). Essential omega-3 fatty acids tune microglial phagocytosis of synaptic elements in the mouse developing brain. Nat. Commun. 11:6133. doi: 10.1038/s41467-020-19861-z
Maeda, S., Sahara, N., Saito, Y., Murayama, M., Yoshiike, Y., Kim, H., et al. (2007). Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. doi: 10.1021/bi061359o
Mandelkow, E. M., and Mandelkow, E. (2012). Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2:a006247. doi: 10.1101/cshperspect.a006247
Mango, D., Saidi, A., Cisale, G. Y., Feligioni, M., Corbo, M., and Nisticò, R. (2019). Targeting synaptic plasticity in experimental models of Alzheimer’s disease. Front. Pharmacol. 10:778. doi: 10.3389/fphar.2019.00778
Mapstone, M., Cheema, A. K., Fiandaca, M. S., Zhong, X., Mhyre, T. R., MacArthur, L. H., et al. (2014). Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 20, 415–418. doi: 10.1038/nm.3466
Marioni, R. E., Harris, S. E., Zhang, Q., McRae, A. F., Hagenaars, S. P., Hill, W. D., et al. (2018). GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8:99. doi: 10.1038/s41398-018-0150-6
Marschallinger, J., Iram, T., Zardeneta, M., Lee, S. E., Lehallier, B., Haney, M. S., et al. (2020). Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208. doi: 10.1038/s41593-019-0566-1
Marshall, S. A., McClain, J. A., Wooden, J. I., and Nixon, K. (2020). Microglia dystrophy following binge-kike alcohol exposure in adolescent and adult male rats. Front. Neuroanat. 14:52. doi: 10.3389/fnana.2020.00052
Martini, A. C., Helman, A. M., McCarty, K. L., Lott, I. T., Doran, E., Schmitt, F. A., et al. (2020). Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement. (Amst) 12, e12113. doi: 10.1002/dad2.12113
Masliah, E., Ellisman, M., Carragher, B., Mallory, M., Young, S., Hansen, L., et al. (1992). Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J. Neuropathol. Exp. Neurol. 51, 404–414. doi: 10.1097/00005072-199207000-00003
Masliah, E., Mallory, M., Hansen, L., DeTeresa, R., Alford, M., and Terry, R. (1994). Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci. Lett. 174, 67–72. doi: 10.1016/0304-3940(94)90121-x
Masliah, E., Mallory, M., Hansen, L., DeTeresa, R., and Terry, R. D. (1993a). Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 43, 192–197. doi: 10.1212/wnl.43.1_part_1.192
Masliah, E., Miller, A., and Terry, R. D. (1993b). The synaptic organization of the neocortex in Alzheimer’s disease. Med. Hypotheses 41, 334–340. doi: 10.1016/0306-9877(93)90078-5
Masliah, E., Terry, R. D., DeTeresa, R. M., and Hansen, L. A. (1989). Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci. Lett. 103, 234–239. doi: 10.1016/0304-3940(89)90582-x
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262. doi: 10.1101/cshperspect.a006262
Matarin, M., Salih, D. A., Yasvoina, M., Cummings, D. M., Guelfi, S., Liu, W., et al. (2015). A genome-wide gene-expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep. 10, 633–644. doi: 10.1016/j.celrep.2014.12.041
Matejuk, A., and Ransohoff, R. M. (2020). Crosstalk between astrocytes and microglia: an overview. Front. Immunol. 11:1416. doi: 10.3389/fimmu.2020.01416
Mathys, H., Adaikkan, C., Gao, F., Young, J. Z., Manet, E., Hemberg, M., et al. (2017). Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380. doi: 10.1016/j.celrep.2017.09.039
Mathys, H., Davila-Velderrain, J., Peng, Z., Gao, F., Mohammadi, S., Young, J. Z., et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. doi: 10.1038/s41586-019-1195-2
Mauch, D. H., Nägler, K., Schumacher, S., Göritz, C., Müller, E. C., Otto, A., et al. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357. doi: 10.1126/science.294.5545.1354
McAleese, K. E., Colloby, S. J., Thomas, A. J., Al-Sarraj, S., Ansorge, O., Neal, J., et al. (2021). Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia. Alzheimers Dement. 17, 1121–1133. doi: 10.1002/alz.12291
McAlpine, C. S., Park, J., Griciuc, A., Kim, E., Choi, S. H., Iwamoto, Y., et al. (2021). Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595, 701–706. doi: 10.1038/s41586-021-03734-6
McGeer, P. L., Akiyama, H., Itagaki, S., and McGeer, E. G. (1989). Immune system response in Alzheimer’s disease. Can. J. Neurol. Sci. 16(4 Suppl), 516–527. doi: 10.1017/s0317167100029863
McQuade, A., Kang, Y. J., Hasselmann, J., Jairaman, A., Sotelo, A., Coburn, M., et al. (2020). Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 11:5370. doi: 10.1038/s41467-020-19227-5
