
- Lab of Toxicology, Katholieke Universiteit Leuven, Leuven, Belgium
Voltage-gated sodium channels (VGSCs) are large transmembrane proteins that conduct sodium ions across the membrane and by doing so they generate signals of communication between many kinds of tissues. They are responsible for the generation and propagation of action potentials in excitable cells, in close collaboration with other channels like potassium channels. Therefore, genetic defects in sodium channel genes can cause a wide variety of diseases, generally called “channelopathies.” The first insights into the mechanism of action potentials and the involvement of sodium channels originated from Hodgkin and Huxley for which they were awarded the Nobel Prize in 1963. These concepts still form the basis for understanding the function of VGSCs. When VGSCs sense a sufficient change in membrane potential, they are activated and consequently generate a massive influx of sodium ions. Immediately after, channels will start to inactivate and currents decrease. In the inactivated state, channels stay refractory for new stimuli and they must return to the closed state before being susceptible to a new depolarization. On the other hand, studies with neurotoxins like tetrodotoxin (TTX) and saxitoxin (STX) also contributed largely to our today’s understanding of the structure and function of ion channels and of VGSCs specifically. Moreover, neurotoxins acting on ion channels turned out to be valuable lead compounds in the development of new drugs for the enormous range of diseases in which ion channels are involved. A recent example of a synthetic neurotoxin that made it to the market is ziconotide (Prialt®, Elan). The original peptide, ω-MVIIA, is derived from the cone snail Conus magus and now FDA/EMA-approved for the management of severe chronic pain by blocking the N-type voltage-gated calcium channels in pain fibers. This review focuses on the current status of research on neurotoxins acting on VGSC, their contribution to further unravel the structure and function of VGSC and their potential as novel lead compounds in drug development.
Voltage-Gated Sodium Channels
Like many other voltage-gated ion channels, VGSCs are transmembrane complexes consisting of a large core protein, the α-subunit (220–260 kDa, ≈2000 amino acids), associated with one or more smaller regulatory β-subunits (22–36 kDa). Alpha-subunits contain the functional ion conduction pore as an aqueous cavity that is selectively permeable for sodium ions. In mammalian cells, nine α-subunit isoforms (classified as Nav 1.1–Nav 1.9) have been characterized so far. Additionally, sodium channel-like proteins, classified as Nax, have been identified but are not yet functionally expressed (Catterall et al., 2005). The VGSC isoforms are distributed differentially throughout electrical excitable cells of the body, which correlates with different functional properties in the corresponding tissues. Nav1.1, 1.2, 1.3, and 1.6 are mainly expressed in the central nervous system (CNS); Nav1.7, 1.8, and 1.9 on the contrary are highly expressed in the peripheral nervous system (PNS) and finally, the Nav1.4 and 1.5 isoforms are present in adult skeletal muscle and heart muscle, respectively (Goldin, 2001).
In contrast to potassium channels, no crystallographic image of a sodium channel could be assessed for a long time and information about the structural composition of the VGSC had to be deduced in an indirect way. Recently however, the group of Catterall enlightened the horizon with the determination of the crystal structure of a bacterial VGSC (Payandeh et al., 2011). In early molecular cloning studies, later confirmed by cryo-electron images (Sato et al., 2001) and the recent crystallographic image, the α-subunit turned out to be composed of four homologous domains, DI–DIV, which all contain six putative transmembrane segments, S1–S6 (Yu and Catterall, 2003). The four domains are connected by three cytoplasmic linker loops of different size and together they form a bell-shaped protein (Sato et al., 2001).
All of the four domains consist of two modules, the first being the voltage-sensing module formed by S1–4, the second being the pore-forming module, formed by S5 and S6 and the connecting loop. How the voltage-sensing module can “sense” voltage and thereby open the channel is a question that can be answered by looking at one particular segment of this module, the voltage sensor S4. These cylindrical α-helical structures display highly conserved positive residues at every third position. In the “sliding helix” (Catterall, 1986) or “helical screw” (Guy and Seetharamulu, 1986) models these positive charges are proposed to be drawn into the membrane by the negative internal resting membrane potential. The positive charges are stabilized by the formation of neutralizing ion pairs with neighboring negative charges in S1, S2, and/or S3. When the membrane depolarizes, the negative membrane potential is relieved and the S4 segments move outward, thereby causing a conformational change that opens the pore and initiates channel activation (Catterall, 2000, 2010).
Another question that has to be answered when discussing VGSC’s function is how ion selectivity can be achieved. The answer lies in the pore region of the channel, formed by S5 and S6 and the connecting pore-loop (P-loop). By dipping into the membrane, the P-loops of DI–DIV form the outer pore of the channels. Herein, two rings determine ion selectivity. The inner selectivity ring is formed by the four amino acids Asp, Glu, Lys, and Ala in DI, DII, DIII, and DIV, respectively (DEKA-ring). The outer selectivity ring is formed in the same way and is represented by the residues Glu, Glu, Asp, and Asp of DI, DII, DIII, and DIV, respectively (EEDD-ring). The first studies that underlie the discovery of the DEKA-ring made use of the historical toxins tetrodotoxin (TTX) and saxitoxin (STX). Their mechanism of action and binding site is discussed in the following chapter. The intracellular part of the pore is formed by all four S6 segments (Yu and Catterall, 2003).
Within a few milliseconds after the opening of the channel, fast inactivation begins. This key feature was the third characteristic set by Hodgkin and Huxley when they first recorded sodium currents (Hodgkin and Huxley, 1952), besides the voltage-dependent activation and selective ion conductance. The part of the channel responsible for fast inactivation is the short, highly conserved intracellular linker that connects domains III and IV. In mutagenesis studies, the three hydrophobic amino acids Ile, Phe, and Met (IFM motif) turned out to be the key sequence necessary for fast inactivation. The inactivation gate receptor for this IFM motif is located within and near the inner mouth of the pore (Smith and Goldin, 1997). Mutations in the S4 segments, and more specifically S4 of DIV revealed that outward movement of S4 DIV initiates fast inactivation, which makes the inactivation process also voltage-dependent. This model was largely confirmed by studies with α-scorpion toxins and sea anemone toxins, of which some will be discussed later. Another mode of VGSC inactivation was described as slow inactivation and occurs in a time scale of seconds after activation, but still remains the topic of further investigations (Ulbricht, 2005).
The cytoplasmic linker loops between the four domains continue to be interesting sites at which the channel’s modulation and regulation of gating processes happen. Several phosphorylation sites were found in the first and third intracellular linkers between DI–DII and DIII–DIV, respectively. Phosphorylation can be carried out by isoforms of protein kinase A (PKA) or C (PKC) and has different results on the channels’ function depending on the kinase or affected VGSC isoform. An ankyrin binding site has also been identified in the linker between DII and DIII, which also may influence the channels’ gating kinetics (Chahine et al., 2005).
The large α-subunit is associated with one or more smaller regulatory β-subunits, which modulate sodium currents and also regulate the cell surface expression of channels (Patino and Isom, 2010). They turned out to be essential pieces of the VGSC complexes as they are needed in the heterologous expression of α-subunits to display full native sodium channel characteristics, such as correct kinetics and voltage-dependence of the gating (Yu and Catterall, 2003). At least five regulatory β-subunits (β1–β4 and β1A) have been identified so far from four different genes (SNC1B–4B), with β1A as a splice variant of the β1-subunit. The β-subunits can be bound either non-covalently (β1- and β4-subunits) or covalently (β2- and β4-subunits) to form a heteromer with the α-subunit (Chahine et al., 2005).
Neurotoxins and VGSC Pharmacology
Neurotoxins
From the early beginning of research on the basic principles of ion channel functioning, neurotoxins fulfilled an important job, especially concerning VGSCs. One particular neurotoxin that deserves special attention is tetrodotoxin (TTX). It was first isolated from the pufferfish (family of Tetraodontidae) and has a close relative: saxitoxin (STX). TTX played a great part in the discovery of the VGSC protein (Agnew et al., 1980) and in the determination of the selectivity filter (Heinemann et al., 1992) and binding site 1 for pore blockers of VGSCs (Noda et al., 1989). Nowadays TTX is still used to classify sodium channels according to their sensitivity to TTX: Nav1.5, Nav1.8, and Nav1.9 are TTX-resistant (TTX-R), the others are TTX-sensitive (TTX-S).
The name “neurotoxin” already reveals at what those natural chemicals and peptides are best: interfering with the normal function of the nervous system. That is exactly what plants, animals, and microorganisms develop them for: to target a possible offender or prey, either by immobilizing or immediately killing it. Neurotoxins produced by those organisms mostly form part of a mixture of peptides and enzymes that act in concert. This mixture can be a venom (peptides and proteins, actively injected and used in defense or prey acquisition) or a poison (secondary products or metabolites, passively used to protect or defend the organism; Mebs, 2002). But their main target is always the CNS, PNS as herein lie the vital functions of the offender/prey. During evolution, venomous and poisonous organisms turned out to be excellent pharmacologists since their toxins evolved to be highly specific for targets in the CNS or PNS. They can act on membrane receptors and ion channels like voltage-gated sodium, potassium, and calcium channels. By doing so they give us outstanding keys to investigate the structure and function of their targets, to define underlying physiological processes, and most importantly, to design new, potent, and selective drugs useful in a wide variety of diseases like pain, diabetes, and multiple sclerosis (Lewis and Garcia, 2003). As an example of a rich and nearly unexplored library of valuable bioactive compounds, one can look at cone snails, which are venomous marine invertebrates. More than 700 species are known to date and it is estimated that a single Conus species may contain up to 200 different venom peptides, implying that a library of more than 140,000 compounds of possible pharmaceutical interest is available in their venom. At least one compound has led to a new therapy for severe chronic pain (ziconotide/Prialt®) and many other lead compounds can be suspected from these and other venomous and poisonous organisms (Terlau and Olivera, 2004).
Neurotoxin Binding
Neurotoxins acting on VGSCs can aim at six different sites in the channels, distinguished not only by matters of localization of the toxin binding place but also by the results of the toxin’s action. Apart from the neurotoxin receptor sites, two other sites are determined. The pyrethroid binding site is the binding site for some insecticidal agents like DDT and pyrethroids. Anticonvulsants and antiarrhythmics bind in a use-dependent manner to the local anesthetics (LA) site (Table 1). The latter two will not be discussed in this review because they are not affected by neurotoxins and therefore, are not in the scope of this review.
