 Peifeng Li1,2†Jia Chai1†Zi Chen3Yang Liu1,4Jie Wei1Yixiong Liu1Danhui Zhao1Jing Ma1Kaijing Wang1Xia Li1Yang Shao5Li Gong6Wei Zhang6Shuangping Guo1Qingguo Yan1Mingyang Li1*‡Linni Fan1*‡
Peifeng Li1,2†Jia Chai1†Zi Chen3Yang Liu1,4Jie Wei1Yixiong Liu1Danhui Zhao1Jing Ma1Kaijing Wang1Xia Li1Yang Shao5Li Gong6Wei Zhang6Shuangping Guo1Qingguo Yan1Mingyang Li1*‡Linni Fan1*‡ Zhe Wang1*‡
Zhe Wang1*‡- 1State Key Laboratory of Cancer Biology, Department of Pathology, Xijing Hospital and School of Basic Medicine, Fourth Military Medical University, Xi'an, China
- 2Department of Pathology, The 960th Hospital of the Chinese People's Liberation Army, Jinan, China
- 3Division of Cellular and Molecular Pathology, Department of Pathology, University of Cambridge, Cambridge, United Kingdom
- 4Department of Oral Mucosa, Affiliated Stomatological Hospital, Fourth Military Medical University, Xi'an, China
- 5Nanjing Geneseeq Technology Inc., School of Public Health, Nanjing Medical University, Nanjing, China
- 6Department of Pathology, Tangdu Hospital, Fourth Military Medical University, Xi'an, China
Primary gastrointestinal diffuse large B-cell lymphoma (GI-DLBCL) is the most common gastrointestinal lymphoma, but its genetic features are poorly understood. We performed whole-exome sequencing of 25 primary tumor samples from patients with GI-DLBCL and 23 matched normal tissue samples. Oncogenic mutations were screened, and the correlations between genetic mutations and clinicopathological characteristics were analyzed. Twenty-five patients with GI-DLBCL were enrolled in the genetic mutation analysis with a median of 184 (range 79–382) protein-altering variants per patient. We identified recurrent oncogenic mutations in GI-DLBCL, including those in TP53, MUC16, B2M, CCND3, HIST1H1C, NEB, and ID3. Compared with nodal DLBCL, GI-DLBCL exhibited an increased mutation frequency of TP53 and reduced mutation frequencies of PIM1, CREBBP, BCL2, KMT2D, and EZH2. Moreover, GI-DLBCL exhibited fewer MYD88 and CD79B mutations than DLBCL in the testis and central nervous system. GI-DLBCLs with HLA-B, MEF2A, RHOA, and NAV3 mutations exhibited a tendency toward a high proliferation index. MUC16 and ETV6 mutations often occurred in tumors with early clinical staging. Our data provide a comprehensive understanding of the landscape of mutations in a small subset of GI-DLBCLs. The genetic mutation profiles of GI-DLBCL differ from those of nodal DLBCL and DLBCL in immune-privileged sites. The different mutated genes are related to the NF-κB and JAK-STAT pathways, and the different pathogenetic mechanisms leading to the development of DLBCL may be influenced by the tissue microenvironment. Differences in genetic alterations might influence the clinicopathological characteristics of GI-DLBCL.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common aggressive non-Hodgkin lymphoma. It is subdivided into multiple morphological variants given its complex and diverse histological characteristics. Based on gene expression profiling, DLBCL is classified into the following two molecular subtypes: activated B-cell-like (ABC) subtype and germinal center B-cell-like (GCB) subtype (1). Therefore, DLBCL is a clinically, morphologically, immunologically, and genetically heterogeneous diagnostic category (2, 3). Its molecular genetic characteristics have been thoroughly studied with the advent of next-generation sequencing (4). Chromosomal copy-number aberrations, gene rearrangements, and mutations promote the occurrence and progression of DLBCL (2, 5).
DLBCL occurs not only in the lymph nodes but also in many extranodal sites (6–8). Approximately 40% of DLBCLs are manifested at primary extranodal sites, and the gastrointestinal tract is the most common extranodal site (1, 7). Previous studies have shown that genetic mutations in gastrointestinal DLBCL (GI-DLBCL) are not identical to those in non-gastrointestinal DLBCL. GI-DLBCL has a significantly reduced frequency of CD79B and MYD88 mutations compared with nodal DLBCL and extranodal DLBCL of immune-privileged sites (9–11). These targeted analyses of select alterations suggest that the pathogenesis of GI-DLBCL might differ from that of non-gastrointestinal DLBCL. However, comprehensive research on the genetic mutation landscape specifically focused on GI-DLBCL remains lacking, and the correlation between its genetic mutation and clinicopathological features is poorly understood.
In this study, we examined 25 primary tumor samples from patients with GI-DLBCL using whole-exome sequencing and analyzed their genetic mutation characteristics. The screened oncogenic mutations were compared with mutation data from other DLBCL cases. In addition, the correlations between gene mutations and clinicopathological features were analyzed. We attempted to elucidate the genomic mutation profile of GI-DLBCL and its clinicopathological significance and provide insights into the mechanisms of lymphomagenesis in GI-DLBCL.
Materials and Methods
Patient Cohort
The institutional ethical committee of Xijing Hospital and Tangdu Hospital approved this study, and written informed consent was obtained from each patient. Twenty-five patients were selected from the clinical database of both hospitals based on a recorded pathological diagnosis of GI-DLBCL. All patients met the criteria for primary gastrointestinal lymphoma as defined by Lewin et al. (12). Given that the combined therapy of surgery and chemotherapy is superior to other treatment strategies for gastrointestinal lymphoma (13–15), many patients with GI-DLBCL underwent surgical treatment in both hospitals. Twenty-three surgical resection specimens and two biopsy specimens with a histopathological diagnosis of GI-DLBCL were used in this study, and all specimens were reviewed by two senior hematopathologists (mostly Z.W., S.G., or W.Z.) according to the criteria published in the 2017 WHO classification (1). All patients had sufficient formalin-fixed, paraffin-embedded tumor tissues, and 23 of 25 patients had available corresponding normal tissue samples. Patient clinicopathological information was collected, and evaluation and scoring of various features were strictly performed in accordance with corresponding international standards (16).
Whole-Exome Sequencing
DNA extraction of formalin-fixed, paraffin-embedded tissues was performed as described previously (17). Whole-exome sequencing was performed using a targeted capture approach with the Agilent SureSelect Human All Exon Kit (Santa Clara, CA, USA) followed by massively parallel sequencing of enriched fragments on the Illumina HiSeq Platform (Geneseeq Technology Inc., Nanjing China). Tumor and normal DNA samples were sequenced to an average depth of 101.2× and 42.2× in targeted exonic regions, respectively. All tumor specimens had an average sequencing depth of the target region >60× and coverage of the target region >90% at 20× (Supplementary Figure 1A). All reads were aligned to the human reference genome (hg19) using BWA version 0.7.12, and duplicate reads were removed using the Picard tool (http://broadinstitute.github.io/picard/).
