 Jingping Fang
Jingping Fang Aiting Lin2
Aiting Lin2 Weijing Qiu
Weijing Qiu Muhammad Umar
Muhammad Umar Ray Ming
Ray Ming- 1Key Lab of Sugarcane Biology and Genetic Breeding, Ministry of Agriculture, Fujian Agriculture and Forestry University, Fuzhou, China
- 2FAFU and UIUC-SIB Joint Center for Genomics and Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, China
- 3Department of Plant Biology, University of Illinois at Urbana-Champaign, Urbana, IL, USA
Papaya is a productive and nutritious tropical fruit. Papaya Ringspot Virus (PRSV) is the most devastating pathogen threatening papaya production worldwide. Development of transgenic resistant varieties is the most effective strategy to control this disease. However, little is known about the genome-wide functional changes induced by particle bombardment transformation. We conducted transcriptome sequencing of PRSV resistant transgenic papaya SunUp and its PRSV susceptible progenitor Sunset to compare the transcriptional changes in young healthy leaves prior to infection with PRSV. In total, 20,700 transcripts were identified, and 842 differentially expressed genes (DEGs) randomly distributed among papaya chromosomes. Gene ontology (GO) category analysis revealed that microtubule-related categories were highly enriched among these DEGs. Numerous DEGs related to various transcription factors, transporters and hormone biosynthesis showed clear differences between the two cultivars, and most were up-regulated in transgenic papaya. Many known and novel stress-induced and disease-resistance genes were most highly expressed in SunUp, including MYB, WRKY, ERF, NAC, nitrate and zinc transporters, and genes involved in the abscisic acid, salicylic acid, and ethylene signaling pathways. We also identified 67,686 alternative splicing (AS) events in Sunset and 68,455 AS events in SunUp, mapping to 10,994 and 10,995 papaya annotated genes, respectively. GO enrichment for the genes displaying AS events exclusively in Sunset was significantly different from those in SunUp. Transcriptomes in Sunset and transgenic SunUp are very similar with noteworthy differences, which increased PRSV-resistance in transgenic papaya. No detrimental pathways and allergenic or toxic proteins were induced on a genome-wide scale in transgenic SunUp. Our results provide a foundation for unraveling the mechanism of PRSV resistance in transgenic papaya.
Introduction
Papaya (Carica papaya L.) is a major tropical fruit crop with outstanding nutritional and medicinal values. Various aspects of genomics and genetics are relatively easy with papaya owing to its small genome (2n = 18, 372 Mb), short generation time, incipient sex chromosome, and established transformation system (Arumuganathan and Earle, 1991; Ming et al., 2008; Wang et al., 2012). The lack of a recent genome wide duplication in papaya helps to study angiosperm genome evolution (Ming et al., 2012). In addition, papaya is in the order Brassicales with the model plant Arabidopsis thaliana that diverged about 72 million years ago, facilitating comparative structural and evolutional genomic research in both species (Wikström et al., 2001). These characteristics make papaya an excellent model for tree fruit species.
Papaya Ringspot Virus (PRSV) is the most devastating threat to year-round production of papaya in the world. PRSV is a single-stranded positive sense RNA virus that belongs to the genus Potyvirus of family Potyviridae (Tripathi et al., 2008). It can be easily transmitted in a non-persistent manner by aphids and mechanical means, but difficult to control due to the lack of naturally resistant germplasm from Carica papaya (Gonsalves, 1998; Ming and Moore, 2014). The infected papaya plants are characterized by abnormally stunted growth, malformed yellowing leaves with mosaic and reduction of fruit. Other symptoms such as water soaked oily streaks on petioles, dark green streaks on trunks, round spots and bumps on fruit are also indicative of this devastating viral disease. The papaya industry in Hawaii has been severely damaged since the onset of PRSV's spread in 1992. Hawaii's production of marketable papaya drastically declined from 55.8 million pounds in 1992 to 33.6 million pounds in 1998. None of the approaches aiming at creating papaya resistant germplasm by crossing it to wild relatives (Manshardt and Wenslaff, 1989) or cross-protection (Yeh and Gonsalves, 1984; Mau et al., 1989) were able to allow the papaya industry to recover. In 1998, the deregulation and commercialization of PRSV-resistant transgenic papaya, SunUp and Rainbow, revived the industry (Tripathi et al., 2008). Today, transgenic papaya acreage is about 85% of the total in the state of Hawaii.
Genetically engineered (GE) papaya plants were developed based on the concept of pathogen-derived resistance (PDR) using PRSV coat protein (cp) gene transformation to protect the host plants against PRSV infection (Sanford and Johnston, 1985; Fitch et al., 1992). Post transcriptional gene silencing (PTGS) or RNA interference (RNAi) is a sequence-specific mRNA degradation mechanism by which plants are able to protect themselves from viral infection by targeting viral transgenes (Vaucheret et al., 1998, 2001). The PRSV-resistant papaya SunUp is essentially a genetically engineered version of the existing Sunset cultivar. Sunset is a pink-fleshed cultivar with a low level of residual heterozygosity due to over 25 generations of inbreeding (Storey et al., 1969). SunUp is derived from the transgenic line 55-1 R0 which was obtained via microprojectile bombardment-mediated transformation of 2,4-D-treated immature zygotic embryos with a plasmid construction containing the neophosphotransferase II (nptII) and β-glucuronidase (uidA; gus) genes flanking a PRV HA 5-1 cp gene expression cassette (Fitch et al., 1990, 1992). SunUp is a cultivar homozygous for the PRSV CP transgene and is highly inbred with a heterozygosity of only 0.06% (Ming et al., 2008). Rainbow is an F1 hybrid hemizygous for the PRSV CP transgene resulting from a cross between SunUp and the yellow-fleshed non-GE cultivar Kapoho (Manshardt, 1999). SunUp and Sunset have separated for more than 20 years, i.e., more than 20 meiosis, and they share lots of phenotypic and functional features. SunUp plants grow phenotypically normal and both cultivars have the same fruit appearance of pink-tinged flesh with a melting texture (Figure S1C). The total soluble solids of SunUp fruit were within the expected range (Ming and Moore, 2014). It is obviously that transgenic papaya showed excellent resistance to PRSV (Figure S1B). However, we also found in our research that even at the initial stage of growth prior to infection with PRSV, seedlings of transgenic papaya SunUp exhibited better growth performance compared with that of its progenitor Sunset under the same conditions (Figure S1A). Their increased growth rate was particularly pronounced from ~3 weeks after germination.
Although transgenic papaya reversed the downward trend of papaya production in Hawaii, papaya production has not yet returned to its 1992 levels. Perhaps the primary reason is that this industry still faces some challenges in bringing transgenic papaya to markets outside the US. Taking biosafety into account, some countries have zero tolerance policies for transgenic papaya. Overall, potential allergenicity of this new product and possible altered nutritional composition are the two main areas of concern in regard to the food safety of GE organisms. There is no publicly available evidence to date that the coat protein of PRSV or other plant viruses is allergenic or detrimental to human health in any way. Detailed studies on the potential of the cp gene as an allergen showed that the transgene-derived PRSV CP does not pose a risk of food allergy following accepted allergenicity assessment criteria (Fermín et al., 2011). In the toxicology assessment, CP was not considered a novel protein due to the history of human consumption of PRSV-infected plants without adverse health effects. Measured amounts of CP in transgenic plants were even much lower than those of infected plants. No differences were observed between GE and non-GE papaya for 36 nutrients at any of the tested fruit ripeness stages (Tripathi et al., 2011). Transgene number or vector elements inserts were characterized as part of a petition to Japan to allow import of these transgenic papaya fruit from Hawaii. Three stably inherited transgene inserts were detected in Rainbow and SunUp by Southern blot analysis (Suzuki et al., 2008). One was a 9789 bp functional insert, coding for the PRSV cp, nptII, and uidA; two were unintended inserts: a partial nptII gene fragment and a partial tetA gene fragment. Intriguingly, five out of six transgene flanking sequences were chloroplast-derived, and one was mitochondria-derived. Four of the flanking sequences were closely associated with topoisomerase I recognition sites. From analysis of the insertion site and flanking genomic DNA, no changes to endogenous gene function and no allergenic or toxic proteins were predicted. Polymerase Chain Reaction (PCR) and Southern blot analysis are the most commonly used methods to detect integration of vector sequences at the genome structure level. However, at the genome function level, how genome-wide gene expression is altered and whether interrupted genes are induced by bombardment remained to be determined. Proving no detrimental functional changes induced at the genome-wide level in GE papaya is an essential criterion in the context of genetically modified organism (GMO) biosafety regulation. Moreover, DNA introduction into a host organism via different methods such as agrobacterium-mediated and particle bombardment transformation can cause varied single nucleotide polymorphism (SNPs), insertions/deletions (InDels), rearrangement and integration positions, affecting both transgene expression and the potential for gene disruption in different ways (Kohli et al., 2003; Latham et al., 2006; Wilson et al., 2006). Therefore, a better understanding of how bombardment affects plant genome will further our understanding to develop more reliable methods for crop improvement.
The last decade has seen revolutionary advances in DNA and RNA sequencing technologies with the advent of Next Generation Sequencing (NGS) techniques (Metzker, 2010). RNA-sequencing (RNA-Seq) is an attractive analytical tool in transcriptomics. It is well established and developed very rapidly, decreasing the running costs and playing a central role for unraveling the complexity of gene expression regulation (Morozova and Marra, 2008). A female SunUp genome was sequenced and assembled using whole-genome shotgun (WGS) sequencing, small-insert libraries and Sanger sequencing approaches with integration of physical and genetic maps in 2008 (Ming et al., 2008). This is the first transgenic crop to be sequenced. This genome spans 372 Mb including embedded gaps, representing 75% of papaya genome, 90% of the euchromatic regions and 92% of the papaya ESTs. The availability of the SunUp draft genome and the development of RNA-Seq technology have enabled us in this study to visualize the landscape of changes occurring in transcriptome of SunUp after transformation of its progenitor Sunset with the aim of unraveling the global impact of particle bombardment-mediated transformation on whole genome function. The comparison of dynamic transcriptome expression profiles between these two cultivars may shed light on the potential gene disruption and changes of expression caused by genome structure variation after transformation and give evidence at the transcriptional level that genetically modified papaya are not harmful concerning the biosafety of GMOs.
Materials and Methods
Plant Materials and Growth Conditions
Seeds of transgenic papaya cultivar SunUp and donor control Sunset were planted and grown in plastic pots filled with organic loam in a greenhouse in March, 2015. Greenhouse temperature was set at 27°C. Sixty pots (one seedling per pot, 30 pots per cultivar) were used in this experiment. Plants were watered every day. After 68 days, when the plants had 5–7 leaves, the plantlets were transplanted to field at Fujian Agriculture and Forestry University (FAFU), Fuzhou, China. Young and healthy leaf tissue from 4-month-old plants was collected for RNA extraction. All plants tested PRSV-negative. Three biological replicates were analyzed for each cultivar.
RNA Extraction and Library Construction
Total RNA was extracted from ground leaf using RNAprep pure Plant Kit (Tiangen, #DP432), according to the manufacturer's protocol. Residual genomic DNA was removed with RNase-Free DNase I (Code No. 2212, Takara). The quality of total RNAs was verified on an Agilent 2100 Bioanalyzer (Plant RNA Nano Chip, Agilent). After quantity and quality determination, a single indexed RNA-Seq library was constructed using NEBNext Ultra™ RNA Library Prep Kit for illumina (NEB, #E7530), and then sequenced by Illumina HiSeq2500 in paired-end 150 nt mode. Prior to downstream processing, raw reads generated by Illumina HiSeq2500 were initially processed to obtain clean reads by removing adaptor sequences, empty reads, and low quality sequences (>50% of the bases with a quality score ≤ 5).
Sequencing Read Mapping and Gene Expression Estimation
Sequences of three SunUp transformation plasmid derived inserts with genomic borders are available at Genbank (accession numbers: FJ467933, FJ467932, and FJ467934). We then concatenate these three sequences with the papaya “SunUp” reference genome (http://www.plantgdb.org/CpGDB/) which does not include the sequences of transgene inserts. The trimmed paired-end reads of each sample were aligned to the new assembled papaya “SunUp” reference genome using TopHat v2.0.14 default settings (Trapnell et al., 2012). The reads for each biological replicate were mapped independently, and reads that mapped to reference sequences from multiple genes were filtered out. The resulting Binary Alignment/Map (BAM) alignment files were provided to Cufflinks v2.2.1 to generate a transcriptome assembly for each replicate. These transcriptome assembly files were then merged with the reference transcriptome annotation into one unified annotation by using the Cufflinks cuffmerge utility for further annotation and differential expression analysis. The RNA-Seq read-mapping result was used to predict gene expression profiles, and the FPKM (Fragments per kilobase of exon per million fragments mapped) value of each sample were estimated by Cufflinks. To avoid false positive estimation of gene expression, transcripts with FPKM values < 1 in both libraries were not subjected to further analysis.
