
- 1Cellular Immunology Unit, Department of Immunology, Infectious Diseases and Transplantation, San Raffaele Scientific Institute, Milan, Italy
- 2Università Vita Salute San Raffaele, Milan, Italy
The tumor is a hostile microenvironment for T lymphocytes. Indeed, irregular blood flow, and endothelial cell (EC) anergy that characterize most solid tumors hamper leukocyte adhesion, extravasation, and infiltration. In addition, hypoxia and reprograming of energy metabolism within cancer cells transform the tumor mass in a harsh environment that limits survival and effector functions of T cells, regardless of being induced in vivo by vaccination or adoptively transferred. In this review, we will summarize on recent advances in our understanding of the characteristics of tumor-associated neo-angiogenic vessels as well as of the tumor metabolism that may impact on T cell trafficking and fitness of tumor infiltrating lymphocytes. In particular, we will focus on how advances in knowledge of the characteristics of tumor ECs have enabled identifying strategies to normalize the tumor-vasculature and/or overcome EC anergy, thus increasing leukocyte-vessel wall interactions and lymphocyte infiltration in tumors. We will also focus on drugs acting on cells and their released molecules to transiently render the tumor microenvironment more suitable for tumor infiltrating T lymphocytes, thus increasing the therapeutic effectiveness of both active and adoptive immunotherapies.
Introduction
Active and adoptive cancer immunotherapies have breached the wall between bench and bedside at last, and have just entered a new golden age. This is the result of several concomitant technological advancements and breakthrough discoveries. On the one hand, powerful technical tools (e.g., tetramers and live imaging) have been made available to more deeply investigate the interactions between the growing tumor and the host, and especially the immune system. Thus, sophisticated genetically engineered animal models have allowed building new theories on the process of cancer immune surveillance (1). Furthermore, high-throughput technologies are making possible investigating the tumor microenvironment as a whole (2), and genomic landscaping, proteomic profiling, and more recently metabolomics and algorithms applied to cancer histochemistry (3–8) are drawing an entirely new picture of the tumor mass. On the other hand, strong efforts from hundreds of laboratories around the word in the last 20 years have defined tumor-associated antigens (TAAs) and adjuvants to such an high degree of knowledge [e.g.,(9)] that active immunotherapy has eventually reached the bedside with the first FDA approved cancer vaccine for metastatic prostate cancer patients (10). Even more importantly, clinical grade in vitro expanded tumor infiltrating lymphocytes (TILs) and genetically engineered T cells have demonstrated the full potential of adoptive immunotherapy (11, 12).
Yet, several hurdles still need to be overcome (Figure 1) to extend such treatments to the majority of cancer patients. Firstly, the tumor mass is characterized by abnormal tumor vessels and interstitium that limit leukocyte adhesion, extravasation, and infiltration (13), and favors hypoxia and reprograming of energy metabolism within cancer cells (14). Metabolic alterations within the tumor mass also limit T cell functions, and the tumor microenvironment eventually becomes a site of immune privilege where several cancer cell intrinsic and extrinsic mechanisms suppress the tumor-specific T cell response (15).
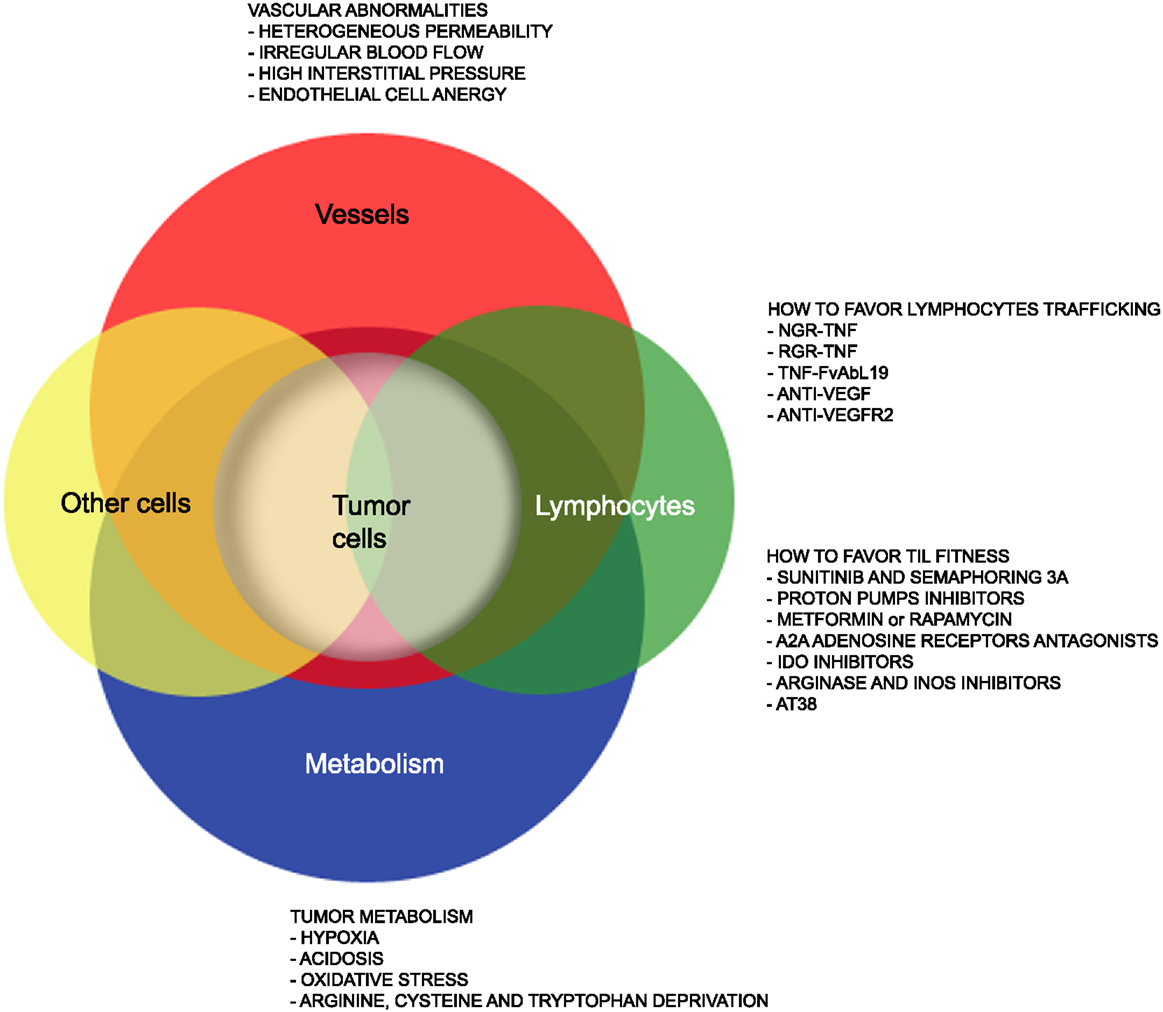
Figure 1. Strategies that favor lymphocyte trafficking into tumors and fitness of TILs. The cartoon highlights abnormalities of tumor-associated vessels and alterations of the metabolism within the tumor microenvironment that limit lymphocyte trafficking into tumor and TIL anti-tumor activities. Strategies to overcome such hurdles are also indicated.
Here, we will summarize on recent advances in our understanding of the characteristics of tumor-associated neo-angiogenic vessels as well as of the tumor metabolism that may impact on T cell trafficking and fitness of TILs. We will also report on drugs acting on cells and their released molecules to transiently render the tumor microenvironment more suitable for tumor TILs (Figure 1), thus increasing T cell trafficking into tumors and the therapeutic effectiveness of both active and adoptive immunotherapies.
T Cell Adhesion to the Endothelium, Extravasation, and Infiltration within Inflamed Tissues
Once a T cell has been activated in secondary lymphoid organs, it reaches the blood flow and navigates within vessels to the site of extravasation, which usually coincides with a site of inflammation. Activated T cells prefer to exit the blood stream at the level of post-capillary venules, where the hemodynamic shear stress is lower than in arteries and capillaries and the endothelium is more prone to extravasation. Activated T cells travel more efficiently than naïve T cells to inflamed tissues because they upregulate adhesion molecules and chemoattractant receptors for inflammation induced ligands. Transendothelial migration involves specific adhesive interactions between T cells and endothelial cells (ECs) that guide the lymphocytes from the vascular compartment to the extravascular tissue. We refer the interested reader to excellent reviews on this topic (16–20). In brief, T cells undergo four distinct adhesion steps during their migration through blood vessels. These include tethering, rolling, activation, and arrest. Tethering and rolling of leukocytes are mediated by interactions between selectins and specific carbohydrate moieties bound to a protein backbone (21), which allow rapid engagement with high tensile strength. The selectins are a family of three C-type lectins expressed by bone marrow-derived cells and ECs. l-selectin (CD62L) is expressed by all myeloid cells, naïve T cells, and some activated and memory cells. P-selectin (CD62P) is found in secretory granules of platelets and ECs and is expressed on the cell surface after activation by inflammatory stimuli. E-selectin (CD62E) is expressed by acutely inflamed ECs in most organs and by non-inflamed skin microvessels. Thus, P-selectin glycoprotein ligand 1 (PSGL-1) and CD43 on activated T cells engage CD62P and CD62E on activated ECs, respectively. Rolling T cells receive signals from chemokines on ECs, which induce modulation of integrins to acquire high avidity for their ligands. Integrins may participate to the rolling phase but are essential for the firm adhesion of leukocytes. In particular, activated T cells depend on lymphocyte function-associated antigen 1 (LFA-1), very late antigen-4 (VLA-4; α4β1), and α4β7 for their interactions with activated ECs that express intracellular adhesion molecule 1 (ICAM-1), intracellular adhesion molecule 2 (ICAM-2), VCAM-1, and mucosal addressin-cell adhesion molecule type 1 (MAdCAM-1), respectively (22).
Quiescent ECs poorly interact with circulating leukocytes. Autacoid mediators released by mast cells and other cells of the innate immunity, upon stimulation by inflammatory signals (e.g., infection and tissue damage), cause rapid enhancement of venular permeability, translocation of integrins, and chemokines from intracellular stores to the cell surface and formation of a provisional matrix (23), all processes that favor T cell-EC interactions.
A very different scenario may characterize T cell-EC interactions in tumor-associated vessels.