Meda, L., Cassatella, M. A., Szendrei, G. I., Otvos, L. Jr., Baron, P., Villalba, M., et al. (1995). Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 374, 647–650. doi: 10.1038/374647a0
Melcangi, R. C., Garcia-Segura, L. M., and Mensah-Nyagan, A. G. (2008). Neuroactive steroids: state of the art and new perspectives. Cell Mol. Life Sci. 65, 777–797. doi: 10.1007/s00018-007-7403-5
Mendiola, A. S., Tognatta, R., Yan, Z., and Akassoglou, K. (2021). ApoE and immunity in Alzheimer’s disease and related tauopathies: low-density lipoprotein receptor to the rescue. Neuron 109, 2363–2365. doi: 10.1016/j.neuron.2021.07.013
Mesulam, M. M. (1999). Neuroplasticity failure in Alzheimer’s disease: bridging the gap between plaques and tangles. Neuron 24, 521–529. doi: 10.1016/s0896-6273(00)81109-5
Miyamoto, A., Wake, H., Ishikawa, A. W., Eto, K., Shibata, K., Murakoshi, H., et al. (2016). Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 7:12540. doi: 10.1038/ncomms12540
Monoranu, C. M., Apfelbacher, M., Grünblatt, E., Puppe, B., Alafuzoff, I., Ferrer, I., et al. (2009). pH measurement as quality control on human post mortem brain tissue: a study of the BrainNet Europe consortium. Neuropathol. Appl. Neurobiol. 35, 329–337. doi: 10.1111/j.1365-2990.2008.01003a.x
Montagna, E., Dorostkar, M. M., and Herms, J. (2017). The role of APP in structural spine plasticity. Front. Mol. Neurosci. 10:136. doi: 10.3389/fnmol.2017.00136
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Mukrasch, M. D., Bibow, S., Korukottu, J., Jeganathan, S., Biernat, J., Griesinger, C., et al. (2009). Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 7:e34. doi: 10.1371/journal.pbio.1000034
Naj, A. C., Jun, G., Beecham, G. W., Wang, L. S., Vardarajan, B. N., Buros, J., et al. (2011). Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 43, 436–441. doi: 10.1038/ng.801
Nakanishi, H., Ni, J., Nonaka, S., and Hayashi, Y. (2021). Microglial circadian clock regulation of microglial structural complexity, dendritic spine density and inflammatory response. Neurochem. Int. 142:104905. doi: 10.1016/j.neuint.2020.104905
Nanjundaiah, S., Chidambaram, H., Chandrashekar, M., and Chinnathambi, S. (2021). Role of microglia in regulating cholesterol and tau pathology in Alzheimer’s disease. Cell Mol. Neurobiol. 41, 651–668. doi: 10.1007/s10571-020-00883-6
Navarro, V., Sanchez-Mejias, E., Jimenez, S., Muñoz-Castro, C., Sanchez-Varo, R., Davila, J. C., et al. (2018). Microglia in Alzheimer’s disease: activated, dysfunctional or degenerative. Front. Aging Neurosci. 10:140. doi: 10.3389/fnagi.2018.00140
Neff, R. A., Wang, M., Vatansever, S., Guo, L., Ming, C., Wang, Q., et al. (2021). Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 7:eabb5398. doi: 10.1126/sciadv.abb5398
Neniskyte, U., Neher, J. J., and Brown, G. C. (2011). Neuronal death induced by nanomolar amyloid β is mediated by primary phagocytosis of neurons by microglia. J. Biol. Chem. 286, 39904–39913. doi: 10.1074/jbc.M111.267583
Nguyen, A. T., Wang, K., Hu, G., Wang, X., Miao, Z., Azevedo, J. A., et al. (2020). APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease. Acta Neuropathol. 140, 477–493. doi: 10.1007/s00401-020-02200-3
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Nitsch, R. M., Blusztajn, J. K., Pittas, A. G., Slack, B. E., Growdon, J. H., and Wurtman, R. J. (1992). Evidence for a membrane defect in Alzheimer disease brain. Proc. Natl. Acad. Sci. U.S.A. 89, 1671–1675. doi: 10.1073/pnas.89.5.1671
Noble, W., Hanger, D. P., Miller, C. C., and Lovestone, S. (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front. Neurol. 4:83. doi: 10.3389/fneur.2013.00083
Nugent, A. A., Lin, K., van Lengerich, B., Lianoglou, S., Przybyla, L., Davis, S. S., et al. (2020). TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e839. doi: 10.1016/j.neuron.2019.12.007
Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., et al. (2003). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39, 409–421. doi: 10.1016/s0896-6273(03)00434-3
Ofengeim, D., Mazzitelli, S., Ito, Y., DeWitt, J. P., Mifflin, L., Zou, C., et al. (2017). RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 114, E8788–E8797. doi: 10.1073/pnas.1714175114
Ohsawa, K., Imai, Y., Kanazawa, H., Sasaki, Y., and Kohsaka, S. (2000). Involvement of Iba1 in membrane ruffling and phagocytosis of macrophages/microglia. J. Cell Sci. 113(Pt 17), 3073–3084.