Identification of Somatic Single Nucleotide Variants (SNVs) and Indels
Variants were identified by UnifiedGenotyper based on hg19 and annotated with dbSNP version 144 (https://www.ncbi.nlm.nih.gov/SNP/). Variants in tumor samples were filtered against pooled normal samples, and synonymous SNVs, and intronic variants were removed. Variants identified in the 1,000 Genomes database (https://www.1000genomes.org/) with a frequency >1% (unless they were in the Catalog of Somatic Mutations in Cancer (COSMIC) database) or in the Exome Aggregation Consortium (http://exac.broadinstitute.org/) with a frequency >0.1% were discarded from the analysis. Variants with an alternate allele depth <2 and a frequency <5% were also excluded. In addition, SNVs and indels were filtered to remove benign changes predicted by ≥5 of the nine predictive software programs, including Polyphen2_HDIV, Polyphen2_HVAR, MutationTaster, Mutation Assessor, FATHMM, Radial SVM, LR, SIFT, and LRT.
Mutational Landscapes of Common DLBCLs
To more clearly define the commonalities and differences between GI-DLBCL and non-gastrointestinal or nodal DLBCL, we analyzed the mutational landscapes of common DLBCL with COSMIC and cBioPortal (https://www.cbioportal.org/). In COSMIC, a filtered file on DLBCL was acquired from genome-wide screens (including whole-exome sequencing), and coding point mutations of 256 samples (https://cancer.sanger.ac.uk/cosmic/) were analyzed, including one case of primary breast tumor, one case in soft tissue, two cases in the abdomen, two cases in the spleen, one case in the tonsil, 23 cases in lymph nodes, and 226 unspecified cases. In cBioPortal, 1,446 profiled samples of DLBCL with mutation data were collected, including seven cohorts of DLBCL patients [DFCI (Dana-Farber Cancer Institute), Nat Med 2018 (n = 135); Broad, PNAS 2012 (n = 58); Duke, Cell 2017 (n = 1,001); TCGA (The Cancer Genome Atlas), PanCancer Atlas (n = 48); BCGSC (BC Cancer Agency's Genome Sciences Center), Blood 2013 (n = 53); MD Anderson Cancer Center (n = 148); and BCGSC, Nature 2011 (n = 10)]. Unfortunately, we could not obtain the primary sites of these 1,446 tumors. Furthermore, Zehir et al. (18) detected mutations in 74 patients with DLBCL using MSK-IMPACT (Memorial Sloan Kettering Cancer Center-Integrated Mutation Profiling of Actionable Cancer Targets), which revealed mutations in 410 or 341 genes. There were 35 cases of nodular DLBCL and 5 cases of GI-DLBCL in MSK-IMPACT. The mutational differences between GI-DLBCL and nodal DLBCL in MSK-IMPACT were analyzed to clarify the unique mutation profile of GI-DLBCL.
Statistical Analysis
Statistical analysis was performed using IBM-SPSS Statistics version 23 (IBM, Armonk, NY, USA). Logistic regression was used to analyze the correlations between gene mutations and clinicopathological characteristics. The Mann–Whitney U-test was employed to analyze the relationship between the number of mutations or mutated genes and clinicopathological characteristics. Genetic mutation differences between the data and those obtained from COSMIC, cBioPortal and MSK-IMPACT were investigated using the Pearson χ2 test. Statistical significance was defined as a P < 0.05.
Results
Clinicopathological Characteristics of GI-DLBCL
Twenty-five patients with GI-DLBCL were enrolled in our study. Twelve (48.0%) of 25 patients were female. The age at the time of diagnosis ranged from 21 to 83 years, and the average age was 48.7 ± 15.3 years (median: 49 years). Tumors were present in the stomach of 12 patients, all of whom received total gastrectomy. Three patients with small intestine tumors underwent partial small intestine resection, and an additional 10 patients with colon DLBCL underwent partial colectomy. All patients received postoperative CHOP-based chemotherapy (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone). Of the 12 gastric cases, four cases showed Helicobacter pylori infection but lacked MALT (extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue) characteristics, including dense infiltration of centrocyte-like cells in the lamina propria and typical lymphoepithelial lesions. Tumors were classified into GCB (n = 6) and non-GCB (n = 19) molecular subtypes based on immunohistochemical features. No statistically significant differences in patient age (<50 or ≥50 years), sex or tumor location (stomach or intestine) were noted between GCB-DLBCL and non-GCB-DLBCL in this study. MYC and BCL2 status was evaluated in 15 and 12 cases of DLBCL by fluorescence in situ hybridization, respectively. MYC translocations were observed only in one case, while BCL2 translocation was not observed. Most samples exhibited a high Ki-67 index (>90% in 5 samples, 60~90% in 18 samples, and <60% in two samples). The clinical stage was stage I in 4 patients (16%), stage II in 4 patients (16%), stage III in 12 patients (48%), and stage IV in 5 patients (20%). The follow-up duration ranged from 1 to 76 months (median: 19 months) (Figure 1). The clinicopathological characteristics of the GI-DLBCL patients are summarized in Table 1 and Supplementary Table 1.
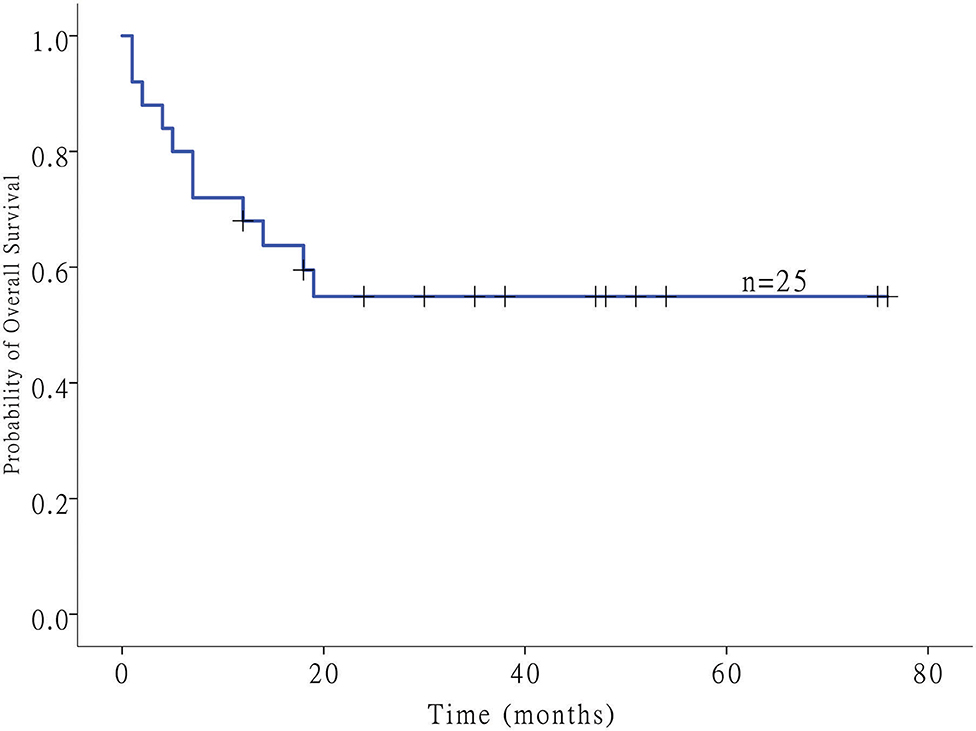
Figure 1. The Kaplan–Meier survival curve showed the overall survival in 25 patients with GI-DLBCL.