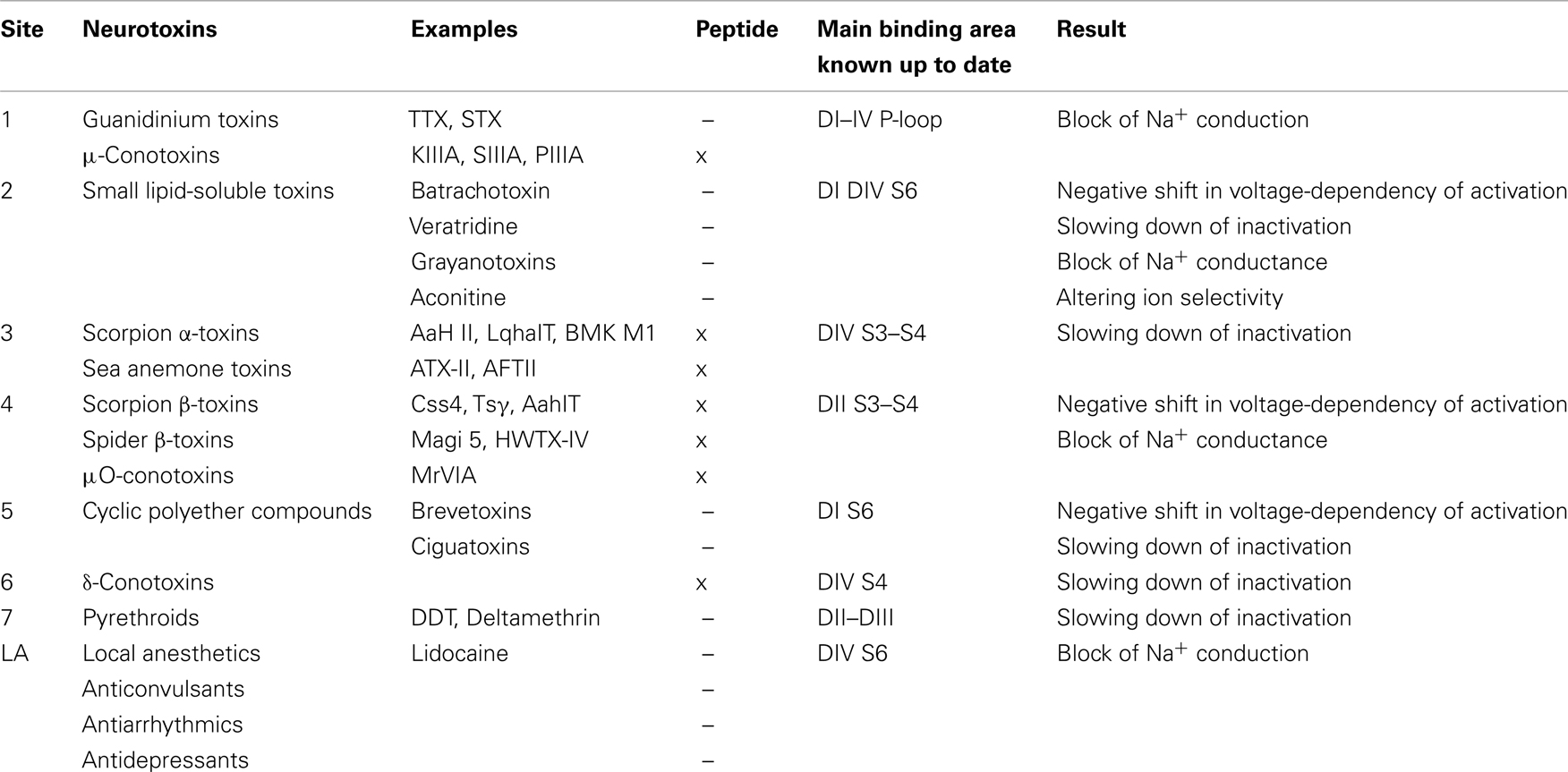
Table 1. Overview of neurotoxin binding sites according to the revised model.
The interaction between neurotoxins and VGSCs can occur in two different ways. It either results in a pore block when the toxin physically occludes the pore and thereby inhibits the sodium conductance, or in a modification of the gating, which leads to altered gating kinetics and voltage-dependence of the channels. Toxins binding on site 1 use the first mechanism. The previously mentioned guanidinium toxins TTX and STX are such site 1 pore blockers; they will form a plug in the outer vestibule of the pore. Site 2 toxins like batrachotoxin and grayanotoxin will prevent inactivation and therefore persistently activate the channel. Scorpion α-toxins and sea anemone toxins are typical examples of site 3 toxins; they will slow or inhibit inactivation. Scorpion β-toxins and β-spider toxins bind to site 4 and shift the voltage-dependence of activation toward more hyperpolarized potentials. Site 5 neurotoxins like brevetoxins and ciguatoxins exhibit a real arsenal of effects upon VGSC binding, e.g., inhibition of activation and a hyperpolarizing shift of the voltage-dependence of activation. Lastly, δ-conotoxins acting on site 6 will cause similar effects as site 3 toxins by slowing or inhibiting inactivation (Figure 1 and Table 1).
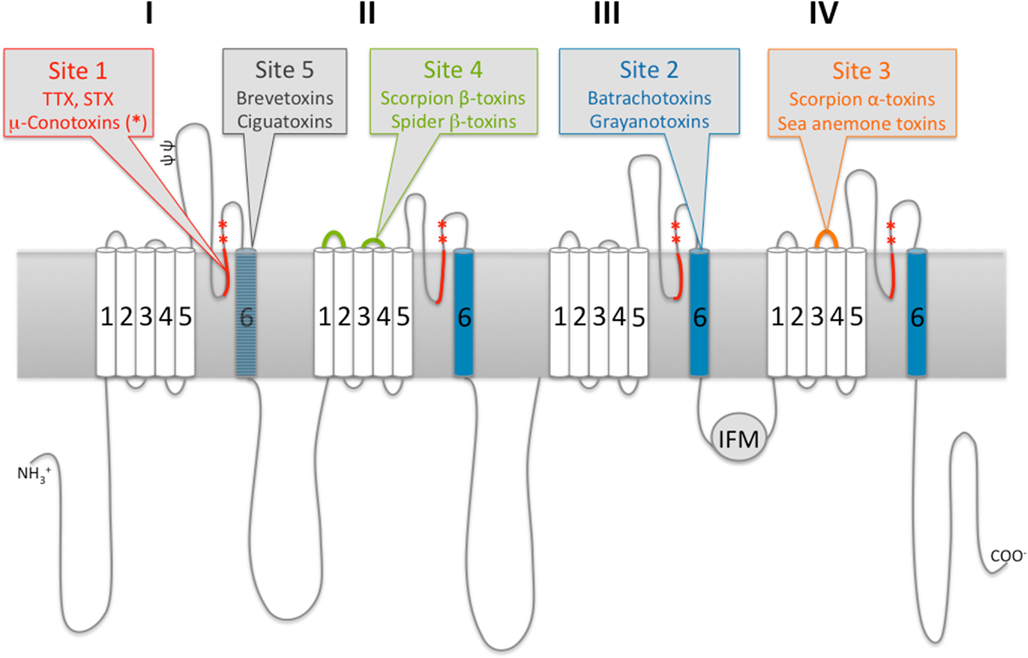
Figure 1. Schematic two-dimensional representation of the functional α-subunit of voltage-gated ion channels and identification of known neurotoxin binding areas.
As mentioned previously, toxins can be differentiated based on their chemical composition. Organic hydrophilic or lipophilic neurotoxin compounds are mostly secondary metabolites, used by the producing organism as defense molecules. Examples of lipophilic compounds include the site 2 grayanotoxins and the brevetoxins acting on site 5. Hydrophilic secondary metabolites include site 1 toxins TTX and STX. On the other hand, organisms can use larger chemical entities like peptides to paralyze or kill their prey, like the peptides found in scorpions, spiders, wasps, cone snails, and others. But although their origin might be different, both kinds of neurotoxins can have the same effect (binding of μ-conotoxins and TTX for both results in block of the Na+ conductance). For other reviews on neurotoxins acting on VGSCs see for example (Catterall et al., 2007; de la Vega and Possani, 2007; Billen et al., 2008; Andavan and Lemmens-Gruber, 2011).
Neurotoxin Binding Site 1
Among all putative binding sites on VGSCs, site 1 is probably the best defined and most straightforward of all sites. It is composed by residues at the reentrant P-loops connecting S5 and S6 of all four domains. The historical guanidinium molecules TTX and STX were the first neurotoxins shown to bind at this site. Upon their binding, Na+ conductance is blocked. They are produced by some bacteria and dinoflagellate species, respectively (Narahashi, 2008; Chau et al., 2011). Later on, the μ-conotoxins found in cone snails, also turned out to bind to this specific site and to cause the same effects. At first it was thought that both types of toxins would interact with exactly the same residues but it seems to be a more complex interplay, as some mutations in Nav1.4 channels affecting TTX binding did only affect binding of a μ-conotoxin to a minor extent. Therefore, it was suggested that TTX and μ-conotoxins share an overlapping but non-identical binding site (Stephan et al., 1994). In this model, the core of the binding site is situated more in the inner side of the pore mouth and can be occupied by TTX or STX as well as μ-conotoxins, the latter interacting with some extra residues laying at the outer vestibule of the pore. Recently, another TTX/μ-conotoxin binding model was proposed in which TTX, STX, and the μ-conotoxins were suggested to be “syntoxins,” acting on adjacent sites in the VGSCs and influencing each other’s binding kinetics. Zhang et al. (2009) first proposed the simultaneous binding of TTX and KIIIA, a μ-conotoxin from Conus kinoshitai. Addition of both toxins to oocytes expressing Nav1.2 leads to the formation of a bi-liganded, ternary complex of Nav·TTX/alkaloid·μ-conotoxin. If KIIIA binds first to the channel, this will slow the subsequent binding of TTX at its specific binding place, further down into the channel; and the other way around. But in another series of experiments done by the same group, this appeared not to be completely true for STX and its sulfated congener GTX2/3. Another, extended model was suggested, wherein the binary complex Nav·μ-conotoxin could flicker between a permissive state, in which the alkaloid can “sneak” by the μ-conotoxin, and an unpermissive state (French et al., 2010; Zhang et al., 2010). Because STX has an extra positive charge compared with TTX, this could lead to electrostatic repulsion. The authors also checked this model with a docking model based on the current homology model for VGSCs. This was developed by Lipkind and Fozzard (2000), based on the crystal structure of the bacterial potassium channel KcsA. Docking of TTX and KIIIA was consistent with their experimental results; both molecules do fit simultaneously in the vestibule.
The VGSC homology model previously turned out to be valuable to define the structure of the outer channel vestibule (Cervenka et al., 2010). Although the outer vestibule was long time believed to be a rigid structure, this does not correspond to the large amount of studies that predict a highly flexible P-loop that even might undergo conformational changes that are linked to gating transitions upon binding of TTX or μ-conotoxins. This can be deduced, amongst other things, from mutagenesis studies examining the effects of mutations in this region. Those mutations mostly correlated with alterations in gating kinetics and more specifically decreased (mutations in the P-loop of DI) or enhanced inactivation (mutations in the P-loop of DIV; for a review see Cervenka et al., 2010). The latter can be explained by the proximity of DIV S6 to the P-loop of DIV, as DIV S6 plays an important role in the inactivation process. Moreover, this DIV S6 segment is supposed to enclose the binding site of LA, which on its turn connects site 1 and 6. Site 1 toxins such as μ-conotoxins can also influence the activation process by interacting with the voltage sensors. μ-Conotoxins such as KIIIA and GIIIA are strongly cationic peptides. Upon binding at site 1, these peptides electrostatically impede the outward movement of the positively charged residues in the S4 voltage sensor segments, which is a necessity for channel activation. The electrostatic repulsion, induced by binding of the toxin at site 1, most probably stabilizes the closed state, resulting in channels which will open at more depolarized membrane potentials (French et al., 1996; Van Der Haegen et al., 2011).
