 Maria Laura De Angelis
Maria Laura De Angelis Federica Francescangeli
Federica Francescangeli Filippo La Torre
Filippo La Torre Ann Zeuner
Ann Zeuner- 1Department of Oncology and Molecular Medicine, Istituto Superiore di Sanità, Rome, Italy
- 2Department of Surgical Sciences Policlinico Umberto I, Sapienza University of Rome, Rome, Italy
Cancer treatment with either standard chemotherapy or targeted agents often results in the emergence of drug-refractory cell populations, ultimately leading to therapy failure. The biological features of drug resistant cells are largely overlapping with those of cancer stem cells and include heterogeneity, plasticity, self-renewal ability, and tumor-initiating capacity. Moreover, drug resistance is usually characterized by a suppression of proliferation that can manifest as quiescence, dormancy, senescence, or proliferative slowdown. Alterations in key cellular pathways such as autophagy, unfolded protein response or redox signaling, as well as metabolic adaptations also contribute to the establishment of drug resistance, thus representing attractive therapeutic targets. Moreover, a complex interplay of drug resistant cells with the micro/macroenvironment and with the immune system plays a key role in dictating and maintaining the resistant phenotype. Recent studies have challenged traditional views of cancer drug resistance providing innovative perspectives, establishing new connections between drug resistant cells and their environment and indicating unexpected therapeutic strategies. In this review we discuss recent advancements in understanding the mechanisms underlying drug resistance and we report novel targeting agents able to overcome the drug resistant status, with particular focus on strategies directed against dormant cells. Research on drug resistant cancer cells will take us one step forward toward the development of novel treatment approaches and the improvement of relapse-free survival in solid and hematological cancer patients.
Introduction
Resistance to chemotherapy and molecularly targeted therapies is a major problem that limits the effectiveness of cancer treatments. While some tumors are intrinsically insensitive to therapies due to pre-existing resistance factors (primary or intrinsic resistance), others become resistant during drug treatment (1). The development of resistance after an initial period of response (acquired resistance) is due to the molecular heterogeneity of tumor cells which, together with their ability to evolve at the genetic, epigenetic, and phenotypic level, is able to overcome the action of cancer therapies. The emergence of resistant cells has been observed upon treatment with chemotherapy, radiotherapy, and targeted therapies, including EGFR tyrosine kinase inhibitors in lung cancer, anti-HER2 therapies in breast cancer, and BRAF inhibitors in melanoma. Even cancer immunotherapies, which exploit a dynamic interaction between the host immune system and tumor cells thus achieving lasting antitumor responses, are linked to the development of resistance and consequent cancer progression (2). Treatment with chemotherapeutic or targeted drugs is increasingly recognized to promote the emergence of resistant cells with features of cancer stem cells (CSCs) (3). This process clearly involves a Darwinian selection of cell populations with novel genetic mutations conferring drug resistance (4–6). However, non-genetic events involving both chromatin remodeling and the activation of stress-related pathways are responsible for the establishment of drug tolerance, a process more rapid, and massive than genetic mutation (7–9). Drug tolerance is habitually associated to a transient state of slow proliferation, thus identifying a population of Drug Tolerant Persisters (DTPs) that are largely quiescent and maintain viability in conditions where other cancer cells are killed (9). Drug tolerance is a temporary condition, which can revert after the cessation of cytotoxic stimuli. Differently, in the presence of continuous drug stimulation or other cellular stresses such as hypoxia, drug tolerance stabilizes into an enduring drug resistant state (9, 10). Besides quiescence, senescence has also been proposed as a process adopted by tumor cells to escape from therapy (11), suggesting that drug resistance is a composite picture of heterogeneous cell states. This picture is further complicated by a plethora of cell-intrinsic and extrinsic factors that contribute to the establishment of drug resistance including hypoxia, cytokines (among which IL-6, IL-8, and TGF-β play a prominent role), cellular composition and stiffness of the extracellular matrix. Drug resistant cells are found not only within bulk tumor populations but are also scattered in distant organs as disseminated tumor cells (DTCs), which have been recognized as the seeds of metastasis. DTCs are in a state of dormancy, which is induced and maintained by interactions with the target organ niche (12). The neutralization of DTCs is a primary goal in patients with cancers subject to late relapses such as breast and prostate cancer: in fact, recent insights on the mechanisms by which DTCs persist and reawaken are paving the way for new therapeutic avenues (13). This review will draw a picture of drug resistant cells in different contexts such as primary tumors or pre-metastatic niches and discuss a surge of recent findings that shed new light on their strengths and weaknesses, making drug resistance one of the most fertile fields of cancer research.
Drug Resistant Cancer Stem Cells: A Concentrate of Robustness and Plasticity
The concept of CSCs originated as a hierarchical model where, in parallel to normal tissues, a small number of undifferentiated elements give rise to intermediate progenitors and finally to a differentiated progeny. While the hierarchical model remains fundamentally valid for normal tissues (with the exception of rare dedifferentiation events occurring during tissue regeneration or artificial reprogramming), it is becoming clear that boundaries between stem and non-stem cells are much weaker in cancer. In fact, in tumors state transitions seem to be very frequent and chaotic, thus generating high levels of heterogeneity that constitute the foundation of drug resistance (14–16). Not surprisingly, state transitions also affect the expression of molecules expressed on the cell membrane, such as surface markers used for CSCs isolation. Expression of surface or intracellular markers can in some cases identify a population of cells with enhanced self-renewal and/or metastatic capacity in several tumors (Supplementary Table 1). However, it should be kept in mind that such expression is transient, dynamic, and variable both among individual tumors (17, 18). In fact, few past studies on the expression of CSCs markers analyzed the expression of such markers over time (particularly upon flow cytometry isolation of positive and negative populations) or the variation of CSCs markers upon microenvironmental stimuli. Consequently, the phenotypic plasticity and dynamic properties of CSCs populations were often overlooked. Functional features such as tumor-repopulating ability in limiting dilution/serial transplantation assays are more suitable to identify CSCs populations. Such assays may nonetheless select for particularly robust cells able to thrive in harsh experimental conditions. Genetic barcoding makes use of lentiviral infection systems to tag human cells and has been employed to analyze and track stem cell hierarchies, particularly in colorectal cancer. In this context, molecular tracking studies revealed a stable functional heterogeneity of the colorectal CSCs population during serial xenografting despite profound changes in genomic subclone contribution (19), thus highlighting the functional robustness of cancer cell hierarchies. In addition to cell-intrinsic features, interactions with the tumor microenvironment are increasingly recognized as crucial determinants of stemness. The fact that soluble molecules released by the tumor microenvironment have the potential to initiate CSC-like programs was first demonstrated in brain tumors, where the self-renewal and proliferation of stem-like cells were shown to crucially depend from their interaction with endothelial cells (20). The tumor endothelium has also been shown to produce nitric oxide, which diffuses to neighboring glioma cells and activates the Notch pathway to induce stem-like characteristics (21). Later studies in colorectal cancer showed that cancer-associated fibroblasts secrete hepatocyte growth factor, osteopontin, and stromal-derived factor 1α, which activate the WNT pathway to promote cancer cell stemness (22, 23). Tumor-associated macrophages play also a role in supporting breast CSCs and brain CSCs, further supporting the importance of the niche in dictating a cancer stem cell phenotype (24, 25). Moreover, exosomes and microvesicles produced by niche cells are increasingly recognized to influence CSCs and drug resistance. For example, microvesicles produced by breast cancer-associated fibroblasts transfer miR-221 to cancer cells thus increasing the drug resistant CD133hi stem cell population (26). In addition to soluble factors, other microenvironmental features such as clone location have been recently shown to determine the self-renewal capacity of colorectal cancer cells (27). In light of these evidences, stemness in cancer can be defined as a transient state of enhanced plasticity and robustness crucially influenced by microenvironmental signals, including interactions with niche elements, tumor, and non-tumoral cells, soluble factors, and anticancer therapies. The link between stemness and drug resistance derives mainly from three observations: (1) CSCs populations are more resistant to therapy (28), (2) cancers with a stemness-related gene expression have a worse prognosis (29–33), and (3) cells with combined features of stemness, drug resistance, and dormancy have been identified in several tumors including pancreatic carcinoma (34, 35), ovarian cancer (36), melanoma (37), lung cancer (38), and CML (39). More recently, dormant/slow cycling CSCs have been identified in acute leukemia (40), glioblastoma (8, 41, 42), breast (43), and colorectal cancer (44, 45). An interesting association between stemness and dormancy, together with enhanced migratory features, has also been reported in early metastatic cells which are largely responsible for tumor dissemination (46–49). While increasing evidences point to the presence of drug resistant CSCs in multiple cancers, the effect of conventional, and targeted therapies is not usually evaluated specifically on the CSCs compartment, rendering difficult to estimate CSCs permanence after therapy. Likewise, current diagnostic and therapeutic approaches include few tools for the identification, quantification, and elimination of drug resistant cells and DTCs. The elucidation of mechanisms of drug resistance and the identification of biomarkers of resistant cells are therefore essential to improve the clinical management of cancer patients.
