 Daisuke Obinata1,2
Daisuke Obinata1,2 Mitchell G. Lawrence2,3,4Kenichi Takayama5
Mitchell G. Lawrence2,3,4Kenichi Takayama5 Nicholas Choo2Gail P. Risbridger2,3,4Satoru Takahashi1
Nicholas Choo2Gail P. Risbridger2,3,4Satoru Takahashi1 Satoshi Inoue5,6*
Satoshi Inoue5,6*- 1Department of Urology, Nihon University School of Medicine, Tokyo, Japan
- 2Monash Biomedicine Discovery Institute Cancer Program, Prostate Cancer Research Group, Department of Anatomy and Developmental Biology, Monash University, Clayton, VIC, Australia
- 3Cancer Research Division, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 4Sir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, VIC, Australia
- 5Department of Systems Aging Science and Medicine, Tokyo Metropolitan Institute of Gerontology, Tokyo, Japan
- 6Research Center for Genomic Medicine, Saitama Medical University, Saitama, Japan
The androgen receptor (AR) is the main therapeutic target in advanced prostate cancer, because it regulates the growth and progression of prostate cancer cells. Patients may undergo multiple lines of AR-directed treatments, including androgen-deprivation therapy, AR signaling inhibitors (abiraterone acetate, enzalutamide, apalutamide, or darolutamide), or combinations of these therapies. Yet, tumors inevitably develop resistance to the successive lines of treatment. The diverse mechanisms of resistance include reactivation of the AR and dysregulation of AR cofactors and collaborative transcription factors (TFs). Further elucidating the nexus between the AR and collaborative TFs may reveal new strategies targeting the AR directly or indirectly, such as targeting BET proteins or OCT1. However, appropriate preclinical models will be required to test the efficacy of these approaches. Fortunately, an increasing variety of patient-derived models, such as xenografts and organoids, are being developed for discovery-based research and preclinical drug screening. Here we review the mechanisms of drug resistance in the AR signaling pathway, the intersection with collaborative TFs, and the use of patient-derived models for novel drug discovery.
Introduction
Prostate cancer is one of the most common causes of cancer-related death among men in Western countries. At diagnosis, most prostate cancers rely on the androgen receptor (AR) signaling for growth and survival. In this pathway, the AR is bound by ligands, such as dihydrotestosterone (DHT), and regulates the expression of target genes (1–5). In addition, the AR collaborates with cofactors, including transcription factors (TFs), which bind to specific DNA elements in regulatory regions of AR-responsive genes. Since AR collaborative TFs fine-tune androgen-responsive gene expression, it is important to further elucidate their role in the progression of prostate cancer.
In normal prostate epithelium, the AR suppresses proliferation and promotes differentiation (6); however, during carcinogenesis prostate cancer cells develop “lineage-addiction,” where the AR promotes tumor progression (7). Given the importance of the AR pathway in prostate cancer, it is the target of most treatments for advanced disease. Androgen deprivation therapy (ADT), through surgical or pharmacological castration, is initially effective at reducing tumor burden. ADT is administered alone or in combination with chemotherapy or AR-signaling inhibitors (8). A subset of cancer cells withstand treatment and eventually develop into castration-resistant prostate cancer (CRPC), which proliferates despite castrate concentrations of circulating androgens. Since AR signaling persists in most cases of CRPC, patients receive further treatment with AR signaling inhibitors (abiraterone acetate, enzalutamide, apalutamide, and darolutamide) based on whether they have metastatic or non-metastatic disease (9–12). Yet, tumors inevitably acquire further resistance, often by reactivating AR signaling. Once patients fail an AR signaling inhibitor, further treatments include another AR-directed therapy, chemotherapy, or if there are genomic defects in homologous recombination repair genes, a PARP inhibitor (13, 14). However, as CRPC is ultimately lethal, there is an ongoing need for new treatments.
An important step in developing novel therapies is testing their effectiveness in preclinical models. Although there is a longstanding paucity of preclinical models of CRPC, larger collections of patient-derived models are providing new tools to validate and prioritize candidate treatments for clinical trials. In this review, we examine mechanisms of castration-resistance involving the AR and collaborating TFs, new strategies for targeting tumors with these features, and the use of different patient-derived models for testing these novel treatments.
Mechanisms of Castration-Resistance Through Alterations of the AR
The AR gene on Xq11-13 consists of 8 exons encoding the N-terminal domain (NTD; 555 amino acids; exon 1), DNA-binding domain (DBD; 68 amino acids; exons 2 and 3), hinge region (40 amino acids; exon 4), and ligand binding domain (LBD; 295 amino acids; exons 4–8) (15, 16). Binding of androgens to the LBD triggers an intramolecular interaction with the NTD, which in turn interacts with AR co-activators (17, 18).
Amplifications of the AR locus are one the most common mechanisms of castration-resistance, and they often encompass an enhancer located ~700 kilobases upstream (19–21). In some tumors, the AR gene and enhancer are amplified independently of each other (21). The AR enhancer is bound by several transcriptional activators, including FOXA1, GATA2, NKX3.1, HOXB13, and the AR itself (20). Amplifications of the AR and its enhancer are associated with higher levels of AR expression, and over-expressing the AR in prostate cancer cell lines causes enzalutamide-resistance (20, 21). Accordingly, patients with amplifications of the AR locus and/or enhancer are often resistant to AR-directed therapies, including enzalutamide and abiraterone acetate (22, 23). In preclinical studies with VCaP cells, which have an AR amplification and express high levels of the AR, darolutamide had a lower IC50 than enzalutamide and apalutamide in suppressing proliferation (24), suggesting that potent inhibition of the AR may be required for tumors with this mechanism of resistance.
The conformation of the AR can be disrupted by point mutations, which commonly arise in CRPC and mediate resistance to AR-directed treatments (25, 26). Occasionally, two AR mutations can occur in the same tumor (27–30). Point mutations often occur in the LBD, causing gain-of-function in ligand binding, so the AR is activated by other steroids, and antagonists, like enzalutamide, are converted into agonists (31–33) (Supplementary Table 1). Since AR mutations confer resistance to particular antagonists, they are potential predictive biomarkers for AR-directed inhibitors. Enzalutamide may not be suitable for tumors with AR mutations that convert it into a partial agonist (F877L, H875Y/T878A, F877L/T878A) (27). Darolutamide might be more effective for these tumors, since it remains an antagonist despite these AR mutations (34). In addition, darolutamide has unique flexibility that allows it to bind the W742C/L mutated ligand-binding pocket, unlike enzalutamide (35). However, the utility of AR mutations as biomarkers needs confirmation in patients. For example, the F877L AR mutation converts apalutamide into a partial agonist in vitro, but neither this mutation nor T878A was a common cause of acquired resistance to apalutamide in a phase I/II trial (36).
In addition to AR mutations, constitutively active AR splice variants (ARVs) can mediate castration resistance (37). Increased expression of ARVs can arise through amplifications or structural rearrangements of the AR gene in CRPC (20, 30, 38, 39). Among numerous ARVs, AR-V7, and ARv567es have been studied in the most detail. AR-V7 includes exons 1/2/3, encoding the NTD, followed by a cryptic exon (Figures 1A,B) (37). ARv567es includes exons 1/2/3/4/8, but skips exons 5/6/7 (40). Since both variants lack the LBD, they are not bound by most AR signaling inhibitors, so can sustain AR-driven gene expression. The lack of the AR hinge region in AR-V7 may also promote therapy resistance. SPOP (speckle type POZ protein), an E3 ubiquitin ligase that is upregulated by enzalutamide treatment, usually binds to the hinge region of the AR and induces its degradation (41). By escaping this ubiquitin degradation pathway, AR-V7 may enable enzalutamide resistance. In addition, the hinge region usually mediates microtubule binding and translocation of the AR into the nucleus (42, 43). Since AR-V7 lacks the hinge region, its transport is independent of microtubules, enabling resistance to taxane chemotherapy, which targets microtubules, unlike ARv567es which still contains the hinge region (43, 44). Cell-cycle or cell-division associated genes such as ubiquitin-conjugating enzyme E2 C (UBE2C) are unique AR-V7 targets, contributing to cell proliferation under androgen-depleted conditions (45, 46). Recent functional analyses demonstrated the importance of various splicing factors, which are highly expressed in CRPC tissues (47–50). Enhanced expression of splicing factors would promote their recruitment to pre-mRNA, facilitating the mRNA splicing process. Thus, altered splicing machinery would result in a dysregulated AR splicing process. Splicing factor, proline- and glutamine-rich (PSF/SFPQ) is responsible for wide-ranging upregulation of spliceosome gene expression in CRPC to activate a broad range of oncogenic pathways, including AR (48). Thus, these studies provide an intriguing insight into prostate cancer progression through splicing machinery.
