 Cristina Sáez1
Cristina Sáez1 Cecilia Martínez2
Cecilia Martínez2 Javier Montero-Pau3
Javier Montero-Pau3 Cristina Esteras1Alicia Sifres
Cristina Esteras1Alicia Sifres José Blanca1
José Blanca1 María Ferriol4
María Ferriol4 Carmelo López1
Carmelo López1 Belén Picó1*
Belén Picó1*- 1Institute for the Conservation and Breeding of Agricultural Biodiversity, Universitat Politècnica de València, Valencia, Spain
- 2Agrifood Campus of International Excellence (ceiA3), Department of Biology and Geology, Universidad de Almería, Almería, Spain
- 3Department of Biochemistry and Molecular Biology, Universitat de València, Valencia, Spain
- 4Instituto Agroforestal Mediterráneo, Universitat Politècnica de València, Valencia, Spain
Tomato leaf curl New Delhi virus (ToLCNDV) is a bipartite whitefly transmitted begomovirus, responsible since 2013 of severe damages in cucurbit crops in Southeastern Spain. Zucchini (Cucurbita pepo) is the most affected species, but melon (Cucumis melo) and cucumber (Cucumis sativus) are also highly damaged by the infection. The virus has spread across Mediterranean basin and European countries, and integrated control measures are not being enough to reduce economic losses. The identification of resistance genes is required to develop resistant cultivars. In this assay, we studied the inheritance of the resistance to ToLCNDV previously identified in two Cucurbita moschata accessions. We generated segregating populations crossing both resistant pumpkins, an American improved cultivar Large Cheese (PI 604506) and an Indian landrace (PI 381814), with a susceptible C. moschata genotype (PI 419083). The analysis of symptoms and viral titers of all populations established the same monogenic recessive genetic control in both resistant accessions, and the allelism tests suggest the occurrence of alleles of the same locus. By genotyping with a single nucleotide polymorphism (SNP) collection evenly distributed along the C. moschata genome, a major quantitative trait locus (QTL) was identified in chromosome 8 controlling resistance to ToLCNDV. This major QTL was also confirmed in the interspecific C. moschata × C. pepo segregating populations, although C. pepo genetic background affected the resistance level. Molecular markers here identified, linked to the ToLCNDV resistance locus, are highly valuable for zucchini breeding programs, allowing the selection of improved commercial materials. The duplication of the candidate region within the C. moschata genome was studied, and genes with paralogs or single-copy genes were identified. Its synteny with the region of chromosome 17 of the susceptible C. pepo revealed an INDEL including interesting candidate genes. The chromosome 8 candidate region of C. moschata was also syntenic to the region in chromosome 11 of melon, previously described as responsible of ToLCNDV resistance. Common genes in the candidate regions of both cucurbits, with high- or moderate-impact polymorphic SNPs between resistant and susceptible C. moschata accessions, are interesting to study the mechanisms involved in this recessive resistance.
Introduction
Tomato leaf curl New Delhi virus (ToLCNDV) is an economically important begomovirus (family Geminiviridae) with two circular single-stranded DNA genome components of ∼2.7 kb, designated as DNA-A and DNA-B (Padidam et al., 1995; Jyothsna et al., 2013). ToLCNDV is transmitted in nature by the whitefly Bemisia tabaci biotypes MEAM1 and MED (Chang et al., 2010; Rosen et al., 2015; Janssen et al., 2017), but some isolates of this virus can also be transmitted by mechanical inoculation (Usharani et al., 2004; Chang et al., 2010; Sohrab et al., 2013; López et al., 2015).
ToLCNDV has a wide host range. It affects crops of the Solanaceae family, such as tomato (Solanum lycopersicum L.), potato (Solanum tuberosum L.), chili pepper (Capsicum annuum L.), and eggplant (Solanum melongena L.) (Padidam et al., 1995; Hussain et al., 2004; Usharani et al., 2004; Pratap et al., 2011). It is also highly damaging to crops of the Cucurbitaceae family, including luffa (Luffa cylindrica M. Roem.), ash gourd [Benincasa hispida (Thunb.) Cogn.], cucumber (Cucumis sativus L.), watermelon (Citrullus lanatus L), melon (C. melo L.), and different types of squashes (Cucurbita spp.) (Sohrab et al., 2003; Ito et al., 2008; Singh et al., 2009; Chang et al., 2010; Roy et al., 2013). Recently, it has been reported to be affecting species of other plant families, such as opium poppy (Papaver somniferum L., Papaveraceae) (Srivastava et al., 2016), cotton (Gossypium hirsutum L., Malvaceae) (Zaidi et al., 2016), soybean (Glycine max, Fabaceae L. Merr.) (Jamil et al., 2017), and firecracker flower (Crossandra infundibuliformis L. Nees, Acanthaceae) (Sundararaj et al., 2019). Furthermore, some weeds as black nightshade (Solanum nigrum L.), thorn apple (Datura stramonium L.), squirting cucumber [Ecballium elaterium (L.) A. Rich], smooth sowthistle (Sonchus oleraceus L.), false daisy [Eclipta prostrata (L.) L.], and apple of Sodom [Calotropis procera (Aiton) Dryand.] (Haider et al., 2006; Moriones et al., 2017; Zaidi et al., 2017; Juárez et al., 2019) have been found to be hosts of the virus, acting as reservoirs during the whole year.
ToLCNDV was first detected in North India in 1995 (Srivastava et al., 1995), from where it spread to South and Southeast Asian countries. It was limited to Asia until 2012, when it was reported affecting cucurbits in different Mediterranean countries, first in Spain (Juárez et al., 2014) and later in Tunisia (Mnari-Hattab et al., 2015), Italy (Panno et al., 2016), Morocco (Sifres et al., 2018), Greece (Orfanidou et al., 2019), and Algeria (Kheireddine et al., 2019). More recently, the virus has been identified in cucurbits plants in Portugal and Estonia (EPPO, 2019), which is indicative of the rapid spread of ToLCNDV through Europe. The most affected crop in European countries is Zucchini squash (Cucurbita pepo L. subsp. pepo). In this crop, the virus causes severe stunting of plants, which exhibit upward and downward curling of the leaves, severe mosaic, and fruit skin roughness (Juárez et al., 2014). Infected plants often present partial or complete yield loss and fruits with lower market value. Zucchini is one of the most widely grown crops and appreciated vegetable in the Mediterranean basin. This region produced nearly 300,000 tm of this vegetable and other species of the Cucurbita genus (pumpkins, squash, and gourds) in FAO (2017), representing almost 24% of world production, excluding China and India. Before the arrival of ToLCNDV, the aphid-borne potyvirus Zucchini yellow mosaic virus (ZYMV) was the major viral pathogen of this crop (Capuozzo et al., 2017). Since 2013, ToLCNDV is the most prevalent virus in the area, where it is an important constraint to zucchini production. In the background of the severe epidemic outbreaks of ToLCNDV in cucurbits, both in greenhouses and in open fields, European and Mediterranean Plant Protection Organization (EPPO) has added this virus to the EPPO Alert List (EPPO, 2017).
Cultural practices, such as the control of the whitefly vector, the elimination of infected plants, and the avoidance of the most susceptible cultivars, are not very effective in preventing ToLCNDV outbreaks (EPPO, 2017). In fact, breeding resistant varieties is considered the most economical and effective method to control virus diseases. Genetic resistance to ToLCNDV has been identified in some accessions of the Cucurbita genus (Sáez et al., 2016). In that work, authors screened for ToLCNDV resistance in a large collection of Cucurbita spp. accessions including landraces and commercial varieties of the cultivated species (C. pepo L., C. moschata Duchesne and C. maxima Duchesne) and wild Cucurbita species. All the C. pepo and C. maxima accessions behaved as highly susceptible, but four C. moschata accessions were highly resistant, two of them after both mechanical and whitefly inoculation, remaining symptomless with a reduced viral accumulation (Sáez et al., 2016).
Genetic resistance to ToLCNDV has also been characterized in some other species belonging to different families. In Solanum habrochaites S. Knapp & D.M. Spooner, a wild species related to tomato, three dominant genes are responsible for the resistance (Rai et al., 2013). In L. cylindrica, a popular cucurbit vegetable in India, a dominant monogenic resistance was reported (Islam et al., 2010, 2011). More recently, in melon, Sáez et al. (2017) found one major locus in chromosome 11 and two additional regions in chromosomes 12 and 2 that control resistance to ToLCNDV. In this context, the purpose of this study was to map the quantitative trait loci (QTL) associated with the resistance to ToLCNDV in C. moschata using segregating populations derived from these resistant sources and a susceptible accession of this species, and to confirm this resistance in interspecific C. moschata × C. pepo populations as the first step to transfer the resistance to zucchini.
Materials and Methods
Plant Material
In this work, we selected two Cucurbita moschata accessions (PI 604506 and PI 381814), previously reported (Sáez et al., 2016) as symptomless or with slight symptoms after whitefly and sap inoculation with ToLCNDV, to study the genetic control of the resistance. PI 604506 is the improved pumpkin cultivar Large Cheese from the United States and PI 381814, an Indian landrace. The Chinese C. moschata accession PI 419083 was used as susceptible control. Seeds of the three accessions were first provided by the United States Department of Agriculture-National Plant Germplasm System (USDA-NPGS) genebank, then fixed by selfing and multiplied by the cucurbits breeding group at the Institute for the Conservation and Breeding of Agricultural Biodiversity (COMAV), and stored at the COMAV genebank.
Virus Source and Mechanical Inoculation
To generate the viral inoculum source, susceptible zucchini plants were agroinfiltrated with an infectious clone based on the Spanish isolate of ToLCNDV (99% nucleotide identity with the sequences of the A and B viral genomic particles: KF749224 and KF749225 (Juárez et al., 2014), following the procedure described in Sáez et al. (2016).
The tissue of symptomatic leaves from 15 days post-ToLCNDV agroinoculation plants was crushed in a mortar together with inoculation buffer [50 mM potassium phosphate (pH 8.0), 1% polyvinylpyrrolidone 10, 1% polyethylene glycol 6000, 10 mM 2-mercaptoethanol and 1% activated charcoal] in a 1:4 (w/v) proportion (López et al., 2015). The homogenate was used to mechanically inoculate all plants at the stage of one true leaf, dusting on the true leaf and on one cotyledon with Carborundum 600 mesh and scratching with a cotton swab dipped in the blend. Inoculated plants were grown in a climatic chamber, and disease progression was monitored. Symptomless plants 15 days after mechanical inoculation (dpi) were reinoculated to avoid escaping to the infection.
Generation of F1 and Segregating Populations
Ten seeds of each C. moschata accession were disinfected and germinated as described by Sáez et al. (2016). Seedlings were transplanted to pots and grown in climatic chamber under controlled conditions (photoperiod of 16 h day at 25°C and 8 h night at 18°C and 70% of relative humidity). Subsequently, plants were moved to a greenhouse and crossed to obtain three F1 progenies: F1 PI 419083 × PI 604506, F1 PI 419083 × PI 381814, and F1 PI 604506 × PI 381814. Eight plants of each parent and the corresponding hybrids were mechanically inoculated with ToLCNDV as described above and phenotyped according to symptomatology and viral accumulation as described below.
Eight additional plants of the C. moschata parents were cultivated in a greenhouse along with eight plants of the F1 progenies. To generate segregating populations, F1 plants were selfed to obtain F2 progenies and backcrossed to plants of PI 604506, PI 381814, and PI 419083 to generate the BC1PI 604506, BC1PI 381814, and BC1PI 419083 populations, respectively. All these segregating populations were screened against ToLCNDV with the same inoculation and phenotyping methodology, using three plants of each C. moschata accession as controls. F2 and BC1 derived from F1 PI 419083 × PI 381814 were obtained later because of the influence of the local climate conditions in PI 381814 vegetative growth, causing late-flowering and slow development of fruits. Hence, we studied first the genetic control of the resistance to ToLCNDV in the segregating populations derived from PI 604506, and results were validated in F2 and BC1 coming from F1 PI 419083 × PI 381814.