Table 1. Clinicopathological characteristics of 25 patients with GI-DLBCL.
Mutational Landscapes of GI-DLBCL Identified by Whole-Exome Sequencing
We performed whole-exome sequencing of 25 patient-derived tumor specimens, and 23 patients had matched normal DNA. A total of 4,999 exonic mutations were identified in 25 patients with GI-DLBCL. Of these, 3,562 were missense variants, 332 were stop-gain mutations, 42 were stop-loss mutations, 164 were non-frameshift deletions, 148 were non-frameshift insertions, 78 were frameshift deletions, 64 were frameshift insertions, 389 were unknown-function mutations, and 220 were splice site mutations. Mutations with the number of base alterations equal to the sequencing depth and mutations with an unknown function were removed, resulting in a median of 184 (range 79–382) protein-altering variants (SNVs and small indels) per patient (Supplementary Figure 1B).
Oncogenic Driver Genes in GI-DLBCL
Known pathogenic genes associated with human tumors were determined with the aid of COSMIC, MDG125 (19), SMG127 (20), CDG291 (21), and a recently published reference (18). A total of 333 mutations of various mutation types were identified in 189 genes. There were 242 missense mutations, 33 non-sense mutations, 21 splice site mutations, 12 in-frame deletions, 6 in-frame insertions, 12 frameshift deletions, and 7 frameshift insertions. The number of mutated genes ranged from 3 to 28 per patient with a median of 11 mutated genes (Figure 2 and Supplementary Table 2). There was no obvious difference in the gene mutations between gastric DLBCL and intestinal DLBCL. KEGG pathway analysis revealed that many of the mutated genes are involved in cancer, signal transduction, infectious disease, and immune system pathways. Crucial signal transduction pathways included the PI3K-AKT, FOXO, MAPK, and JAK-STAT signaling pathways (Figure 3). The mutated genes potentially promote tumor cell proliferation and evasion of apoptosis through cascade reactions. No obvious differential pathways were noted between GCB-DLBCL and non-GCB-DLBCL.
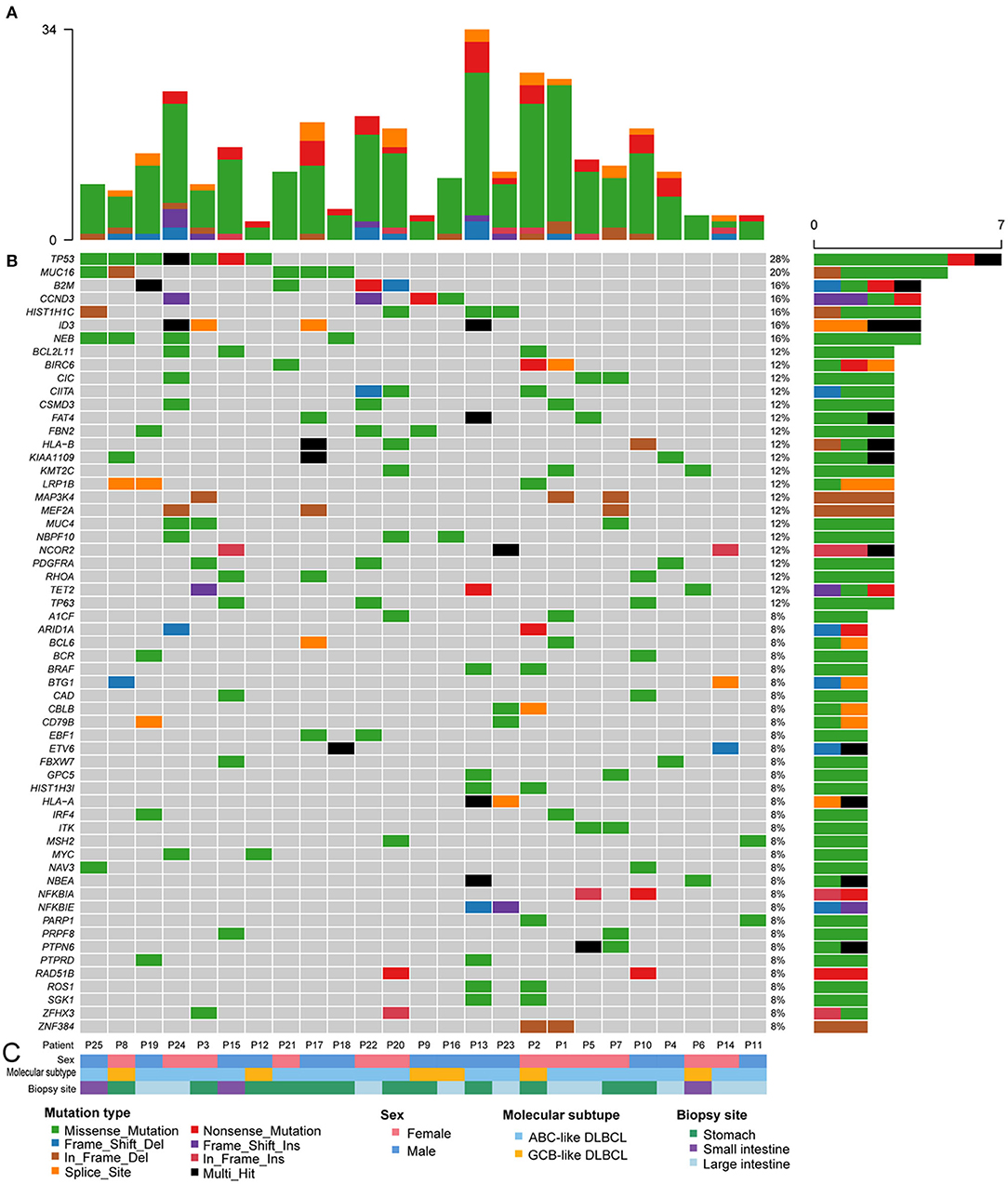
Figure 2. Recurrently mutated oncogenes in 25 patients with GI-DLBCL. (A) The absolute number of oncogenic mutations in each patient. (B) Fifty-nine recurrently and significantly mutated genes constitute the individual rows and are sorted according to their mutational frequencies. The heatmap represents individual mutations in 25 patient samples color-coded by the mutation type. Right: histogram shows the number of mutations in each gene. Percentages represent the fraction of tumors with at least one mutation in the specified gene. (C) Tracks at the bottom of the plot provide information on sex, the molecular subtype and biopsy site. Bottom: the mutation type, sex, molecular subtype, and biopsy site are color-coded as indicated in the legend.