Ohsawa, K., Imai, Y., Sasaki, Y., and Kohsaka, S. (2004). Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem. 88, 844–856. doi: 10.1046/j.1471-4159.2003.02213.x
Olah, M., Menon, V., Habib, N., Taga, M. F., Ma, Y., Yung, C. J., et al. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer’s disease. Nat. Commun. 11:6129. doi: 10.1038/s41467-020-19737-2
Otero-Garcia, M., Xue, Y.-Q., Shakouri, T., Deng, Y., Morabito, S., Allison, T., et al. (2020). Single-soma transcriptomics of tangle-bearing neurons in Alzheimer’s disease reveals the signatures of tau-associated synaptic dysfunction. bioRxiv [Preprint] doi: 10.1101/2020.05.11.088591
Overk, C. R., and Masliah, E. (2014). Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem. Pharmacol. 88, 508–516. doi: 10.1016/j.bcp.2014.01.015
Paasila, P. J., Davies, D. S., Kril, J. J., Goldsbury, C., and Sutherland, G. T. (2019). The relationship between the morphological subtypes of microglia and Alzheimer’s disease neuropathology. Brain Pathol. 29, 726–740. doi: 10.1111/bpa.12717
Paasila, P. J., Davies, D. S., Sutherland, G. T., and Goldsbury, C. (2020). Clustering of activated microglia occurs before the formation of dystrophic neurites in the evolution of Aβ plaques in Alzheimer’s disease. Free Neuropathol. 1:20. doi: 10.17879/freeneuropathology-2020-2845
Paasila, P. J., Fok, S. Y. Y., Flores-Rodriguez, N., Sajjan, S., Svahn, A. J., Dennis, C. V., et al. (2021). Ground state depletion microscopy as a tool for studying microglia-synapse interactions. J. Neurosci. Res. 99, 1515–1532. doi: 10.1002/jnr.24819
Palop, J. J., Chin, J., Roberson, E. D., Wang, J., Thwin, M. T., Bien-Ly, N., et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. doi: 10.1016/j.neuron.2007.07.025
Paolicelli, R. C., Bolasco, G., Pagani, F., Maggi, L., Scianni, M., Panzanelli, P., et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. doi: 10.1126/science.1202529
Parkhurst, C. N., Yang, G., Ninan, I., Savas, J. N., Yates, J. R. III, Lafaille, J. J., et al. (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. doi: 10.1016/j.cell.2013.11.030
Pascoal, T. A., Benedet, A. L., Ashton, N. J., Kang, M. S., Therriault, J., Chamoun, M., et al. (2021). Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 27, 1592–1599. doi: 10.1038/s41591-021-01456-w
Peng, Q., Malhotra, S., Torchia, J. A., Kerr, W. G., Coggeshall, K. M., and Humphrey, M. B. (2010). TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci. Signal. 3:ra38. doi: 10.1126/scisignal.2000500
Perlmutter, L. S., Barron, E., and Chui, H. C. (1990). Morphologic association between microglia and senile plaque amyloid in Alzheimer’s disease. Neurosci. Lett. 119, 32–36. doi: 10.1016/0304-3940(90)90748-x
Petrov, A. M., Kasimov, M. R., and Zefirov, A. L. (2016). Brain cholesterol metabolism and its defects: linkage to neurodegenerative diseases and synaptic dysfunction. Acta Nat. 8, 58–73. doi: 10.32607/20758251-2016-8-1-58-73
Petrov, A. M., Kasimov, M. R., and Zefirov, A. L. (2017). Cholesterol in the pathogenesis of Alzheimer’s, Parkinson’s diseases and autism: link to synaptic dysfunction. Acta Nat. 9, 26–37.
