Modulation of GSK-3 as a therapeutic strategy on tau pathologies
- 1 Research Department, Noscira S.A., Tres Cantos, Madrid, Spain
- 2 Departamento de Neurobiología Celular, Molecular y del Desarrollo, Instituto Cajal, Consejo Superior de Investigaciones Cientificas, Madrid, Spain
- 3 Centro de Biología Molecular “Severo Ochoa,” CSIC-UAM-CIBERNED, Cantoblanco, Madrid, Spain
Glycogen synthase kinase-3 (GSK-3) is ubiquitously expressed and unusually active in resting, non-stimulated cells. In mammals, at least three proteins (α, β1, and β2), generated from two different genes, gsk-3α and gsk-3β, are widely expressed at both the RNA and protein levels although some tissues show preferential expression of some of the three proteins. Control of GSK-3 activity occurs by complex mechanisms that depend on specific signaling pathways, often controlling the inhibition of the kinase activity. GSK-3 appears to integrate different signaling pathways from a wide selection of cellular stimuli. The unique position of GSK-3 in modulating the function of a diverse series of proteins and its association with a wide variety of human disorders has attracted significant attention as a therapeutic target and as a means to understand the molecular basis of brain disorders. Different neurodegenerative diseases including frontotemporal dementia, progressive supranuclear palsy, and Alzheimer’s disease, present prominent tau pathology such as tau hyperphosphorylation and aggregation and are collectively referred to as tauopathies. GSK-3 has also been associated to different neuropsychiatric disorders, like schizophrenia and bipolar disorder. GSK-3β is the major kinase to phosphorylate tau both in vitro and in vivo and has been proposed as a target for therapeutic intervention. The first therapeutic strategy to modulate GSK-3 activity was the direct inhibition of its kinase activity. This review will focus on the signaling pathways involved in the control of GSK-3 activity and its pathological deregulation. We will highlight different alternatives of GSK-3 modulation including the direct pharmacological inhibition as compared to the modulation by upstream regulators.
Tau Hyperphosphorylation as a Common Marker in Tau Pathologies
A number of neurodegenerative disorders present intraneuronal proteinaceous aggregates, denoted as neurofibrillary tangles (NFT), as common markers of disease. NFT are made up of hyperphosphorylated, aggregated tau. Thus, they are collectively referred as tauopathies, and include disorders such as frontotemporal dementia (FTP), progressive supranuclear palsy (PSP), and Alzheimer’s disease (AD), among others.
The mechanisms responsible for tau aggregation are still unknown. Therefore, understanding how tau is regulated may be essential to identify the origin of tau aggregate formation. The regulation of tau takes place predominantly through post-translational modifications where tau phosphorylation has been suggested to play a central role in tau aggregation. It is generally accepted that an increase in tau phosphorylation reduces its affinity for microtubules, resulting in neuronal cytoskeleton instability. Even though abnormal tau phosphorylation occurs in many of the tau pathologies mentioned above, tau phosphorylation appears not to be sufficient to induce tau aggregation (Buee et al., 2000; Hernandez et al., 2009). There are at least 85 putative phosphorylation sites in tau, 45 serines, and 35 threonines. It has been shown that tau phosphorylation is regulated by many different kinases, including GSK-3β, CDK5, MAPKs, PKA, PKB/Akt, PKC, PKN, and CaMKII (Hanger et al., 1992; Harr et al., 1996; Yu et al., 2009). The precise role of some of these kinases is still under investigation with particular interest on glycogen synthase kinase-3 (GSK-3). However, we do not discard the important role of other kinases such as CDK5, p38, etc. In fact, the role of CDK5 in tau phosphorylation and NFT formation has been clearly established in transgenic mice (Noble et al., 2003; Piedrahita et al., 2010).
Tau hyperphosphorylation could result from the inhibition of phosphatases. Indeed, earlier work indicates that phosphatase activity is decreased in AD brains (Gong et al., 1993; Rahman et al., 2005). In summary, a combination of kinases and phosphatases may be postulated to play a central role in tauopathies.
Role and Regulation of GSK-3 Activity
Glycogen synthase kinase-3 is a serine/threonine protein kinase initially described to phosphorylate and inhibit glycogen synthase (Rylatt et al., 1980). GSK-3 has been evolutionary conserved and homolog genes have been identified in practically every eukaryotic genome examined including Xenopus laevis, Drosophila melanogaster, and Dictyostelium discoideum (Ruel et al., 1993; Itoh et al., 1995; Kim et al., 1999).
In mammals, GSK-3 is encoded by two genes known as gsk-3α and gsk-3β (Woodgett, 1990; Frame and Cohen, 2001; Grimes and Jope, 2001) that encode three different proteins, GSK-3α and two GSK-3β proteins (β1 and β2; Mukai et al., 2002; Schaffer et al., 2003). These isoforms are almost identical (98%) but differ in their N- and C-terminal domains (Woodgett, 1990). It has been extensively established that GSK-3 plays an important role in various essential physiological processes, including cell cycle or survival-apoptosis (Pap and Cooper, 1998; Lucas et al., 2001a; Ferkey and Kimelman, 2002). It is well known that GSK-3 has a plethora of substrates, identified in many cell types and in all cellular compartments; i.e., metabolic proteins (Alessi et al., 1996), cytoskeletal proteins (Hanger et al., 1992), as well as transduction (Cohen, 1999) and transcription factors (Hart et al., 1998).
Glycogen synthase kinase-3 is a kinase differentially regulated by tyrosine and serine/threonine phosphorylation (Wang et al., 1994). One of the main characteristics of GSK-3 is that its activity is relatively high in resting, non-stimulated cells. For many years, it was believed to be a constitutively active kinase. However, it has become apparent that the activity of GSK-3 may be regulated by a variety of signals (Sayas et al., 2002) and many extracellular signals may induce a rapid and reversible decrease in enzymatic activity. Moreover, the regulation of GSK-3 activity has turned out to be much more complex than originally thought (Medina and Wandosell, 2011). The regulatory mechanisms of GSK-3 activity may be classified as follows.
Regulation by Phosphorylation
Two general mechanism are involved in the regulation of GSK-3 activity, Ser/Thr phosphorylation of GSK-3 specific residues by other kinases, and Tyr phosphorylation, through auto-phosphorylation (Frame and Cohen, 2001; Harwood, 2001). Different regions can be identified in the GSK-3 structure that can be modified by phosphorylation: the first one corresponds to the amino-terminal domain and contains a serine residue at positions 21 in GSK-3α and 9 in GSK-3β. It has been undoubtedly demonstrated that phosphorylation of serine 21 or 9, correlates with inhibition of the kinase activity (Sutherland et al., 1993; Stambolic and Woodgett, 1994; Frame et al., 2001). Many protein kinases including Akt, ILK, PKA, p90Rsk are competent at phosphorylating GSK-3 at residues 21 and 9 in vitro and in vivo (Cross et al., 1995; Delcommenne et al., 1998; Fang et al., 2000).
Two other regulatory sites have been described: threonine 43, only present in the GSK-3β isoform, may be phosphorylated by Erk (Ding et al., 2005) whereas p38 MAPK phosphorylates serine 389 and threonine 390 present in GSK-3β (Thornton et al., 2008). In both cases, the data suggests that these phosphorylations may facilitate the competence of serine 9 to be phosphorylated rather than support a direct GSK-3 inhibition.
Conversely, an increase in tyrosine phosphorylation at residues 216 (GSK-3β) or 279 (GSK-3α) is in clear correlation with an increase on GSK-3 activity in neuronal cells, following exposure to lysophosphatidic acid (LPA; Sayas et al., 1999) or after neurotoxic insults such as β-amyloid or PrP (Munoz-Montano et al., 1997; Takashima et al., 1998; Perez et al., 2003b). Different candidates have been reported to phosphorylate GSK-3 on tyrosine 216/279 including Pyk-2 and Fyn kinases in vitro or MEK1/2 in fibroblasts (Lesort et al., 1999; Hartigan et al., 2001). These data are in contrast with those reported in D. discoideum where there is convincing evidence demonstrating that ZAK 1 (Kim et al., 1999, 2002) is the kinase responsible for this tyrosine phosphorylation of GSK-3. However, no homolog of ZAK1 has been found in mammals.