Identification of Differentially Expressed Genes
Differentially expressed gene and isoform analysis of the mapping results was analyzed and calculated using the Cufflinks cuffdiff program. Cuffdiff is an independent, widely used tool which takes a nonparametric, annotation-guided approach to estimate the means and variances of transcript FPKM values under different conditions, using Student's t-tests to identify differentially expressed transcripts. Cuffdiff allows taking multiple technical or biological replicate sequencing libraries per condition (Trapnell et al., 2012). The BAM alignment files and the merged annotation are fed to cuffdiff to calculate expression levels in two samples and test the statistical significance of observed changes in expression between them. An absolute log2 (FPKMSU−NP/FPKMSS−NP) >2 (fold change >2) and an adjusted p-value (FDR, false discovery rate) < 0.05 were used as thresholds to identify significant differences in gene expression. CummeRbund (http://compbio.mit.edu/cummeRbund/), an extension R package of the cufflinks, was used for visualization of results and read dispersion.
Functional Annotation
Sequence-similarity Blast searches of all papaya predicted protein sequences were conducted with a typical cut-off E-value of 10−5 against several publicly available protein databases: the National Center for Biotechnology Information (NCBI) non-redundant (Nr) protein database, Clusters of Orthologous Groups (COGs), and Kyoto Encyclopedia of Genes and Genomes (KEGG). Gene Ontology (GO) terms describing biological processes, molecular functions and cellular components were assigned to the predicted genes by Blast2GO program (Conesa et al., 2005) based on the Nr blastp output. Functional classification of GO and COG for all genes was performed by in-house Perl scripts.
For the comparative analysis of DEGs between PRSV susceptible and resistant transgenic papaya cultivars, all DEGs identified in this pairwise comparison were used for GO and KO enrichment analysis. Single enrichment analysis (SEA) was performed in agriGO program (Du et al., 2010) using the gene models of papaya reference genome as background. The Fisher statistical test was applied to test for enrichment of functional categories with Bonferroni's correction (FDR ≤ 0.05). KEGG metabolic pathway annotation and enrichment of the DEGs were performed by using KOBAS (KEGG Orthology Based Annotation System) (Xie et al., 2011). The hypergeometric test was applied with Benjamini and Hochberg's correction method at an FDR of 5%.
Differential expression genes were classified as genes encoding transcription factors (TFs), transporter proteins (TPs) and hormone-related genes according to PlantTFDB v3.0 (Jin et al., 2014), TransportDB (Ren et al., 2007), and the Arabidopsis Hormone Database 2.0 (Jiang et al., 2011). The log2-transformed (FPKM) values for each transcription factor, transporter, and hormone-related gene were used to generate heat maps in R version 3.2.1 statistical package (www.CRAN.R-project.org).
Validation of Differentially Expressed Genes by qRT-PCR
A total of 21 candidate DEGs between Sunset and SunUp were selected for quantitative real-time PCR (qRT-PCR) assays (Table S7). Approximately 1 μg of total RNA per sample was treated with DNase I (Takara, Dalian, China), and then reverse-transcribed using a Prime-Script 1st Strand cDNA Synthesis Kit (Takara, Dalian, China) following the manufacturer's protocol. A total of 1 μl of 10-fold diluted cDNAs were used as template in 20 μl PCR reaction mixture containing 10 μl of 2 × SYBR® Green I Master Mix (Takara, Dalian, China), 0.4 μM of each primer. Gene specific qRT-PCR Primers (See Table S8) were designed from coding sequences of all candidate genes using IDT SciTools® Web Tools (Owczarzy et al., 2008). The qRT-PCR reactions were performed on a CFX Connect Real-Time System (Bio-Rad, Singapore) under the following conditions: one cycle at 95°C for 30 s, followed by 42 cycles at 95°C for 5 s and 60°C for 30 s. Melting curve analysis was added at the end of 42 cycles to check the amplification specificity of target fragments. Three biological replicates were performed for each gene in order to exclude sampling errors. The reference housekeeping gene EIF (Eukaryotic initiation factor 4A) (Zhu et al., 2012) was used as the internal control. Relative expression levels of the selected DEGs were calculated using the 2−ΔΔCt method (Schmittgen and Livak, 2008), and EIF was used to normalize the amount of template cDNA in each reaction.
Alternative Splicing Identification and Classification
To identify alternative mRNA processing events and their corresponding types in each cultivar, in-house Perl scripts (available upon request) were used to predict AS events and determine the types of the events from the exon-exon splice junctions' bed files output by TopHat. Three biological replicates were combined and aligned against the reference genome in order to find all possible splice junctions between annotated exons for each gene in a cultivar. Six different alternative splicing types were identified by comparing RNA-Seq reads with annotated gene models, namely skipped exon (SE), retained intron (RI), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), alternative 5′ UTR splice (5UTR), and alternative 3′ UTR splice (3UTR). They were extracted for further analyses.
Results
Analysis and Mapping of Illumina Sequencing Reads
Sunset is a pink-tinged flesh cultivar that is susceptible to PRSV, and SunUp is a PRSV-resistant papaya cultivar that is essentially a transgenic Sunset. SunUp plants showed vigorous growth compared to Sunset plants (Figure S1A). No abnormal phenotypes were observed in SunUp. To characterize transcriptomic changes at the genome scale induced by microprojectile bombardment, we used the Illumina2500 sequencing platform to sequence SunUp and Sunset transcriptomes, including three PRSV-negative Sunset leaf samples (Sunset non-PRSV leaves, SS-NP) and three PRSV-negative SunUp leaf samples (SunUp non-PRSV leaves, SU-NP). Sequencing generated 342.7 million reads, a total raw dataset of 51.8 Gb. After removing low-quality sequences and trimming adapter sequences, over 342.1 million paired-end clean reads were retained from all libraries, with the number of RNA-Seq reads per library ranging from 45.9 to 64.1 million (Table 1). Mapping to the available papaya SunUp genome, transcript assembly, and quantification were performed by using TopHat and Cufflinks. After mapping the sequence reads against the reference genome, approximately 80% of the total reads matched unique genomic locations (74.9–76%), while the remainders had either multiple matches (3.2–4.5%) or remained unaligned (20.5–20.8%). Only uniquely mapped reads were used in the analysis of gene expression in different cultivars. Before downstream analysis, the correlation coefficient between each pair of three biological replicates was evaluated by comparing their RNA-Seq expression profiles (Figure 1A). This shows that the estimated levels of gene expression were highly consistent between any replicate pair of each cultivar (r = 0.81 − 0.96). In total, 20,700 transcripts were identified from both conditions, accounting for 74.1% of the annotated genes in papaya. In both Sunset and SunUp transcriptomes, the number of mapped genes in Sunset was found to be similar to that in SunUp (20,102 vs. 20,060) (Table 1). Among these genes, more than 74% had FPKM values in the range of 1–100 for each cultivar (Figures 1B,C), and it was observed that the FPKM ranges in two cultivars were almost similar. The log10FPKM shows a primary peak of high expression genes, with two shoulders of low-expression transcripts (Figure 1B). We further compared the mapped genes between Sunset and SunUp transcriptomes, and found that 93.81% of mapped genes were common in two cultivars, but up to 662 and 620 genes were specifically expressed in Sunset and SunUp, respectively (Figure 1D).
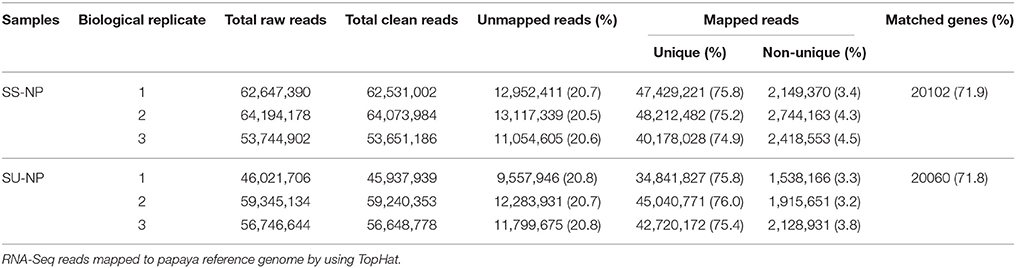
Table 1. Summary of the Illumina 2500 sequencing reads and their matches in the papaya genome.
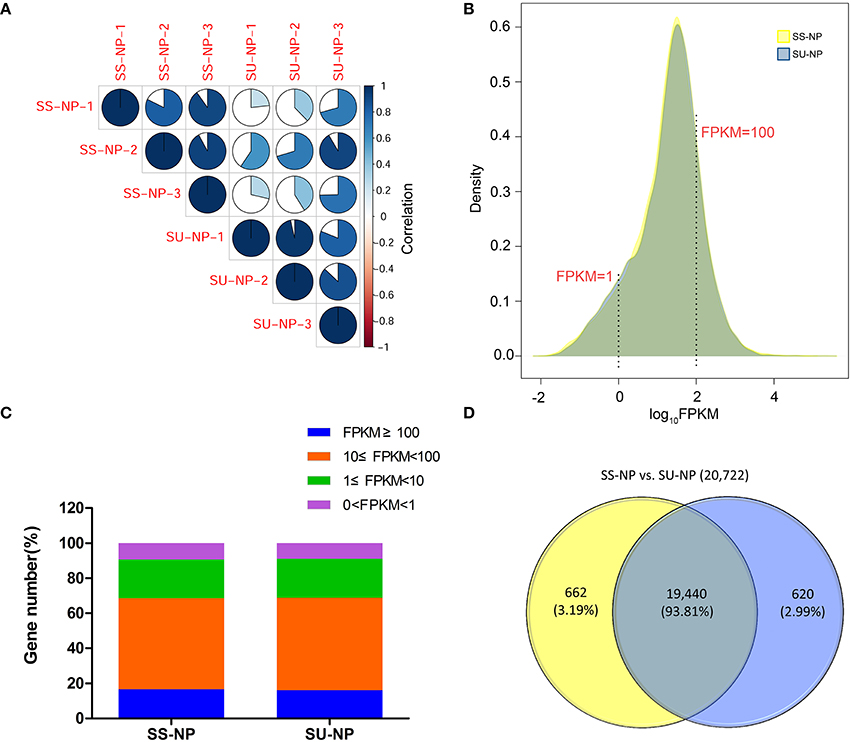
Figure 1. Overview of papaya PRSV-negative SunUp and Sunset transcriptomes. (A) Pairwise correlation of different biological replicates from SS-NP and SU-NP using FPKM values. The color intensities (scale in the side bar) and the numbers indicate the degree of pairwise correlation. (B) log10FPKM distributions of genes for SS-NP and SU-NP. Yellow and purple represent Sunset and SunUp, respectively. (C) Percentage of genes expressed in each variety. (D) Venn diagrams showing mapped genes expressed in SS-NP and SU-NP.
Annotation, Functional Classification, and KEGG Analysis of DEGs
To identify global transcriptional changes occurring after bombardment of the transgene, we applied cuffdiff to identify genes that were differentially expressed between PRSV-negative SunUp and its progenitor cultivar Sunset. Expression profiles of differentially expressed genes (DEGs) are expected to meet the following three criteria: (i) the FPKM value is ≥1 in either of the libraries, (ii) log2 (FPKMSU−NP/FPKMSS−NP) is >2 or < −2, and (iii) the adjusted p-value is < 0.05. In this study, DEGs with higher expression levels in SunUp than in Sunset were defined “up-regulated genes,” whereas those with lower expression levels in SunUp were referred to as “down-regulated genes.” As shown in the scatter plot (Figure 2A), transcript abundance in both Sunset and SunUp are similar for most of the transcripts, which is consistent with Figures 1B,C. To determine how many genes are significantly regulated, a volcano plot was constructed by plotting the fold change values against the negative log10 of adjusted p-values (Figure 2B). The higher the negative log-transformed FDR, the more significant the regulation. A fold change of zero is in the middle of the volcano. On the left side, negative fold change values indicate down-regulation, while on the other side are the positive fold change values thereby indicating up-regulation. In total, 842 DEGs were identified showing up- or down- regulation between Sunset and SunUp, in which 475 genes were up-regulated, and 367 were down-regulated. The DEGs were found to be randomly distributed amongst the 9 papaya chromosomes (Table 2). Chromosome 3 and 8 displayed a relatively higher rate of DEGs than others, while chromosome 2 and 9 contained the least.