Pettegrew, J. W., Panchalingam, K., Hamilton, R. L., and McClure, R. J. (2001). Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem. Res. 26, 771–782. doi: 10.1023/a:1011603916962
Piccioni, G., Mango, D., Saidi, A., Corbo, M., and Nisticò, R. (2021). Targeting microglia-synapse interactions in Alzheimer’s disease. Int. J. Mol. Sci. 22:2342. doi: 10.3390/ijms22052342
Pimenova, A. A., Marcora, E., and Goate, A. M. (2017). A tale of two genes: microglial Apoe and Trem2. Immunity 47, 398–400. doi: 10.1016/j.immuni.2017.08.015
Pimenova, A. A., Raj, T., and Goate, A. M. (2018). Untangling genetic risk for Alzheimer’s disease. Biol. Psychiatry 83, 300–310. doi: 10.1016/j.biopsych.2017.05.014
Poderoso, J. J., Helfenberger, K., and Poderoso, C. (2019). The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 88, 61–72. doi: 10.1016/j.niox.2019.04.005
Podleśny-Drabiniok, A., Marcora, E., and Goate, A. M. (2020). Microglial phagocytosis: a disease-associated process emerging from Alzheimer’s disease genetics. Trends Neurosci. 43, 965–979. doi: 10.1016/j.tins.2020.10.002
Prelli, F., Castaño, E., Glenner, G. G., and Frangione, B. (1988). Differences between vascular and plaque core amyloid in Alzheimer’s disease. J. Neurochem. 51, 648–651. doi: 10.1111/j.1471-4159.1988.tb01087.x
Prieto, G. A., Trieu, B. H., Dang, C. T., Bilousova, T., Gylys, K. H., Berchtold, N. C., et al. (2017). Pharmacological rescue of long-term potentiation in Alzheimer diseased synapses. J. Neurosci. 37, 1197–1212. doi: 10.1523/jneurosci.2774-16.2016
Priller, C., Bauer, T., Mitteregger, G., Krebs, B., Kretzschmar, H. A., and Herms, J. (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 26, 7212–7221. doi: 10.1523/jneurosci.1450-06.2006
Prinz, M., Jung, S., and Priller, J. (2019). Microglia biology: one century of evolving concepts. Cell 179, 292–311. doi: 10.1016/j.cell.2019.08.053
Proitsi, P., Kim, M., Whiley, L., Pritchard, M., Leung, R., Soininen, H., et al. (2015). Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer’s disease. Transl. Psychiatry 5:e494. doi: 10.1038/tp.2014.127
Rahimi, J., and Kovacs, G. G. (2014). Prevalence of mixed pathologies in the aging brain. Alzheimers Res. Ther. 6:82. doi: 10.1186/s13195-014-0082-1
Reddy, P. H., Mani, G., Park, B. S., Jacques, J., Murdoch, G., Whetsell, W. Jr., et al. (2005). Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J. Alzheimers Dis. 7, 103–117; discussion 173–180. doi: 10.3233/jad-2005-7203
Rodriguez, G. A., Tai, L. M., LaDu, M. J., and Rebeck, G. W. (2014). Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J. Neuroinflammation 11:111. doi: 10.1186/1742-2094-11-111
Ronaldson, P. T., and Davis, T. P. (2020). Regulation of blood-brain barrier integrity by microglia in health and disease: a therapeutic opportunity. J. Cereb. Blood Flow Metab. 40(1_suppl), S6–S24. doi: 10.1177/0271678x20951995
Saito, T., Suemoto, T., Brouwers, N., Sleegers, K., Funamoto, S., Mihira, N., et al. (2011). Potent amyloidogenicity and pathogenicity of Aβ43. Nat. Neurosci. 14, 1023–1032. doi: 10.1038/nn.2858
Sáiz-Vazquez, O., Puente-Martínez, A., Ubillos-Landa, S., Pacheco-Bonrostro, J., and Santabárbara, J. (2020). Cholesterol and Alzheimer’s disease risk: a meta-meta-analysis. Brain Sci. 10:386. doi: 10.3390/brainsci10060386
Salloway, S. P., Sevingy, J., Budur, K., Pederson, J. T., DeMattos, R. B., Von Rosenstiel, P., et al. (2020). Advancing combination therapy for Alzheimer’s disease. Alzheimers Dement. (N. Y.) 6:e12073. doi: 10.1002/trc2.12073
Salminen, A., Ojala, J., Kauppinen, A., Kaarniranta, K., and Suuronen, T. (2009). Inflammation in Alzheimer’s disease: amyloid-β oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 87, 181–194. doi: 10.1016/j.pneurobio.2009.01.001
Sanchez-Mejias, E., Navarro, V., Jimenez, S., Sanchez-Mico, M., Sanchez-Varo, R., Nuñez-Diaz, C., et al. (2016). Soluble phospho-tau from Alzheimer’s disease hippocampus drives microglial degeneration. Acta Neuropathol. 132, 897–916. doi: 10.1007/s00401-016-1630-5
Sasaki, A., Yamaguchi, H., Ogawa, A., Sugihara, S., and Nakazato, Y. (1997). Microglial activation in early stages of amyloid β protein deposition. Acta Neuropathol. 94, 316–322. doi: 10.1007/s004010050713
Sasaki, Y., Ohsawa, K., Kanazawa, H., Kohsaka, S., and Imai, Y. (2001). Iba1 is an actin-cross-linking protein in macrophages/microglia. Biochem. Biophys. Res. Commun. 286, 292–297. doi: 10.1006/bbrc.2001.5388