More recently, an alternative hypothesis has been proposed for the regulation of GSK-3 tyrosine phosphorylation. This hypothesis suggests that phosphotyrosine 279/216 in GSK-3 corresponds to an intra-molecular auto-phosphorylation event (Cole et al., 2004). However, this hypothesis still lacks a cellular demonstration. Data generated in our laboratory indicated that not all pharmacological inhibitors of GSK-3 decrease the level of phosphotyrosine (Simon et al., 2008). Thus, in view of the tantalizing autoregulatory system proposed and taken all data together, we hypothesize that some as-yet-unidentified tyrosine kinases and phosphatases may also regulate GSK-3 activity.
Regulation by Protein Complex Association
It is well known that GSK-3 forms part of a multiprotein complex formed by axin and adenomatous polyposis coli (APC) among others. This protein complex is the core of the canonical Wnt signaling (for review see Moon et al., 2004). In the absence of ligand, β-catenin is phosphorylated by GSK-3 in this multiprotein complex and targeted for proteasome degradation (Aberle et al., 1997). In addition, different GSK-3-binding proteins have been described.
The first GSK-3-binding protein was denoted as FRAT (Li et al., 1999; Fraser et al., 2002) and three different FRATs have been characterized since then. Surprisingly, FRAT1 appears to act as an inhibitory system, whereas FRAT2 appears to increase GSK-3 activity (Yost et al., 1998; Stoothoff et al., 2005). More recently, a GSK-3 interacting protein symbolized by GSKIP, has been cloned and characterized. GSKIP can block phosphorylation of different substrates and functions as a negative regulator of GSK-3β (Chou et al., 2006).
Regulation by Priming/Substrate Specificity
As a general rule, the specificity of many kinases is governed by a consensus sequence of amino acids present in their substrates. However, the crystal structure of human GSK-3β has provided a model for the binding of pre-phosphorylated substrates to the kinase (Dajani et al., 2001; ter Haar et al., 2001; Noble et al., 2005). It is now evident that some GSK-3 substrates require a previous (primed) phosphorylation by a priming kinase. This primed residue (Ser or Thr) is usually located four amino acids, C-terminal to the Ser or Thr residue to be modified by GSK-3 (Dajani et al., 2001; ter Haar et al., 2001). Several priming kinases have been identified, such as cdk-5, PAR-1, casein kinase I, PKC, or PKA (Sengupta et al., 1997; Amit et al., 2002; Liu et al., 2003; Noble et al., 2003; Nishimura et al., 2004; Alonso Adel et al., 2006). However, is not completely established whether a second set of “non-primed” substrates may define a different group of functions (Twomey and McCarthy, 2006). In addition, different GSK-3 isoforms appear to exhibit distinct substrate preferences in the brain (Soutar et al., 2010).
Regulation by Subcellular Localization
The presence of GSK-3 appears to be developmentally regulated in the brain (Takahashi et al., 1994) and, in fact, the developmental profile of GSK-3α and GSK-3β is different and the putative differential role of each isoform has been explored (Garrido et al., 2007). It is important to indicate that a portion of GSK-3, mostly β, has been associated with the growth cone. This GSK-3 pool appears to respond rapidly, being modified by phosphorylation and/or re-located in the growth cone, by external signals such as semaphorins (Eickholt et al., 2002) or NGF (Zhou et al., 2004). The presence of GSK-3α and β in primary neurons have been reported in different neuronal compartments, like in axon and dendrites, as well as in the cytoplasm, endoplasmic reticulum (ER; Meares et al., 2011), nucleus, and the mitochondria (Grimes and Jope, 2001; Watcharasit et al., 2002; Bijur and Jope, 2003). With respect to the nuclear localization, GSK-3 may be involved in phosphorylation of many transcription factors such as: Cyclin D1, β-catenin, HSF-1, NFAT, and cAMP-response element-binding protein, among others (for review see Cohen and Frame, 2001; Harwood, 2001; Xu et al., 2009). Moreover, the nuclear presence of GSK-3 in the nucleus may also play a role in alternative splicing (Avila et al., 2004).
Regulation by Proteolytic Cleavage
A new mechanism of GSK-3 regulation has been recently proposed. This regulation involves the removal by calpain of a fragment from the N-terminal region of GSK-3, including the regulatory serines 9/21 (Goni-Oliver et al., 2007). Interestingly, GSK-3β has also been recently shown to be cleaved at the N-terminus (and subsequently activated) by matrix metallo-proteinase 2 (MMP-2) in cardiomyoblasts (Kandasamy and Schulz, 2009).
Physiological and Pathological Regulation of GSK-3
GSK-3 Physiology
In neuronal development, GSK-3 has been reported to control many different aspects of neuronal and glial physiology, like morphogenesis and axonal polarity (Shi et al., 2004; Garrido et al., 2007), synaptogenesis (Lucas and Salinas, 1997), and cell survival (Pap and Cooper, 1998; Lucas et al., 2001a). Some of these cellular activities of GSK-3 are regulated by signals orchestrated by a wide set of extracellular ligands including growth factors, neurotrophic factors, neurotransmitters, and hormones but also by cell–cell interactions. Most of these extracellular factors trigger signaling pathways which correlate with the inhibition, or transient inhibition, of GSK-3 activity. In the case of insulin/IGF1 (Cross et al., 1995; Delcommenne et al., 1998; Fang et al., 2000) and NGF (Zhou et al., 2004) the regulatory role of Akt and/or ILK has been reported to be responsible for the increased inhibitory serine phosphorylation. More recently, we and others have shown that estradiol may also inhibit GSK-3 by a pathway modulated by PI3K-Akt (Cardona-Gomez et al., 2004; Mendez et al., 2005; Varea et al., 2009).
An alternative system of GSK-3 regulation is represented by the Wnt signaling pathway in which, in the absence of ligand, GSK-3β phosphorylates β-catenin, as a part of a degradation signal (for review see, i.e., Moon et al., 2004; Rosso et al., 2005). When Wnt (at least some Wnt’s) binds the Wnt receptor frizzled (fz), disheveled (Dsh) inhibits the activity of GSK-3β in a manner that is not fully understood. This system appears to be specific to GSK-3β as no counterpart has been described for GSK-3α to date. Some recent data suggests that this complex may be specific for the GSK-3β2 isoform (Castaño et al., 2010).
In addition, Reelin and Netrin signaling have been shown to regulate GSK-3 activity even though the exact mechanism is far from being fully elucidated. The binding of Reelin appears to induce signals which may activate GSK-3 (Gonzalez-Billault et al., 2005). In this pathway, the function of GSK-3 appears to be associated with CDK5, however, the specific system that controls GSK-3 activity has yet to be defined (Gonzalez-Billault et al., 2005). Similarly, Netrin 1 regulates, both in vivo and in vitro, the phosphorylation of neuronal MAP’s, in a signaling pathway that depends essentially on the kinases GSK-3 and CDK5 (Del Rio et al., 2004).
As mentioned, GSK-3 is regulated by a plethora of membrane receptors, some of which show a specific subcellular localization in neurons. Among these receptors, some purinergic ATP/ADP receptors, such as P2 × 7, P2Y1, or P2Y13, show a localized subcellular expression in neurons (Diaz-Hernandez et al., 2008; del Puerto et al., 2011). Purinergic receptors are expressed in glial and neuronal cells in the central and peripheral nervous system and are activated by purines and pyrimidines. The ATP-gated P2 × 7 ionotropic receptor is expressed in the distal region of the axon of hippocampal neurons during development (Diaz-Hernandez et al., 2008) and has been identified in presynaptic terminals in adult brain. Inhibition of P2 × 7 with brilliant blue G (BBG) increases GSK-3 phosphorylation, reducing its activity, and favors axonal elongation and branching in cortical and hippocampal neurons (Diaz-Hernandez et al., 2008), similarly to activation of P2Y1-ADP receptors (del Puerto et al., 2011). Inhibition of GSK-3 decreases PHF-I levels (phosphorylated Tau) and increases Tau-1 levels (dephosphorylated Tau). Moreover, this effect is also produced by GSK-3 inhibition with AR-A014418 (Garrido et al., 2007), which can counteract the action of ATP activation of the P2 × 7 receptor (Diaz-Hernandez et al., 2008).