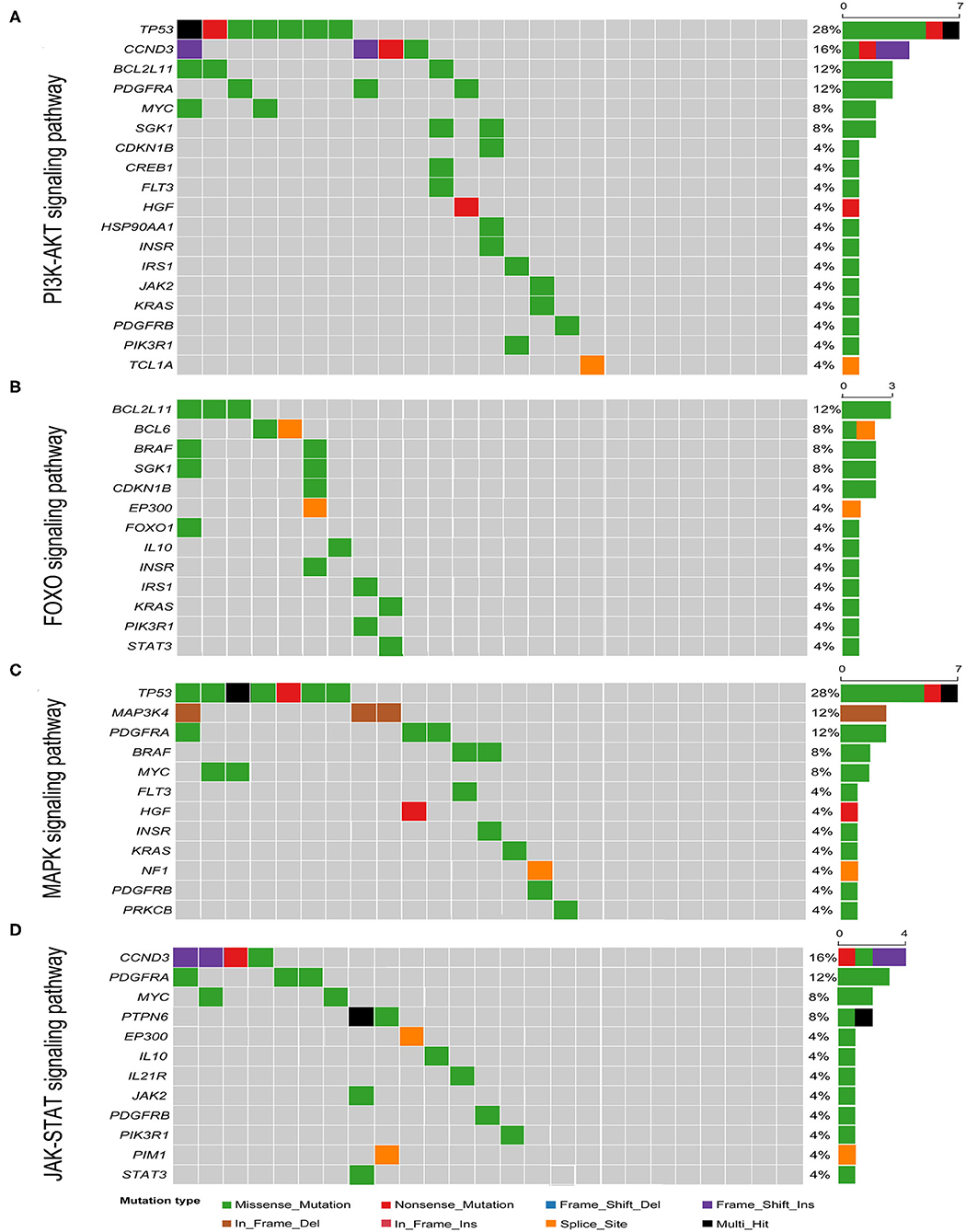
Figure 3. Heatmap of pathways most commonly affected by genetic mutations in GI-DLBCL. (A) PI3K-AKT signaling pathway; (B) FOXO signaling pathway; (C) MAPK signaling pathway; (D) JAK-STAT signaling pathway. Right: histogram shows the number of mutations in each gene. Percentages represent the fraction of tumors with at least one mutation in the specified gene.
Among the 189 mutated genes, 59 recurrent genes were identified as candidate oncogenes in 25 patients with GI-DLBCL, and the mutational frequencies, mutation types and clinical/molecular features are displayed in Figure 2. At least two recurrent oncogenic mutations were detected in each tumor. These gene-sets prominently included members of the PI3K-AKT (TP53, CCND3, BCL2L11, PDGFRA, MYC, and SGK1), T cell receptor (RHOA, CBLB, ITK, NFKBIA, NFKBIE, and PTPN6) and MAPK (TP53, MAP3K4, PDGFRA, BRAF, and MYC) signaling pathways. TP53 (n = 7) was the most frequently mutated gene, and MUC16 (n = 5), B2M (n = 4), CCND3 (n = 4), HIST1H1C (n = 4), NEB (n = 4), and ID3 (n = 4) were also commonly mutated. The mutation characteristics and possible effects on their proteins are illustrated in Supplementary Figure 2. Interestingly, CCND3 and ID3 typically exhibited truncating mutations, which may lead to loss of function of these proteins.
Contrasting Mutational Landscapes to Non-gastrointestinal DLBCL
To investigate whether genetic alterations in GI-DLBCL are similar to those in common DLBCL, the mutational frequencies of oncogenic driver genes in GI-DLBCL were compared with those in common DLBCL in cBioPortal and COSMIC (Supplementary Table 3). In cBioPortal, genetic mutations in 1,446 patients with DLBCL were analyzed. KMT2D (24.48%), MYD88 (16.53%), PIM1 (16.46%), CREBBP (13.49%), BCL2 (11.89%), TP53 (11.69%), HIST11E (11.07%), and CARD11 (10.93%) exhibited mutation frequencies >10%. The TP53 gene showed more mutations in our cohort (P = 0.023), but mutations in MYD88, CREBBP, BCL2, and CARD11 were not found. Our study also revealed a lower mutation frequency for the KMT2D gene (P = 0.001; Figure 4A). Similar results were obtained in COSMIC. BCL2, KMT2D, CREBBP, and EZH2 all exhibited high mutation frequencies in COSMIC, but few or no mutations in these genes were observed in 25 patients with GI-DLBCL (P < 0.05; Figure 4A). Furthermore, we analyzed the mutation differences between GI-DLBCL in this study and nodal DLBCL in COSMIC (n = 23). GI-DLBCL exhibited a higher mutation frequency in TP53 than nodal DLBCL (P = 0.010) and a lower mutation frequency in KMT2D (P = 0.020), BCL2 (P = 0.008), CREBBP (P = 0.008), and EZH2 (P = 0.008; Figure 4B). Moreover, compared with nodal DLBCL in MSK-IMPACT, GI-DLBCL also exhibited a significantly lower mutation frequency of KMT2D (P = 0.003), PIM1 (P = 0.035), BCL2 (P = 0.016), CREBBP (P = 0.000) and EZH2 (P = 0.036) in our study (Supplementary Table 3). These comparisons indicate that GI-DLBCL has a mutation profile that differs from that of non-gastrointestinal and nodal DLBCLs. GI-DLBCL exhibited an increased mutation frequency in TP53, MUC16, CCND3, HIST1H1C, NEB, and ID3 and a reduced mutation frequency in MYD88, CREBBP, BCL2, KMT2D, PIM1, and EZH2.