Heterogeneity of Therapy Resistant Cancer Stem Cells
Previously thought to be a quite homogeneous condition, drug resistance is emerging as a surprisingly heterogeneous state that includes quiescent, drug-tolerant, and persister cells (50). This scenario is further complicated by the recent addition of post-senescent cells to drug-resistant cells responsible for disease recurrence (11). In this section we propose a functional distinction of drug resistant cells based on their origin, location, and cellular state. Figure 1 illustrates schematically three main populations associated with drug resistance such as (1) “spontaneous” drug-resistant cells in untreated tumors, (2) stress-induced drug resistant cells (including drug-tolerant persisters, post-senescent cells, and cells resident in hypoxic tumor areas), and (3) DTCs. As mentioned previously, a small population of drug-resistant cells is already present in tumors before any kind of treatment (32, 34, 37–40, 42, 44, 45). Recent studies showed that populations of endogenous drug resistant cells transiently arise as the result of stochastic state transitions that induce a high expression of resistance factors (9, 51). Such factors have been identified in melanoma cells and include EGFR, NGFR, WNT5A, AXL, PDGFRB, and JUN, with cells expressing more than one factor displaying a higher level of resistance (51). The emergence of drug tolerant cells has been detected and quantified in multiple tumors upon treatment with chemotherapeutics or targeted agents including cisplatin (in NSCLC), erlotinib, and gefitinib (NSCLC), lapatinib (breast cancer), the RAF kinase inhibitor AZ628 (melanoma and colorectal cancer), and the MET inhibitor PF-2341066 (MET-amplified gastric cancer) (9). DTPs represent a variable percentage of the parental cell population ranging approximately from 0.2 to 5% and have been identified as largely quiescent cells, although a minor part of them can resume proliferation even in the presence of the drug (9). A particularly interesting mechanism that leads to the emergence of DTPs has been recently elucidated in melanoma cells that develop resistance to BRAF inhibitors (BRAFi). A subset of melanoma cells constitutively activates the Aryl hydrocarbon Receptor (AhR), a basic helix-loop-helix transcription factor responsible for the de-differentiation of melanoma cells and the expression of BRAFi- resistance genes. Treatment with BRAFi results in the enrichment of a small subpopulation of AhR-activated BRAFi-persister cells, responsible for melanoma relapse (52). While spontaneous resistant cells and DTPs can be appropriately defined as “quiescent” as their cell cycle interruption is transient and programmed to last until the subsequent change of gene expression or drug concentration. However, such quiescent state can be stabilized by a protracted environmental stresses like hypoxia, thus shifting the balance from a short-term quiescent state to medium- or long-term dormancy. Interestingly, cells deriving from a hypoxic tumor microenvironment have been shown to activate a dormancy program giving rise to chemoresistant DTCs, further strengthening the link between dormancy and drug resistance (10). Recently, senescence has emerged as another key cellular response to drug resistance, further contributing to the heterogeneous picture of drug resistant cells. Chemotherapy and targeted therapies can induce senescence in tumor cells, intended as a stable form of growth arrest (11, 53, 54). Senescent cells are characterized by peculiar morphological features, by the expression of senescent markers (mainly SA-β-Gal and H3K9me3) and by the so-called Senescence-Associated Secretory Phenotype (11, 55). Moreover, senescent cells are arrested in the G1 or G2/M phase of the cell cycle, differently from quiescent cells that are in G0 or G0/G1 transition. Importantly, an estimated 1/106 cells can escape from senescence and re-enter the cell cycle, gaining increased aggressiveness and tumor-initiating potential (56). Cells able to escape from senescence express nuclear β-catenin and stem cell markers, indicating they underwent a process of cellular reprogramming that rendered them a fully functional cancer stem cell population (56). Finally, DTCs represent a category of drug resistant stem cells located in lymph nodes or distant organs that can persist for decades after removal of the primary tumor. DTCs are crucially dependent from niche interactions and can be resistant to chemotherapy, targeted therapies, and hormonal therapy (57–59). Thus, understanding the biology of DTCs is crucial for devising alternative strategies aimed at eradicating DTCs while dormant or preventing their awakening (12, 60). Many efforts are currently dedicated to answer key unresolved questions regarding DTCs, such as which pathways are involved in maintaining DTCs dormancy, how DTCs evade immune surveillance and what triggers their awakening (12, 60, 61). However, it is also of note that physiologic models for cancer cell dissemination are represented by orthotopic/metastatic tumors in mice, which are relatively straightforward in the case of breast cancer but less feasible in other tumors. An increased use of orthotopic/metastatic models is therefore, warranted to improve the knowledge on tumor cell dissemination, dormancy, and reawakening. A provocative contribution to the field of DTCs came from recent studies showing that disseminated breast cancer cells are protected from chemotherapy through integrin-mediated interactions irrespectively from their cell cycle status (57). Disrupting the interactions between DTCs and the perivascular niche with integrin inhibitors results in DTCs chemosensitization and may represent a clinical strategy to eradicate minimal residual disease (57). In summary, it appears that both cell-intrinsic factors and cell-extrinsic signals (either local or systemic) crucially contribute to drug resistance (Figure 2). Therefore, integrated approaches able to interfere with the establishment of drug resistance at multiple levels are urgently needed to increase the life expectancy of cancer patients.
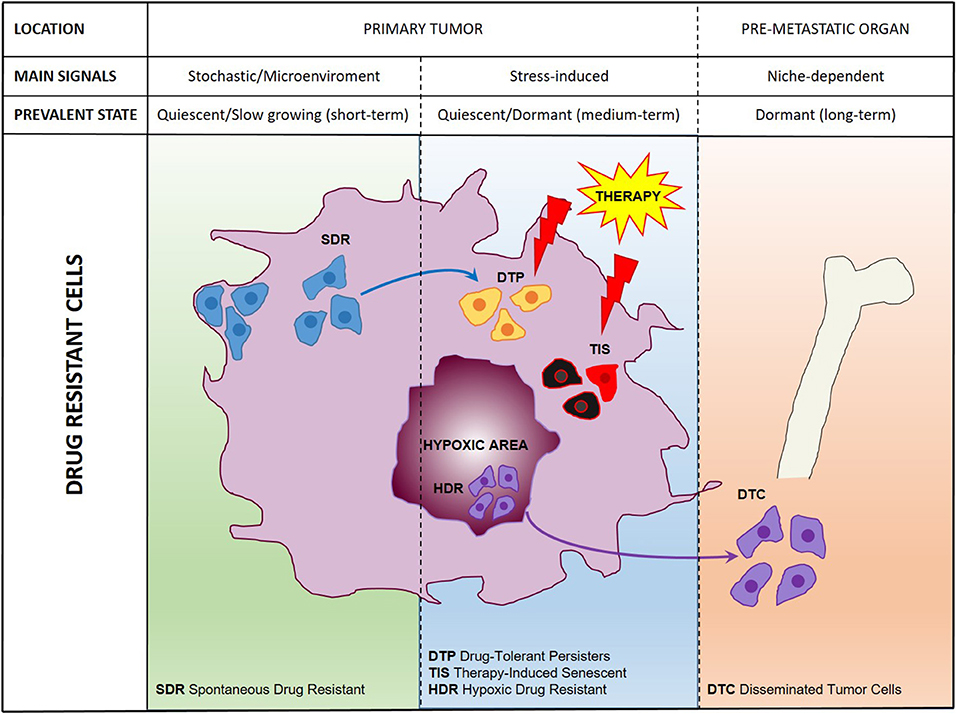
Figure 1. A comprehensive scheme of drug resistant cancer stem cells. Representation of main cell populations responsible for drug resistance present in the untreated tumor (SDR, Spontaneous Drug Resistant) induced by therapeutic treatment (DTPs, Drug-Tolerant Persisters; TIS, Therapy-Induced Senescent; HDR, Hypoxic Drug Resistant) or disseminated in pre-metastatic organs (DTCs, Disseminated Tumor Cells).
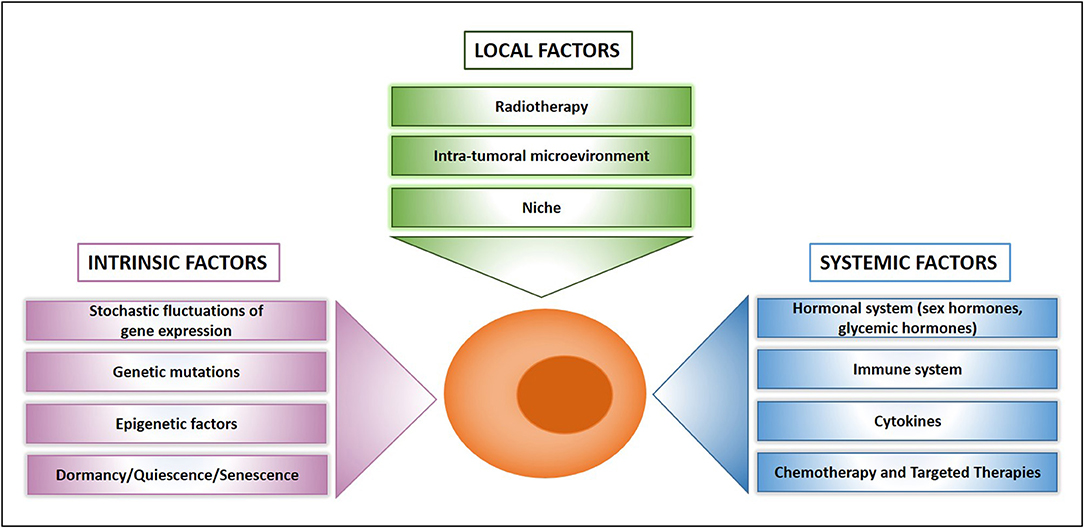
Figure 2. What makes a drug resistant cell. Schematic presentation of main factors that influence drug resistance at the cellular, local, and systemic level.
Non-Genetic Pathways Involved in Drug Resistance
Pioneer studies on dormant cells and their microenvironment have led to a deeper understanding of drug resistance (62), thus paving the way for a flurry of recent studies in this field. Here, we will discuss the main categories of factors crucially involved in the determination of a dormant/drug resistant status with particular focus on recent discoveries. It is important to acknowledge that factors responsible for dormancy/drug resistance are not mutually exclusive, but many of them are likely active at the same time and crosstalk to reinforce each other.