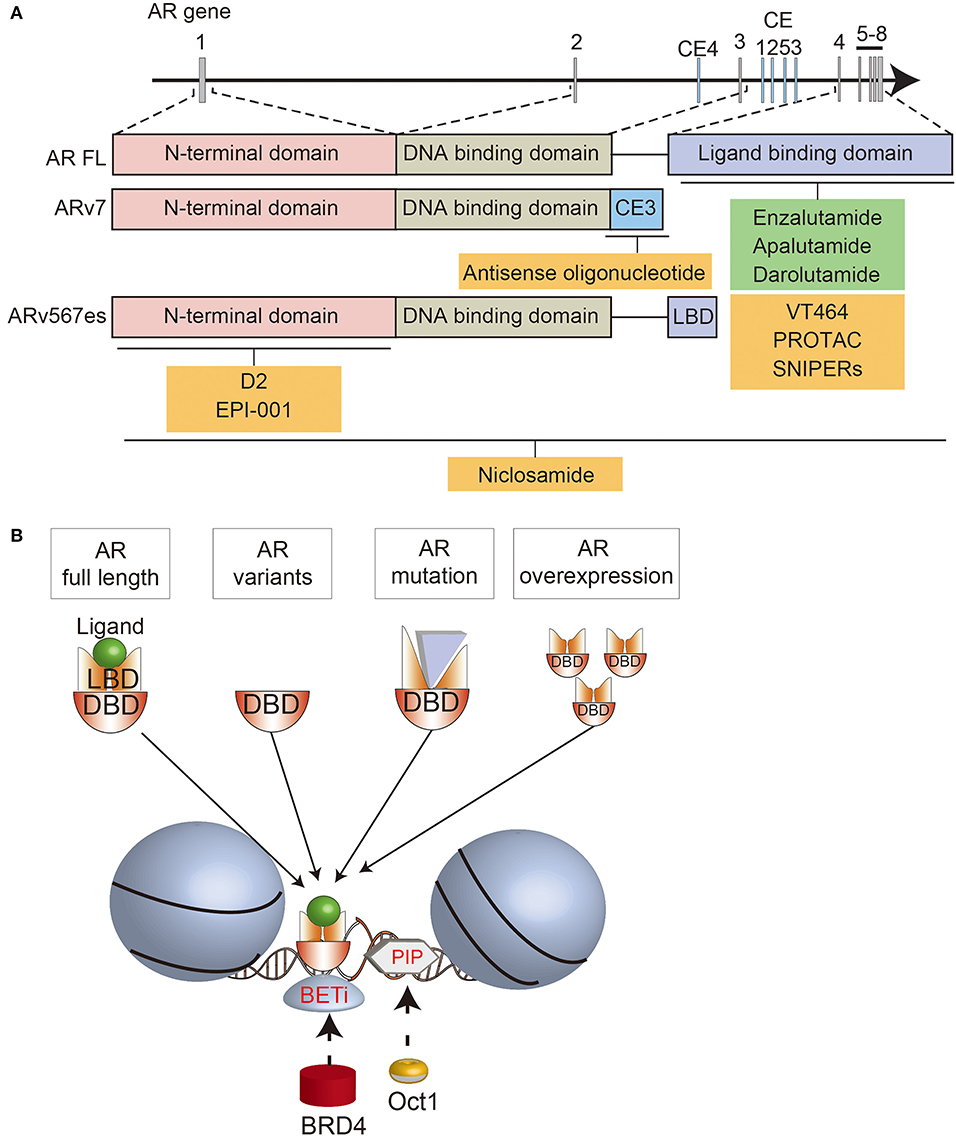
Figure 1. Schematic summary of the AR structure, AR-directed treatments, and interactions with BRD4 and OCT1. (A) Overview of the AR locus, the structure of full length (FL) and variant (AR-V7, ARv567es) forms of the AR, and various AR-targeted treatments that are approved (green) or in development (yellow). (B) Summary of the interactions between different forms of the AR, BRD4, and OCT1 on chromatin.
New Strategies for Directly Targeting the AR in CRPC
In an effort to overcome resistance to current treatments, new therapies are being developed to target the AR. Some compounds have a similar mechanisms-of-action to existing AR-directed treatments. Like abiraterone actetate, the new compound VT464 (seviteronel) is a CYP17A1 inhibitor that suppresses androgen biosynthesis (51). Unlike abiraterone acetate, VT464, selectively inhibits the 17,20-lyase rather than 17α-hydroxylase reactions, so it is proposed that the combination with prednisone is not necessary. However, phase 1 testing of VT464 suggested that there is minor inhibition of CYP17 hydroxylase (52), so low-dose dexamethasone is being administered with VT464 in ongoing trials with prostate cancer patients (53, 54). Abiraterone acetate and VT464 both also function as competitive AR antagonists, including of AR mutants, with VT464 more potent than abiraterone in cells with the T878A AR mutation (55–58).
An alternative strategy is to deplete the AR in prostate cancer cells. This may be done by blocking gene expression with antisense oligonucleotides targeting different regions of AR pre-mRNA transcripts (59–62). Antisense oligonucleotides against exon 1 reduce full-length AR and ARV expression, while antisense oligonucleotides against cryptic splicing signals specifically downregulate AR-V7 expression (60, 62). The AR can also be depleted using selective AR degraders (SARD), which bind to the AR and induce proteasome-mediated degradation (63). Some SARDs bind to both the N- and C-termini of the AR, so also promote degradation of ARVs (64). Preclinical studies suggested that niclosamide, an approved treatment for parasitic worms, could be repurposed as a SARD, since one of its effects is degradation of ARVs. Although the combination of niclosamide with enzalutamide or abiraterone significantly reduced the growth of castrate-resistant cells (65–67), a phase I trial showed that inhibitory concentrations of niclosamide could not be achieved in patients (68). Therefore, this approach must rely on newer generations of SARDs being developed (64).
Another way of inducing AR degradation is with proteolysis targeting chimeras (PROTAC) or SNIPERs (specific and non-genetic inhibitor of apoptosis protein [IAP]-dependent protein erasers). These heterobifunctional small molecules contain a ligand that binds to the target protein, such as an AR antagonist, linked to another ligand that engages the ubiquitin ligase complex (69–71). Since current AR-targeted PROTACs bind to the LBD, they induce degradation of full-length AR, but not ARVs. Nevertheless, they still inhibit the growth of enzalutamide-resistant cells, emphasizing the ongoing importance of full-length AR in many cases of CRPC (71).