Tumor-Associated Modifications of the Endothelium Hamper T Cell Adhesion, Extravasation, and Tumor Infiltration
While acute inflammation is an efficacious means by which the organism repairs a tissue that has been damaged by a physical insult or infection, chronic inflammation has emerged has indispensable requisite for chronic diseases including cancer (24). Indeed, tumor-promoting inflammation has been recently recognized has as an enabling characteristic that allows cancer cells to acquire multiple hallmark capabilities including sustaining proliferative signaling, resisting cell death, avoiding immune destruction, activating invasion, and metastasis and inducing angiogenesis (25). Virtually any neoplastic lesion contains immune inflammatory cells although with variable densities (26). Yet, gene expression profiling of the total cellular composition of tumors has evidenced at least two subsets of tumors. The first “inflamed” subset is characterized by transcripts encoding innate immune cell molecules, chemokines that can contribute to effector T cell recruitment, various T lineage-specific markers, and, paradoxically, immune inhibitory mechanisms. Conversely, the “not-inflamed” phenotype is distinguished for high expression of angiogenesis-associated factors as well as macrophages and fibroblasts (2). Thus, it has been hypothesized that TILs effectively extravasate in inflamed tumors but are inhibited by immunosuppressive mechanisms, including indoleamine-2,3-dioxygenase (IDO), programed cell death 1 ligand 1 (PD-L1), and forkhead box P3 (FoxP3)+ regulatory T cells (Tregs), whereas, T cell migration is defective in not-inflamed tumors (2).
More in general, vessels are irregularly distributed within the tumor mass that, when reaches 1–2 mm in diameter, presents a patchy distribution of less-perfused and hypoxic areas (27). Hypoxia is one of the strongest stimulators of angiogenesis, largely through the expression of hypoxia inducible transcription factors [HIFs; (28)]. Tumor vessels that sprout from existing ones are disorganized, tortuous, dilated, saccular, and leaker then the normal ones. Also the composition of the vessel is abnormal, and ECs may acquire aberrant morphology, pericytes may be absent or loosely attached, and the vessel may lack basement membrane or have it unusually thick (13, 29). In addition, tumor cells may mimic ECs and generate vascular conduits, which however, are even more abnormal (30). All together, these vascular abnormalities render tumor vessels leaker then normal ones, may increase the interstitial pressure, cause heterogeneous permeability, and promote irregular blood flow, therefore making leukocyte trafficking within the tumor mass difficult. Interstitial pressure is also increased by the extrinsic compression of tumor vessels by proliferating cancer cells. In addition, angiogenic factors such as vascular EC growth factors (VEGFs) and fibroblast growth factors (FGFs) cause down-regulation of ICAM-1/2, VCAM-1, and CD34 on ECs, a phenomenon defined as “EC anergy” (31–33). Thus, the few effector T cells that circulate in tumor vessels, regardless of being induced in vivo by vaccination or adoptively transferred (34, 35), can hardly interact with ECs and begin their migration through blood vessels (Figure 2A). In line with this view, gene expression profiling and in situ immunohistochemical staining of large cohorts of cancer patients have shown that more aggressive tumors are characterized by peritumoral immune infiltrates (36), whereas a strong in situ accumulation of T cells both in the center of the tumor and the invading margin correlates with a favorable prognosis regardless of the local extent of the tumor and of metastasis (37).
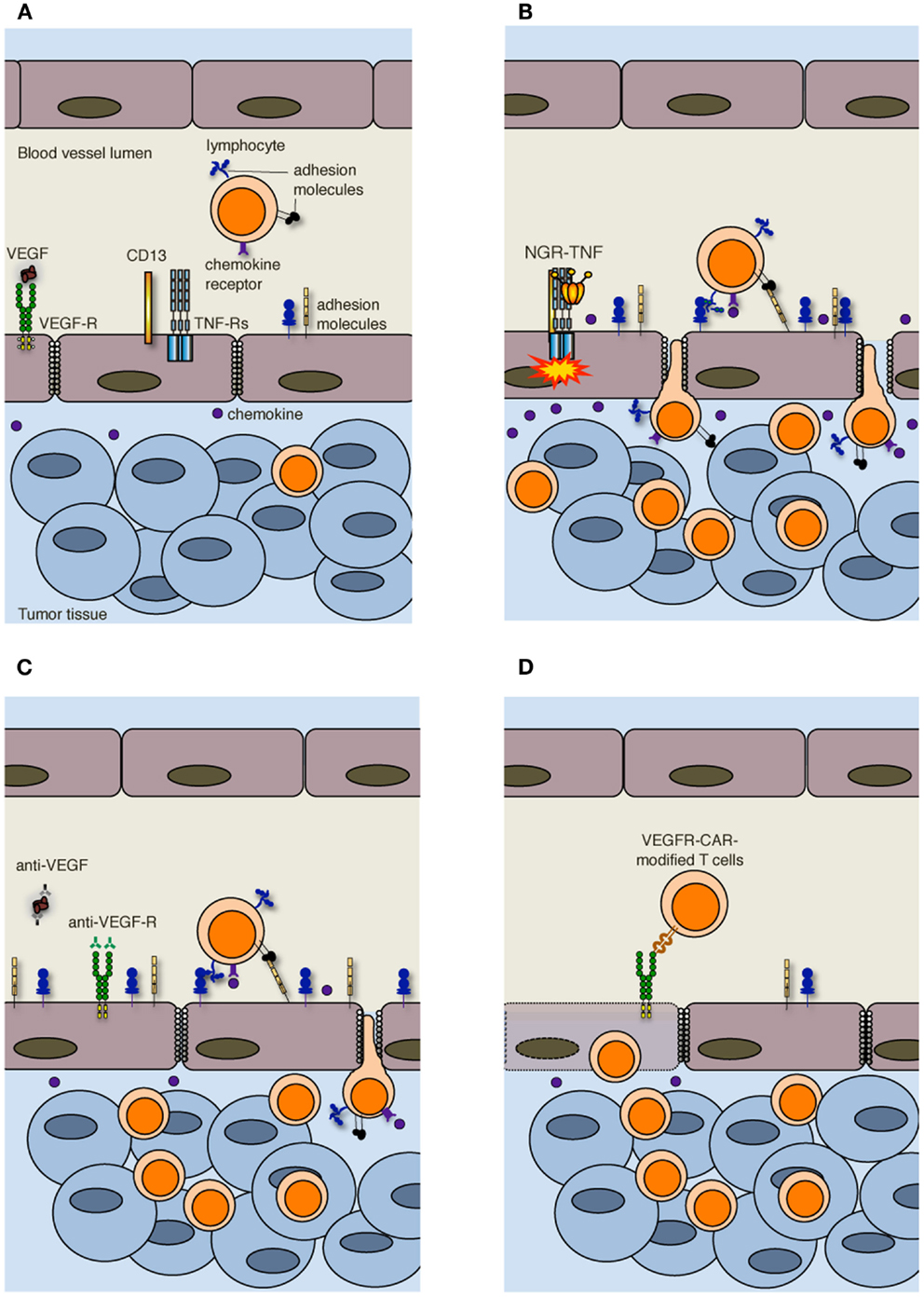
Figure 2. Strategies to increase T cell infiltration into tumors. (A) Increased interstitial pressure, heterogeneous permeability and irregular blood flow, together with reduced expression of adhesion molecules on ECs, limit lymphocyte penetration in tumors. (B) NGR-TNF, which selectively binds CD13 expressed in ECs of neo-angiogenic vessels and favors the interaction of TNF with TNF receptors (TNF-Rs), alters tumor vessel permeability by loosening VE-cadherin dependent adherence junctions, induces up-regulation of adhesion molecules in ECs, and elicits the release of pro-inflammatory cytokines and chemokines, thereby favoring the recruitment and extravasation of T lymphocytes. (C) Anti-VEGF and anti-VEGF-R antibodies both transiently normalize the tumor-vasculature and overcome EC anergy, thus favoring T cell trafficking within tumors. (D) Also immunization against VEGF-R2 or the adoptive transfer of autologous T cells genetically engineered to express chimeric antigen receptor targeted against VEGF-R2 (VEGFR-CAR) favor tumor infiltration by T cells, although the mechanism has not yet been clarified. It has been proposed that VEGF-R-specific T cells kill both ECs and MDSCs and Tregs (not shown) that express VEGF-R.
Ways to Favor T Cell Adhesion to Tumor-Associated Endothelial Cells, Extravasation, and Tumor Infiltration
Crossing the abnormal tumor vessel barrier and interstitium is one major hurdle for tumor-specific T cells that have reached the tumor mass (Figure 1). Few years ago, we proposed that delivery of vasoactive inflammatory cytokines like tumor necrosis factor α (TNF) to neo-angiogenic vessels might represent a good strategy to induce selective activation of ECs in tumor tissues, thereby enhancing T cell extravasation and tumor infiltration (38). TNF is produced in the tumor microenvironment mainly by macrophages, but also by smooth muscle cells, ECs and tumor cells (39), and it affects primarily the tumor-associated vasculature (40). Indeed, most tumor cells and vessels of normal tissues are resistant to TNF (41). Depending on the amount of TNF that reaches the tumor mass, its effects range from EC activation, to increased vessel permeability, EC damage, and massive hemorrhagic necrosis (42). The in vivo effects of TNF have been well characterized both in pre-clinical models and in humans undergoing isolated limb perfusion, a regional cancer therapy used to deliver high doses of a drug into the bloodstream of a limb avoiding severe systemic side effects (42). An alternative strategy to avoid TNF-induced systemic toxicity is indeed to selectively target minute amounts of the cytokine to the tumor vessels. Selective delivery of TNF to tumor vessels has been achieved by fusing this cytokine with a tumor-vasculature-homing peptide that contains the Cys-Asn-Gly-Arg-Cys (NGR) sequence, a ligand of a CD13 isoform expressed by neo-angiogenic vessels (43, 44). The new moiety called NGR-TNF was shown to transiently enhance tumor vessel permeability (45), thus increasing the penetration of chemotherapy agents in murine models of lymphoma, melanoma, and spontaneous prostate cancer without TNF-related systemic toxicity (46, 47). NGR-TNF is currently under clinical investigation in various clinical studies in cancer patients (48).
In accordance with our original hypothesis (38), we have recently shown that extremely low doses of NGR-TNF (5 ng/Kg) are sufficient to induce the up-regulation of VCAM-1 and ICAM-2 on the endothelial lining of tumor vessels as well as the release, in the tumor microenvironment, of chemokines that favor T-cell trafficking (Figure 2B). Rapid and transient modification of the tumor microenvironment can enhance the infiltration of either fully activated endogenous or adoptively transferred T cells in transplantable melanoma and autochthonous prostate cancer (49). Additionally, we have demonstrated that NGR-TNF can increase the therapeutic efficacy of tumor vaccines and adoptive immunotherapy with no evidence of toxic reactions (49). The effects of NGR-TNF on tumor infiltration by leukocytes go beyond the transient activation of tumor-associated ECs (50). Indeed, NGR-TNF transiently modifies the endothelial barrier function by loosening VE-cadherin dependent adherence junctions (51), thus favoring T cell extravasation (Figure 2B). It can also transiently reduce hypoxic areas of the tumor (52) and favor TIL proliferation and survival (50).