Sassi, C., Guerreiro, R., Gibbs, R., Ding, J., Lupton, M. K., Troakes, C., et al. (2014). Investigating the role of rare coding variability in Mendelian dementia genes (APP, PSEN1, PSEN2, GRN, MAPT, and PRNP) in late-onset Alzheimer’s disease. Neurobiol. Aging 35, 2881.e2881–2881.e2881. doi: 10.1016/j.neurobiolaging.2014.06.002
Savage, J. C., Picard, K., González-Ibáñez, F., and Tremblay, M. (2018). A brief History of microglial ultrastructure: distinctive features, phenotypes, and functions discovered over the past 60 years by electron microscopy. Front. Immunol. 9:803. doi: 10.3389/fimmu.2018.00803
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Scheff, S. W., and Price, D. A. (1993). Synapse loss in the temporal lobe in Alzheimer’s disease. Ann. Neurol. 33, 190–199. doi: 10.1002/ana.410330209
Scheff, S. W., and Price, D. A. (1998). Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 1146–1153. doi: 10.1097/00005072-199812000-00006
Scheff, S. W., and Price, D. A. (2001). Alzheimer’s disease-related synapse loss in the cingulate cortex. J. Alzheimers Dis. 3, 495–505. doi: 10.3233/jad-2001-3509
Scheff, S. W., and Price, D. A. (2003). Synaptic pathology in Alzheimer’s disease: a review of ultrastructural studies. Neurobiol. Aging 24, 1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002
Scheff, S. W., and Price, D. A. (2006). Alzheimer’s disease-related alterations in synaptic density: neocortex and hippocampus. J. Alzheimers Dis. 9(3 Suppl), 101–115. doi: 10.3233/jad-2006-9s312
Scheff, S. W., DeKosky, S. T., and Price, D. A. (1990). Quantitative assessment of cortical synaptic density in Alzheimer’s disease. Neurobiol. Aging 11, 29–37. doi: 10.1016/0197-4580(90)90059-9
Scheff, S. W., Neltner, J. H., and Nelson, P. T. (2014). Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem. Pharmacol. 88, 517–528. doi: 10.1016/j.bcp.2013.12.028
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Scheff, S. W., Price, D. A., Schmitt, F. A., DeKosky, S. T., and Mufson, E. J. (2007). Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68, 1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f
Scheff, S. W., Price, D. A., Schmitt, F. A., Roberts, K. N., Ikonomovic, M. D., and Mufson, E. J. (2013). Synapse stability in the precuneus early in the progression of Alzheimer’s disease. J. Alzheimers Dis. 35, 599–609. doi: 10.3233/jad-122353
Scheff, S. W., Price, D. A., Schmitt, F. A., Scheff, M. A., and Mufson, E. J. (2011). Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 24, 547–557. doi: 10.3233/jad-2011-101782
Scheff, S. W., Sparks, D. L., and Price, D. A. (1996). Quantitative assessment of synaptic density in the outer molecular layer of the hippocampal dentate gyrus in Alzheimer’s disease. Dementia 7, 226–232. doi: 10.1159/000106884
Scheff, S. W., Sparks, L., and Price, D. A. (1993). Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer’s disease. Ann. Neurol. 34, 356–361. doi: 10.1002/ana.410340309
Schetters, S. T. T., Gomez-Nicola, D., Garcia-Vallejo, J. J., and Van Kooyk, Y. (2017). Neuroinflammation: microglia and T cells get ready to tango. Front. Immunol. 8:1905. doi: 10.3389/fimmu.2017.01905
Schneider, J. A., Arvanitakis, Z., Bang, W., and Bennett, D. A. (2007). Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69, 2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24
Schwabe, T., Srinivasan, K., and Rhinn, H. (2020). Shifting paradigms: The central role of microglia in Alzheimer’s disease. Neurobiol. Dis. 143:104962. doi: 10.1016/j.nbd.2020.104962
Schweers, O., Schönbrunn-Hanebeck, E., Marx, A., and Mandelkow, E. (1994). Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for β-structure. J. Biol. Chem. 269, 24290–24297. doi: 10.1016/s0021-9258(19)51080-8
Sekino, Y., Koganezawa, N., Mizui, T., and Shirao, T. (2017). Role of drebrin in synaptic plasticity. Adv. Exp. Med. Biol. 1006, 183–201. doi: 10.1007/978-4-431-56550-5_11
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. doi: 10.15252/emmm.201606210
Seyfried, N. T., Dammer, E. B., Swarup, V., Nandakumar, D., Duong, D. M., Yin, L., et al. (2017). A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst. 4, 60–72.e64. doi: 10.1016/j.cels.2016.11.006
Sheffield, L. G., Marquis, J. G., and Berman, N. E. (2000). Regional distribution of cortical microglia parallels that of neurofibrillary tangles in Alzheimer’s disease. Neurosci. Lett. 285, 165–168. doi: 10.1016/s0304-3940(00)01037-5
Shi, Y., and Holtzman, D. M. (2018). Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 18, 759–772. doi: 10.1038/s41577-018-0051-1
Shi, Y., Andhey, P. S., Ising, C., Wang, K., Snipes, L. L., Boyer, K., et al. (2021). Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 109, 2413–2426.e2417. doi: 10.1016/j.neuron.2021.05.034
