 Katie L. Bailey
Katie L. Bailey Mark A. Carlson
Mark A. Carlson- 1Department of Surgery, University of Nebraska Medical Center, Omaha, NE, United States
- 2Department of Surgery and Department of Genetics, Cell Biology and Anatomy, University of Nebraska Medical Center, Omaha, NE, United States
- 3Department of Surgery, VA Nebraska-Western Iowa Health Care System, Omaha, NE, United States
Pancreatic cancer is the fourth most common cause of cancer-related deaths in both men and women. The 5-year survival rate for metastatic pancreatic cancer is only 8%. There remains a need for improved early diagnosis and therapy for pancreatic cancer. Murine models are the current standard for preclinical study of pancreatic cancer. However, mice may not accurately reflect human biology because of a variety of differences between the two species. Remarkably, only 5–8% of anti-cancer drugs that have emerged from preclinical studies and entered clinical studies have ultimately been approved for clinical use. The cause of this poor approval rate is multi-factorial, but may in part be due to use of murine models that have limited accuracy with respect to human disease. Murine models also have limited utility in the development of diagnostic or interventional technology that require a human-sized model. So, at present, there remains a need for improved animal models of pancreatic cancer. The rationale for a porcine model of pancreatic cancer is (i) to enable development of diagnostic/therapeutic devices for which murine models have limited utility; and (ii) to have a highly predictive preclinical model in which anti-cancer therapies can be tested and optimized prior to a clinical trial. Recently, pancreatic tumors were induced in transgenic Oncopigs and porcine pancreatic ductal cells were transformed that contain oncogenic KRAS and p53-null mutations. Both techniques to induce pancreatic tumors in pigs are undergoing further refinement and expansion. The Oncopig currently is commercially available, and it is conceivable that other porcine models of pancreatic cancer may be available for general use in the near future.
Background: Pancreatic Cancer
Pancreatic cancer (PC) is the twelfth most common cancer worldwide, with 460,000 new cases reported in 2018 (1). In the United States alone, it is estimated there will be 55,000 new cases of PC diagnosed in 2018, and 44,000 people with succumb to the disease (1). Over the last 40 years the demographic most affected by PC has been white men over the age of 60 (2). One of the main risk factors associated with development of PC is smoking, which is associated with a two-fold increase in incidence (2). Even with advances in our understanding of PC, the incidence has been rising ~0.5% each year over the last 10 years (2), and the 5-year survival rates in localized, regional (nodal spread), or metastatic disease have been 29, 11, and 2.6%, respectively (1–3). By 2030, PC is expected to be the second-leading cause of cancer mortality, which primarily is due to late presentation of symptoms and typically advanced disease stage at the time of diagnosis (2). Therefore, we need to improve our methods for diagnosing, detecting, and treating pancreatic cancer.
Current and Emerging Treatment Trends for PC
The current treatment paradigm for PC involves surgery, radiotherapy, and chemotherapy (2, 4). Operative resection is still the preferred treatment for resectable tumors. Advancement in surgical and imaging technology likely contributed to a slight decrease in PC mortality in the early 2010's (2). In 1996, the first line treatment for patients with metastatic PC included gemcitabine (5). Combinational studies using gemcitabine with other agents failed to improve survival further until nab-paclitaxel was added (6, 7), which increased the median overall survival by 1.7 months compared to gemcitabine alone. However, this combination regimen has toxicity which excludes PC patients that have a poor performance status (6, 7). Another treatment option for PC is FOLFIRINOX (5-fluorouracil, irinotecan, and oxaliplatin), which resulted in a 4.3-month survival benefit compared to gemcitabine alone (8). These two treatment options, FOLFIRNOX and gem/nab-p, are the current best therapies until disease progression. Second-line treatment options include nanoliposomal irinotecan and 5-FU (approved in 2015), which improved median overall survival by 1.9 months compared to 5-FU alone (9).
Emerging treatment options for PC patients includes tumor microenvironment targeting (including immunotherapies), gene therapy, and PARP inhibitors. All immunotherapies are still in the clinical trial phase, with the most advanced trial involving CXCessoR4, a combination study with anti-CXCR4 (chemokine receptor) and anti-PD-1 (programmed cell death protein, an immune checkpoint inhibitor) (6, 10). In an open-label phase 1b study in patients that had disease progression while under treatment, combinatory therapy with a CC-chemokine receptor 2 (CCR2) kinase antagonist and FOLFIRINOX produced a tumor response in 49% of patients (6). A gene delivery system to deliver wild type p53 (SGT-53) into tumor cells is currently being tested in combination with gem/nab-p (6, 11). PARP inhibitors inactivate the repair mechanism for single-stranded DNA breaks (12, 13). These inhibitors induce cell death in tumors, and are given in combination with DNA-damaging agents. Clinical trials are currently underway for all of these emerging treatments for PC. For many of these novel therapeutic regimens, a highly-predictive preclinical model of PC might be helpful to assess and/or optimize the regimen prior to a clinical trial, which theoretically could reduce the risk of a failed clinical trial, thus decreasing (i) cost of drug development and (ii) strain on clinical resources. That is, a highly-predictive preclinical model of PC could streamline the drug development pipeline.