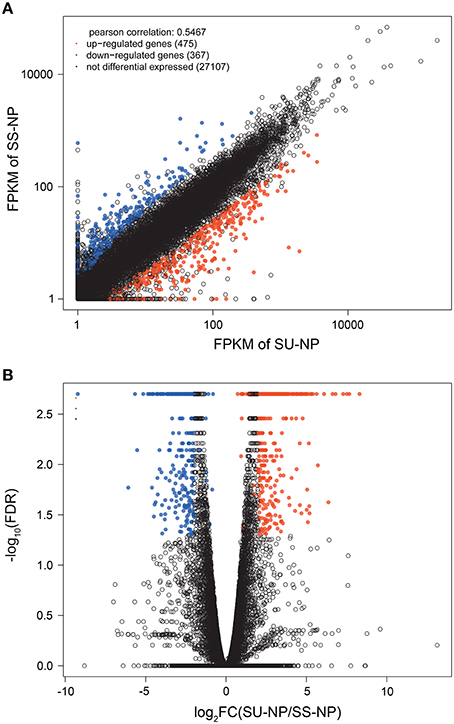
Figure 2. Visualization of expression changes in different libraries. (A) Scatter plot and (B) Volcano plot of the transcriptomes of PRSV-negative Sunset and SunUp. In the scatter plot, FPKM values in donor control (Y-axis) have been plotted against FPKM values of transgenic papaya (X-axis). In the volcano plot, statistical significance (−log10 of adjusted p-value; Y-axis) have been plotted against log2 fold change (X-axis). Significantly up-regulated genes are represented by red dots, while down-regulated genes are represented by blue dots.
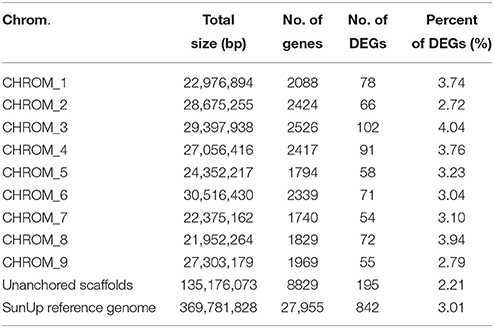
Table 2. The distribution of differentially expressed genes along papaya chromosomes.
For the global functional analysis of DEGs, GO annotation was performed using Blast2GO. Out of the 842 DEGs, 507 corresponding proteins were associated with at least one GO term. The up- and down- regulated DEGs annotated in GO were grouped into 45 and 37 groups based on GO level2 classification, respectively (Figure 3A, Figure S2, and Tables S1A,B). The assigned GO terms belonged to three main ontologies: molecular function, biological process and cellular component. In the molecular function, the most common categories were “catalytic activity” (154 up-regulated, 135 down-regulated) and “binding” (123 and 125, respectively), followed by “transporter activity” and “transcription factor activity.” In the category of cellular component ontology, membrane and cell acted as two primary places. Within biological process, “metabolic process,” “cellular process,” and “single-organism process” were predominant. We further applied GO category enrichment analysis to elucidate the functional enrichment of the DEGs, using Fisher's exact test with an FDR cutoff ≤ 0.05. A total of 45 GO terms were enriched in biological processes, molecular function and cellular components (See Table S2, Figure S3). The biological process GO term “microtubule-based movement” was the most significantly and specifically overrepresented term, followed by “polysaccharide catabolic process,” “sucrose metabolic process,” and “cell wall organization or biogenesis.” The enrichment in the biological process category was also reflected by enrichment in the molecular function GO terms “microtubule motor activity,” “microtubule binding,” and “tubulin binding” and by enrichment in the cellular component GO terms “extracellular region,” “cell wall,” “external encapsulating structure,” and “microtubule cytoskeleton.”
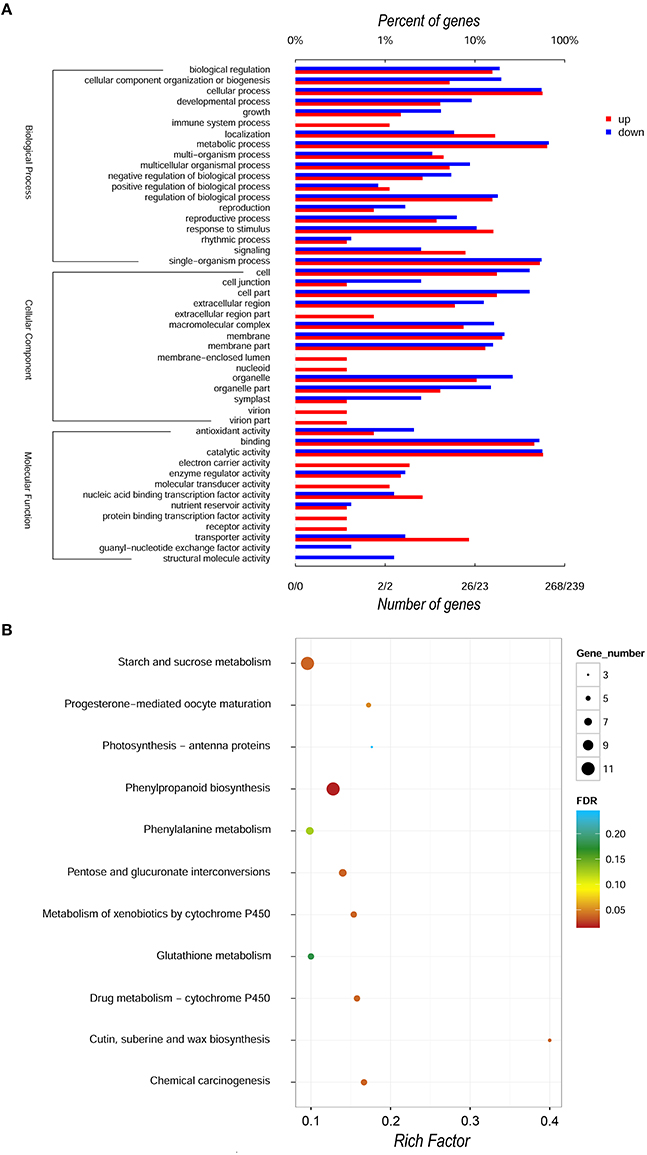
Figure 3. GO and KEGG annotation of DEGs comparison of SunUp and Sunset transcriptomes. (A) Level 2 GO annotation of up-regulated and down-regulated genes. We divided the sets into three major GO ontologies: biological process, cellular component and molecular function. Red and blue bars represent the number and percentage of up-regulated and down-regulated genes, respectively. (B) A KEGG pathway enrichment scatter diagram of DEGs. Only the top 11 most strongly represented pathways were displayed in the diagram. The degree of KEGG pathway enrichment was represented by a rich factor, the FDR, and the number of DEGs enriched in a KEGG pathway. The rich factor indicates the ratio of DEGs enriched in this pathway to the total number of papaya predict genes in this pathway.
All detected DEGs were blasted to STRING 9.0 for further annotation based on Cluster of Orthologous Groups (COG) of protein categories. A total of 161 differentially expressed genes with at least one COG annotations were grouped into 22 of 25 functional categories (Figure S4). The largest category was “General function prediction only” (36 COG annotations), followed by “Signal transduction mechanisms” (20), “Cytoskeleton” (16) and “Secondary metabolites biosynthesis, transport and catabolism” (16). Only 7 COG annotations belonged to the “Function unknown.”
In addition, prediction of the biochemical pathways associated with the DEGs was performed by the Kyoto Encyclopedia of Genes and Genomes (KEGG) identifiers. We used hypergeometric test to identify those pathways that were significantly affected using an FDR ≤ 0.05, relative to the whole papaya transcriptome background. Of the 842 DEGs, 95 genes were assigned at least a KO ID per gene and categorized into 125 pathways. Seven pathways, phenylpropanoid biosynthesis (PATHWAY: ko00940), cutin, suberine, and wax biosynthesis (PATHWAY: ko00073), starch and sucrose metabolism (PATHWAY: ko00500), chemical carcinogenesis (PATHWAY: ko05204), pentose and glucuronate interconversions (PATHWAY: ko00040), drug metabolism-cytochrome P450 (ko00982) and metabolism of xenobiotics by cytochrome P450 (ko00980) were significantly overrepresented with corrected p-values smaller than 0.05 among the differentially expressed genes between two cultivars (Figure 3B, Table S3). However, for the FDR threshold less than 0.01 no significant KEGG pathways have been detected.
DEGs Related to Transcription Factors, Transporter Proteins and Hormones
Expression profiles of the DEGs classified as genes encoding transcription factors (TFs), transporter proteins (TPs) and hormone-related genes (HRGs) between non-GE and GE cultivars were further analyzed on the basis of annotations in Arabidopsis thaliana. In total, we identified 118, 59 and 66 DEGs encoding TFs, TPs and HRGs (Figure 4, Tables S4–S6). Heat map results indicated that most of the DEGs between two cultivars, or between each of the four clusters, showed distinct expression dynamics. Most DEGs belonging to these three types were up-regulated in the transgenic cultivar.
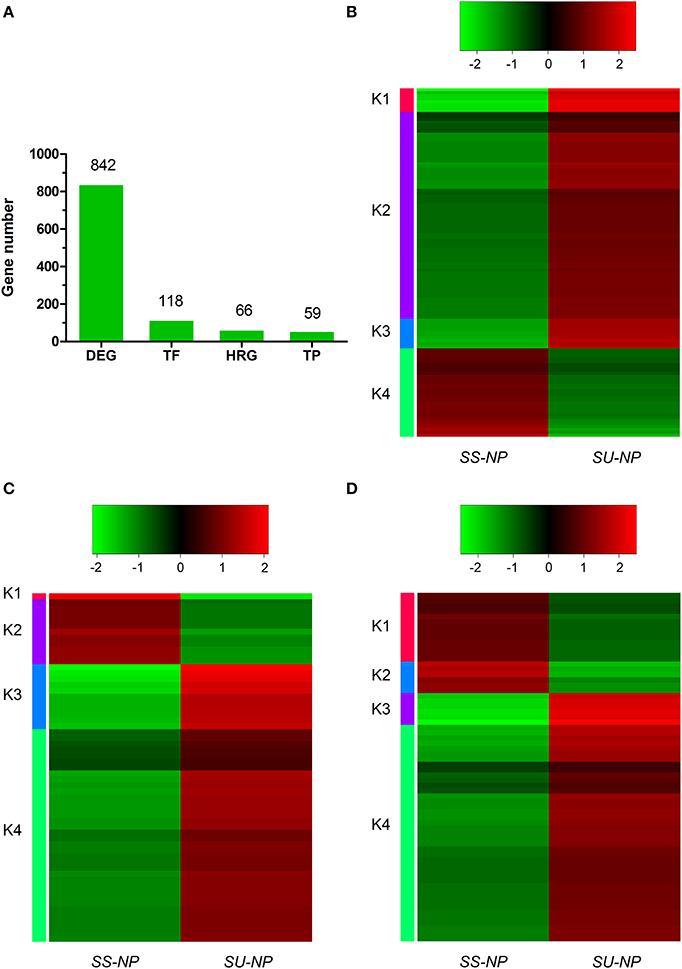
Figure 4. Differentially expressed gene numbers and expression patterns of transcription factors (TFs), transporter proteins (TPs) and hormone-related genes (HRGs). (A) Numbers of total DEGs and those annotated as TFs, TPs and HRGs. (B–D) Heat maps showing log2 FPKM values of 118 TFs, 59 TPs and 66 HRGs annotated DEGs.
Numerous transcription factor families, including AP2/ERF (ethylene response factor), MYB, WRKY, bZIP, bHLH, NAC, DREB, kinase and C2H2-type Zn-finger, were identified to be induced or suppressed upon introduction of the PRSV CP transgene in this study (Figure 4B). Of the 118 TFs, 88 genes were up-regulated and 30 were down-regulated in response to the introduction of the PRSV CP transgene. There were 115 TFs expressed in both cultivars, but one TF (evm.TU.supercontig_55.62) was specifically expressed in SunUp and two TFs (evm.TU.supercontig_19.100 and evm.TU.supercontig_14.16) were specifically expressed in Sunset (Table S4). These three genes were all annotated as basic helix-loop-helix (bHLH) DNA-binding superfamily protein in Arabidopsis thaliana. In terms of the most abundantly differentially expressed TFs (FPKM≥2, absolute fold change >2.5, FDR < 0.01), a total of 35 genes encoding TFs in Arabidopsis were identified, of which 33 were up-regulated in SunUp and only 2 down-regulated (Table 3).