Sierra, A., Abiega, O., Shahraz, A., and Neumann, H. (2013). Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell Neurosci. 7:6. doi: 10.3389/fncel.2013.00006
Smith, A. M., Davey, K., Tsartsalis, S., Khozoie, C., Fancy, N., Tang, S. S., et al. (2021). Diverse human astrocyte and microglial transcriptional responses to Alzheimer’s pathology. Acta Neuropathol. doi: 10.1007/s00401-021-02372-6
Snowden, S. G., Ebshiana, A. A., Hye, A., An, Y., Pletnikova, O., O’Brien, R., et al. (2017). Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 14:e1002266. doi: 10.1371/journal.pmed.1002266
Song, S., Wang, X., Sava, V., Weeber, E. J., and Sanchez-Ramos, J. (2014). In vivo administration of granulocyte colony-stimulating factor restores long-term depression in hippocampal slices prepared from transgenic APP/PS1 mice. J. Neurosci. Res. 92, 975–980. doi: 10.1002/jnr.23378
Spangenberg, E. E., Lee, R. J., Najafi, A. R., Rice, R. A., Elmore, M. R., Blurton-Jones, M., et al. (2016). Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139(Pt 4), 1265–1281. doi: 10.1093/brain/aww016
Spangenberg, E., Severson, P. L., Hohsfield, L. A., Crapser, J., Zhang, J., Burton, E. A., et al. (2019). Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 10:3758. doi: 10.1038/s41467-019-11674-z
Stephan, A. H., Barres, B. A., and Stevens, B. (2012). The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389. doi: 10.1146/annurev-neuro-061010-113810
Stephen, T. L., Cacciottolo, M., Balu, D., Morgan, T. E., LaDu, M. J., Finch, C. E., et al. (2019). APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice. Acta Neuropathol. Commun. 7:82. doi: 10.1186/s40478-019-0729-z
Stevens, B., Allen, N. J., Vazquez, L. E., Howell, G. R., Christopherson, K. S., Nouri, N., et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. doi: 10.1016/j.cell.2007.10.036
Streit, W. J., Braak, H., Xue, Q. S., and Bechmann, I. (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 118, 475–485. doi: 10.1007/s00401-009-0556-6
Streit, W. J., Graeber, M. B., and Kreutzberg, G. W. (1988). Functional plasticity of microglia: a review. Glia 1, 301–307. doi: 10.1002/glia.440010502
Streit, W. J., Khoshbouei, H., and Bechmann, I. (2020). Dystrophic microglia in late-onset Alzheimer’s disease. Glia 68, 845–854. doi: 10.1002/glia.23782
Streit, W. J., Xue, Q. S., Tischer, J., and Bechmann, I. (2014). Microglial pathology. Acta Neuropathol. Commun. 2:142. doi: 10.1186/s40478-014-0142-6
Sultana, R., Banks, W. A., and Butterfield, D. A. (2010). Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J. Neurosci. Res. 88, 469–477. doi: 10.1002/jnr.22227
Suzuki, N., Iwatsubo, T., Odaka, A., Ishibashi, Y., Kitada, C., and Ihara, Y. (1994). High tissue content of soluble β1-40 is linked to cerebral amyloid angiopathy. Am. J. Pathol. 145, 452–460.
Sze, C. I., Troncoso, J. C., Kawas, C., Mouton, P., Price, D. L., and Martin, L. J. (1997). Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J. Neuropathol. Exp. Neurol. 56, 933–944. doi: 10.1097/00005072-199708000-00011
Tabira, T., Chui, D. H., Nakayama, H., Kuroda, S., and Shibuya, M. (2002). Alzheimer’s disease with spastic paresis and cotton wool type plaques. J. Neurosci. Res. 70, 367–372. doi: 10.1002/jnr.10392
Takata, K., Ginhoux, F., and Shimohama, S. (2021). Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem. Pharmacol. 192:114754. doi: 10.1016/j.bcp.2021.114754
Tarkowski, E., Andreasen, N., Tarkowski, A., and Blennow, K. (2003). Intrathecal inflammation precedes development of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 74, 1200–1205. doi: 10.1136/jnnp.74.9.1200
Tay, T. L., Béchade, C., D’Andrea, I., St-Pierre, M. K., Henry, M. S., Roumier, A., et al. (2017). Microglia gone rogue: impacts on psychiatric disorders across the lifespan. Front. Mol. Neurosci. 10:421. doi: 10.3389/fnmol.2017.00421
Terry, R. D. (2000). Cell death or synaptic loss in Alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 1118–1119. doi: 10.1093/jnen/59.12.1118
Terry, R. D., and Davies, P. (1980). Dementia of the Alzheimer type. Annu. Rev. Neurosci. 3, 77–95. doi: 10.1146/annurev.ne.03.030180.000453
Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., et al. (1991). Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. doi: 10.1002/ana.410300410
Thal, D. R., Rüb, U., Schultz, C., Sassin, I., Ghebremedhin, E., Del Tredici, K., et al. (2000). Sequence of Aβ-protein deposition in the human medial temporal lobe. J. Neuropathol. Exp. Neurol. 59, 733–748. doi: 10.1093/jnen/59.8.733