Current Animal Modeling of PC
Similar to many human diseases, the study of PC has been aided by the use of genetically-edited murine models. Hallmark genetic mutations that drive the progression of PC have been well characterized (14–19). Oncogenic KRAS activation has been observed in 95% of PC patients, with 99% of point mutations occurring at the G12 position (20). Murine models have been utilized to study KRAS and other genes involved with PC progression, including TP53, SMAD4, and CDKN2A (14, 18, 19, 21). Expression of the mutant KRASG12D in mice produced metastatic pancreatic tumors; duration of survival in these subjects decreased further with TP53 antagonism (22). TP53 is a well-known tumor suppressor that promotes apoptosis in response to cellular stress and DNA damage, and is mutated in 70% of PC patients (20). Furthermore, deletion of tumor suppressor genes (SMAD4 or CDKN2A) enhanced tumor growth in a KRASG12D murine pancreatic cancer model (23, 24).
Despite the progress in genetically-edited murine PC models, a basic issue persists in regard to the mouse's relative ability to recapitulate human disease, including progression of PC and response to therapy. The magnitude of this issue is difficult to quantity using the current biomedical literature, in which many laboratories are heavily invested in the utilization of murine models. To be clear, it is not the intent of this article to criticize or discourage the use of mice in biomedical research, but rather to echo other voices which have questioned the predictive ability of murine models (25–27), and to propose alternative solutions. There has been some indirect evidence of murine fallibility in modeling human disease in the low regulatory approval rate for therapeutics that actually have reached the clinical trial stage, which has been in the range of 5–8% (28, 29). There are many factors that contribute to this low drug approval rate, but one likely reason is the less-than-optimal predictive ability of some murine models (e.g., tumor xenografting into immunosuppressed mice) to determine the efficacy of various therapeutics in humans (30–37).
Rodents may not accurately reflect human biology due to differences in physiology, anatomy, immune response, and genetic sequence (26, 30, 31, 36). For example, there are a number of genes for which the genotype-phenotype correlation is different between mice and humans (Table 1). One of these genes is APC+/−, in which the human phenotype includes colorectal polyposis (leading to colorectal cancer); the murine APC+/− mutant, however, develops small intestinal polyps. In addition, current genetically-edited murine models of cancer have limited tumor heterogeneity and low intratumor mutation rates (43–45), which could limit the clinical relevance of these models and their ability to study tumor immunity and immunotherapy (45, 46). And finally, there is a practical limitation to using murine models in preclinical research: size. Specifically, the development of clinically-relevant diagnostic or interventional technology often is not feasible with murine models due to their small size.
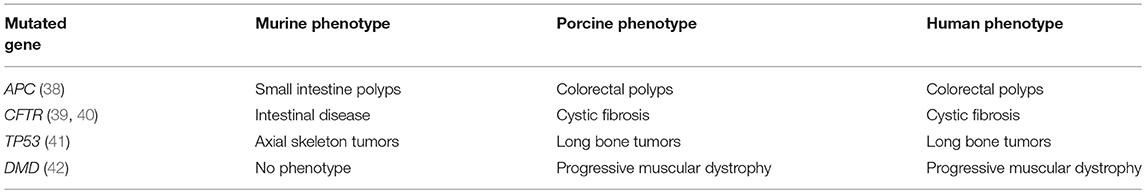
Table 1. Comparison of phenotypes from the same genetic mutations between mice, pigs, and humans.
In fairness, murine models are being continually refined for cancer research, including genetically-engineered mouse models (GEMMs) as described above, mice with humanized immune systems (i.e., immunodeficient mice engrafted with human hematopoietic stem cells), and in vivo site-directed CRISPR/Cas9 gene-edited mice (25, 31, 47–49). Bacterial microbiota models also have been utilized to demonstrate the effects of bacteria on cancer development and progression in murine models; however the role of the microbiome has not yet been studied in large animal models of cancer (50). Though promising, these more sophisticated murine models come with increased cost and complexity, and experience with them is still early. There remains a need for improved animal models of PC, including potential alternatives to mice, to better predict the human response to anti-cancer therapy. In addition, possession of an animal model of PC with human-sized organs would be helpful in regards to developing specific diagnostic and/or interventional technologies.
Rationale for a Large Animal Model of PC
As implied above, the rationale for utilizing a large animal model to study PC is to (i) have a platform for research and development of diagnostic/ therapeutic technologies that would not be feasible in murine models, and (ii) to have a highly-predictive preclinical model in which emerging anti-cancer therapies could be vetted and optimized prior to clinical trial. Some current large animal models that are used for biomedical research include non-human primates, dogs, and pigs. Non-human primates are the most “human-like,” but there are societal and ethical concerns involved with the use of these animals for research (51, 52). Similarly, utilization of dogs in biomedical research also can bring up social concerns due to their role as companion animals (53). However, secondary to their relatively long life expectancy as companions, dogs have had some utility in the study of treatments for natural/inherent (i.e., age associated) tumors, including mammary carcinoma, prostate carcinoma, lymphoma, and various sarcomas (54).
