 Jaclyn P. Kerr
Jaclyn P. Kerr Christopher W. Ward
Christopher W. Ward Robert J. Bloch
Robert J. Bloch- 1Department of Physiology, University of Maryland School of Medicine, Baltimore, MD, USA
- 2Department of Organizational Systems and Adult Health, University of Maryland School of Nursing, Baltimore, MD, USA
The class of muscular dystrophies linked to the genetic ablation or mutation of dysferlin, including Limb Girdle Muscular Dystrophy 2B (LGMD2B) and Miyoshi Myopathy (MM), are late-onset degenerative diseases. In lieu of a genetic cure, treatments to prevent or slow the progression of dysferlinopathy are of the utmost importance. Recent advances in the study of dysferlinopathy have highlighted the necessity for the maintenance of calcium handling in altering or slowing the progression of muscular degeneration resulting from the loss of dysferlin. This review highlights new evidence for a role for dysferlin at the transverse (t-) tubule of striated muscle, where it is involved in maintaining t-tubule structure and function.
Introduction
The class of muscular dystrophies linked to the genetic ablation or mutation of dysferlin, including Limb Girdle Muscular Dystrophy 2B (LGMD2B) and Miyoshi Myopathy (MM), are degenerative diseases of skeletal muscle that typically appear in the teen years and ultimately lead to loss of mobility. In the absence of a genetic cure, individuals with these myopathies would benefit from treatments that slow the dystrophic progression and improve quality of life. Understanding the role of dysferlin within the myofiber and how its loss affects muscle function may speed the development of therapeutics designed to prevent or ameliorate the pathogenic events that occur in its absence.
Dysferlin is a member of the ferlin subgroup, a family of proteins comprised of multiple Ca2+-sensitive C2 domains that are implicated in vesicle fusion, trafficking, and membrane repair (Lek et al., 2012). The seven C2 domains of dysferlin have variant affinity for Ca2+ and phospholipids (Davis et al., 2002; Therrien et al., 2009; Marty et al., 2013; Fuson et al., 2014) and regulate the association of dysferlin with multiple protein complexes (Huang et al., 2007; Azakir et al., 2010; Di Fulvio et al., 2011). In adult skeletal muscle cells, the early identification of dysferlin at the sarcolemma led to its assignment as a protein important for the repair of sarcolemmal damage (Bansal et al., 2003; Bansal and Campbell, 2004). However, an increasing body of evidence indicates an association of dysferlin with the transverse (t)-tubule membrane (Ampong et al., 2005; Roche et al., 2011; Flix et al., 2013; Demonbreun et al., 2014), where it is involved in maintaining Ca2+ homeostasis following cellular stress (Kerr et al., 2013).
Here, we review our recent evidence for dysferlin's preferential localization within the t-tubules of mature myofibers and its role in maintaining Ca2+ homeostasis (Roche et al., 2011; Kerr et al., 2013). Consistent with its localization at the t-tubule and its association with the L-type Ca2+ channel (LTCC), we showed that dysferlin contributes to the maintenance of Ca2+ homeostasis during mechanical stress. In dysferlin-deficient muscle fibers, acute mechanical stress disrupted Ca2+ homeostasis, resulting in localized t-tubule damage. As these effects were abrogated by both low external Ca2+ and the LTCC inhibitor diltiazem, these results are consistent with an increase of stress-dependent Ca2+ influx through the LTCC. Importantly, we showed that in vivo treatment of dysferlin-deficient mice with diltiazem provided protection from the enhanced contraction-induced damage characteristic of dysferlin-deficient muscle (Kerr et al., 2013). Taken together, our results demonstrated a novel role for dysferlin as a modulator of stress-dependent LTCC activity and identified the LTCC as a therapeutic target for LGMD2B and MM.
Dysferlin is a T-Tubule Protein
Dysferlin's large, modular structure, comprised of multiple C2 domains in tandem with structural domains common to the ferlin superfamily and a single transmembrane domain (Lek et al., 2012), makes it an attractive scaffold for structural and signaling proteins at the cytoplasmic surface of the membrane. Its role in staunching membrane damage in cultured muscle cells injured by laser illumination and its apparent translocation from internal structures to the sarcolemma led to the hypothesis that dysferlin was primarily involved in repair of the sarcolemmal membrane following Ca2+ influx (Bansal et al., 2003).