Thomas, D. X., Bajaj, S., McRae-McKee, K., Hadjichrysanthou, C., Anderson, R. M., and Collinge, J. (2020). Association of TDP-43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer’s disease neuropathology. Sci. Rep. 10:14579. doi: 10.1038/s41598-020-71305-2
Touboul, D., and Gaudin, M. (2014). Lipidomics of Alzheimer’s disease. Bioanalysis 6, 541–561. doi: 10.4155/bio.13.346
Tozzi, A., Sclip, A., Tantucci, M., de Iure, A., Ghiglieri, V., Costa, C., et al. (2015). Region- and age-dependent reductions of hippocampal long-term potentiation and NMDA to AMPA ratio in a genetic model of Alzheimer’s disease. Neurobiol. Aging 36, 123–133. doi: 10.1016/j.neurobiolaging.2014.07.002
Trettenbrein, P. C. (2016). The demise of the synapse as the locus of memory: a looming paradigm shift? Front. Syst. Neurosci. 10:88. doi: 10.3389/fnsys.2016.00088
Trinchese, F., Fa, M., Liu, S., Zhang, H., Hidalgo, A., Schmidt, S. D., et al. (2008). Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 118, 2796–2807. doi: 10.1172/jci34254
Tsatsanis, A., McCorkindale, A. N., Wong, B. X., Patrick, E., Ryan, T. M., Evans, R. W., et al. (2021). The acute phase protein lactoferrin is a key feature of Alzheimer’s disease and predictor of Aβ burden through induction of APP amyloidogenic processing. Mol. Psychiatry doi: 10.1038/s41380-021-01248-1
Tzioras, M., Daniels, M. J. D., King, D., Popovic, K., Holloway, R., Stevenson, A., et al. (2019). Altered synaptic ingestion by human microglia in Alzheimer’s disease. bioRxiv [Preprint] doi: 10.1101/795930
Ulland, T. K., and Colonna, M. (2018). TREM2 – a key player in microglial biology and Alzheimer disease. Nat. Rev. Neurol. 14, 667–675. doi: 10.1038/s41582-018-0072-1
Ulrich, J. D., Ulland, T. K., Mahan, T. E., Nyström, S., Nilsson, K. P., Song, W. M., et al. (2018). ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058. doi: 10.1084/jem.20171265
Valdearcos, M., Robblee, M. M., Benjamin, D. I., Nomura, D. K., Xu, A. W., and Koliwad, S. K. (2014). Microglia dictate the impact of saturated fat consumption on hypothalamic inflammation and neuronal function. Cell Rep. 9, 2124–2138. doi: 10.1016/j.celrep.2014.11.018
van der Kant, R., Langness, V. F., Herrera, C. M., Williams, D. A., Fong, L. K., Leestemaker, Y., et al. (2019). Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24, 363–375.e369. doi: 10.1016/j.stem.2018.12.013
van Meer, G., Voelker, D. R., and Feigenson, G. W. (2008). Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. doi: 10.1038/nrm2330
Varma, V. R., Büşra Lüleci, H., Oommen, A. M., Varma, S., Blackshear, C. T., Griswold, M. E., et al. (2021). Abnormal brain cholesterol homeostasis in Alzheimer’s disease-a targeted metabolomic and transcriptomic study. NPJ Aging Mech. Dis. 7:11. doi: 10.1038/s41514-021-00064-9
Varma, V. R., Oommen, A. M., Varma, S., Casanova, R., An, Y., Andrews, R. M., et al. (2018). Brain and blood metabolite signatures of pathology and progression in Alzheimer disease: a targeted metabolomics study. PLoS Med. 15:e1002482. doi: 10.1371/journal.pmed.1002482
Vogrinc, D., Goričar, K., and Dolžan, V. (2021). Genetic variability in molecular pathways implicated in Alzheimer’s disease: a comprehensive review. Front. Aging Neurosci. 13:646901. doi: 10.3389/fnagi.2021.646901
Wadhwa, M., Kumari, P., Chauhan, G., Roy, K., Alam, S., Kishore, K., et al. (2017). Sleep deprivation induces spatial memory impairment by altered hippocampus neuroinflammatory responses and glial cells activation in rats. J. Neuroimmunol. 312, 38–48. doi: 10.1016/j.jneuroim.2017.09.003
Wakabayashi, K., Honer, W. G., and Masliah, E. (1994). Synapse alterations in the hippocampal-entorhinal formation in Alzheimer’s disease with and without Lewy body disease. Brain Res. 667, 24–32. doi: 10.1016/0006-8993(94)91709-4
Wake, H., Moorhouse, A. J., Miyamoto, A., and Nabekura, J. (2013). Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 36, 209–217. doi: 10.1016/j.tins.2012.11.007
Wallace, W., Ahlers, S. T., Gotlib, J., Bragin, V., Sugar, J., Gluck, R., et al. (1993). Amyloid precursor protein in the cerebral cortex is rapidly and persistently induced by loss of subcortical innervation. Proc. Natl. Acad. Sci. U.S.A. 90, 8712–8716. doi: 10.1073/pnas.90.18.8712
Wan, Y. W., Al-Ouran, R., Mangleburg, C. G., Perumal, T. M., Lee, T. V., Allison, K., et al. (2020). Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 32:107908. doi: 10.1016/j.celrep.2020.107908