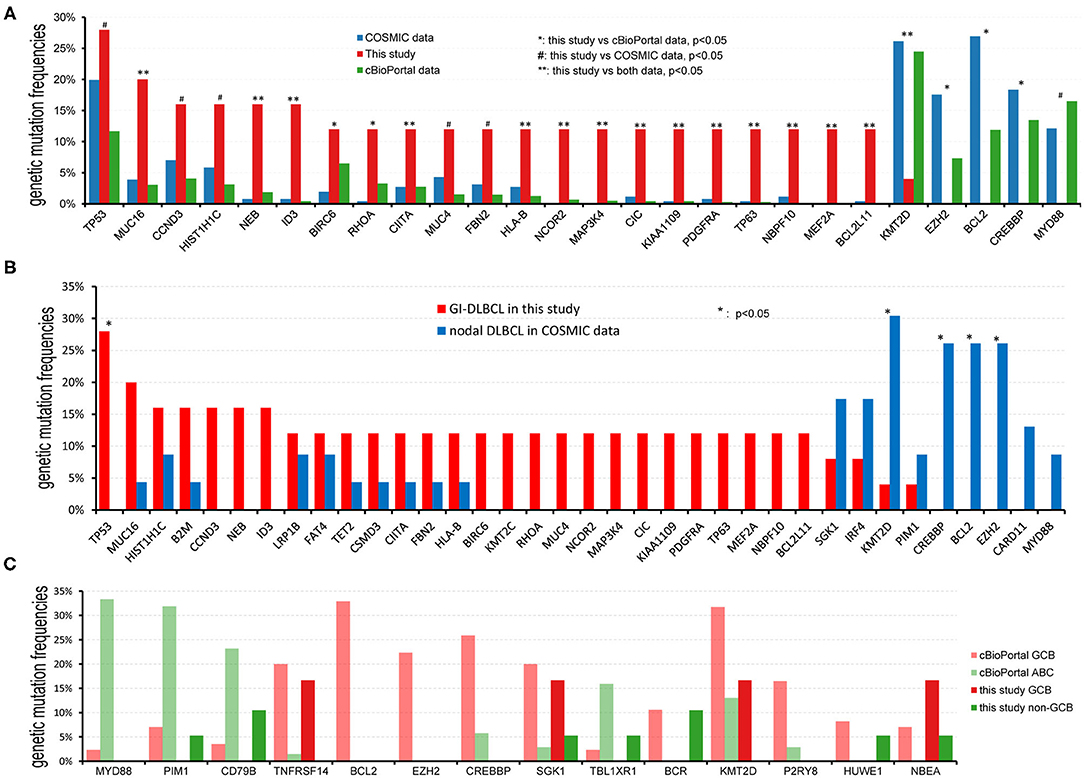
Figure 4. Comparison of the mutational frequencies of the most commonly mutated oncogenes in DLBCL. (A) Comparison of mutation frequencies between GI-DLBCL and DLBCL in cBioPortal and COSMIC. GI-DLBCL exhibited different mutation profiles compared with common DLBCL in cBioPortal and COSMIC. (B) Comparison of mutation frequencies between GI-DLBCL and nodal DLBCL in COSMIC. (C) Comparison of mutation frequencies for different oncogenic mutations between GCB-DLBCL and non-GCB-DLBCL identified in cBioPortal; no gene exhibited different mutation frequencies between GCB-DLBCL and non-GCB-DLBCL in this study.
DLBCL has GCB and ABC molecular subtypes, and these subtypes have different mutation cohorts. In cBioPortal, the mutation features of 85 patients with GCB-DLBCL were compared to those of 69 patients with ABC-DLBCL, and many oncogenic driver genes revealed different mutation frequencies (Figure 4C). GCB-DLBCL tumors harbored more mutations in BCL2, EZH2, CREBBP, KMT2D, SGK1, BCR, NBEA, P2RY8, HUWE1, and TNFRSF14, while ABC-DLBCL tumors harbored more mutations in MYD88, PIM1, CD79B, and TBL1XR1. However, the mutation frequencies and variants observed in 6 patients with GCB-DLBCL and 19 patients with non-GCB-DLBCL in our study were not different. The most likely cause of this finding is that some genetic mutations, such as those in MYD88, CREBBP, BCL2, KMT2D, PIM1, and EZH2, rarely occur in GI-DLBCL. In addition, the incidence of genetic mutations in TNFRSF14, TBL1XR1, and P2RY8 was reduced in GI-DLBCL compared with those in non-gastrointestinal DLBCL; however, the differences between the two were not statistically significant (Supplementary Table 3). Furthermore, we analyzed the mutation differences between non-GCB-DLBCL in this study (n = 19) and ABC-DLBCL in cBioPortal (n = 69). MYD88 and PIM1 mutations were significantly reduced in non-GCB-type GI-DLBCL (P = 0.002 and 0.019, respectively), but mutations in ID3, CIC, PDGFRA, MEF2A, RHOA, MAP3K4, TP63, and NCOR2 were increased in these GI-DLBCLs. In addition, no differences were noted in the mutation profiles of GCB-DLBCL in this study (n = 6) and GCB-DLBCL in cBioPortal (n = 85). The small number of patients in our study may be one reason for the statistical homogeneity.
Genetic Mutations and Clinicopathological Characteristics
We analyzed the correlation between the number of genetic mutations or oncogenic mutations and clinicopathological characteristics, such as age, sex, tumor site, Ki-67 proliferation index, Eastern Cooperative Oncology Group (ECOG) score, B symptoms, International Prognostic Index (IPI), tumor stage and molecular subtype. The number of gene mutations and the number of mutant genes were not related to the abovementioned clinicopathological indicators. In addition, logistic regression analysis also revealed that patient age, sex, B symptoms, ECOG score, and tumor site were not related to oncogenic mutations. However, some mutations exhibited a tendency toward a high proliferation index (Ki-67 index >90%), such as HLA-B (P = 0.031), MEF2A (P = 0.031), RHOA (P = 0.031) and NAV3 (P = 0.003). LRPIB mutations occurred in tumors with low IPI scores (P = 0.037). MUC16 and ETV6 mutations occurred in early-stage tumors (P = 0.010 and 0.032, respectively).