Additional hypotheses for dysferlin's function arose following the identification of a number of its binding partners. These binding partners include tubulin, annexins, caveolin 3, Bin1, and AHNAK, consistent with a role for dysferlin in trafficking and membrane repair (Matsuda et al., 2001; Lee et al., 2002; Lennon et al., 2003; Ampong et al., 2005; Turk et al., 2006; Huang et al., 2007; Rezvanpour and Shaw, 2009; Waddell et al., 2011; Flix et al., 2013; McDade and Michele, 2013). However, other work identified the LTCC (also referred to as the dihydropyridine receptor, or DHPR) and the ryanodine receptor (RyR) (Ampong et al., 2005; Flix et al., 2013), implicating dysferlin in Ca2+-dependent signaling, consistent with limited reports of dysferlin localization at or near the t-tubule during muscle maturation and stress (Roche et al., 2011; Waddell et al., 2011; Demonbreun et al., 2014).
Our evidence for dysferlin's association with the t-tubule membrane stems from improvements in the immunolabeling of frozen sections of muscle tissue and isolated muscle fibers in vitro (Roche et al., 2011; Kerr et al., 2013). With these improved techniques, we found a predominant association of dysferlin at the A-I junction of mature myofibers, where the triad junctions are formed between the t-tubules and the terminal cisternae of the sarcoplasmic reticulum. This localization was consistent with reports suggesting that dysferlin was involved in early t-tubule development (Klinge et al., 2010) as well as those that indicated that dysferlin could translocate to and from the t-tubules following sarcolemmal damage or extreme stretch (Klinge et al., 2007; Waddell et al., 2011). However, our results indicated that dysferlin's localization to the t-tubule was not injury-dependent and was maintained at the t-tubule following muscle maturation.
Despite these advancements, it was impossible to determine whether dysferlin localized specifically to the t-tubule membrane using only immunofluorescence and confocal light microscopy. Therefore, we developed an expression construct that contained a specialized pH-sensitive fluorescent protein (pHluorin). When attached pHluorin to the C-terminus of dysferlin, an acute change in extracellular pH was sensed by pHluorin within 30 s, indicating its exposure to the extracellular milieu of the t-tubule lumen. In contrast, dysferlin with N-terminal pHluorin was not responsive to acute changes in external pH in this time frame (Kerr et al., 2013), consistent with the ability of the cytoplasm of mammalian striated muscles to buffer intracellular pH (Arus and Barany, 1986; Portman and Ning, 1990; Westerblad et al., 1997; Chin and Allen, 1998; Zaniboni et al., 2003; Capellini et al., 2013). We conclude from these results that dysferlin localizes in the membrane of the t-tubule, oriented with its N-terminal C2 domains in the cytoplasm and its C-terminal sequence in the lumen. Our identification of dysferlin within the t-tubule membrane is consistent with previously reported binding partners within the triad junction, noted above (Figure 1). Combined with our previous immunofluorescence studies and other reports of dysferlin's involvement in the development and maintenance of the t-tubule structure (Klinge et al., 2010; Roche et al., 2011; Waddell et al., 2011; Demonbreun et al., 2014), these studies point to a role for dysferlin in the normal function of the t-tubule.
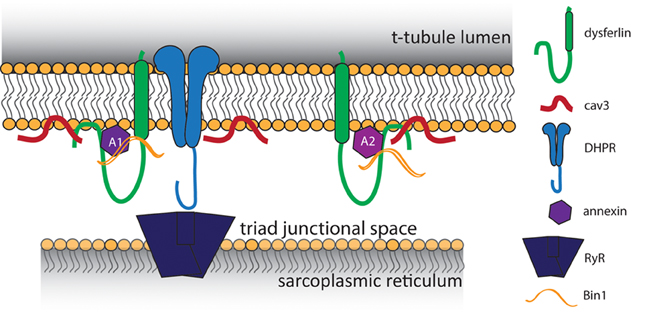
Figure 1. Proposed model of t-tubule dysferlin. Dysferlin is anchored in the t-tubule membrane by its transmembrane domain, with its extreme C-terminus exposed to the lumen of the t-tubule. In close proximity to dysferlin are proteins of the triad junction, the L-type Ca2+ channel (DHPR) in the t-tubule and the ryanodine receptor (RyR) in the sarcoplasmic reticulum. Caveolin 3 (Cav3) and Bin1, both important for the development of t-tubules, are known binding partners of dysferlin. Dysferlin also associates with annexins, which respond to changes in intracellular Ca2+ to promote wound repair.