Conversely, some ligands such as LPA may trigger GSK-3 kinase activity. LPA is a bioactive lipid that can act as a growth factor and binds to specific G-protein-coupled seven transmembrane domain receptors (GPCR; Chun et al., 2002). In neurons, LPA has been shown to promote growth cone collapse and neurite retraction (Tigyi et al., 1996; Sayas et al., 1999). We have demonstrated this LPA-induced GSK-3 activation event, not only inferred from hyperphosphorylation of tau, but also directly measured from neuronal cell extracts (Sayas et al., 2002). Similarly, semaphorin 3A (Sema 3A), a molecule that inhibits axonal growth or neurite retraction, activates GSK-3 at the leading edge of neuronal growth cones. The inhibition of GSK-3 activity can prevent the growth cone collapse response induced by Sema 3A, suggesting that GSK-3 activity might play a role in coupling Sema 3A signaling to changes in cell motility, axonal growth and/or maintenance of axonal extension (Eickholt et al., 2002).
Other membrane receptors that regulate GSK-3 function are only expressed in brain regions associated to specific brain diseases. For example, D2 dopamine receptors are mainly expressed in striatum and can also regulate the activity of GSK-3. Activation of D2-like receptors inhibits the phosphorylation of Akt, leading to activation (dephosphorylation) of GSK-3 through a G protein-independent mechanism involving a complex of Akt:PP2A scaffolded by β-arrestin-2. Lithium inhibits the behavioral actions of dopamine via disruption of the D2 receptor-mediated complex of Akt:βArr2:PP2A (Pawlak and Kerr, 2008). Similarly, other neuropeptide receptors, such as those of serotonin, regulate GSK-3 activity. Two serotonin receptors, 5-HT1A and 5-HT2A, appear to play antagonistic roles in regulating GSK-3β activity. Administration of a 5-HT1A agonist or a 5-HT2A antagonist to mice results in increased brain GSK-3β phosphorylation (Beaulieu et al., 2009). Moreover, drugs acting on 5-HT neurotransmission such as the selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase (MAO) inhibitors, and tricyclic antidepressants have been shown to inhibit GSK-3β in many brain regions, including the frontal cortex, hippocampus, and striatum of normal mice.
The acetylcholinesterase inhibitor physostigmine, the muscarinic agonist pilocarpine, and the N-methyl-D-aspartate (NMDA) receptors antagonist memantine, also modulate the phosphorylation state of the two isoforms of GSK-3 in mouse hippocampus, cerebral cortex, and striatum (De Sarno et al., 2006). Metabotropic glutamate receptor 5 (mGluR5) can modulate the PI3K/Akt/GSK-3 pathway in the hippocampus, and that modulation of this signaling pathway can reverse β-amyloid-induced neuronal toxicity (Liu et al., 2005).
Glycogen synthase kinase-3 or GSK-3-upstream elements can be selectively regulated in specific neuronal domains, such as the pre- or post-synaptic domains or the axon initial segment (place of neuronal action potential generation), in order to maintain or generate changes in neuronal function. For example, prolonged stimulation of neurons by depolarization results in GSK-3 phosphorylation and inactivation. Thus, GSK-3 activity could be acutely regulated by action potential stimulation (Smillie and Cousin, 2011). In conclusion, it is of great interest to identify proteins specifically located in domains that can regulate GSK-3 activity and the generation of selective pharmacological inhibitors to control GSK-3 activity at the subcellular level. Dendritic spines express GSK-3 regulated through NMDA receptors by two different stimuli (LTP stimulus or LTD stimulus), that, in turn, can activate or inhibit GSK-3 activity (Peineau et al., 2007).
Is Deregulation or Dysfunction of GSK-3 the Cause Neurodegenerative Diseases?
Aberrantly phosphorylated tau is a common factor in tauopathies but the pathological mechanisms responsible may be diverse (Trojanowski and Lee, 2005). The presence of a FTDP-17 mutation in tau may result in altered splicing or may correspond with missense mutations that modify the microtubule binding capacity and subsequently the phosphorylation/dephosphorylation kinetics (Ballatore et al., 2007; Hernandez and Avila, 2008). In fact, early tau-related deficits could be the result of synaptic abnormalities caused by the accumulation of hyperphosphorylated tau within intact dendritic spines (Hoover et al., 2010). Indeed, alterations in the expression and/or the activity of tau kinases (i.e., GSK-3β; Pei et al., 1997; Swatton et al., 2004; Hye et al., 2005) have been reported in affected brains, suggesting that one or several kinases could be involved in tau hyperphosphorylation (for review see, i.e., Hernandez and Avila, 2008). In addition, in transgenic mice the over-expression of some kinases such as GSK-3 increase tau phosphorylation and neurodegeneration (Lucas et al., 2001b). Conversely, inhibition of GSK-3β reduces tau phosphorylation and neurodegeneration and may block NFT formation (Nakashima et al., 2005; Noble et al., 2005; Engel et al., 2006b). Without excluding that more than one kinase may be important in the pathological phosphorylation of tau, these data suggests that inhibition of GSK-3 is probably an excellent therapeutic strategy for the treatment of tauopathies.
Interestingly, the P2 × 7 receptor has also been related to Alzheimer’s disease and β-amyloid production or involved in the regulation of a β-amyloid effect (Sanz et al., 2009). The purinergic receptor P2 × 7 triggers α-secretase-dependent processing of the amyloid precursor protein (APP; Delarasse et al., 2010). BBG treatment is also able to reduce inflammation after spinal cord injury in rats, reduce neuronal death, and it is a promising drug for axonal regeneration (Wang et al., 2004).
As mentioned, blockade of the mGluR5-mediated pathways inhibits GSK-3. The NMDA receptor antagonist memantine confers neuroprotection to Aβ peptides (Liu et al., 2005). Moreover, memantine, physostigmine, or pilocarpine, have been used as therapeutic agents in AD due to their capacity to regulate GSK-3 activity (De Sarno et al., 2006). When using GSK-3 inhibitors, one may also need to consider the different cell types involved in GSK-3 associated diseases. In some tauopathies glial cells and/or neuronal cells are an essential component. Thus, inhibition of GSK-3 through cell specific receptors may help avoid alteration of GSK-3 in other cell types.
Besides its association with AD (Ishiguro et al., 1993; Lovestone et al., 1994) GSK-3 dysfunction has also been associated with different pathological conditions or brain damage; such as Ischemia (Bhat et al., 2000), prion neurotoxicity (Perez et al., 2003b), or with some psychiatric disorders such as schizophrenia (Beaulieu et al., 2009). In fact in Disrupted in schizophrenia 1 mutant (DISC1) neuronal progenitor proliferation is altered and the inhibition of GSK-3 restore this proliferation (Mao et al., 2009). Thus, GSK-3 is emerging as a promising therapeutic target in several neurologic disorders.
Therapeutic Approaches: Direct Inhibition Versus Pharmacological Down-Regulation
As mentioned above, GSK-3 unique position in modulating the function of a diverse series of proteins in combination with its association with a wide variety of human disorders has attracted significant attention to the protein both as a therapeutic target and as a tool to understand the molecular bases of these disorders. Furthermore, GSK-3 appears to be a cellular nexus, integrating several signaling systems, including numerous second messengers and a wide selection of cellular stimulants.
If we consider GSK-3 as an example of a kinase associated with tauopathies, two different approaches would be considered as therapeutic strategies: direct inhibition of GSK-3 activity or a more generic modulation of GSK-3 acting upstream of the kinase. The first strategy, has been widely considered and indeed, some general inhibitors such as lithium have been tested in several AD animal models (Engel et al., 2008). A second approach is the search for candidates taking into account that neurodegeneration may reflect “pleiotropic” neuronal cell dysfunction. Accordingly, we have considered here some of the pathways that may control neural morphogenesis and survival including the PI3K-Akt, PKA, and Wnt pathways and their implication in neurodegeneration. Also, we propose alternative and complementary “non-target-based” approaches, that may also be useful for the study of neurodegeneration (Simon et al., 2011). Using cellular based assays we may identify molecules that regardless of their ability to cross the cellular membrane may be identified by a cellular/neuronal modification downstream of a selected kinase, i.e., the tau phosphorylation level. Activation of serotonergic 5-HT4 receptors have been shown to improve memory processes in preclinical cognition models, mainly through the stimulation of cholinergic neurotransmission (Lezoualc’h, 2007).