Symptoms Evaluation and Quantification of the Viral Accumulation
Symptomatology was evaluated in all plants at 15 and 30 dpi using the visual scale described by López et al. (2015). Symptoms score ranged from 0 (absence of symptoms) to 4 (highly severe symptoms), classifying as resistant those plants with symptoms scored 0 or 1 and as susceptible those with symptoms scored from 2 to 4. The goodness-of-fit between the expected and observed segregation ratios resistant/susceptible plants was analyzed by chi-squared (χ2) test (p < 0.05) in the F2 and BC1-segregating populations.
The relative ToLCNDV accumulation in each plant was determined at 30 dpi by quantitative PCR (qPCR). Total DNA from apical leaves was extracted using the cetyltrimethyl ammonium bromide (CTAB) method (Doyle and Doyle, 1990) and quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Waltham, MA, United States). DNA was diluted with sterile-deionized water to a final concentration of 5 ng μl–1. Three biological replicates were done for each parental genotype, and all plants of the assay were analyzed in three technical replicates using a LightCycler® 480 System (Roche). In each qPCR reaction, 15 ng of genomic DNA were used as templates, in a final volume of 15 μl. We used 7.5 μl of 2 × iTaqTM universal SYBR® Green Supermix (BIO-RAD) and 1.5 μl (100 nM) of each primer and 1.5 μl of H2O. Primers ToLCNDVF1 (5′-AATGCCGACT ACACCAAGCAT-3′, positions 1145–1169) and ToLCNDVR1 (5′-GGATCGAGCAGAGAGTGGCG-3′, positions 1399–1418) were used for the amplification of a 273-bp fragment of viral DNA-A. The single-copy gene CpACS2 was amplified in all samples as internal control using the primers CpACS2F (5′-ACT CGATCAACTTCGAGCAAA-3′) and CpACS2R (5′-GCCTA TCCAAAGACCTCGGCCTTCCC-3′). Both ToLCNDVF1/R1 and CpACS2 primers were used in previous works by Sáez et al. (2016). Cycling conditions consisted of incubation at 95°C for 5 min, 45 cycles of 95°C for 5 s and 60°C for 30 s. Relative ToLCNDV levels were calculated using the 2–ΔCt expression, a variation of the Livak method (Livak and Schmittgen, 2001; Bio-Rad Laboratories, 2006), where ratio (target/reference) = 2–ΔCt = 2–[Ct (viral target) – Ct (reference gen)].
QTL Analysis in C. moschata F2 Population Derived From PI 419083 × PI 604506
PI 604506 and PI 419083 accessions were included in an RNAseq analysis, performed in the frame of a de novo assembly of the zucchini genome project (Montero-Pau et al., 2018), and their transcriptome sequences were used to generate the single nucleotide polymorphism (SNP) panel used here. SNPs were selected by aligning each sequence to the version 1 of the C. moschata cv. Rifu genome (Sun et al., 2017), available at the Cucurbit Genomics Database1. We used the Bowtie2 tool with the very-sensitive-local argument. Variant calling was performed using Freebayes version 1.0.2 (Garrison and Marth, 2012), excluding alignments from the analysis if they had a mapping quality < 40, alleles with quality under 20, and filtering SNPs with minimum count of 10. A set of 137 SNPs evenly distributed throughout the C. moschata genome (Supplementary Table 1) were selected and used to genotype PI 604506, PI 419083, their derived F1, and 134 plants of the corresponding F2 population.
All plants were genotyped using the Agena Bioscience iPLEX® Gold MassARRAY (Agena Biosciences) system at the Epigenetic and Genotyping unit of the University of Valencia [Unitat Central d’Investigació en Medicina (UCIM), Faculty of Medicine, Spain]. Total DNA was extracted from the tissue of young leaves, using the protocol described above, and quantified and adjusted to 15 ng μl–1. F2 genotyping results were run in MAPMAKER 3.0 (Lander et al., 1987; Lincoln et al., 1992) with the Kosambi map function, obtaining the genetic position of each marker.
To identify markers linked to the resistance to ToLCNDV derived from the PI 604506 accession, a QTL analysis was performed using symptoms at 15 and 30 dpi and ToLCNDV relative accumulation at 30 dpi as quantitative traits, and a qualitative score of resistance (0 susceptible phenotype and 1 resistant phenotype) assigned to each plant according to symptoms and viral accumulation. We used the Kolmogorov–Smirnov test to check the normality assumption of traits distribution. Since the traits were not normally distributed, Kruskal–Wallis non-parametric test was used for QTL detection using the MapQTL version 4.1 software (Van Ooijen, 2009), considering as significant associations those with p < 0.05. Since 2(–ΔCt) values have a skewed distribution, we used the original ΔCt data for QTL analysis. The binary qualitative trait of resistance was also analyzed by logistic regression model, with a significance level of α = 0.05.
In addition, a composite interval mapping approach (CIM, Zeng, 1994) was applied in Qgene 4.0 (Joehanes and Nelson, 2008), using the genetic map previously generated with this F2. The logarithm of the odds ratio (LOD) threshold was calculated using a 1,000 permutations test per trait, for p < 0.05. The percentage of phenotypic variance explained (R2), the additive and dominance effects, degree of dominance, and the interval position of the QTL in accordance with a 2-U LOD drop was estimated for the highest significant peak LOD. Loci identified with both methods (Kruskal–Wallis and CIM) were considered true QTLs of putative interest.
Validation of the QTL of Chromosome 8 in Additional C. moschata Segregating Populations Derived From PI 419083, PI 604506, and PI 381814
The previous analysis allowed detecting a major QTL responsible for the resistance in chromosome 8. To confirm this QTL in additional C. moschata-segregating populations and to introgress the candidate region in chromosome 8 of C. moschata in the zucchini (C. pepo) background (the cucurbit crop more severely affected by ToLCNDV), a new set of 19 SNPs of the chromosome 8 candidate region was implemented in a new Agena Bioscience platform. These new SNPs were selected to be useful for both purposes. The transcriptomic sequences of PI 604506, PI 381814, and PI 419083 (obtained in the RNAseq analysis by Montero-Pau et al., 2018) were aligned to the C. pepo genome (Zucchini accession MU-CU-16), available at the Cucurbit Genomics Database2, using Bowtie2. Integrative Genomics Viewer (IGV) (Thorvaldsdóttir et al., 2013) was used to detect variations between sequences, and those polymorphic SNPs between resistant (PI 604506 and PI 381814) and susceptible (PI 419083 and MU-CU-16) genotypes were selected. This Agena platform was employed to genotype a subset of 131 plants of the previously genotyped F2 (PI 419083 × PI 604506), 121 of F2 (PI 419083 × PI 381814), 31 BC1PI 604506, and 73 of BC1PI 381814.
For further saturation of the candidate region, five additional SNPs, not integrated in the new Agena Bioscience set, were designed with the same requirements and used to genotype the F2 (PI 419083 × PI 604506) population by high-resolution melting (HRM) (Vossen et al., 2009). PRIMER3 software (Untergasser et al., 2012) was employed to design the oligonucleotides for the HRM analysis. The genomic positions of all these new SNPs (Agena Bioscience platform and HRM markers) and their flanking sequences are shown in Supplementary Table 2.
A new map of chromosome 8 was constructed with 24 SNP markers (3 and 16 SNPs from the first and second Agena platforms, respectively, and 5 HRM), using genotyping results of F2 (PI 419083 × PI 604506). MAPMAKER 3.0 (Lander et al., 1987; Lincoln et al., 1992) software and the Kosambi map function were employed to generate the new map. The genetic distances of the new map were used in a second QTL analysis, with the F2 (PI 419083 × PI 604506) population, following the same procedure described above. Means of symptom scores at 30 dpi of plants from F2 (PI 419083 × PI 381814), BC1PI 604506, and BC1PI 381814 populations classified according to the marker classes (a, b, and h for F2 and h and a for BC1) were analyzed by ANOVA and Bonferroni multiple range tests using STATGRAPHIC Centurion XVI.I statistic software, to evaluate differences between means, considering statistically significant differences when p ≤ 0.01.
Validation of the QTL in the Interspecific Cross C. pepo × C. moschata
An interspecific cross between the ToLCNDV-susceptible C. pepo accession MU-CU-16 (Sáez et al., 2016) and the resistant C. moschata accession PI 604506 provided five F1 seeds that were germinated as described above. Four seedlings were moved to a greenhouse and selfed to obtain F2 (MU-CU-16 × PI 604506) generation. The remaining F1 seedling and 176 plants of F2 (MU-CU-16 × PI 604506) were screened by mechanical inoculation of ToLCNDV. Symptoms and viral titers were determined by the same procedure described above.
This Cucurbita-interspecific F2 population was genotyped with the new Agena Bioscience platform and the five HRM SNP markers of chromosome 8. The genotyping results were used to construct a new genetic map of chromosome 8 and to perform an additional QTL analysis as described above.
Genomic Variation, Structural Variants, and Synteny
To obtain a more detailed view of the underlying genomic variation in the candidate region, both C. moschata resistant and susceptible parents (PI 604506 and PI 419083) were fully sequenced. Raw reads are deposited in the National Center for Biotechnology Information (NCBI) under BioProject PRJNA604046. Genomic DNA was obtained from fresh tissue using CTAB extraction, and a pair-end library (2 × 150 bp) was built for each accession. Libraries were sequenced as part of an Illumina HiSeq 2000 lane by Polar Genomics (Ithaca, NY, United States). Reads were cleaned using the ngs_crumbs software3 to eliminate adapters, low-quality bases (Phred quality < 25 in a 5-bp window), reads shorter than 50 bp, and duplicated sequences. Clean reads were mapped against the reference C. moschata genome using bwa-mem (Li, 2013), and variant calling was performed using Freebayes version 1.1.0 (Garrison and Marth, 2012) after filtering reads with a mapping quality cutoff MAPQ lower than 57. To study the potential effect of the genetic changes, SNPs were annotated based on its predicted effect on the gene using SNPEff v4.3 (Cingolani et al., 2012). Differences in sequencing genome coverage between both accessions were studied to explore possible genomic deletions. Read coverage along the candidate region was calculated using samtools v.1.9 (Li et al., 2009), and we checked if coverage deviated from the 99% confidence interval of the observed coverage for each accession assuming a log-normal distribution. Confidence interval for the log-normal distribution was calculated using the function elnorm of R package “EnvStats” (Millard, 2013). In addition to that, the structural variant caller Manta v.1.6 (Chen et al., 2016) was used to check for differential large insertion/deletions. Identification of putative paralogs of the genes in the candidate region was done with OrthoMCL (Li et al., 2003).
Identification of syntenic regions between C. moschata and C. pepo and C. melo was done by nucleotide basic local alignment search tool (BLAST) of each gene within the candidate region of C. moschata against the other two genomes. BLAST hits were filtered using an E value cutoff of 10–20 and a minimum overlap between sequences of 70%. For C. pepo, to inspect for possible insertion/deletions, a dot plot comparing chromosome 17 region of C. pepo and chromosome 8 of C. moschata was built based on the alignment of both sequences using LAST (Kielbasa et al., 2011). For C. melo, the module of Tripal “SyntenyViewer,” available in cucurbitgenomics.com, was used to visualize the synteny.
New C. moschata and C. pepo genome assemblies have become recently available4,5 (online availability since November 2019), but after finishing the analysis that we showed here. Our results were checked through alignments with the new assemblies to avoid misinformation.
In addition to the analysis of the genomic sequences, SNPs discovered using the available RNAseq data (Montero-Pau et al., 2018) from the three C. moschata accessions used as parentals in the previous crosses and six additional C. moschata from different origins that exhibited susceptibility to ToLCNDV in previous works (López et al., 2015; Sáez et al., 2016) were also annotated using SNPEff.
Results
Response to ToLCNDV of F1 Progenies
The inoculation assay showed that the two C. moschata accessions resistant to ToLCNDV, PI 604506 and PI 381814, remained totally symptomless or with only slight symptoms (score from 0 to 1) at 30 dpi, contrasting with the severe mosaic developed in the susceptible control (score 4), PI 419083 (Figure 1). F1 plants of the two susceptible × resistant crosses were highly susceptible, displaying a similar symptomatology as PI 419083 at 30 dpi. Conversely, F1 progeny derived from the cross between the two C. moschata-resistant accessions remained symptomless throughout the essay (Figure 1).