Stress-Induced Pathways Part 1: The p38 Hub
Early studies pointed to a key role for p38 stress-activated protein kinase activation in dormant cells indicating that the balance between proliferation and dormancy is determined by the ratio between the activity of p38 and ERK1/2 (63, 64). Since then, p38 has emerged as a hub in the control of multiple pathways involved in both drug resistance and cellular stress and has itself reported to induce chemoresistance in a variety of tumors. Further insights into p38-activated pathways led to the key discovery that the orphan nuclear receptor NR2F1 upon activation by p38 induces dormancy through SOX9, RARβ, CDK inhibitors, and global chromatin repression in head and neck squamous cell carcinoma (65). Recently, NR2F1 has emerged as a clinical marker of dormancy, its expression being able to discriminate breast cancer patients with short term systemic relapse from those with long disease-free intervals (66). Most interestingly, factors involved in the p38/NR2F1/retinoic acid receptors pathway are possibly emerging as part of a general program of dormancy/drug resistance active across several types of cancer. In line with this hypothesis another retinoic acid-binding nuclear receptor, RORγ, emerged during a recent mapping of molecular traits related to stemness and drug resistance in pancreatic cancer (67). RORγ, which is also known for its role in immune modulation ad inflammation, was correlated with the aggressiveness of pancreatic cancer and its inhibition led to a striking defect in tumor growth (67). A current clinical trial based on the use of 5-azacytidine/all-trans retinoic acid aimed at inhibiting DTCs reawakening in prostate cancer patients (NCT03572387) will provide clinical evidence on the efficacy of epigenetic therapies that induce histone demethylation and NR2F1 activation. SOX9 is another factor downstream of p38 that has recently been involved in drug resistance of CSCs in breast and esophageal cancer (68, 69) and in chemoresistance of cholangiocarcinoma (70). Interestingly, SOX9 expression is regulated by the SCFFBW7 (Skp1/Cul1/F-box), a component of the ubiquitin ligase complex (71, 72), which has been recently shown to regulate dormancy/drug resistance in breast cancer. In fact, Fbxw7 ablation sensitizes disseminated breast tumor cells to chemotherapy, arguing for a key role of the ubiquitination pathway in dictating drug resistance by selective substrate degradation (72). SOX2 and SOX9 transcription factors are also typically expressed by dormant CSCs in breast and lung cancer. Interestingly, these cells maintain dormancy in an autocrine fashion by inhibiting Wnt signals through expression of the Wnt inhibitor DKK1 (73). Other tumors have been recently reported to modulate Wnt signaling in order to maintain dormancy: prostate cancer cells receive Wnt5a from the osteoblastic niche and activate a non-canonical signaling that represses canonical Wnt3a/β-catenin signaling (74). Notably, osteoblast-produced Wnt5a acts by inducing the Siah E3 Ubiquitin Protein Ligase expression, further supporting the role of E3 ligases in dormancy/drug resistance (75). Wnt signals have also been implicated in the survival of dormant tumor cells in colorectal cancer, which have been identified as a population of partially differentiated cells characterized by high clonogenic capacity and chemoresistance (45).
Stress-Induced Pathways Part 2: Hypoxia, Endoplasmic Reticulum Stress, and Autophagy
Global stress responses such as the hypoxia, unfolded protein response (UPR), endoplasmic reticulum stress and autophagy have all been implicated in drug resistance and pre-metastatic dormancy. Hypoxia has been habitually linked to tumor aggressiveness and poor survival but the underlying mechanisms are still under elucidation. Cells in low oxygen microenvironments activate hypoxia-inducible factors and increase the expression of key dormancy genes such as N2RF1, p27, and MIG6, inducing a combined state of dormancy and drug resistance (10, 76). Hypoxic responses can be triggered by hypoxic microenvironments in primary tumors but can also be induced by chemotherapy, which promotes a signaling cascade involving calcium release from the endoplasmic reticulum and expression of pluripotency genes, leading to an enrichment of stem cells in breast cancer (77). The endoplasmic reticulum and the related UPR, which is responsible for re-establishing endoplasmic reticulum homeostasis following cellular stress, are implicated in several steps of the drug resistance process (78). The endoplasmic reticulum stress sensor GRP78, previously shown to be downstream of activated p38 (79), seems to play a central role in the induction of drug resistance and has been particularly investigated in pancreatic cancer, where it has been involved in both chemoresistance and maintenance of the stem cell population (80, 81). Besides chemoresistance, endoplasmic reticulum stress has been demonstrated to be involved also in resistance to tyrosine kinase inhibitors in lung cancer, where drug persister cells activate the recently described ufmylation pathway and downstream UPR to upregulate key survival signals such as Bcl-xL (82). An interesting crosstalk occurs between endoplasmic reticulum stress/UPR and autophagy, which occur simultaneously and are both implicated in tumorigenesis and chemoresistance. In fact, GRP78, PERK, and ATF6 lie at the crossroads between UPR and autophagy, being able to modulate both pathways (83). Autophagy was recognized a decade ago as being implicated in the regulation of tumor cell survival and dormancy in ovarian and gastrointestinal tumors (84, 85). In fact, autophagy is required during quiescence for recycling of aminoacids and nucleotides (86), but new evidence adds to a specific role of autophagy in dictating chemoresistance in colorectal cancer (87), liver cancer (88), brain tumors (89), and melanoma (90). Recently, autophagy has been shown to be essential for the survival of disseminated dormant breast cancer cells and its inhibition with antimalarial hydroxychloroquine eliminates DTCs while dormant (91). In KRAS-dependent tumors such as pancreatic adenocarcinomas (PDAC), KRAS inhibition has demonstrated to increase autophagic signaling resulting in autophagy dependance. Removing this protective mechanism through the combined use of MEK/ERK inhibitors and autophagy inhibitors may be therapeutically beneficial in patients with PDAC, NRAS-driven melanoma, and BRAF-mutant colorectal cancer (92). By contrast, the activation of autophagic/lysosomal pathways can occur as the consequence of anticancer therapies, as has been demonstrated in melanoma treated with anti-BRAF targeted agents (93). In this case, autophagy blockade has detrimental effects, resulting in enhanced tumor progression, metastatic dissemination, and chemoresistance. Thus, autophagy may play different roles in multiple contexts and further studies are needed to clarify the potential utility of autophagy modulators in cancer therapy.
Metabolic Reprogramming of Drug Resistant Cells
Metabolic deregulation is recognized as a hallmark of cancer, and increasing evidences suggest that it can be exploited by neoplastic cells in order to acquire a drug resistant phenotype. Intuitively, since chemotherapy kills highly proliferative cells that rely on aerobic glycolysis, it also induces a selective pressure toward the emergence of slow growing cells switched to OXPHOS metabolism (94). However, this apparently straightforward hypothesis is contradicted by very different metabolic patterns found in resistant tumor cells (95). On one hand, several studies indicate that chemotherapy-resistant cells become OXPHOS-dependent (96–99). However, other reports showed that chemoresistant cells rely on high ATP levels (100) and express glycolytic markers (101–104). A possible explanation for such divergences can be found in the timing of data collection relatively to drug treatment: during therapy, resistant cells may activate a survival program based on proliferative slowdown and switch to OXPHOS, while some time after therapy cessation cells may recover a high proliferative rate associated to aerobic glycolysis. However, the main explanation for the different metabolic patterns found in drug resistant cells probably resides in the high plasticity of the metabolic response to cytotoxic challenges. A recent study confirmed this hypothesis by showing that cancer cells are able to switch between OXPHOS and glycolysis to circumvent the inhibition of either process (105). A therapeutic strategy targeting metabolic plasticity based on intermittent fasting (to reduce glucose availability) plus the OXPHOS inhibitor metformin effectively restrained tumor growth by activating PP2A, GSK3β, and the pro-apoptotic protein Mcl-1 (105). These results also suggest that an optimization of metformin administration schedules may potentiate its ability on tumor metabolism and increase its therapeutic efficacy. Metabolic stress is a condition often encountered by tumor cells, particularly by the quiescent population resident in poorly vascularized/hypoxic areas. The combination of hypoxia and reduced nutrient availability limits the metabolic plasticity of tumor cells, which become more sensitive to drugs that target mitochondrial respiration. In fact, drugs targeting mitochondrial bioenergetics have been proposed to eliminate metabolically stressed quiescent cells, alone or in combination with autophagy inhibitors (106). Interesting insights into the mechanisms of drug-induced metabolic reprogramming have come from the study of estrogen receptor (ER)-positive breast cancers. In these cancers, hormonal therapy has been shown to result in the emergence of dormant CD133high/ERlow cells responsible for metastatic progression and to induce an OXPHOS metabolic editing of breast cancer cells through IL6/Notch3 signaling (107). Hormonal therapy resistance and the generation of breast CSCs have been correlated to microvesicle-mediated horizontal transfer of microRNAs from host stromal cells (26). Recently, extracellular microRNAs have been further implicated in the metabolic crosstalk between tumor cells and their microenvironment by showing that cancer cell-secreted miR-105 instructs cancer-associated fibroblasts to display different metabolic features, thus helping the tumor to face changes in the metabolic environment (108). Finally, metabolic reprogramming related to the development of drug resistance has been also shown to occur during antiangiogenic therapies (109). Interestingly, overexpression of the glucose transporter 3 (GLUT3) recapitulates all the metabolic features of bevacizumab-resistant cells indicating GLUT3 as a potential metabolic target in glioblastoma (110). In summary, it appears increasingly clear that metabolic heterogeneity can be driven by both intrinsic (either genetic or epigenetic) mechanisms or as an adaptation to environmental changes and plays a key role in the development of drug resistance, representing a potential avenue for targeted therapies (111).