Another neurotoxin believed to affect site 1 is Tx1 from Phoneutria nigriventer (Martin-Moutot et al., 2006). Though binding of labeled Tx1 was not inhibited by TTX, it was inhibited by the μ-conotoxin GIIIB (Conus geographus). Therefore it was suggested that Tx1, like TTX, binds to a micro site and that the binding site of μ-conotoxins overlaps the micro sites of TTX and Tx1. So far, only one other spider toxin is known to interact with binding site 1, that is Hainantoxin-I (HNTX-I) from Ornithoctonus hainana (Li et al., 2003). Other spider toxins like Huwentoxin-IV (HWTX-IV) from Ornithoctonus huwena and some Hainantoxins (HNTX III-V) were at first considered to be potential site 1 neurotoxins as they inhibit Na+ conductance, an effect concerned to be typical for site 1 (Li et al., 2003; Nicholson, 2007). The same is valid for μO-conotoxins; but this hypothesis had to be adjusted as evidence occurred that these toxins do not bind to residues at site 1 but at residues in S3–S4 of DII (Leipold et al., 2007). Intriguingly, these residues form part of what is generally determined as “site 4.” Together with other recent similar findings concerning the other “traditional” neurotoxin binding sites, this prompted us to consider a revision of the binding sites. Therefore the latter toxins will be discussed into more detail in the section about the revision of the binding sites.
Neurotoxin Binding Site 2
This site is targeted by a wide array of lipid-soluble toxins with greatly diverting chemical structures. Their structural non-relatedness is mirrored in their diverse source of origin as they can be found in plants, animals, and bacteria. Well-known examples of site 2 toxins from plants are alkaloids like veratridine (VTD; from Liliaceae) and aconitine (Aconitum napellus) and grayanotoxins (GTX) from Ericaceae. Batrachotoxin (BTX) and homologs are site 2 toxins produced by animals such as frogs (Phyllobates spp.) and birds (Pitohui and Ifrita spp.; Dumbacher et al., 2000). Recently, antillatoxin and hoiamide were isolated from some cyanobacteria (Lyngbya majuscula) and were found to bind to neurotoxin receptor site 2 (Pereira et al., 2009; Cao et al., 2010).
Site 2 toxins are known as activators as they modulate sodium channels in such a way that the channels open more easily and stay open longer. Activators preferentially bind to channels in the open state and their binding leads to Nav channels with a unique and complex behavior (Hille, 2001). Several channel properties are altered upon site 2 binding: (i) the voltage-dependence of activation is shifted toward more negative potentials causing channels to open at resting potentials; (ii) the inactivation is slowed down or inhibited resulting in sustained, non-inactivating currents; (iii) the sodium conductance through toxin-bound channels is reduced; (iv) the ion selectivity of modified channels is altered due to a decreased discrimination for permeating ions (Tikhonov and Zhorov, 2005; Du et al., 2011). Numerous studies have been conducted to map the neurotoxin site 2 and to provide a better understanding of the molecular determinants responsible for these intriguing channel gating alterations upon toxin binding. Studies using photo-labeled BTX identified the first residues within the inner helixes of DI S6 (I433, N434, and L437) and DIV S6 (F1579 and N1584) that are crucial for BTX binding in Nav1.4 (Wang and Wang, 1998, 1999). The key role of these residues in BTX binding was confirmed as point mutations of the equivalent residues in Nav1.5 rendered these channels insensitive to BTX (Wang et al., 2007). Although site 2 is believed to be localized mainly at the S6 of DI and DIV, site-directed mutagenesis studies have shown that specific amino acid residues among all four S6 segments are contributing to the neurotoxin receptor site 2 (Wang et al., 2000, 2001). The exact location of all known molecular determinants contributing to the BTX binding site has recently been well summarized (Du et al., 2011).
The observed channel gating alterations upon binding to site 2 used to be explained by an allosteric model in which the lipid-soluble toxins bind at lipid-exposed sites distinct from the pore or the voltage sensors (Catterall et al., 2007). Even though this model of allosteric interactions provided a reasonable interpretation of modifications in ion selectivity, channel gating, and conductance at the time, an increasing number of studies have emerged the necessity to revise this model. Although the exact molecular mechanism of action is still poorly understood, there is increasing evidence suggesting that these toxins bind within the pore rather than at the lipid-exposed channel interface (Tikhonov and Zhorov, 2005; Du et al., 2011). Recently, a new model, which is consistent with most studies on BTX binding, has been proposed (Du et al., 2011). In this model, activators such as BTX bind within the inner pore with residues in the S6 segments of all four domains, exposing the activator directly to the permeation pathway. This model confirms the previous hypothesis that the observed inhibition of inactivation might be due to BTX interaction with the above-described residues in S6 of DIV. The DIV S6 segment is not only involved in fast inactivation, as it can also be seen that altered movements of toxin-bound S6 segments influence the movement of adjacent segments. Therefore, it is possible that BTX binding alters the voltage-dependent movement of the DIV S4 voltage sensor and thereby modifies channel activation and its coupling to inactivation (Linford et al., 1998; Catterall et al., 2007). The reduced sodium conductance can be seen as the outcome of a narrower Na+ binding site due to the presence of a BTX molecule in the inner pore. In contrast, the altered ion selectivity could be a direct consequence of a wider selectivity filter in modified channels. It has been well established that the DEKA locus within the selectivity filter determines the sodium channel selectivity. Moreover, K1422 can be seen as a key residue since a point mutation of this residue into a glutamic acid conferred calcium conducting characteristics onto sodium channels (Heinemann et al., 1992). The new BTX binding model suggests that BTX does not interact directly with the DEKA locus but rather causes a deficiency of water molecules in the proximity of the selectivity filter. The displacement of water molecules may lead to a shift in the PKA of the ion selectivity-determining residue K1422 and in this way, lower the discrimination in permeating ions (Du et al., 2011). Although this model is consistent with most available experimental data, it should be noted that there are limitations to homology models and that more experiments are required to confirm the theoretical analysis of this BTX binding model.
Several studies have contributed in locating the binding site for the lipid-soluble grayanotoxins. Similar to the BTX binding residues, it was found that S251, I433, N434, L437, I1575, F1579 on Nav1.4 and their equivalents on the cardiac sodium channel Nav1.5 are involved in the binding of GTXs (Ishii et al., 1999; Kimura et al., 2001). Mutation of one specific residue at position 1586 in DIV S6 completely abolished grayanotoxin-induced effects on Nav1.4 channels. Remarkably, the same mutation did not alter BTX binding (Kimura et al., 2000). Altogether it can be concluded that the GTX binding site is not completely identical to the BTX binding site but is overlapping as they share numerous molecular determinants.
In contrast with BTX, the channel modulation by veratridine is less investigated. In general it is assumed that VTD binds at the same site as BTX since VTD induces channel alterations that are similar to these of BTX (Wang and Wang, 2003). However, contrary to BTX, which does not dissociate from its receptor, VTD binding is reversible and it does dissociate from its receptor upon membrane hyperpolarization (Ulbricht, 1998). More recently, a bell-shaped relationship was described between the concentration of veratridine and the sodium current peak amplitude in murine vas deferens myocytes. It was observed that increasing concentrations of VTD enhance the peak amplitude, reaching a maximum around 10 μM while higher concentrations of VTD reduced the sodium conductance (Zhu et al., 2009).
The effects of aconitine still remain poorly studied. However, it is known that aconitine binding causes an incompletion of inactivation and an alteration of the ion selectivity in muscle VGSCs but not in nerve fibers (Campbell, 1982; Wang and Wang, 2003).
Neurotoxin Binding Site 3
Neurotoxins binding to site 3 include members of different phyla of the kingdom of Animalia, among which major players are toxins from scorpions, sea anemones, and spiders. In fact, binding site 3 was first determined by radiolabeling and mutagenesis studies performed with α-scorpion and sea anemone toxins (Tejedor and Catterall, 1988; Rogers et al., 1996). Scorpion toxins affecting the gating of VGSCs are historically classified into α- and β-toxins according to the effects that they cause. β-Scorpion toxins cause a strong hyperpolarizing shift in the voltage-dependence of activation, which is linked to neurotoxin binding site 4 and will be discussed in the following chapter. α-Scorpion toxins are long polypeptides of 60–70 amino acids and can be further subdivided into three groups according to their phylogenetic specificity (Gordon et al., 1996; Hamon et al., 2002). Classical α-scorpion toxins or anti-mammalian toxins will inhibit inactivation of mammalian VGSCs and have low affinity for insect neuronal membranes. Well-known examples are AaH II (Androctonus australis Hector; Jover et al., 1978) and Lqh II (Leiurus quinquestriatus hebraeus; Sautiere et al., 1998). Anti-insect α-scorpion toxins like LqhaIT (Eitan et al., 1990) only show minor activity against mammalian brain preparations but do show significantly higher inhibition of insect VGSCs. Finally, α-like scorpion toxins bind to both rat brain and insect VGSCs (e.g., BMK M1 (Hamon et al., 2002; Sun et al., 2003).
The second class of toxins that are famous for inhibiting VGSC inactivation is found in the venom of sea anemones. Sea anemone toxins targeting VGSCs are subdivided into several classes, but unlike α-scorpion toxins, this classification is not based on their phylogenetic preferences but on their amino acid sequences. Norton (1991) first proposed three classes to which a sea anemone toxin can belong. Type 1 and 2 toxins include larger peptides, composed of 46–49 amino acids and 3 disulfide bridges. Type 3 toxins are shorter peptides, composed of 27–30 amino acids only (Honma and Shiomi, 2006; Bosmans and Tytgat, 2007; Shiomi, 2009). Besides toxins from scorpions and sea anemones, site 3 is also targeted by toxins from other animals like spiders (e.g., δ-atracotoxins, Nicholson et al., 2004; Tx4(6-1), de Lima et al., 2002; and Magi 4, Corzo et al., 2003) and wasps (e.g., β-PMTX, Schiavon et al., 2010).