How dysferlin arrives at the t-tubule remains an open question. Recent efforts have been directed at determining the ability of truncated dysferlin to mediate membrane repair (Azakir et al., 2012), though their focus has been on myoblasts and myotubes, rather than mature myofibers with well-organized and functional t-tubules. In that regard, dysferlin mutations causing truncation of the protein were shown to disrupt its association with Bin1 (Ampong et al., 2005), indicating that some mutations in dysferlin may reduce its association with the t-tubule and would likely affect dysferlin's function at that structure. Furthermore, the more N-terminal C2 domains of dysferlin may play an important role in its trafficking to the t-tubule, as dysferlin's C2A and C2B domains mediate microtubule binding (Azakir et al., 2010; Di Fulvio et al., 2011). Variations in dysferlin's association with the microtubule network may also affect its function at the t-tubule. Identifying the effects of disease-causing mutations of dysferlin on its targeting to t-tubules will be a critical step in uncovering the mechanisms underlying dysferlin's function in mature muscle.
Dysferlin Protects the T-Tubule from Damage by Mechanical Stress
Our group previously demonstrated an increase in contraction-induced damage in dysferlin-deficient muscle (Roche et al., 2008, 2012). As dysregulated Ca2+ signaling at the t-tubule is known to underscore contraction-induced damage in Duchenne muscular dystrophy (Yeung et al., 2005; Fanchaouy et al., 2009; Shkryl et al., 2009) and dysferlin is preferentially localized within the t-tubule, we hypothesized that the t-tubules may be especially susceptible to damage in dysferlinopathy, and that a Ca2+ -dependent process may play a critical role in disease progression.
Our recent work implicates dysferlin in regulating Ca2+ signaling and homeostasis. Using a mild osmotic shock injury on isolated adult myofibers, we found increased structural disruption of the t-tubule in dysferlin-deficient myofibers. In addition, osmotic shock of dysferlin-null myofibers lead to an immediate decrease in the amplitudes of Ca2+ transients that was concomitant with a dramatic rise in cytosolic Ca2+. These effects were mitigated by blocking the LTCC with diltiazem.
Previously, we demonstrated that following damage by eccentric injury, dysferlin-deficient muscle exhibits a depressed rate of functional recovery (Roche et al., 2008; Lovering et al., 2011). Extending our in vitro findings, we demonstrated that diltiazem treatment in vivo improved the recovery of function. Examination of muscle 3 days post-injury revealed that diltiazem limited both necrosis and inflammation, and decreased the number of centrally nucleated fibers. A protection of t-tubule structure 3 h post-injury implicated diltiazem's action on the LTCC as proximate to the enhanced recovery (Kerr et al., 2013).
Although our results indicate that dysferlin protects the t-tubule from damage by mechanical stress, how it does so is unclear. Early hypotheses proposed dysferlin as a membrane repair protein, and in this capacity dysferlin may contribute to maintaining the integrity of the t-tubule membrane during mechanical stress. Consistent with this possibility, dysferlin binds to annexins A1 and A2 (Figure 1), which associate with t-tubules following injury (Waddell et al., 2011; Voigt et al., 2013). Annexins A1 and A2 are highly upregulated in patients with LGMD2B (Lennon et al., 2003), and A1 is involved in membrane repair events following membrane injury (McNeil et al., 2006; Voigt et al., 2013). An elegant study by Lek et al. (2013), demonstrated the reliance of the membrane repair mechanism on cleavage of dysferlin by calpain. In wild type cells, cellular injury results in rapid Ca2+ influx and cleavage of dysferlin to a synaptotagmin-like product that accumulates at the area of injury. Dysferlin's cleavage and accumulation are both blocked by inhibition of LTCC-mediated Ca2+ influx, indicating that Ca2+ influx upon injury is crucial for repair. However, as we demonstrated, influxes of Ca2+ in dysferlin-deficient muscle are dysregulated and sustained, resulting in secondary deficits in EC-coupling that eventually degrade the muscle fiber (Kerr et al., 2013). The pathogenic role of dysregulated Ca2+ signaling in other muscular dystrophies has been noted (Millay et al., 2009; Goonasekera et al., 2011).