For many years, the mood stabilizing drug lithium has been considered as the reference GSK-3 inhibitor. Lithium ions directly inhibit GSK-3 (Klein and Melton, 1996), most likely by competing with magnesium but it also inhibits at least four phosphomonoesterases (including inositol monophosphatase; York et al., 1995), and phosphoglucomutase (Ray et al., 1978; Stambolic and Woodgett, 1994). Although the mechanism of action by which lithium exerts its therapeutic effects is unknown, GSK-3 is significantly inhibited at therapeutic lithium concentrations (Shaldubina et al., 2001; Gould and Manji, 2002; Phiel et al., 2003). It has been generally assumed that a significant proportion of the therapeutic action of lithium in bipolar disorders results from the inhibition of GSK-3 (Li et al., 2002; Rowe et al., 2007).
Inhibition of GSK-3 by lithium prevents Aβ-induced neurodegeneration of cortical and hippocampal primary cultures (Alvarez et al., 1999). With regards to tau metabolism, lithium is able to prevent tau phosphorylation in several mouse models of tauopathies (Perez et al., 2003a; Nakashima et al., 2005; Noble et al., 2005; Engel et al., 2006a; Caccamo et al., 2007). In AD models, lithium has been shown to block the accumulation of Aβ peptides in mice that overproduce human APP (Phiel et al., 2003). Furthermore, it has been suggested that lithium could also be effective in a mouse model of FTDP-17 (Perez et al., 2003a).
Only a few observational studies have attempted to address the clinical effect of lithium in patients with AD. A retrospective study with a large sample of patients with dementia resulted in an increased risk of AD in patients who had been treated with lithium within 4 years prior to diagnosis (Ayuso-Mateos et al., 2001). This effect might be partially accounted for by the increased occurrence of depression associated with AD. Moreover, a single case study reported in a dementia patient showed that lithium treatment alleviated symptoms of aggression and agitation, while cognition persisted after 1.5 years of treatment (Havens and Cole, 1982). Furthermore, a significantly increased global cognitive ability, as measured by MMSE in non-demented patients, appears to be associated with lithium intake (Terao et al., 2006). However, the study design and low sample size precludes drawing any causative conclusion from these studies.
In addition, some pilot studies have been carried out to directly address the effect of lithium treatment in AD patients. An open label feasibility and tolerability study with a small cohort of 22 subjects receiving a low dose of lithium was carried out in the UK and reported a high discontinuation rate despite few, relatively mild, and reversible side effects (Macdonald et al., 2008). A second randomized, single-blind, placebo-controlled, parallel group, multicenter 10-week study was carried out in Germany as a proof-of-principle (Hampel et al., 2009). A total of 71 patients with mild AD (MMSE scores between 21 and 26) were treated with lithium or placebo for 10 weeks after which neuropsychological and neuropsychiatric assessment was performed together with some biomarker determinations in plasma (Aβ1-42), lymphocytes (GSK-3 activity), and CSF (total tau, phospho-tau, and Aβ1-42). In spite of the fact that lithium plasma levels were within the therapeutic range, no treatment effect was observed in any of the cognition assessment scales or the selected biomarkers used. Given the short time of treatment of this study, the possibility that lithium has long-term effects on cognition or any other biomarker in AD remains to be tested.
Very recently, a randomized, double-blind, placebo-controlled study on 45 people with amnestic mild cognitive impairment (aMCI) treated with lithium for 12 months was reported (Forleza et al., 2011). Lithium treatment was associated with a significant decrease in CSF concentrations of pTau and with a better performance in cognitive tasks, strongly suggesting that lithium (most likely through GSK-3 inhibition) might have disease-modifying properties in AD and perhaps other tauopathies.
Use of lithium in clinical practice is problematic since it has a narrow therapeutic window (blood serum levels 0.6–1.2 mM) above which side effects are intolerable requiring titration. An overdose can lead to severe neurological dysfunction and in some cases death. Non-CNS side effects of lithium (common within therapeutic levels) include tremor, polyuria, polydipsia, nausea, and weight gain. Moreover, lithium can have adverse reactions with other drug classes including diuretics, NSAIDS, and other drugs that alter kidney function (Gould and Manji, 2006).
The unique position of GSK-3 as a pivotal and central player in the pathogenesis of both sporadic and familial forms of AD has attracted significant attention to this enzyme as a therapeutic target and has led to the synthesis of a high number of GSK-3 inhibitors, some of which are currently being tested in phase II proof-of-concept clinical trials (Mangialasche et al., 2010; Medina and Avila, 2010). Inhibition of GSK-3 with small-molecules would be expected to slow down progression of neurodegeneration in AD and perhaps other tauopathies as well.
A number of novel potent and relatively selective small-molecule inhibitors of GSK-3 activity from different chemical families have been recently described, including hymenialdisine, indirubins, paullones, maleimides, amino pyrazoles, thiazoles, and 2,4-disubstituted thiadiazolidinones (TDZD; reviewed in Medina and Castro, 2008). Most of them are ATP-competitive inhibitors, although more recently, new small-molecule derivatives that exhibit substrate competitive inhibition activity toward GSK-3 have been shown. Since different GSK-3 isoforms display a high degree of homology within the ATP-binding site, inhibitors are unable to exhibit isoform selectivity, as they all show similar potencies toward purified GSK-3α and GSK-3β. Thus, the development of isoform-specific inhibitors or the inhibition of signaling pathways specific for one of the isoforms is necessary to selectively target the different substrates involved in different neurodegenerative diseases. Although ATP-competitive inhibitors occupy the general area of the highly conserved ATP-binding site, they also explore other available nearby space, depending on their structure. Thus, it may be possible to identify selective inhibitors by taking advantage of the small differences in primary sequence that exist between the different kinases. The crystal structures of GSK-3β complexed with a variety of ligands (ter Haar, 2006), together with molecular modeling approaches (Gadakar et al., 2007), provide the necessary clues for enhancing selectivity toward GSK-3 (ter Haar et al., 2001; Patel et al., 2007).
Besides small-molecule inhibitors, some physiological peptides act as GSK-3 inhibitors, including GBP, a maternal Xenopus GSK-3-binding protein homologous to a mammalian T cell proto-oncogene (Yost et al., 1998) and p24, a heat resistant GSK-3-binding protein (Martin et al., 2002). That finding led to a synthetic strategy to develop new inhibitors, such as L803-mts, a peptidic inhibitor that binds to the substrate site (Plotkin et al., 2003). L803-mts has been more recently used to examine the impact of long-term in vivo inhibition of GSK-3 and its effects in specific tissues (Kaidanovich-Beilin and Eldar-Finkelman, 2006).
The last few years have seen the synthesis of quite a number of relatively selective, potent GSK-3 inhibitors with relative efficacy in vivo in a diverse array of animal models of human diseases, including AD. Despite the challenges faced by this approach with respect to safety and specificity, a number of efforts are underway to develop kinase inhibitors and, in fact, Noscira’s tideglusib (NP-12; see Table 1), is already in phase II clinical trials for the treatment of both AD and PSP, a tauopathy (Medina and Castro, 2008).
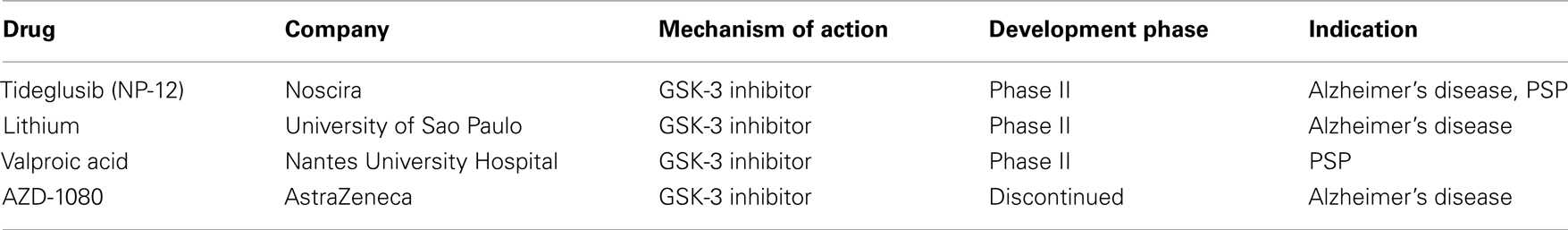
Table 1. Selected GSK-3 kinase inhibitor drugs in clinical development for the treatment of tauopathies.