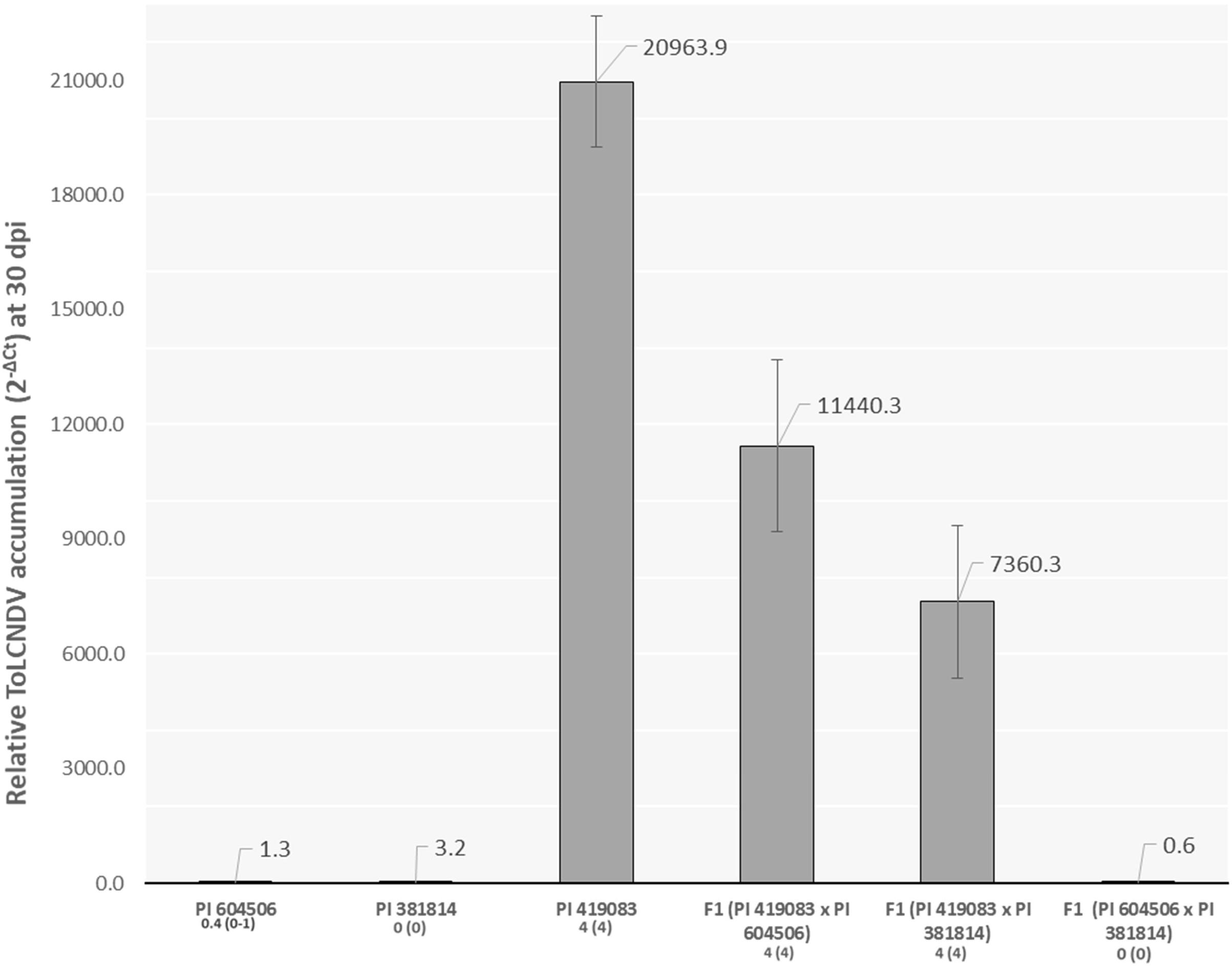
Figure 1. Relative tomato leaf curl New Delhi virus (ToLCNDV) accumulation (calculated as 2– ΔCt) at 30 days after mechanical inoculation (dpi) with ToLCNDV in the resistant Cucurbita moschata accessions PI 604506 and PI 381814, the susceptible control PI 419083, and their respective hybrids. Mean and range of symptoms scores at 30 dpi are indicated in the x-axis legend.
Strong correlation between symptom severity and viral titers was observed (r2 = 0.73, p = 0.030) after measuring relative ToLCNDV accumulation by qPCR. In accordance with their resistant behavior, PI 604506, PI 381814, and the F1 (PI 604506 × PI 381814) had viral titers, on average, 7.8 × 103 times lower than those of the susceptible control PI 419083 and the two F1 derived from it (Figure 1).
The fact that F1 progenies derived from the two susceptible × resistant crosses were susceptible, while the F1 derived from the resistant × resistant cross was resistant, suggests a recessive genetic control of the resistance in both accessions, controlled by common genes. A further analysis of the genetic control of the resistance was performed in F2 and BC1 populations.
Response to ToLCNDV of Segregating Populations Derived From the Cross Between Resistant and Susceptible C. moschata Accessions
F2 and BC1PI 604506 populations, derived from the F1 PI 419083 × PI 604506, segregated for symptoms severity. Table 1 shows resistant:susceptible plants segregation, according to symptomatology at 30 dpi. At the end of the assay, 38 plants of F2 remained symptomless (score 0), and 96 showed severe symptomatology (scores 2–4). The X2 test indicated that this segregation fitted to a 1:3 (resistant:susceptible) ratio expected for a single recessive gene for resistance (p = 0.43) (Table 1). To further characterize the response to ToLCNDV, virus accumulation was estimated in the segregating population F2 (PI 419083 × PI 604506) by qPCR (Figure 2). On average, viral titer strongly correlated to symptoms severity following an exponential model (r2 = 0.82, p = 0.035). All plants developing mosaic, deformation, or short internodes had high viral titers, whereas in the symptomless plants, ToLCNDV accumulation was detected at very low concentrations. On average, the viral accumulation (2–ΔCt) in susceptible plants was 2.2 × 103 times higher than in resistant plants. Since viral titer is in concordance with symptoms development, symptom scores were used to phenotype the response to ToLCNDV in plants of the remaining F2 and BC1 populations. In BC1PI 604506, 33 plants were resistant (score 0) and 26 were susceptible (scores 2–4). This segregation also fitted to a 1:1 ratio expected for a single recessive gene (p = 0.44) (Table 1). In accordance with the occurrence of a single recessive gene controlling the resistance, all plants of the BC1PI 419083 generation had severe symptoms at the end of the assay.
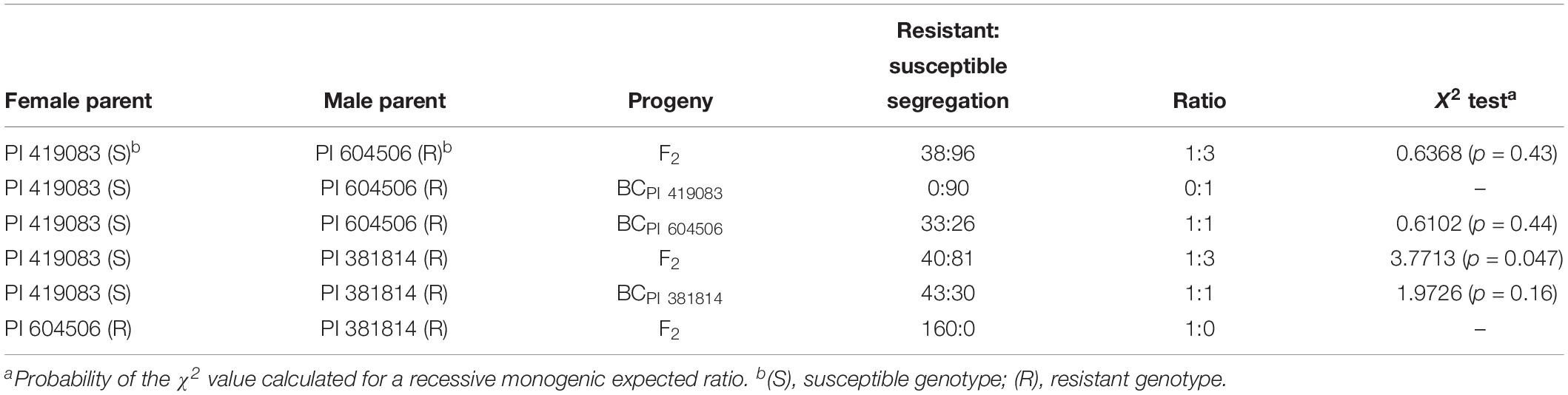
Table 1. Segregation of resistant/susceptible plants in F2 and BC progenies at 30 days after mechanical inoculation with tomato leaf curl New Delhi virus (ToLCNDV).
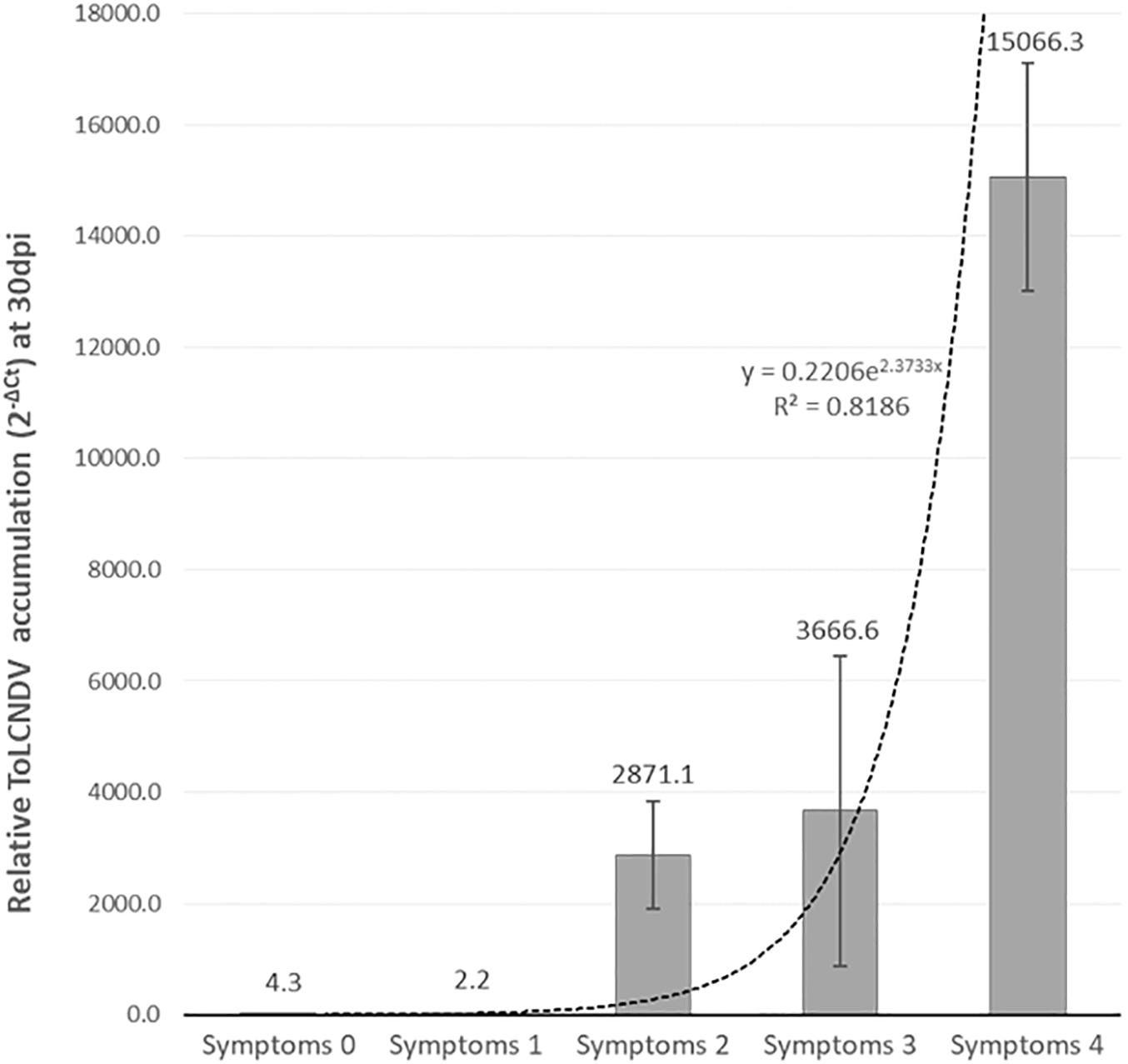
Figure 2. Mean of relative tomato leaf curl New Delhi virus (ToLCNDV) accumulation (calculated as 2– ΔCt) in plants within each symptomatic class in F2 (PI 419083 × PI 604506) at 30 days after mechanical inoculation. Dot line represents the exponential relationship between both variables, which was statistically significant for a confident level of α = 0.05.