Recent work proposes that mechanical stress-induces the production of reactive oxygen species (ROS) by NADPH oxidase 2 (termed X-ROS). This mechano-activated ROS sensitizes the activation of mechano-sensitive Ca2+ channels in the t-tubule. In dystrophic muscle, X-ROS is enhanced (Prosser et al., 2011; Khairallah et al., 2012). Recently, we identified amplified X-ROS signaling in dysferlin-deficient muscle (Prosser et al., 2013; Kombairaju et al., 2014) consistent with another recent report of enhanced muscle oxidation in the same model (Terrill et al., 2013). The increased production of X-ROS in several dystrophic models suggests that it arises secondary to functional deficits linked to mutations in essential muscle genes. The contribution of X-ROS to the enhanced sensitivity to mechanical stress experienced by dysferlin-null muscle could further contribute to the muscle degeneration and myopathy that occur in dysferlinopathies (Figure 2). These pathways are all attractive targets for therapeutics, as their interactions indicate that mitigating one is likely to dampen the others.
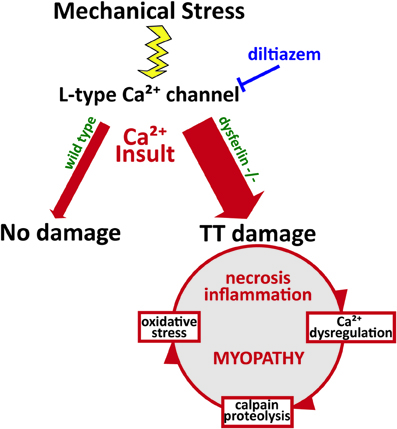
Figure 2. Pathophysiology of dysferlin deficiency. Dysferlin is hypothesized to respond to influxes of Ca2+ and promote wound repair of the t-tubule membrane. Mechanical stress or membrane injury results in influx of Ca2+, mediated by the L-type Ca2+ channel, and this Ca2+ influx does not cause significant muscle injury in wild type muscle cells. However, in the absence of dysferlin, Ca2+ influx to the cytosol is greatly exaggerated, disrupting Ca2+ homeostasis and EC-coupling. This activates a cascade of Ca2+-mediated events that promote further damage to the muscle fiber, including Ca2+-induced proteolysis and oxidative stress. Together, these processes contribute to the eventual myopathy, spurring increased necrosis and inflammation.
Conclusions
In lieu of a genetic cure, treatments that prevent or slow the progression of LGMD2B and MM are of the utmost importance. Our demonstration of altered t-tubule structure and the elevated Ca2+-sensitive pathways in dysferlin-null muscle is consistent with reports that both are disrupted in dysferlinopathies (Selcen et al., 2001; Campanaro et al., 2002; Suzuki et al., 2005; Demonbreun et al., 2014). Our results indicate that targeting LTCC-dependent Ca2+ influx is likely to have significant therapeutic benefit for patients with dysferlinopathy.
Our recent findings and those of others support a model of dysferlin as a Ca2+-sensitive signaling scaffold localized to the t-tubule membrane (Figure 1), that is designed to respond to changes in intracellular Ca2+ caused by t-tubule membrane stress and damage. We propose that this scaffold is uniquely positioned near the triad junctions of muscle, where Ca2+ homeostasis is tightly regulated to facilitate contraction and mediate downstream signaling cascades that maintain the normal functions of muscle. In the absence of dysferlin, the myofiber lacks the ability to maintain Ca2+ homeostasis during stress, resulting in abnormally high cytosolic Ca2+ and the activation of myriad processes that result in proteolysis and oxidative stress, and eventually, necrosis, inflammation, and the progression of the myopathy (Figure 2).