Due to their size similarity with humans, various strains of pig have been used for years in biomedical research to develop and refine surgical equipment, instrumentation, and techniques (55). In addition, swine have greater similarity to humans with respect to genomic, epigenetic, physiological, metabolic, and immunological characteristics when compared to the mouse-human similarities (56–60). Generally speaking, the homology between the human and porcine genome is greater than the homology between the human and murine genome. A quantitative indicator of this genomic homology is difficult to generate and depends on the chosen endpoints, a discussion of which is beyond the scope of this review (55). However, these homologies have been estimated at 80–90% (human-porcine) and 60–70% (human-murine) (56, 61–63). Porcine models have been utilized to study a wide range of fields, including physiology, trauma, wound healing, and atherosclerosis (55, 59, 64). Along with primates, swine have been a favored model to study transplantation (65). Human-pig concordance with regard to genotype-phenotype correlation is generally better than human-mouse concordance (Table 1). For example, the CFTR−/− and APC+/− mutants have the same basic phenotype in swine as in humans (38–40). Of note, a porcine genome map was generated in 2012, and further coverage, annotation, and confirmation is ongoing (60, 63, 66). Genetic manipulation of pigs (including knockouts, tissue-specific transgenics, inducible expression, and CRISPR editing), formerly done mostly in mice, has become more routine, with new gene-edited porcine models emerging for diseases such as atherosclerosis, cystic fibrosis, Duchenne muscular dystrophy, and ataxia telangiectasia (67–70).
Use of porcine models would offer other specific advantages. An animal research as large and robust as a pig would permit the testing of multiple, concurrent, clinically-relevant interventions, such as surgery, catheter-directed therapy, systemic chemotherapy, and/or radiotherapy; such combinatory interventions would have questionable feasibility in mice. Regarding the potential to study tumor biomarkers, the relatively large blood volume of a porcine PC model would allow for multiple blood samples to be drawn from the same pig during tumor development (a luxury not possible with the mouse), so precise timing and quantification of biomarker appearance could be correlated with tumor stage. This capability is not possible with a rodent model. On a similar note, immunotherapy study in a porcine PC model would be facilitated by the ability to obtain sufficient quantities of tumor-exposed immune cells that could be conditioned for re-infusion, e.g., as an autologous tumor-specific immunotherapy (71, 72). Furthermore, a porcine PC model could provide clinically-relevant tumor size/burden that would enable development and refinement of technologies to image and localize tumor for diagnosis, treatment, and surveillance (73). The relative size of the porcine subjects also would facilitate the sharing of tissue and blood sample with other investigators to a greater degree that could be accomplished with rodents. This effect would increase the potential number of investigators that could participate, the number of research protocols that could benefit, and the total amount of data that could be produced per research subject.
Of course, there are some caveats in using pigs to study PC. Specifically, the disadvantages of using a porcine model of PC with respect to a murine model include: (i) Husbandry and Cost. Depending on the swine strain utilized, the research subject could become quite large (>100 kg) if a prolonged (>1 year) latency is required for tumor development. Specialized equipment and experience would be necessary to handle such subjects. Husbandry is generally more cumbersome and expensive with swine as compared to mice. (ii) Biosafety. Biosafety issues, particularly when working with recombinant DNA technology, become more complex when the subject is a pig that is house in a pen, as opposed to a mouse inside a microisolator. (iii) Aged Subject Availability. While it is possible to work with aged murine subjects, and even elderly canine companion subjects, this is not really practical with swine, which potentially have a 20–30 year lifespan. Housing pigs for decades would be impractical, costly, and difficult, primarily due to the relatively large size of the mature subject (>150 kg for many strains). (iv) Reagents and Tools. Although use of swine in biomedical research has been growing, the availability of reagents and molecular tools specific for swine is not at the same level of availability that exists for mice. For example, the general availability of antibodies specific for porcine antigens is less than that for murine and human antigens. While difficult to quantify, in general this deficiency in porcine research is slowly improving. Of note, some anti-human antibodies will cross-react with porcine antibodies, but this has to be determined on a case-by-case basis. Secondary to these and/or other issues, it may not be practical or desirable for some research laboratories to utilize porcine models.