Parts of the VGSCs that were first identified to be involved in the binding of site 3 neurotoxins were located in the extracellular loops between S5 and S6 in DI and DIV (Thomsen and Catterall, 1989). Later on, residues in the extracellular loop between S3 and S4 in DIV also turned out to be involved in the binding of site 3 neurotoxins (Rogers et al., 1996). Fluorescence labeling studies indicated that site 3 neurotoxins stabilize the voltage sensor S4 of DIV in its deactivated position, thereby inhibiting its movement (Campos et al., 2008). As S4 of DIV is known to be involved in the voltage-dependent coupling between activation and fast inactivation (Chahine et al., 1994; Sheets and Hanck, 1995), it is logic that neurotoxin binding to this site causes an impairment of the fast inactivation. The inactivation can be slowed or even completely abolished and these effects can be associated with a minor hyperpolarizing shift in the activation. It was also shown that site 3 neurotoxins enhance the recovery from inactivation (e.g., work with the α-scorpion toxin Ts3 from Tityus serrulatus, Campos et al., 2004; and Anthopleurin B from the sea anemone Anemonia xanthogrammica, Sheets and Hanck, 1995). Another important characteristic is the voltage-dependency of the binding of these neurotoxins, such that they bind to a lesser extent at more depolarizing potentials (Catterall, 1977).
Site 3 neurotoxins turned out to be interesting tools for the investigation of gating currents, which are small transient currents that occur by movement of gating charges, mostly located on the S4 segments of the channel (Bezanilla, 2000). Via measurements of gating currents one can obtain much information about the S4 voltage sensors in ion channels and their contribution in sensing membrane potential. Sea anemone toxins ATX-II from Anemonia sulcata (Neumcke et al., 1985) and Anthopleurin A from Anemonia xanthogrammica (Sheets and Hanck, 1995) both reduced the maximum gating charge of VGSC and via mutagenesis studies it could nicely been demonstrated that this reduction resulted specifically from the inhibition of S4 of DIV, indicating clearly the main binding site of these toxins (Sheets et al., 1999).
The prior mentioned α-scorpion toxins, sea anemone toxins and spider toxins have all been shown to bind to the conventional site 3 and exert their conventional site 3 action: slowing or inhibiting the VGSC’s fast inactivation. Recently however, this traditional view on site 3 was samewise challenged as with site 1, as the δ-palutoxins (from the spider Paracoelotes luctuosus) were suggested to bind to site 4, although they exhibit actions that are typical for site 3 neurotoxins (Corzo et al., 2005; Ferrat et al., 2005). Another study with some other spider toxins gaining similar unexpected findings related with binding sites is the electrophysiological characterization of β/δ-agatoxins, originating from Agelena orientalis (Billen et al., 2010). It has been shown by Billen et al. that these toxins affect, in a voltage-dependent manner, both the inactivation process (site 3 effect) and the activation process (site 4 effect). These results even more questioned the generally accepted definition of “site 3” and the concepts of binding sites in general. The latter toxins will be discussed in the section about the revision of the binding sites, together with the unconventional site 1 neurotoxins.
Neurotoxin Binding Site 4
Receptor site 4 is recognized by the class of β-scorpion toxins targeting voltage-gated sodium channels (βNaScTxs) and by several spider toxins. These toxins exert their toxicity by acting as gating modifiers. Toxin binding at site 4 causes a shift in the voltage-dependence of activation toward more hyperpolarized membrane potentials and reduces the peak sodium current amplitude (Vijverberg and Lazdunski, 1984; Cestele et al., 2006). These alterations in channel gating are believed to be a direct result of toxin binding at site 4, hereby trapping the voltage sensor in its outward, activated position (Cestele et al., 2001, 2006). Receptor site 4 has been primarily defined to specific residues in the extracellular loops connecting the S1–S2 and S3–S4 segments of DII (Catterall et al., 2007). However, using the scorpion β-toxin Tz1 (Tityus zulianus) it was shown that three residues in the pore-loop of DIII are determining for the specificity of β-toxin for different sodium channel isoforms (Leipold et al., 2006). A recent report showed that specific mutations in the voltage sensor of DIII enhance the binding of β-toxins to S4 of DII, providing evidence for the involvement of the DIII voltage sensor in the action mechanism of βNaScTxs (Song et al., 2011).
Toxins belonging to the class of βNaScTxs are long chain peptides composed of 58–76 amino acids, cross-linked by four disulfide bridges. They belong to the structural superfamily of cysteine stabilized α/β motif containing proteins. This spatially conserved scaffold provides βNaScTxs with a high stability and a strong resistance against mutations in their sequence (Possani et al., 1999; de la Vega and Possani, 2007; Gurevitz et al., 2007). βNaScTxs are, similar to their αNaScTxs counterparts, classified into three groups according to their pharmacological properties exemplified by their preference for mammalian or insect sodium channels: mammalian-selective, β-like or insect-selective. (i) Mammalian-selective β-toxins such as Css4 (Centruroides suffusus suffusus) are highly toxic to mammals (Martin et al., 1987). (ii) β-like toxins are capable of competing for binding sites on both insect and mammalian Nav channels. Tsγ, also known as Ts1 or Ts VII (Tityus serrulatus) and Lqhβ1 (Leiurus quinquestriatus hebraeus) are well-studied examples of such βNaScTxs acting on both insects and mammals (Possani et al., 1999; Gordon and Gurevitz, 2003). (iii) Insect-selective β-toxins fail to exert any affinity whatsoever for mammalian sodium channels, even in very high concentrations (de Dianous et al., 1987). Exactly this complete lack of mammal activity combined with their strong insect specificity and potency makes these insect-selective βNaScTxs interesting lead compounds in the design of new insecticides (Gurevitz et al., 2007).
The insect-selective β-toxins can be further subdivided into excitatory and depressant toxins according to the symptoms they evoke in vivo. Injection of excitatory toxins induces a fast repetitive activity of motor nerves that results in a reversible contraction paralysis. AahIT (Androctonus australis Hector), LqqIT1 (Leiurus quinquestriatus quinquestriatus) and Bj-xtrIT (Hottentotta judaicus, formerly known as Buthotus judaicus) belong to this group (Froy et al., 1999; Billen et al., 2008). These excitatory toxins differ from the other β-toxins as one disulfide bridge is located differently and furthermore they display extra secondary structural elements. The depressant toxins cause a transient contraction followed by a slow depressant and flaccid paralysis (Zlotkin et al., 1991; Karbat et al., 2007). Current-clamp experiments have shown that peptides belonging to this group suppress the evoked action potentials as a result of strong depolarization of the membrane (Strugatsky et al., 2005). Representatives of this group are LqqIT2 (L. q. quinquestriatus), BjIT2 (H. judaicus), and the highly potent toxin Lqh-dprIT3 (L. q. hebraeus; Zlotkin et al., 1993). It is interesting to note that LqqIT2 did not only cause a hyperpolarizing shift in the activation of channels but also affected the inactivation and the ion selectivity (Bosmans et al., 2005). Remarkably, when mammalian channels are excited by a long, preconditioning and depolarizing prepulse, insect-selective depressant β-toxins are given the opportunity to affect those channels. The same phenomenon is observed in the case of simultaneous binding of an α-toxin to site 3 (Cohen et al., 2007). As such, it can be seen that the presence of depressant β-toxins in the scorpion venom may still contribute significantly to the toxicity toward mammals.
Spiders are, similar to scorpions, capable of producing toxins that recognize the neurotoxin receptor site 4. Magi 5 (Macrothele gigas) was the first spider toxin shown to compete with the radioiodinated scorpion β-toxin 125I-CssIV for site 4 (Corzo et al., 2003). Another group of site 4 spider toxins is constituted by the δ-palutoxins (P. luctuosus), curtatoxins (Hololena curta), μ-agatoxins (Agelenopsis aperta), and the recently characterized β/δ-agatoxins (A. orientalis; Stapleton et al., 1990; Corzo et al., 2000; Billen et al., 2010). All peptides belonging to this group are structurally related as they are composed of 36–37 residues and cross-linked by four disulfide bridges forming an inhibitor cystine knot (ICK) motif (Nicholson, 2007). Little is known about the mechanism of action of the curtatoxin but the highly homologous μ- and β/δ-agatoxins and δ-palutoxins have been well-studied. It was reported that the μ-agatoxins shift the voltage-activation curve toward more hyperpolarized potentials, like scorpion β-toxins do. However, these toxins also slowed down the inactivation process of the sodium channels, resulting in a non-inactivating persistent current (Adams, 2004). The same observations were made for the β/δ-agatoxins and a thorough electrophysiological characterization of the action of these agatoxins was performed. This indicated that β/δ-agatoxins induce a bell-shaped voltage-dependent modulation of both the activation and the inactivation, suggesting no strict correlation between the toxin binding site and its effect on channel gating (Billen et al., 2010). The δ-palutoxins compete with the depressant scorpion β-toxin Bj-xtrIT for site 4 but they fail to displace the binding of α-toxin LqhαIT from site 3 (Corzo et al., 2005). However, these toxins act as insect-selective modulators of sodium channels by slowing down the inactivation, a modulation typically seen upon toxin binding at site 3 (Corzo et al., 2000). All together these results indicate that βNaScTxs and their spider homologs should be considered as macromolecular ligands that may have additional contact points with the extracellular domains of VGSCs, in addition to the neurotoxin receptor site 4. More details can be found in the section about the revision of the binding sites.
Neurotoxin Binding Site 5
Marine dinoflagellates produce highly lipophilic, cyclic polyether compounds which target the neurotoxin receptor site 5. Brevetoxins (Karenia brevis) and ciguatoxins Gambierdiscus toxicus) are such multi-ring polyether ladder toxins acting at site 5. Brevetoxins (PbTxs) consist of 11 transfused rings, 23 stereocenters and an overall linear low-energy conformation (Jeglitsch et al., 1998). PbTx-1 and PbTx-2 are two most potent brevetoxins and they are considered to be the parent toxins. Up to date at least 14 brevetoxins have been described. The two most potent brevetoxins are PbTx-1 and PbTx-2 that slightly differ from each other in their backbone structure. PbTx-1 and PbTx-2 are considered parent toxins since all other brevetoxins can be seen as derivates from one of these two structural backbones. All PbTxs possess a lactone in the A-ring and a strictly rigid region that forms a ladder structure and which is separated from the A-ring by a spacer region with limited flexibility (Gawley et al., 1995). Furthermore, they all possess a side chain that allows modification at the molecules’ termini (Baden et al., 2005). PbTxs interact with VGSCs by intercalating in the membrane in a head-down orientation. Several studies have indicated that these toxins position themselves across the plasma membrane, parallel with the transmembrane segments, with the A-ring toward the intracellular side and the tail end of the molecule facing the extracellular side (Trainer et al., 1994; Jeglitsch et al., 1998). Experiments, in which a photoreactive PbTx-3 derivate was used as probe, could identify S6 of DI and S5 of DIV to participate in the formation of neurotoxin receptor 5 (Trainer et al., 1994). However, the key residues involved in brevetoxin activity still remain unknown. PbTx binding at site 5 leads to distinct alteration in channel gating: (i) the activation potential is shifted toward hyperpolarized potentials; (ii) channels remain longer in the open configuration which results in a longer mean open time; (iii) the inactivation is slowed down or inhibited; and (iv) brevetoxins have, among all known voltage-gated sodium channel modifying toxins, the unique capability to stabilize more than one conductance level. As such brevetoxin binding induces distinct sodium ion subconductance states in addition to the normal 21 pS rate (Schreibmayer and Jeglitsch, 1992; Jeglitsch et al., 1998; Baden et al., 2005). It is believed that the terminal, rigid four ring system is involved in channel binding while the functional lactone A-ring is responsible for the alterations in channel inactivation and prolongation of the mean open time (Jeglitsch et al., 1998; Purkerson-Parker et al., 2000).