In summary, there is a significant effort being made to validate a number of tau-related targets and to develop novel alternatives for the treatment of tauopathies such as AD or PSP (reviewed in Medina and Wandosell, 2011) including GSK-3 inhibition. We hope that future clinical trials will reveal the safety and efficacy of potential treatment strategies that are being tested in translational laboratories across the world.
Conclusion
Three decades after its discovery as a protein kinase involved in glycogen metabolism, GSK-3 has revealed itself as a common link, integrating several signaling systems, including second messengers and a wide selection of cellular stimulants. Modulation of its activity has also turned out to be much more complex than originally thought, as GSK-3 regulation occurs by complex mechanisms that are dependent on specific signaling pathways, including post-translational modifications, protein complex formation, and subcellular localization. Although there seems to be a good degree of functional overlapping between different isoforms, some tissue- and isoform-specific functions and substrates are beginning to emerge. Thus, new data will open the possibility to design better and more specific inhibitors.
Deregulation or abnormal GSK-3 activity appears to be associated with various relevant pathologies, including AD and other tauopathies, as the enzyme is uniquely positioned as central player in AD pathogenesis. GSK-3 plays a critical role in key events such as tau phosphorylation, Aβ formation, and neurotoxicity, microtubule dynamics, synaptic plasticity, neuritic dystrophy, cognition, neuronal survival, and neurodegeneration.
In the last two decades, drug discovery and development efforts for AD have primarily focused on targets defined by the amyloid cascade hypothesis with disappointing results, underscoring the need for novel therapeutic targets. In contrast, tau-based approaches have received little attention until recently, despite the fact that tau pathology is paramount in all tauopathies including AD (Medina and Wandosell, 2011). A significant effort has being made in the last few years to synthesize a high number of relatively selective and potent GSK-3 inhibitors, while some of them have shown in vivo efficacy in various animal models of AD. Some of the known drug discovery and development challenges should be met, including: lack of good predictive animal models, the need for validated biomarkers of disease progression, appropriate clinical trial design, early diagnosis, and treatment, definition of target populations, difficulties in demonstrating disease-modifying effects, etc. Despite the challenges faced by this approach with respect to safety and specificity, a number of efforts are underway to develop GSK-3 inhibitors as potential drugs for the treatment of AD. Some agents have already reached phase II clinical trials and some proof-of-concept studies are currently ongoing or planned.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. M. Medina is a full employee of Noscira S.A.
Acknowledgments
Miguel Medina acknowledges grant support from EU-FP7 (Neuro.GSK3, project no. 223276) and from CDTI’s CENIT program (DENDRIA, project no. CEN-20101023). Juan Jose Garrido acknowledges grant support from Ministerio de Ciencia y Tecnología (project no. SAF2009-12249-C02-02). Francisco G. Wandosell acknowledges grant support from CDTI’s program “CENIT-INGENIO 2010”: “Consorcio MELIUS” (2006-2010), and “Consorcio MIND” (2006-2010), Ref-050106090004; MICIN-ref no. SAF2009-12249-C02-01 and EU-FP7-2009-ref no. CT222887. In addition, Francisco G. Wandosell was supported by Grants from CIBERNED (which was an initiative of ISCIII), and by an Institutional grant from the “Fundación Areces.”
References
Aberle, H., Bauer, A., Stappert, J., Kispert, A., and Kemler, R. (1997). beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804.
Alessi, D. R., Caudwell, F. B., Andjelkovic, M., Hemmings, B. A., and Cohen, P. (1996). Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 399, 333–338.
Alonso Adel, C., Li, B., Grundke-Iqbal, I., and Iqbal, K. (2006). Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc. Natl. Acad. Sci. U.S.A. 103, 8864–8869.
Alvarez, G., Munoz-Montano, J. R., Satrustegui, J., Avila, J., Bogonez, E., and Diaz-Nido, J. (1999). Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 453, 260–264.
Amit, S., Hatzubai, A., Birman, Y., Andersen, J. S., Ben-Shushan, E., Mann, M., Ben-Neriah, Y., and Alkalay, I. (2002). Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 16, 1066–1076.
Avila, J., Lucas, J. J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384.
Ayuso-Mateos, J. L., Vazquez-Barquero, J. L., Dowrick, C., Lehtinen, V., Dalgard, O. S., Casey, P., Wilkinson, C., Lasa, L., Page, H., Dunn, G., and Wilkinson, G. (2001). Depressive disorders in Europe: prevalence figures from the ODIN study. Br. J. Psychiatry 179, 308–316.
Ballatore, C., Lee, V. M., and Trojanowski, J. Q. (2007). Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 8, 663–672.
Beaulieu, J. M., Gainetdinov, R. R., and Caron, M. G. (2009). Akt/GSK3 signaling in the action of psychotropic drugs. Annu. Rev. Pharmacol. Toxicol. 49, 327–347.
Bhat, R. V., Shanley, J., Correll, M. P., Fieles, W. E., Keith, R. A., Scott, C. W., and Lee, C. M. (2000). Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 11074–11079.
Bijur, G. N., and Jope, R. S. (2003). Glycogen synthase kinase-3 beta is highly activated in nuclei and mitochondria. Neuroreport 14, 2415–2419.
Buee, L., Bussiere, T., Buee-Scherrer, V., Delacourte, A., and Hof, P. R. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130.
Caccamo, A., Oddo, S., Tran, L. X., and LaFerla, F. M. (2007). Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am. J. Pathol. 170, 1669–1675.
Cardona-Gomez, P., Perez, M., Avila, J., Garcia-Segura, L. M., and Wandosell, F. (2004). Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol. Cell. Neurosci. 25, 363–373.
Castaño, Z., Gordon-Weeks, P., and Kypta, R. (2010). The neuron-specific isoform of GSK-3b2 is required for axon growth. J. Neurochem. 113, 117–130.
Chou, H. Y., Howng, S. L., Cheng, T. S., Hsiao, Y. L., Lieu, A. S., Loh, J. K., Hwang, S. L., Lin, C. C., Hsu, C. M., Wang, C., Lee, C. I., Lu, P. J., Chou, C. K., Huang, C. Y., and Hong, Y. R. (2006). GSKIP is homologous to the Axin GSK3beta interaction domain and functions as a negative regulator of GSK3beta. Biochemistry 45, 11379–11389.
Chun, J., Goetzl, E. J., Hla, T., Igarashi, Y., Lynch, K. R., Moolenaar, W., Pyne, S., and Tigyi, G. (2002). International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 54, 265–269.
Cohen, P. (1999). The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 485–495.
Cole, A., Frame, S., and Cohen, P. (2004). Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem. J. 377, 249–255.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789.
Dajani, R., Fraser, E., Roe, S. M., Young, N., Good, V., Dale, T. C., and Pearl, L. H. (2001). Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732.
De Sarno, P., Bijur, G. N., Zmijewska, A. A., Li, X., and Jope, R. S. (2006). In vivo regulation of GSK3 phosphorylation by cholinergic and NMDA receptors. Neurobiol. Aging 27, 413–422.
del Puerto, A., Díaz-Hernández, J.-I., Tapia, M., Benitez, M. J., Zhang, J., Díaz-Hernández, M., Wandosell, F., and Garrido, J.-J. (2011). Adenylate cyclase 5 coordinates the action of ADP, P2Y1, P2Y13, and ATP-gated P2 × 7 receptors on axonal elongation. J. Cell Sci. (in press).
Del Rio, J. A., Gonzalez-Billault, C., Urena, J. M., Jimenez, E. M., Barallobre, M. J., Pascual, M., Pujadas, L., Simo, S., La Torre, A., Wandosell, F., Avila, J., and Soriano, E. (2004). MAP1B is required for Netrin 1 signaling in neuronal migration and axonal guidance. Curr. Biol. 14, 840–850.
Delarasse, C., Auger, R., Gonnord, P., Fontaine, B., and Kanellopoulos, J. M. (2010). The purinergic receptor P2 × 7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J. Biol. Chem. 286, 2596–2606.
Delcommenne, M., Tan, C., Gray, V., Rue, L., Woodgett, J., and Dedhar, S. (1998). Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. U.S.A. 95, 11211–11216.