A Transgenic Approach to Porcine PC Modeling: the Oncopig Cancer Model
In 2012, the University of Illinois and the NSRRC (National Swine Resource and Research Center, nsrrc.missouri.edu) engineered a Cre-inducible swine model (the “Oncopig;” mini-pig background) (74) which carries an LSL-cassette containing dominant negative TP53 (R167H mutation) and activated KRAS (G12D mutation); i.e., the porcine analog of the KRAS/p53 mouse (22). This Cre-inducible system allows for the expression of both mutations in any cell within the pig. Upon addition of adenovirus expressing Cre recombinase (AdCre) to cultured Oncopig fibroblasts, expression of both mutant KRAS and TP53 was noted (74). The transformed fibroblasts had a shorter cell cycle length and demonstrated in vitro “tumorigenic” properties (increased cell migration, soft agar colony formation) and formation of tumors when injected into immunocompromised mice (74). Injection of AdCre into the subcutaneous/intramuscular regions of the Oncopig resulted in tumor formation with pleomorphic features (74). This transgenic pig hence became known as the Oncopig Cancer Model (OCM).
Primary pancreatic ductal cells were cultured from the OCM and then infected with AdCre; these epithelial cells also displayed a transformed phenotype in vitro, and expressed mutant KRAS and TP53 (75). These transformed epithelial cells were injected into SCID mice and formed subcutaneous tumors that were histologically and phenotypically similar to human pancreatic ductal adenocarcinoma (PDAC) (75). In vivo injection of AdCre directly into the main pancreatic duct of an Oncopig resulted in several nodular tumors after 12 months. Comparison of tumor induced in the OCM pancreas with human PDAC revealed similar morphological features, including a dense desmoplastic stromal reaction that is one key hallmark features of human PDAC (75). In addition, increased expression of proliferative markers (ERK and PCNA) was present in the OCM pancreatic tumor (75).
Key features of modeling PC with the OCM include: (1) the initial tumor induction is genetically defined; (2) the induced tumor is autochthonous; (3) the host has an intact immune system, which is capable of producing an anti-tumor immune response similar to humans, for studying immunotherapies (76); and (4) the tumor induction procedure (AdCre injection) is relatively simple and safe. However, there are some potential issues, such as specificity. Injection of AdCre theoretically could result in non-specific infection of multiple cell types, producing a pleomorphic tumor which could detract from the clinical relevance of the model. There also may an issue of tumor latency with pancreatic tumor in the OCM; in the initial report (75), pancreatic tumor formation required 12 months, and this was not visible on computed tomography nor was it clinically apparent. So, further refinement of the OCM for PC studies might be beneficial.
Orthotopic Approach: Transformed Porcine PDECs
In contrast to the autochthonous mechanism of tumor induction that the OCM provides, an orthotopic method of tumor induction involves seeding of tumorigenic cells into the pancreas, preferably into an immunocompetent host. In pursuit of this model type, primary cultures of porcine pancreatic ductal epithelial cells (PDECs) were established from explants of normal pancreatic tissue; IHC for cytokeratin-19 in early-passage strains were consistent with epithelial origin of the cultured cells (77). Strains of PDECs subsequently were infected with a lentiviral vector containing GFP, TP53R167H, and KRASG12D (LV-GKP; generated using porcine sequences), producing clones with demonstrable expression of mutant p53 and KRAS; refer to Table 2 (77). Initial in vitro tumorigenic assays of these clones (denoted as PGKP, for PDECs transformed with LV-GKP) demonstrated increases in migration and soft agar colony formation relative to primary PDECs (77). To further increase the transformed phenotype of the PGKP cells, RNAi of SMAD4 and CDKN2A were added using additional LV vectors, with ~70–90% knockdown (77). Relative to primary cells, these secondary clones (PKGPS and PGKPSC) also displayed increased proliferation, soft agar colony formation, invasion, and migration, i.e., evidence of in vitro “tumorigenicity” (77), with perhaps enhanced capabilities compared to the primary clone (PGKP cells). The three types of transformed PDECs (summarized in Table 2) were then implanted subcutaneously in nude mice; all three cell lines formed tumors and demonstrated equivalent in vivo tumorigenicity (77). In summary, PDEC-derived tumorigenic cell lines were established, which currently are undergoing orthotopic implantation into syngeneic, immunocompetent domestic swine.
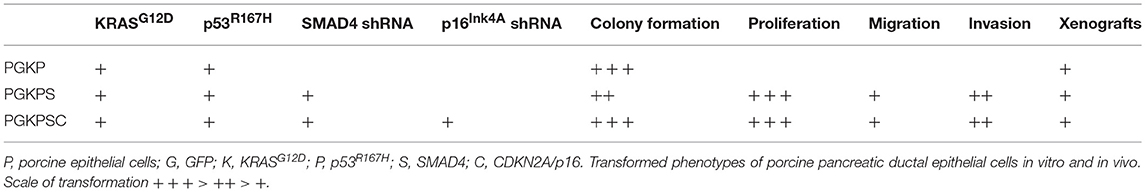
Table 2. Characteristics of transformed porcine ductal epithelial cells [data published as preprint (77)].