Wang, C. F., Zhao, C. C., Liu, W. L., Huang, X. J., Deng, Y. F., Jiang, J. Y., et al. (2020). Depletion of microglia attenuates dendritic spine loss and neuronal apoptosis in the acute stage of moderate traumatic brain injury in mice. J. Neurotrauma 37, 43–54. doi: 10.1089/neu.2019.6460
Wang, C., Wang, L., and Gu, Y. (2021). Microglia, synaptic dynamics and forgetting. Brain Res. Bull. 174, 173–183. doi: 10.1016/j.brainresbull.2021.06.005
Ward, S. M., Himmelstein, D. S., Lancia, J. K., and Binder, L. I. (2012). Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 40, 667–671. doi: 10.1042/bst20120134
Weinhard, L., di Bartolomei, G., Bolasco, G., Machado, P., Schieber, N. L., Neniskyte, U., et al. (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 9:1228. doi: 10.1038/s41467-018-03566-5
Wendeln, A. C., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. doi: 10.1038/s41586-018-0023-4
Whiley, L., Sen, A., Heaton, J., Proitsi, P., García-Gómez, D., Leung, R., et al. (2014). Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol. Aging 35, 271–278. doi: 10.1016/j.neurobiolaging.2013.08.001
Wilkins, J. M., and Trushina, E. (2017). Application of metabolomics in Alzheimer’s disease. Front. Neurol. 8:719. doi: 10.3389/fneur.2017.00719
Williams, C., Mehrian Shai, R., Wu, Y., Hsu, Y. H., Sitzer, T., Spann, B., et al. (2009). Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer’s disease. PLoS One 4:e4936. doi: 10.1371/journal.pone.0004936
Williams, J. B., Cao, Q., and Yan, Z. (2021). Transcriptomic analysis of human brains with Alzheimer’s disease reveals the altered expression of synaptic genes linked to cognitive deficits. Brain Commun. 3:fcab123. doi: 10.1093/braincomms/fcab123
Wisniewski, H. M., Sadowski, M., Jakubowska-Sadowska, K., Tarnawski, M., and Wegiel, J. (1998). Diffuse, lake-like amyloid-β deposits in the parvopyramidal layer of the presubiculum in Alzheimer disease. J. Neuropathol. Exp. Neurol. 57, 674–683. doi: 10.1097/00005072-199807000-00004
Wood, P. L. (2012). Lipidomics of Alzheimer’s disease: current status. Alzheimers Res. Ther. 4:5. doi: 10.1186/alzrt103
Wood, P. L., Medicherla, S., Sheikh, N., Terry, B., Phillipps, A., Kaye, J. A., et al. (2015). Targeted lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer’s disease: validation of DAG accumulation early in the pathophysiology of Alzheimer’s disease. J. Alzheimers Dis. 48, 537–546. doi: 10.3233/jad-150336
Wu, Y., Dissing-Olesen, L., MacVicar, B. A., and Stevens, B. (2015). Microglia: dynamic mediators of synapse development and plasticity. Trends Immunol. 36, 605–613. doi: 10.1016/j.it.2015.08.008
Xu, Y., Propson, N. E., Du, S., Xiong, W., and Zheng, H. (2021). Autophagy deficiency modulates microglial lipid homeostasis and aggravates tau pathology and spreading. Proc. Natl. Acad. Sci. U.S.A. 118:e2023418118. doi: 10.1073/pnas.2023418118
Yang, W., Zhou, X., Zimmermann, H. R., Cavener, D. R., Klann, E., and Ma, T. (2016). Repression of the eIF2α kinase PERK alleviates mGluR-LTD impairments in a mouse model of Alzheimer’s disease. Neurobiol. Aging 41, 19–24. doi: 10.1016/j.neurobiolaging.2016.02.005
Yeh, F. L., Wang, Y., Tom, I., Gonzalez, L. C., and Sheng, M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-β by microglia. Neuron 91, 328–340. doi: 10.1016/j.neuron.2016.06.015
Yoshiyama, Y., Higuchi, M., Zhang, B., Huang, S. M., Iwata, N., Saido, T. C., et al. (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. doi: 10.1016/j.neuron.2007.01.010
Zhang, B., Gaiteri, C., Bodea, L. G., Wang, Z., McElwee, J., Podtelezhnikov, A. A., et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. doi: 10.1016/j.cell.2013.03.030
Zhang, X., Liu, W., Zan, J., Wu, C., and Tan, W. (2020). Untargeted lipidomics reveals progression of early Alzheimer’s disease in APP/PS1 transgenic mice. Sci. Rep. 10:14509. doi: 10.1038/s41598-020-71510-z
Keywords: Alzheimer’s disease, APOE, lipids, microglia, synapses, TREM2
Citation: Paasila PJ, Aramideh JA, Sutherland GT and Graeber MB (2022) Synapses, Microglia, and Lipids in Alzheimer’s Disease. Front. Neurosci. 15:778822. doi: 10.3389/fnins.2021.778822
Received: 17 September 2021; Accepted: 06 December 2021;
Published: 12 January 2022.
Edited by:
Bente Finsen, University of Southern Denmark, DenmarkReviewed by:
Dan Frenkel, Tel Aviv University, IsraelGareth R. Howell, The Jackson Laboratory, United States
Copyright © 2022 Paasila, Aramideh, Sutherland and Graeber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick J. Paasila, patrick.paasila@sydney.edu.au; Manuel B. Graeber, manuel.graeber@sydney.edu.au