Epigenetic Plasticity in the Regulation of Drug Resistance
Epigenetic deregulation is a feature of virtually all human cancers (112). Tumors exhibit a continuously changing epigenetic landscape that includes altered modifications of DNA promoter regions, deregulated acetylation, or methylation of histone proteins or inappropriate expression of repetitive regions, contributing to tumors biological properties (113). The involvement of epigenetic (rather than genetic) mechanisms in drug resistance is particularly evident when drug resistant states are transient, rapidly emerging, and functionally heterogeneous. A number of past studies have demonstrated a contribution of epigenetic modifiers such as histone deacetylases (HDACs) to oncogenesis with different mechanisms strongly depending on the cellular context (114). Recent observations point to a crucial role of histone demethylases, and in particular KDM2, KDM3, KDM5, KDM6, and KDM7, in generating a drug resistant state and often a concomitant slow-dividing and stem cell-like state (8, 9, 115–121). Additional interesting insights into the epigenetic mechanisms of drug resistance came from the observation that drug treatment induces a rapid reprogramming of spontaneous resistant cells in primary tumors, converting the transient quiescent state into a stably resistant state and generating DTPs, the cells that actually survive drug-induced toxicity (Figure 1) (51). In breast cancer, multiple epigenetic enzymes including KDM5B, bromodomain, and extraterminal (BET) proteins and the histone methyltransferase EZH2 have been shown to be responsible for the generation of persister cells through a dynamic remodeling of the chromatin architecture, and such state transitions can be counteracted with inhibitors of chromatin-modifying enzymes (117). Recently, BET inhibitors were found to revert drug resistance and to block the pro-tumorigenic activity exerted by YAP/TAZ binding to the epigenetic coactivator bromodomain-containing protein 4 (BRD4) (122). However, cancer cells can develop also resistance to epigenetic inhibitors. In neuroblastoma, PI3K pathway activation and transcriptional reprogramming can confer resistance to BET inhibitors, indicating that sequential or combination therapies will likely be required to achieve durable antitumor effects (123). In this regard, the combined inhibition of BET proteins and HDACs is increasingly regarded as a strategy to improve the effectiveness of these drugs in cancer (124). A novel link between epigenetic regulation and chemoresistance has come from colorectal cancer, where the epigenetic dioxygenase TET2 has been shown to control a population of slow cycling cells responsible for chemoresistance and tumor recurrence (44). Slow cycling cell populations generated by epigenetic factors in multiple tumor settings likely represent a reservoir for the subsequent emergence of heterogeneous proliferating drug resistant cells. In fact, further epigenetic rearrangements and even genetic mutations can occur in quiescent cells giving rise to a variety of survival strategies (125). In line with this hypothesis, a single lung cancer persister cell was shown to generate a variety of colonies with different mechanisms of erlotinib resistance (126). Finally, recent discoveries suggest that repetitive transposable elements may be involved in the epigenetic determination of drug resistance. Repetitive elements constitute nearly half of the human genome and in normal cells they are tightly regulated to avoid dangerous inappropriate activation events. In cancer cells repetitive elements are often aberrantly activated, in part due to decreases in DNA methylation (127). Chemotherapeutic and targeted drugs can also induce a strong activation of repeated elements in cancer cells that results in cell death (116, 128). By contrast, DTPs within the heterogeneous cancer cell population are able to maintain the epigenetic repression of repetitive elements through increased histone H3K9 and H3K27 methylation even during drug treatment, exploiting this strategy to survive drug exposure (116). In line with these observations, a new generation of drugs targeting epigenetic modulators are finding their way to the clinic, in the attempt to exploit cancer-associated epigenetic traits for therapeutic intervention (129). Clinically induced derepression of genomic repeat elements also harbors the potential to enhance the immunogenicity of cancer cells and enhance the response to immunotherapeutic approaches (127), fostering further investigations on the mechanisms that deregulate repeat element expression in tumor cells.
Interactions Between the Immune System and Therapy Resistant Cells
The importance of the immune system in controlling tumor growth, metastatization, and relapse is undisputed, as witnessed by the expanding role of immunotherapies in cancer treatment. New challenges concerning the use of immunotherapeutic drugs are often related to properties of CSCs, which have been reported to have a low immunogenic profile and peculiar interactions with immune cells (130). A striking example of interactions between CSCs and immune cells resulting in immunotherapy resistance has been recently highlighted in squamous cell carcinoma. Here, a population of tumor-initiating cells responsive to TGF-β acquires the expression of CD80 (a molecule previously identified on cells of the immune system) and hinders cytotoxic T cell activity leading to tumor relapse (131). The relationships between drug resistant cancer cells and the immune system are the object of particularly intense investigations in the pre-metastatic context. In fact, while tumor cells in the primary tumor are often surrounded by an immune-suppressive microenvironment (132), DTCs are theoretically more vulnerable to immune attack, providing a window for therapeutic interventions aimed at preventing metastasis formation. Recent reviews have addressed in detail the role of the immune system in cancer (133) and the mechanisms of DTCs immune escape (60, 134). Here, we will focus our discussion to a limited number of recent breakthroughs in the relationships between drug resistant cells and the immune system. New insights on how DTCs evade immune recognition came from the observation that dormant cells activate the UPR, which in turn causes the downregulation of major histocompatibility complex class I (MHC I) molecules required for antigen presentation to CD8+ T cells. This mechanism rendered DTCs undetectable by CD8+ T cells, while targeting the UPR led to MHC I re-expression and reversal of the immune-evasive phenotype (135). The most important soluble factors in cancer-immune system interactions are interferons, and particularly IFNγ produced by T cells and NK cells (136). IFNγ has been shown to induce cancer cell dormancy through multiple pathways and, interestingly, to exert different effects in indolent (Ki67low) cells, and in dormant (Ki67−) cells (137). Besides its established role in contributing to anti-tumor immunity, IFNγ is also implicated in mediating resistance to various cancer therapies, including anti-PD1 therapies via downregulation of MHC I molecules (138, 139). An interesting link between IFNγ and CSCs metabolism came from the observation that IFNγ triggers cancer dormancy through indolamine 2,3 dioxygenase (IDO1), an enzyme that catalyzes tryptophan metabolism. Blocking IDO1 metabolic circuitry abrogates dormancy and induces apoptosis of tumor-repopulating cells (140). The same metabolic pathway was found to be involved in IFNβ-induced dormancy in melanoma (141). The Stimulator of Interferon Genes (STING) is a central component of the intracellular DNA sensing pathway and has been initially characterized for its capacity to mediate type I interferon inflammatory responses in immune cells during infections. Recent breakthroughs indicate that the STING pathway has much broader functions, being implicated also in fundamental cancer-related processes such as cellular transformation (142, 143), metastasis (144), and response to radio- and chemotherapy (145, 146). STING has been recently identified as an activator of autophagy downstream of the ancestral cyclic GMP-AMP synthase (cGAS) pathway (147) and may also be implicated in chemoresistance-related autophagy. Additional evidences indicate a direct link between STING and LKB1, which is a crucial regulator of stem cell quiescence, metabolism, and anti-tumor immunity (148). Specifically, loss of LKB1 leads to the suppression of STING and insensitivity to cytoplasmic double-strand DNA detection, resulting in resistance of lung cancer to immunotherapy (149). Therefore, therapies that reactivate LKB1 or the STING pathway may boost anticancer immune response in cancers with resistance to immune-checkpoint blockade (150). Finally, immune cells have been recently implicated in DTC reawakening from dormancy in a study showing a key role for Neutrophil Extracellular Traps (NETs) produced by neutrophils in the lung parenchyma upon inflammation. NETs trigger integrin-mediated activation of focal-adesion kinase in DTCs and subsequent exit from dormancy, while integrin-blocking antibodies prevent DTC reactivation in NET-enriched lungs (151). The latter study confirms the involvement of integrins in chemo- and radiotherapy resistance of multiple cancers, raising hopes for the future development of effective therapeutic agents blocking integrin signaling (152).