A similar compound, consisting of TNF fused to another tumor-vasculature-homing peptide (RGR) has been recently shown to stabilize tumor vessels and to enhance active immunotherapy in experimental pancreatic neuroendocrine tumors (52). Notably, a recent phase II study of NGR-TNF (0.8 μg/m2) in combination with doxorubicin in relapsed ovarian cancer patients showed that patients with baseline peripheral blood lymphocyte count higher than the first quartile had improved progression-free survival and overall survival (53), therefore suggesting that a similar effect may occur in humans.
Other strategies have been pursed to target TNF to the tumor. As an example, TNF has been fused with the single chain Fv Ab L19, which is specific for the extradomain B of fibronectin expressed by the tumor neovasculature (54). However, the location of the target molecules in tumor vessels and their level of expression are different from that of CD13, and additional studies are necessary to investigate whether this compound acts in synergy with active or adoptive immunotherapy.
Leukocyte infiltration in tumors can also be favored by the use of classic anti-angiogenic drugs. VEGF is the focus of most of these approaches (55). The importance of VEGF-mediated mechanisms in cancer is underlined by clinical data showing that the expression of VEGF in tumor tissue is negatively correlated with the presence of TILs. This was reported to be one of the strongest prognostic factors in ovarian carcinoma (56). In addition, VEGF negatively regulates functional maturation of and antigen presentation by dendritic cells (DCs), favors the accrual and activity of cell populations with immunosuppressive functions including myeloid derived suppressor cells [MDSCs; (57)] and regulatory T cells [Tregs; (58)], and induces T cell apoptosis, therefore contributing to the immunosuppressive tumor microenvironment (59). Over the last decades several therapeutic approaches have been proposed to counteract VEGF and neoangiogenesis, such as anti-VEGF antibodies and tyrosine kinase inhibitors of multiple pro-angiogenic growth factor receptors (13). Inhibition of VEGF interaction with its receptors has been also reported to be at the basis of vessel “normalization” (29). Anti-angiogenic drugs transiently normalize the tumor-vasculature, pruning away immature and leaky vessels and remodeling the remaining vasculature. As a result, the enhanced oncotic pressure gradient together with decreased interstitial fluid pressure and hydrostatic pressure gradient facilitate delivery of oxygen, nutrients, and also chemotherapeutic agents into the tumor microenvironment (13). Some of these strategies can also overcome EC anergy and promote leukocyte infiltration in tumors [Figure 2C; (60–62)]. In addition, it has been reported that lower-dose of anti-VEGF (DC101; 10 mg/Kg), when compared with the standard high dose (40 mg/Kg), normalizes the tumor-vasculature, favors extravasation of T cells, reduces the fraction of MDSCs, and polarizes macrophages toward an M1 phenotype within the tumor mass (63). Thus, anti-angiogenic drugs and TNF targeting are conceptually different approaches, as the former aims at vessel normalization, whereas the latter exploits the cytokine as an inflammatory agent that induces vascular activation.
Alternative approaches to target the VEGF-VEGF receptor (VEGFR) pathway are immunization against VEGFR-2 (64) or the adoptive transfer of autologous T cells genetically engineered to express a chimeric antigen receptor targeted against VEGFR-2 [Figure 2D; (65)]. The simultaneous targeting of VEGFR-2 and TAAs by a mixture of genetically engineered T cells expressing a chimeric antigen receptor targeting VEGFR-2 and T cells expressing a TCR specific for a melanoma-associated TAA synergistically eradicated established melanoma tumors in mice and prolonged their tumor free survival (66). The mechanism behind this synergy is still under investigation, and the transduction of anti-VEGFR-2 CAR into TCR transgenic T cells did not enhance the therapeutic efficacy of adoptively transferred cells (66). Because of the extensive tumor necrosis induced by the adoptive transfer of T cells, vessels could not be investigated in these tumors (66). The authors favor the hypothesis that anti-VEGFR-2 T cells not only target ECs but also suppressor cell populations including MDSCs and Tregs that express VEGFR-2 (67, 68).
In general, anti-VEGF-mediated transient normalization of tumor vessels lasts between few days to a month (29). Unfortunately, the anti-angiogenic drugs available to date are not sufficiently selective in damaging only neo-angiogenic vessels. Risks of sustained and/or aggressive anti-angiogenic therapies are the unselected recruitment of pro-angiogenic inflammatory cells, and excessive trimming of vessels with inadequate delivery of oxygen and drugs. The latter effect may be dangerous also for highly vascularized tissues, including the cardiovascular, endocrine, and nervous systems (69).
As mentioned before, targeting TNF to the tumor vessels enhances tumor permeability to chemotherapeutic agents (48). We have recently reported that the combination of active or adoptive immunotherapy, vascular targeting, and chemotherapy act in synergy against melanoma (49). Our preliminary results also suggest that in the context of adoptive T cell therapy after hematopoietic stem cell transplantation (70, 71), NGR-TNF dramatically increases the infiltration of TILs into the prostate of mice affected by autochthonous prostate cancer (49) and contributes to tumor debulking (Mondino A. Personal communication).
Interestingly, chemotherapeutic agents, beside their effects in promoting anti-tumor immunity by inducing a more immunogenic death of cancer cells, increasing their sensitivity to immune effectors or depleting the tumor microenvironment of Treg cells and MDSCs (72, 73) have been shown to promote intratumor expression of chemokines attracting T cells (74).
Taken together, these findings support the concept that increasing T-cell traffic to the tumor, possibly in association with immunogenic chemotherapy, may be a valid strategy to enhance response to immunotherapy in cancer patients.
Reprograming Energy Metabolism in Cancer
Reprograming energy metabolism is an emerging hallmark of cancer closely linked to hypoxia and neoangiogenesis (25). Indeed, uncontrolled cell proliferation requires a continuous adjustment of energy metabolism in order to fuel cell growth and division also in the absence of adequate tumor perfusion (14). As early as in 1930, Otto Warburg showed that cancer cells craving for energy take up much more glucose than normal cells and mainly process it through aerobic glycolysis, the so-called “Warburg effect” (75). Curiously enough, also T cells that differentiate from the naïve to the effector state upregulate genes encoding glycolytic enzymes (76), but tumor cells incorporate 10- to 100-fold greater glucose than T cells over a fixed time period (77), thus suggesting a biased competition for glucose between cancer cells and activated T cells within the same microenvironment.
As summarized in Figure 3, a direct consequence of aerobic glycolysis is the production of lactate from pyruvate, and acidic metabolites that cause drop in extracellular pH (78), which may select for more aggressive acid-resistant clones and favor tumor invasion (14). Pyruvate decarboxylation within mitochondria causes the generation and subsequent release of CO2, which favors increased expression of carbonic anhydrase IX (CA-IX), a cancer-associated membrane bound isoform of the enzyme carbonic anhydrase that catalyzes the hydration of CO2 to bicarbonate and H+, thus contributing to acidify the extracellular microenvironment of tumors (79, 80). A low extracellular pH triggers the activation on tumor cell membranes of transporters that protect the cytosol from acidosis. In addition, hypoxia stabilizes the heterodimer HIF-1, which in turn induces the up-regulation of glucose transporters and CA-IX, thereby increasing acidity within the tumor microenvironment (80). As a result, while in normal tissues the extracellular pH is maintained around 7.4, in malignant tumors the pH can drop to values of 6.0 and less, with averages of 0.2–0.6 units lower than in normal tissues (81). The tumor-supporting role of low pH has been recently corroborated by the observation that pharmacologic inhibition of CA-IX or of the vacuolar H+-ATPases display antineoplastic effects (79, 82).
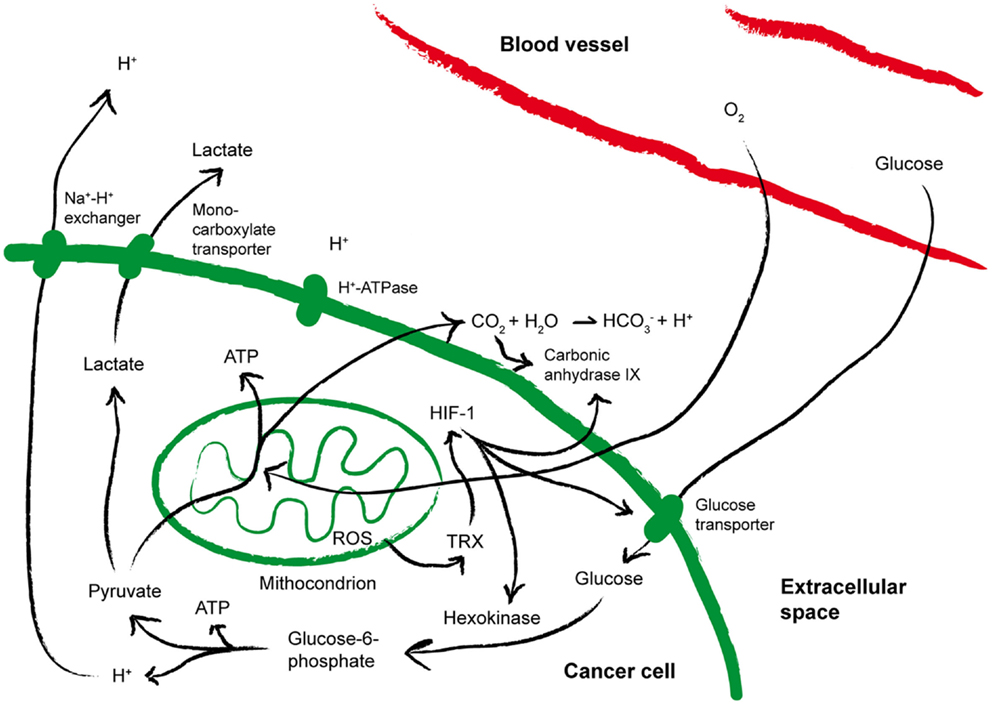
Figure 3. Metabolic alterations within the tumor microenvironment. The cartoon summarizes the metabolic alterations often found within the tumor microenvironment that may impact on T cell fitness. See the text for more details. ATP, Adenosine-5′-triphosphate; HIF-1, hypoxia inducible factor 1; ROS, reactive oxygen species; TRX, thioredoxin.