Discussion
Several studies have examined the mutational landscape of DLBCL, and many recurrent genetic mutations have been detected. The most commonly mutated genes include KMT2D, TP53, B2M, EZH2, GNA13, MEF2B, SGK1, CREBBP, CD79B, and MYD88 (1). In our study, GI-DLBCL exhibited high-frequency mutations in B2M, CCND3, HIST1H1C, BIRC6, TET2, KMT2C, LRP1B, CSMD3, FAT4, RHOA, MUC4, and FBN2, revealing consistent mutation frequencies and variants in common with DLBCL in cBioPortal and COSMIC. Furthermore, we rediscovered the previously discovered important drivers of DLBCL, such as SGK1, BRAF, and BTG1 (22, 23); however, these genes did not exhibit high mutation frequencies. The mutations mentioned above are involved in many cellular processes and pathways, including cell growth, proliferation, migration, differentiation, metabolism, survival, apoptosis, and immunoregulation (3).
Genetic classification based on multiple analytic platforms, including mutations, translocations, and/or copy-number alterations, divides DLBCL into distinct clusters with discrete genetic signatures and different clinical characteristics (24). Chapuyg et al. (25) discovered six robust subsets (C0–C5) by applying non-negative matrix factorization consensus. Schmitz et al. (2) identified four prominent genetic subtypes termed MCD, BN2, N1, and EZB. Recurrently, seven genetic subtypes are related to distinct indolent lymphoma types, including MCD, BN2, N1, EZB, A53, ST2, and others (26). In our study, TP53 mutations were detected in 7 cases of GI-DLBCL, and SGK1 and TET2 mutations were observed in four cases. Two cases had CD79B mutations, but no MYD88 mutation was detected. Mutation of EZH2, NOTCH1, or NOTCH2 was not detected. Simultaneously, BCL2 translocation was not detected in 13 cases of GI-DLBCL by fluorescence in situ hybridization. Although GI-DLBCLs cannot be accurately classified given the incompleteness of our data, most GI-DLBCL cases in this study may have the characteristics of A53 and ST2 genetic subtypes, and a few cases were classified as MCD, EZB and N1 subtypes. However, in the study by Wright et al., the A53 and ST2 subtypes accounted for only 6.6 and 4.7% of DLBCL cases, respectively. This finding suggests that GI-DLBCL may exhibit different molecular genetic changes from other DLBCL.
The driver genes responsible for the development of gastrointestinal DLBCL differed from those of nodal or non-gastrointestinal DLBCL (27). First, TP53 mutations demonstrated a significantly increased mutation frequency compared with that in nodal DLBCL in COSMIC and common DLBCL in cBioPortal. Similarly, Voropaeva et al. (28) reported a TP53 mutation frequency of 16.22% in 74 patients with DLBCL, and nodal DLBCL and extranodal DLBCL exhibited mutation frequencies of 9.52 and 25.00%, respectively. Furthermore, in our study, most mutations (6/7) in TP53 occurred in the DNA binding domain (Supplementary Figure 2), which is believed to have an adverse prognostic impact (29, 30). Second, the mutation frequencies of MUC16, NEB, ID3, CIITA, HLB-A, NCOR2, MAP3K4, CIC, KIAA1109, PDGFRA, TP63, NBPF10, MEF2A, and BCL2L11 in this study were also increased compared with those in common DLBCL in cBioPortal and COSMIC. Third, mutations in the BCL2, KMT2D, CREBBP, EZH2, and MYD88 genes are considered common genetic alterations in DLBCL with mutation frequencies of >10%; however, except for one mutation in the KMT2D gene, mutations in any of the above genes were not detected in our study. Consistent with our study, the MSK-IMPACT Clinical Sequencing Cohort reported a mutation frequency of 37.84% for KMT2D in 74 patients with DLBCL, but KMT2D mutation was not detected in 5 cases of GI-DLBCL (18). Frick et al. (9) and Nagakita et al. (31) reported that MYD88 mutations exhibited less involvement in GI-DLBCL than in nodal DLBCL and other extranodal DLBCLs. In our previous study, the EZH2 Y641 mutation was successfully detected in one of 94 patients with GI-DLBCL (including 33 patients with GCB-DLBCL and 61 patients with non-GCB-DLBCL) by Sanger sequencing, and the mutation frequency was significantly reduced compared with that in non-gastrointestinal DLBCL (16). Therefore, we hypothesized that the mutation profile of GI-DLBCL differed from that of nodal DLBCL and non-gastrointestinal DLBCL.
The different mutation profiles between GI-DLBCL and non-gastrointestinal DLBCL suggest different pathogenesis. In addition to mutations in the KMT2D, MYD88, CREBBP, BCL2 and EZH2 genes, PIM1 and CARD11 gene mutations were also observed at a lower frequency in GI-DLBCL than in common or nodal DLBCL (32, 33); however, the difference was not statistically significant in this study (Figure 4 and Supplementary Table 3). Some of these gene mutations are related to the NF-κB pathway (BCL2, MYD88, and CARD11) and JAK-STAT pathway (BCL2, CREBBP, and PIM1). Gene mutations leading to overactivation of the NF-κB-related pathway is an important molecular mechanism of DLBCL lymphomagenesis. Gene mutations in CARD11, CD79A, CD79B, and other genes activate the NF-κB pathway through the B-cell receptor signaling pathway (2, 34), and MYD88 mutations promote IRAK4 and IRAK1 phosphorylation and activate the NF-κB pathway in an antigen-independent manner (35). Moreover, in barrier-protected tissues, such as the testis (59–61%) and central nervous system (75–94%), MYD88 mutations are frequently detected (10, 11, 25, 36). Studies have reported missense mutations in the CARD11 gene in 9.6% of ABC-DLBCLs (34). However, MYD88 and CARD11 mutations were not detected in 25 patients with GI-DLBCL. Intriguingly, although CD79B mutations were detected in two patients (8%) with GI-DLBCL, the frequency was significantly reduced compared with that in the testes (18.9~71.4%) (9, 11, 37) and central nervous system (31.6~61.1%) DLBCLs (36, 38, 39). Moreover, only one case had an activating mutation at p.Y197 of CD79B gene (Supplementary Table 2). These results are indicative of different pathogenetic mechanisms leading to the development of DLBCL, which may be influenced by the tissue microenvironment. The gastrointestinal tract has immunologically specific environments where antigen stimuli are fulminant, and the NF-κB pathway is activated by antigen stimulation in the chronic inflammatory environment. Interestingly, 4 of 12 patients with gastric DLBCL exhibited Helicobacter pylori infection in our study. However, in barrier-protected tissues where tumor cells experience less antigenic stimulation, NF-κB pathway activation is particularly dependent on chronic B-cell receptor signaling and Toll-like receptor signaling maintained by CARD11, CD79B, and MYD88 mutations (31). In addition, many low-frequency oncogenic mutations also promote lymphomagenesis (Supplementary Table 3), and KEGG pathway analysis of the mutant genes in this study revealed that the PI3K-AKT pathway might play an important role in promoting GI-DLBCL tumor cell growth. Genetic mutations were detected in CCND3, MYC, PDGFRA, SGK1, BCL2L11, PDGFRB, and PIK3R1, all of which perform cellular functions through the PI3K-AKT pathway (2). Therefore, DLBCLs in different primary sites have various mutation characteristics due to their unique microenvironment. Primary DLBCL in immune-privileged sites exhibits high-frequency mutations in MYD88 and CD79B, and nodal DLBCL exhibits high-frequency mutations in KMT2D, PIM1, BCL2, CREBBP, and EZH2. However, these mutations rarely occur in GI-DLBCL.