Considerations on Targeting Therapy Resistant Cancer Stem Cells
Therapies directed against drug resistant cells resident in either primary tumors, pre-metastatic niches, or metastatic cancers have to face an array of genetic and epigenetic survival strategies exploited by cancer cells. Conventional therapies such as radio- and chemotherapy, although representing the mainstay of cancer therapy, are intrinsically limited in their capacity to face drug resistance and may themselves promote the emergence of more aggressive cells. In recent years the mechanisms underlying cancer cell plasticity, heterogeneity, stress responses, and dormancy have begun to be elucidated, indicating new routes of therapeutic intervention. At the same time, new therapeutic approaches should undergo careful pre-clinical evaluation for their effects on the CSCs compartment, which would provide indications on the development of resistant cell populations. New therapeutic strategies directed against dormant/drug resistant cells in primary tumors are progressively focusing on epigenetic modulators such as inhibitors of KDMs, HDACs, or BET proteins. By contrast, therapeutic strategies directed against pre-metastatic DTCs are divided in three main workstreams: the so-called “sleeping strategies” include drugs that suppress proliferative signals such as anti-estrogen therapies (153), inhibitors of CDK4/6 (154), and inhibitors of ERK or Src (155). Prolonging dormant states can also be obtained with drugs that increase the expression of dormancy factors such as p38, DYRK1A, and N2RF1 (63, 65, 156, 157). Sleeping strategies such as anti-estrogen therapies for breast and prostate cancer had a profound impact in the clinical setting. However, sleeping strategies must be long-lasting (or even lifetime long) and therefore must deal with unwanted side effects that can limit their long-term usage and reduce patient compliance. In this regard, the use of retinoic acid or fenretinide derivatives to induce cancer cell quiescence appear as a feasible and relatively non-toxic therapeutic approach (12, 158). An additional problem related to sleeping strategies is that not all tumor cells are responsive, as ER-positive breast tumors can give rise to metastases even during hormonal therapy. Thus, the combination or sequential administration of “sleeping” therapeutics should be explored, with the caution of avoiding toxic side effects. A second type of strategies directed against dormant cells is represented by cell cycle reactivation, classically with G-CSF or IFNα (159). However, treatment with reactivating agents may not be effective on all the tumor cells, leaving behind some dormant persisters. Also, therapeutic reactivation may render tumor cells more aggressive and potentially resistant to subsequent chemotherapy. The third strategy consists in eliminating dormant/drug resistant cells while dormant, a difficult but not impossible challenge. Elimination of dormant cells has been achieved in experimental brain tumors with mithramycin (160), HDACs or KDMs inhibition (8, 9, 116, 119), while in pancreatic tumors resistant cells were eliminated by IGF1 inhibition (161). Activators of ferroptosis, a form of cell death characterized by the accumulation of lipid peroxidation products and lethal reactive oxygen species derived from iron metabolism, were shown to kill drug tolerant cells in multiple tumors (162, 163). Mitochondrial respiration is also considered a promising target (106, 164–167), although metabolic plasticity could result in the unresponsiveness of some resistant cells. Besides the inhibition of specific cellular proteins/pathways, other strategies with a broader mechanism of action may be useful to cope with drug resistance. First, immunotherapy alone or in combination with targeted agents is proving effective in several cancers even in the metastatic setting (168, 169). Adoptive transfer of tumor-reactive lymphocytes has led to striking anti-tumor immune responses in breast cancer and other tumors (170–174), indicating that totally drug resistant cells such as those in advanced metastatic tumors can still be eliminated by the immune system. Also, emerging evidences suggest that micro- and macro-environmental signals can profoundly impact on the biology of drug resistant cells. While inflammation has been shown to facilitate metastatic outgrowth (151, 175, 176), anti-inflammatory agents such as NSAIDs seem to dramatically decrease the risk of metastatic relapse, possibly by preventing the reawakening of dormant cells caused by niche alterations that occur during inflammation (177, 178). Finally, therapeutic strategies that include lifestyle-related factors such as exercise and nutrition are emerging as an important tool not only in cancer prevention but also in managing established cancers (179). Diet and lifestyle likely act through reinforcing the immune system, modulating hormone levels, shaping gut microbiota, preventing inflammatory conditions, and influencing the pre-metastatic niche to become less favorable to DTCs awakening (179, 180). A link between diet-related factors and therapy has recently emerged by studies showing that a hypoglycemic diet improves the effectiveness of PI3K inhibitors (181). Likewise, fasting or fasting-mimicking diets are increasingly considered as valid supports in cancer therapy due to their ability to induce wide alterations in growth factors and metabolite levels that generate unfavorable environments for cancer cells and improve the effects of cancer therapies (182). Finally, physical exercise is being explored for its ability to promote and restore antitumor immunity. In fact, the infiltration and antitumor activity of immune cells are limited by a scarcely oxygenated and acidic tumor microenvironment. Exercise has been reported to modulate oxygen concentration and pH in the tumor bed and to directly stimulate tumor cell killing by immune cells, thus appearing as a potential tool to improve the effectiveness of immunotherapy (183). In summary, drug resistance increasingly appears as a multifactorial process (Figure 2) where each class of factors can be considered as a target for novel therapeutic strategies.
Conclusions
Understanding the mechanisms of drug resistance is mandatory in order to improve the effectiveness of cancer therapies. While novel and unexpected mechanisms of drug resistance continue to emerge, translational research is moving toward new therapeutic approaches involving not only cancer cells and peritumoral cells but also other components of the body such as the immune and the hormonal system. As a result of the discoveries made in the last decade, drug resistant cancer cells in their different contexts are starting to appear as a treatable target. However, increasing efforts are required to explore the mechanisms that regulate drug resistance of cancer cells either in primary tumors, pre-metastatic niches, and overt metastases in order to find new therapeutic avenues.
Author Contributions
AZ, MD, and FF wrote the manuscript. FL provided essential expertise.
Funding
This work was supported by an ERA-NET TRANSCAN grant TRS-2015-00000096 to AZ, by Italian Association for Cancer Research (AIRC) Investigator Grant 20744 to AZ, and by Awards Sapienza University of Rome grant C26H15ZKWL to FL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2019.00626/full#supplementary-material
Abbreviations
SA-β-Gal, senescence- associated-β-galactosidase; H3K9me3, trimethylated lysine 9 at histone H3; CML, chronic myeloid leukemia; p38 MAPK, p38 mitogen-activated protein kinase; ERK 1/2, extracellular signal-regulated kinase ½; RARβ, retinoic acid receptor beta; RORγ, nuclear hormone receptor retinoic acid receptor-related orphan receptor gamma; SOX9 (sex-determining region Y [SRY]–containing box 9); GRP78, glucose regulatory protein 78; EZH2, enhancer of Zeste homolog 2; OXPHOS, oxidative phosphorylation; NSAIDs, non-steroidal antinflammatory drugs; IGF1, insulin growth factor 1; EGFR, epidermal growth factor receptor; NGFR, nerve growth factor receptor; NSCLC, non-small cell lung cancer.
References
1. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. (2013) 13:714–26. doi: 10.1038/nrc3599
2. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
3. Martins-Neves SR, Cleton-Jansen AM, Gomes CMF. Therapy-induced enrichment of cancer stem-like cells in solid human tumors: where do we stand? Pharmacol Res. (2018) 137:193–204. doi: 10.1016/j.phrs.2018.10.011
4. Bhang HE, Ruddy DA, Krishnamurthy Radhakrishna V, Caushi JX, Zhao R, Hims MM, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat Med. (2015) 21:440–8. doi: 10.1038/nm.3841
5. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. (2008) 451:1116–20. doi: 10.1038/nature06633
6. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. (2010) 17:77–88. doi: 10.1016/j.ccr.2009.11.022
7. Knoechel B, Roderick JE, Williamson KE, Zhu J, Lohr JG, Cotton MJ, et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat Genet. (2014) 46:364–70. doi: 10.1158/1538-7445.AM2014-4782
8. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell. (2017) 20:233–46.e7. doi: 10.1016/j.stem.2016.11.003
9. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. (2010) 141:69–80. doi: 10.1016/j.cell.2010.02.027
10. Fluegen G, Avivar-Valderas A, Wang Y, Padgen MR, Williams JK, Nobre AR, et al. Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nat Cell Biol. (2017) 19:120–32. doi: 10.1038/ncb3465
11. Saleh T, Tyutyunyk-Massey L, Gewirtz DA. Tumor cell escape from therapy-induced senescence as a model of disease recurrence after dormancy. Cancer Res. (2019) 79:1044–1046. doi: 10.1158/0008-5472.CAN-18-3437
12. Aguirre-Ghiso JA, Sosa MS. Emerging topics on disseminated cancer cell dormancy and the paradigm of metastasis. Annu Rev Cancer Biol. (2018) 2:377–93. doi: 10.1146/annurev-cancerbio-030617-050446
13. Aguirre-Ghiso JA. How dormant cancer persists and reawakens. Science. (2018) 361:1314–15. doi: 10.1126/science.aav0191
14. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. (2011) 146:633–44. doi: 10.1016/j.cell.2011.07.026
15. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
16. Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell. (2014) 15:692–705. doi: 10.1016/j.stem.2014.11.012
17. Francescangeli F, Contavalli P, De Angelis ML, Baiocchi M, Gambara G, Pagliuca A, et al. Dynamic regulation of the cancer stem cell compartment by Cripto-1 in colorectal cancer. Cell Death Differ. (2015) 22:1700–13. doi: 10.1038/cdd.2015.19
18. Bragado P, Estrada Y, Sosa MS, Avivar-Valderas A, Cannan D, Genden E, et al. Analysis of marker-defined HNSCC subpopulations reveals a dynamic regulation of tumor initiating properties. PLoS ONE. (2012) 7:e29974. doi: 10.1371/journal.pone.0029974
19. Giessler KM, Kleinheinz K, Huebschmann D, Balasubramanian GP, Dubash TD, Dieter SM, et al. Genetic subclone architecture of tumor clone-initiating cells in colorectal cancer. J Exp Med. (2017) 214:2073–88. doi: 10.1084/jem.20162017
20. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. (2007) 11:69–82. doi: 10.1016/j.ccr.2006.11.020
21. Charles N, Ozawa T, Squatrito M, Bleau AM, Brennan CW, Hambardzumyan D, et al. Perivascular nitric oxide activates notch signaling and promotes stem-like character in PDGF-induced glioma cells. Cell Stem Cell. (2010) 6:141–52. doi: 10.1016/j.stem.2010.01.001
22. Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. (2010) 12:468–76. doi: 10.1038/ncb2048
23. Todaro M, Gaggianesi M, Catalano V, Benfante A, Iovino F, Biffoni M, et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell. (2014) 14:342–56. doi: 10.1016/j.stem.2014.01.009
24. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. (2014) 16:1105–17. doi: 10.1038/ncb3041
25. Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
26. Sansone P, Berishaj M, Rajasekhar VK, Ceccarelli C, Chang Q, Strillacci A, et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. (2017) 77:1927–41. doi: 10.1158/0008-5472.CAN-16-2129
27. van der Heijden M, Miedema DM, Waclaw B, Veenstra VL, Lecca MC, Nijman LE, et al. Spatiotemporal regulation of clonogenicity in colorectal cancer xenografts. Proc Natl Acad Sci USA. (2019) 116:6140–5. doi: 10.1073/pnas.1813417116
28. Makena MR, Ranjan A, Thirumala V, Reddy A. Cancer stem cells: road to therapeutic resistance and strategies to overcome resistance. Biochim Biophys Acta Mol Basis Dis. (2018). doi: 10.1016/j.bbadis.2018.11.015. [Epub ahead of print].