A different strategy for directly targeting the AR is to disrupt its interactions with other molecules. D2 is a peptidomimetic that disrupts the interaction between the AR and a co-regulator, PELP1 (proline, glutamate and leucine rich protein 1), by mimicking the LXXLL motif in the AF2 domain of the AR C-terminus (72). By blocking this interaction, D2 inhibits nuclear translocation of the AR and reduces the growth of prostate cancer cells. EPI-001 also blocks the interactions between the AR and coactivators, and is notable because it binds the NTD (73). Thus, EPI-001 also inhibits ARVs (74). EPI-001 inhibits the growth of prostate cancer cell lines in vitro and in vivo, and had increased activity in combination with docetaxel (74, 75). However, off-target effects have been identified, highlighting the difficulty of targeting the AR NTD (76). EPI-506 was developed as a successor to EPI-001 (77), but was required at high doses in a phase I trial due to low potency and a short half-life. Therefore, the development of N-terminal AR inhibitors is ongoing (78).
Compounds are also being developed to block the interaction of the AR with DNA. This could target both full-length and variant forms of the AR, which contain the DBD. For example, AR binding to specific androgen response element sequences can be blocked using PI polyamides, N-methylimidazole (Im) and N-methylpyrrole (Py) amino acids that bind to the minor groove of DNA with high affinity and sequence specificity (79–81). PI polyamides that bind particular AREs can suppress androgen-responsive gene expression (82), and inhibit binding of RNA polymerase II to the transcription start site of AR-driving genes (83).
Indirectly Targeting AR Signaling via Cofactors and Collaborating Transcription Factors
Cofactors, including coregulators and TFs, are also necessary for AR-regulated gene expression. Whilst coregulators directly bind to activation function (AF) 1 or 2 domains of the AR, TFs bind to DNA elements near AR binding sites (84). Some TFs are also pioneer factors that facilitate AR recruitment to target regions through chromatin remodeling (85). Dysregulation of TFs can dramatically change the pattern of AR responsive gene expression. Indeed, there are differences in AR binding regions and coordinating TFs between treatment-naïve and castration-resistant prostate cancer (86). AR binding sites that are unique to CRPC were not AR-regulated in treatment-naïve prostate cancer cells or enriched in binding of common AR collaborative TFs, such as MYC (86). MYC is a oncogenic transcription factor that plays a critical role in prostate cancer progression by influencing diverse molecular mechanisms (87).
The importance of cofactors and collaborative TFs makes them potential therapeutic targets for indirectly targeting the AR. There are numerous strategies for targeting different AR interacting proteins, so here we focus on two notable examples, bromodomain and extra terminal domain (BET) proteins and OCT1 (POU2F1; POU class 2 homeobox 1) that collaborate with MYC.
BET Proteins
The BET family of epigenetic readers, including BRD2/3/4 (bromodomain containing 2, 3, and 4) and BRDT (bromodomain testis associated), bind to acetylated histones and regulate the expression of downstream genes such as MYC (88).
BET proteins are therapeutic targets in different tumor types, but are of particular interest in prostate cancer because they affect the expression and activity of the AR pathway (89). BET proteins directly interact with the NTD of the AR (90). Moreover, BRD4 has numerous shared DNA binding loci with full-length AR and AR-V7 (90, 91). With FOXA1, BRD4 and AR-V7 bind to canonical AR target genes, but with ZFX they bind to non-canonical genes related to cell cycle, autophagy, and WNT signaling (91). Accordingly, BET inhibitors downregulate the expression of AR target genes, as well as MYC (90, 91). BET inhibitors also decrease AR-V7 levels by regulating alternative splicing (92, 93). This culminates in reduced growth of prostate cancer cell lines, organoids and xenografts treated with BET inhibitors, including enzalutamide-resistant models (90, 92, 94, 95).
The promising preclinical data for BET inhibitors suggests that they are potential new treatments for CRPC, functioning in part by indirectly targeting the AR. Numerous BET inhibitors are clinical development and some are in phase I/II clinical trials enrolling men with CRPC, such as ABBV-075 (mivebresib) and MK-8628/OTX015 (birabresib) (89). So far, prostate cancer patients in these trials have still had progressive or stable disease, although a partial response has been reported (89, 96, 97). Ongoing trials are also testing combination treatments of BET inhibitors with AR-directed treatments, PARP inhibitors, chemotherapy and immunotherapy (89, 98). For example, a phase 1b/2a trial of the BET inhibitor ZEN-3694 in combination with enzalutamide demonstrated that the treatment had acceptable tolerability in men with metastatic CRPC who had previously failed abiraterone or enzalutamide (99). Encouragingly, a subset of these patients had prolonged progression-free survival with the combination therapy, including those with tumors with low AR activity.
A challenge in the clinical development of BET inhibitors is overcoming toxicity and off-target effects, so new forms of BET inhibitors are being developed. Using the PROTAC approach, BET degraders target BET proteins for ubiquitination and proteasomal destruction (71, 100). In addition, compounds have been developed to selectively target one of the two bromodomains (BD1 and BD2) within BET proteins (101, 102). BET degraders and selective bromodomain inhibitors both inhibit the growth of prostate cancer cells in vitro and in vivo (71, 100, 101). Therefore, ongoing trials, combination treatments and new compounds, may provide opportunities to treat CRPC by targeting BET proteins.
OCT1
Another canonical AR collaborative TF is OCT1. Of the eight OCT proteins, OCT1 is most widely expressed, and is related to the pluripotency master regulator OCT4 (103, 104). OCT1 acts downstream of pioneer factors that make histone modifications to support AR binding to target regions. GATA2 (GATA binding protein 2) and OCT1 work in a hierarchical network where GATA2 is recruited with AR, followed by OCT1 binding to its motifs. Increased immunoreactivity of OCT1 is correlated with worse prognosis of localized prostate cancer (105). OCT1 is also highly expressed in other cancers, including gastric and colorectal cancer (106, 107). Interestingly, in MYC-driven lung adenocarcinoma, OCT1 binding sites were enriched in a set of genes regulated by MYC (108), suggesting that OCT1 and MYC may also co-regulate a subset of androgen responsive genes in prostate cancer. Furthermore, OCT1 interacts with PARP-1 and BRCA1 (109, 110). OCT1 enhances breast cancer aggressiveness, and BRCA1 catalyzes OCT1 degradation to inhibit tumorigenicity (110). PARP inhibitors are often effective for cancers with BRCA1 mutations, however some tumors are resistant (111). These findings suggest that OCT1 may have a significant effect when used in combination with PARP inhibitors.
Of the genes that are jointly regulated by OCT1 and the AR in prostate cancer, acyl-CoA synthetase long-chain family member 3 (ACSL3) is the mostly highly differentially expressed (112). ACSL3 in turn increases AKR1C3 (aldo-keto reductase family 1 member C3) expression, enhancing the backdoor pathway of androgen synthesis that confers resistance to abiraterone (113, 114). Beyond ACSL3, the genome-wide network of OCT1 target genes in CRPC is enriched in factors such as anillin actin binding protein (ANLN) and DLG associated protein 5 (DLGAP5) that regulate proliferation and migration (115, 116).
Although there are few drugs targeting OCT1, PI polyamides have been developed that block the interaction between OCT1 and specific DNA binding sites. A PI polyamide targeting the OCT1 binding sites of ACSL3 suppresses ACSL3 expression and inhibits the growth of CRPC by repressing global OCT1 chromatin association and AR signaling (112). These preclinical data support further development of compounds targeting OCT1 in CRPC.