Symptom segregation ratios observed in the F2 (PI 419083 × PI 381814) and BC1PI 381814 populations also fitted to one recessive gene for resistance null hypothesis in X2 test (Table 1). Forty and 43 plants of F2 and BC1PI 381814, respectively, remained symptomless (score 0), and 81 and 30 plants showed severe symptoms (scores 2–4), with p = 0.047 and p = 0.16, in both respective populations (Table 1).
In accordance with the F1 results, the 160 plants of the F2 derived from the resistant × resistant cross PI 604506 × PI 381814 remained totally symptomless along all the assay.
QTL Analysis in F2 (PI 419083 × PI 604506) Population
The F2 (PI 419083 × PI 604506) population was genotyped with the 137 SNPs markers evenly distributed throughout C. moschata genome and used to construct a linkage map that included 20 linkage groups and spanned a total of 2,681.5 cM of genetic distance, with an average of 22.92 cM between markers (Supplementary Table 1). The linkage map was used to identify QTLs involved in ToLCNDV resistance in C. moschata, based on genotyping and phenotyping results (symptoms scores at 15 and 30 dpi, virus titer at 30 dpi, and the qualitative resistance score) of F2 (PI 419083 × PI 604506) population. QTL analysis, performed using non-parametric Kruskal–Wallis test (KW) followed by CIM, resulted in the detection of a major QTL in chromosome 8 (Table 2), validated by logistic regression of the qualitative trait of resistance (data not shown). Four QTLs, all located in almost the same genetic position, showed significant association with all the traits evaluated, explaining a proportion between 29.0 and 45.0% of the observed phenotypic variance. All QTLs (ToLCNDVCm_Sy15-8, ToLCNDVCm_Sy30-8, ToLCNDVCm_VT30-8, and ToLCNDVCm_Re-8) were located close to D133 (physical position, 1,366,729 bp), with LOD peaks between 10.06 and 17.31.
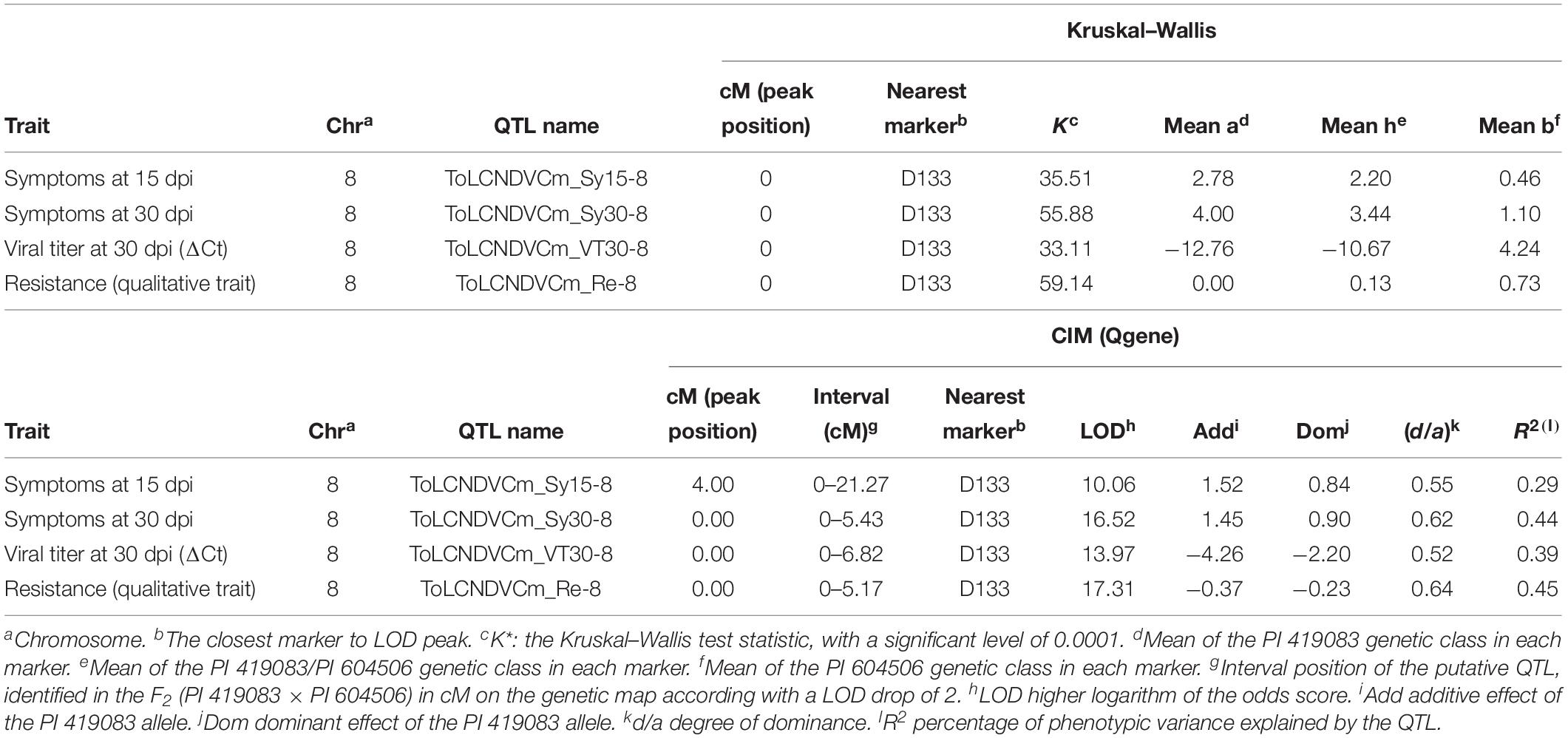
Table 2. Quantitative trait loci (QTLs) identified in the F2 (PI 419083 × PI 604506) segregating population genotyped with the set of 137 single nucleotide polymorphisms (SNPs) evenly distributed through the C. moschata genome, using the non-parametric Kruskal–Wallis test and composite interval mapping method.
Narrowing the Candidate Region in Chromosome 8
To validate the major QTL identified in the previous analysis and to increase marker density in the candidate region, F2 (PI 419083 × PI 604506) population was genotyped with the new Agena Bioscience-HRM SNPs set of chromosome 8. Twenty-one out of the 24 new markers (Supplementary Table 2) were polymorphic in this population, despite all of them were selected in silico as SNP variants between both parents using the IGV software. Genotyping results were employed to generate a new linkage map in this region (Supplementary Table 2), covering 72.5 cM, with an average distance between consecutive markers of 3.15 cM, and two clusters of linked markers at 0 and 11.7 cM genetic positions. The QTL analysis was performed using the new map and the new genotyping results (using one selected marker of each of the two clusters of completely linked SNPs) (Table 3). ToLCNDVCm_Sy15-8 QTL, associated to the variation of symptoms at 15 dpi, was identified again with both non-parametric Kruskal–Wallis and CIM analysis, near D133 (located at 18.8 cM in this new map) and with similar explained variance, LOD peak, additive, and dominant effects. However, ToLCNDVCm_Sy30-8, ToLCNDVCm_VT30-8, and ToLCNDVCm_Re-8 QTLs, corresponding to traits measured at the end of the assay (30 dpi), when differences are clearer between resistant and susceptible plants, were closely linked to a new marker with both analysis methods. The closest markers (those linked at 11.7 cM DPM37, DMP39, DMP11, DMP10, DMP42, DMP43, DMP44, DMP41, and snp_8202510 markers) are included in the interval position of the same QTLs identified with Kruskal–Wallis and CIM (between 8 and 14 cM) (Figure 3A) and validated with logistic regression of the qualitative trait of resistance, according to their physical and genetic position. The interval of the four QTLs was flanked by DPM34 and D133 markers, with physical positions 561,788 and 1,366,729 bp, respectively. After this further QTL analysis of chromosome 8, the proportion of explained variance was increased, with percentages between 29.5 and 66.0% of R2.
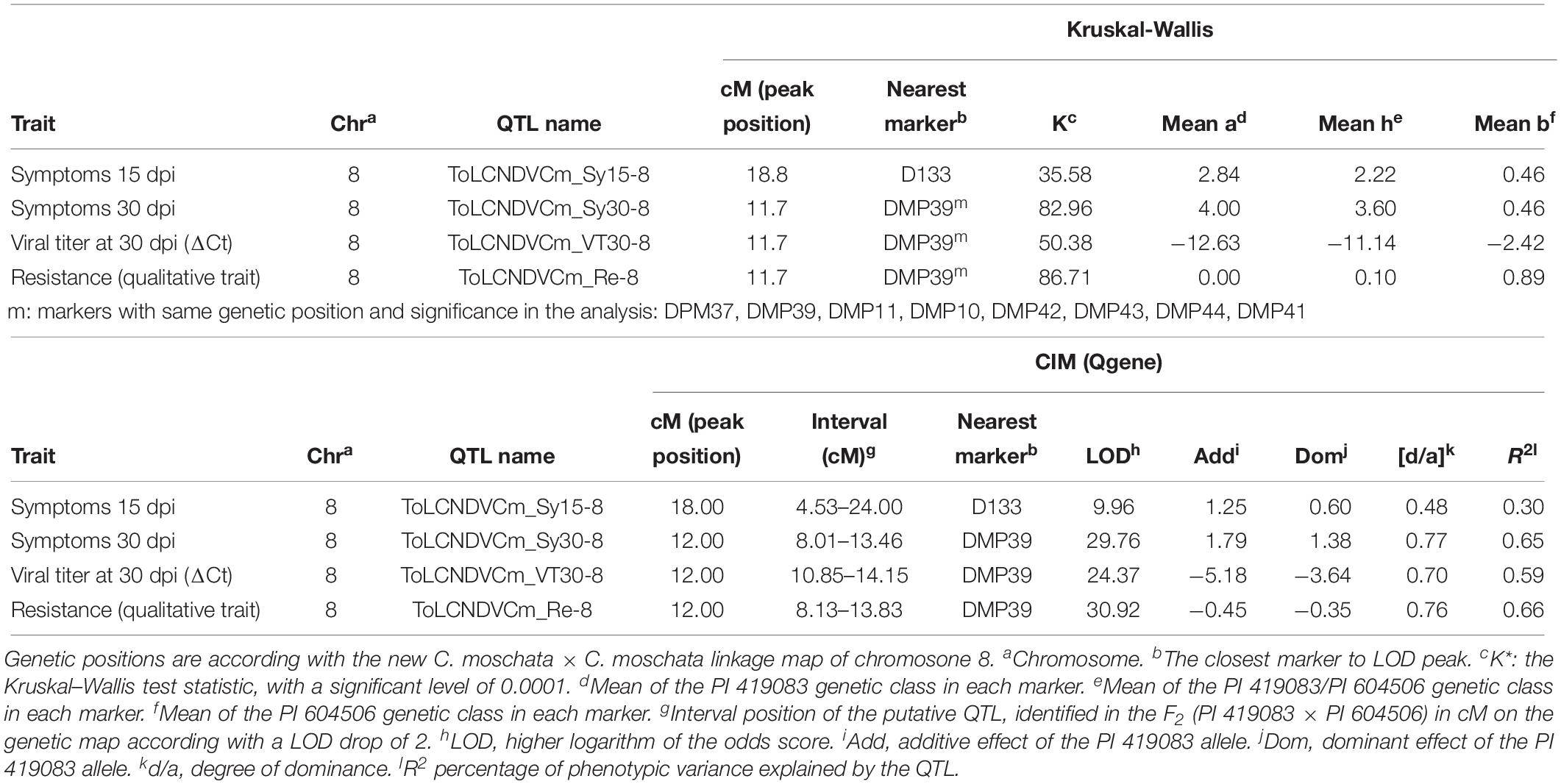
Table 3. Quantitative trait loci (QTLs) identified in the F2 (PI419083 × PI604506) segregating population genotyped with markers of chromosome 8 of C. moschata, using the non-parametric Kruskal–Wallis test and composite interval mapping (CIM).