Hypoxia and pH are also strongly tangled with reduction-oxidation (redox) reactions (Figure 3). Already at the earliest stages of tumor development, free radicals, HIF-1-induced gene expression and hypoxia are strictly interconnected (83). Indeed, reactive oxygen species (ROS) are generated in mitochondria of cells exposed to low oxygen (84), and the phenomenon is further amplified by cyclic reoxygenation. Also the anti-oxidant systems upregulated by tumor cells to counterbalance oxidative stress contribute to the altered redox of the tumor microenvironment and to tumor progression. Overexpression of reducing enzymes such as thioredoxin (TRX) has been found in many tumors and correlated to poor prognosis (85, 86). TRX induces and stabilizes HIF-1α (87), and co-localizes with both HIF-1α and CA-IX in hypoxic areas of the tumors (79). In addition, proton pumps have been proposed to de-toxify tumor cells from microenvironmental ROS (88). Thus, hypoxia, acidosis, redox-remodeling can cooperate to establish a more aggressive malignant phenotype, and possibly to promote the derangement of immune functions (77).
Alterations of the Tumor Metabolism That Impact on T Cell Fitness
The immune system has been proposed as sensor of the metabolic state (89). Bidirectional communication and coordination between metabolism and immunity, while effective in maintaining and defending the internal environment from the environment around us, may result in inhibition of immune functions and may favor chronic inflammation and cancer. A well-known example of metabolism-mediated limitation of the function and survival of TILs is tryptophan consumption by tumor cells and antigen presenting cells (APCs) producing IDO (90). This mechanism can also restrain the therapeutic efficacy of checkpoint blockade strategies such as targeting of cytotoxic T lymphocyte antigen-4 (CTLA-4), glucocorticoid-induced TNFR family related gene (GITR), and the PD-1/PD-L1 axis (91).
More specifically, hypoxia, acidosis, and redox-remodeling are all perceived as sensors by the immune system. Thus, as summarized in Table 1, hypoxia inhibits TCR-triggered signaling, proliferation and cytokine production by T cells (92, 93). Intracellular HIF-1α appears to have a direct role in T cell inhibition, since HIF-1α is induced upon TCR triggering (94, 95), it is further increased in hypoxic conditions, and knocking down HIF-1α in T cells increases their cytokine production potential both in vitro and in vivo (94). T cells are also inhibited by hypoxia-driven accumulation of extracellular adenosine (96).
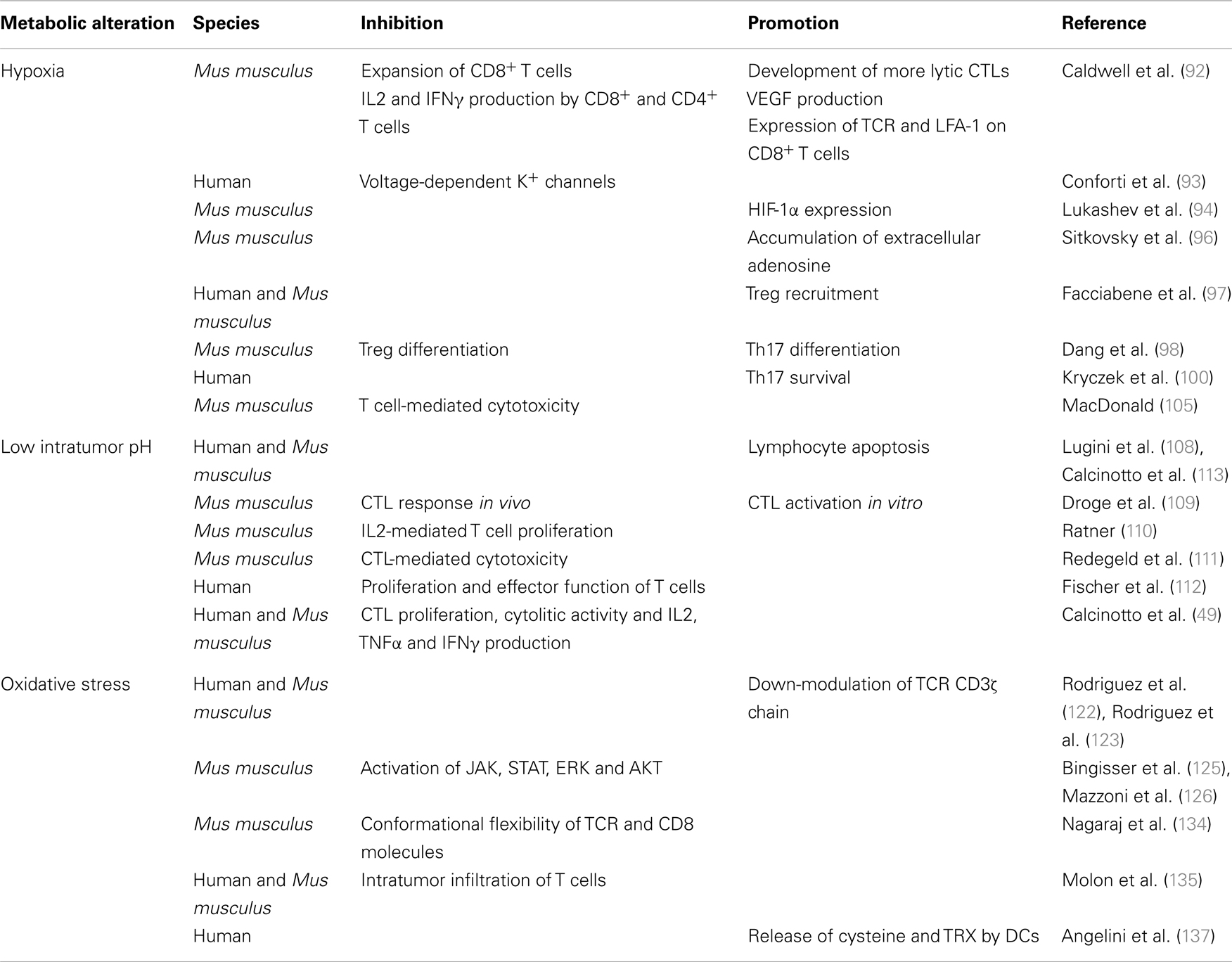
Table 1. Effects of the tumor metabolism on TILs.
More recently it has been reported that hypoxia within the tumor microenvironment promotes Treg recruitment through the induction of CC-chemokine ligand 28 (97). Conversely, up-regulation of HIF-1α under hypoxic conditions inhibits Treg differentiation through FoxP3 degradation, and favors the differentiation of Th17 cells by directly inducing RAR-related orphan receptor gamma t (RORγt) transcription (98) and glycolytic genes (99). HIF-1α also induces several survival promoting genes in Th17 cells, thus preventing their apoptosis (100). Th17 cells are a subpopulation of T helper cells producing IL17, IL17F, and IL22, which play a critical role in immunity to certain pathogens and autoimmune inflammation (101). The role of Th17 cells in cancer is more debated. Indeed, Th17 exert anti-tumorigenic activities, likely by facilitating the recruitment of other effector immune cells (102), and pro-tumorigenic activities by inducing tumor vascularization and the release of tumor-promoting factors by tumor and stromal cells (103). Thus, the effects of hypoxia on the tumor microenvironment are rather complex, and the use of HIF inhibitors for therapeutic purposes should be carefully balanced to avoid the dominance of pro-tumorigenic over anti-tumorigenic mechanisms.
Hypoxia may also render the tumor cells more resistant to cytotoxic T lymphocyte (CTL)-mediated lysis through HIF-1α-dependent induction in cancer cells of miR-210, which downregulates the expression of PTPN1, HOXA1, and TP53I11 genes (104). It remains to be defined how coordinated silencing of these three genes affects cancer cell susceptibility to CTL lysis. The effect of hypoxia on CTLs is still debated (77). Interestingly, simultaneous glucose deprivation and hypoxia block T cell-mediated cytotoxicity in vitro (105), therefore suggesting an additional mechanism of immunosuppression within the tumor microenvironment.
There are relatively few reports on the impact of low intratumor pH on T cells (106). Clinical evidence suggests that metabolic acidosis is often associated with immunodeficiency (107). Indeed, both leukocyte activation and the bactericidal capacity of leukocytes are generally impaired at reduced pH (106) suggesting that T cells could be extremely sensitive to pH variations. Lymphocytes also die at the same acidic pH malignant tumor cells perfectly remain alive (108). Droge et al. (109) studied the effect of lactate on murine T-cell populations and found that lactate is able to suppress the CTL response in vivo, whereas activation of CTLs in vitro is increased. Few years later, it was reported that the proliferation of IL-2-stimulated T cells is inhibited at pH 6.7 (110), and the cytolytic activity against cancer cells of CTLs is markedly reduced when T cells are exposed to acidic pH (111). More recently, Fischer and colleagues demonstrated that high lactic acid concentrations, as the ones found in the tumor environment, block lactic acid export in human T cells, thereby disturbing proliferation and effector functions (112).
We have found that lowering the pH in vitro to values most frequently detected within tumors (pH 6–6.5) induces hyporesponsiveness in both human and mouse tumor-specific CTLs, which is characterized by impaired proliferation, cytolytic activity, and cytokine secretion (113). Interestingly, buffering of culture pH to physiologic values associates with the complete recovery of T cell functions, although longer exposure or lower pH values causes permanent damage and T cell death (113), arguing that a portion of T cell immunity might be lost at tumor site when extreme metabolic alterations are present. From a molecular standpoint, TCR triggering at low pH associates with reduced expression of IL-2Rα (CD25) and TCR, and diminished activation of signal transducer and activator of transcription 5 (STAT5) and extracellular signal-regulated kinase (ERK) (113), signaling alterations frequently found in anergic T cells (114, 115). Interestingly, similar characteristics were found in tumor-specific CTLs infiltrating melanoma lesions, whose pH was 6.5 (113). Thus, acidity per se is a novel tumor cell extrinsic mechanism of immune escape (116).