29. Felipe De Sousa EM, Wang X, Jansen M, Fessler E, Trinh A, De Rooij LP, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. (2013) 19:614. doi: 10.1038/nm.3174
30. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. (2008) 40:499–507. doi: 10.1038/ng.127
31. Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. (2011) 8:511–24. doi: 10.1016/j.stem.2011.02.020
32. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. (2010) 140:62–73. doi: 10.1016/j.cell.2009.12.007
33. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. (2011) 17:1086–93. doi: 10.1038/nm.2415
34. Dembinski JL, Krauss S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin Exp Metastasis. (2009) 26:611–23. doi: 10.1007/s10585-009-9260-0
35. Lin WC, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. (2013) 73:1821–30. doi: 10.1158/0008-5472.CAN-12-2067
36. Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. (2010) 29:2672–80. doi: 10.1038/onc.2010.35
37. Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. (2010) 141:583–94. doi: 10.1016/j.cell.2010.04.020
38. Zeuner A, Francescangeli F, Contavalli P, Zapparelli G, Apuzzo T, Eramo A, et al. Elimination of quiescent/slow-proliferating cancer stem cells by Bcl-XL inhibition in non-small cell lung cancer. Cell Death Differ. (2014) 21:1877–88. doi: 10.1038/cdd.2014.105
39. Holtz MS, Forman SJ, Bhatia R. Nonproliferating CML CD34+ progenitors are resistant to apoptosis induced by a wide range of proapoptotic stimuli. Leukemia. (2005) 19:1034–41. doi: 10.1038/sj.leu.2403724
40. Ebinger S, Ozdemir EZ, Ziegenhain C, Tiedt S, Castro Alves C, Grunert M, et al. Characterization of rare, dormant, and therapy-resistant cells in acute lymphoblastic leukemia. Cancer Cell. (2016) 30:849–62. doi: 10.1016/j.ccell.2016.11.002
41. Lan X, Jorg DJ, Cavalli FMG, Richards LM, Nguyen LV, Vanner RJ, et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature. (2017) 549:227–32. doi: 10.1038/nature23666
42. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. (2012) 488:522–6. doi: 10.1038/nature11287
43. Kabraji S, Sole X, Huang Y, Bango C, Bowden M, Bardia A, et al. AKT1(low) quiescent cancer cells persist after neoadjuvant chemotherapy in triple negative breast cancer. Breast Cancer Res. (2017) 19:88. doi: 10.1186/s13058-017-0877-7
44. Puig I, Tenbaum SP, Chicote I, Arques O, Martinez-Quintanilla J, Cuesta-Borras E, et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J Clin Invest. (2018) 128:3887–905. doi: 10.1172/JCI96393
45. Buczacki SJA, Popova S, Biggs E, Koukorava C, Buzzelli J, Vermeulen L, et al. Itraconazole targets cell cycle heterogeneity in colorectal cancer. J Exp Med. (2018) 215:1891–912. doi: 10.1084/jem.20171385
46. Harper KL, Sosa MS, Entenberg D, Hosseini H, Cheung JF, Nobre R, et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature. (2016) 540:588–92. doi: 10.1038/nature20609
47. Werner-Klein M, Scheitler S, Hoffmann M, Hodak I, Dietz K, Lehnert P, et al. Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat Commun. (2018) 9:595. doi: 10.1038/s41467-017-02674-y
48. Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. (2015) 526:131. doi: 10.1038/nature15260
49. Hosseini H, Obradović MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, et al. Early dissemination seeds metastasis in breast cancer. Nature. (2016) 540:552–8. doi: 10.1038/nature20785
50. Vallette FM, Olivier C, Lézot F, Oliver L, Cochonneau D, Lalier L, et al. Dormant, quiescent, tolerant and persister cells: four synonyms for the same target in cancer. Biochem Pharmacol. (2019) 162:169–76. doi: 10.1016/j.bcp.2018.11.004
51. Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. (2017) 546:431–5. doi: 10.1038/nature22794
52. Corre S, Tardif N, Mouchet N, Leclair HM, Boussemart L, Gautron A, et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat Commun. (2018) 9:4775. doi: 10.1038/s41467-018-06951-2
53. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. (2005) 65:2795–803. doi: 10.1158/0008-5472.CAN-04-1270
54. Francica P, Aebersold DM, Medova M. Senescence as biologic endpoint following pharmacological targeting of receptor tyrosine kinases in cancer. Biochem Pharmacol. (2017) 126:1–12. doi: 10.1016/j.bcp.2016.08.022
55. Saleh T, Tyutynuk-Massey L, Cudjoe EK Jr, Idowu MO, Landry JW, Gewirtz DA. Non-cell autonomous effects of the senescence-associated secretory phenotype in cancer therapy. Fron Oncol. (2018) 8:164. doi: 10.3389/fonc.2018.00164
56. Milanovic M, Fan DNY, Belenki D, Dabritz JHM, Zhao Z, Yu Y, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. (2018) 553:96–100. doi: 10.1038/nature25167
57. Carlson P, Dasgupta A, Grzelak CA, Kim J, Barrett A, Coleman IM, et al. Targeting the perivascular niche sensitizes disseminated tumour cells to chemotherapy. Nat Cell Biol. (2019) 21:238–50. doi: 10.1038/s41556-018-0267-0
58. Ogba N, Manning NG, Bliesner BS, Ambler SK, Haughian JM, Pinto MP, et al. Luminal breast cancer metastases and tumor arousal from dormancy are promoted by direct actions of estradiol and progesterone on the malignant cells. Breast Cancer Res. (2014) 16:489. doi: 10.1186/s13058-014-0489-4
59. Polzer B, Klein CA. Metastasis awakening: the challenges of targeting minimal residual cancer. Nat Med. (2013) 19:274–5. doi: 10.1038/nm.3121
60. Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol. (2018) 20:1240–9. doi: 10.1038/s41556-018-0214-0
61. Linde N, Fluegen G, Aguirre-Ghiso JA. The relationship between dormant cancer cells and their microenvironment. Adv Cancer Res. (2016) 132:45–71. doi: 10.1016/bs.acr.2016.07.002
62. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. (2014) 14:611–22. doi: 10.1038/nrc3793
63. Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. (2003) 63:1684–95. doi: 10.1016/j.urolonc.2003.12.012
64. Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. (2001) 12:863–79. doi: 10.1091/mbc.12.4.863
65. Sosa MS, Parikh F, Maia AG, Estrada Y, Bosch A, Bragado P, et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat Commun. (2015) 6:6170. doi: 10.1038/ncomms7170
66. Borgen E, Rypdal MC, Sosa MS, Renolen A, Schlichting E, Lonning PE, et al. NR2F1 stratifies dormant disseminated tumor cells in breast cancer patients. Breast Cancer Res. (2018) 20:120. doi: 10.1186/s13058-018-1049-0
67. Lytle NK, Ferguson LP, Rajbhandari N, Gilroy K, Fox RG, Deshpande A, et al. A multiscale map of the stem cell state in pancreatic adenocarcinoma. Cell. (2019) 177:572–86.e22. doi: 10.1016/j.cell.2019.03.010
68. Domenici G, Aurrekoetxea-Rodriguez I, Simoes BM, Rabano M, Lee SY, Millan JS, et al. A Sox2-Sox9 signalling axis maintains human breast luminal progenitor and breast cancer stem cells. Oncogene. (2019) 38:3151–69. doi: 10.1038/s41388-018-0656-7
69. Wang L, Zhang Z, Yu X, Huang X, Liu Z, Chai Y, et al. Unbalanced YAP-SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene. (2019) 38:2042–55. doi: 10.1038/s41388-018-0476-9
70. Yuan X, Li J, Coulouarn C, Lin T, Sulpice L, Bergeat D, et al. SOX9 expression decreases survival of patients with intrahepatic cholangiocarcinoma by conferring chemoresistance. Br J Cancer. (2018) 119:1358–66. doi: 10.1038/s41416-018-0338-9
71. Rahmanto AS, Savov V, Brunner A, Bolin S, Weishaupt H, Malyukova A, et al. FBW7 suppression leads to SOX9 stabilization and increased malignancy in medulloblastoma. EMBO J. (2016) 35:2192–212. doi: 10.15252/embj.201693889
72. Shimizu H, Takeishi S, Nakatsumi H, Nakayama KI. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight. (2019) 4:e125138. doi: 10.1172/jci.insight.125138
73. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
74. Ren D, Dai Y, Yang Q, Zhang X, Guo W, Ye L, et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J Exp Med. (2019) 216:428–49. doi: 10.1084/jem.20180661
75. Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer. (2018) 18:69–88. doi: 10.1038/nrc.2017.105
76. Endo H, Okami J, Okuyama H, Nishizawa Y, Imamura F, Inoue M. The induction of MIG6 under hypoxic conditions is critical for dormancy in primary cultured lung cancer cells with activating EGFR mutations. Oncogene. (2017) 36:2824–34. doi: 10.1038/onc.2016.431
77. Lu H, Chen I, Shimoda LA, Park Y, Zhang C, Tran L, et al. Chemotherapy-induced Ca(2+) release stimulates breast cancer stem cell enrichment. Cell Rep. (2017) 18:1946–57. doi: 10.1016/j.celrep.2017.02.001
78. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: from oncogenesis to chemoresistance. Biol Cell. (2019) 111:1–17. doi: 10.1111/boc.201800050
79. Ranganathan AC, Zhang L, Adam AP, Aguirre-Ghiso JA. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. (2006) 66:1702–11. doi: 10.1158/0008-5472.CAN-05-3092
80. Dauer P, Sharma NS, Gupta VK, Durden B, Hadad R, Banerjee S, et al. ER stress sensor, glucose regulatory protein 78 (GRP78) regulates redox status in pancreatic cancer thereby maintaining “stemness”. Cell Death Dis. (2019) 10:132. doi: 10.1038/s41419-019-1408-5
81. Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, et al. Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol Cancer Ther. (2016) 15:1043–52. doi: 10.1158/1535-7163.MCT-15-0774
82. Terai H, Kitajima S, Potter DS, Matsui Y, Quiceno LG, Chen T, et al. ER stress signaling promotes the survival of cancer “persister cells” tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. (2018) 78:1044–57. doi: 10.1158/0008-5472.CAN-17-1904
83. Yan MM, Ni JD, Song D, Ding M, Huang J. Interplay between unfolded protein response and autophagy promotes tumor drug resistance. Oncol Lett. (2015) 10:1959–69. doi: 10.3892/ol.2015.3508
84. Gupta A, Roy S, Lazar AJ, Wang WL, McAuliffe JC, Reynoso D, et al. Autophagy inhibition and antimalarials promote cell death in gastrointestinal stromal tumor (GIST). Proc Natl Acad Sci USA. (2010) 107:14333–8. doi: 10.1073/pnas.1000248107
85. Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. (2008) 118:3917–29. doi: 10.1172/JCI35512
86. Rabinowitz JD, White E. Autophagy and metabolism. Science. (2010) 330:1344–8. doi: 10.1126/science.1193497
87. Ou J, Peng Y, Yang W, Zhang Y, Hao J, Li F, et al. ABHD5 blunts the sensitivity of colorectal cancer to fluorouracil via promoting autophagic uracil yield. Nat Commun. (2019) 10:1078. doi: 10.1038/s41467-019-08902-x
88. Huang X, Gan G, Wang X, Xu T, Xie W. The HGF-MET axis coordinates liver cancer metabolism and autophagy for chemotherapeutic resistance. Autophagy. (2019) 15:1258–79. doi: 10.1080/15548627.2019.1580105
89. Moeckel S, LaFrance K, Wetsch J, Seliger C, Riemenschneider MJ, Proescholdt M, et al. ATF4 contributes to autophagy and survival in sunitinib treated brain tumor initiating cells (BTICs). Oncotarget. (2019) 10:368–82. doi: 10.18632/oncotarget.26569
90. Ojha R, Leli NM, Onorati A, Piao S, Verginadis II, Tameire F, et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF-mutant melanoma. Cancer Discov. (2019) 9:396–415. doi: 10.1158/2159-8290.CD-18-0348
91. Vera-Ramirez L, Hunter KW. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Res. (2017) 6:2134. doi: 10.12688/f1000research.12174.1
92. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. (2019) 25:620–27. doi: 10.1038/s41591-019-0367-9
93. Li S, Song Y, Quach C, Guo H, Jang GB, Maazi H, et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat Commun. (2019) 10:1693. doi: 10.1038/s41467-019-09634-8
94. Zeuner A. The secret life of quiescent cancer stem cells. Mol Cell Oncol. (2015) 2:e968067. doi: 10.4161/23723548.2014.968067
95. Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. (2017) 1868:1–6. doi: 10.1016/j.bbcan.2016.12.004
96. Giannoni E, Taddei ML, Morandi A, Comito G, Calvani M, Bianchini F, et al. Targeting stromal-induced pyruvate kinase M2 nuclear translocation impairs oxphos and prostate cancer metastatic spread. Oncotarget. (2015) 6:24061–74. doi: 10.18632/oncotarget.4448
97. Ippolito L, Marini A, Cavallini L, Morandi A, Pietrovito L, Pintus G, et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells. Oncotarget. (2016) 7:61890–904. doi: 10.18632/oncotarget.11301
98. Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. (2016) 23:1542–54. doi: 10.1038/cdd.2016.39
99. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. (2014) 514:628–32. doi: 10.1038/nature13611
100. Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J, et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. (2012) 72:304–14. doi: 10.1158/0008-5472.CAN-11-1674
101. Bacci M, Giannoni E, Fearns A, Ribas R, Gao Q, Taddei ML, et al. miR-155 drives metabolic reprogramming of ER+ breast cancer cells following long-term estrogen deprivation and predicts clinical response to aromatase inhibitors. Cancer Res. (2016) 76:1615–26. doi: 10.1158/0008-5472.CAN-15-2038
102. Botzer LE, Maman S, Sagi-Assif O, Meshel T, Nevo I, Yron I, et al. Hexokinase 2 is a determinant of neuroblastoma metastasis. Br J Cancer. (2016) 114:759–66. doi: 10.1038/bjc.2016.26
103. Bovenzi CD, Hamilton J, Tassone P, Johnson J, Cognetti DM, Luginbuhl A, et al. Prognostic indications of elevated MCT4 and CD147 across Cancer types: a meta-analysis. Biomed Res Int. (2015) 2015:242437. doi: 10.1155/2015/242437
104. Doyen J, Trastour C, Ettore F, Peyrottes I, Toussant N, Gal J, et al. Expression of the hypoxia-inducible monocarboxylate transporter MCT4 is increased in triple negative breast cancer and correlates independently with clinical outcome. Biochem Biophys Res Commun. (2014) 451:54–61. doi: 10.1016/j.bbrc.2014.07.050
105. Elgendy M, Cirò M, Hosseini A, Weiszmann J, Mazzarella L, Ferrari E, et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3β-MCL-1 axis. Cancer Cell. (2019) 35:798–815.e5. doi: 10.1016/j.ccell.2019.03.007
106. Zhang X, De Milito A, Demiroglu-Zergeroglu A, Gullbo J, D'Arcy P, Linder S. Eradicating quiescent tumor cells by targeting mitochondrial bioenergetics. Trends Cancer. (2016) 2:657–63. doi: 10.1016/j.trecan.2016.10.009
107. Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nat Commun. (2016) 7:10442. doi: 10.1038/ncomms10442
108. Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol. (2018) 20:597–609. doi: 10.1038/s41556-018-0083-6
109. Rapisarda A, Melillo G. Role of the hypoxic tumor microenvironment in the resistance to anti-angiogenic therapies. Drug Resist Updat. (2009) 12:74–80. doi: 10.1016/j.drup.2009.03.002
110. Kuang R, Jahangiri A, Mascharak S, Nguyen A, Chandra A, Flanigan PM, et al. GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight. (2017) 2:e88815. doi: 10.1172/jci.insight.88815
111. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
112. Pfister SX, Ashworth A. Marked for death: targeting epigenetic changes in cancer. Nat Rev Drug Discov. (2017) 16:241–63. doi: 10.1038/nrd.2016.256
113. Flavahan WA, Gaskell E, Bernstein BE. Epigenetic plasticity and the hallmarks of cancer. Science. (2017) 357:eaal2380. doi: 10.1126/science.aal2380
114. Wawruszak A, Kalafut J, Okon E, Czapinski J, Halasa M, Przybyszewska A, et al. Histone deacetylase inhibitors and phenotypical transformation of cancer cells. Cancers. (2019) 11:148. doi: 10.3390/cancers11020148
115. Dalvi MP, Wang L, Zhong R, Kollipara RK, Park H, Bayo J, et al. Taxane-platin-resistant lung cancers co-develop hypersensitivity to jumonjic demethylase inhibitors. Cell Rep. (2017) 19:1669–84. doi: 10.1016/j.celrep.2017.04.077
116. Guler GD, Tindell CA, Pitti R, Wilson C, Nichols K, KaiWai Cheung T, et al. Repression of stress-induced LINE-1 expression protects cancer cell subpopulations from lethal drug exposure. Cancer Cell. (2017) 32:221–37.e13. doi: 10.1016/j.ccell.2017.07.002
117. Risom T, Langer EM, Chapman MP, Rantala J, Fields AJ, Boniface C, et al. Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer. Nat Commun. (2018) 9:3815. doi: 10.1038/s41467-018-05729-w
118. Staberg M, Rasmussen RD, Michaelsen SR, Pedersen H, Jensen KE, Villingshoj M, et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol Oncol. (2018) 12:406–20. doi: 10.1002/1878-0261.12174
119. Vinogradova M, Gehling VS, Gustafson A, Arora S, Tindell CA, Wilson C, et al. An inhibitor of KDM5 demethylases reduces survival of drug-tolerant cancer cells. Nat Chem Biol. (2016) 12:531–8. doi: 10.1038/nchembio.2085
120. Hinohara K, Wu HJ, Vigneau S, McDonald TO, Igarashi KJ, Yamamoto KN, et al. KDM5 histone demethylase activity links cellular transcriptomic heterogeneity to therapeutic resistance. Cancer Cell. (2018) 34:939–53.e9. doi: 10.1016/j.ccell.2018.10.014
121. Leadem BR, Kagiampakis I, Wilson C, Cheung TK, Arnott D, Trojer P, et al. A KDM5 inhibitor increases global H3K4 trimethylation occupancy and enhances the biological efficacy of 5-Aza-2'-deoxycytidine. Cancer Res. (2018) 78:1127–39. doi: 10.1158/0008-5472.CAN-17-1453
122. Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. (2018) 24:1599–610. doi: 10.1038/s41591-018-0158-8
123. Iniguez AB, Alexe G, Wang EJ, Roti G, Patel S, Chen L, et al. Resistance to epigenetic-targeted therapy engenders tumor cell vulnerabilities associated with enhancer remodeling. Cancer Cell. (2018) 34:922–38.e7. doi: 10.1016/j.ccell.2018.11.005
124. Manzotti G, Ciarrocchi A, Sancisi V. Inhibition of BET proteins and histone deacetylase (HDACs): crossing roads in cancer therapy. Cancers. (2019) 11:304. doi: 10.3390/cancers11030304
125. Roche B, Arcangioli B, Martienssen R. Transcriptional reprogramming in cellular quiescence. RNA Biol. (2017) 14:843–53. doi: 10.1080/15476286.2017.1327510
126. Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. (2016) 7:10690. doi: 10.1038/ncomms10690
127. Ishak CA, Classon M, De Carvalho DD. Deregulation of retroelements as an emerging therapeutic opportunity in cancer. Trends Cancer. (2018) 4:583–97. doi: 10.1016/j.trecan.2018.05.008
128. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature. (2017) 548:471–5. doi: 10.1038/nature23465
129. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. (2019) 25:403–18. doi: 10.1038/s41591-019-0376-8
130. Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. (2018) 53:189–200. doi: 10.1016/j.semcancer.2018.09.006
131. Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, et al. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. (2019) 177:1172–86.e14. doi: 10.1016/j.cell.2019.03.025
132. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol. (2013) 25:214–21. doi: 10.1016/j.coi.2012.12.003
133. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
134. Mohme M, Riethdorf S, Pantel K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol. (2017) 14:155–67. doi: 10.1038/nrclinonc.2016.144
135. Pommier A, Anaparthy N, Memos N, Kelley ZL, Gouronnec A, Yan R, et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science. (2018) 360:eaao4908. doi: 10.1126/science.aao4908
136. Aqbi HF, Wallace M, Sappal S, Payne KK, Manjili MH. IFN-gamma orchestrates tumor elimination, tumor dormancy, tumor escape, and progression. J Leukoc Biol. (2018) 103:1219–23. doi: 10.1002/JLB.5MIR0917-351R
137. Payne KK, Keim RC, Graham L, Idowu MO, Wan W, Wang XY, et al. Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. J Leukoc biol. (2016) 100:625–35. doi: 10.1189/jlb.5A1215-580R
138. Budhwani M, Mazzieri R, Dolcetti R. Plasticity of type I interferon-mediated responses in cancer therapy: from anti-tumor immunity to resistance. Front Oncol. (2018) 8:322. doi: 10.3389/fonc.2018.00322
139. Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. (2017) 77:839–50. doi: 10.1158/0008-5472.CAN-15-3142
140. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. (2017) 8:15207. doi: 10.1038/ncomms15207
141. Liu Y, Lv J, Liu J, Liang X, Jin X, Xie J, et al. STAT3/p53 pathway activation disrupts IFN-beta-induced dormancy in tumor-repopulating cells. J Clin Invest. (2018) 128:1057–73. doi: 10.1172/JCI96329
142. Ranoa DRE, Widau RC, Mallon S, Parekh AD, Nicolae CM, Huang X, et al. STING promotes homeostasis via regulation of cell proliferation and chromosomal stability. Cancer Res. (2019) 79:1465–79. doi: 10.1158/0008-5472.CAN-18-1972
143. Nassour J, Radford R, Correia A, Fuste JM, Schoell B, Jauch A, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. (2019) 565:659–63. doi: 10.1038/s41586-019-0885-0
144. Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. (2018) 553:467–72. doi: 10.1038/nature25432
145. Hou Y, Liang H, Rao E, Zheng W, Huang X, Deng L, et al. Non-canonical NF-kappaB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity. (2018) 49:490–503.e4. doi: 10.1016/j.immuni.2018.07.008
146. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A, et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. (2016) 533:493–8. doi: 10.1038/nature18268
147. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
148. Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. (2010) 468:701–4. doi: 10.1038/nature09595
149. Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. (2019) 9:34–45. doi: 10.1158/2159-8290.CD-18-0689
150. Cheng N, Watkins-Schulz R, Junkins RD, David CN, Johnson BM, Montgomery SA, et al. A nanoparticle-incorporated STING activator enhances antitumor immunity in PD-L1-insensitive models of triple-negative breast cancer. JCI Insight. (2018) 3:e120638. doi: 10.1172/jci.insight.120638
151. Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. (2018) 361:eaao4227. doi: 10.1126/science.aao4227
152. Cooper J, Giancotti FG. Integrin signaling in cancer: mechanotransduction, stemness, epithelial plasticity, and therapeutic resistance. Cancer Cell. (2019) 35:347–67. doi: 10.1016/j.ccell.2019.01.007
153. Abderrahman B, Jordan VC. Rethinking extended adjuvant antiestrogen therapy to increase survivorship in breast cancer. JAMA Oncol. (2018) 4:15–6. doi: 10.1001/jamaoncol.2017.3510
154. O'Leary B, Finn RS, Turner NC. Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol. (2016) 13:417–30. doi: 10.1038/nrclinonc.2016.26
155. El Touny LH, Vieira A, Mendoza A, Khanna C, Hoenerhoff MJ, Green JE. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J Clin Invest. (2014) 124:156–68. doi: 10.1172/JCI70259
156. Chen JY, Lin JR, Tsai FC, Meyer T. Dosage of dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol cell. (2013) 52:87–100. doi: 10.1016/j.molcel.2013.09.009
157. Soppa U, Schumacher J, Florencio Ortiz V, Pasqualon T, Tejedor FJ, Becker W. The down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle. (2014) 13:2084–100. doi: 10.4161/cc.29104
158. Orienti I, Francescangeli F, De Angelis ML, Fecchi K, Bongiorno L, Signore M, et al. A new bioavailable fenretinide formulation with antiproliferative, antimetabolic and cytotoxic effects on solid tumors. Cell Death Disease. (in press).
159. Essers MA, Trumpp A. Targeting leukemic stem cells by breaking their dormancy. Mol Oncol. (2010) 4:443–50. doi: 10.1016/j.molonc.2010.06.001
160. Vanner RJ, Remke M, Gallo M, Selvadurai HJ, Coutinho F, Lee L, et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell. (2014) 26:33–47. doi: 10.1016/j.ccr.2014.05.005
161. Rajbhandari N, Lin WC, Wehde BL, Triplett AA, Wagner KU. Autocrine IGF1 signaling mediates pancreatic tumor cell dormancy in the absence of oncogenic drivers. Cell Rep. (2017) 18:2243–55. doi: 10.1016/j.celrep.2017.02.013
162. Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. (2017) 551:247–50. doi: 10.1038/nature24297
163. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
164. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med. (2017) 23:1234–40. doi: 10.1038/nm.4399
165. Roesch A, Vultur A, Bogeski I, Wang H, Zimmermann KM, Speicher D, et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell. (2013) 23:811–25. doi: 10.1016/j.ccr.2013.05.003
166. Goff DJ, Court Recart A, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. (2013) 12:316–28. doi: 10.1016/j.stem.2012.12.011
167. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. (2013) 12:329–41. doi: 10.1016/j.stem.2012.12.013
168. Carreau N, Pavlick A. Revolutionizing treatment of advanced melanoma with immunotherapy. Surg Oncol. (2019). doi: 10.1016/j.suronc.2019.01.002. [Epub ahead of print].
169. Esteva FJ, Hubbard-Lucey VM, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. (2019) 20:e175–86. doi: 10.1016/S1470-2045(19)30026-9
170. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
171. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. (2017) 356:200–5. doi: 10.1126/science.aak9510
172. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. (2015) 350:1387–90. doi: 10.1126/science.aad1253
173. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
174. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. (2014) 344:641–5. doi: 10.1126/science.1251102
175. De Cock JM, Shibue T, Dongre A, Keckesova Z, Reinhardt F, Weinberg RA. Inflammation triggers Zeb1-dependent escape from tumor latency. Cancer Res. (2016) 76:6778–84. doi: 10.1158/0008-5472.CAN-16-0608
176. El Rayes T, Catena R, Lee S, Stawowczyk M, Joshi N, Fischbach C, et al. Lung inflammation promotes metastasis through neutrophil protease-mediated degradation of Tsp-1. Proc Natl Acad Sci USA. (2015) 112:16000–5. doi: 10.1073/pnas.1507294112
177. Retsky M, Demicheli R, Hrushesky WJ, Forget P, De Kock M, Gukas I, et al. Reduction of breast cancer relapses with perioperative non-steroidal anti-inflammatory drugs: new findings and a review. Curr Med Chem. (2013) 20:4163–76. doi: 10.2174/09298673113209990250
178. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. (2012) 379:1591–601. doi: 10.1016/S0140-6736(12)60209-8
179. Song M, Chan AT. The potential role of exercise and nutrition in harnessing the immune system to improve colorectal cancer survival. Gastroenterology. (2018) 155:596–600. doi: 10.1053/j.gastro.2018.07.038
180. Grzelak CA, Ghajar CM. Metastasis ‘systems’ biology: how are macro-environmental signals transmitted into microenvironmental cues for disseminated tumor cells? Curr Opin Cell Biol. (2017) 48:79–86. doi: 10.1016/j.ceb.2017.06.002
181. Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D, et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. (2018) 560:499–503. doi: 10.1038/s41586-018-0343-4
182. Nencioni A, Caffa I, Cortellino S, Longo VD. Fasting and cancer: molecular mechanisms and clinical application. Nat Rev Cancer. (2018) 18:707–19. doi: 10.1038/s41568-018-0061-0
Keywords: cancer stem cells, chemoresistance, dormancy, quiescence, plasticity, drug resistance, target therapies
Citation: De Angelis ML, Francescangeli F, La Torre F and Zeuner A (2019) Stem Cell Plasticity and Dormancy in the Development of Cancer Therapy Resistance. Front. Oncol. 9:626. doi: 10.3389/fonc.2019.00626
Received: 08 May 2019; Accepted: 24 June 2019;
Published: 10 July 2019.
Edited by:
Alexandre Prieur, ECS-Screening SA, SwitzerlandReviewed by:
Loredana Bergandi, University of Turin, ItalyBenjamin Le Calvé, University of Namur, Belgium
Copyright © 2019 De Angelis, Francescangeli, La Torre and Zeuner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ann Zeuner, ann.zeuner@iss.it