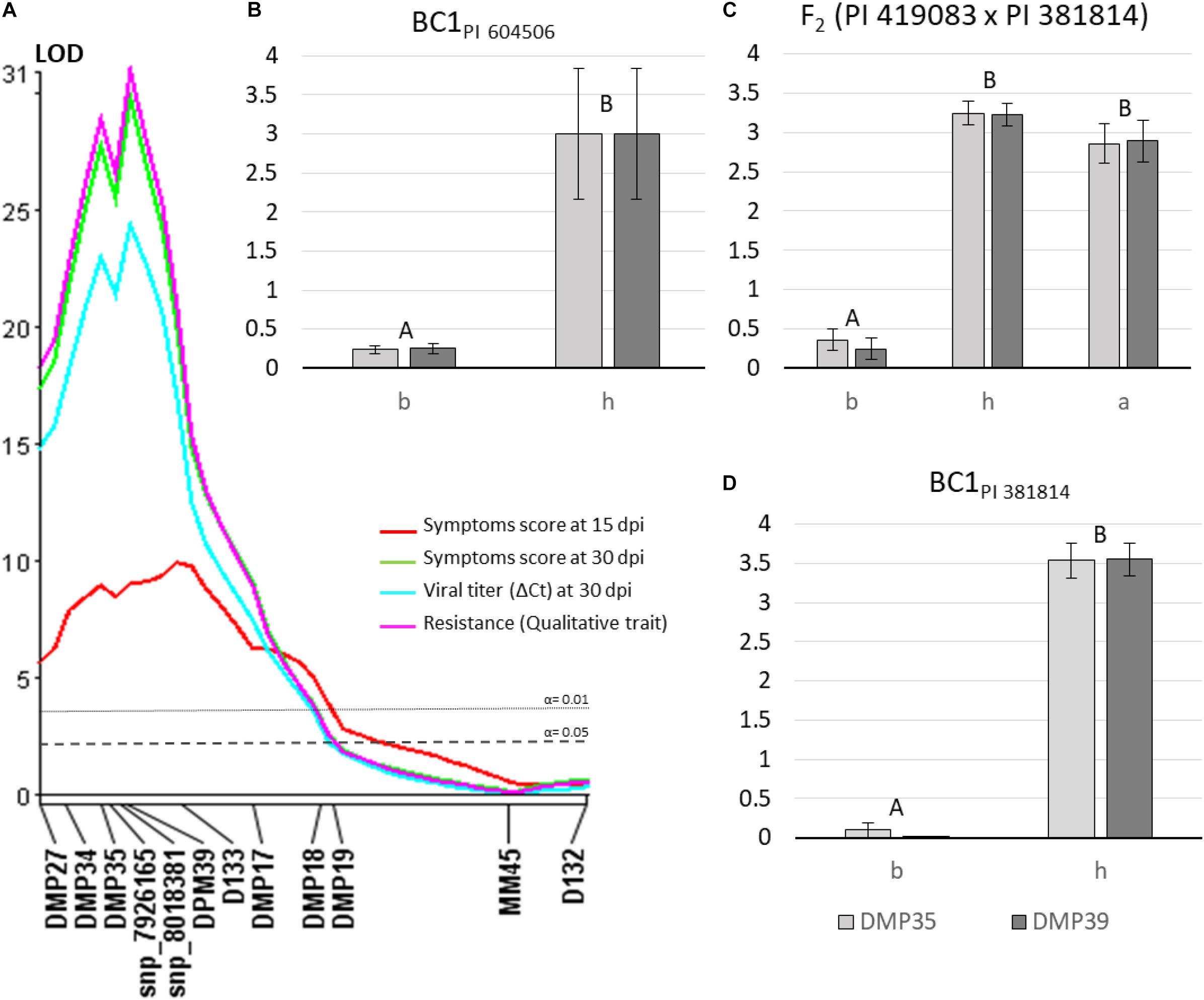
Figure 3. (A) Molecular markers linked to quantitative trait loci (QTLs) for the four traits [symptoms at 15 and 30 days after mechanical inoculation (dpi), virus titer at 30 dpi and the qualitative resistance trait] associated with ToLCNDV resistance. QTL location was obtained by composite interval mapping (CIM) method using F2 (PI 419083 × PI 604506). (B–D) Mean of symptom score at 30 dpi in BCPI 604506, F2 (PI 419083 × PI 381814) and BCPI 381814 populations, respectively, according to each genotypic class DMP35 (light gray bars) and DMP39 (dark gray bars) markers (chromosome 8). PI 604506 or PI 381814 homozygous genotype is represented as “b,” heterozygous as “h” and PI 419083 homozygous plants as “a.” Bars with same capitals letters are not significantly different at p ≤ 0.01.
Validation of the Major QTL in Chromosome 8 in BC1PI 604506, F2 (PI 419083 × PI 381814) and BC1PI 381814 Segregating Populations
The Agena Bioscience SNPs panel of chromosome 8 was used to genotype the BC1PI 604506 derived from the PI 419083 × PI 604506 cross, and the F2 and BC1PI 381814 populations derived from the PI 419083 × PI 381814 cross. Mean of symptoms scores at 30 dpi were calculated for each genotypic class of selected SNPs located within the defined QTL interval (DMP35 and DMP39) and compared in Figures 3B–D. The lowest level of symptoms was observed when plants in the three populations had the PI 604506 or PI 381814 homozygous genotype (b), in DPM35 or DPM39 indistinctly. Plants heterozygous (h) or PI 419083 homozygous (a) in both markers displayed significantly more severe symptomatology.
QTL Analysis and Validation of the Candidate Region in C. pepo
Consistently with the results obtained in F1 from susceptible × resistant C. moschata crosses, severe symptoms were developed by F1 C. pepo MU-CU-16 × C. moschata PI 604506 plants at 15 and 30 dpi (Figure 4). This result supports that resistance in PI 604506 has a recessive genetic control. F2 (MU-CU-16 × PI 604506) plants segregated for symptomatology and viral accumulation. Symptoms, including upward and downward curling and severe mosaic of young leaves, short internodes, and bad distorted development, were observed in 124 and 151 F2 (MU-CU-16 × PI 604506) plants at 15 and 30 dpi, respectively. The number of resistant plants decreased from 52 to 25 between 15 and 30 dpi. Nine plants had bad development or died in the course of the infection. On average, virus titers determined by qPCR at 30 dpi were in concordance with symptoms development, with mean of relative viral accumulation expressed as 2(–ΔCt) of 1.04 ± 0.31 and 49,571.67 ± 9,670.31 in resistant and susceptible plants, respectively. The observed segregation proportion was adjusted to the expected ratio resistant/susceptible plants, in case of one recessive gene responsible on the genetic control of resistance to ToLCNDV at 15 dpi (X2 = 2.1894, p = 0.14), but not at 30 dpi (X2 = 10.312, p = 0.0014).

Figure 4. Plants of the interspecific F1 resulting from the cross C. pepo MU-CU-16 × C. moschata PI 604506. (A) Typical symptoms of tomato leaf curl New Delhi virus including curling, severe mosaic, and short internodes. (B) Healthy plant used to obtain F2 progeny by selfing in a greenhouse. (C) Detail of F1 (MU-CU-16 × PI 604506) fruit obtained by selfing.
The genetic map of chromosome 8 generated with the genotyping results of the Agena Bioscience-HRM SNPs in the F2 (MU-CU-16 × PI 604506) gave a total genetic length of 21.4 cM, with an average genetic distance between successive markers of 0.98 cM (Supplementary Table 2).
The QTL analysis performed in this population show that the QTLs identified in the C. moschata populations were stable in the cross with the C. pepo accession MU-CU-16. ToLCNDVCm_Sy15-8, ToLCNDVCm_Sy30-8, ToLCNDVCm_ VT30-8, and ToLCNDVCm_Re-8 were located in the same region that in C. moschata (Table 4), physically mapped in chromosome 17. The highest R2 value (65%) was explained by the ToLCNDVCm_Sy15-8, associated to DMP39 as the nearest marker to the peak LOD. R2 values were lower in QTLs related to advanced stages of the ToLCNDV infection, mainly in the viral titer at 30 dpi (ΔCt) trait. In these cases, the nearest markers to the LOD peaks were DMP39 and snp_7926165 in ToLCNDVCm_Sy30-8 (Kruskal–Wallis and CIM tests, respectively), snp_7926165 in ToLCNDVCm_VT30-8 and DMP35, and snp_7926165 in ToLCNDVCm_Re-8 (Kruskal–Wallis and CIM tests, respectively). Logistic regression validate the occurrence of ToLCNDVCm_Re-8 QTL. According to the 2-LOD drop confidence intervals, the position interval where the four QTLs are comapping in chromosome 17 of C. pepo genome (v.4.1) is delimited between DMP34 (7,658,175 bp) and DMP41 (8,165,929).
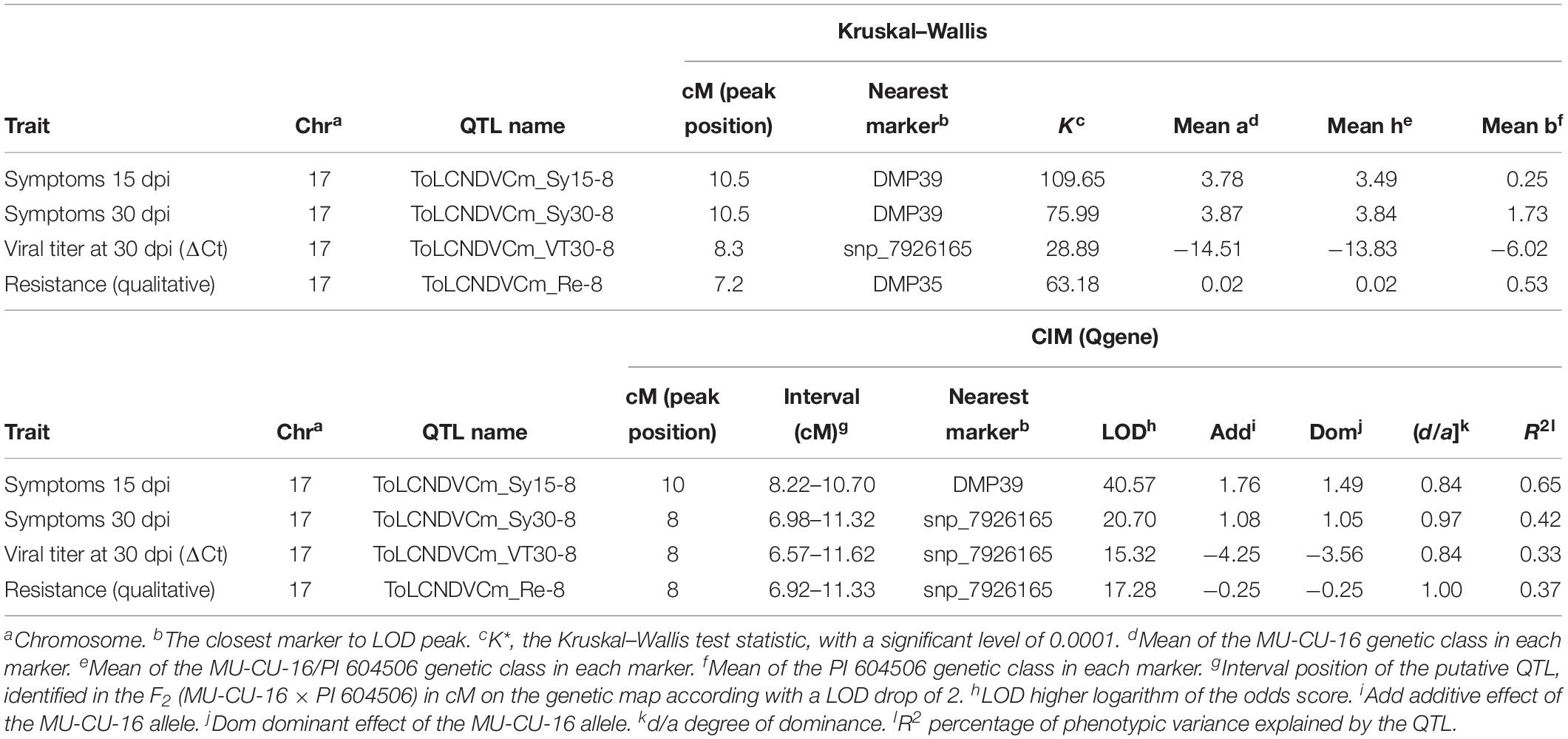
Table 4. Quantitative trait loci (QTLs) identified in the F2 (MU-CU-16 × PI 604506) segregating population genotyped with markers evenly distributed in chromosome 8 of C. moschata, using the genetic map obtained with this population, using the non-parametric Kruskal–Wallis test and composite interval mapping (CIM).