In terms of generating pancreatic tumor, the theoretical advantages of transformed PDEC implantation over AdCre injection in the OCM include: (i) Specificity. the former technique only involves transformed pancreatic ductal cells, meaning that tumor induced with transformed PDEC implantation would be more likely to originate from a specific cell type than tumor induced with AdCre injection in the OCM. (ii) Target Flexibility. Cell implantation permits the investigator to choose the targets by which transformation will be accomplished, instead of being restricted to mutant KRAS and TP53, as in the OCM. (iii) Host Flexibility. The investigator can choose the background strain of pig (or another species altogether) with cell implantation, while the OCM by definition involves one transgenic genotype. (iv) Cost. The purchase price of OCM subjects likely will be greater compared to most strains of research-quality pigs (though this cost differential becomes less of an issue in the face of multiple months of housing that these experiments would require).
On the other hand, the potential disadvantages of transformed PDEC implantation with respect to AdCre injection in the OCM include: (i) Immune Rejection. If allogeneic transformed PDECs are implanted, then there is the possibility that the host would reject the transplanted material (this issue might be minimized by utilizing syngeneic or autologous PDECs). (ii) Simplicity. AdCre injection into the OCM is straightforward and has potentially fewer Biosafety issues, as compared to pancreatic harvest, primary cell culture, and numerous viral transformations required for the PDEC implantation technique. (iii) Local Environment. As discussed above, tumor induction in the OCM is autochthonous, and likely does not involve local traumatic disruption of tissue architecture which presumably ensues when a cellular suspension is injected. However, the amount and biological relevance of local architecture disruption in these models is not known at this time.
Applications and Impact
The availability of a validated, genetically-defined porcine model of PC would have multiple potential applications, including (in no particular order):
1. Development and refinement of catheter-based technologies for diagnosis and/or intervention.
2. Discovery and study of serum tumor biomarkers (“liquid biopsy” technology).
3. A preclinical trial tool: a penultimate platform to test novel chemotherapeutic agents that were screened in murine models, prior to pushing a nascent therapy into an expensive clinical trial.
4. A platform for the testing of multiple, concurrent, clinically-relevant interventions, such as surgery, catheter-directed therapy, systemic chemotherapy, and/or radiotherapy (as described under the Rationale section).
5. Study of early events in tumor initiation and progression in an animal subject with a relatively high degree of genetic, physiological, metabolic, immune, and anatomic similarity with humans.
6. Detailed study of tumor heterogeneity (facilitated by a relatively large tumor specimen).
7. Study of the interactions and effects of the microbiome on tumor biology.
8. Development and refinement of tumor-visualization aids (such as fluorescent tumor agents) to assist with R0 resection in surgery.
9. Development and refinement of tools for open and minimally invasive surgery.
10. Refinement of existing imaging tools (such as MRI-based technologies) to diagnosis early stage tumors.
11. Development of novel tumor imaging tools.
12. An educational tool to instruct trainees in surgical resection techniques.
The primary impact of such a porcine PC model would be to increase the efficiency and safety at which impactful technologies and therapies could be brought into the clinical realm. For example, the anti-tumor effect and toxicity of a new chemotherapeutic regimen could be vetted in the porcine model, which could promote (or eliminate) the regimen's introduction into a clinical trial; this screening step likely would increase the probability of success for the human study. As another example, the feasibility, safety, and utility of a catheter-directed energy source in the treatment of PC could be accomplished in a porcine model without ever having to place a patient at risk. Another impact of a porcine model of PC would be an increased understanding of the molecular and cellular biology of the disease in an animal model that would have more relevance than the mouse.
Conclusion and Future Directions
Current murine models of PC have been tremendously helpful in the progression of understanding and treatment for this disease, but there is an ongoing issue of the relative predictive ability of these murine models. The issue of modeling accuracy likely has contributed in part to an unacceptably high failure rate of experimental therapeutics in clinical trials. Utilizing pigs to model PC has potential benefits, including relevant subject size, increased genetic homology, and better immunological/metabolic mimicry with respect to humans. Specifically, the size of pigs allows for improvement upon imaging and surgical techniques which is not possible with rodents. The OCM has already demonstrated that pancreatic tumor can be induced in the pig with histopathological features similar to human PC. This PDAC model will provide ways for improving early detection, imaging, and surgical techniques of PDAC by following the disease after a defined induction point. Even though the current OCM does have some limitations due to the amount of time it takes to develop tumors, this model potentially could be refined to accelerate tumor growth; for example, by introducing additional edits within the Cre-recombinated cells that would inhibit DNA repair and promote genomic instability, or by generating a tissue-specific inducible promoter for targeted initiation of cellular transformation upon AdCre administration. Another approach to generate a porcine PC model has been orthotopic implantation of transformed PDECs into the pancreas of the syngeneic, immunocompetent pigs. Additional approaches to pancreatic tumor induction in the pig might include direct pancreatic infection with viral vectors containing key tumor-associated gene sequences, in vivo CRISPR editing, or combinations of two or more of the technologies described herein. To address the issue of tumor induction in relatively young subjects, diet-induced metabolic syndrome could be used as an adjunctive measure, which likely would increase the physiological age of the subject (and mimic a common clinical co-morbidity). Work remains to be done in the development and validation of a tractable porcine model of PC. Once established, however, a porcine PC model should be a useful addition to the armamentarium of the PC researcher, and should be able to augment and/or complement work done with established murine models.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the National Cancer Institute and from the Fred and Pamela Buffett Cancer Center, and also with funds from the UNMC Department of Surgery.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. American Cancer Society. Key Statistics in Pancreatic Cancer. Atlanta: American Cancer Society. (2018). Available online at: https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html (Accessed October 29, 2018).