Whereas redox-activated signaling events are physiologically needed both as antimicrobial defense and to guarantee the correct spatial and temporal extension of the immune reaction, redox-remodeling within the tumor microenvironment negatively affects immune surveillance. Indeed, oxygen ions and peroxides are potent antibacterial agents produced by phagocytic cells including macrophages and neutrophils (117). ROS are also implicated in NLRP3 inflammasome activation in myeloid cells (118). It has also been increasingly appreciated that endogenous ROS are required for optimal T cell activation (119). Yet, exogenous oxidative stress may dramatically suppress T cell activation and effector functions. As an example, macrophages within the tumor microenvironment express inducible nitric oxide synthase (iNOS) and can induce tumor killing by generating large amounts of nitric oxide (NO). However, iNOS is also expressed by MDSCs, a heterogeneous population of cells of myeloid origin that include immature macrophages, granulocytes, DCs and other myeloid cells (57). MDSCs also express arginase 1 (Arg1) that together with iNOS metabolizes the essential aminoacid arginine to either l-ornithine and urea, or to l-citrulline and NO (120, 121). Depletion of arginine from the microenvironment induces T cell dysfunction because of loss of CD3ζ chain expression (122, 123), and prevents the up-regulation of cell cycle regulators by these cells (124), thus blocking their proliferation. In addition, NO blocks the activation of Janus-activated kinase 1 (JAK1), JAK3, STAT5, ERK, and AKT (125, 126), thus suppressing several T cell functions (125–129).
Depletion of l-arginine may also trigger superoxide generation from iNOS (130, 131), which is eventually converted to hydrogen peroxide. ROS contribute to the MDSC-mediated suppression of tumor-specific T cell responses in tumor-bearing mice (57, 132).
Finally, the reaction between NO and generates reactive nitrogen-oxide species (RNOS), among which peroxynitrite (ONOO−) (133). ONOO− mediated nitration of tyrosine residues in the TCR and CD8 co-receptor causes a decreased conformational flexibility of these molecules and failure in proper T cell activation (134). Nitration of chemokines also prevents intratumoral infiltration of antigen-specific T cells (135).
Also Tregs modulate the redox of the microenvironment by subtracting cysteine necessary to effector T cell, function (136). Indeed, DCs within the tumor microenvironment may have additional nutritional and redox-remodeling roles, since they reduce the extracellular microenvironment required for T cell activation by releasing cysteine and TRX (137). In the same vein, Tregs diminish glutathione synthesis in DCs and consume extracellular cysteine (138), thus remodeling extracellular redox.
Additional hypoxia-driven metabolic dysfunctions, leading to the accumulation of extracellular adenosine, further increased by Tregs (139, 140), could act in synergy with acidic pH in dampening T cell function through A2A adenosine receptor-driven cAMP intracellular accumulation (96).
All together these findings sustain the concept that hypoxia, nutrient deprivation, abnormal glycolysis, and low pH act in synergy crippling immune surveillance (Figure 1).
Also alterations of the lipid metabolism that occur in the tumor microenvironment might affect T cell functions [e.g., (141)], but direct in vivo evidences of this phenomenon are poor.
Strategies That Impact on Tumor Metabolism and Promote Fitness of Tumor Infiltrating Lymphocytes
Different therapeutic approaches have been proposed to modulate hypoxia, tumor acidity or redox, which directly or indirectly affect TIL viability and effector functions (Figure 1). Being the tumor microenvironment so complex and redundant, the risk remains that interfering with one metabolic pathway, thus inhibiting one pro-tumoral mechanism, may favor another. For the sake of brevity, we will touch upon some clarifying examples.
It has been reported that the anti-VEGF monoclonal antibody bevacizumab induces intratumoral hypoxia, likely through excessive vessel remodeling (142), thus increasing the population of cancer stem cells (CSCs) in human breast cancer xenografts (143), and promoting epithelial to mesenchymal transition (EMT) in a murine model of bevacizumab-resistant pancreatic cancer (144). Angiogenesis inhibitors targeting the VEGF pathway may also elicit tumor adaptation and progression to stages of greater malignancy, with heightened invasiveness and in some cases increased lymphatic and distant metastasis (145). Bevacizumab has also been shown to induce malignant traits through induction of paracrine factors, which recruited pro-angiogenic myeloid cells (146), whose phenotype is reminiscent of MDSCs. Thus, anti-angiogenic compounds while cutting nutritional support to tumor cells, may favor local hypoxia and MDSC accumulation.
Also sunitinib, a receptor tyrosine kinase inhibitor with anti-angiogenic properties (147), which has been recently approved for the treatment of metastatic renal cell carcinoma (148), induces hypoxia, through a yet undefined mechanism, and increase in CSCs (143). Interestingly, semaphoring 3A (Sema3A), an endogenous anti-angiogenic agent, counteracts sunitinib-induced tumor hypoxia, and Sema3A and sunitinib synergize to enhance survival of tumor-bearing mice (149). In addition, one cycle of treatment with sunitinib is sufficient to increase the proportion of type 1 T cells (150), likely by reducing MDSCs (151). These findings have been confirmed in mouse models of cancer, in which sunitinib reduced viability and proliferation of MDSCs (152) and their accumulation in tumors (153).
Several drugs have been identified that target HIF-1α, thus inhibiting angiogenesis (154). However, HIF-1 inhibitors may impact on balance between Treg and Th17 cells favoring the former (99, 155). Thus, further investigation is needed to fully elucidate the therapeutic potential of HIF-1 inhibitors in cancer patients.
Conversely, hypoxia can be skillfully utilized to selectively target TILs. Indeed, hypoxia induces expression of CD137 (4-1BB) on TILs, and low-dose intratumoral injections of agonist anti-CD137 monoclonal antibodies avoid systemic toxicity while achieving anti-tumor systemic effects (156). In addition, intratumoral anti-CD137 antibodies synergized with systemic blockade of PD-L1 (156).
Several strategies have been proposed to neutralize intratumor acidity and therefore affect TILs. Robey et al. (157) reported that oral treatment with NaHCO3 increased the extracellular pH of spontaneous metastases, inhibited cancer cell extravasation and colonization in mouse models of breast and prostate cancer. However, no information is available on the effects of systemic administration of bicarbonate on T cells and there is some concern related to the risk of metabolic alkalosis.
We obtained evidence that systemic administration of the PPI esomeprazole (12.5 mg/Kg) to tumor-bearing mice caused a rapid (within 60 min) increase in tumor pH, which associated with enhanced IFNγ production by TILs (113). Indeed, on a per cell basis, TILs in the tumor of PPI-treated mice produced more IFNγ than TILs from mice treated with vehicle (113). PPI treatment also increased phosphorylated ERK in TILs, thus giving molecular support to the PPI-mediated effect. As expected for a drug that is administered as a pro-drug and requires protonation at low pH (158), IFNγ production by T cells isolated from the spleen, lung, and kidney of mice treated either with PPIs or vehicle did not differ (113), thus suggesting that also the effects of PPIs on T cells are restricted to area of acidosis. PPIs also affected adoptively transferred T cells that reached the tumor, and PPI treatment increased the therapeutic potential of both active and adoptive immunotherapies (113). Because of the high selectivity for an acidic milieu and instability, PPIs can be safely used at high doses (158) as the one tested by us (113), which also affect tumor cells in vivo (159). Thus, PPI treatment may represent a promising strategy for recovering specific immunity and improving the efficacy of T cell-based cancer treatments.
PPIs are also known for their anti-oxidant and anti-inflammatory activities (160). Vacuolar proton pumps are expressed in the membrane of phagolysosomes of neutrophils, and lysosomal acidification is relevant for neutrophil oxidative burst. Thus, PPIs reduce release of ROS by neutrophils further impacting on the tumor microenvironment. Whiles the mechanism of action of PPIs on leukocytes is still under investigation (160), our data suggest that in vivo PPIs enhance anti-tumor activities of TILs (113, 116).
Cancer cells promote chronic autophagy as survival adaptation to the acidic microenvironment (161). Because at least in vitro autophagy can also be induced in tumor cells by PPIs (162), strategies might be devised to inhibit autophagy during PPI treatment, yet taking into account the potentially negative effects of autophagy inhibitors on TILs (163).
Regarding redox, it has been reported that the anti-diabetic drug metformin or the mTOR inhibitor rapamycin, restore catabolic mitochondrial fatty acid oxidation and favor the induction of memory T cells, thus increasing the therapeutic efficacy of cancer vaccines (141, 164, 165).
Several pre-clinical studies also support the use of A2A adenosine receptors (A2ARs) antagonists to increase T cell activity within the tumor microenvironment (166). As an example, the compounds ZM241385 or 1,3,7-trimethylxanthine (caffeine) showed to increase the anti-tumor activity of adoptively transferred T cells in mice bearing large tumors (167). Curiously, drinking coffee was found to correlate with significant decreased risk of cutaneous malignant melanoma only in women (167, 168), suggesting that caffeine may also impact on cancer immune surveillance.
Finally, therapeutic strategies targeting either Tregs (169, 170) or MDSCs (171, 172); (173, 174), collaborate in making the tumor microenvironment more permissive for TIL survival and anti-tumor activities. Interestingly, MDSCs impair T cell trafficking through down-regulation of CD62L on CD4 and CD8 T cells (175) and chemokine nitration (135). Thus, therapeutic strategies that block MDSCs accrual at the tumor site and their immunosuppressive function, and more specifically drugs interfering with chemokine nitration, are expected to significantly improve the efficacy of both active and adoptive immunotherapies.
Conclusion and Perspectives
Despite considerable progress over the last decade, the tumor microenvironment is an area of research that remains ripe for further investigation, especially with regard to the relentless and dynamic modifications in its cellular composition and metabolism.
Accumulating experimental evidence lends weight to the concept that the most effective therapeutic strategies against cancer will be the ones that consider the tumor and its microenvironment as a whole, and yet simultaneously and coordinately address several individual aspects of this complex system. So far, either chemotherapy, surgery or radiotherapy have been combined with either one of active immunotherapy and/or adoptive T cell therapy, checkpoint blockade strategies or drugs that modify the vascularization and metabolism of the tumor (38, 50, 116, 176–178), thus improving distribution and synergistic anti-cancer activity of drugs and T cells. A step forward will be to carefully devise multiple targeted therapies that simultaneously or subsequently attack tumor cells and the diverse aspects of the tumor microenvironment, and yet preserve the function of organs not involved by the neoplasm. Thus, it can be anticipated that adoptive and active immunotherapy given together with treatments that transiently normalize and/or activate tumor-associated ECs and drugs that impact on tumor metabolism and reduce the local immunosuppressive environment would greatly benefit cancer patients without causing relevant systemic toxic effects. As an example, in TRAMP mice affected by advanced prostate cancer, the combination of non-myeloablative total body irradiation, hematopoietic stem cell transplantation, infusion of donor mature lymphocytes, and tumor-specific vaccination overcomes tumor-specific T cell tolerance, prompts tumor debulking, and induces long-lasting tumor-specific memory response that protects mice from tumor recurrence (70). Interestingly, the addition of NGR-TNF at the peak of the vaccination-induced immune response favors penetration of activated T cells within the transformed prostate epithelium (49) and guarantees an even stronger anti-tumor activity (Mondino A. Personal communication). We are investigating the possibility to add PPIs to this already complex combined therapy to favor the anti-tumor activity of adoptively transferred and vaccine-induced T cells that have reached the prostate.