Patient-Derived Models for Testing New Treatments for CRPC
As novel compounds are developed to directly and indirectly target the AR, their efficacy must be tested with appropriate preclinical models. Unfortunately, the development of preclinical models of CRPC lags behind the evolving understanding of CRPC and changes in clinical practice. Most studies use a small collection of cell lines, including LNCaP, PC3, DU145, VCaP, 22RV1, and LAPC4 cells (117). These cells are very well-characterized and have been used for important discoveries. They have different mechanisms of castration-resistance, such AR amplification and expression of AR-V7 in VCaP cells, and an intragenic duplication of the AR gene and expression of several AR isoforms in 22Rv1 cells (40, 47, 118, 119). An important use of prostate cancer cell lines is high-throughput screening, including in the NCI-60 panel (120, 121). With this approach, cell lines can be used to identify drug targets with genome-wide genetic screens, such as with siRNA or CRISPR-Cas9, and treated with large compound libraries to identify candidate drugs for further evaluation (50, 122). Nevertheless, this small number of cell lines does not encompass the heterogeneity of CRPC. To address this challenge, there are ongoing efforts to develop new in vivo, ex vivo, and in vitro models from patient specimens.
The ability to establish patient-derived xenografts (PDXs) from patient tumors has advanced with the use of more highly immune-deficient strains of mice. Yet, PDXs are often more difficult to establish from prostate cancer compared to other malignancies, due to low take rates (10–40%) and long latency periods (up to 12 months) (123). Nevertheless, several groups have established collections of serially transplantable prostate cancer PDXs (30, 124–128). At least 51 PDXs of CRPC have been established, primarily from patients who failed ADT, but fewer from men treated with second generation AR-directed therapies (125). To simulate androgen deprivation, PDXs of CRPC are often grafted in castrated mice, with circulating androgen levels equivalent to patients treated with abiraterone (129). By increasing the number of models of CRPC, PDXs provide new opportunities to study the mechanisms of castration resistance, including mutations and ARVs. PDXs are also useful for testing whether candidate therapeutics are effective at reducing the growth of tumors with diverse alterations in the AR pathway. The typical endpoint to determine whether drug treatment reduces the growth rate of PDXs is decreased tumor volume, or ideally regression.
Like all experimental models, PDXs have limitations, so they can be integrated with other patient-derived models to maximize the advantages and offset the limitations of each approach (Figures 2A–C). PDXs provide a rigorous way to evaluate in vivo drug responses, but the experiments are expensive, labor-intensive, low throughput and have long timeframes. Explants and organoids are complementary models that address these limitations. Explants are intact pieces of tissue maintained for several days ex vivo on filters or gelatin sponges, so they retain the native tissue architecture and microenvironment (130, 131). Organoids are digested prostate tissue grown in extracellular matrix solutions, such as Matrigel. Explants and organoids can be established from fresh patient specimens or PDXs, which are renewable sources of tissue (30, 132, 133). These patient-derived models are higher throughput and can be used to rapidly test whether compounds affect proliferation and apoptosis. These ex vivo and in vitro cultures are also useful for testing tool compounds that have poor bioavailability or are not available in sufficient quantities for in vivo experiments. Explants and organoids can also be used for experiments that are challenging with PDXs, including large-scale dose responses of single or combination treatments, genetic manipulation, and short-term time points for mechanistic studies (132, 134, 135). This bridges the gap between high-throughput experiments with cell lines, and in vivo treatments with PDXs. Therefore, by combining different patient-derived models established from different cases of CRPC, it will be possible to test the next generation of therapies with greater rigor and efficiency to help prioritize them for further clinical trials.
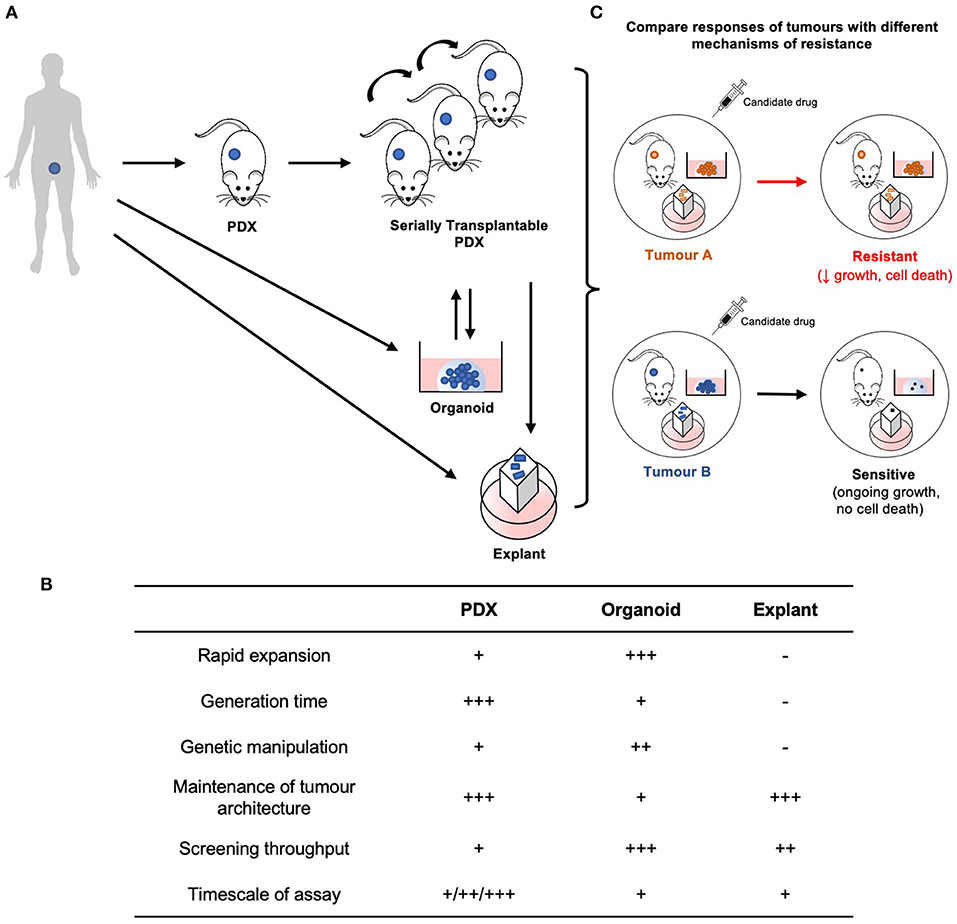
Figure 2. Establishment and application of patient-derived models for preclinical testing of new treatments for CRPC. (A) PDXs are established from human patient tumor tissue, and are considered serially transplantable when repassaged into additional host mice and expanded. Explants and organoids can be established directly from fresh patient specimens or from PDXs. PDXs may also be established from organoids. (B) Each model is unique with its own advantages and limitations. (C) Therefore, by integrating these models established from tumors with different resistance mechanisms, preclinical therapeutic evaluation can be performed with greater rigor and efficiency.
Conclusion
Over the last decade, the introduction of new treatments for CRPC has extended patient survival, but tumors still eventually fail treatment. The increasingly detailed understanding of the underlying mechanisms of resistance has facilitated the development of novel compounds that use alternative approaches to target the AR pathway, directly or indirectly. Two examples of drug targets are BET proteins, with BET inhibitors in ongoing clinical trials for prostate cancer, and OCT1, with novel compounds in preclinical development. Whether these compounds are effective as monotherapies, or should be used in combination with other treatments is still under investigation. Nevertheless, growing collections of patient-derived models, spanning xenografts, organoids and explants, are providing ways to test the efficacy of these candidate drugs across a wider spectrum of tumors. Collectively, this ongoing effort will provide a rich pipeline of new treatments for further validation in clinical trials.
Author Contributions
DO, ST, and SI: conceived the concepts. DO, ML, and NC: wrote the first draft of the manuscript. KT and GR: revise the first draft. All authors reviewed and approved of the final manuscript.
Funding
The authors are supported by funding from the National Health and Medical Research Council, Australia (fellowship to GR 1102752, project grants 1138242, 1156570), the Department of Health and Human Services acting through the Victorian Cancer Agency (fellowship to ML MCRF18017), the EJ Whitten Foundation, the Peter and Lyndy White Foundation, P-CREATE (Grant No. JP18ck0106194 from AMED, Japan), Takeda Science Foundation, and JSPS KAKENHI (Grant Nos. JP19H03793 and JP19K09740).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2020.581515/full#supplementary-material
References
1. Hobisch A, Culig Z, Radmayr C, Bartsch G, Klocker H, Hittmair A. Distant metastases from prostatic carcinoma express androgen receptor protein. Cancer Res. (1995) 55:3068–72.