Ciguatoxins (CTXs) are, like brevetoxins, lipid-soluble compounds with a structural backbone composed of 13 ether rings (Yasumoto, 2001). Although 29 ciguatoxin derivates have been identified, the information on their biological activity remains scarce, mainly due to difficulties in obtaining pure toxin (Perez et al., 2011). They compete with brevetoxins for site 5 and upon binding CTXs do cause similar modifications of channel gating. CTXs shift the activation of channels toward more negative potentials and they suppress the fast inactivation. Even though they bind at the same neurotoxin receptor, PbTxs and CTXs possibly differ from each other in their mechanism of action. This was first suggested by the observation that CTXs were, unlike PbTxs, capable of producing Nav dependent oscillations in neuronal membrane potential (Hogg et al., 2002). More recently it was shown that CTX causes a concentration-dependent decrease of the sodium current amplitude in mammalian sensory neurons. This observation is in contrast to the observed amplitude increase after PbTx application (Cohen et al., 2008; Yamaoka et al., 2009; Perez et al., 2011).
Neurotoxin Binding Site 6
Among all sites, site 6 still is the most speculative and yet undefined site. A first proposal for this site arose when TxVIA and GmVIA, δ-conotoxins from the cone snails Conus textile and Conus gloriamaris respectively, were characterized (Fainzilber et al., 1994; Shon et al., 1994). As they turned out to slow down VGSC inactivation without the typical voltage-dependency that is seen with classical site 3 toxins, it was suggested that they bind to a novel, unidentified site (Fainzilber et al., 1994). To date, 19 δ-conotoxins sequences can be found in the ConoServer database (Kaas et al., 2008), but for none of them the exact binding site has been reported. Only for δ-SVIE an interaction with conserved residues in the linker between S3–S4 of DIV was shown (Leipold et al., 2005). This indicates that at least the S3–S4 of DIV is involved in the interaction of the toxin and VGSC. It remains to be determined if this is a general phenomenon and if δ-SVIE is a good representative of all δ-conotoxins, or if a difference exists between the two distinct groups that exist in δ-conotoxins. Those two groups or “clades” are based on the prey targeted by the according cone snail: namely the fish-hunting clade and the mollusk-hunting clade, as structural differences can be recognized between both groups (Bulaj et al., 2001). However, data on δ-conotoxins are still very limited and further studies are needed to elucidate the exact binding site(s) and mechanism of action of δ-conotoxins and to determine if other toxins apart from δ-conotoxins bind to this site.
Revision of Neurotoxin Binding Sites
In the past, the classical nomenclature of neurotoxin binding sites has been a valuable tool for the investigation and classification of neurotoxins. However, more and more evidence emerges that these sites can not be as strictly delineated as once was believed (Catterall, 1980) and therefore the need for a revision of the binding sites comes forth. We would like to suggest a preliminary revision of the nomenclature of the neurotoxin binding sites, but are aware of the new insights that soon will be extracted from the recently discovered bacterial sodium channel crystal structure (Payandeh et al., 2011) which will definitely help in further revisions of the binding sites.
The revised binding site model can specifically signify a huge simplification of the conventional site 3 and site 4 since their current pharmacological profiles become more and more complex (Billen et al., 2010; Bosmans and Swartz, 2010; Liu et al., 2011). In general, the location of the site 4 binding area in the revised model can remain identical to the conventional site 4, located at DII, comprising the extracellular linkers between S1–S3 and S3–S4. The same is valid for the revised site 3, remaining located at the conventional S3–S4 linker of DIV. Because of the increasing number of toxins displaying a structure–function relationship incompatible with the conventional binding site model, we propose an adjusted model that can provide a more straightforward and definite description of the binding sites, which will help us to further clarify the missing parts in the puzzle of VGSC’s structure and function.
As mentioned in the section about site 1, μO-conotoxins (e.g., MrVIA and MrVIB) and some spider toxins (e.g., HWTX-IV) were at first considered to be site 1 toxins, as they exhibit the same effects as the conventional site 1 toxins. Nevertheless, these toxins are nowadays believed to bind at site 4 or close by. For μO-conotoxins for example, it was proven that they do not compete with STX for site 1 (Terlau et al., 1996). On the contrast, it was shown that they compete functionally with some classical β-scorpion toxins for modification of VGSCs and therefore are suggested to share, at least partially, a binding site similar to the classical defined site 4. However, in contrast to β-scorpion toxins that trap the S4 of DII in its outward position, μO-conotoxins will rather bind to the S4 voltage sensor in its inward position. As such they prevent the outward movement of the voltage sensor upon membrane depolarization and hence prevent the channels from opening, causing an inhibition of the Na+ conductance (Leipold et al., 2007). Another example of a toxin interacting in the same way with site 4, is represented by the spider toxin Huwentoxin-IV (Xiao et al., 2008).
In the conventional definition, a site 4 neurotoxin is a toxin that (1) binds to an extracellular region on the channel, formed by the S3–S4 loop of DII, and (2) causes a hyperpolarizing shift in the voltage-dependence of the activation. However, the two examples mentioned in the previous paragraph, serve as prove that it makes sense to uncouple these two conditions. Instead, a site 4 neurotoxin could be defined by using only the first condition, indicating the toxin’s main “binding area.” The effect that a site 4 neurotoxin can cause could be the classical site 4 effect, a hyperpolarizing shift in the activation, or the μO-conotoxin/HWTX-IV effect of inhibiting Na+ conductance. This effect will depend on specific residues or core regions that the toxin contains.
To define the main binding area one should be aware of the many contact points that a toxin can have on the VGSC, while only a few of them will be responsible for the exposed effects. To which binding area or “site” a toxin is assigned, should therefore only depend on which contact point causes the essential functional effect. Other contact points on the VGSC can rather be seen as anchor points, leading to non-functional binding of the toxin or serving as subtype-selective recognition sites for the toxin. Nevertheless, these non-functional contact points can also be a requirement for the full activity of the toxin. He et al. (2011) recently described this for binding of BmK IT2, a site 4 insect β-toxin from Buthus martensii Karsch, to insect DmNav VGSCs. Mutations in the site 4 binding area (DII S3–S4) largely abolished the functional action of BmK IT2 on DmNav. However, some anchor points in DIII turned out to be responsible for the selective recognition of the insect channel over the rat Nav1.2 channel. In addition, the N-terminal part of the S5–S6 linker (SS2–S6) in this DIII greatly influenced the potency of the BmK IT2 binding to DmNav (He et al., 2011).
In competitive binding experiments, the occupancy of the functional binding area of a radiolabeled toxin can be diminished by another toxin that also recognizes this contact point, as it is one of its anchor points but not its functional binding area. On a macroscopic level this may raise the impression that the non-labeled toxin has an identical functional effect as the labeled toxin. In the proposed model however, the competitive binding of the displacing non-labeled toxin, does not necessarily correlates this contact point on the VGSC with the functional binding area of the non-labeled toxin. This can explain the apparent paradox as is the case for site 3 toxins like BmK I and BmK αIV (α-like scorpion toxins from B. martensii Karsch) that can displace radiolabeled site 4 toxins like BmK AS (β-like scorpion toxin) and BmK IT2 (depressant insect β-scorpion toxin) respectively (Li et al., 2000; Chai et al., 2006). For a comprehensive review on BmK toxins and another binding model for the complex sites 3 and 4 (see Billen and Tytgat, 2009; Liu et al., 2011).
Another group of toxins pleading for the revision of VGSC binding sites is that of the spider toxins β/δ-agatoxins, mentioned in the section about site 4. Thorough electrophysiological characterization of these toxins showed that both activation and inactivation of an insect VGSC is affected when the toxin is applied. The voltage-dependence of activation is shifted towards more hyperpolarized potentials and a non-inactivating persistent current is induced. In the classical view these effects suggest toxin binding to site 4 and site 3, respectively. Therefore it is difficult to “match” the toxin to one of those binding sites as both effects can never correlate with one of these classical defined sites. This problem is solved in the revised binding site model, as this does not couple the effects of a toxin to its binding site. Therefore, as long as other experimental evidence is lacking, β/δ-agatoxins could eventually be stated to be site 4 toxins based on their high sequence homology with site 4 δ-palutoxins (Billen et al., 2010). Moreover, it is more likely that the effect on the inactivation is a result of toxin biding at site 4 rather than that the effect on activation is an effect of toxin binding at site 3. As experimental data are still lacking at this point, these suggestions still have be confirmed in future experiments.
The revised binding model also provides additional insights in the recently developed innovative concept of transferring voltage paddles of the VGSC Nav1.2 into fourfold symmetric potassium (Kv) channels (Bosmans et al., 2008). With these chimeric channels, the individual contribution of all four voltage paddles to toxin binding can be elucidated. Binding of the α-scorpion AaHII to the chimeric channels clearly involves only the paddle motif of DIV, which fits with the (revised) definition of site 3. Binding of the β-scorpion toxin revealed a less conventional pattern in which DII, III, and IV are all involved in toxin binding. Nevertheless, with the revised binding site model this can easily be explained and a distinction can be made between the toxin’s functional binding area, located at the voltage paddle of DII, and its additional anchor points, located at DIII and DIV. If only one of the toxin’s contact points is transplanted into a Kv channel to prove the original VGSC’s sensitivity to the toxin, it makes sense to suggest that sensitivity of the chimeric channel to the applied toxin will be induced anyway, no matter if the contact point is the functional binding area, or an additional anchor point.
In the same article, the interesting spider toxins ProTx-I and ProTx-II (Thrixopelma pruriens) are found to interact with DII and DIV of Nav1.2 and ProTx-I also displayed an interaction with DI of Nav1.2. Which domain comprises the main binding area for these toxins and which can serve as anchor point still remains the topic of discussion. Recently, Smith et al. (2007) conducted a thorough mutagenesis study of several predicted key residues in site 3 and 4 of the Nav1.5 VGSC isoform. Contrary to the results in Nav1.2, no main binding area could be detected. At the same time, they investigated which residues in ProTx-II were essential for its activity. The most important residues were hydrophobic and basic amino acids, making the bioactive surface amphiphatic in nature. This raises the exciting possibility that a yet undefined binding area or contact point exists, not located on the VGSC itself but somewhere in the surrounding membrane. This possibility was previously explored for Kv channels in several studies, examining for example partitioning of several toxins into model membranes (Milescu et al., 2007). All together, it was shown that a number of spider toxins do partition into membranes and that the composition of membrane lipids influences toxin effects. On the other hand, Cohen et al. (2006) demonstrated for several β-scorpion toxins that they do not interact with any binding area at the phospholipid bilayer. It should be noted however, that the tested spider toxins are members of a growing class of promiscuous toxins that not only target a particular class of ion channels but instead seem to bind to the voltage sensors of several classes of ion channels (e.g., Hanatoxin binds to several members of the Kv, Cav, and Nav ion channel families, a detailed list of other promiscuous toxins can be found in Bosmans and Swartz, 2010). This could indicate that the mechanism of action and the binding area of promiscuous spider toxins differ from β-scorpion toxins and that an unidentified contact point in the lipid membrane may be responsible for at least part of the full activity of the ProTx-I and ProTx-II spider toxins. Interaction between the spider toxin and the VGSC may then result in a tri-molecular complex, comprising the toxin, and contact points at the voltage sensor and at the lipid membrane (Bosmans and Swartz, 2010).