Given the outstanding results obtained with immune checkpoint blockers in cancer patients (179), it will be particularly interesting to investigate the therapeutic efficacy of their combination with metabolism and vessel modulators.
It is also important to underline that more is not always better (180). One example is the recent failure of a well-designed and carefully analyzed multi-institutional clinical trial in which 732 patients with previously untreated metastatic colorectal cancer were randomly assigned to receive the combination of capecitabine, oxaliplatin, and bevacizumab (i.e., a monoclonal antibody against VEGF) or the same three drugs with cetuximab (i.e., a monoclonal antibody directed against the epidermal growth factor receptor; EGFR) (181). The four-drug combination resulted in significantly shorter progression-free survival and inferior quality of life. A similarly negative effect was obtained when another anti-EGFR monoclonal antibody (i.e., panitumumab) was added to the combination of folinic acid, fluorouracil, oxaliplatin, and bevacizumab in previously untreated metastatic colorectal cancer patients (182).
Thus, additional investigation is needed to define the best settings for each combination approach. In this perspective, reliable animal models of human diseases remain instrumental.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Catia Traversari (MolMed, Milan, Italy), and Angelo Corti and Vincenzo Russo (San Raffaele Scientific Institute, Milan, Italy) for comments on the manuscript. Funding: This work was supported by grants from the Associazione Italiana per la Ricerca sul Cancro (AIRC; AIRC 5 × 1000) and the Ministero della Salute. Author contributions: Matteo Bellone and Arianna Calcinotto were both responsible for all aspects of the manuscript – from conception to approval of the final manuscript. We apologize to any investigators whose work has not been underlined in this review due to oversight or space limitations. Arianna Calcinotto conducted this study as partial fulfillment of her PhD in Molecular Medicine, Program in Molecular Oncology, Università Vita-Salute San Raffaele, Milan, Italy.
References
1. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi:10.1146/annurev.immunol.22.012703.104803
2. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol (2011) 23:286–92. doi:10.1016/j.coi.2010.11.013
3. Ogino S, Galon J, Fuchs CS, Dranoff G. Cancer immunology – analysis of host and tumor factors for personalized medicine. Nat Rev Clin Oncol (2011) 8:711–9. doi:10.1038/nrclinonc.2011.122
4. Benjamin DI, Cravatt BF, Nomura DK. Global profiling strategies for mapping dysregulated metabolic pathways in cancer. Cell Metab (2012) 16:565–77. doi:10.1016/j.cmet.2012.09.013
5. Charoentong P, Angelova M, Efremova M, Gallasch R, Hackl H, Galon J, et al. Bioinformatics for cancer immunology and immunotherapy. Cancer Immunol Immunother (2012) 61:1885–903. doi:10.1007/s00262-012-1354-x
6. Galon J, Pages F, Marincola FM, Thurin M, Trinchieri G, Fox BA, et al. The immune score as a new possible approach for the classification of cancer. J Transl Med (2012) 10:1. doi:10.1186/1479-5876-10-1
7. Klinke DJ II. An evolutionary perspective on anti-tumor immunity. Front Oncol (2012) 2:202. doi:10.3389/fonc.2012.00202
8. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi:10.1126/science.1235122
9. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med (2013) 19:747–52. doi:10.1038/nm.3161
10. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363:411–22. doi:10.1056/NEJMoa1001294
11. Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol (2012) 12:269–81. doi:10.1038/nri3191
12. Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med (2012) 4:132ra153. doi:10.1126/scitranslmed.3003761
13. Chung AS, Lee J, Ferrara N. Targeting the tumour vasculature: insights from physiological angiogenesis. Nat Rev Cancer (2010) 10:505–14. doi:10.1038/nrc2868
14. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
15. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol (2008) 8:74–80. doi:10.1038/nri2233
16. von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med (2000) 343:1020–34.
17. Ley K, Kansas GS. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat Rev Immunol (2004) 4:325–35. doi:10.1038/nri1351
18. Luster AD, Tager AM. T-cell trafficking in asthma: lipid mediators grease the way. Nat Rev Immunol (2004) 4:711–24. doi:10.1038/nri1438
19. Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity (2007) 26:17–27. doi:10.1016/j.immuni.2007.01.002
20. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol (2013) 13:309–20. doi:10.1038/nri3442
21. Lasky LA, Singer MS, Dowbenko D, Imai Y, Henzel WJ, Grimley C, et al. An endothelial ligand for l-selectin is a novel mucin-like molecule. Cell (1992) 69:927–38. doi:10.1016/0092-8674(92)90612-G
22. Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol (1993) 11:767–804. doi:10.1146/annurev.iy.11.040193.004003
23. Pober JS, Kluger MS, Schechner JS. Human endothelial cell presentation of antigen and the homing of memory/effector T cells to skin. Ann N Y Acad Sci (2001) 941:12–25. doi:10.1111/j.1749-6632.2001.tb03706.x
24. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi:10.1038/nature07201
25. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
26. Pages F, Galon J, Dieu-Nosjean MC, Tartour E, Sautes-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene (2010) 29:1093–102. doi:10.1038/onc.2009.416
27. Huang H, Bhat A, Woodnutt G, Lappe R. Targeting the ANGPT-TIE2 pathway in malignancy. Nat Rev Cancer (2010) 10:575–85. doi:10.1038/nrc2894
28. Dayan F, Mazure NM, Brahimi-Horn MC, Pouyssegur J. A dialogue between the hypoxia-inducible factor and the tumor microenvironment. Cancer Microenviron (2008) 1:53–68. doi:10.1007/s12307-008-0006-3
29. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science (2005) 307:58–62. doi:10.1126/science.1104819
30. Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer (2003) 3:411–21. doi:10.1038/nrc1092
31. Piali L, Fichtel A, Terpe HJ, Imhof BA, Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med (1995) 181:811–6. doi:10.1084/jem.181.2.811
32. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Res (1996) 56:1111–7.
33. Hellwig SM, Damen CA, Van Adrichem NP, Blijham GH, Groenewegen G, Griffioen AW. Endothelial CD34 is suppressed in human malignancies: role of angiogenic factors. Cancer Lett (1997) 120:203–11. doi:10.1016/S0304-3835(97)00310-8
34. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer (2008) 8:299–308. doi:10.1038/nrc2355
35. Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol (2009) 27:83–117. doi:10.1146/annurev.immunol.021908.132544
36. Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res (2011) 71:5601–5. doi:10.1158/0008-5472.CAN-11-1316
37. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi:10.1126/science.1129139
38. Bellone M, Mondino A, Corti A. Vascular targeting, chemotherapy and active immunotherapy: teaming up to attack cancer. Trends Immunol (2008) 29:235–41. doi:10.1016/j.it.2008.02.003
39. Bemelmans MH, Van Tits LJ, Buurman WA. Tumor necrosis factor: function, release and clearance. Crit Rev Immunol (1996) 16:1–11. doi:10.1615/CritRevImmunol.v16.i1.10
40. Manda T, Shimomura K, Mukumoto S, Kobayashi K, Mizota T, Hirai O, et al. Recombinant human tumor necrosis factor-alpha: evidence of an indirect mode of antitumor activity. Cancer Res (1987) 47:3707–11.
41. McIntosh JK, Mule JJ, Travis WD, Rosenberg SA. Studies of effects of recombinant human tumor necrosis factor on autochthonous tumor and transplanted normal tissue in mice. Cancer Res (1990) 50:2463–9.
42. ten Hagen TLM, Seynhaeve ALB, Eggermont AMM. Tumor necrosis factor-mediated interactions between inflammatory response and tumor vascular bed. Immunol Rev (2008) 222:299–315. doi:10.1111/j.1600-065X.2008.00619.x
43. Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res (2000) 60:722–7.
44. Curnis F, Arrigoni G, Sacchi A, Fischetti L, Arap W, Pasqualini R, et al. Differential binding of drugs containing the NGR motif to CD13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res (2002) 62:867–74.
45. Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13). Nat Biotechnol (2000) 18:1185–90. doi:10.1038/81183
46. Curnis F, Sacchi A, Corti A. Improving chemotherapeutic drug penetration in tumors by vascular targeting and barrier alteration. J Clin Invest (2002) 110:475–82. doi:10.1172/JCI200215223
47. Bertilaccio MT, Grioni M, Sutherland BW, Degl’Innocenti E, Freschi M, Jachetti E, et al. Vasculature-targeted tumor necrosis factor-alpha increases the therapeutic index of doxorubicin against prostate cancer. Prostate (2008) 68:1105–15. doi:10.1002/pros.20775
48. Marcucci F, Bellone M, Rumio C, Corti A. Approaches to improve tumor accumulation and interactions between monoclonal antibodies and immune cells. MAbs (2013) 5:34–46. doi:10.4161/mabs.22775
49. Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-alpha to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol (2012) 188:2687–94. doi:10.4049/jimmunol.1101877
50. Bellone M, Calcinotto A, Corti A. Won’t you come on in? How to favor lymphocyte infiltration in tumors. Oncoimmunology (2012) 1:986–8. doi:10.4161/onci.20213
51. Dondossola E, Gasparri AM, Colombo B, Sacchi A, Curnis F, Corti A. Chromogranin A restricts drug penetration and limits the ability of NGR-TNF to enhance chemotherapeutic efficacy. Cancer Res (2011) 71:5881–90. doi:10.1158/0008-5472.CAN-11-1273
52. van Laarhoven HW, Gambarota G, Heerschap A, Lok J, Verhagen I, Corti A, et al. Effects of the tumor vasculature targeting agent NGR-TNF on the tumor microenvironment in murine lymphomas. Invest New Drugs (2006) 24:27–36. doi:10.1007/s10637-005-4540-2
53. Lorusso D, Scambia G, Amadio G, Di Legge A, Pietragalla A, De Vincenzo R, et al. Phase II study of NGR-hTNF in combination with doxorubicin in relapsed ovarian cancer patients. Br J Cancer (2012) 107:37–42. doi:10.1038/bjc.2012.233
54. Borsi L, Balza E, Carnemolla B, Sassi F, Castellani P, Berndt A, et al. Selective targeted delivery of TNFalpha to tumor blood vessels. Blood (2003) 102:4384–92. doi:10.1182/blood-2003-04-1039
55. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov (2004) 3:391–400. doi:10.1038/nrd1381
56. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med (2003) 348:203–13. doi:10.1056/NEJMoa020177
57. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol (2012) 12:253–68. doi:10.1038/nri3175
58. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6:295–307. doi:10.1038/nri1806
59. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
60. Dirkx AEM, Oude Egbrink MGA, Castermans K, Van Der Schaft DWJ, Thijssen VLJL, Dings RPM, et al. Anti-angiogenesis therapy can overcome endothelial cell anergy and promote leukocyte-endothelium interactions and infiltration in tumors. FASEB J (2006) 20:621–30. doi:10.1096/fj.05-4493com
61. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF-secreting cancer immunotherapy. Clin Cancer Res (2006) 12:6808–16. doi:10.1158/1078-0432.CCR-06-1558
62. Shrimali RK, Yu Z, Theoret MR, Chinnasamy D, Restifo NP, Rosenberg SA. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer Res (2010) 70:6171–80. doi:10.1158/0008-5472.CAN-10-0153
63. Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A (2012) 109:17561–6. doi:10.1073/pnas.1215397109
64. Wei YQ, Wang QR, Zhao X, Yang L, Tian L, Lu Y, et al. Immunotherapy of tumors with xenogeneic endothelial cells as a vaccine. Nat Med (2000) 6:1160–6. doi:10.1038/80506
65. Chinnasamy D, Yu Z, Theoret MR, Zhao Y, Shrimali RK, Morgan RA, et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest (2010) 120:3953–68. doi:10.1172/JCI43490
66. Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res (2013) 73:3371–80. doi:10.1158/0008-5472.CAN-12-3913
67. Yang L, Debusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6:409–21. doi:10.1016/j.ccr.2004.08.031
68. Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, et al. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. Eur J Immunol (2010) 40:197–203. doi:10.1002/eji.200939887
69. Choueiri TK, Schutz FA, Je Y, Rosenberg JE, Bellmunt J. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J Clin Oncol (2010) 28:2280–5. doi:10.1200/JCO.2009.27.2757
70. Hess Michelini R, Freschi M, Manzo T, Jachetti E, Degl’Innocenti E, Grioni M, et al. Concomitant tumor and minor histocompatibility antigen-specific immunity initiate rejection and maintain remission from established spontaneous solid tumors. Cancer Res (2010) 70:3505–14. doi:10.1158/0008-5472.CAN-09-4253
71. Hess Michelini R, Manzo T, Sturmheit T, Basso V, Rocchi M, Freschi M, et al. Vaccine-instructed intratumoral IFN-gamma enables regression of autochthonous mouse prostate cancer in allogeneic T cell transplantation. Cancer Res (2013) 73(15):4641–52. doi:10.1158/0008-5472.CAN-12-3464
72. Lake RA, Robinson BW. Immunotherapy and chemotherapy – a practical partnership. Nat Rev Cancer (2005) 5:397–405. doi:10.1038/nrc1613
73. Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G. Immunological aspects of cancer chemotherapy. Nat Rev Immunol (2008) 8:59–73. doi:10.1038/nri2216
74. Abastado JP. The next challenge in cancer immunotherapy: controlling T-cell traffic to the tumor. Cancer Res (2012) 72:2159–61. doi:10.1158/0008-5472.CAN-11-3538
75. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8:519–30. doi:10.1085/jgp.8.6.519
76. Cham CM, Xu H, O’Keefe JP, Rivas FV, Zagouras P, Gajewski TF. Gene array and protein expression profiles suggest post-transcriptional regulation during CD8+ T cell differentiation. J Biol Chem (2003) 278:17044–52. doi:10.1074/jbc.M212741200
77. Cham CM, Gajewski TF. Metabolic mechanisms of tumor resistance to T cell effector function. Immunol Res (2005) 31:107–18. doi:10.1385/IR:31:2:107
78. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4:891–9. doi:10.1038/nrc1478
79. Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov (2008) 7:168–81. doi:10.1038/nrd2467
80. Chiche J, Ilc K, Laferriere J, Trottier E, Dayan F, Mazure NM, et al. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res (2009) 69:358–68. doi:10.1158/0008-5472.CAN-08-2470
81. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med (1997) 3:177–82. doi:10.1038/nm0297-177
82. Fais S, De Milito A, You H, Qin W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res (2007) 67:10627–30. doi:10.1158/0008-5472.CAN-07-1805
83. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer (2008) 8:425–37. doi:10.1038/nrc2397
84. Pani G, Giannoni E, Galeotti T, Chiarugi P. Redox-based escape mechanism from death: the cancer lesson. Antioxid Redox Signal (2009) 11:2791–806. doi:10.1089/ars.2009.2739
85. Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol (2006) 16:420–6. doi:10.1016/j.semcancer.2006.10.009
86. Ceccarelli J, Delfino L, Zappia E, Castellani P, Borghi M, Ferrini S, et al. The redox state of the lung cancer microenvironment depends on the levels of thioredoxin expressed by tumor cells and affects tumor progression and response to prooxidants. Int J Cancer (2008) 123:1770–8. doi:10.1002/ijc.23709
87. Csiki I, Yanagisawa K, Haruki N, Nadaf S, Morrow JD, Johnson DH, et al. Thioredoxin-1 modulates transcription of cyclooxygenase-2 via hypoxia-inducible factor-1alpha in non-small cell lung cancer. Cancer Res (2006) 66:143–50. doi:10.1158/0008-5472.CAN-05-1357
88. De Milito A, Iessi E, Logozzi M, Lozupone F, Spada M, Marino ML, et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res (2007) 67:5408–17. doi:10.1158/0008-5472.CAN-06-4095
89. Odegaard JI, Chawla A. The immune system as a sensor of the metabolic state. Immunity (2013) 38:644–54. doi:10.1016/j.immuni.2013.04.001
90. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol (2013) 34:137–43. doi:10.1016/j.it.2012.10.001
91. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J Exp Med (2013) 210:1389–402. doi:10.1084/jem.20130066
92. Caldwell CC, Kojima H, Lukashev D, Armstrong J, Farber M, Apasov SG, et al. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J Immunol (2001) 167:6140–9.
93. Conforti L, Petrovic M, Mohammad D, Lee S, Ma Q, Barone S, et al. Hypoxia regulates expression and activity of Kv1.3 channels in T lymphocytes: a possible role in T cell proliferation. J Immunol (2003) 170:695–702.
94. Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, et al. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol (2006) 177:4962–5.
95. Thiel M, Caldwell CC, Kreth S, Kuboki S, Chen P, Smith P, et al. Targeted deletion of HIF-1alpha gene in T cells prevents their inhibition in hypoxic inflamed tissues and improves septic mice survival. PLoS ONE (2007) 2:e853. doi:10.1371/journal.pone.0000853
96. Sitkovsky MV, Kjaergaard J, Lukashev D, Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin Cancer Res (2008) 14:5947–52. doi:10.1158/1078-0432.CCR-08-0229
97. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature (2011) 475:226–30. doi:10.1038/nature10169
98. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146:772–84. doi:10.1016/j.cell.2011.07.033
99. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208:1367–76. doi:10.1084/jem.20110278
100. Kryczek I, Wu K, Zhao E, Wei S, Vatan L, Szeliga W, et al. IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. J Immunol (2011) 186:4388–95. doi:10.4049/jimmunol.1003251
101. Weaver CT, Elson CO, Fouser LA, Kolls JK. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol (2013) 8:477–512. doi:10.1146/annurev-pathol-011110-130318
102. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity (2009) 31:787–98. doi:10.1016/j.immuni.2009.09.014
103. Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol (2010) 10:248–56. doi:10.1038/nri2742
104. Noman MZ, Buart S, Romero P, Ketari S, Janji B, Mari B, et al. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res (2012) 72:4629–41. doi:10.1158/0008-5472.CAN-12-1383
105. MacDonald HR. Energy metabolism and T-cell-mediated cytolysis. II. Selective inhibition of cytolysis by 2-deoxy-d-glucose. J Exp Med (1977) 146:710–9. doi:10.1084/jem.146.3.710
107. Kellum JA. Metabolic acidosis in patients with sepsis: epiphenomenon or part of the pathophysiology? Crit Care Resusc (2004) 6:197–203.
108. Lugini L, Matarrese P, Tinari A, Lozupone F, Federici C, Iessi E, et al. Cannibalism of live lymphocytes by human metastatic but not primary melanoma cells. Cancer Res (2006) 66:3629–38. doi:10.1158/0008-5472.CAN-05-3204
109. Droge W, Roth S, Altmann A, Mihm S. Regulation of T-cell functions by l-lactate. Cell Immunol (1987) 108:405–16. doi:10.1016/0008-8749(87)90223-1
110. Ratner S. Lymphocytes stimulated with recombinant human interleukin-2: relationship between motility into protein matrix and in vivo localization in normal and neoplastic tissues of mice. J Natl Cancer Inst (1990) 82:612–6. doi:10.1093/jnci/82.7.612
111. Redegeld F, Filippini A, Sitkovsky M. Comparative studies of the cytotoxic T lymphocyte-mediated cytotoxicity and of extracellular ATP-induced cell lysis. Different requirements in extracellular Mg2+ and pH. J Immunol (1991) 147:3638–45.
112. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi:10.1182/blood-2006-07-035972
113. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72:2746–56. doi:10.1158/0008-5472.CAN-11-1272
114. Grundstrom S, Dohlsten M, Sundstedt A. IL-2 unresponsiveness in anergic CD4+ T cells is due to defective signaling through the common gamma-chain of the IL-2 receptor. J Immunol (2000) 164:1175–84.
115. Wells AD, Walsh MC, Sankaran D, Turka LA. T cell effector function and anergy avoidance are quantitatively linked to cell division. J Immunol (2000) 165:2432–43.
116. Bellone M, Calcinotto A, Filipazzi P, De Milito A, Fais S, Rivoltini L. The acidity of the tumor microenvironment is a mechanism of immune escape that can be overcome by proton pump inhibitors. Oncoimmunology (2013) 2:e22058. doi:10.4161/onci.22058
117. Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest (1973) 52:741–4. doi:10.1172/JCI107236
118. Rubartelli A. Redox control of NLRP3 inflammasome activation in health and disease. J Leukoc Biol (2012) 92:951–8. doi:10.1189/jlb.0512265
119. Williams MS, Kwon J. T cell receptor stimulation, reactive oxygen species, and cell signaling. Free Radic Biol Med (2004) 37:1144–51. doi:10.1016/j.freeradbiomed.2004.05.029
120. Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J (1998) 336(Pt 1):1–17.