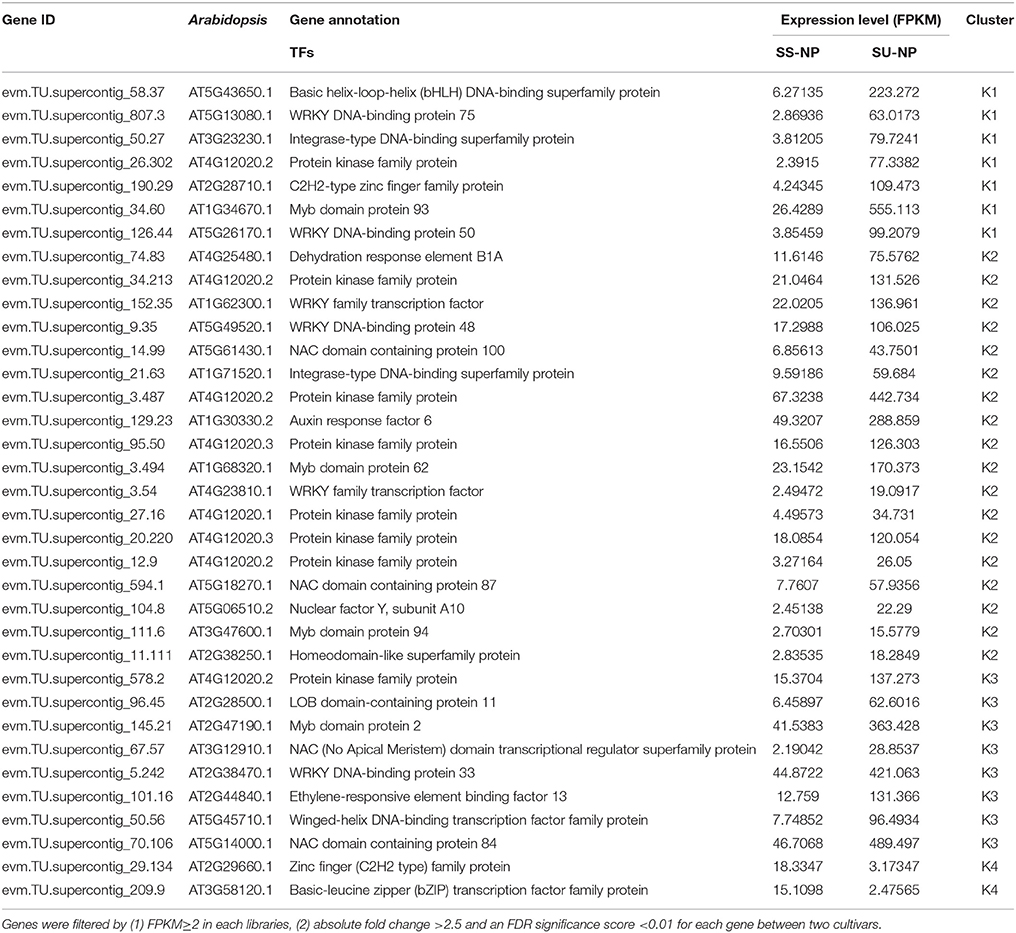
Table 3. The most abundantly differentially expressed transcription factor (TF) genes between two cultivars.
Hierarchical clustering of 59 differentially expressed TPs showed extensive differences between Sunset and SunUp. Among these DEGs, 47 were found to be up-regulated while only 12 were down-regulated in SunUp (Figure 4C, Table S5). We found that genes annotated as ABC-2 type transporter, tonoplast intrinsic protein, laccase 11, and plant L-ascorbate oxidase in Arabidopsis thaliana were down-regulated in SunUp, while genes such as nitrate transporter, zinc transporter, mechanosensitive channel of small conductance-like were significantly up-regulated in SunUp (Table 4).
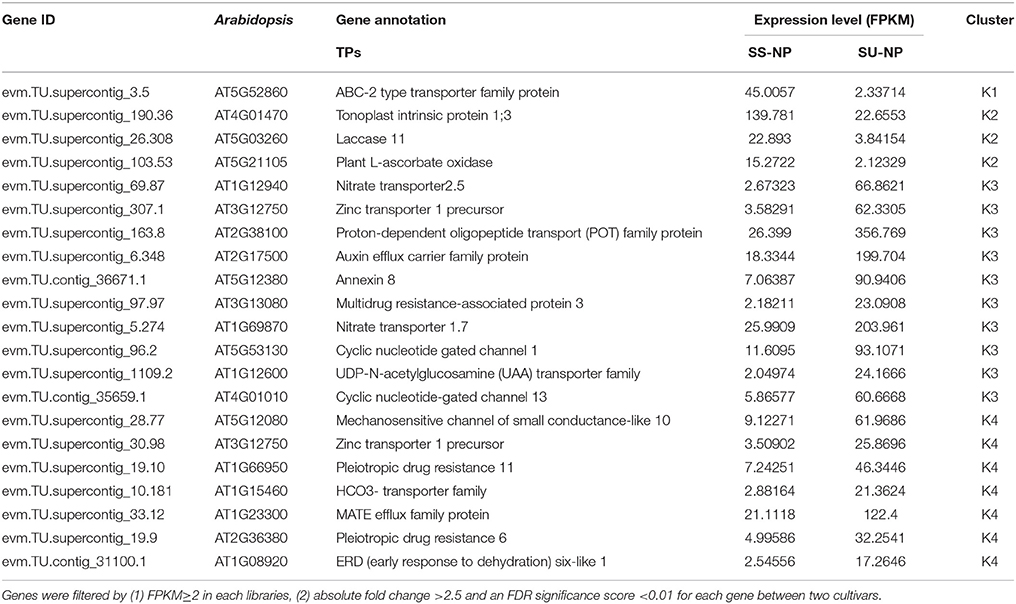
Table 4. The most abundantly differentially expressed transporter (TP) genes between two cultivars.
With respect to hormone-related genes, 66 of these genes were differentially regulated between Sunset and SunUp, consisting of 47 up-regulated and 19 down-regulated DEGs (Figure 4D, Table S6). Regarding the most abundantly differentially expressed hormone-related genes, there were only 3 down-regulated genes consisting of 2 abscisic acid (ABA) related genes and 1 gibberellin related gene (Table 5). By contrast, 22 genes were found to be up-regulated in SunUp, including 10 ABA, 9 salicylic acid (SA), 6 ethylene, 5 auxin, 1 JA and 1 brassinosteroid HRGs. Interestingly, two auxin-responsive genes (evm.TU.supercontig_37.203, evm.TU.supercontig_37.215) were found to be only expressed in Sunset, whereas two gibberellin-related genes (evm.TU.contig_47211.1, evm.TU.supercontig_133.24) were exclusively expressed in SunUp (Table S6).
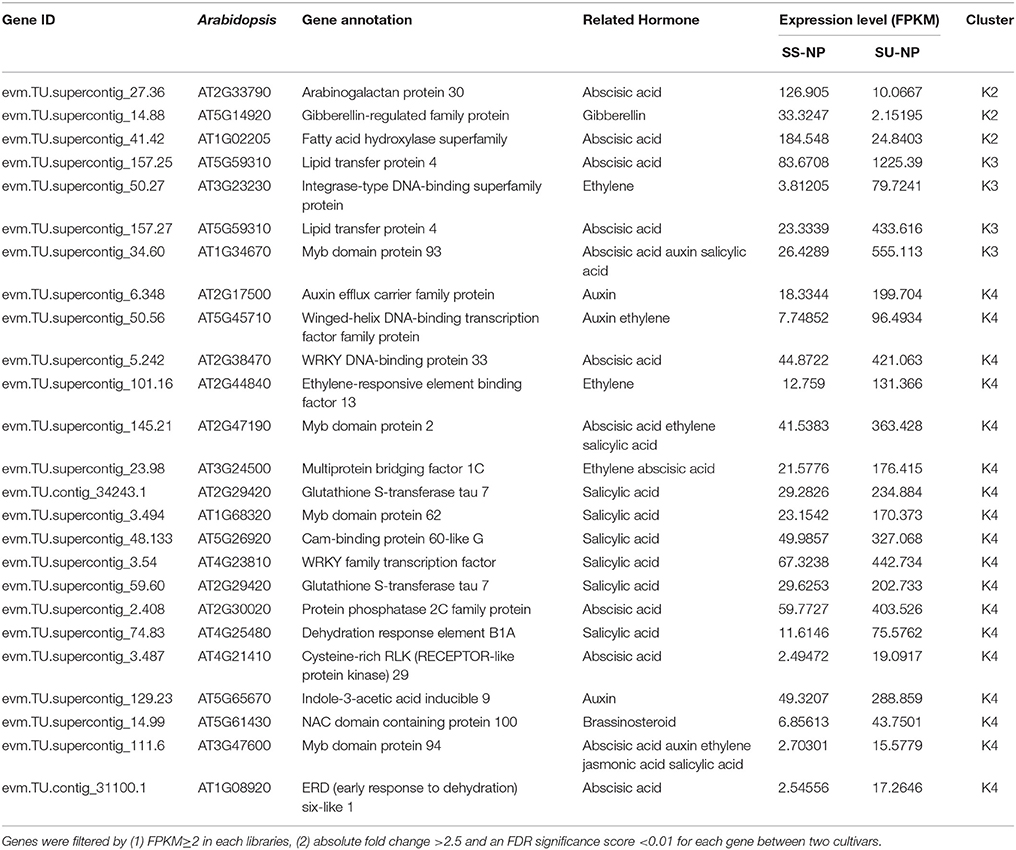
Table 5. The most abundantly differentially expressed hormone-related genes between two cultivars.
Confirmation of Candidate DEGs by qRT-PCR Analysis
To confirm the accuracy of Illumina RNA-Seq results, a subset of 21 genes, which were differentially expressed between non-GE and GE papaya, was chosen for quantitative real-time PCR analysis. The primer sequences, gene annotations, and FPKM and qRT-PCR expression values were listed in Tables S7–S9. Although the fold-changes in transcript abundance determined by RNA-Seq and qRT-PCR did not exactly match, all of the 21 qRT-PCR analyses showed trends of expression (up- or down-regulation) similar to those revealed by RNA-Seq results (Figure 5). The expression patterns from the two platforms were largely consistent, with a Pearson's correlation coefficient of 0.9247.
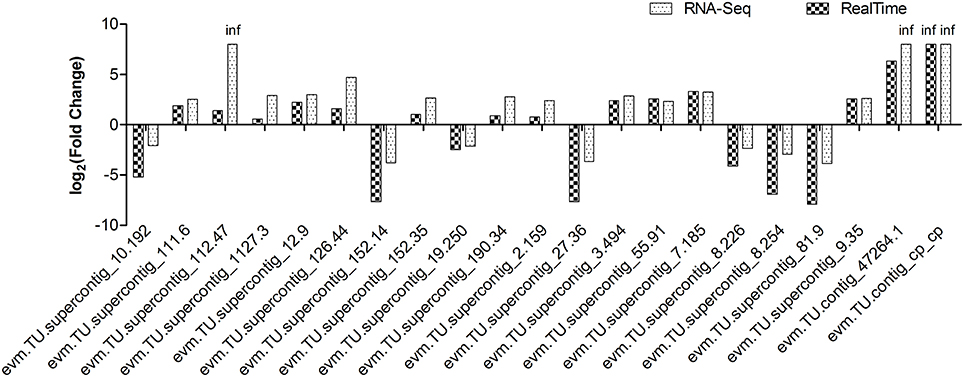
Figure 5. Expression pattern validation of 21 selected DEGs by qRT-PCR in transgenic SunUp relative to the donor control Sunset. The transcriptional level of candidate genes was examined by real-time PCR with three biological replicates. EIF was used as an internal control. RNA-Seq data were highly consistent with qRT-PCR results (r = 0.9247). The Y-axis indicates the fold change of transcript abundance in SU-NP relative to the control SS-NP.
Alternative Splicing of Transcripts in the Two Papaya Cultivars
To investigate the patterns of AS in non-GE and GE papaya, we developed a series of in-house Perl scripts that could detect the variations in the splicing structure and identify AS events by extracting information from the output of TopHat. In this study, we focused on six types of AS events: SE, RI, A5SS, A3SS, 5UTR, and 3UTR (Figure 6, Table 6), by comparing transcripts with the papaya annotated gene model. Among the detected AS events, SE was the most abundant type (53%), followed by A3SS (17%), A5SS (10%), 3UTR (10%), 5UTR (7%), and RI (3%) (Table 6). In total, we identified 67,686 AS events in Sunset and 68,455 AS events in SunUp. However, the number of genes exhibiting AS events in both cultivars was similar, mapping to 10,994 and 10,995 papaya annotated genes, respectively. Around 610 genes were specifically alternatively spliced in Sunset and 611 genes in SunUp (Figure 7A).