An important, unresolved question in the study of dysferlinopathy is the mechanistic underpinning for its delayed onset. Typically, symptoms do not appear until the second or third decade of life, and sometimes only much later (Klinge et al., 2008). Work by our laboratory and others has shown that, while pre-clinical animal models exhibit minor, overt functional deficits, dysferlin deficiency is clearly associated with delayed recovery from muscle injury induced by eccentric exercise (Roche et al., 2008, 2012; Biondi et al., 2013). Recent studies point to a temporal progression of altered molecular signaling and histopathology, suggesting that the phenotypic appearance in pre-clinical models and the clinical appearance of disease in patients may only be revealed after a threshold of cellular dysfunction is reached (Biondi et al., 2013). This concept is consistent with the observation that patients who participated more in sports showed more rapid disease progression (Angelini et al., 2011).
The recent re-examination of the cellular function of dysferlin highlighted in this review has provided a number of possible strategies for therapeutics. Further, these advances have expanded our understanding of dysferlin's potential roles in striated muscle. They have also stimulated new questions about the protein, how it traffics to t-tubules, and how it protects t-tubules from structural damage when muscle is stressed. Studies of the role of dysferlin's C2 domains and its effects on Ca2+ homeostasis and signaling may reveal an array of therapeutic options for individuals with LGMD2B and MM that will likely include both drug and genetic approaches.
Author Contributions
Jaclyn P. Kerr, Christopher W. Ward, and Robert J. Bloch wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Jaclyn P. Kerr is supported by the Integrative Training Grant in Muscle Biology (T32 AR07592). Further funding provided by The Jain Foundation (Robert J. Bloch), the Muscular Dystrophy Association (Robert J. Bloch), and RO1-AR062554 (Christopher W. Ward).
References
Ampong, B. N., Imamura, M., Matsumiya, T., Yoshida, M., and Takeda, S. (2005). Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 24, 134–144. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/16550931
Angelini, C., Peterle, E., Gaiani, A., Bortolussi, L., and Borsato, C. (2011). Dysferlinopathy course and sportive activity: clues for possible treatment. Acta Myol. 30, 127–132. Available online at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3235880/
Arus, C., and Barany, M. (1986). Application of high-field 1H-NMR spectroscopy for the study of perifused amphibian and excised mammalian muscles. Biochim. Biophys. Acta 886, 411–424. doi: 10.1016/0167-4889(86)90177-1
Azakir, B. A., Di, F. S., Salomon, S., Brockhoff, M., Therrien, C., and Sinnreich, M. (2012). Modular dispensability of dysferlin C2 domains reveals rational design for mini-dysferlin molecules. J. Biol. Chem. 287, 27629–27636. doi: 10.1074/jbc.M112.391722
Azakir, B. A., Di, F. S., Therrien, C., and Sinnreich, M. (2010). Dysferlin interacts with tubulin and microtubules in mouse skeletal muscle. PLoS ONE 5:e10122. doi: 10.1371/journal.pone.0010122
Bansal, D., and Campbell, K. P. (2004). Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 14, 206–213. doi: 10.1016/j.tcb.2004.03.001
Bansal, D., Miyake, K., Vogel, S. S., Groh, S., Chen, C. C., Williamson, R., et al. (2003). Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 423, 168–172. doi: 10.1038/nature01573
Biondi, O., Villemeur, M., Marchand, A., Chretien, F., Bourg, N., Gherardi, R. K., et al. (2013). Dual effects of exercise in dysferlinopathy. Am. J. Pathol. 182, 2298–2309. doi: 10.1016/j.ajpath.2013.02.045