2. Hobisch A, Culig Z, Radmayr C, Bartsch G, Klocker H, Hittmair A. Androgen receptor status of lymph node metastases from prostate cancer. Prostate. (1996) 28:129–35. doi: 10.1002/(SICI)1097-0045(199602)28:2<129::AID-PROS9>3.0.CO;2-B
3. Sadi MV, Walsh PC, Barrack ER. Immunohistochemical study of androgen receptors in metastatic prostate cancer. Comparison of receptor content and response to hormonal therapy. Cancer. (1991) 67:3057–64. doi: 10.1002/1097-0142(19910615)67:12<3057::AID-CNCR2820671221>3.0.CO;2-S
4. Tilley WD, Lim-Tio SS, Horsfall DJ, Aspinall JO, Marshall VR, Skinner JM. Detection of discrete androgen receptor epitopes in prostate cancer by immunostaining: measurement by color video image analysis. Cancer Res. (1994) 54:4096–102.
5. van der Kwast TH, Tetu B. Androgen receptors in untreated and treated prostatic intraepithelial neoplasia. Eur Urol. (1996) 30:265–8. doi: 10.1159/000474179
6. Vander Griend DJ, Litvinov IV, Isaacs JT. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int J Biol Sci. (2014) 10:627–42. doi: 10.7150/ijbs.8756
7. Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer. (2006) 6:593–602. doi: 10.1038/nrc1947
8. Sathianathen NJ, Koschel S, Thangasamy IA, Teh J, Alghazo O, Butcher G, et al. Indirect comparisons of efficacy between combination approaches in metastatic hormone-sensitive prostate cancer: a systematic review and network meta-analysis. Eur Urol. (2020) 77:365–72. doi: 10.1016/j.eururo.2019.09.004
9. Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. (2012) 367:1187–97. doi: 10.1056/NEJMoa1207506
10. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. (2014) 371:424–33. doi: 10.1056/NEJMoa1405095
11. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. (2011) 364:1995–2005. doi: 10.1056/NEJMoa1014618
12. Small EJ, Saad F, Chowdhury S, Oudard S, Hadaschik BA, Graff JN, et al. Apalutamide and overall survival in non-metastatic castration-resistant prostate cancer. Ann Oncol. (2019) 30:1813–20. doi: 10.1093/annonc/mdz397
13. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair defects and olaparib in metastatic prostate cancer. N Engl J Med. (2015) 373:1697–708. doi: 10.1056/NEJMoa1506859
14. Cornford P, Bellmunt J, Bolla M, Briers E, De Santis M, Gross T, et al. EAU-ESTRO-SIOG guidelines on prostate cancer. Part II: treatment of relapsing, metastatic, and castration-resistant prostate cancer. Eur Urol. (2017) 71:630–42. doi: 10.1016/j.eururo.2016.08.002
15. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. (1994) 63:451–86. doi: 10.1146/annurev.bi.63.070194.002315
16. Hu DG, McKinnon RA, Hulin JA, Mackenzie PI, Meech R. Novel nine-exon ar transcripts (exon 1/exon 1b/exons 2-8) in normal and cancerous breast and prostate cells. Int J Mol Sci. (2016) 18:40. doi: 10.3390/ijms18010040
17. Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. (1995) 270:7341–6. doi: 10.1074/jbc.270.13.7341
18. Schaufele F, Carbonell X, Guerbadot M, Borngraeber S, Chapman MS, Ma AA, et al. The structural basis of androgen receptor activation: intramolecular and intermolecular amino-carboxy interactions. Proc Natl Acad Sci USA. (2005) 102:9802–7. doi: 10.1073/pnas.0408819102
19. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell. (2018) 174:758–69 e9. doi: 10.1016/j.cell.2018.06.039
20. Takeda DY, Spisak S, Seo JH, Bell C, O'Connor E, Korthauer K, et al. A somatically acquired enhancer of the androgen receptor is a noncoding driver in advanced prostate cancer. Cell. (2018) 174:422–32 e13. doi: 10.1016/j.cell.2018.05.037
21. Viswanathan SR, Ha G, Hoff AM, Wala JA, Carrot-Zhang J, Whelan CW, et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell. (2018) 174:433–7.e19. doi: 10.1016/j.cell.2018.05.036
22. Dang HX, Chauhan PS, Ellis H, Feng W, Harris PK, Smith G, et al. Cell-Free DNA alterations in the ar enhancer and locus predict resistance to ar-directed therapy in patients with metastatic prostate cancer. JCO Precision Oncol. (2020) 4:680–713. doi: 10.1200/PO.20.00047
23. Romanel A, Gasi Tandefelt D, Conteduca V, Jayaram A, Casiraghi N, Wetterskog D, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. (2015) 7:312re10. doi: 10.1126/scitranslmed.aac9511
24. Moilanen A-M, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. (2015) 5:12007. doi: 10.1038/srep12007
25. Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. (2013) 63:920–6. doi: 10.1016/j.eururo.2012.08.053
26. Steinkamp MP, O'Mahony OA, Brogley M, Rehman H, Lapensee EW, Dhanasekaran S, et al. Treatment-dependent androgen receptor mutations in prostate cancer exploit multiple mechanisms to evade therapy. Cancer Res. (2009) 69:4434–42. doi: 10.1158/0008-5472.CAN-08-3605
27. Lallous N, Volik SV, Awrey S, Leblanc E, Tse R, Murillo J, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. (2016) 17:10. doi: 10.1186/s13059-015-0864-1
28. Prekovic S, van den Broeck T, Linder S, van Royen ME, Houtsmuller AB, Handle F, et al. Molecular underpinnings of enzalutamide resistance. Endocr Relat Cancer. (2018) 25:R545–57. doi: 10.1530/ERC-17-0136
29. Prekovic S, van Royen ME, Voet AR, Geverts B, Houtman R, Melchers D, et al. The effect of f877l and t878a mutations on androgen receptor response to enzalutamide. Mol Cancer Ther. (2016) 15:1702–12. doi: 10.1158/1535-7163.MCT-15-0892
30. Lawrence MG, Obinata D, Sandhu S, Selth LA, Wong SQ, Porter LH, et al. Patient-derived models of abiraterone- and enzalutamide-resistant prostate cancer reveal sensitivity to ribosome-directed therapy. Eur Urol. (2018) 74:562–72. doi: 10.1016/j.eururo.2018.06.020
31. Schrecengost R, Knudsen KE. Molecular pathogenesis and progression of prostate cancer. Semin Oncol. (2013) 40:244–58. doi: 10.1053/j.seminoncol.2013.04.001
32. Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. (1997) 3:1383–8.
33. Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, et al. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. (2000) 6:703–6. doi: 10.1038/76287
34. Borgmann H, Lallous N, Ozistanbullu D, Beraldi E, Paul N, Dalal K, et al. Moving towards precision urologic oncology: targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201). Eur Urol. (2018) 73:4–8. doi: 10.1016/j.eururo.2017.08.012
35. Sugawara T, Baumgart SJ, Nevedomskaya E, Reichert K, Steuber H, Lejeune P, et al. Darolutamide is a potent androgen receptor antagonist with strong efficacy in prostate cancer models. Int J Cancer. (2019) 145:1382–94. doi: 10.1002/ijc.32242
36. Rathkopf DE, Smith MR, Ryan CJ, Berry WR, Shore ND, Liu G, et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann Oncol. (2017) 28:2264–71. doi: 10.1093/annonc/mdx283
37. Ho Y, Dehm SM. Androgen receptor rearrangement and splicing variants in resistance to endocrine therapies in prostate cancer. Endocrinology. (2017) 158:1533–42. doi: 10.1210/en.2017-00109
38. Li Y, Yang R, Henzler CM, Ho Y, Passow C, Auch B, et al. Diverse AR gene rearrangements mediate resistance to androgen receptor inhibitors in metastatic prostate cancer. Clin Cancer Res. (2020) 26:1965–76. doi: 10.1158/1078-0432.CCR-19-3023
39. Henzler C, Li Y, Yang R, McBride T, Ho Y, Sprenger C, et al. Truncation and constitutive activation of the androgen receptor by diverse genomic rearrangements in prostate cancer. Nat Commun. (2016) 7:13668. doi: 10.1038/ncomms13668
40. Li Y, Hwang TH, Oseth LA, Hauge A, Vessella RL, Schmechel SC, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. (2012) 31:4759–67. doi: 10.1038/onc.2011.637
41. An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep. (2014) 6:657–69. doi: 10.1016/j.celrep.2014.01.013
42. Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer Res. (2010) 70:7992–8002. doi: 10.1158/0008-5472.CAN-10-0585
43. Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, et al. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer Res. (2014) 74:2270–82. doi: 10.1158/0008-5472.CAN-13-2876
44. Tagawa ST, Antonarakis ES, Gjyrezi A, Galletti G, Kim S, Worroll D, et al. Expression of AR-V7 and ARv(567es) in circulating tumor cells correlates with outcomes to taxane therapy in men with metastatic prostate cancer treated in TAXYNERGY. Clin Cancer Res. (2019) 25:1880–8. doi: 10.1158/1078-0432.CCR-18-0320
45. Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. (2012) 72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892
46. Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. (2009) 138:245–56. doi: 10.1016/j.cell.2009.04.056
47. Liu LL, Xie N, Sun S, Plymate S, Mostaghel E, Dong X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene. (2014) 33:3140–50. doi: 10.1038/onc.2013.284
48. Takayama KI, Suzuki T, Fujimura T, Yamada Y, Takahashi S, Homma Y, et al. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc Natl Acad Sci USA. (2017) 114:10461–6. doi: 10.1073/pnas.1706076114
49. Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, et al. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci USA. (2018) 115:E4584–93. doi: 10.1073/pnas.1802415115
50. Fei T, Chen Y, Xiao T, Li W, Cato L, Zhang P, et al. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci USA. (2017) 114:E5207–15. doi: 10.1073/pnas.1617467114
51. Maity SN, Titus MA, Gyftaki R, Wu G, Lu JF, Ramachandran S, et al. Targeting of CYP17A1 Lyase by VT-464 inhibits adrenal and intratumoral androgen biosynthesis and tumor growth of castration resistant prostate cancer. Sci Rep. (2016) 6:35354. doi: 10.1038/srep35354
52. Gupta S, Nordquist LT, Fleming MT, Berry WR, Zhang J, Ervin SL, et al. Phase I study of seviteronel, a selective CYP17 lyase and androgen receptor inhibitor, in men with castration-resistant prostate cancer. Clin Cancer Res. (2018) 24:5225–32. doi: 10.1158/1078-0432.CCR-18-0564
53. Madan RA, Schmidt KT, Karzai F, Peer CJ, Cordes LM, Chau CH, et al. Phase 2 study of seviteronel (Ino-464) in patients with metastatic castration-resistant prostate cancer after enzalutamide treatment. Clin Genitourin Cancer. (2020) 18:258–67 e1. doi: 10.1016/j.clgc.2019.11.002
54. Peer CJ, Schmidt KT, Kindrick JD, Eisner JR, Brown VV, Baskin-Bey E, et al. A population pharmacokinetic analysis of the oral CYP17 lyase and androgen receptor inhibitor seviteronel in patients with advanced/metastatic castration-resistant prostate cancer or breast cancer. Cancer Chemother Pharmacol. (2019) 84:759–70. doi: 10.1007/s00280-019-03908-0
55. Norris JD, Ellison SJ, Baker JG, Stagg DB, Wardell SE, Park S, et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J Clin Invest. (2017) 127:2326–38. doi: 10.1172/JCI87328
56. Suzman DL, Antonarakis ES. Castration-resistant prostate cancer: latest evidence and therapeutic implications. Ther Adv Med Oncol. (2014) 6:167–79. doi: 10.1177/1758834014529176
57. Alex AB, Pal SK, Agarwal N. CYP17 inhibitors in prostate cancer: latest evidence and clinical potential. Ther Adv Med Oncol. (2016) 8:267–75. doi: 10.1177/1758834016642370
58. Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ. Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg Med Chem Lett. (2014) 24:2444–7. doi: 10.1016/j.bmcl.2014.04.024
59. Zhang Y, Castaneda S, Dumble M, Wang M, Mileski M, Qu Z, et al. Reduced expression of the androgen receptor by third generation of antisense shows antitumor activity in models of prostate cancer. Mol Cancer Ther. (2011) 10:2309–19. doi: 10.1158/1535-7163.MCT-11-0329
60. Yamamoto Y, Loriot Y, Beraldi E, Zhang F, Wyatt AW, Al Nakouzi N, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res. (2015) 21:1675–87. doi: 10.1158/1078-0432.CCR-14-1108
61. De Velasco MA, Kura Y, Sakai K, Hatanaka Y, Davies BR, Campbell H, et al. Targeting castration-resistant prostate cancer with androgen receptor antisense oligonucleotide therapy. JCI Insight. (2019) 4:e122688. doi: 10.1172/jci.insight.122688
62. Luna Velez MV, Verhaegh GW, Smit F, Sedelaar JPM, Schalken JA. Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene. (2019) 38:3696–709. doi: 10.1038/s41388-019-0696-7
63. Beretta GL, Zaffaroni N. Androgen Receptor-directed molecular conjugates for targeting prostate cancer. Front Chem. (2019) 7:369. doi: 10.3389/fchem.2019.00369
64. Ponnusamy S, Coss CC, Thiyagarajan T, Watts K, Hwang DJ, He Y, et al. Novel selective agents for the degradation of androgen receptor variants to treat castration-resistant prostate cancer. Cancer Res. (2017) 77:6282–98. doi: 10.1158/0008-5472.CAN-17-0976
65. Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. (2014) 20:3198–210. doi: 10.1158/1078-0432.CCR-13-3296
66. Liu C, Armstrong C, Zhu Y, Lou W, Gao AC. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget. (2016) 7:32210–20. doi: 10.18632/oncotarget.8493