Diaz-Hernandez, M., del Puerto, A., Diaz-Hernandez, J. I., Diez-Zaera, M., Lucas, J. J., Garrido, J. J., and Miras-Portugal, M. T. (2008). Inhibition of the ATP-gated P2 × 7 receptor promotes axonal growth and branching in cultured hippocampal neurons. J. Cell. Sci. 121, 3717–3728.
Ding, Q., Xia, W., Liu, J. C., Yang, J. Y., Lee, D. F., Xia, J., Bartholomeusz, G., Li, Y., Pan, Y., Li, Z., Bargou, R. C., Qin, J., Lai, C. C., Tsai, F. J., Tsai, C. H., and Hung, M. C. (2005). Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19, 159–170.
Eickholt, B. J., Walsh, F. S., and Doherty, P. (2002). An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J. Cell Biol. 157, 211–217.
Engel, T., Goni-Oliver, P., Gomez de Barreda, E., Lucas, J. J., Hernandez, F., and Avila, J. (2008). Lithium, a potential protective drug in Alzheimer’s disease. Neurodegener. Dis. 5, 247–249.
Engel, T., Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2006a). Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J. Neurochem. 99, 1445–1455.
Engel, T., Hernandez, F., Avila, J., and Lucas, J. J. (2006b). Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 26, 5083–5090.
Fang, X., Yu, S. X., Lu, Y., Bast, R. C. Jr., Woodgett, J. R., and Mills, G. B. (2000). Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. U.S.A. 97, 11960–11965.
Ferkey, D. M., and Kimelman, D. (2002). Glycogen synthase kinase-3 beta mutagenesis identifies a common binding domain for GBP and Axin. J. Biol. Chem. 277, 16147–16152.
Forleza, O., Diniz, B., Radanovic, M., Santos, F., Talib, L., and Gattaz, W. (2011). Disease-modifying properties of long-term lithium treatment for amnesic milsd cognitive impairment:randomised controlled trial. Br. J. Psychol. 198, 351–356.
Frame, S., and Cohen, P. (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 359, 1–16.
Frame, S., Cohen, P., and Biondi, R. M. (2001). A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol. Cell 7, 1321–1327.
Fraser, E., Young, N., Dajani, R., Franca-Koh, J., Ryves, J., Williams, R. S., Yeo, M., Webster, M. T., Richardson, C., Smalley, M. J., Pearl, L. H., Harwood, A., and Dale, T. C. (2002). Identification of the Axin and Frat binding region of glycogen synthase kinase-3. J. Biol. Chem. 277, 2176–2185.
Gadakar, P., Phukan, S., Dattatreya, P., and Balaji, V. (2007). Pose prediction accuracy in docking studies and enrichment of actives in the active site of GSK-3beta. J. Chem. Inf. Model. 47, 446–459.
Garrido, J. J., Simon, D., Varea, O., and Wandosell, F. (2007). GSK3 alpha and GSK3 beta are necessary for axon formation. FEBS Lett. 581, 1579–1586.
Gong, C. X., Singh, T. J., Grundke-Iqbal, I., and Iqbal, K. (1993). Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 61, 921–927.
Goni-Oliver, P., Lucas, J. J., Avila, J., and Hernandez, F. (2007). N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J. Biol. Chem. 282, 22406–22413.
Gonzalez-Billault, C., Del Rio, J. A., Urena, J. M., Jimenez-Mateos, E. M., Barallobre, M. J., Pascual, M., Pujadas, L., Simo, S., Torre, A. L., Gavin, R., Wandosell, F., Soriano, E., and Avila, J. (2005). A role of MAP1B in Reelin-dependent neuronal migration. Cereb. Cortex 15, 1134–1145.
Gould, T., and Manji, H. (2006). “Glycogen synthase kinase 3: a target for novel mood disorder treatments,” Chapter 7, in Glycogen Synthase Kinase 3 (GSK-3) and its Inhibitors-Drug Discovery and Development, ed. B. Wang (New Jersey, NJ: Wiley), 125–154.
Gould, T. D., and Manji, H. K. (2002). The Wnt signaling pathway in bipolar disorder. Neuroscientist 8, 497–511.
Grimes, C. A., and Jope, R. S. (2001). The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 65, 391–426.
Hampel, H., Ewers, M., Burger, K., Annas, P., Mortberg, A., Bogstedt, A., Frolich, L., Schroder, J., Schonknecht, P., Riepe, M. W., Kraft, I., Gasser, T., Leyhe, T., Moller, H. J., Kurz, A., and Basun, H. (2009). Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 70, 922–931.
Hanger, D. P., Hughes, K., Woodgett, J. R., Brion, J. P., and Anderton, B. H. (1992). Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 147, 58–62.
Harr, S. D., Hollister, R. D., and Hyman, B. T. (1996). Glycogen synthase kinase 3 alpha and 3 beta do not colocalize with neurofibrillary tangles. Neurobiol. Aging 17, 343–348.
Hart, M. J., de los Santos, R., Albert, I. N., Rubinfeld, B., and Polakis, P. (1998). Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr. Biol. 8, 573–581.
Hartigan, J. A., Xiong, W. C., and Johnson, G. V. (2001). Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem. Biophys. Res. Commun. 284, 485–489.
Havens, W. W. II, and Cole, J. (1982). Successful treatment of dementia with lithium. J. Clin. Psychopharmacol. 2, 71–72.
Hernandez, F., and Avila, J. (2008). Tau aggregates and tau pathology. J. Alzheimers Dis. 14, 449–452.
Hernandez, F., de Barreda, E. G., Fuster-Matanzo, A., Goni-Oliver, P., Lucas, J. J., and Avila, J. (2009). The role of GSK3 in Alzheimer disease. Brain Res. Bull. 80, 248–250.
Hoover, B. R., Reed, M. N., Su, J., Penrod, R. D., Kotilinek, L. A., Grant, M. K., Pitstick, R., Carlson, G. A., Lanier, L. M., Yuan, L. L., Ashe, K. H., and Liao, D. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081.
Hye, A., Kerr, F., Archer, N., Foy, C., Poppe, M., Brown, R., Hamilton, G., Powell, J., Anderton, B., and Lovestone, S. (2005). Glycogen synthase kinase-3 is increased in white cells early in Alzheimer’s disease. Neurosci. Lett. 373, 1–4.
Ishiguro, K., Shiratsuchi, A., Sato, S., Omori, A., Arioka, M., Kobayashi, S., Uchida, T., and Imahori, K. (1993). Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–172.
Itoh, K., Tang, T. L., Neel, B. G., and Sokol, S. Y. (1995). Specific modulation of ectodermal cell fates in Xenopus embryos by glycogen synthase kinase. Development 121, 3979–3988.
Kaidanovich-Beilin, O., and Eldar-Finkelman, H. (2006). Long-term treatment with novel glycogen synthase kinase-3 inhibitor improves glucose homeostasis in ob/ob mice: molecular characterization in liver and muscle. J. Pharmacol. Exp. Ther. 316, 17–24.
Kandasamy, A. D., and Schulz, R. (2009). Glycogen synthase kinase-3beta is activated by matrix metalloproteinase-2 mediated proteolysis in cardiomyoblasts. Cardiovasc. Res. 83, 698–706.
Kim, L., Harwood, A., and Kimmel, A. R. (2002). Receptor-dependent and tyrosine phosphatase-mediated inhibition of GSK3 regulates cell fate choice. Dev. Cell 3, 523–532.
Kim, L., Liu, J., and Kimmel, A. R. (1999). The novel tyrosine kinase ZAK1 activates GSK3 to direct cell fate specification. Cell 99, 399–408.
Klein, P. S., and Melton, D. A. (1996). A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 93, 8455–8459.
Lesort, M., Jope, R. S., and Johnson, G. V. (1999). Insulin transiently increases tau phosphorylation: involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J. Neurochem. 72, 576–584.
Lezoualc’h, F. (2007). 5-HT4 receptor and Alzheimer’s disease: the amyloid connection. Exp. Neurol. 205, 325–329.
Li, L., Yuan, H., Weaver, C. D., Mao, J., Farr, G. H. III, Sussman, D. J., Jonkers, J., Kimelman, D., and Wu, D. (1999). Axin and Frat1 interact with dvl and GSK, bridging Dvl to GSK in Wnt-mediated regulation of LEF-1. EMBO J. 18, 4233–4240.