121. Bogdan C. Nitric oxide and the immune response. Nat Immunol (2001) 2:907–16. doi:10.1038/35098532
122. Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC. Regulation of T cell receptor CD3zeta chain expression by l-arginine. J Biol Chem (2002) 277:21123–9. doi:10.1074/jbc.M110675200
123. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64:5839–49. doi:10.1158/0008-5472.CAN-04-0465
124. Rodriguez PC, Quiceno DG, Ochoa AC. l-arginine availability regulates T-lymphocyte cell-cycle progression. Blood (2007) 109:1568–73. doi:10.1182/blood-2006-06-031856
125. Bingisser RM, Tilbrook PA, Holt PG, Kees UR. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J Immunol (1998) 160:5729–34.
126. Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol (2002) 168:689–95.
127. Lejeune P, Lagadec P, Onier N, Pinard D, Ohshima H, Jeannin JF. Nitric oxide involvement in tumor-induced immunosuppression. J Immunol (1994) 152:5077–83.
128. Bobe P, Benihoud K, Grandjon D, Opolon P, Pritchard LL, Huchet R. Nitric oxide mediation of active immunosuppression associated with graft-versus-host reaction. Blood (1999) 94:1028–37.
129. Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, et al. Nitric oxide plays a critical role in suppression of T-cell proliferation by mesenchymal stem cells. Blood (2007) 109:228–34. doi:10.1182/blood-2006-02-002246
130. Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem (1998) 273:25804–8. doi:10.1074/jbc.273.40.25804
131. Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol (2003) 170:270–8.
132. Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother (2006) 55:237–45. doi:10.1007/s00262-005-0048-z
133. Squadrito GL, Pryor WA. The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem Biol Interact (1995) 96:203–6. doi:10.1016/0009-2797(94)03591-U
134. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828–35. doi:10.1038/nm1609
135. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208:1949–62. doi:10.1084/jem.20101956
136. Yan Z, Garg SK, Kipnis J, Banerjee R. Extracellular redox modulation by regulatory T cells. Nat Chem Biol (2009) 5:721–3. doi:10.1038/nchembio.212
137. Angelini G, Gardella S, Ardy M, Ciriolo MR, Filomeni G, Di Trapani G, et al. Antigen-presenting dendritic cells provide the reducing extracellular microenvironment required for T lymphocyte activation. Proc Natl Acad Sci U S A (2002) 99:1491–6. doi:10.1073/pnas.022630299
138. Yan Z, Garg SK, Banerjee R. Regulatory T cells interfere with glutathione metabolism in dendritic cells and T cells. J Biol Chem (2010) 285:41525–32. doi:10.1074/jbc.M110.189944
139. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5′-adenosine monophosphate to adenosine. J Immunol (2006) 177:6780–6.
140. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med (2007) 204:1257–65. doi:10.1084/jem.20062512
141. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi:10.1038/nature08097
142. Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A (2011) 108:3749–54. doi:10.1073/pnas.1014480108
143. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A (2012) 109:2784–9. doi:10.1073/pnas.1018866109
144. Carbone C, Moccia T, Zhu C, Paradiso G, Budillon A, Chiao PJ, et al. Anti-VEGF treatment-resistant pancreatic cancers secrete proinflammatory factors that contribute to malignant progression by inducing an EMT cell phenotype. Clin Cancer Res (2011) 17:5822–32. doi:10.1158/1078-0432.CCR-11-1185
145. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15:220–31. doi:10.1016/j.ccr.2009.01.027
146. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol (2007) 25:911–20. doi:10.1038/nbt1323
147. Christensen JG. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Ann Oncol (2007) 18(Suppl 10):x3–10. doi:10.1093/annonc/mdm408
148. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol (2009) 27:3584–90. doi:10.1200/JCO.2008.20.1293
149. Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, et al. Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest (2012) 122:1832–48. doi:10.1172/JCI58976
150. Finke JH, Rini B, Ireland J, Rayman P, Richmond A, Golshayan A, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res (2008) 14:6674–82. doi:10.1158/1078-0432.CCR-07-5212
151. van Cruijsen H, Van Der Veldt AA, Vroling L, Oosterhoff D, Broxterman HJ, Scheper RJ, et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin Cancer Res (2008) 14:5884–92. doi:10.1158/1078-0432.CCR-08-0656
152. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15:2148–57. doi:10.1158/1078-0432.CCR-08-1332
153. Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, et al. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer (2011) 129:2158–70. doi:10.1002/ijc.25863
154. Pan F, Barbi J, Pardoll DM. Hypoxia-inducible factor 1: A link between metabolism and T cell differentiation and a potential therapeutic target. Oncoimmunology (2012) 1:510–5. doi:10.4161/onci.19457
155. Kryczek I, Zhao E, Liu Y, Wang Y, Vatan L, Szeliga W, et al. Human TH17 cells are long-lived effector memory cells. Sci Transl Med (2011) 3:104ra100. doi:10.1126/scitranslmed.3002949
156. Palazon A, Martinez-Forero I, Teijeira A, Morales-Kastresana A, Alfaro C, Sanmamed MF, et al. The HIF-1alpha hypoxia response in tumor-infiltrating T lymphocytes induces functional CD137 (4-1BB) for immunotherapy. Cancer Discov (2012) 2:608–23. doi:10.1158/2159-8290.CD-11-0314
157. Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res (2009) 69:2260–8. doi:10.1158/0008-5472.CAN-07-5575
158. Mullin JM, Gabello M, Murray LJ, Farrell CP, Bellows J, Wolov KR, et al. Proton pump inhibitors: actions and reactions. Drug Discov Today (2009) 14:647–60. doi:10.1016/j.drudis.2009.03.014
159. De Milito A, Canese R, Marino ML, Borghi M, Iero M, Villa A, et al. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int J Cancer (2010) 127:207–19. doi:10.1002/ijc.25009
160. Kedika RR, Souza RF, Spechler SJ. Potential anti-inflammatory effects of proton pump inhibitors: a review and discussion of the clinical implications. Dig Dis Sci (2009) 54:2312–7. doi:10.1007/s10620-009-0951-9
161. Wojtkowiak JW, Rothberg JM, Kumar V, Schramm KJ, Haller E, Proemsey JB, et al. Chronic autophagy is a cellular adaptation to tumor acidic pH microenvironments. Cancer Res (2012) 72:3938–47. doi:10.1158/0008-5472.CAN-11-3881
162. Marino ML, Fais S, Djavaheri-Mergny M, Villa A, Meschini S, Lozupone F, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis (2010) 1:e87. doi:10.1038/cddis.2010.67
163. Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev (2012) 249:176–94. doi:10.1111/j.1600-065X.2012.01141.x
164. Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity (2011) 34:541–53. doi:10.1016/j.immuni.2011.04.006
165. Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W, et al. Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol (2012) 188:3080–7. doi:10.4049/jimmunol.1103365
166. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A (2006) 103:13132–7. doi:10.1073/pnas.0605251103
167. Stensvold I, Jacobsen BK. Coffee and cancer: a prospective study of 43,000 Norwegian men and women. Cancer Causes Control (1994) 5:401–8. doi:10.1007/BF01694753
168. Veierod MB, Thelle DS, Laake P. Diet and risk of cutaneous malignant melanoma: a prospective study of 50,757 Norwegian men and women. Int J Cancer (1997) 71:600–4. doi:10.1002/(SICI)1097-0215(19970516)71:4<600::AID-IJC15>3.3.CO;2-C
169. Colombo MP, Piconese S. Regulatory-T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer (2007) 7:880–7. doi:10.1038/nrc2250
170. Chaudhry A, Rudensky AY. Control of inflammation by integration of environmental cues by regulatory T cells. J Clin Invest (2013) 123:939–44. doi:10.1172/JCI57175
171. Ugel S, Delpozzo F, Desantis G, Papalini F, Simonato F, Sonda N, et al. Therapeutic targeting of myeloid-derived suppressor cells. Curr Opin Pharmacol (2009) 9:470–81. doi:10.1016/j.coph.2009.06.014
172. Filipazzi P, Huber V, Rivoltini L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother (2012) 61:255–63. doi:10.1007/s00262-011-1161-9
173. Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi:10.1111/j.1600-065X.2008.00608.x
174. Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer J (2010) 16:348–53. doi:10.1097/PPO.0b013e3181eb3358
175. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate l-selectin expression on CD4+ and CD8+ T cells. J Immunol (2009) 183:937–44. doi:10.4049/jimmunol.0804253
176. Terme M, Ullrich E, Delahaye NF, Chaput N, Zitvogel L. Natural killer cell-directed therapies: moving from unexpected results to successful strategies. Nat Immunol (2008) 9:486–94. doi:10.1038/ni1580
177. Rigamonti N, Bellone M. Prostate cancer, tumor immunity and a renewed sense of optimism in immunotherapy. Cancer Immunol Immunother (2012) 61:453–68. doi:10.1007/s00262-012-1216-6
178. Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol (2013) 3:154. doi:10.3389/fonc.2013.00154
179. Lipson EJ. Re-orienting the immune system: durable tumor regression and successful re-induction therapy using anti-PD1 antibodies. Oncoimmunology (2013) 2:e23661. doi:10.4161/onci.23661
180. Mayer RJ. Targeted therapy for advanced colorectal cancer – more is not always better. N Engl J Med (2009) 360:623–5. doi:10.1056/NEJMe0809343
181. Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med (2009) 360:563–72. doi:10.1056/NEJMoa0808268
Keywords: cytotoxic T lymphocyte, vaccine, adoptive T cell therapy, pH, redox, proton pump inhibitor, NGR-TNF, combination therapy
Citation: Bellone M and Calcinotto A (2013) Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 3:231. doi: 10.3389/fonc.2013.00231
Received: 27 June 2013; Paper pending published: 12 July 2013;
Accepted: 23 August 2013; Published online: 11 September 2013.
Edited by:
Ronald Berenson, Compliment Corporation, USAReviewed by:
Carine Michiels, University of Namur, BelgiumMarc Poirot, Institut National de la Santé et de la Recherche Médicale, France
Copyright: © 2013 Bellone and Calcinotto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Bellone, Cellular Immunology Unit, San Raffaele Scientific Institute, Via Olgettina 58, Milan 20132, Italy e-mail: bellone.matteo@hsr.it