Campanaro, S., Romualdi, C., Fanin, M., Celegato, B., Pacchioni, B., Trevisan, S., et al. (2002). Gene expression profiling in dysferlinopathies using a dedicated muscle microarray. Hum. Mol. Genet. 11, 3283–3298. doi: 10.1093/hmg/11.26.3283
Capellini, V. K., Restini, C. B., Bendhack, L. M., Evora, P. R., and Celotto, A. C. (2013). The effect of extracellular pH changes on intracellular pH and nitric oxide concentration in endothelial and smooth muscle cells from rat aorta. PLoS ONE 8:e62887. doi: 10.1371/journal.pone.0062887
Chin, E. R., and Allen, D. G. (1998). The contribution of pH-dependent mechanisms to fatigue at different intensities in mammalian single muscle fibres. J. Physiol. 512, 831–840. doi: 10.1111/j.1469-7793.1998.831bd.x
Davis, D. B., Doherty, K. R., Delmonte, A. J., and McNally, E. M. (2002). Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J. Biol. Chem. 277, 22883–22888. doi: 10.1074/jbc.M201858200
Demonbreun, A. R., Rossi, A. E., Alvarez, M. G., Swanson, K. E., Kieran Deveaux, H., Earley, J. U., et al. (2014). Dysferlin and myoferlin regulate transverse tubule formation and glycerol sensitivity. Am. J. Pathol. 184, 248–259. doi: 10.1016/j.ajpath.2013.09.009
Di Fulvio, S., Azakir, B. A., Therrien, C., and Sinnreich, M. (2011). Dysferlin interacts with histone deacetylase 6 and increases alpha-tubulin acetylation. PLoS ONE 6:e28563. doi: 10.1371/journal.pone.0028563
Fanchaouy, M., Polakova, E., Jung, C., Ogrodnik, J., Shirokova, N., and Niggli, E. (2009). Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium 46, 114–121. doi: 10.1016/j.ceca.2009.06.002
Flix, B., de la Torre, C., Castillo, J., Casal, C., Illa, I., and Gallardo, E. (2013). Dysferlin interacts with calsequestrin-1, myomesin-2 and dynein in human skeletal muscle. Int. J. Biochem. Cell Biol. 45, 1927–1938. doi: 10.1016/j.biocel.2013.06.007
Fuson, K., Rice, A., Mahling, R., Snow, A., Nayak, K., Shanbhogue, P., et al. (2014). Alternate splicing of dysferlin C2A confers Ca2+-dependent and Ca2+-independent binding for membrane repair. Structure 22, 104–115. doi: 10.1016/j.str.2013.10.001
Goonasekera, S. A., Lam, C. K., Millay, D. P., Sargent, M. A., Hajjar, R. J., Kranias, E. G., et al. (2011). Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Invest. 121, 1044–1052. doi: 10.1172/JCI43844
Huang, Y., Laval, S. H., Van, R. A., Baudier, J., Benaud, C., Anderson, L. V., et al. (2007). AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 21, 732–742. doi: 10.1096/fj.06-6628com
Kerr, J. P., Ziman, A. P., Mueller, A. L., Muriel, J. M., Kleinhans-Welte, E., Gumerson, J. D., et al. (2013). Dysferlin stabilizes stress-induced Ca2+ signaling in the transverse tubule membrane. Proc. Natl. Acad. Sci. U.S.A. 110, 20831–20836. doi: 10.1073/pnas.1307960110
Khairallah, R. J., Shi, G., Sbrana, F., Prosser, B. L., Borroto, C., Mazaitis, M. J., et al. (2012). Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci. Signal. 5, ra56. doi: 10.1126/scisignal.2002829
Klinge, L., Dean, A. F., Kress, W., Dixon, P., Charlton, R., Muller, J. S., et al. (2008). Late onset in dysferlinopathy widens the clinical spectrum. Neuromuscul. Disord. 18, 288–290. doi: 10.1016/j.nmd.2008.01.004
Klinge, L., Harris, J., Sewry, C., Charlton, R., Anderson, L., Laval, S., et al. (2010). Dysferlin associates with the developing t-tubule system in rodent and human skeletal muscle. Muscle Nerve 41, 166–173. doi: 10.1002/mus.21166