Li, X., Bijur, G. N., and Jope, R. S. (2002). Glycogen synthase kinase-3beta, mood stabilizers, and neuroprotection. Bipolar Disord. 4, 137–144.
Liu, F., Gong, X., Zhang, G., Marquis, K., Reinhart, P., and Andree, T. H. (2005). The inhibition of glycogen synthase kinase 3beta by a metabotropic glutamate receptor 5 mediated pathway confers neuroprotection to Abeta peptides. J. Neurochem. 95, 1363–1372.
Liu, S. J., Zhang, A. H., Li, H. L., Wang, Q., Deng, H. M., Netzer, W. J., Xu, H., and Wang, J. Z. (2003). Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J. Neurochem. 87, 1333–1344.
Lovestone, S., Reynolds, C. H., Latimer, D., Davis, D. R., Anderton, B. H., Gallo, J. M., Hanger, D., Mulot, S., Marquardt, B., Stabel, S., Woodgett, J. R., and Miller, C. C. J. (1994). Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr. Biol. 4, 1077–1086.
Lucas, F. R., and Salinas, P. C. (1997). WNT-7a induces axonal remodeling and increases synapsin I levels in cerebellar neurons. Dev. Biol. 192, 31–44.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001a). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
Lucas, J. J., Hernandez, F., Gomez-Ramos, P., Moran, M. A., Hen, R., and Avila, J. (2001b). Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 20, 27–39.
Macdonald, A., Briggs, K., Poppe, M., Higgins, A., Velayudhan, L., and Lovestone, S. (2008). A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 23, 704–711.
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P., and Kivipelto, M. (2010). Alzheimer’s disease: clinical trials and drug development. Lancet Neurol. 9, 702–716.
Mao, Y., Ge, X., Frank, C. L., Madison, J. M., Koehler, A. N., Doud, M. K., Tassa, C., Berry, E. M., Soda, T., Singh, K. K., Biechele, T., Petryshen, T. L., Moon, R. T., Haggarty, S. J., and Tsai, L. H. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136, 1017–1031.
Martin, C. P., Vazquez, J., Avila, J., and Moreno, F. J. (2002). P24, a glycogen synthase kinase 3 (GSK 3) inhibitor. Biochim. Biophys. Acta 1586, 113–122.
Meares, G. P., Mines, M. A., Beurel, E., Eom, T. Y., Song, L., Zmijewska, A. A., and Jope, R. S. (2011). Glycogen synthase kinase-3 regulates endoplasmic reticulum (ER) stress-induced CHOP expression in neuronal cells. Exp. Cell Res. 317, 1621–1628.
Medina, M., and Avila, J. (2010). Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer’s disease. Curr. Pharm. Des. 16, 2790–2798.
Medina, M., and Castro, A. (2008). Glycogen synthase kinase-3 (GSK-3) inhibitors reach the clinic. Curr. Opin. Drug Discov. Dev. 11, 533–543.
Medina, M., and Wandosell, F. (2011). Deconstructing GSK-3: the fine regulation of its activity. Int. J. Alzheimers Dis. 2011, 479249.
Mendez, P., Azcoitia, I., and Garcia-Segura, L. M. (2005). Interdependence of oestrogen and insulin-like growth factor-I in the brain: potential for analysing neuroprotective mechanisms. J. Endocrinol. 185, 11–17.
Moon, R. T., Kohn, A. D., De Ferrari, G. V., and Kaykas, A. (2004). WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701.
Mukai, F., Ishiguro, K., Sano, Y., and Fujita, S. C. (2002). Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3beta. J. Neurochem. 81, 1073–1083.
Munoz-Montano, J. R., Moreno, F. J., Avila, J., and Diaz-Nido, J. (1997). Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett. 411, 183–188.
Nakashima, H., Ishihara, T., Suguimoto, P., Yokota, O., Oshima, E., Kugo, A., Terada, S., Hamamura, T., Trojanowski, J. Q., Lee, V. M., and Kuroda, S. (2005). Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 110, 547–556.
Nishimura, I., Yang, Y., and Lu, B. (2004). PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116, 671–682.
Noble, W., Olm, V., Takata, K., Casey, E., Mary, O., Meyerson, J., Gaynor, K., LaFrancois, J., Wang, L., Kondo, T., Davies, P., Burns, M., Veeranna, Nixon, R., Dickson, D., Matsuoka, Y., Ahlijanian, M., Lau, L. F., and Duff, K. (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565.
Noble, W., Planel, E., Zehr, C., Olm, V., Meyerson, J., Suleman, F., Gaynor, K., Wang, L., LaFrancois, J., Feinstein, B., Burns, M., Krishnamurthy, P., Wen, Y., Bhat, R., Lewis, J., Dickson, D., and Duff, K. (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 6990–6995.
Pap, M., and Cooper, G. M. (1998). Role of glycogen synthase kinase-3 in the phosphatidylinositol 3- Kinase/Akt cell survival pathway. J. Biol. Chem. 273, 19929–19932.
Patel, D. S., Dessalew, N., Iqbal, P., and Bharatam, P. V. (2007). Structure-based approaches in the design of GSK-3 selective inhibitors. Curr. Protein Pept. Sci. 8, 352–364.
Pawlak, V., and Kerr, J. N. (2008). Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J. Neurosci. 28, 2435–2446.
Pei, J. J., Tanaka, T., Tung, Y. C., Braak, E., Iqbal, K., and Grundke-Iqbal, I. (1997). Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J. Neuropathol. Exp. Neurol. 56, 70–78.
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., Lo, E., Wu, D., Saule, E., Bouschet, T., Matthews, P., Isaac, J. T., Bortolotto, Z. A., Wang, Y. T., and Collingridge, G. L. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717.
Perez, M., Hernandez, F., Lim, F., Diaz-Nido, J., and Avila, J. (2003a). Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J. Alzheimers Dis. 5, 301–308.
Perez, M., Rojo, A. I., Wandosell, F., Diaz-Nido, J., and Avila, J. (2003b). Prion peptide induces neuronal cell death through a pathway involving glycogen synthase kinase 3. Biochem. J. 372, 129–136.
Phiel, C. J., Wilson, C. A., Lee, V. M., and Klein, P. S. (2003). GSK-3alpha regulates production of Alzheimer’s disease amyloid-beta peptides. Nature 423, 435–439.
Piedrahita, D., Hernandez, I., Lopez-Tobon, A., Fedorov, D., Obara, B., Manjunath, B. S., Boudreau, R. L., Davidson, B., Laferla, F., Gallego-Gomez, J. C., Kosik, K. S., and Cardona-Gomez, G. P. (2010). Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J. Neurosci. 30, 13966–13976.
Plotkin, B., Kaidanovich, O., Talior, I., and Eldar-Finkelman, H. (2003). Insulin mimetic action of synthetic phosphorylated peptide inhibitors of glycogen synthase kinase-3. J. Pharmacol. Exp. Ther. 305, 974–980.
Rahman, A., Grundke-Iqbal, I., and Iqbal, K. (2005). Phosphothreonine-212 of Alzheimer abnormally hyperphosphorylated tau is a preferred substrate of protein phosphatase-1. Neurochem. Res. 30, 277–287.
Ray, W. J. Jr., Szymanki, E. S., and Ng, L. (1978). The binding of lithium and of anionic metabolites to phosphoglucomutase. Biochim. Biophys. Acta 522, 434–442.
Rosso, S. B., Sussman, D., Wynshaw-Boris, A., and Salinas, P. C. (2005). Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8, 34–42.
Rowe, M. K., Wiest, C., and Chuang, D. M. (2007). GSK-3 is a viable potential target for therapeutic intervention in bipolar disorder. Neurosci. Biobehav. Rev. 31, 920–931.
Ruel, L., Bourouis, M., Heitzler, P., Pantesco, V., and Simpson, P. (1993). Drosophila shaggy kinase and rat glycogen synthase kinase-3 have conserved activities and act downstream of Notch. Nature 362, 557–560.
Rylatt, D. B., Aitken, A., Bilham, T., Condon, G. D., Embi, N., and Cohen, P. (1980). Glycogen synthase from rabbit skeletal muscle. Amino acid sequence at the sites phosphorylated by glycogen synthase kinase-3, and extension of the N-terminal sequence containing the site phosphorylated by phosphorylase kinase. Eur. J. Biochem. 107, 529–537.