After both QTL analysis of chromosome 8, a consensus candidate region considered as responsible for ToLCNDV resistance in C. moschata, was established between DMP34 (561,788) and snp_8202510 (1,116,660).
Genomic Variation, Structural Variants, and Synteny
The alignment between the reference assemblies of C. moschata and C. pepo used for mapping purposes in the current paper6 and the new assemblies available in November 20197 showed no significant effect on the QTL region studied here (Supplementary Figure 1). Consequently, we keep working with the previous reference versions of both genomes.
A total of 53.2 and 31.5 million genomic clean reads were obtained from PI 604506 and PI 419083, respectively, and approximately more than 97% of them mapped against the C. moschata v.1 reference genome. No large structural variants were found between both accessions, and the read genome coverage was similar among them (Figure 5), which indicates that there are no deletions causing the observed phenotype. Some genomic positions show significant deviations for the expected coverage in both accessions (Figure 5), which could indicate some assembly errors on the reference genome.
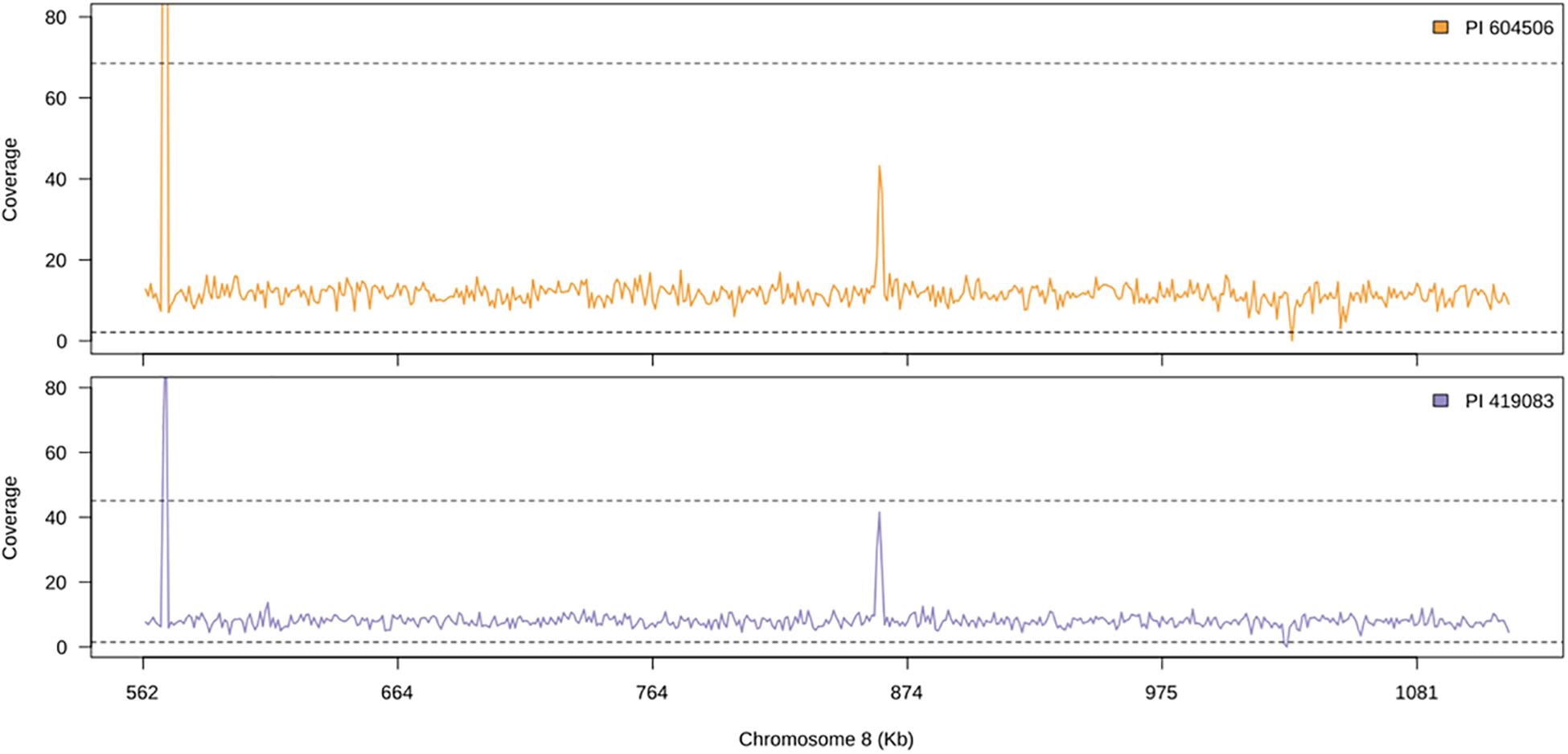
Figure 5. Genomic coverage along candidate region of chromosome 8 of the two accession used as parents for quantitative trait locus (QTL) mapping. Solid line shows the average coverage for 1-kb windows. Dashed line shows the upper and lower 99% confidence interval for the observed coverage for the whole genome.
After filtering for mapping quality, 28.2 and 18.6 million reads were kept. A total of 1,220,940 SNPs were found to be variable between both parental accessions, and 2,748 were located in the candidate region in chromosome 8. Out of them, nine SNPs had a predicted high impact (either a frameshift or missense variant, a stop codon gain/loss, or a splice site variant) and located within six genes (Supplementary Table 3). Two of these markers are located in the same genes where SNPs used in mapping (snp_7926165 and DMP44) were detected to be linked to ToLCNDV resistance [CmoCh08G001470 encoding a BZIP transcription factor bZIP80 (835,327 to 841,749 bp) and CmoCh08G001770 encoding an unknown protein (1,047,526–1,051,835 bp)]. The remaining seven SNPs with predicted high impact were located in three additional genes of this interval [CmoCh08G001130 encoding a Ribosome inactivating protein (583,200–588,238 bp), CmoCh08G001780 encoding a putative transmembrane protein (1,051,479–1,053,847 bp) and CmoCh08G001880 coding a IQ-DOMAIN 14-like protein (1,097,864–1,102,974)]. In addition, some other SNPs with low, moderate, or unknown modifying effect are placed in genes related to plant virus resistance (Supplementary Table 3).
In addition to the genomic SNPs, the transcriptomic sequences of the three parentals and the six additional susceptible C. moschata accessions provided 731 SNPs in the candidate region, 94 of them were fixed for different alleles in the PI 604506-resistant accession and in the seven susceptible accessions (Supplementary Table 3). PI 381814 transcriptomic sequence had a low coverage in the candidate region, and it was not possible to identify common polymorphisms between the two resistant accessions, PI 604506 and PI 381814. Three SNPs were detected with high predicted effect, all of them were common to those found in the genomic sequences analyzed and were located in three genes (Supplementary Table 3) (CmoCh08G001130 encoding a ribosome-inactivating protein, CmoCh08G001470 encoding a BZIP transcription factor bZIP80 and, CmoCh08G001770 encoding an unknown protein).
The structure of the candidate region was studied in more detail. A whole genome duplication likely occurred in the species that originated the Cucurbita genus (Montero-Pau et al., 2018). In fact, the search for putative paralogs of the genes in the chromosome 8 region indicated that 68 out of the 86 genes in the chromosome 8 candidate region could be assigned to an orthogroup, and 58 of them presented at least one paralog gene. These paralog genes are widespread along the genome (Figure 6), although it seems that there is a conserved duplicated region of chromosome 8 on chromosome 17. Interestingly, some genes of the candidate region have been identified as single copy in chromosome 8 (Supplementary Table 4), without paralog genes in other chromosomes, which is consistent with a major QTL responsible of ToLCNDV resistance.
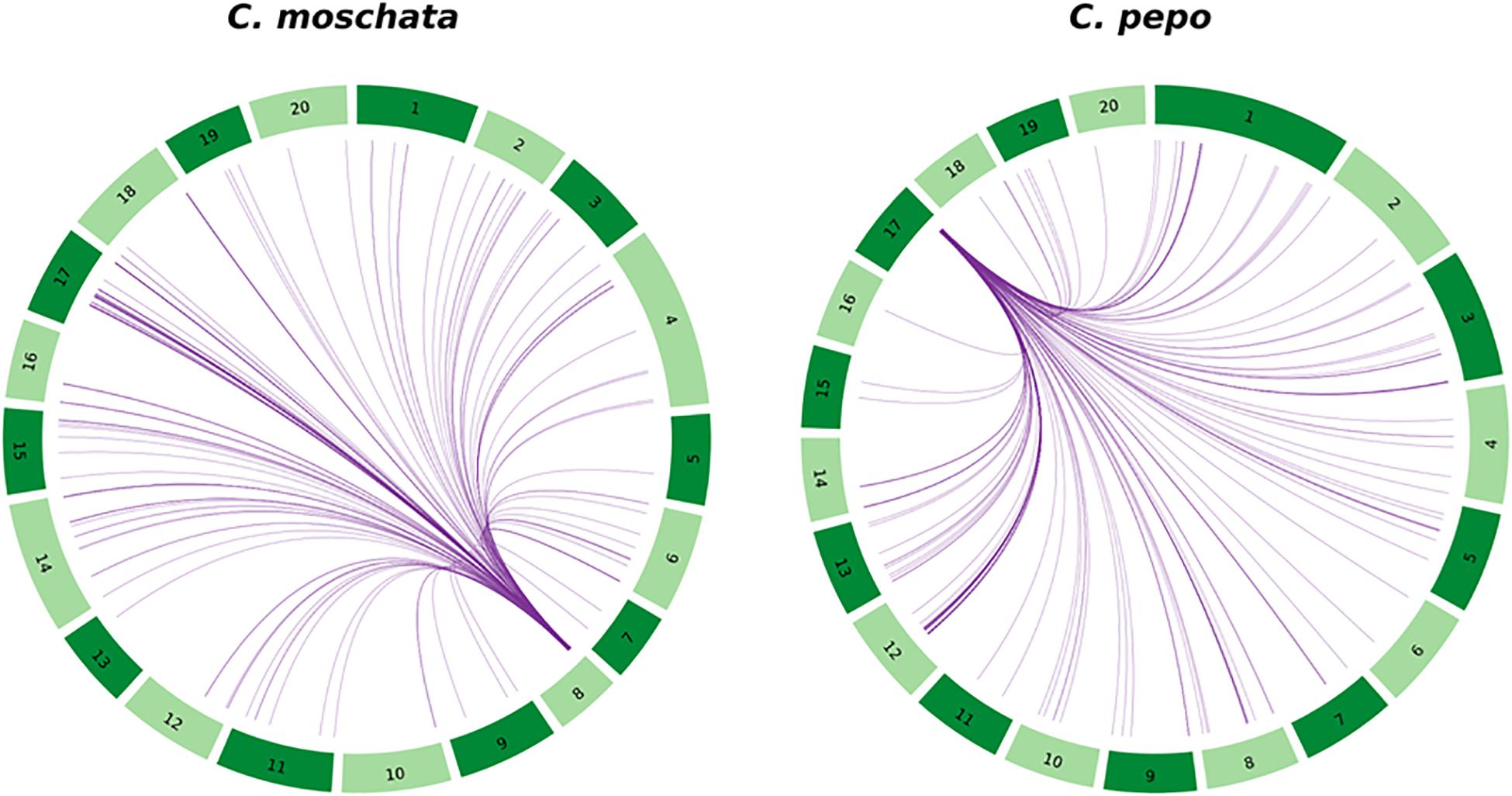
Figure 6. Circos plot showing the location of the duplicated genes located in C. moschata’s candidate region of chromosome 8 and in C. pepo’s syntenic region to C. moschata located in chromosome 17.