Klinge, L., Laval, S., Keers, S., Haldane, F., Straub, V., Barresi, R., et al. (2007). From t-tubule to sarcolemma: damage-induced dysferlin translocation in early myogenesis. FASEB J. 21, 1768–1776. doi: 10.1096/fj.06-7659com
Kombairaju, P., Kerr, J. P., Roche, J. A., Pratt, S. J., Lovering, R. M., Sussan, T. E., et al. (2014). Genetic silencing of Nrf2 enhances X-ROS in dysferlin-deficient muscle. Front. Physiol. 5:57. doi: 10.3389/fphys.2014.00057
Lee, E., Marcucci, M., Daniell, L., Pypaert, M., Weisz, O. A., Ochoa, G.-C., et al. (2002). Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science 297, 1193–1196. doi: 10.1126/science.1071362
Lek, A., Evesson, F. J., Lemckert, F. A., Redpath, G. M. I., Lueders, A.-K., Turnbull, L., et al. (2013). Calpains, cleaved mini-dysferlin C72, and L-type channels underpin calcium-dependent muscle membrane repair. J. Neurosci. 33, 5085–5094. doi: 10.1523/JNEUROSCI.3560-12.2013
Lek, A., Evesson, F. J., Sutton, R. B., North, K. N., and Cooper, S. T. (2012). Ferlins: regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair. Traffic 13, 185–194. doi: 10.1111/j.1600-0854.2011.01267.x
Lennon, N. J., Kho, A., Bacskai, B. J., Perlmutter, S. L., Hyman, B. T., and Brown, R. H. Jr. (2003). Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 278, 50466–50473. doi: 10.1074/jbc.M307247200
Lovering, R. M., Roche, J. A., Goodall, M. H., Clark, B. B., and McMillan, A. (2011). An in vivo rodent model of contraction-induced injury and non-invasive monitoring of recovery. J. Vis. Exp. 51: 2782. doi: 10.3791/2782
Marty, N. J., Holman, C. L., Abdullah, N., and Johnson, C. P. (2013). The C2 domains of otoferlin, dysferlin, and myoferlin alter the packing of lipid bilayers. Biochemistry 52, 5585–5592. doi: 10.1021/bi400432f
Matsuda, C., Hayashi, Y. K., Ogawa, M., Aoki, M., Murayama, K., Nishino, I., et al. (2001). The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum. Mol. Genet. 10, 1761–1766. doi: 10.1093/hmg/10.17.1761
McDade, J. R., and Michele, D. E. (2013). Membrane damage induced vesicle-vesicle fusion of dysferlin-containing vesicles in muscle cells requires microtubules and kinesin. Hum. Mol. Genet. doi: 10.1093/hmg/ddt557. [Epub ahead of print].
McNeil, A. K., Rescher, U., Gerke, V., and McNeil, P. L. (2006). Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 281, 35202–35207. doi: 10.1074/jbc.M606406200
Millay, D. P., Goonasekera, S. A., Sargent, M. A., Maillet, M., Aronow, B. J., and Molkentin, J. D. (2009). Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 19023–19028. doi: 10.1073/pnas.0906591106
Portman, M. A., and Ning, X. H. (1990). Developmental adaptations in cytosolic phosphate content and pH regulation in the sheep heart in vivo. J. Clin. Invest. 86, 1823–1828. doi: 10.1172/JCI114912
Prosser, B. L., Khairallah, R. J., Ziman, A. P., Ward, C. W., and Lederer, W. J. (2013). X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 58, 172–181. doi: 10.1016/j.yjmcc.2012.11.011
Prosser, B. L., Ward, C. W., and Lederer, W. J. (2011). X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333, 1440–1445. doi: 10.1126/science.1202768
Rezvanpour, A., and Shaw, G. S. (2009). Unique S100 target protein interactions. Gen. Physiol. Biophys. 28, F39–F46. Available online at: http://www.gpb.sav.sk/FI-2009/F39.pdf
Roche, J. A., Lovering, R. M., and Bloch, R. J. (2008). Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport 19, 1579–1584. doi: 10.1097/WNR.0b013e328311ca35
Roche, J. A., Ru, L. W., and Bloch, R. J. (2012). Distinct effects of contraction-induced injury in vivo on four different murine models of dysferlinopathy. J. Biomed. Biotechnol. 2012, 134031. doi: 10.1155/2012/134031