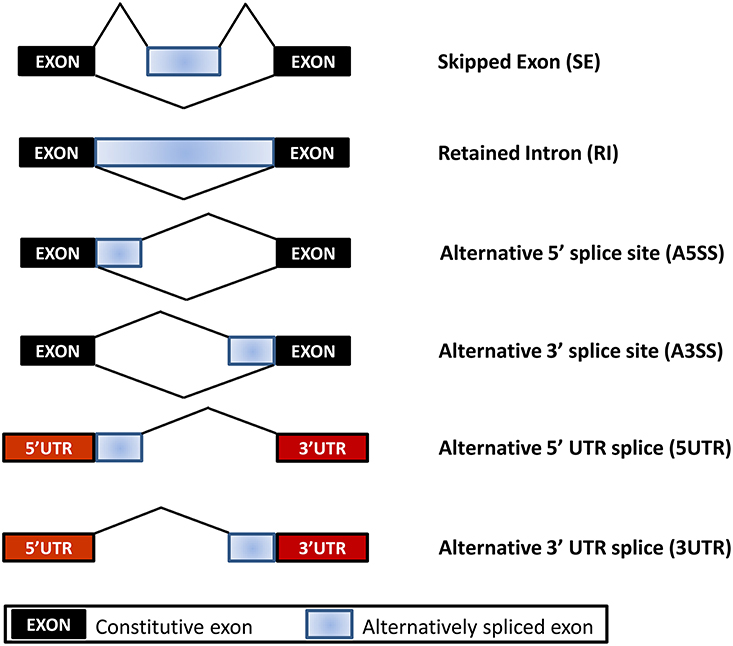
Figure 6. Types of Alternative Splicing Events. Skipped Exon (SE): one or more exons are spliced out of the mature messenger RNA (mRNA). Retained Intron (RI): one or more introns are included in the mature mRNA; Alternative 5′ (or 3′) splice site (A5SS or A3SS): distinct 5′ (or 3′) splice site corresponds to a longer or shorter exon, which are particularly difficult to interrogate by microarray analysis due to the small variably included region; 5UTR or 3UTR: alternative promoter use results in shorter or longer 5′ UTR or 3′ UTR isoforms.
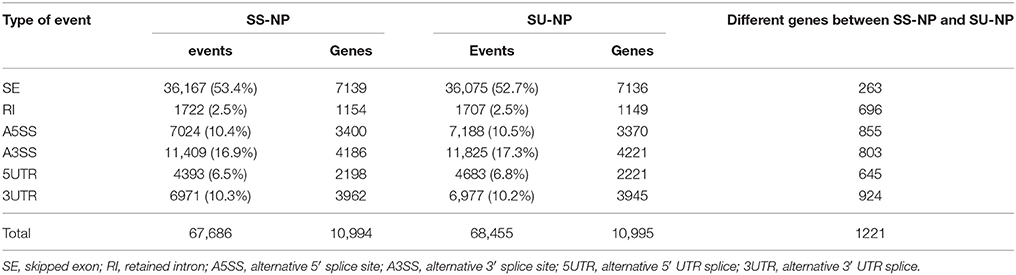
Table 6. Summary of the six types of alternative splicing events of papaya Sunset and SunUp.
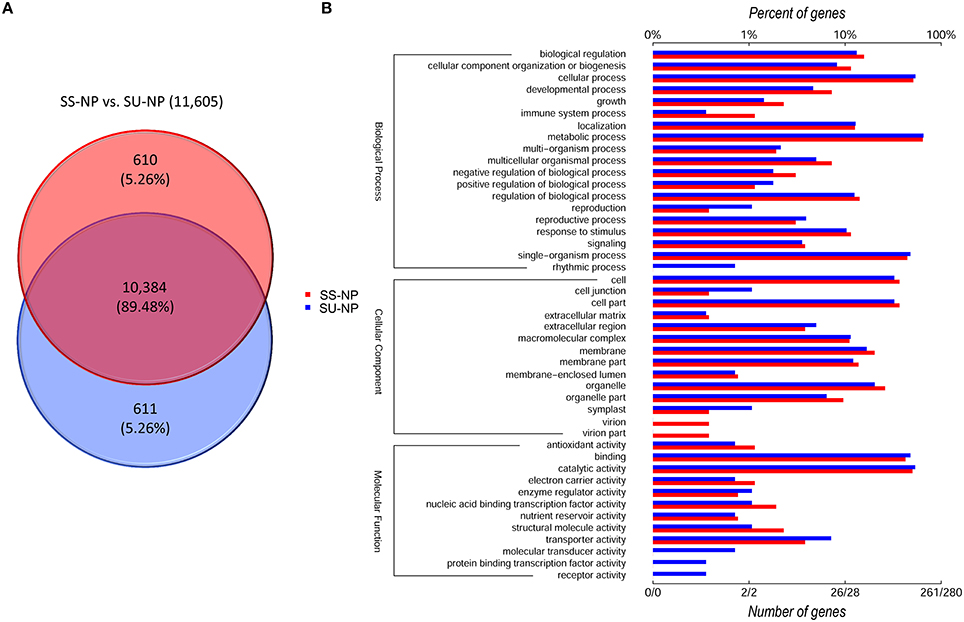
Figure 7. Venn diagram and functional enrichment analysis of genes showing AS events between PRSV-negative Sunset and SunUp. (A) Unique and shared genes between two varieties. (B) Level 2 GO annotation of variety-specific genes undergoing AS events comparison between Sunset and SunUp. We divided the sets into three major GO ontologies: biological process, cellular component and molecular function. Red and blue bars represent the number and percentage of genes specific in Sunset and SunUp, respectively.
We further classified those genes that underwent AS events on the basis of gene ontology annotation into three component ontologies: molecular function (MF), biological process (BP) and cellular component (CC) (Figure 7B, Tables S10A,B). In both cultivars, the molecular function ontology included a high portion of catalytic activity (50.19%, 53.57%) and protein binding (42.53%, 47.86%). With regard to the cellular component, 73.56% (Sunset) and 65% (SunUp) were assigned to cell or cell part followed by membrane, and organelle. In the biological process ontology, metabolic process (64.37%, 65.36%), cellular process (51.34%, 53.93%) and single-organism process (44.44%, 47.86) were the top three most dominant groups. It is noteworthy that some GO annotation for the genes displaying AS events exclusively in Sunset was significantly different from that in SunUp. For instance, together 0.77% (evm.TU.supercontig_4800.1) was assigned to virion or virion part in Sunset while no corresponding GO terms were assigned in SunUp. Genes involved in immune system process (BP), structural molecule activity (MF) and nucleic acid binding transcription factor activity (MF) in Sunset were much more abundant than those in SunUp. Conversely, compared to assigned GO terms in Sunset, four additional annotations were found in SunUp: protein binding transcription factor activity, molecular transducer activity, and receptor activity in category of molecular function, and rhythmic process in biological process. Terms such as reproduction (BP), cell junction (CC), symplast (CC), and transporter activity (MF) enriched a much higher percentage of genes in SunUp than that in its progenitor cultivar Sunset.
Discussion
The primary objective of this study was to identify genome-wide perturbations of steady state levels of transcripts in young healthy leaves of transgenic papaya subjected to bombardment and expression of the PRSV CP transgene. Here, we carried out paired end sequencing of RNA-Seq libraries prepared from mRNA isolated from 4-month old leaves of SunUp and its progenitor Sunset grown under the same conditions prior to infection with PRSV. High throughput sequencing generated more than 342 million filtered reads and nearly 80% of the total reads were uniquely mapped to the papaya reference genome. Subsequently, resultant transcriptome of unique reads were used in the downstream analysis of expression (FPKM values), gene ontologies and other functional categories. Results showed that the expression profiles of multiple genes are altered in transgenic cultivar SunUp and these transcripts are components of important disease resistant processes, hormonal signaling pathways and regulatory networks. The detected expression patterns from two platforms, RNA-Seq and qRT-PCR, were highly consistent (Figure 5), confirming the accuracy and reliability of the RNA-Seq results.
Several Factors Responsible for Variable Gene Expression after Transgene Insertion
RNA-Seq expression profiles in SunUp and Sunset were similar, consistently with previous RNA-Seq studies, showing that gene expression follows a bimodal distribution of high and low expression genes (Hebenstreit et al., 2011). Studies have demonstrated that highly expressed genes are active genes performing the necessary cellular processes while other genes are likely to be the by-products of biological or experimental noise (Hebenstreit et al., 2011; Hart et al., 2013). Approximately 94% of mapped genes were shared by the transcriptomes of Sunset and SunUp with other 620 and 662 specifically expressed genes, respectively (Figure 1D). After strict filtering criteria, out of all mapped genes, only 842 DEGs were identified, 475 of which were up-regulated in SunUp and 367 down-regulated (Figure 2). Several factors were responsible for the identified variations in gene expression between transgenic cultivar SunUp and its progenitor Sunset.
The introduction of PRSV CP transgene and other unexpected DNA fragments was considered to be a major causal effect of differential gene expression. The faithful execution of cp gene expression might require a precise and carefully orchestrated set of steps that depend on the proper cooperation of the molecular machinery (general transcription factors, activators and coactivators). On the other hand, transgene integration position within the genome is associated with variable gene expression (Matzke and Matzke, 1998). The insertions of the exogenous gene or the unintended DNA fragments could probably cause a mutation and altered expression of the gene into which it inserts. Or the insertions might muck up the various classes of transcriptional regulatory elements adjacent to the genes that they regulate, such as core/proximal promoters, distal enhancers, silencers, insulators/boundary elements, and locus control regions (LCR) (Maston et al., 2006), hence affecting the controlled patterns of gene expression. Three transgenic insertions with flanking border sequences were able to be identified from corresponding (bacterial artificial chromosome) BAC clones or sequences reads from the whole-genome shotgun (WGS) sequence of SunUp papaya, and the number and types of inserts were subsequently confirmed by Southern analysis using probes spanning the entire transformation plasmid (Suzuki et al., 2008). However, the organelle-like borders of three transgenic insertions make them difficult to be assembled and located into SunUp reference genome because nuclear organelle DNA (norgDNA) are abundance in nuclear genomes (Martin et al., 2002; Shahmuradov et al., 2003). In present study, the lack of information on the precise location of the integrated region kept us from thorough and systematical examination of their impact on the function of interrupted genes and neighboring genes. From this point of view, a completely sequenced papaya genome in the near future will complement the insufficiency of the present study. In addition to these three large insertions, multiple copies of the three insertions or some other small insertions of a few base pairs (< 20 bp) induced by particle bombardment may exist but they are below the detection limit of PCR and Southern blot analysis which are the standard methods used to detect integration of vector sequences (Endo et al., 2015). Increased transgene copy number can result in higher or lower expression level (Sijen et al., 1996; Wang and Waterhouse, 2000; Altpeter et al., 2005), and the existence of small undetected exogenous sequences may also have some profound effects on gene expression levels and/or protein function.
Besides transgene copy numbers and integration position effects, other factors such as somaclonal variations (Phillips et al., 1994; Duncan, 1996; Kaeppler et al., 2000) and spontaneous mutations during meiosis (Magni, 1963; Golubovskaya et al., 1992; Lercher and Hurst, 2002) of over 20 generations might induce segregated SNPs, InDels, and rearrangement, which would lead to the divergence of gene expression between GE and non-GE papaya cultivars. Additionally, sources of bias and error, such as technical variability during library preparation and sequencing (Hansen et al., 2010; Roberts et al., 2011), biological variability between replicates of the same sample (Trapnell et al., 2012), and the inevitable error rates during reconstruction of all full-length transcripts from short reads (Li et al., 2010; Grabherr et al., 2011), could also account for this difference. The random distribution of DEGs amongst papaya chromosomes (Table 2) suggests that bombardment-mediated transformation might have a genome-wide effect on the papaya genome, not just specifically affecting the flanking sequences of insertion sites.