2. Saad AM, Turk T, Al-Husseini MJ, Abdel-Rahman O. Trends in pancreatic adenocarcinoma incidence and mortality in the United States in the last four decades; a SEER-based study. BMC Cancer. (2018) 18:688. doi: 10.1186/s12885-018-4610-4
3. SEER (Surveillance Epidemiology and End Results Program) Stat Fact Sheets: Pancreas Cancer. National Cancer Institute. Available online at: https://www.seer.cancer.gov/statfacts/html/pancreas.html
4. National Comprehensive Cancer Network. Pancreatic Adenocarcinoma, Version 1.2018. In: NCCN Clinical Practice Guidelines (NCCN Guidelines®). (2018). Available online at: www.nccn.org (Accessed April 27 2018).
5. Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. (1997) 15:2403–13. doi: 10.1200/JCO.1997.15.6.2403
6. Manji GA, Olive KP, Saenger YM, Oberstein P. Current and emerging therapies in metastatic pancreatic cancer. Clin Cancer Res. (2017) 23:1670–8. doi: 10.1158/1078-0432.CCR-16-2319
7. Rajeshkumar NV, Yabuuchi S, Pai SG, Tong Z, Hou S, Bateman S, et al. Superior therapeutic efficacy of nab-paclitaxel over cremophor-based paclitaxel in locally advanced and metastatic models of human pancreatic cancer. Br J Cancer. (2016) 115:442–53. doi: 10.1038/bjc.2016.215
8. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
9. Wang-Gillam A, Li CP, Bodoky G, Dean A, Shan YS, Jameson G, et al. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial. Lancet. (2016) 387:545–57. doi: 10.1016/S0140-6736(15)00986-1
10. Tavor SWI, Weiss I, Beider K, Wald H, Eizenber O, Pereg Y, et al. The CXCR4 antagnoist BL-8040 efficiently induces apoptosis and inhibits the survival of AML cells. Blood. (2013) 122:3939. doi: 10.1038/leu.2017.82
11. Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. (2013) 21:1096–103. doi: 10.1038/mt.2013.32
12. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. (2015) 33:244–50. doi: 10.1200/JCO.2014.56.2728
13. Melisi D, Ossovskaya V, Zhu C, Rosa R, Ling J, Dougherty PM, et al. Oral poly(ADP-ribose) polymerase-1 inhibitor BSI-401 has antitumor activity and synergizes with oxaliplatin against pancreatic cancer, preventing acute neurotoxicity. Clin Cancer Res. (2009) 15:6367–77. doi: 10.1158/1078-0432.CCR-09-0910
14. Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. (2009) 15:4674–9. doi: 10.1158/1078-0432.CCR-09-0227
15. Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. (1994) 8:27–32. doi: 10.1038/ng0994-27
16. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. (2008) 321:1801–6. doi: 10.1126/science.1164368
17. Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol. (2013) 15:2–8. doi: 10.1038/ncb2641
18. Oliver MH, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect Biol. (2010) 2:1. doi: 10.1101/cshperspect.a001008
19. Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. (1988) 16:7773–82. doi: 10.1093/nar/16.22.10952
20. Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte-Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers. (2017) 9:5. doi: 10.3390/cancers9050042
21. Donghui LKX, Wolff R, Abbruzzese JL. Pancreatic cancer. Lancet. (2004) 363:1049–57. doi: 10.1016/S0140-6736(04)15841-8
22. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
23. Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci USA. (2006) 103:5947–52. doi: 10.1073/pnas.0601273103
24. Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. (2006) 20:3130–46. doi: 10.1101/gad.1478706
25. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. (2015) 163:39–53. doi: 10.1016/j.cell.2015.08.068
26. Le Magnen C, Dutta A, Abate-Shen C. Optimizing mouse models for precision cancer prevention. Nat Rev Cancer. (2016) 16:187–96. doi: 10.1038/nrc.2016.1
27. Gould SE, Junttila MR, de Sauvage FJ. Translational value of mouse models in oncology drug development. Nat Med. (2015) 21:431–9. doi: 10.1038/nm.3853