Additional evidence for the influence of other contact points in the near environment of VGSC α-subunits resulted from recent experiments, investigating the influence of the expressed β-subunit on the action of μO-conotoxin MrVIB. Wilson et al. (2011) discovered that expression of Nav1.8 channels with four different β-subunits resulted in a higher-affinity block of MrVIB compared to the α-subunit on its own. Contrary to this finding, the affinity of STX turned out to be independent of coexpression with β-subunits. One of their suggested mechanistic explanations for the influence of β-subunit expression on toxin function is that MrVIB could have a certain contact point on these β-subunits that contributes to its action. Further structure–function studies are necessary to confirm if the interaction between toxin and β-subunit is specific for μO-conotoxins or rather is the case for other toxins – apart from the guanidinium toxins, as well.
Conclusion and Further Perspectives
In the past, neurotoxins already have proven to be indispensible tools for the exploration of the structure and function of VGSCs. The long awaited elucidation of the crystal structure of the bacterial VGSC NavAb (Payandeh et al., 2011) certainly inaugurates a new era in VGSC research and will help to provide new insights into neurotoxin binding to VGSCs. Further examination of toxin–lipid interactions and toxin–β-subunit interactions, determination of the bioactive core of neurotoxins and detailed mutagenesis studies with possible key residues for neurotoxin binding are just a grasp out of the possible experiments of which we may gain interesting new concepts for VGSCs.
With our revised model for neurotoxin binding sites on VGSC we try to address the growing problems that arise with the traditional classification of neurotoxin binding sites. The uncoupling of the toxin’s functional outcome from its binding area should help to get a more clear-cut classification of neurotoxins. However, to distinguish the toxin’s main binding area from additional anchor points, one should not make preliminary conclusions by linking the effects of the toxin immediately to its binding area, nor should one deduct site classification or function of new toxins solely based on homology with other toxins. Therefore all available techniques (e.g., mutagenesis studies, chimeric channels, electrophysiological characterization) should be optimally used to characterize a toxin and its putative binding area on the channel.
Naturally occurring neurotoxins can be considered as valuable and promising tools, not only to further unravel structure–function of VGSCs but they also can serve as lead compounds in the development of novel drugs. Considering the fact that malfunctioning of VGSCs underlies a large range of diseases like epilepsy, neuropathic pain, and long QT syndrome, there is an urgent need for molecules that can selectively and potently target these membrane proteins. The quest for such molecules can start at the venom and poisons of organisms, providing us an almost endless library of possible lead compounds.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adams, M. E. (2004). Agatoxins: ion channel specific toxins from the American funnel web spider, Agelenopsis aperta. Toxicon 43, 509–525.
Agnew, W. S., Moore, A. C., Levinson, S. R., and Raftery, M. A. (1980). Identification of a large molecular weight peptide associated with a tetrodotoxin binding protein from the electroplax of Electrophorus electricus. Biochem. Biophys. Res. Commun. 92, 860–866.
Andavan, G. S., and Lemmens-Gruber, R. (2011). Voltage-gated sodium channels: mutations, channelopathies and targets. Curr. Med. Chem. 18, 377–397.
Baden, D. G., Bourdelais, A. J., Jacocks, H., Michelliza, S., and Naar, J. (2005). Natural and derivative brevetoxins: historical background, multiplicity, and effects. Environ. Health Perspect. 113, 621–625.
Bezanilla, F. (2000). The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80, 555–592.
Billen, B., Bosmans, F., and Tytgat, J. (2008). Animal peptides targeting voltage-activated sodium channels. Curr. Pharm. Des. 14, 2492–2502.
Billen, B., and Tytgat, J. (2009). “The venom of the Asian scorpion Buthus martensii Karsch: an overview of the toxins and their biological targets,” in Animal Toxins: State of the Art. Perspectives on Health and Biotechnology, ed. M. E. de Lima (Belo Horizonte: Editora UFMG), 137–166.
Billen, B., Vassilevski, A., Nikolsky, A., Debaveye, S., Tytgat, J., and Grishin, E. (2010). Unique bell-shaped voltage-dependent modulation of Na+ channel gating by novel insect-selective toxins from the spider Agelena orientalis. J. Biol. Chem. 285, 18545–18554.
Bosmans, F., Martin-Eauclaire, M. F., and Swartz, K. J. (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208.
Bosmans, F., Martin-Eauclaire, M. F., and Tytgat, J. (2005). The depressant scorpion neurotoxin LqqIT2 selectively modulates the insect voltage-gated sodium channel. Toxicon 45, 501–507.
Bosmans, F., and Swartz, K. J. (2010). Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 31, 175–182.
Bosmans, F., and Tytgat, J. (2007). Sea anemone venom as a source of insecticidal peptides acting on voltage-gated Na+ channels. Toxicon 49, 550–560.
Bulaj, G., DeLaCruz, R., Azimi-Zonooz, A., West, P., Watkins, M., Yoshikami, D., and Olivera, B. M. (2001). Δ-conotoxin structure/function through a cladistic analysis. Biochemistry 40, 13201–13208.
Campbell, D. T. (1982). Modified kinetics and selectivity of sodium channels in frog skeletal muscle fibers treated with aconitine. J. Gen. Physiol. 80, 713–731.
Campos, F. V., Chanda, B., Beirao, P. S., and Bezanilla, F. (2008). α-scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J. Gen. Physiol. 132, 251–263.
Campos, F. V., Coronas, F. I., and Beirao, P. S. (2004). Voltage-dependent displacement of the scorpion toxin Ts3 from sodium channels and its implication on the control of inactivation. Br. J. Pharmacol. 142, 1115–1122.
Cao, Z., Gerwick, W. H., and Murray, T. F. (2010). Antillatoxin is a sodium channel activator that displays unique efficacy in heterologously expressed rNav1.2, rNav1.4 and rNav1.5 a subunits. BMC Neurosci. 11, 154. doi:10.1186/1471-2202-11-154
Catterall, W. A. (1977). Activation of the action potential Na+ ionophore by neurotoxins. An allosteric model. J. Biol. Chem. 252, 8669–8676.
Catterall, W. A. (1980). Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu. Rev. Pharmacol. Toxicol. 20, 15–43.
Catterall, W. A. (1986). Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55, 953–985.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Catterall, W. A. (2010). Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67, 915–928.
Catterall, W. A., Cestele, S., Yarov-Yarovoy, V., Yu, F. H., Konoki, K., and Scheuer, T. (2007). Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409.
Cervenka, R., Zarrabi, T., Lukacs, P., and Todt, H. (2010). The outer vestibule of the Na+ channel-toxin receptor and modulator of permeation as well as gating. Mar. Drugs 8, 1373–1393.
Cestele, S., Scheuer, T., Mantegazza, M., Rochat, H., and Catterall, W. A. (2001). Neutralization of gating charges in domain II of the sodium channel a subunit enhances voltage-sensor trapping by a ß-scorpion toxin. J. Gen. Physiol. 118, 291–302.
Cestele, S., Yarov-Yarovoy, V., Qu, Y., Sampieri, F., Scheuer, T., and Catterall, W. A. (2006). Structure and function of the voltage sensor of sodium channels probed by a ß-scorpion toxin. J. Biol. Chem. 281, 21332–21344.
Chahine, M., George, A. L. Jr., Zhou, M., Ji, S., Sun, W., Barchi, R. L., and Horn, R. (1994). Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron 12, 281–294.
Chahine, M., Ziane, R., Vijayaragavan, K., and Okamura, Y. (2005). Regulation of Na v channels in sensory neurons. Trends Pharmacol. Sci. 26, 496–502.
Chai, Z. F., Zhu, M. M., Bai, Z. T., Liu, T., Tan, M., Pang, X. Y., and Ji, Y. H. (2006). Chinese-scorpion (Buthus martensii Karsch) toxin BmK aIV, a novel modulator of sodium channels: from genomic organization to functional analysis. Biochem. J. 399, 445–453.
Chau, R., Kalaitzis, J. A., and Neilan, B. A. (2011). On the origins and biosynthesis of tetrodotoxin. Aquat. Toxicol. 104, 61–72.
Cohen, L., Gilles, N., Karbat, I., Ilan, N., Gordon, D., and Gurevitz, M. (2006). Direct evidence that receptor site-4 of sodium channel gating modifiers is not dipped in the phospholipid bilayer of neuronal membranes. J. Biol. Chem. 281, 20673–20679.
Cohen, L., Lipstein, N., Karbat, I., Ilan, N., Gilles, N., Kahn, R., Gordon, D., and Gurevitz, M. (2008). Miniaturization of scorpion ß-toxins uncovers a putative ancestral surface of interaction with voltage-gated sodium channels. J. Biol. Chem. 283, 15169–15176.
Cohen, L., Troub, Y., Turkov, M., Gilles, N., Ilan, N., Benveniste, M., Gordon, D., and Gurevitz, M. (2007). Mammalian skeletal muscle voltage-gated sodium channels are affected by scorpion depressant “insect-selective” toxins when preconditioned. Mol. Pharmacol. 72, 1220–1227.
Corzo, G., Escoubas, P., Stankiewicz, M., Pelhate, M., Kristensen, C. P., and Nakajima, T. (2000). Isolation, synthesis and pharmacological characterization of d-palutoxins IT, novel insecticidal toxins from the spider Paracoelotes luctuosus (Amaurobiidae). Eur. J. Biochem. 267, 5783–5795.
Corzo, G., Escoubas, P., Villegas, E., Karbat, I., Gordon, D., Gurevitz, M., Nakajima, T., and Gilles, N. (2005). A spider toxin that induces a typical effect of scorpion a-toxins but competes with ß-toxins on binding to insect sodium channels. Biochemistry 44, 1542–1549.