Enrichment of Disease Resistance Genes in DEGs
With respect to gene ontologies, 10 GO terms were assigned exclusively to down-regulated genes and two terms to up-regulated genes (Figure 3A, and Tables S1A,B). This finding revealed that unique cellular functions have been created in the transgenic cultivar in acquiring resistance to PRSV, and these two cultivars could already have developed different genetic pathways for adaptation to the environment even at an early growth stage. For instance, the up-regulated gene in the SunUp genome associated with the “virion part” or “virion” was found to be a coat protein (cp) gene, which was the target gene derived from PRSV HA 5-1 that was bombarded into the papaya genome using gene gun DNA particle bombardment. Previous studies on virus-derived resistance in transgenic tobacco plants revealed that transgenic tobacco plants expressed high levels of the TMV CP and were more resistant to TMV virions than to TMV RNA inoculations (Abel et al., 1986). CP-mediated protection (CPMP) against TMV occurs through the inhibition of virion disassembly in the originally infected cells (Register and Beachy, 1988). There is limited information regarding the developmental stages where a transgenic plant is capable of performing RNA silencing. An intriguing finding has shown that high levels of transgene-derived short interfering RNAs (siRNAs), the key constituents in PTGS/RNAi triggering for the process which leads to degradation of homologous RNAs (Hutvágner and Zamore, 2002), were generated in mature transgenic potato plants (Solanum tuberosum L.) (at least 2 months old) prior to potato virus Y (PVY) inoculation, and the concentration further increased as the plants grew (Missiou et al., 2004). We used 4-month-old plants in this study, allowing sufficient time for the siRNAs to be generated and have initiated biological processes that differed from those in non-transgenic plants. Three up-regulated genes, evm.TU.supercontig_120.27, evm.TU.supercontig_1.397, and evm.TU.supercontig_48.133, were involved exclusively in the assigned term “immune system process” in SunUp transcriptomes. Their functions are ammonium transporter 2 (AMT2), protein of unknown function (DUF506), and calmodulin-binding protein 60-like (CBP60g) annotated in Arabidopsis and Pfam database, respectively (data not shown), suggesting those genes play roles in defending the PRSV disease. CBP60g, which belongs to a new family of transcription factors triggered by microbe-associated molecular patterns (Vlot et al., 2009; Wang et al., 2009; Wan et al., 2012) was found in many studies to be a positive regulator of disease resistance via the SA signaling pathway. GO enrichment analysis reflected that microtubule-related categories were highly enriched among these DEGs, followed by polysaccharide catabolic and sucrose metabolic processes (Table S2). These enriched processes mostly take place in the “extracellular region” and “cell wall.” Microtubules are one of the major components of the cytoskeleton, and found to carry out a wide range of functions in structural support, intracellular transport, cell motility and DNA segregation and other fundamental biological functions (Vale, 1987; Mandelkow and Mandelkow, 1995; Nogales, 2001; Wittmann and Waterman-Storer, 2001; Garner et al., 2004). Microtubules often interact with three main classes: the microtubule-associated proteins (MAPs), the motor proteins and proteins that are not normally called MAPs but often found associated with microtubules and may even copurify with them (Mandelkow and Mandelkow, 1995). Representatives are MAP1a and 1b, MAP2a, 2b, and 2c, MAP4, tau protein, kinesin, dynein, glycolytic enzymes and kinases. The finding of enriched microtubule-related categories prompted a hypothesis that microtubule-associated proteins (MAPs), motor proteins and other related proteins are activated in response to the structural changes caused by particle bombardment to stabilize cellular structures. Numerous genetic studies have provided new lines of evidence implicating that cell wall polysaccharides function as latent signal molecules during pathogen infection and elicit defense responses by the plant (Vorwerk et al., 2004). A report has also revealed that sucrose accumulated at higher levels in leaves of disease resistance transgenic rice (Oryza sativa L.), and pretreatment of rice plants with sucrose was able to enhance resistance to M. oryzae infection, supporting a sucrose-mediated priming of defense responses in transgenic rice which results in broad-spectrum protection against pathogens (Gómez-Ariza et al., 2007). The enrichment in GO terms “polysaccharide catabolic process” and “sucrose metabolic process” suggested this potential relationship between disease resistance and sucrose metabolism in the transgenic cultivar SunUp.
Based on the KEGG enrichment analysis, no significant pathways were detected with the FDR threshold less than 0.01, indicating that the transgenic cultivar SunUp did not undergo major changes upon DNA particle bombardment. Whereas seven pathways including phenylpropanoid biosynthesis, cutin, suberine and wax biosynthesis, and starch and sucrose metabolism and others were overrepresented for the FDR cut-off of 0.05 (Figure 3B, Table S3). Some previous observations provided direct evidence that natural products synthesized by plants such as phenylpropanoid products contribute to their resistance to pathogens, and this increased resistance is a pathogen-induced response but dependent on developmental accumulation of phenylpropanoid products (Maher et al., 1994). Hence, the overrepresented pathway “phenylpropanoid biosynthesis” pathway could be explained by the increased disease susceptibility of non-transgenic cultivar via synthesizing lower levels of phenylpropanoid products during development compared with its transgenic cultivar.
The Key Regulons (TFs, TPs, and HRGs) Induce Defense Response
The activities of transporters and transcription factors are the two main categories in the molecular function based on GO analysis of DEGs. The increased vigor and growth and the nature of high resistance to PRSV in SunUp indicate that SunUp and Sunset cultivars must sense and respond to their environment differently in a complex manner, through signaling and regulatory pathways mediated by phytohormones such as jasmonic acid (JA) and salicylic acid, generally resulting in altered expression of transcription factors and in enhanced or repressed expression of genes encoding related proteins. Hence, the expression patterns of TFs, TPs, and HRGs were dissected in this study to detect their changes in expression after bombardment transformation and under years of the continuous challenge of virus inoculations by the natural aphid vector.
The highly variable expression patterns of TFs, TPs, and HRGs reflected the significant changes of expression that occurred upon transgene insertion and that may play critical roles in plant resistance to pathogens. Transcription factors, interacting with the transcriptional regulatory elements (enhancers, promoters, silencers, insulators, and LCR regions) adjacent to the genes that they regulate, have the ability to control the expression of many downstream genes to regulate diverse biological processes. A slight alteration in transcript abundance of TFs can trigger a cascade of reactions implicated in many physiological processes resulting in a substantial change in downstream gene expression (Ramirez and Basu, 2009; Wang et al., 2013b). Hence, the transcriptional level of the gene is either up- or down-regulated depending on the adjacent transcription factor. Numerous transcription factor families were detected to be induced or suppressed after transgene insertion (Figure 4B). Several families of the TFs, such as MYB, WRKY, ERF, NAC, kinase and zinc-finger, which are known key regulators in the defense response (Xie et al., 1999; Gutterson and Reuber, 2004; Liu et al., 2004; Ryu et al., 2006; Eulgem and Somssich, 2007; Guo et al., 2009; Nuruzzaman et al., 2010, 2013), were most highly expressed in transgenic papaya (Table 3). Silencing of genes encoding the mitogen-activated protein kinase (MAPK) NTF6 or the MAPK kinase MEK1 attenuated tobacco N-mediated resistance to TMV, where the defense response NbWRKY1-NbWRKY3 and NbMYB1 transcription factors are also involved (Liu et al., 2004). A large number of WRKY DNA-binding proteins are implicated in the transcriptional activation of defense related genes in response to pathogens (Ryu et al., 2006; Eulgem and Somssich, 2007). A systematic expression analysis of WRKYs in rice revealed that the expression of 15 out of 45 WRKY genes significantly increased in an incompatible rice-pathogen interaction (Ryu et al., 2006). The expression of two and three WRKY genes increased in response to SA- and JA- treatments, respectively. CAZFP1, a C2H2-type zinc-finger transcription factor, functions as a pathogen-induced early-defense gene to enhance disease resistance (Kim et al., 2004). Overexpression of the CAZFP1 in stably transformed Arabidopsis plants enhanced the resistance against infection by Pseudomonas syringae pv. tomato. A novel CCCH-type zinc finger protein GhZFP1 from cotton also enhances fungal disease resistance in transgenic tobacco by interacting with two other proteins (Guo et al., 2009). The expression of ERF and NAC genes is regulated by plant hormones, such as JA, SA and ethylene, and several lines of research have confirmed that they participate in the regulation of disease resistance pathways (Gutterson and Reuber, 2004; He et al., 2005; Xia et al., 2010a,b; Nuruzzaman et al., 2012, 2013; Zheng et al., 2012). In rice seedlings, 19 NAC genes displayed higher expression levels after PSV infection, and 13 NAC genes upon RTSV infection (Nuruzzaman et al., 2010). Several NAC proteins can act as repressors as well as activators in virus replication by directly interacting with virus-encoded proteins (Xie et al., 1999; Ren et al., 2000; Selth et al., 2005). The above findings imply that different types of transcription factors have developed a novel significant way in regulating the expression of a large set of immune-related genes, which are activated by defense responses to PRSV in transgenic papaya after years of plant-pathogen interactions. Numerous DEGs were also predicted to be hormone-related genes in this study. This finding corroborates earlier findings, which showed that the phytohormones ethylene, SA and JA play a central role in the regulation of plant immune responses (Vlot et al., 2009; Robert-Seilaniantz et al., 2011). Suppression of the JA signaling component COI1 ortholog affected tobacco N resistance (Liu et al., 2004). Other plant hormones that have been thoroughly described to function in the regulation of plant growth, development and the response to abiotic stresses, such as auxins, ABA, brassinosteroid, gibberellin, and cytokinin, have recently emerged as crucial regulators of plant immunity (Mauch-Mani and Mauch, 2005; Kazan and Manners, 2009; Ton et al., 2009; Wang and Fu, 2011). All these lines of evidence might suggest that hormonal signaling involving crosstalk between ABA, SA, ethylene, auxin and other hormones may be essential in the papaya pathogen response.
Transport proteins, embedded within plasma membrane, cytoplasm or nucleus, function in both active and passive transport to regulate molecules moving in and out of cell. Some ABC transporters are known to play a role in resistance to pathogens (Krattinger et al., 2009; Wang et al., 2013a; Goyer et al., 2015). In barley, ABC transporters are highly expressed upon inoculation with barley yellow dwarf virus (Wang et al., 2013a). The down-regulation of ABC transporters in the papaya transgenic cultivar SunUp might suggest that fewer ABC transporters were implicated in transgenic papaya since its divergence from non-transgenic papaya Sunset due to the inherent resistance of SunUp. Many nitrate and zinc transporters were highly expressed in SunUp compared to that in Sunset. Nitrogen is the mineral nutrient needed in greatest abundance by plants because it is a major component of amino acids, nucleic acids, ATP (adenosine triphosphate) and chlorophyII (Crawford, 1995). Zinc is needed in small but if its amount is not adequate, plants will suffer from physiological stress brought about by the dysfunction of several enzyme systems and other metabolic functions in which zinc plays a vital part (Alloway, 2004). Both nitrate and zinc are essential for the normal healthy growth of plants. Therefore, the better growth of SunUp plants (Figure S1A) could be easily explained by the up-regulation of nitrate and zinc transporters. Notably, two genes, homologous to the Arabidopsis thaliana zinc transporter 5 precursor and nodulin MtN21, were found to be exclusively expressed in SunUp, suggesting these transgenic plant lines may have induced new transporters for environmental adaptation. The role of nodulin MtN21-like genes is not yet known. The maize nodulin MtN21-like gene is probably involved in transport of a component related to vascular tissue assembly (Guillaumie et al., 2008).
Increased Rate of as in the PRSV CP Transgene in Stress Response
Alternative splicing is a regulated process during gene expression that results in the production of multiple mRNAs from a single gene by variable selection of splice sites in the precursor-mRNA. RNA-Seq is a proven high-throughput and cost-saving technology that identifies AS events. In our analysis, we focused on six types of AS events: SE, RI, A5SS, A3SS, 5UTR, and 3UTR, among which, SE was the most abundant type, accounting for more than 50% of total AS events. A greater number of AS events were found in SunUp compared to Sunset (68,455 vs. 67,686), while the number of genes undergoing AS in both cultivars was similar. These findings suggest that ~39% of papaya genes potentially undergo the AS process, but AS events occurring in papaya increased upon PRSV CP transgene insertion. This observed percentage of AS events in papaya (39%) is comparable to that observed in A. thaliana (42%) and much higher than rice (33%) (Wang and Brendel, 2006; Simpson et al., 2008; Syed et al., 2012). 610 genes in Sunset and 611 genes in SunUp were exclusively alternatively spliced (Figure 7A), indicating that transgenic papaya diverged from its progenitor to undergo exclusive alternatively splicing events in responses to biotic and abiotic stresses. The biological role of AS in photosynthesis, defense responses and grain quality in rice have been verified (Reddy, 2007). More than 60% of intron-containing genes undergo alternative splicing in plants (Syed et al., 2012). Nevertheless, little is known of the alternative splicing of papaya genes at the genome-wide level. Despite having a significantly larger genome than Arabidopsis thaliana, it is estimated that papaya has fewer genes than other sequenced genomes of flowering plants (Ming et al., 2008, 2012). This is partly due to the absence of further genome wide duplication detected in papaya since the ancient triplication event shared by eudicots. The small set of genes in papaya implies that papaya may have a relatively higher frequency of AS events to synthesize various types of proteins to maintain basic functions during development and in response to environmental signals compared to other plants. A nonlinear correlation between NBS gene number and total gene number revealed that papaya has significantly fewer NBS genes (~0.2% of total genes), which could explain the susceptibility of papaya to multiple pathogens (Nishijima, 2002; Porter et al., 2009). Intriguingly, numerous splice variants were identified in the papaya NBS gene family, suggesting that alternative splicing may play a vital role in contributing to the diversity of NBS-encoding genes.
Conclusions
This study is the first large scale transcriptome RNA-Seq analysis comparing a PRSV resistant transgenic papaya SunUp and its progenitor cultivar Sunset. Genome-wide transcriptional profiling and analysis of alternative splicing events in the present work indicated that most biological processes remain shared by the two cultivars SunUp and Sunset. Numerous DEGs were identified, of which up-regulated genes were mainly related to stress tolerance and pathogen resistance, including various TFs, TPs as well as genes involved in hormone signaling pathways. These findings demonstrated that transgenic papaya has the potential to improve the abiotic and biotic tolerance in addition to PRSV resistance. The differential expressions of candidate DEGs inferred from RNA-Seq were confirmed by qRT-PCR, demonstrating the accuracy and reliability of the RNA-Seq data. GO and KEGG enrichment analysis established that two cultivars trigger a different cascade of molecular changes, deepening the understanding of the complex molecular and cellular events during various biological pathways in these two cultivars. Further investigations will be needed to elucidate the specific mechanisms of genes associated with TFs, TPs, and HRGs and of genes belonging to GO terms/KEGG pathways significantly enriched. These genes involved in pathways of interest may be important targets for biotechnological manipulation to improve papaya stress tolerance and disease resistance, particularly in the early development stages.