28. Reichert JM, Wenger JB. Development trends for new cancer therapeutics and vaccines. Drug Discov Today. (2008) 13:30–7. doi: 10.1016/j.drudis.2007.09.003
29. Sharpless NE, Depinho RA. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat Rev Drug Discov. (2006) 5:741–54. doi: 10.1038/nrd2110
30. Begley CG, Ellis LM. Drug development: raise standards for preclinical cancer research. Nature. (2012) 483:531–3. doi: 10.1038/483531a
31. Cook N, Jodrell DI, Tuveson DA. Predictive in vivo animal models and translation to clinical trials. Drug Discov Today. (2012) 17:253–60. doi: 10.1016/j.drudis.2012.02.003
32. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. (2011) 8:210–21. doi: 10.1038/nrclinonc.2011.21
33. Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nat Rev Cancer. (2011) 11:135–41. doi: 10.1038/nrc3001
34. O'Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. (2006) 59:467–77. doi: 10.1002/ana.20741
35. Scott S, Kranz JE, Cole J, Lincecum JM, Thompson K, Kelly N, et al. Design, power, and interpretation of studies in the standard murine model of ALS. Amyotroph Lateral Scler. (2008) 9:4–15. doi: 10.1080/17482960701856300
36. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. (2013) 110:3507–12. doi: 10.1073/pnas.1222878110
37. Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. (2007) 170:793–804. doi: 10.2353/ajpath.2007.060929
38. Flisikowska T, Merkl C, Landmann M, Eser S, Rezaei N, Cui X, et al. A porcine model of familial adenomatous polyposis. Gastroenterology. (2012) 143:1173–5 e7. doi: 10.1053/j.gastro.2012.07.110
39. Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. (2012) 487:109–13. doi: 10.1038/nature11130
40. Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science. (2008) 321:1837–41. doi: 10.1126/science.1163600
41. Saalfrank A, Janssen KP, Ravon M, Flisikowski K, Eser S, Steiger K, et al. A porcine model of osteosarcoma. Oncogenesis. (2016) 5:e210. doi: 10.1038/oncsis.2016.19
42. Selsby JT, Ross JW, Nonneman D, Hollinger K. Porcine models of muscular dystrophy. ILAR J. (2015) 56:116–26. doi: 10.1093/ilar/ilv015
43. Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. (2015) 21:846–53. doi: 10.1038/nm.3915
44. McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. (2014) 156:1298–311. doi: 10.1016/j.cell.2014.02.031
45. McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci USA. (2016) 113:E6409–17. doi: 10.1073/pnas.1613601113
46. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
47. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature. (2014) 516:423–7. doi: 10.1038/nature13902
48. Singh M, Murriel CL, Johnson L. Genetically engineered mouse models: closing the gap between preclinical data and trial outcomes. Cancer Res. (2012) 72:2695–700. doi: 10.1158/0008-5472.CAN-11-2786
49. Zitvogel L, Pitt JM, Daillere R, Smyth MJ, Kroemer G. Mouse models in oncoimmunology. Nat Rev Cancer. (2016) 16:759–73. doi: 10.1038/nrc.2016.91
50. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. (2013) 13:800–12. doi: 10.1038/nrc3610
51. Abee CMK, Tardiff S, Morris T. Nonhuman Primates In Biomedical Research, Vol1: Biology and Management. 2nd ed. Amsterdam: Elsevier (2012).
52. Phillips KA, Bales KL, Capitanio JP, Conley A, Czoty PW, 't Hart BA, et al. Why primate models matter. Am J Primatol. (2014) 76:801–27. doi: 10.1002/ajp.22281
53. Hasiwa N, Bailey J, Clausing P, Daneshian M, Eileraas M, Farkas S, et al. Critical evaluation of the use of dogs in biomedical research and testing in Europe. ALTEX. (2011) 28:326–40. doi: 10.14573/altex.2011.4.326
54. Khanna C, Lindblad-Toh K, Vail D, London C, Bergman P, Barber L, et al. The dog as a cancer model. Nat Biotechnol. (2006) 24:1065–6. doi: 10.1038/nbt0906-1065b
55. Swindle MM, Smith AC. Swine in the Laboratory: Surgery, Anesthesia, Imaging, and Experimental Techniques. 3rd ed. Boca Raton, FL: CRC Press (2016).