We also studied the synteny of this region with the susceptible C. pepo, which is phylogenetically closely related to C. moschata. BLAST alignment showed synteny between chromosome 8 region and chromosome 17 from 7,658,023 to 8,205,474 bp of C. pepo (see Figure 7 and Supplementary Table 4). Gene order and orientation is preserved for most genes, but there is one region showing INDELs. Interestingly, the region with a major insertion in C. pepo, from 8,108,962 to 8,113,419 bp, is the region in which the MAD-box transcription factor CmoCh08G001760 maps. This region correspond to position 1,024,011 bp of C. moschata, located between the 5′ untranslated region (UTR) and the first exonic region of this gene. Specific analysis of this C. pepo insertion sequence allowed to detect a long terminal repeats (LTR) retrotransposon of Ty1-copia Retrofit/Ale kind, of 3,692 bp length located from 8,109,186 to 8,113,548 bp. This transposable sequence was previously annotated using the annotation procedure for repetitive sequences described in Montero-Pau et al. (2018). Supplementary Table 6 shows the annotation results and the fasta sequence of the region. Although this insertion is absent in both resistant (PI 604506) and susceptible (PI 419083) C. moschata accessions (Figure 5), many polymorphic SNPs between them are located in this gene, including 5′ UTR and 3′ UTR variants (1,023,872 and 1,047,775 bp), and a missense variant with moderate effect (1,043,369).
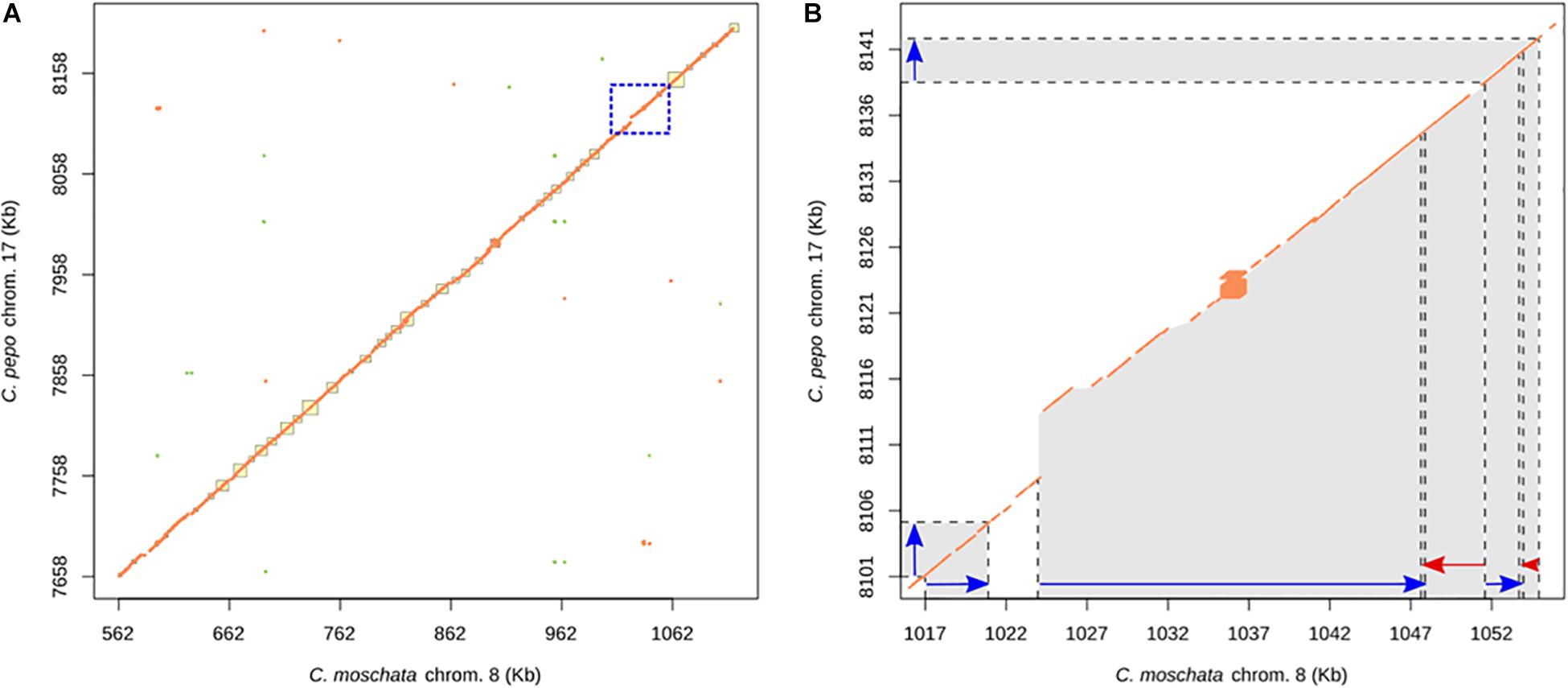
Figure 7. (A) Dot plot showing the alignment between the chromosome 8 of C. moschata assembly v.1 and chromosome 17 of C. pepo v.4.1. (B) Expanded syntenic region where large INDELs have been detected. Blue and red arrows points genes sense.
BLAST search of the C. moschata QTL region against C. melo found several syntenic regions. In the case of C. melo, highly significant alignments were obtained against chromosome 11 where a major QTL associated with resistance to ToLCNDV is located (Sáez et al., 2017). Results show inversions in SNPs positions between both species, with at least two points of inversion events and loss of information regions (Supplementary Table 2). This syntenic relationship was confirmed with the information displayed by the SyntenyViewer of cucurbitgenomics.org tool. Using chromosome 8 of C. moschata as query genome and location, circular representation showed regions of synteny with eight chromosomes of melon DHL92 (v3.6.1), including the candidate region of chromosome 11 (Figure 8A). Figures 8B,C show the syntenic blocks where ToLCNDV resistance-linked QTLs are located (coded as cmomedB906 and cmomedB910 in the database), the genomic position covered, and the graphic synteny relationship in both blocks. Furthermore, statistical significance of synteny between homologous genes in the candidate region of C. moschata and C. melo is presented in Supplementary Table 5. Seventeen genes are shared by both candidate regions, including the MAD-box transcription factor CmoCh08G001760 and the transmembrane protein CmoCh08G001780 where INDELs or high-effect SNPs have been identified.
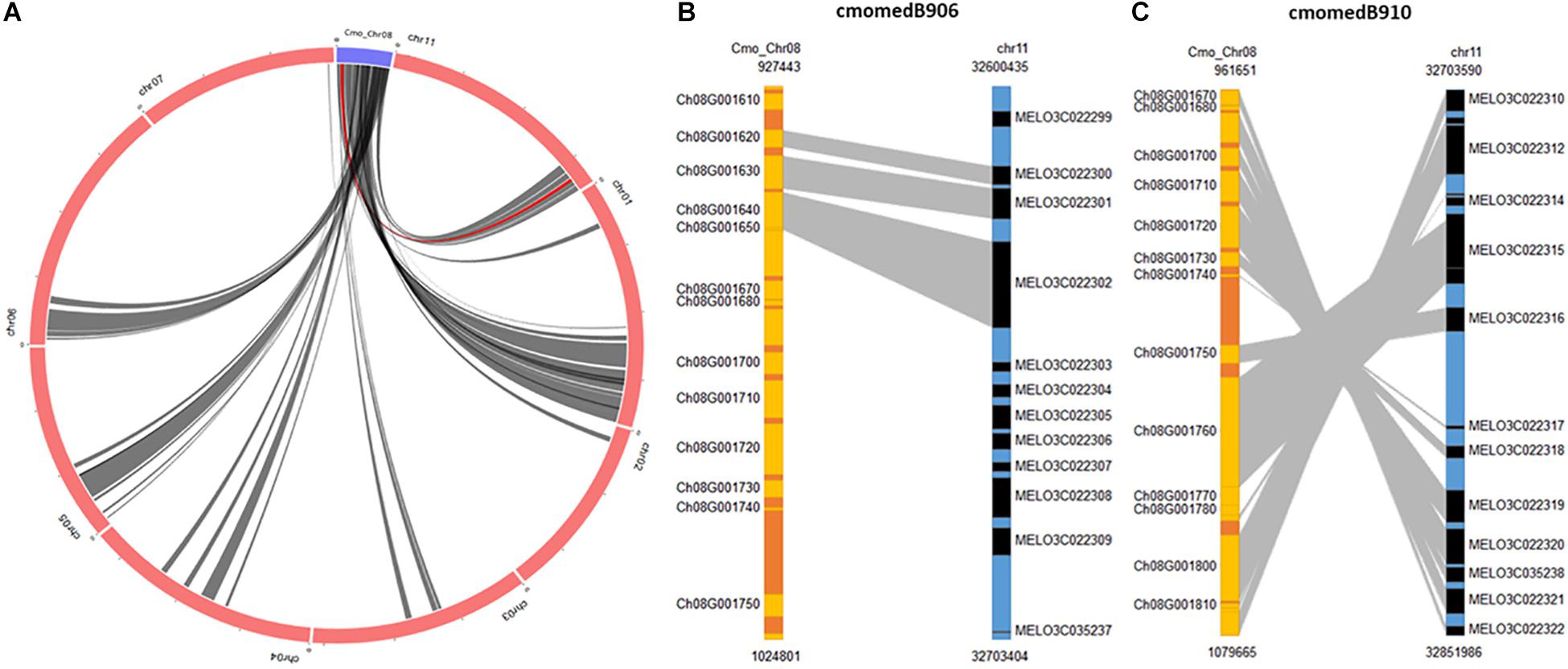
Figure 8. Synteny between the interval regions of quantitative trait loci (QTLs) detected in chromosome 8 of C. moschata and the major QTL in chromosome 11 of C. melo. (A) Circular representation showing synteny with genome of melon DHL92 (v3.6.1). (B,C) Synteny blocks where QTLs linked to tomato leaf curl New Delhi virus (ToLCNDV) resistance are located (coded as cmomedB906 and cmomedB910 in the database).
Discussion
In this work, we evaluated the resistance to ToLCNDV previously described in the two C. moschata accessions PI 604506 and PI 381814 using mechanical inoculation (Sáez et al., 2016). Our results confirmed that both genotypes remain symptomless after inoculation assays. The Large Cheese improved cultivar PI 604506 originated in the United States (Burpee Company). Even though the primary center of C. moschata diversity is located in Northern South America and Central America, it spread soon to Mexico and later to the Caribbean area and the United States, where it diversified (Decker-Walters and Walters, 2000). The landrace PI 381814 was collected in India, a secondary center of C. moschata variation, where resistance to ToLCNDV was found in melon accessions (López et al., 2015). This fact can be related with the coevolution of host and pathogen in this area, in which ToLCNDV was detected for the first time infecting cucurbits many years ago. Indian cucurbits germplasm has been previously used as source of resistances to viral and fungal pathogens (Dhillon et al., 2012; McCreight et al., 2017). Mendelian analysis of symptom segregation in F2 and BC1s populations derived from PI 604506 and PI 381814, as well as QTL results, suggested the presence of a major recessive gene in chromosome 8 of C. moschata controlling symptoms development and virus titer. Allelism test results, which show resistance in all plants of F2 (PI 604506 × PI 381814), suggests that alleles of the same locus control ToLCDV resistance in both accessions.