Sanz, J. M., Chiozzi, P., Ferrari, D., Colaianna, M., Idzko, M., Falzoni, S., Fellin, R., Trabace, L., and Di Virgilio, F. (2009). Activation of microglia by amyloid {beta} requires P2 × 7 receptor expression. J. Immunol. 182, 4378–4385.
Sayas, C. L., Avila, J., and Wandosell, F. (2002). Regulation of neuronal cytoskeleton by lysophosphatidic acid: role of GSK-3. Biochim. Biophys. Acta 1582, 144–153.
Sayas, C. L., Moreno-Flores, M. T., Avila, J., and Wandosell, F. (1999). The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J. Biol. Chem. 274, 37046–37052.
Schaffer, B., Wiedau-Pazos, M., and Geschwind, D. H. (2003). Gene structure and alternative splicing of glycogen synthase kinase 3 beta (GSK-3beta) in neural and non-neural tissues. Gene 302, 73–81.
Sengupta, A., Wu, Q., Grundke-Iqbal, I., Iqbal, K., and Singh, T. J. (1997). Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol. Cell. Biochem. 167, 99–105.
Shaldubina, A., Agam, G., and Belmaker, R. H. (2001). The mechanism of lithium action: state of the art, ten years later. Prog. Neuropsychopharmacol. Biol. Psychiatry 25, 855–866.
Shi, S. H., Cheng, T., Jan, L. Y., and Jan, Y. N. (2004). APC and GSK-3beta are involved in mPar3 targeting to the nascent axon and establishment of neuronal polarity. Curr. Biol. 14, 2025–2032.
Simon, D., Benitez, M. J., Gimenez-Cassina, A., Garrido, J. J., Bhat, R. V., Diaz-Nido, J., and Wandosell, F. (2008). Pharmacological inhibition of GSK-3 is not strictly correlated with a decrease in tyrosine phosphorylation of residues 216/279. J. Neurosci. Res. 86, 668–674.
Simon, D., Medina, M., Avila, J., and Wandosell, F. (2011). Overcoming cell death and tau phosphorylation mediated by PI3K-inhibition: a cell assay to measure neuroprotection. CNS Neurol. Disord. Drug Targets 10, 208–214.
Smillie, K. J., and Cousin, M. A. (2011). The role of GSK3 in presynaptic function. Int. J. Alzheimers Dis. 263673.
Soutar, M. P., Kim, W. Y., Williamson, R., Peggie, M., Hastie, C. J., H. McLauchlan, Snider, W. D., Gordon-Weeks, P. R., and Sutherland, C. (2010). Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J. Neurochem. 115, 974–983.
Stambolic, V., and Woodgett, J. R. (1994). Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 303(Pt 3), 701–704.
Stoothoff, W. H., Cho, J. H., McDonald, R. P., and Johnson, G. V. (2005). FRAT-2 preferentially increases glycogen synthase kinase 3 beta-mediated phosphorylation of primed sites, which results in enhanced tau phosphorylation. J. Biol. Chem. 280, 270–276.
Sutherland, C., Leighton, I. A., and Cohen, P. (1993). Inactivation of glycogen synthase kinase-3 beta by phosphorylation: new kinase connections in insulin and growth-factor signalling. Biochem. J. 296, 15–19.
Swatton, J. E., Sellers, L. A., Faull, R. L., Holland, A., Iritani, S., and Bahn, S. (2004). Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 19, 2711–2719.
Takahashi, M., Tomizawa, K., Kato, R., Sato, K., Uchida, T., Fujita, S. C., and Imahori, K. (1994). Localization and developmental changes of tau protein kinase I/glycogen synthase kinase-3 beta in rat brain. J. Neurochem. 63, 245–255.
Takashima, A., Honda, T., Yasutake, K., Michel, G., Murayama, O., Murayama, M., Ishiguro, K., and Yamaguchi, H. (1998). Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25-35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 31, 317–323.
ter Haar, E. (2006). “The crystal structure of glycogen synthase kinase 3,” Chapter 5, in Glycogen Synthase Kinase 3 (GSK-3) and its Inhibitors-Drug Discovery and Development, ed. B. Wang (New Jersey, NJ: Wiley), 61–82.
ter Haar, E., Coll, J. T., Austen, D. A., Hsiao, H. M., Swenson, L., and Jain, J. (2001). Structure of GSK3beta reveals a primed phosphorylation mechanism. Nat. Struct. Biol. 8, 593–596.
Terao, T., Nakano, H., Inoue, Y., Okamoto, T., Nakamura, J., and Iwata, N. (2006). Lithium and dementia: a preliminary study. Prog. Neuropsychopharmacol. Biol. Psychiatry 30, 1125–1128.
Thornton, T. M., Pedraza-Alva, G., Deng, B., Wood, C. D., Aronshtam, A., Clements, J. L., Sabio, G., Davis, R. J., Matthews, D. E., Doble, B., and Rincon, M. (2008). Phosphorylation by p38 MAPK as an alternative pathway for GSK3beta inactivation. Science 320, 667–670.
Tigyi, G., Fischer, D. J., Sebok, A., Marshall, F., Dyer, D. L., and Miledi, R. (1996). Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite- protective effects of cyclic AMP signaling. J. Neurochem. 66, 549–558.
Trojanowski, J. Q., and Lee, V. M. (2005). Pathological tau: a loss of normal function or a gain in toxicity? Nat. Neurosci. 8, 1136–1137.
Twomey, C., and McCarthy, J. V. (2006). Presenilin-1 is an unprimed glycogen synthase kinase-3beta substrate. FEBS Lett. 580, 4015–4020.
Varea, O., Garrido, J. J., Dopazo, A., Mendez, P., Garcia-Segura, L. M., and Wandosell, F. (2009). Estradiol activates beta-catenin dependent transcription in neurons. PLoS ONE 4, e5153. doi: 10.1371/journal.pone.0005153
Wang, Q. M., Fiol, C. J., DePaoli-Roach, A. A., and Roach, P. J. (1994). Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J. Biol. Chem. 269, 14566–14574.
Wang, X., Arcuino, G., Takano, T., Lin, J., Peng, W. G., Wan, P., Li, P., Xu, Q., Liu, Q. S., Goldman, S. A., and Nedergaard, M. (2004). P2 × 7 receptor inhibition improves recovery after spinal cord injury. Nat. Med. 10, 821–827.
Watcharasit, P., Bijur, G. N., Zmijewski, J. W., Song, L., Zmijewska, A., Chen, X., Johnson, G. V., and Jope, R. S. (2002). Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc. Natl. Acad. Sci. U.S.A. 99, 7951–7955.
Woodgett, J. R. (1990). Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438.
Xu, C., Kim, N. G., and Gumbiner, B. M. (2009). Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8, 4032–4039.
York, J. D., Ponder, J. W., and Majerus, P. W. (1995). Definition of a metal-dependent/Li(+)-inhibited phosphomonoesterase protein family based upon a conserved three-dimensional core structure. Proc. Natl. Acad. Sci. U.S.A. 92, 5149–5153.
Yost, C., Farr, G. H. III, Pierce, S. B., Ferkey, D. M., Chen, M. M., and Kimelman, D. (1998). GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell 93, 1031–1041.
Yu, Y., Run, X., Liang, Z., Li, Y., Liu, F., Liu, Y., Iqbal, K., Grundke-Iqbal, I., and Gong, C. X. (2009). Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 108, 1480–1494.
Keywords: GSK-3, kinases modulation, tau pathologies, Alzheimer
Citation: Medina M, Garrido JJ and Wandosell FG (2011) Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Front. Mol. Neurosci. 4:24. doi: 10.3389/fnmol.2011.00024
Received: 26 July 2011; Accepted: 30 August 2011;
Published online: 05 October 2011.
Edited by:
Richard Scott Jope, University of Alabama at Birmingham, USAReviewed by:
Jesus Avila, Centro de Biología Molecular Severo Ochoa CSIC-UAM, SpainMathieu Lesort, University of Alabama at Birmingham, USA
Copyright: © 2011 Medina, Garrido and Wandosell. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Francisco G. Wandosell, Centro de Biología Molecular “Severo Ochoa,” CSIC-UAM-CIBERNED, Nicolás Cabrera 1, 28049 Madrid, Spain. e-mail: fwandosell@cbm.csic.es