67. Liu C, Armstrong CM, Lou W, Lombard AP, Cucchiara V, Gu X, et al. Niclosamide and bicalutamide combination treatment overcomes enzalutamide- and bicalutamide-resistant prostate cancer. Mol Cancer Ther. (2017) 16:1521–30. doi: 10.1158/1535-7163.MCT-16-0912
68. Schweizer MT, Haugk K, McKiernan JS, Gulati R, Cheng HH, Maes JL, et al. A phase I study of niclosamide in combination with enzalutamide in men with castration-resistant prostate cancer. PLoS ONE. (2018) 13:e0198389. doi: 10.1371/journal.pone.0198389
69. Han X, Wang C, Qin C, Xiang W, Fernandez-Salas E, Yang CY, et al. Discovery of ARD-69 as a highly potent proteolysis targeting chimera (protac) degrader of androgen receptor (ar) for the treatment of prostate cancer. J Med Chem. (2019) 62:941–64. doi: 10.1021/acs.jmedchem.8b01631
70. Shibata N, Nagai K, Morita Y, Ujikawa O, Ohoka N, Hattori T, et al. Development of Protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J Med Chem. (2018) 61:543–75. doi: 10.1021/acs.jmedchem.7b00168
71. Kregel S, Wang C, Han X, Xiao L, Fernandez-Salas E, Bawa P, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. (2020) 22:111–9. doi: 10.1016/j.neo.2019.12.003
72. Ravindranathan P, Lee TK, Yang L, Centenera MM, Butler L, Tilley WD, et al. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. (2013) 4:1923. doi: 10.1038/ncomms2912
73. Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. (2010) 17:535–46. doi: 10.1016/j.ccr.2010.04.027
74. Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. (2013) 123:2948–60. doi: 10.1172/JCI66398
75. Martin SK, Banuelos CA, Sadar MD, Kyprianou N. N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Mol Oncol. (2014) doi: 10.1016/j.molonc.2014.10.014
76. Brand LJ, Olson ME, Ravindranathan P, Guo H, Kempema AM, Andrews TE, et al. EPI-001 is a selective peroxisome proliferator-activated receptor-gamma modulator with inhibitory effects on androgen receptor expression and activity in prostate cancer. Oncotarget. (2015) 6:3811–24. doi: 10.18632/oncotarget.2924
77. Le Moigne R, Zhou H-J, Obst JK, Banuelos CA, Jian K, Williams D, et al. Lessons learned from the metastatic castration-resistant prostate cancer phase I trial of EPI-506, a first-generation androgen receptor N-terminal domain inhibitor. J Clin Oncol. (2019) 37(7_suppl):257. doi: 10.1200/JCO.2019.37.7_suppl.257
78. Moigne RL, Banuelos CA, Mawji NR, Tam T, Wang J, Jian K, et al. Abstract B117: Treatment of castrated resistant prostate cancer with EPI-7386, a second generation N-terminal domain androgen receptor inhibitor. Mol Cancer Ther. (2019) 18(12 Suppl):B117. doi: 10.1158/1535-7163.TARG-19-B117
79. Trauger JW, Baird EE, Dervan PB. Recognition of DNA by designed ligands at subnanomolar concentrations. Nature. (1996) 382:559–61. doi: 10.1038/382559a0
80. Kielkopf CL, Bremer RE, White S, Szewczyk JW, Turner JM, Baird EE, et al. Structural effects of DNA sequence on T.A recognition by hydroxypyrrole/pyrrole pairs in the minor groove. J Mol Biol. (2000) 295:557–67. doi: 10.1006/jmbi.1999.3364
81. Dervan PB, Edelson BS. Recognition of the DNA minor groove by pyrrole-imidazole polyamides. Curr Opin Struct Biol. (2003) 13:284–99. doi: 10.1016/S0959-440X(03)00081-2
82. Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci USA. (2007) 104:10418–23. doi: 10.1073/pnas.0704217104
83. Yang F, Nickols NG, Li BC, Marinov GK, Said JW, Dervan PB. Antitumor activity of a pyrrole-imidazole polyamide. Proc Natl Acad Sci USA. (2013) 110:1863–8. doi: 10.1073/pnas.1222035110
84. Takayama K, Inoue S. Transcriptional network of androgen receptor in prostate cancer progression. Int J Urol. (2013) 20:756–68. doi: 10.1111/iju.12146
85. Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. (2011) 25:2227–41. doi: 10.1101/gad.176826.111
86. Sharma NL, Massie CE, Ramos-Montoya A, Zecchini V, Scott HE, Lamb AD, et al. The androgen receptor induces a distinct transcriptional program in castration-resistant prostate cancer in man. Cancer Cell. (2013) 23:35–47. doi: 10.1016/j.ccr.2012.11.010
87. Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and prostate cancer. Genes Cancer. (2010) 1:617–28. doi: 10.1177/1947601910379132
88. Stathis A, Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov. (2018) 8:24–36. doi: 10.1158/2159-8290.CD-17-0605
89. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. (2019) 25:403–18. doi: 10.1038/s41591-019-0376-8
90. Asangani Ia, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. (2014) 510:278–82. doi: 10.1038/nature13229
91. Cai L, Tsai YH, Wang P, Wang J, Li D, Fan H, et al. ZFX mediates non-canonical oncogenic functions of the androgen receptor splice variant 7 in castrate-resistant prostate cancer. Mol Cell. (2018) 72:341–54 e6. doi: 10.1016/j.molcel.2018.08.029
92. Welti J, Sharp A, Yuan W, Dolling D, Nava Rodrigues D, Figueiredo I, et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin Cancer Res. (2018) 24:3149–62. doi: 10.1158/1078-0432.CCR-17-3571
93. Chan SC, Selth LA, Li Y, Nyquist MD, Miao L, Bradner JE, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. (2015) 43:5880–97. doi: 10.1093/nar/gkv262
94. Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to ar antagonists in the treatment of prostate cancer. Mol Cancer Res. (2016) 14:324–31. doi: 10.1158/1541-7786.MCR-15-0472
95. Wyce A, Degenhardt Y, Bai Y, Le B, Korenchuk S, Crouthame MC, et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget. (2013) 4:2419–29. doi: 10.18632/oncotarget.1572
96. Massard C, Soria JC, Stathis A, Delord JP, Awada A, Peters S, et al. A phase Ib trial with MK-8628/OTX015, a small molecule inhibitor of bromodomain (BRD) and extra-terminal (BET) proteins, in patients with selected advanced solid tumors. Eur J Cancer. (2016) 69:S2–S3. doi: 10.1016/S0959-8049(16)32609-0
97. Piha-Paul SA, Sachdev JC, Barve M, LoRusso P, Szmulewitz R, Patel SP, et al. First-in-human study of mivebresib (abbv-075), an oral pan-inhibitor of bromodomain and extra terminal proteins, in patients with relapsed/refractory solid tumors. Clin Cancer Res. (2019) 25:6309–19. doi: 10.1158/1078-0432.CCR-19-0578
98. Vaishampayan UN, Narayan V, Wise D, Lang JM, Lowentritt BH, Mellado B, et al. A phase Ib open-label, dose escalation and expansion study to investigate the safety, pharmacokinetics, pharmacodynamics and clinical activity of GSK525762 in combination with abiraterone or enzalutamide in metastatic castrate-resistant prostate cancer. J Clin Oncol. (2018) 36(6_suppl):TPS391-TPS. doi: 10.1200/JCO.2018.36.6_suppl.TPS391
99. Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A phase 1b/2a study of the pan-bet bromodomain inhibitor zen-3694 in combination with enzalutamide in patients with metastatic castration resistant prostate cancer. Clin Cancer Res. (2020) doi: 10.1158/1078-0432.CCR-20-1707. [Epub ahead of print].
100. Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA. (2016) 113:7124–9. doi: 10.1073/pnas.1521738113
101. Gilan O, Rioja I, Knezevic K, Bell MJ, Yeung MM, Harker NR, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. (2020) 368:387–94. doi: 10.1126/science.aaz8455
102. Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. (2020) 578:306–10. doi: 10.1038/s41586-020-1930-8
103. Vazquez-Arreguin K, Tantin D. The Oct1 transcription factor and epithelial malignancies: old protein learns new tricks. Biochim Biophys Acta. (2016) 1859:792–804. doi: 10.1016/j.bbagrm.2016.02.007
104. Tantin D. Oct transcription factors in development and stem cells: insights and mechanisms. Development. (2013) 140:2857–66. doi: 10.1242/dev.095927
105. Obinata D, Takayama K, Urano T, Murata T, Kumagai J, Fujimura T, et al. Oct1 regulates cell growth of LNCaP cells and is a prognostic factor for prostate cancer. Int J Cancer. (2012) 130:1021–8. doi: 10.1002/ijc.26043
106. Xu SH, Huang JZ, Xu ML, Yu G, Yin XF, Chen D, et al. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. (2015) 236:175–85. doi: 10.1002/path.4515
107. Li Y, Dong M, Kong F, Zhou J. Octamer transcription factor 1 mediates epithelial-mesenchymal transition in colorectal cancer. Tumour Biol. (2015) 36:9941–6. doi: 10.1007/s13277-015-3766-7
108. Reymann S, Borlak J. Transcription profiling of lung adenocarcinomas of c-myc-transgenic mice: identification of the c-myc regulatory gene network. BMC Syst Biol. (2008) 2:46. doi: 10.1186/1752-0509-2-46
109. Nie J, Sakamoto S, Song D, Qu Z, Ota K, Taniguchi T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. (1998) 424:27–32. doi: 10.1016/S0014-5793(98)00131-8
110. Vázquez-Arreguín K, Maddox J, Kang J, Park D, Cano RR, Factor RE, et al. BRCA1 through its e3 ligase activity regulates the transcription factor oct1 and carbohydrate metabolism. Mol Cancer Res. (2018) 16:439–52. doi: 10.1158/1541-7786.MCR-17-0364
111. Wang Y, Bernhardy AJ, Nacson J, Krais JJ, Tan YF, Nicolas E, et al. BRCA1 intronic alu elements drive gene rearrangements and PARP inhibitor resistance. Nat Commun. (2019) 10:5661. doi: 10.1038/s41467-019-13530-6
112. Obinata D, Takayama K, Fujiwara K, Suzuki T, Tsutsumi S, Fukuda N, et al. Targeting Oct1 genomic function inhibits androgen receptor signaling and castration-resistant prostate cancer growth. Oncogene. (2016) 35:6350–8. doi: 10.1038/onc.2016.171
113. Migita T, Takayama KI, Urano T, Obinata D, Ikeda K, Soga T, et al. ACSL3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci. (2017) 108:2011–21. doi: 10.1111/cas.13339
114. Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, Gao AC. Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol Cancer Ther. (2017) 16:35–44. doi: 10.1158/1535-7163.MCT-16-0186
115. Takayama KI, Suzuki Y, Yamamoto S, Obinata D, Takahashi S, Inoue S. Integrative genomic analysis of oct1 reveals coordinated regulation of androgen receptor in advanced prostate cancer. Endocrinology. (2019) 160:463–72. doi: 10.1210/en.2018-00923
116. Yamamoto S, Takayama KI, Obinata D, Fujiwara K, Ashikari D, Takahashi S, et al. Identification of new octamer transcription factor 1-target genes upregulated in castration-resistant prostate cancer. Cancer Sci. (2019) 110:3476–85. doi: 10.1111/cas.14183
117. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. (2015) 15:701–11. doi: 10.1038/nrc4016
118. Korenchuk S, Lehr JE, L MC, Lee YG, Whitney S, Vessella R, et al. VCaP, a cell-based model system of human prostate cancer. In Vivo. (2001) 15:163–8.
119. Li Y, Alsagabi M, Fan D, Bova GS, Tewfik AH, Dehm SM. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. (2011) 71:2108–17. doi: 10.1158/0008-5472.CAN-10-1998
120. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. (2006) 6:813–23. doi: 10.1038/nrc1951
121. Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res. (2014) 74:2377–84. doi: 10.1158/0008-5472.CAN-13-2971
122. Zhang P, Xia JH, Zhu J, Gao P, Tian YJ, Du M, et al. High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat Commun. (2018) 9:2022. doi: 10.1038/s41467-018-04451-x
123. Risbridger GP, Lawrence MG. Towards Best Practice in Establishing Patient-Derived Xenografts. In: Wang Y, Lin D, Gout PW, editors. Patient-Derived Xenograft Models of Human Cancer. Cham: Springer International Publishing. (2017). p. 11–28. doi: 10.1007/978-3-319-55825-7_2
124. Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS, et al. LuCaP prostate cancer patient-derived xenografts reflect the molecular heterogeneity of advanced disease an–d serve as models for evaluating cancer therapeutics. Prostate. (2017) 77:654–71. doi: 10.1002/pros.23313
125. Navone NM, van Weerden WM, Vessella RL, Williams ED, Wang Y, Isaacs JT, et al. Movember GAP1 PDX project: an international collection of serially transplantable prostate cancer patient-derived xenograft (PDX) models. Prostate. (2018) 78:1262–82. doi: 10.1002/pros.23701
126. Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. (2014) 74:1272–83. doi: 10.1158/0008-5472.CAN-13-2921-T
127. Marques RB, van Weerden WM, Erkens-Schulze S, de Ridder CM, Bangma CH, Trapman J, et al. The human PC346 xenograft and cell line panel: a model system for prostate cancer progression. Eur Urol. (2006) 49:245–57. doi: 10.1016/j.eururo.2005.12.035
128. Palanisamy N, Yang J, Shepherd PDA, Li-Ning-Tapia EM, Labanca E, Manyam G, et al. The MD Anderson prostate cancer patient-derived xenograft series (MDA PCa PDX) captures the molecular landscape of prostate cancer and facilitates marker-driven therapy development. Clin Cancer Res. (2020). doi: 10.1158/1078-0432.CCR-20-0479. [Epub ahead of print].
129. Michiel Sedelaar JP, Dalrymple SS, Isaacs JT. Of mice and men–warning: intact versus castrated adult male mice as xenograft hosts are equivalent to hypogonadal versus abiraterone treated aging human males, respectively. Prostate. (2013) 73:1316–25. doi: 10.1002/pros.22677
130. Davies EJ, Dong M, Gutekunst M, Narhi K, van Zoggel HJ, Blom S, et al. Capturing complex tumour biology in vitro: histological and molecular characterisation of precision cut slices. Sci Rep. (2015) 5:17187. doi: 10.1038/srep17187
131. Centenera MM, Hickey TE, Jindal S, Ryan NK, Ravindranathan P, Mohammed H, et al. A patient-derived explant (PDE) model of hormone-dependent cancer. Mol Oncol. (2018) 12:1608–22. doi: 10.1002/1878-0261.12354
132. Beshiri ML, Tice CM, Tran C, Nguyen HM, Sowalsky AG, Agarwal S, et al. A PDX/Organoid biobank of advanced prostate cancers captures genomic and phenotypic heterogeneity for disease modeling and therapeutic screening. Clin Cancer Res. (2018) 24:4332–45. doi: 10.1158/1078-0432.CCR-18-0409
133. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. (2014) 159:176–87. doi: 10.1016/j.cell.2014.08.016
134. Risbridger GP, Toivanen R, Taylor RA. Preclinical models of prostate cancer: patient-derived xenografts, organoids, and other explant models. Cold Spring Harb Perspect Med. (2018) 8:a030536. doi: 10.1101/cshperspect.a030536
Keywords: androgen receptor, castration-resistant prostate cancer, transcription factors, octamer transcription factor 1, preclinical models
Citation: Obinata D, Lawrence MG, Takayama K, Choo N, Risbridger GP, Takahashi S and Inoue S (2020) Recent Discoveries in the Androgen Receptor Pathway in Castration-Resistant Prostate Cancer. Front. Oncol. 10:581515. doi: 10.3389/fonc.2020.581515
Received: 09 July 2020; Accepted: 27 August 2020;
Published: 08 October 2020.
Edited by:
Marzia Di Donato, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Alice Zamagni, Romagnolo Scientific Institute for the Study and Treatment of Tumors (IRCCS), ItalyMasaki Shiota, Kyushu University, Japan
Kouji Izumi, Kanazawa University, Japan
Alessio Bruni, University Hospital of Modena, Italy
William Douglas Figg, National Cancer Institute (NCI), United States
Steven Balk, Beth Israel Deaconess Medical Center and Harvard Medical School, United States
Copyright © 2020 Obinata, Lawrence, Takayama, Choo, Risbridger, Takahashi and Inoue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Satoshi Inoue, sinoue@tmig.or.jp