Some high-frequency mutations, such as mutations in KMT2D, TP53, CD79B, CARD11, EZH2, and MYD88, play important roles in lymphomagenesis (23); presumably, however, additional alterations are also indispensable for the development and characterization of DLBCL (5). We analyzed the correlation between the number of mutations and clinicopathological characteristics. Neither the number of somatic mutations nor the number of oncogenic mutations was associated with the following clinicopathological characteristics: sex, age, tumor site, molecular subtype, Ki-67 proliferation index, B symptoms, ECOG score, IPI score, and clinical stage. Similarly, Cascione et al. (40) analyzed 72 patients with MALT lymphomas by next-generation sequencing, and there was no association between the mutational status and clinical stage. However, some genetic mutations, such as those in HLA-B, MEF2A, RHOA, and NAV3, were associated with a high tumor proliferation index in this study. In addition, patients with ETV6 mutations are associated with early clinical stages of GI-DLBCL. In DLBCL, the ETV6 mutation is a common gene alteration, with a frequency of 3.25% and a preference for the ABC subtype (in cBioPortal), especially in primary central nervous system DLBCL (41, 42). In a recent study, the ETV6 gene was identified as a driver gene of DLBCL (43).
Conclusion
GI-DLBCL exhibited low genetic mutation frequencies of the BCL2, KMT2D, CREBBP, EZH2, PIM1, MYD88, and CD79B genes. The different mutated genes are related to the NF-κB and JAK-STAT pathways and suggest that the different pathogenetic mechanisms of GI-DLBCL and non-gastrointestinal DLBCL depend, in part, on the microenvironment. In addition, we also found that some genetic mutations are associated with a high tumor proliferation index and an early clinical stage. Therefore, the identification of these mutations may be useful for the prognostic prediction of GI-DLBCL. This study will help to extend the utility of cancer genome studies and accelerate the pace at which genetic findings are translated into therapeutic impacts.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: GSA-Human (the Genome Sequence Archive for Human) [HRA000533] (https://bigd.big.ac.cn/gsa-human/browse/HRA000533).
Ethics Statement
The studies involving human participants were reviewed and approved by Xijing Hospital Ethics Committee and Tangdu Hospital Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
PL and ZW conceived and designed the study. PL, JC, YaL, JW, YiL, DZ, JM, KW, LG, and WZ performed the research. PL, ZC, XL, SG, QY, and ML performed the data analysis. PL and JC wrote the manuscript. ZW, LF, and ML reviewed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant from the State Key Laboratory of Cancer Biology (CBSKL2019ZDKF02).
Conflict of Interest
YS was employed by the company Nanjing Geneseeq Technology Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to Mingqing Du (Division of Cellular and Molecular Pathology, Department of Pathology, University of Cambridge, Cambridge) for his technical and intellectual input and advice. The authors also thank the scientists at American Journal Experts (Durham, North Carolina).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2021.622648/full#supplementary-material
References
1. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: Inernational Agency for Research on Cancer (2017).
2. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. (2018) 378:1396–407. doi: 10.1056/NEJMoa1801445
3. Li S, Young KH, Medeiros LJ. Diffuse large B-cell lymphoma. Pathology. (2018) 50:74–87. doi: 10.1016/j.pathol.2017.09.006
4. Ollila TA, Kurt H, Waroich J, Vatkevich J, Sturtevant A, Patel NR, et al. Genomic subtypes may predict the risk of central nervous system recurrence in diffuse large B-cell lymphoma. Blood. (2020). doi: 10.1182/blood.2020007236. [Epub ahead of print].
5. Bolen CR, Klanova M, Trneny M, Sehn LH, He J, Tong J, et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: data from the phase III GOYA study. Haematologica. (2020) 105:2298–307. doi: 10.3324/haematol.2019.227892
6. Nanthakwang N, Rattarittamrong E, Rattanathammethee T, Chai-Adisaksopha C, Tantiworawit A, Norrasethada L, et al. Clinicopathological study and outcomes of primary extranodal lymphoma. Hematology reports. (2019) 11:8227. doi: 10.4081/hr.2019.8227
7. Yin X, Xu A, Fan F, Huang Z, Cheng Q, Zhang L, et al. Incidence and mortality trends and risk prediction nomogram for extranodal diffuse large B-cell lymphoma: an analysis of the surveillance, epidemiology, and end results database. Front Oncol. (2019) 9:1198. doi: 10.3389/fonc.2019.01198
8. Hanafy AK, Morani AC, Menias CO, Pickhardt PJ, Shaaban AM, Mujtaba B, et al. Hematologic malignancies of the gastrointestinal luminal tract. Abdom Radiol. (2020) 10:3007–27. doi: 10.1007/s00261-019-02278-8
9. Frick M, Bettstetter M, Bertz S, Schwarz-Furlan S, Hartmann A, Richter T, et al. Mutational frequencies of CD79B and MYD88 vary greatly between primary testicular DLBCL and gastrointestinal DLBCL. Leuk Lymphoma. (2018) 59:1260–3. doi: 10.1080/10428194.2017.1370546
10. Chapuy B, Roemer MG, Stewart C, Tan Y, Abo RP, Zhang L, et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood. (2016) 127:869–81. doi: 10.1182/blood-2015-10-673236
11. Kraan W, van Keimpema M, Horlings HM, Schilder-Tol EJ, Oud ME, Noorduyn LA, et al. High prevalence of oncogenic MYD88 and CD79B mutations in primary testicular diffuse large B-cell lymphoma. Leukemia. (2014) 28:719–20. doi: 10.1038/leu.2013.348
12. Lewin KJ, Ranchod M, Dorfman RF. Lymphomas of the gastrointestinal tract: a study of 117 cases presenting with gastrointestinal disease. Cancer. (1978) 42:693–707. doi: 10.1002/1097-0142(197808)42:2<693::AID-CNCR2820420241>3.0.CO;2-J
13. Yang H, Wu M, Shen Y, Lei T, Mi L, Leng X, et al. Treatment strategies and prognostic factors of primary gastric diffuse large B cell lymphoma: a retrospective multicenter study of 272 cases from the China Lymphoma Patient Registry. Int J Med Sci. (2019) 16:1023–31. doi: 10.7150/ijms.34175
14. Hong YW, Kuo IM, Liu YY, Yeh TS. The role of surgical management in primary small bowel lymphoma: a single-center experience. Eur J Surg Oncol. (2017) 43:1886–93. doi: 10.1016/j.ejso.2017.06.016