Deposited Data
The Illumina RNA-sequencing raw reads of PRSV susceptible cultivar Sunset and PRSV resistant transgenic papaya cultivar SunUp are available from the NCBI Sequence Read Archive database (SRA; http://www.ncbi.nlm.nih.gov/sra/) under project Accession number of SRP075196.
Author Contributions
RM and RC conceived the study and designed the experiments. JF, AL, WQ, HC, and MU carried out the experiments and analyzed the data. JF and RM wrote the manuscript. All authors read and approved the final paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a startup fund from Fujian Agriculture and Forestry University to RM and Fujian Provincial Key Laboratory of Haixia Applied Plant Systems Biology.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.00855
References
Abel, P. P., Nelson, R. S., De, B., Hoffmann, N., Rogers, S. G., Fraley, R. T., et al. (1986). Delay of disease development in transgenic plants that express the tobacco mosaic virus coat protein gene. Science 232, 738–743. doi: 10.1126/science.3457472
Altpeter, F., Baisakh, N., Beachy, R., Bock, R., Capell, T., Christou, P., et al. (2005). Particle bombardment and the genetic enhancement of crops: myths and realities. Mol. Breed. 15, 305–327. doi: 10.1007/s11032-004-8001-y
Arumuganathan, K., and Earle, E. (1991). Nuclear DNA content of some important plant species. Plant Mol. Biol. Rep. 9, 208–218. doi: 10.1007/BF02672069
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Crawford, N. M. (1995). Nitrate: nutrient and signal for plant growth. Plant Cell 7, 859–868. doi: 10.1105/tpc.7.7.859
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Duncan, R. (1996). Tissue culture-induced variation and crop improvement. Adv. Agron. 58, 201–240. doi: 10.1016/S0065-2113(08)60256-4
Endo, M., Kumagai, M., Motoyama, R., Sasaki-Yamagata, H., Mori-Hosokawa, S., Hamada, M., et al. (2015). Whole-genome analysis of herbicide-tolerant mutant rice generated by Agrobacterium-mediated gene targeting. Plant Cell Physiol. 56, 116–125. doi: 10.1093/pcp/pcu153
Eulgem, T., and Somssich, I. E. (2007). Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 10, 366–371. doi: 10.1016/j.pbi.2007.04.020
Fermín, G., Keith, R. C., Suzuki, J. Y., Ferreira, S. A., Gaskill, D. A., Pitz, K. Y., et al. (2011). Allergenicity assessment of the papaya ringspot virus coat protein expressed in transgenic rainbow papaya. J. Agric. Food Chem. 59, 10006–10012. doi: 10.1021/jf201194r
Fitch, M. M., Manshardt, R. M., Gonsalves, D., Slightom, J. L., and Sanford, J. C. (1990). Stable transformation of papaya via microprojectile bombardment. Plant Cell Rep. 9, 189–194. doi: 10.1007/BF00232177
Fitch, M. M., Manshardt, R. M., Gonsalves, D., Slightom, J. L., and Sanford, J. C. (1992). Virus resistant papaya plants derived from tissues bombarded with the coat protein gene of papaya ringspot virus. Nat. Biotechnol. 10, 1466–1472. doi: 10.1038/nbt1192-1466
Garner, E. C., Campbell, C. S., and Mullins, R. D. (2004). Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science 306, 1021–1025. doi: 10.1126/science.1101313
Golubovskaya, I., Avalkina, N. A., and Sheridan, W. F. (1992). Effects of several meiotic mutations on female meiosis in maize. Dev. Genet. 13, 411–424. doi: 10.1002/dvg.1020130605
Gómez-Ariza, J., Campo, S., Rufat, M., Estop,à, M., Messeguer, J., Segundo, B. S., et al. (2007). Sucrose-mediated priming of plant defense responses and broad-spectrum disease resistance by overexpression of the maize pathogenesis-related PRms protein in rice plants. Mol. Plant Microbe Interact. 20, 832–842. doi: 10.1094/MPMI-20-7-0832
Gonsalves, D. (1998). Control of papaya ringspot virus in papaya: a case study. Annu. Rev. Phytopathol. 36, 415–437. doi: 10.1146/annurev.phyto.36.1.415
Goyer, A., Hamlin, L., Crosslin, J. M., Buchanan, A., and Chang, J. H. (2015). RNA-Seq analysis of resistant and susceptible potato varieties during the early stages of potato virus Y infection. BMC Genomics 16:472. doi: 10.1186/s12864-015-1666-2
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guillaumie, S., Goffner, D., Barbier, O., Martinant, J.-P., Pichon, M., and Barrière, Y. (2008). Expression of cell wall related genes in basal and ear internodes of silking brown-midrib-3, caffeic acid O-methyltransferase (COMT) down-regulated, and normal maize plants. BMC Plant Biol. 8:71. doi: 10.1186/1471-2229-8-71
Guo, Y. H., Yu, Y. P., Wang, D., Wu, C. A., Yang, G. D., Huang, J. G., et al. (2009). GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 183, 62–75. doi: 10.1111/j.1469-8137.2009.02838.x
Gutterson, N., and Reuber, T. L. (2004). Regulation of disease resistance pathways by AP2/ERF transcription factors. Curr. Opin. Plant Biol. 7, 465–471. doi: 10.1016/j.pbi.2004.04.007
Hansen, K. D., Brenner, S. E., and Dudoit, S. (2010). Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 38:e131. doi: 10.1093/nar/gkq224
Hart, T., Komori, H. K., Lamere, S., Podshivalova, K., and Salomon, D. R. (2013). Finding the active genes in deep RNA-seq gene expression studies. BMC Genomics 14:778. doi: 10.1186/1471-2164-14-778
He, X. J., Mu, R. L., Cao, W. H., Zhang, Z. G., Zhang, J. S., and Chen, S. Y. (2005). AtNAC2, a transcription factor downstream of ethylene and auxin signaling pathways, is involved in salt stress response and lateral root development. Plant J. 44, 903–916. doi: 10.1111/j.1365-313X.2005.02575.x
Hebenstreit, D., Fang, M., Gu, M., Charoensawan, V., Van Oudenaarden, A., and Teichmann, S. A. (2011). RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol. Syst. Biol. 7:497. doi: 10.1038/msb.2011.28
Hutvágner, G., and Zamore, P. D. (2002). RNAi: nature abhors a double-strand. Curr. Opin. Genet. Dev. 12, 225–232. doi: 10.1016/S0959-437X(02)00290-3
Jiang, Z., Liu, X., Peng, Z., Wan, Y., Ji, Y., He, W., et al. (2011). AHD2. 0: an update version of Arabidopsis Hormone Database for plant systematic studies. Nucleic Acids Res. 39, D1123–D1129. doi: 10.1093/nar/gkq1066
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Kaeppler, S. M., Kaeppler, H. F., and Rhee, Y. (2000). Epigenetic aspects of somaclonal variation in plants. Plant Mol. Biol. 43, 179–188. doi: 10.1023/A:1006423110134
Kazan, K., and Manners, J. M. (2009). Linking development to defense: auxin in plant-pathogen interactions. Trends Plant Sci. 14, 373–382. doi: 10.1016/j.tplants.2009.04.005
Kim, S. H., Hong, J. K., Lee, S. C., Sohn, K. H., Jung, H. W., and Hwang, B. K. (2004). CAZFP1, Cys2/His2-type zinc-finger transcription factor gene functions as a pathogen-induced early-defense gene in Capsicum annuum. Plant Mol. Biol. 55, 883–904. doi: 10.1007/s11103-005-2151-0
Kohli, A., Twyman, R. M., Abranches, R., Wegel, E., Stoger, E., and Christou, P. (2003). Transgene integration, organization and interaction in plants. Plant Mol. Biol. 52, 247–258. doi: 10.1023/A:1023941407376
Krattinger, S. G., Lagudah, E. S., Spielmeyer, W., Singh, R. P., Huerta-Espino, J., Mcfadden, H., et al. (2009). A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science 323, 1360–1363. doi: 10.1126/science.1166453
Latham, J. R., Wilson, A. K., and Steinbrecher, R. A. (2006). The mutational consequences of plant transformation. BioMed. Res. Intern. 2006, 7. doi: 10.1155/jbb/2006/25376
Lercher, M. J., and Hurst, L. D. (2002). Human SNP variability and mutation rate are higher in regions of high recombination. Trends Genet. 18, 337–340. doi: 10.1016/S0168-9525(02)02669-0
Li, B., Ruotti, V., Stewart, R. M., Thomson, J. A., and Dewey, C. N. (2010). RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500. doi: 10.1093/bioinformatics/btp692
Liu, Y., Schiff, M., and Dinesh-Kumar, S. (2004). Involvement of MEK1 MAPKK, NTF6 MAPK, WRKY/MYB transcription factors, COI1 and CTR1 in N-mediated resistance to tobacco mosaic virus. Plant J. 38, 800–809. doi: 10.1111/j.1365-313X.2004.02085.x
Magni, G. (1963). The origin of spontaneous mutations during meiosis. Proc. Natl. Acad. Sci. U.S.A. 50, 975–980. doi: 10.1073/pnas.50.5.975
Maher, E. A., Bate, N. J., Ni, W., Elkind, Y., Dixon, R. A., and Lamb, C. J. (1994). Increased disease susceptibility of transgenic tobacco plants with suppressed levels of preformed phenylpropanoid products. Proc. Natl. Acad. Sci. U.S.A. 91, 7802–7806. doi: 10.1073/pnas.91.16.7802
Mandelkow, E., and Mandelkow, E.-M. (1995). Microtubules and microtubule-associated proteins. Curr. Opin. Cell Biol. 7, 72–81. doi: 10.1016/0955-0674(95)80047-6
Manshardt, R. M. (1999). UH Rainbow' Papaya. New plants for Hawaii-1: University of Hawaii, College of Tropical Agriculture and Human Resources 1, 2.
Manshardt, R., and Wenslaff, T. (1989). Interspecific hybridization of papaya with other Carica species. J. Am. Soc. Horticult. Sci. 114, 689–694.
Martin, W., Rujan, T., Richly, E., Hansen, A., Cornelsen, S., Lins, T., et al. (2002). Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. U.S.A. 99, 12246–12251. doi: 10.1073/pnas.182432999
Maston, G. A., Evans, S. K., and Green, M. R. (2006). Transcriptional regulatory elements in the human genome. Annu. Rev. Genomics Hum. Genet. 7, 29–59. doi: 10.1146/annurev.genom.7.080505.115623
Matzke, A. J., and Matzke, M. A. (1998). Position effects and epigenetic silencing of plant transgenes. Curr. Opin. Plant Biol. 1, 142–148. doi: 10.1016/S1369-5266(98)80016-2
Mau, R., Gonsalves, D., and Bautista, R. (1989). “Use of cross protection to control papaya ringspot virus at Waianae”, in Proc. 25th Annual Papaya Industry Association Conference (Hilo, HI), 77–84.