Corzo, G., Gilles, N., Satake, H., Villegas, E., Dai, L., Nakajima, T., and Haupt, J. (2003). Distinct primary structures of the major peptide toxins from the venom of the spider Macrothele gigas that bind to sites 3 and 4 in the sodium channel. FEBS Lett. 547, 43–50.
de Dianous, S., Hoarau, F., and Rochat, H. (1987). Re-examination of the specificity of the scorpion Androctonus australis hector insect toxin towards arthropods. Toxicon 25, 411–417.
de la Vega, R. C., and Possani, L. D. (2007). Novel paradigms on scorpion toxins that affects the activating mechanism of sodium channels. Toxicon 49, 171–180.
de Lima, M. E., Stankiewicz, M., Hamon, A., de Figueiredo, S. G., Cordeiro, M. N., Diniz, C. R., Martin-Eauclaire, M., and Pelhate, M. (2002). The toxin Tx4(6-1) from the spider Phoneutria nigriventer slows down Na+ current inactivation in insect CNS via binding to receptor site 3. J. Insect Physiol. 48, 53–61.
Du, Y., Garden, D. P., Wang, L., Zhorov, B. S., and Dong, K. (2011). Identification of new batrachotoxin-sensing residues in segment IIIS6 of the sodium channel. J. Biol. Chem. 286, 13151–13160.
Dumbacher, J. P., Spande, T. F., and Daly, J. W. (2000). Batrachotoxin alkaloids from passerine birds: a second toxic bird genus (Ifrita kowaldi) from New Guinea. Proc. Natl. Acad. Sci. U.S.A. 97, 12970–12975.
Eitan, M., Fowler, E., Herrmann, R., Duval, A., Pelhate, M., and Zlotkin, E. (1990). A scorpion venom neurotoxin paralytic to insects that affects sodium current inactivation: purification, primary structure, and mode of action. Biochemistry 29, 5941–5947.
Fainzilber, M., Kofman, O., Zlotkin, E., and Gordon, D. (1994). A new neurotoxin receptor site on sodium channels is identified by a conotoxin that affects sodium channel inactivation in molluscs and acts as an antagonist in rat brain. J. Biol. Chem. 269, 2574–2580.
Ferrat, G., Bosmans, F., Tytgat, J., Pimentel, C., Chagot, B., Gilles, N., Nakajima, T., Darbon, H., and Corzo, G. (2005). Solution structure of two insect-specific spider toxins and their pharmacological interaction with the insect voltage-gated Na+ channel. Proteins 59, 368–379.
French, R. J., Prusak-Sochaczewski, E., Zamponi, G. W., Becker, S., Kularatna, A. S., and Horn, R. (1996). Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron 16, 407–413.
French, R. J., Yoshikami, D., Sheets, M. F., and Olivera, B. M. (2010). The tetrodotoxin receptor of voltage-gated sodium channels – perspectives from interactions with µ-conotoxins. Mar. Drugs 8, 2153–2161.
Froy, O., Zilberberg, N., Gordon, D., Turkov, M., Gilles, N., Stankiewicz, M., Pelhate, M., Loret, E., Oren, D. A., Shaanan, B., and Gurevitz, M. (1999). The putative bioactive surface of insect-selective scorpion excitatory neurotoxins. J. Biol. Chem. 274, 5769–5776.
Gawley, R. E., Rein, K. S., Jeglitsch, G., Adams, D. J., Theodorakis, E. A., Tiebes, J., Nicolaou, K. C., and Baden, D. G. (1995). The relationship of brevetoxin “length” and A-ring functionality to binding and activity in neuronal sodium channels. Chem. Biol. 2, 533–541.
Gordon, D., and Gurevitz, M. (2003). The selectivity of scorpion a-toxins for sodium channel subtypes is determined by subtle variations at the interacting surface. Toxicon 41, 125–128.
Gordon, D., Martin-Eauclaire, M. F., Cestele, S., Kopeyan, C., Carlier, E., Khalifa, R. B., Pelhate, M., and Rochat, H. (1996). Scorpion toxins affecting sodium current inactivation bind to distinct homologous receptor sites on rat brain and insect sodium channels. J. Biol. Chem. 271, 8034–8045.
Gurevitz, M., Karbat, I., Cohen, L., Ilan, N., Kahn, R., Turkov, M., Stankiewicz, M., Stuhmer, W., Dong, K., and Gordon, D. (2007). The insecticidal potential of scorpion ß-toxins. Toxicon 49, 473–489.
Guy, H. R., and Seetharamulu, P. (1986). Molecular model of the action potential sodium channel. Proc. Natl. Acad. Sci. U.S.A. 83, 508–512.
Hamon, A., Gilles, N., Sautiere, P., Martinage, A., Kopeyan, C., Ulens, C., Tytgat, J., Lancelin, J. M., and Gordon, D. (2002). Characterization of scorpion a-like toxin group using two new toxins from the scorpion Leiurus quinquestriatus hebraeus. Eur. J. Biochem. 269, 3920–3933.
He, H., Liu, Z., Dong, B., Zhang, J., Shu, X., Zhou, J., and Ji, Y. (2011). Localization of receptor site on insect sodium channel for depressant ß-toxin BmK IT2. PLoS ONE 6, e14510. doi:10.1371/journal.pone.0014510
Heinemann, S. H., Terlau, H., Stuhmer, W., Imoto, K., and Numa, S. (1992). Calcium channel characteristics conferred on the sodium channel by single mutations. Nature 356, 441–443.
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544.
Hogg, R. C., Lewis, R. J., and Adams, D. J. (2002). Ciguatoxin-induced oscillations in membrane potential and action potential firing in rat parasympathetic neurons. Eur. J. Neurosci. 16, 242–248.
Honma, T., and Shiomi, K. (2006). Peptide toxins in sea anemones: structural and functional aspects. Mar. Biotechnol. (NY) 8, 1–10.
Ishii, H., Kinoshita, E., Kimura, T., Yakehiro, M., Yamaoka, K., Imoto, K., Mori, Y., and Seyama, I. (1999). Point-mutations related to the loss of batrachotoxin binding abolish the grayanotoxin effect in Na+ channel isoforms. Jpn. J. Physiol. 49, 457–461.
Jeglitsch, G., Rein, K., Baden, D. G., and Adams, D. J. (1998). Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J. Pharmacol. Exp. Ther. 284, 516–525.
Jover, E., Martin-Moutot, N., Couraud, F., and Rochat, H. (1978). Scorpion toxin: specific binding to rat synaptosomes. Biochem. Biophys. Res. Commun. 85, 377–382.
Kaas, Q., Westermann, J. C., Halai, R., Wang, C. K., and Craik, D. J. (2008). ConoServer, a database for conopeptide sequences and structures. Bioinformatics 24, 445–446.
Karbat, I., Turkov, M., Cohen, L., Kahn, R., Gordon, D., Gurevitz, M., and Frolow, F. (2007). X-ray structure and mutagenesis of the scorpion depressant toxin LqhIT2 reveals key determinants crucial for activity and anti-insect selectivity. J. Mol. Biol. 366, 586–601.
Kimura, T., Kinoshita, E., Yamaoka, K., Yuki, T., Yakehiro, M., and Seyama, I. (2000). On site of action of grayanotoxin in domain 4 segment 6 of rat skeletal muscle sodium channel. FEBS Lett. 465, 18–22.
Kimura, T., Yamaoka, K., Kinoshita, E., Maejima, H., Yuki, T., Yakehiro, M., and Seyama, I. (2001). Novel site on sodium channel a-subunit responsible for the differential sensitivity of grayanotoxin in skeletal and cardiac muscle. Mol. Pharmacol. 60, 865–872.
Leipold, E., DeBie, H., Zorn, S., Borges, A., Olivera, B. M., Terlau, H., and Heinemann, S. H. (2007). µO conotoxins inhibit Nav channels by interfering with their voltage sensors in domain-2. Channels (Austin) 1, 253–262.
Leipold, E., Hansel, A., Borges, A., and Heinemann, S. H. (2006). Subtype specificity of scorpion ß-toxin Tz1 interaction with voltage-gated sodium channels is determined by the pore loop of domain 3. Mol. Pharmacol. 70, 340–347.
Leipold, E., Hansel, A., Olivera, B. M., Terlau, H., and Heinemann, S. H. (2005). Molecular interaction of d-conotoxins with voltage-gated sodium channels. FEBS Lett. 579, 3881–3884.
Lewis, R. J., and Garcia, M. L. (2003). Therapeutic potential of venom peptides. Nat. Rev. Drug Discov. 2, 790–802.
Li, D., Xiao, Y., Hu, W., Xie, J., Bosmans, F., Tytgat, J., and Liang, S. (2003). Function and solution structure of hainantoxin-I, a novel insect sodium channel inhibitor from the Chinese bird spider Selenocosmia hainana. FEBS Lett. 555, 616–622.
Li, Y. J., Tan, Z. Y., and Ji, Y. H. (2000). The binding of BmK IT2, a depressant insect-selective scorpion toxin on mammal and insect sodium channels. Neurosci. Res. 38, 257–264.
Linford, N. J., Cantrell, A. R., Qu, Y., Scheuer, T., and Catterall, W. A. (1998). Interaction of batrachotoxin with the local anesthetic receptor site in transmembrane segment IVS6 of the voltage-gated sodium channel. Proc. Natl. Acad. Sci. U.S.A. 95, 13947–13952.
Lipkind, G. M., and Fozzard, H. A. (2000). KcsA crystal structure as framework for a molecular model of the Na+ channel pore. Biochemistry 39, 8161–8170.
Liu, Z. R., Ye, P., and Ji, Y. H. (2011). Exploring the obscure profiles of pharmacological binding sites on voltage-gated sodium channels by BmK neurotoxins. Protein Cell 2, 437–444.
Martin, M. F., Garcia y Perez, L. G., el Ayeb, M., Kopeyan, C., Bechis, G., Jover, E., and Rochat, H. (1987). Purification and chemical and biological characterizations of seven toxins from the Mexican scorpion, Centruroides suffusus suffusus. J. Biol. Chem. 262, 4452–4459.
Martin-Moutot, N., Mansuelle, P., Alcaraz, G., Dos Santos, R. G., Cordeiro, M. N., De Lima, M. E., Seagar, M., and Van Renterghem, C. (2006). Phoneutria nigriventer toxin 1: a novel, state-dependent inhibitor of neuronal sodium channels that interacts with µ conotoxin binding sites. Mol. Pharmacol. 69, 1931–1937.
Milescu, M., Vobecky, J., Roh, S. H., Kim, S. H., Jung, H. J., Kim, J. I., and Swartz, K. J. (2007). Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 130, 497–511.
Narahashi, T. (2008). Tetrodotoxin: a brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 84, 147–154.
Neumcke, B., Schwarz, W., and Stampfli, R. (1985). Comparison of the effects of Anemonia toxin II on sodium and gating currents in frog myelinated nerve. Biochim. Biophys. Acta 814, 111–119.
Nicholson, G. M. (2007). Insect-selective spider toxins targeting voltage-gated sodium channels. Toxicon 49, 490–512.
Nicholson, G. M., Little, M. J., and Birinyi-Strachan, L. C. (2004). Structure and function of d-atracotoxins: lethal neurotoxins targeting the voltage-gated sodium channel. Toxicon 43, 587–599.