56. Dawson HMP, Dayan A, Ganderup N, Hastings K. A comparative assessment of the pig, mouse and human genomes. Minipig Biomed Res. (2011) 2011:323–42. doi: 10.1201/b11356-28
57. Kuzmuk KS, Schook LB. Pigs as a Model for Biomedical Sciences. In: Rothschild MF, Ruvinsky A, editors. Genetics of the Pig. 2nd ed. Wallingford: CABI Publishing (2011). p. 426–38. doi: 10.1079/9781845937560.0426
58. Spurlock ME, Gabler NK. The development of porcine models of obesity and the metabolic syndrome. J Nutr. (2008) 138:397–402. doi: 10.1093/jn/138.2.397
59. Swindle MM, Makin A, Herron AJ, Clubb FJ Jr, Frazier KS. Swine as models in biomedical research and toxicology testing. Vet Pathol. (2012) 49:344–56. doi: 10.1177/0300985811402846
60. Walters EM, Wolf E, Whyte JJ, Mao J, Renner S, Nagashima H, et al. Completion of the swine genome will simplify the production of swine as a large animal biomedical model. BMC Med Genomics. (2012) 5:55. doi: 10.1186/1755-8794-5-55
61. Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. (2012) 20:50–7. doi: 10.1016/j.tim.2011.11.002
62. Suzuki Y, Yamashita R, Shirota M, Sakakibara Y, Chiba J, Mizushima-Sugano J, et al. Sequence comparison of human and mouse genes reveals a homologous block structure in the promoter regions. Genome Res. (2004) 14:1711–8. doi: 10.1101/gr.2435604
63. Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature. (2012) 491:393–8. doi: 10.1038/nature11622
64. Vodicka P, Smetana K, Dvoránková B, Emerick T, Xu YZ, Ourednik J, et al. The miniature pig as an animal model in biomedical research. Ann N Y Acad Sci. (2005) 1049:161–71. doi: 10.1196/annals.1334.015
65. Llore NP, Bruestle KA, Griesemer A. Xenotransplantation tolerance: applications for recent advances in modified swine. Curr Opin Organ Transplant. (2018) 23:642–8. doi: 10.1097/MOT.0000000000000585
66. National Center for Biotechnology Information. New pig (Sus scrofa) Genome Annotation in RefSeq. (2018). Available online at: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Sus_scrofa/106/ (Accessed November 6, 2018).
67. Adam SJ, Rund LA, Kuzmuk KN, Zachary JF, Schook LB, Counter CM. Genetic induction of tumorigenesis in swine. Oncogene. (2007) 26:1038–45. doi: 10.1038/sj.onc.1209892
68. Beraldi R, Meyerholz DK, Savinov A, Kovács AD, Weimer JM, Dykstra JA, et al. Genetic ataxia telangiectasia porcine model phenocopies the multisystemic features of the human disease. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:2862–70. doi: 10.1016/j.bbadis.2017.07.020
69. Leuchs S, Saalfrank A, Merkl C, Flisikowska T, Edlinger M, Durkovic M, et al. Inactivation and inducible oncogenic mutation of p53 in gene targeted pigs. PLoS ONE. (2012) 7:e43323. doi: 10.1371/journal.pone.0043323
70. Prather RS, Lorson M, Ross JW, Whyte JJ, Walters E. Genetically engineered pig models for human diseases. Annu Rev Anim Biosci. (2013) 1:203–19. doi: 10.1146/annurev-animal-031412-103715
71. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. (2014) 32:579–86. doi: 10.1200/JCO.2012.45.2011
72. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
73. Lee JW, Kang CM, Choi HJ, Lee WJ, Song SY, Lee JH, et al. Prognostic value of metabolic tumor volume and total lesion glycolysis on preoperative (1)(8)F-FDG PET/CT in patients with pancreatic cancer. J Nucl Med. (2014) 55:898–904. doi: 10.2967/jnumed.113.131847
74. Schook LB, Collares TV, Hu W, Liang Y, Rodrigues FM, Rund LA, et al. A genetic porcine model of cancer. PLoS ONE. (2015) 10:e0128864. doi: 10.1371/journal.pone.0128864
75. Principe DR, Overgaard NH, Park AJ, Diaz AM, Torres C, McKinney R, et al. KRAS(G12D) and TP53(R167H) cooperate to induce pancreatic ductal adenocarcinoma in sus scrofa pigs. Sci Rep. (2018) 8:12548. doi: 10.1038/s41598-018-30916-6
76. Overgaard NH, Principe DR, Schachtschneider KM, Jakobsen JT, Rund LA, Grippo PJ, et al. Genetically induced tumors in the oncopig model invoke an antitumor immune response dominated by cytotoxic CD8beta(+) T cells and differentiated gammadelta T cells alongside a regulatory response mediated by FOXP3(+) T cells and immunoregulatory molecules. Front Immunol. (2018) 9:1301. doi: 10.3389/fimmu.2018.01301
Keywords: pancreatic cancer, swine, porcine, transgenic, KRAS, p53
Citation: Bailey KL and Carlson MA (2019) Porcine Models of Pancreatic Cancer. Front. Oncol. 9:144. doi: 10.3389/fonc.2019.00144
Received: 20 November 2018; Accepted: 20 February 2019;
Published: 12 March 2019.
Edited by:
Lawrence Schook, University of Illinois at Urbana-Champaign, United StatesReviewed by:
Gabriele Multhoff, Technische Universität München, GermanyKyle Schachtschneider, University of Illinois at Chicago, United States
Paul Grippo, University of Illinois at Chicago, United States
Copyright © 2019 Bailey and Carlson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark A. Carlson, macarlso@unmc.edu