Mauch-Mani, B., and Mauch, F. (2005). The role of abscisic acid in plant-pathogen interactions. Curr. Opin. Plant Biol. 8, 409–414. doi: 10.1016/j.pbi.2005.05.015
Metzker, M. L. (2010). Sequencing technologies - the next generation. Nat. Rev. Genet. 11, 31–46. doi: 10.1038/nrg2626
Ming, R., Hou, S., Feng, Y., Yu, Q., Dionne-Laporte, A., Saw, J. H., et al. (2008). The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996. doi: 10.1038/nature06856
Ming, R., and Moore, P. H. (2014). Genetics and Genomics of Papaya. New York, NY: Springer. doi: 10.1007/978-1-4614-8087-7
Ming, R., Yu, Q., Moore, P. H., Paull, R. E., Chen, N. J., Wang, M.-L., et al. (2012). Genome of papaya, a fast growing tropical fruit tree. Tree Genet. Genomes 8, 445–462. doi: 10.1007/s11295-012-0490-y
Missiou, A., Kalantidis, K., Boutla, A., Tzortzakaki, S., Tabler, M., and Tsagris, M. (2004). Generation of transgenic potato plants highly resistant to potato virus Y (PVY) through RNA silencing. Mol. Breed. 14, 185–197. doi: 10.1023/B:MOLB.0000038006.32812.52
Morozova, O., and Marra, M. A. (2008). Applications of next-generation sequencing technologies in functional genomics. Genomics 92, 255–264. doi: 10.1016/j.ygeno.2008.07.001
Nogales, E. (2001). Structural insights into microtubule function. Annu. Rev. Biophys. Biomol. Struct. 30, 397–420. doi: 10.1146/annurev.biophys.30.1.397
Nuruzzaman, M., Manimekalai, R., Sharoni, A. M., Satoh, K., Kondoh, H., Ooka, H., et al. (2010). Genome-wide analysis of NAC transcription factor family in rice. Gene 465, 30–44. doi: 10.1016/j.gene.2010.06.008
Nuruzzaman, M., Sharoni, A. M., and Kikuchi, S. (2013). Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front. Microbiol. 4:248. doi: 10.3389/fmicb.2013.00248
Nuruzzaman, M., Sharoni, A. M., Satoh, K., Moumeni, A., Venuprasad, R., Serraj, R., et al. (2012). Comprehensive gene expression analysis of the NAC gene family under normal growth conditions, hormone treatment, and drought stress conditions in rice using near-isogenic lines (NILs) generated from crossing Aday Selection (drought tolerant) and IR64. Mol. Genet. Genomics 287, 389–410. doi: 10.1007/s00438-012-0686-8
Owczarzy, R., Tataurov, A. V., Wu, Y., Manthey, J. A., Mcquisten, K. A., Almabrazi, H. G., et al. (2008). IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 36, W163–W169. doi: 10.1093/nar/gkn198
Phillips, R. L., Kaeppler, S. M., and Olhoft, P. (1994). Genetic instability of plant tissue cultures: breakdown of normal controls. Proc. Natl. Acad. Sci. 91, 5222–5226. doi: 10.1073/pnas.91.12.5222
Porter, B. W., Paidi, M., Ming, R., Alam, M., Nishijima, W. T., and Zhu, Y. J. (2009). Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Mol. Genet. Genomics 281, 609–626. doi: 10.1007/s00438-009-0434-x
Ramirez, S. R., and Basu, C. (2009). Comparative analyses of plant transcription factor databases. Curr. Genomics 10, 10–17. doi: 10.2174/138920209787581253
Reddy, A. S. (2007). Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 58, 267–294. doi: 10.1146/annurev.arplant.58.032806.103754
Register, J. C., and Beachy, R. N. (1988). Resistance to TMV in transgenic plants results from interference with an early event in infection. Virology 166, 524–532. doi: 10.1016/0042-6822(88)90523-5
Ren, Q., Chen, K., and Paulsen, I. T. (2007). TransportDB: a comprehensive database resource for cytoplasmic membrane transport systems and outer membrane channels. Nucleic Acids Res. 35, D274–D279. doi: 10.1093/nar/gkl925
Ren, T., Qu, F., and Morris, T. J. (2000). HRT gene function requires interaction between a NAC protein and viral capsid protein to confer resistance to turnip crinkle virus. Plant Cell 12, 1917–1925. doi: 10.1105/tpc.12.10.1917
Roberts, A., Trapnell, C., Donaghey, J., Rinn, J. L., and Pachter, L. (2011). Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 12, R22. doi: 10.1186/gb-2011-12-3-r22
Robert-Seilaniantz, A., Grant, M., and Jones, J. D. (2011). Hormone crosstalk in plant disease and defense: more than just jasmonate-salicylate antagonism. Annu. Rev. Phytopathol. 49, 317–343. doi: 10.1146/annurev-phyto-073009-114447
Ryu, H.-S., Han, M., Lee, S.-K., Cho, J.-I., Ryoo, N., Heu, S., et al. (2006). A comprehensive expression analysis of the WRKY gene superfamily in rice plants during defense response. Plant Cell Rep. 25, 836–847. doi: 10.1007/s00299-006-0138-1
Sanford, J., and Johnston, S. A. (1985). The concept of parasite-derived resistance-Deriving resistance genes from the parasite's own genome. J. Theor. Biol. 113, 395–405. doi: 10.1016/S0022-5193(85)80234-4
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Selth, L. A., Dogra, S. C., Rasheed, M. S., Healy, H., Randles, J. W., and Rezaian, M. A. (2005). A NAC domain protein interacts with tomato leaf curl virus replication accessory protein and enhances viral replication. Plant Cell 17, 311–325. doi: 10.1105/tpc.104.027235
Shahmuradov, I. A., Akbarova, Y. Y., Solovyev, V. V., and Aliyev, J. A. (2003). Abundance of plastid DNA insertions in nuclear genomes of rice and Arabidopsis. Plant Mol. Biol. 52, 923–934. doi: 10.1023/A:1025472709537
Sijen, T., Wellink, J., Hiriart, J.-B., and Van Kammen, A. (1996). RNA-mediated virus resistance: role of repeated transgenes and delineation of targeted regions. Plant Cell 8, 2277–2294. doi: 10.1105/tpc.8.12.2277
Simpson, C., Lewandowska, D., Fuller, J., Maronova, M., Kalyna, M., Davidson, D., et al. (2008). Alternative splicing in plants. Biochem. Soc. Trans. 36, 508–510. doi: 10.1042/BST0360508
Storey, W., Ferwerda, F., and Wit, F. (1969). Outlines of Perennial Crop Breeding in the Tropics. Wageningen: H Veenman & Zonen. 389–408.
Suzuki, J. Y., Tripathi, S., Fermín, G. A., Jan, F.-J., Hou, S., Saw, J. H., et al. (2008). Characterization of insertion sites in Rainbow papaya, the first commercialized transgenic fruit crop. Trop. Plant Biol. 1, 293–309. doi: 10.1007/s12042-008-9023-0
Syed, N. H., Kalyna, M., Marquez, Y., Barta, A., and Brown, J. W. (2012). Alternative splicing in plants-coming of age. Trends Plant Sci. 17, 616–623. doi: 10.1016/j.tplants.2012.06.001
Ton, J., Flors, V., and Mauch-Mani, B. (2009). The multifaceted role of ABA in disease resistance. Trends Plant Sci. 14, 310–317. doi: 10.1016/j.tplants.2009.03.006
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Tripathi, S., Suzuki, J. Y., Carr, J. B., Mcquate, G. T., Ferreira, S. A., Manshardt, R. M., et al. (2011). Nutritional composition of Rainbow papaya, the first commercialized transgenic fruit crop. J. Food Comp. Analysis 24, 140–147. doi: 10.1016/j.jfca.2010.07.003
Tripathi, S., Suzuki, J. Y., Ferreira, S. A., and Gonsalves, D. (2008). Papaya ringspot virus-P: characteristics, pathogenicity, sequence variability and control. Mol. Plant Pathol. 9, 269–280. doi: 10.1111/j.1364-3703.2008.00467.x
Vale, R. D. (1987). Intracellular transport using microtubule-based motors. Annu. Rev. Cell Biol. 3, 347–378. doi: 10.1146/annurev.cb.03.110187.002023
Vaucheret, H., Béclin, C., Elmayan, T., Feuerbach, F., Godon, C., Morel, J. B., et al. (1998). Transgene-induced gene silencing in plants. Plant J. 16, 651–659. doi: 10.1046/j.1365-313x.1998.00337.x
Vaucheret, H., Béclin, C., and Fagard, M. (2001). Post-transcriptional gene silencing in plants. J. Cell Sci. 114, 3083–3091.
Vlot, A. C., Dempsey, D. M. A., and Klessig, D. F. (2009). Salicylic acid, a multifaceted hormone to combat disease. Annu. Rev. Phytopathol. 47, 177–206. doi: 10.1146/annurev.phyto.050908.135202
Vorwerk, S., Somerville, S., and Somerville, C. (2004). The role of plant cell wall polysaccharide composition in disease resistance. Trends Plant Sci. 9, 203–209. doi: 10.1016/j.tplants.2004.02.005
Wan, D., Li, R., Zou, B., Zhang, X., Cong, J., Wang, R., et al. (2012). Calmodulin-binding protein CBP60g is a positive regulator of both disease resistance and drought tolerance in Arabidopsis. Plant Cell Rep. 31, 1269–1281. doi: 10.1007/s00299-012-1247-7
Wang, B.-B., and Brendel, V. (2006). Genomewide comparative analysis of alternative splicing in plants. Proc. Natl. Acad. Sci. 103, 7175–7180. doi: 10.1073/pnas.0602039103
Wang, J., Na, J.-K., Yu, Q., Gschwend, A. R., Han, J., Zeng, F., et al. (2012). Sequencing papaya X and Yh chromosomes reveals molecular basis of incipient sex chromosome evolution. Proc. Natl. Acad. Sci. 109, 13710–13715. doi: 10.1073/pnas.1207833109
Wang, L., Tsuda, K., Sato, M., Cohen, J. D., Katagiri, F., and Glazebrook, J. (2009). Arabidopsis CaM binding protein CBP60g contributes to MAMP-induced SA accumulation and is involved in disease resistance against Pseudomonas syringae. PLoS Pathog. 5:e1000301. doi: 10.1371/journal.ppat.1000301
Wang, M.-B., and Waterhouse, P. M. (2000). High-efficiency silencing of a β-glucuronidase gene in rice is correlated with repetitive transgene structure but is independent of DNA methylation. Plant Mol. Biol. 43, 67–82. doi: 10.1023/A:1006490331303
Wang, S., and Fu, J. (2011). Insights into auxin signaling in plant-pathogen interactions. Front. Plant Sci. 2:74. doi: 10.3389/fpls.2011.00074
Wang, X., Liu, Y., Chen, L., Zhao, D., Wang, X., and Zhang, Z. (2013a). Wheat resistome in response to barley yellow dwarf virus infection. Funct. Integr. Genomics 13, 155–165. doi: 10.1007/s10142-013-0309-4
Wang, Y., Tao, X., Tang, X.-M., Xiao, L., Sun, J.-L., Yan, X.-F., et al. (2013b). Comparative transcriptome analysis of tomato (Solanum lycopersicum) in response to exogenous abscisic acid. BMC Genomics 14:841. doi: 10.1186/1471-2164-14-841
Wikström, N., Savolainen, V., and Chase, M. W. (2001). Evolution of the angiosperms: calibrating the family tree. Proc. R. Soc. Lond. B 268, 2211–2220. doi: 10.1098/rspb.2001.1782
Wilson, A. K., Latham, J. R., and Steinbrecher, R. A. (2006). Transformation-induced mutations in transgenic plants: Analysis and biosafety implications. Biotechnol. Genetic Eng. Rev. 23, 209–238. doi: 10.1080/02648725.2006.10648085
Wittmann, T., and Waterman-Storer, C. M. (2001). Cell motility: can Rho GTPases and microtubules point the way? J. Cell Sci. 114, 3795–3803.
Xia, N., Zhang, G., Liu, X.-Y., Deng, L., Cai, G.-L., Zhang, Y., et al. (2010a). Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Mol. Biol. Rep. 37, 3703–3712. doi: 10.1007/s11033-010-0023-4
Xia, N., Zhang, G., Sun, Y.-F., Zhu, L., Xu, L.-S., Chen, X.-M., et al. (2010b). TaNAC8, a novel NAC transcription factor gene in wheat, responds to stripe rust pathogen infection and abiotic stresses. Physiol. Mol. Plant Pathol. 74, 394–402. doi: 10.1016/j.pmpp.2010.06.005
Xie, C., Mao, X., Huang, J., Ding, Y., Wu, J., Dong, S., et al. (2011). KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. doi: 10.1093/nar/gkr483
Xie, Q., Sanz-Burgos, A. P., Guo, H., García, J. A., and Gutiérrez, C. (1999). GRAB proteins, novel members of the NAC domain family, isolated by their interaction with a geminivirus protein. Plant Mol. Biol. 39, 647–656. doi: 10.1023/A:1006138221874
Yeh, S., and Gonsalves, D. (1984). Evaluation of induced mutants of papaya ringspot virus for control by cross protection. Phytopathology 74, 1086–1091. doi: 10.1094/Phyto-74-1086
Zheng, X.-Y., Spivey, N. W., Zeng, W., Liu, P.-P., Fu, Z. Q., Klessig, D. F., et al. (2012). Coronatine promotes Pseudomonas syringae virulence in plants by activating a signaling cascade that inhibits salicylic acid accumulation. Cell Host Microbe 11, 587–596. doi: 10.1016/j.chom.2012.04.014
Keywords: Carica papaya L., papaya ringspot virus (PRSV), transgenic papaya, differentially expressed gene, alternative splicing
Citation: Fang J, Lin A, Qiu W, Cai H, Umar M, Chen R and Ming R (2016) Transcriptome Profiling Revealed Stress-Induced and Disease Resistance Genes Up-Regulated in PRSV Resistant Transgenic Papaya. Front. Plant Sci. 7:855. doi: 10.3389/fpls.2016.00855
Received: 15 April 2016; Accepted: 31 May 2016;
Published: 16 June 2016.
Edited by:
Changbin Chen, University of Minnesota, USAReviewed by:
Rafael Gomez Kosky, Instituto de Biotecnología de las Plantas, CubaHideo Matsumura, Shinshu University, Japan
Copyright © 2016 Fang, Lin, Qiu, Cai, Umar, Chen and Ming. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ray Ming, rming@life.uiuc.edu