Noda, M., Suzuki, H., Numa, S., and Stuhmer, W. (1989). A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 259, 213–216.
Norton, R. S. (1991). Structure and structure-function relationships of sea anemone proteins that interact with the sodium channel. Toxicon 29, 1051–1084.
Patino, G. A., and Isom, L. L. (2010). Electrophysiology and beyond: multiple roles of Na+ channel ß subunits in development and disease. Neurosci. Lett. 486, 53–59.
Payandeh, J., Scheuer, T., Zheng, N., and Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 7356, 353–358.
Pereira, A., Cao, Z., Murray, T. F., and Gerwick, W. H. (2009). Hoiamide a, a sodium channel activator of unusual architecture from a consortium of two papua new Guinea cyanobacteria. Chem. Biol. 16, 893–906.
Perez, S., Vale, C., Alonso, E., Alfonso, C., Rodriguez, P., Otero, P., Alfonso, A., Vale, P., Hirama, M., Vieytes, M. R., and Botana, L. M. (2011). A comparative study of the effect of ciguatoxins on voltage-dependent Na+ and K+ channels in cerebellar neurons. Chem. Res. Toxicol. 24, 587–596.
Possani, L. D., Becerril, B., Delepierre, M., and Tytgat, J. (1999). Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 264, 287–300.
Purkerson-Parker, S. L., Fieber, L. A., Rein, K. S., Podona, T., and Baden, D. G. (2000). Brevetoxin derivatives that inhibit toxin activity. Chem. Biol. 7, 385–393.
Rogers, J. C., Qu, Y., Tanada, T. N., Scheuer, T., and Catterall, W. A. (1996). Molecular determinants of high affinity binding of a-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel a subunit. J. Biol. Chem. 271, 15950–15962.
Sato, C., Ueno, Y., Asai, K., Takahashi, K., Sato, M., Engel, A., and Fujiyoshi, Y. (2001). The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature 409, 1047–1051.
Sautiere, P., Cestele, S., Kopeyan, C., Martinage, A., Drobecq, H., Doljansky, Y., and Gordon, D. (1998). New toxins acting on sodium channels from the scorpion Leiurus quinquestriatus hebraeus suggest a clue to mammalian vs insect selectivity. Toxicon 36, 1141–1154.
Schiavon, E., Stevens, M., Zaharenko, A. J., Konno, K., Tytgat, J., and Wanke, E. (2010). Voltage-gated sodium channel isoform-specific effects of pompilidotoxins. FEBS J. 277, 918–930.
Schreibmayer, W., and Jeglitsch, G. (1992). The sodium channel activator Brevetoxin-3 uncovers a multiplicity of different open states of the cardiac sodium channel. Biochim. Biophys. Acta 1104, 233–242.
Sheets, M. F., and Hanck, D. A. (1995). Voltage-dependent open-state inactivation of cardiac sodium channels: gating current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106, 617–640.
Sheets, M. F., Kyle, J. W., Kallen, R. G., and Hanck, D. A. (1999). The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77, 747–757.
Shon, K. J., Hasson, A., Spira, M. E., Cruz, L. J., Gray, W. R., and Olivera, B. M. (1994). Δ-conotoxin GmVIA, a novel peptide from the venom of Conus gloriamaris. Biochemistry 33, 11420–11425.
Smith, M. R., and Goldin, A. L. (1997). Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys. J. 73, 1885–1895.
Smith, J. J., Cummins, T. R., Alphy, S., and Blumenthal, K. M. (2007). Molecular interactions of the gating modifier toxin ProTx-II with Nav1.5: implied existence of a novel toxin binding site coupled to activation. J. Biol. Chem. 282, 12687–12697.
Song, W., Du, Y., Liu, Z., Luo, N., Turkov, M., Gordon, D., Gurevitz, M., Goldin, A. L., and Dong, K. (2011). Substitutions in the domain III voltage-sensing module enhance the sensitivity of an insect sodium channel to a scorpion ß-toxin. J. Biol. Chem. 286, 15781–15788.
Stapleton, A., Blankenship, D. T., Ackermann, B. L., Chen, T. M., Gorder, G. W., Manley, G. D., Palfreyman, M. G., Coutant, J. E., and Cardin, A. D. (1990). Curtatoxins. Neurotoxic insecticidal polypeptides isolated from the funnel-web spider Hololena curta. J. Biol. Chem. 265, 2054–2059.
Stephan, M. M., Potts, J. F., and Agnew, W. S. (1994). The microI skeletal muscle sodium channel: mutation E403Q eliminates sensitivity to tetrodotoxin but not to µ-conotoxins GIIIA and GIIIB. J. Membr. Biol. 137, 1–8.
Strugatsky, D., Zilberberg, N., Stankiewicz, M., Ilan, N., Turkov, M., Cohen, L., Pelhate, M., Gilles, N., Gordon, D., and Gurevitz, M. (2005). Genetic polymorphism and expression of a highly potent scorpion depressant toxin enable refinement of the effects on insect Na channels and illuminate the key role of Asn-58. Biochemistry 44, 9179–9187.
Sun, H. Y., Zhu, H. F., and Ji, Y. H. (2003). BmK I, an a-like scorpion neurotoxin, specifically modulates isolated rat cardiac mechanical and electrical activity. Sheng Li Xue Bao 55, 530–534.
Tejedor, F. J., and Catterall, W. A. (1988). Site of covalent attachment of a-scorpion toxin derivatives in domain I of the sodium channel a subunit. Proc. Natl. Acad. Sci. U.S.A. 85, 8742–8746.
Terlau, H., and Olivera, B. M. (2004). Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 84, 41–68.
Terlau, H., Stocker, M., Shon, K. J., McIntosh, J. M., and Olivera, B. M. (1996). µO-conotoxin MrVIA inhibits mammalian sodium channels, but not through site I. J. Neurophysiol. 76, 1423–1429.
Thomsen, W. J., and Catterall, W. A. (1989). Localization of the receptor site for a-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc. Natl. Acad. Sci. U.S.A. 86, 10161–10165.
Tikhonov, D. B., and Zhorov, B. S. (2005). Sodium channel activators: model of binding inside the pore and a possible mechanism of action. FEBS Lett. 579, 4207–4212.
Trainer, V. L., Baden, D. G., and Catterall, W. A. (1994). Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J. Biol. Chem. 269, 19904–19909.
Ulbricht, W. (1998). Effects of veratridine on sodium currents and fluxes. Rev. Physiol. Biochem. Pharmacol. 133, 1–54.
Ulbricht, W. (2005). Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 85, 1271–1301.
Van Der Haegen, A., Peigneur, S., and Tytgat, J. (2011). Importance of position 8 in µ-conotoxin KIIIA for voltage-gated sodium channel selectivity. FEBS J. 278, 3408–3418.
Vijverberg, H. P., and Lazdunski, M. (1984). A new scorpion toxin with a very high affinity for sodium channels. An electrophysiological study. J. Physiol. (Paris) 79, 275–279.
Wang, G. K., and Wang, S. Y. (2003). Veratridine block of rat skeletal muscle Nav1.4 sodium channels in the inner vestibule. J. Physiol. 548(Pt 3), 667–675.
Wang, S. Y., Barile, M., and Wang, G. K. (2001). Disparate role of Na+ channel D2-S6 residues in batrachotoxin and local anesthetic action. Mol. Pharmacol. 59, 1100–1107.
Wang, S. Y., Nau, C., and Wang, G. K. (2000). Residues in Na+ channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophys. J. 79, 1379–1387.
Wang, S. Y., Tikhonov, D. B., Mitchell, J., Zhorov, B. S., and Wang, G. K. (2007). Irreversible block of cardiac mutant Na+ channels by batrachotoxin. Channels (Austin) 1, 179–188.
Wang, S. Y., and Wang, G. K. (1998). Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proc. Natl. Acad. Sci. U.S.A. 95, 2653–2658.
Wang, S. Y., and Wang, G. K. (1999). Batrachotoxin-resistant Na+ channels derived from point mutations in transmembrane segment D4-S6. Biophys. J. 76, 3141–3149.
Wilson, M. J., Zhang, M. M., Azam, L., Olivera, B. M., Bulaj, G., and Yoshikami, D. (2011). Nav β-subunits modulate the inhibition of Nav1.8 by the analgesic gating modifier µO-conotoxin MrVIB. J. Pharmacol. Exp. Ther. 338, 687–693.
Xiao, Y., Bingham, J. P., Zhu, W., Moczydlowski, E., Liang, S., and Cummins, T. R. (2008). Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain II voltage sensor in the closed configuration. J. Biol. Chem. 283, 27300–27313.
Yamaoka, K., Inoue, M., Miyazaki, K., Hirama, M., Kondo, C., Kinoshita, E., Miyoshi, H., and Seyama, I. (2009). Synthetic ciguatoxins selectively activate Nav1.8-derived chimeric sodium channels expressed in HEK293 cells. J. Biol. Chem. 284, 7597–7605.
Yasumoto, T. (2001). The chemistry and biological function of natural marine toxins. Chem. Rec. 1, 228–242.
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4, 207.
Zhang, M. M., Gruszczynski, P., Walewska, A., Bulaj, G., Olivera, B. M., and Yoshikami, D. (2010). Cooccupancy of the outer vestibule of voltage-gated sodium channels by µ-conotoxin KIIIA and saxitoxin or tetrodotoxin. J. Neurophysiol. 104, 88–97.
Zhang, M. M., McArthur, J. R., Azam, L., Bulaj, G., Olivera, B. M., French, R. J., and Yoshikami, D. (2009). Synergistic and antagonistic interactions between tetrodotoxin and µ-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 3, 32–38.
Zhu, H. L., Wassall, R. D., Takai, M., Morinaga, H., Nomura, M., Cunnane, T. C., and Teramoto, N. (2009). Actions of veratridine on tetrodotoxin-sensitive voltage-gated Na currents, Nav1.6, in murine vas deferens myocytes. Br. J. Pharmacol. 157, 1483–1493.
Zlotkin, E., Eitan, M., Bindokas, V. P., Adams, M. E., Moyer, M., Burkhart, W., and Fowler, E. (1991). Functional duality and structural uniqueness of depressant insect-selective neurotoxins. Biochemistry 30, 4814–4821.
Keywords: voltage-gated sodium channel, neurotoxin, binding site
Citation: Stevens M, Peigneur S and Tytgat J (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2:71. doi: 10.3389/fphar.2011.00071
Received: 03 August 2011;
Accepted: 24 October 2011;
Published online: 09 November 2011.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Motohiro Nishida, Kyushu University, JapanBaron Chanda, University of Wisconsin–Madison, USA
Copyright: © 2011 Stevens, Peigneur and Tytgat. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jan Tytgat, Lab of Toxicology, Katholieke Universiteit Leuven, Campus Gasthuisberg O&N 2, Herestraat 49, Box 922, 3000 Leuven, Belgium. e-mail: jan.tytgat@pharm.kuleuven.be