15. Cai YB, Chen HY, He JJ, Hu YT, Yang Q, Chen LB, et al. The role of surgical intervention in primary colorectal lymphoma: a SEER population-based analysis. Oncotarget. (2016) 7:72263–75. doi: 10.18632/oncotarget.12344
16. Liu Y, Yu K, Li M, Zeng K, Wei J, Li X, et al. EZH2 overexpression in primary gastrointestinal diffuse large B-cell lymphoma and its association with the clinicopathological features. Hum Pathol. (2017) 64:213–21. doi: 10.1016/j.humpath.2017.04.011
17. Li P, Li M, Wang K, Liu Y, Wang Y, Zhao D, et al. Genetic alterations in cell cycle regulation-associated genes may promote primary progression of gastrointestinal stromal tumors. Lab Invest J Tech Methods Pathol. (2020) 100:426–37. doi: 10.1038/s41374-019-0322-x
18. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. (2017) 23:703–13. doi: 10.1038/nm.4333
19. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. (2013) 339:1546–58. doi: 10.1126/science.1235122
20. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature. (2013) 502:333–9. doi: 10.1038/nature12634
21. Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. (2013) 3:2650. doi: 10.1038/srep02952
22. Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. (2011) 476:298–303. doi: 10.1038/nature10351
23. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. (2012) 109:3879–84. doi: 10.1073/pnas.1121343109
24. Minderman M, Pals ST. Towards genomic-based prognostication and precision therapy for diffuse large B-cell lymphoma. Haematologica. (2020) 105:2194–6. doi: 10.3324/haematol.2020.255448
25. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. (2018) 24:679–90. doi: 10.1038/s41591-018-0016-8
26. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. (2020) 37:551–68. doi: 10.1016/j.ccell.2020.03.015
27. Hori Y, Yamamoto H, Nozaki Y, Torisu T, Fujiwara M, Taguchi K, et al. Colorectal diffuse large B-cell lymphoma: molecular subclassification and prognostic significance of immunoglobulin gene translocation. Hum Pathol. (2019). doi: 10.1016/j.humpath.2019.09.003
28. Voropaeva EN, Pospelova TI, Voevoda MI, Maksimov VN, Orlov YL, Seregina OB. Clinical aspects of TP53 gene inactivation in diffuse large B-cell lymphoma. BMC medical genomics. (2019) 12: 35. doi: 10.1186/s12920-019-0484-9
29. Young KH, Weisenburger DD, Dave BJ, Smith L, Sanger W, Iqbal J, et al. Mutations in the DNA-binding codons of TP53, which are associated with decreased expression of TRAILreceptor-2, predict for poor survival in diffuse large B-cell lymphoma. Blood. (2007) 110:4396–05. doi: 10.1182/blood-2007-02-072082
30. Karube K, Enjuanes A, Dlouhy I, Jares P, Martin-Garcia D, Nadeu F, et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia. (2018) 32:675–84. doi: 10.1038/leu.2017.251
31. Nagakita K, Takata K, Taniguchi K, Miyata-Takata T, Sato Y, Tari A, et al. Clinicopathological features of 49 primary gastrointestinal diffuse large B-cell lymphoma cases; comparison with location, cell-of-origin, and frequency of MYD88 L265P. Pathol Int. (2016) 66:444–52. doi: 10.1111/pin.12439
32. Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. (2015) 21:1199–208. doi: 10.1038/nm.3943
33. Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. (2015) 21:1190–8. doi: 10.1038/nm.3940
34. Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. (2008) 319:1676–9. doi: 10.1126/science.1153629
35. Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathways in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. (2013) 23:422–30. doi: 10.1016/j.semcancer.2013.04.002
36. Yamada S, Ishida Y, Matsuno A, Yamazaki K. Primary diffuse large B-cell lymphomas of central nervous system exhibit remarkably high prevalence of oncogenic MYD88 and CD79B mutations. Leuk Lymphoma. (2015) 56:2141–5. doi: 10.3109/10428194.2014.979413
37. Chen YP, Ke LF, Lu JP, Wang JC, Zhu WF, Chen FF, et al. Prevalence and clinical significance of oncogenic CD79B and MYD88 mutations in primary testicular diffuse large B-cell lymphoma: a retrospective study in China. Onco Targets Therapy. (2019) 12:10165–75. doi: 10.2147/OTT.S222189
38. Nakamura T, Tateishi K, Niwa T, Matsushita Y, Tamura K, Kinoshita M, et al. Recurrent mutations of CD79B and MYD88 are the hallmark of primary central nervous system lymphomas. Neuropathol Appl Neurobiol. (2016) 42:279–90. doi: 10.1111/nan.12259
39. Zheng M, Perry AM, Bierman P, Loberiza F Jr, Nasr MR, et al. Frequency of MYD88 and CD79B mutations, and MGMT methylation in primary central nervous system diffuse large B-cell lymphoma. Neuropathology. (2017) 37:509–16. doi: 10.1111/neup.12405
40. Cascione L, Rinaldi A, Bruscaggin A, Tarantelli C, Arribas AJ, Kwee I, et al. Novel insights into the genetics and epigenetics of MALT lymphoma unveiled by next generation sequencing analyses. Haematologica. (2019) 104:e558–e61. doi: 10.3324/haematol.2018.214957
41. Bruno A, Boisselier B, Labreche K, Marie Y, Polivka M, Jouvet A, et al. Mutational analysis of primary central nervous system lymphoma. Oncotarget. (2014) 5:5065–75. doi: 10.18632/oncotarget.2080
42. Liu Z, Meng J, Li X, Zhu F, Liu T, Wu G, et al. Identification of Hub genes and key pathways associated with two subtypes of diffuse large B-cell lymphoma based on gene expression profiling via integrated bioinformatics. BioMed Res Int. (2018) 2018:3574534. doi: 10.1155/2018/3574534
Keywords: diffuse large B-cell lymphoma, DLBCL, gastrointestinal tract, whole-exome sequencing, genetic mutation
Citation: Li P, Chai J, Chen Z, Liu Y, Wei J, Liu Y, Zhao D, Ma J, Wang K, Li X, Shao Y, Gong L, Zhang W, Guo S, Yan Q, Li M, Fan L and Wang Z (2021) Genomic Mutation Profile of Primary Gastrointestinal Diffuse Large B-Cell Lymphoma. Front. Oncol. 11:622648. doi: 10.3389/fonc.2021.622648
Received: 29 October 2020; Accepted: 08 February 2021;
Published: 05 March 2021.
Edited by:
Shimin Hu, University of Texas MD Anderson Cancer Center, United StatesReviewed by:
Narendranath Epperla, The Ohio State University, United StatesAndres Quesada, University of Texas MD Anderson Cancer Center, United States
Copyright © 2021 Li, Chai, Chen, Liu, Wei, Liu, Zhao, Ma, Wang, Li, Shao, Gong, Zhang, Guo, Yan, Li, Fan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingyang Li, limingyang1108@sina.com; Linni Fan, linni.fan@hotmail.com; Zhe Wang, zhwang@fmmu.edu.cn
†These authors have contributed equally to this work and share co-first authorship
‡These authors have contributed equally to this work and share co-senoir authorship