Roche, J. A., Ru, L. W., O'Neill, A. M., Resneck, W. G., Lovering, R. M., and Bloch, R. J. (2011). Unmasking potential intracellular roles for dysferlin through improved immunolabeling methods. J. Histochem. Cytochem. 59, 964–975. doi: 10.1369/0022155411423274
Selcen, D., Stilling, G., and Engel, A. G. (2001). The earliest pathologic alterations in dysferlinopathy. Neurology 56, 1472–1481. doi: 10.1212/WNL.56.11.1472
Shkryl, V. M., Martins, A. S., Ullrich, N. D., Nowycky, M. C., Niggli, E., and Shirokova, N. (2009). Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 458, 915–928. doi: 10.1007/s00424-009-0670-2
Suzuki, N., Aoki, M., Hinuma, Y., Takahashi, T., Onodera, Y., Ishigaki, A., et al. (2005). Expression profiling with progression of dystrophic change in dysferlin-deficient mice (SJL). Neurosci. Res. 52, 47–60. doi: 10.1016/j.neures.2005.01.006
Terrill, J. R., Radley-Crabb, H. G., Iwasaki, T., Lemckert, F. A., Arthur, P. G., and Grounds, M. D. (2013). Oxidative stress and pathology in muscular dystrophies: focus on protein thiol oxidation and dysferlinopathies. FEBS J. 17, 4149–4164. doi: 10.1111/febs.12142
Therrien, C., Di, F. S., Pickles, S., and Sinnreich, M. (2009). Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry 48, 2377–2384. doi: 10.1021/bi802242r
Turk, R., Sterrenburg, E., van der Wees, C. G., de Meijer, E. J., de Menezes, R. X., Groh, S., et al. (2006). Common pathological mechanisms in mouse models for muscular dystrophies. FASEB J. 20, 127–129. doi: 10.1096/fj.05-4678fje
Voigt, T., Sebald, H. J., Schoenauer, R., Levano, S., Girard, T., Hoppeler, H. H., et al. (2013). Annexin A1 is a biomarker of t-tubular repair in skeletal muscle of nonmyopathic patients undergoing statin therapy. FASEB J. 27, 2156–2164. doi: 10.1096/fj.12-219345
Waddell, L. B., Lemckert, F. A., Zheng, X. F., Tran, J., Evesson, F. J., Hawkes, J. M., et al. (2011). Dysferlin, annexin A1, and mitsugumin 53 are upregulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 70, 302–313. doi: 10.1097/NEN.0b013e31821350b0
Westerblad, H., Bruton, J. D., and Lannergren, J. (1997). The effect of intracellular pH on contractile function of intact, single fibers of mouse muscle declines with increasing temperature. J. Physiol. 500, 193–204.
Yeung, E. W., Whitehead, N. P., Suchyna, T. M., Gottlieb, P. A., Sachs, F., and Allen, D. G. (2005). Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 562, 367–380. doi: 10.1113/jphysiol.2004.075275
Zaniboni, M., Swietach, P., Rossini, A., Yamamoto, T., Spitzer, K. W., and Vaughan-Jones, R. D. (2003). Intracellular proton mobility and buffering power in cardiac ventricular myocytes from rat, rabbit, and guinea pig. Am. J. Physiol. Heart Circ. Physiol. 285, H1236–H1246. doi: 10.1152/ajpheart.00277.2003
Keywords: muscular dystrophy, calcium, excitation-contraction coupling, myopathy
Citation: Kerr JP, Ward CW and Bloch RJ (2014) Dysferlin at transverse tubules regulates Ca2+ homeostasis in skeletal muscle. Front. Physiol. 5:89. doi: 10.3389/fphys.2014.00089
Received: 06 December 2013; Paper pending published: 13 January 2014;
Accepted: 15 February 2014; Published online: 06 March 2014.
Edited by:
Aikaterini Kontrogianni-Konstantopoulos, University of Maryland School of Medicine, USAReviewed by:
Nagomi Kurebayashi, Juntendo University School of Medicine, JapanBradley Launikonis, University of Queensland, Australia
Joshua Zimmerberg, National Institutes of Health, USA
Copyright © 2014 Kerr, Ward and Bloch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. Bloch, Department of Physiology, University of Maryland School of Medicine, 655 W. Baltimore St., Baltimore, MD 21201, USA e-mail: rbloch@umaryland.edu