The occurrence of a major gene controlling ToLCNDV resistance derived from C. moschata sources is consistent with the existence of a major QTL reported to control the resistance to ToLCNDV in melon, derived from the wild Indian accession of Cucumis melo subsp. agrestis WM-7 (Sáez et al., 2017). Resistance to whitefly transmission of ToLCNDV in sponge gourd (L. cylindrica), a cucurbit crop widely cultivated in India (Islam et al., 2010), has also been described to be regulated by a main dominant gene, for which two linked sequence-related amplified polymorphism (SRAP) markers were reported (Islam et al., 2011).
Even though the major QTL linked to the resistance in C. moschata was stable in the C. pepo × C. moschata interspecific progeny, the mendelian segregation of symptoms only fitted to one recessive gene at 15 dpi. The effect of additional minor genes contributing to ToLCNDV resistance that are segregating in this interspecific population could account for these differences. In fact, in melon, besides the major QTL of chromosome 11, two additional minor regions in chromosomes 12 and 2 modifying the resistant response were identified (Sáez et al., 2017). In a recent publication (Romay et al., 2019), one recessive (bgm-1) and two dominant (Bgm-2 and Tolcndv) genes were also found controlling resistance to ToLCNDV in the same Indian accession WM-7. A similar oligogenic control, three dominant genes, has been reported in S. habrochaites S. Knapp & D. M. Spooner, a wild species related to tomato, after ToLCNDV agroinoculation (Rai et al., 2013).
The role of the genetic background in resistance to plant viruses is considered determinant in breeding programs when transferring QTLs from one species to another. Gallois et al. (2018) have studied and reviewed the effect of epistatic relationship with QTL analysis on virus resistance, suggesting that a major-effect QTL (proportion of phenotypic variance explained by the QTL R2 > 0.60) could be more susceptible to genetic background influence than minor-effect QTLs. This statement supports the incomplete penetrance obtained when we tried to transfer the QTL conferring resistance to ToLCNDV from chromosome 8 of C. moschata into C. pepo background. In this work, R2 percentages of QTLs detected in the F2 (PI 419083 × PI 604506) at 30 dpi ranged between 53 and 64% (Table 3), while in F2 (MU-CU-16 × PI 604506), the R2 percentages of QTLs linked to the same candidate region decreased from 15 dpi (R2 = 65%) to 30 dpi (R2 = 33–42%). These results suggest the requirement of other loci fixed in the C. moschata genetic background needed in the mechanism of resistance to ToLCNDV, but segregating in C. pepo. With this information, it is recommended to select for resistance at 30 dpi in populations coming from interspecific crosses, as it is the final stage of infection that better reflect the final response of the plants to the virus.
Resistance to ZYMV was found in different C. moschata accessions (Munger and Provvidenti, 1987; Paris et al., 1988; Wessel-Beaver, 2005). The resistance in the Portuguese C. moschata accession, Menina, is conferred by one dominant gene, Zym-1, in the cross with the susceptible C. moschata Waltham Butternut (Paris et al., 1988). However, when the resistance from Menina was introgressed into the C. pepo, segregation did not adjust to a single-gene ratio, and other additional dominant genes, Zym-2 and Zym-3, seemed to be involved in the resistance (Paris and Cohen, 2000). According to Pachner et al. (2011), even Zym-1, Zym-2, and Zym-3 together in C. pepo do not confer the same level of resistance seen in “Menina.” Studies of inheritance of ZYMV resistance showed that the presence of Zym-1 is essential, but must be combined with other six genes to obtain different levels of expression and durability of resistance in C. pepo (Pachner et al., 2015; Capuozzo et al., 2017). In accordance with these works, future QTL analysis of F2 (MU-CU-16 × PI 604506), including genotyping with SNPs covering the whole Cucurbita genome, are crucial to reveal epistatic effects of other loci affecting ToLCNDV resistance.
The major locus for resistance to ToLCNDV in chromosome 8 of both C. moschata sources, PI 604506 and PI 381814, is recessively inherited. Recessive resistance genes, or susceptibility genes, because their presence conditions virus susceptibility (Garcia-Ruiz, 2018), are a common defense strategy against plant viruses (Diaz-Pendón et al., 2004; Kang et al., 2005). In cucurbits, recessive resistance genes have been reported in several viruses. Translation initiation factors eIF(iso)4E and eIF4G confer recessive resistance against a subset of viruses in several crop species (Hashimoto et al., 2016). The nsv recessive gene, encoding an eIF4E factor, confers resistance to melon necrotic spot virus (Nieto et al., 2006), preventing the accumulation of viral RNA at the single-cell level (Díaz et al., 2002). In potyvirus-infected Nicotiana benthamiana leaf tissues, DEAD-box RNA helicase RH8, which share sequence homology with eIF4A, a component of the eIF4F multiprotein complex, is involved in viral genome translation and replication (Huang et al., 2010). We searched for putative eIF4E and eIF4F at the candidate region for ToLCNDV resistance of C. moschata annotation reference genome and found that two genes (CmoCh08G001290 and CmoCh08G001490) encoding an ATP-dependent RNA helicase and chromodomain-helicase-DNA-binding protein 1-like protein, respectively, mapped on the candidate region of chromosome 8. Concretely, CmoCh08G001490 is a single-copy gene in C. moschata, with a 3′ UTR SNP variant in PI 604506 sequence and is syntenic with a basic leucine zipper (BZIP) domain class transcription factor gene (MELO3C022278) of the chromosome 11 of C. melo.
In addition, other strategies have been reported for recessive resistance against viruses. The recessive cmv1 gene that confers resistance to cucumber mosaic virus in melon encodes a vacuolar protein sorting 41 (VPS41) (Giner et al., 2017) involved in membrane trafficking to the vacuole. Membrane components are key factors required for plant infection success, and viral replication is associated with host intracellular membranes (Nicaise, 2014). In the case of tom1 and tom2A Arabidopsis mutants, tobacco mosaic virus (TMV) accumulation is suppressed in single cells. Both genes encode transmembrane proteins localized in the tonoplast that are required for tobamovirus replication (Ishibashi et al., 2012). Among the annotated genes within the C. moschata candidate region here identified, several genes are related with membrane components. CmoCh08G001420 encodes a vesicle transport protein and CmoCh08G001500 an autophagy-related protein 3. Interestingly, two of the genes where high-impact SNPs have been detected are annotated as putative transmembrane protein (CmoCh08G001780 and CmoCh08G001790), included in the syntenic region between both candidate regions with resistance to ToLCNDV in C. moschata and C. melo.
Comparative physical mapping revealed a high level of synteny between the candidate regions with the major QTLs controlling ToLCNDV resistance of chromosomes 8 and 11 of C. moschata and C. melo, respectively (Sáez et al., 2017). The interval of ∼118 kb encompasses genes from CmoCh08G001670 to CmoCh08G001830 of C. moschata. Comparing the orientations of this syntenic block, the physical positions of genes in both genomes are reversed. Inversions are believed to play an important role in speciation and local adaptation by reducing recombination and protecting genomic regions from introgression (Yang L. et al., 2014). The cluster of genes within this syntenic region contains transcription factors that have been described to confer resistance to viruses in different crops.
Genes of the same family of the WRKY transcription factor-like protein of C. moschata (CmoCh08G001670) appears to be involved in defense responses upon TMV infection in C. annuum (Huh et al., 2012). In PI 604506, six 3′ UTR variants are affecting this gene. Moreover, a BZIP transcription factor gene (CmoCh08G001710) is placed close to SNP_8061105. Although CmoCh08G001470 is not placed in the syntenic region with C. melo, it also encodes a BZIP transcription factor gene. Particularly, a stop codon lost has been detected in this gene of PI 604506, which could alter the primary structure of the protein.
Two genes encoding MADS-box transcription factors are in this same region (CmoCh08G001750 and CmoCh08G001760). This gene family has been associated to different virus-resistance mechanisms. A MADS-box transcription factor was described as the Ty-2 candidate, involved in the tomato resistance to tomato yellow leaf curl virus (TYLCV) (Yang X. et al., 2014), and recently, a MADS-box gene has been reported to be upregulated in the Sw-7 resistance to tomato spotted wilt tospovirus (TSWV) (Padmanabhan et al., 2019). No SNPs with high-impact predicted effect were identified in CmoCh08G001760 between C. moschata accessions, but changes in 5′ and 3′ UTRs and a missense mutation with predicted moderate effect were polymorphic between resistant and susceptible accessions. This gene has no ortholog in C. pepo chromosome 17, likely due to the insertion affecting this region of the genome. The possible involvement of this gene in ToLCNDV resistance would explain the total susceptibility to ToLCNDV found within C. pepo species (Sáez et al., 2016) and the difficulties to introgress the resistance locus from C. moschata to C. pepo.
The CmoCh08G001760 gene has paralogs in different chromosomes of C. moschata. OrthoMCL detected eight putative paralogs in different chromosomes of C. moschata (Chr1, Chr8, Chr12, Chr14, Chr17, and Chr18). The alignment of the aa sequences of the C. moschata paralogs and all MADs-box genes of Arabidopsis thaliana shows that CmoCH08G001760.1 is most similar to the C. moschata paralog located in Chr17, CmoCh17G013780.1. Both genes clustered together and apart from A. thaliana genes (Supplementary Figure 2). The detailed comparison of the aa sequences of both genes showed significant length differences (169 aa versus 71 aa, for CmoCH08G001760 and CmoCh17G013780.1 respectively). Both proteins have a common MADs motif at the N-terminus of the protein but differ in the rest of the sequence. These results are consistent with a different function of both genes. The sequence comparison of the CmoCh17G013780.1 gene of both parentals, PI 604506 and PI 419083 (done using the genomic sequences available at NCBI under BioProject PRJNA604046), does not provide SNP variants between them, also supporting the absence of a role of this paralog in ToLNDV resistance.
Molecular markers located close to the QTLs detected here can be used in marker-assisted selection in breeding ToLCNDV-resistant pumpkins and squash. Further genetic and transcriptomic studies of the candidate genes for resistance to ToLCNDV in the different cucurbit sources of resistance analyzed to date, are needed to develop strategies to control virus useful in different species of this crop family.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
CS, BP, and CL conceived and designed the research. CS, CL, and AS performed the tests with ToLCNDV. CS, CM, CE, and BP conducted the marker development and mapping analysis. JM-P, JB, and CS performed the bioinformatics analysis of the genomic variation and synteny. CS and BP conducted and wrote the manuscript with important contributions from JM-P, MF, and CL. All authors read and approved the final manuscript.
Funding
This work was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades, cofunded with FEDER funds [Project Nos. AGL2017-85563-C2-1-R and RTA2017-00061-C03-03 (INIA)] and by PROMETEO project 2017/078 (to promote excellence groups) by the Conselleria d’Educació, Investigació, Cultura i Esports (Generalitat Valenciana). CS is a recipient of a predoctoral fellowship from Generalitat Valenciana, cofunded by the Operational Program of the European Social Fund (FSECV 2014-2020) (Grant No. ACIF/2016/188). CM was a recipient of a postdoctoral Juan de la Cierva Formation (2014) fellowship from Spanish Ministerio de Ciencia, Innovación y Universidades (Grant No. FJCI-2014-19817).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Eva Martínez and Gorka Perpiñá for their technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00207/full#supplementary-material
FIGURE S1 | Dot plot showing the alignment between the QTL region in the previous assembly and the new assembly. (A) Chromosome 8 of C. moschata assembly v.1 vs. scaffold 15 of the new assembly, (B) chromosome 17 of C. pepo v.4.1 and scaffold 17 of the new assembly and (C) syntenic region for C. moschata and C. pepo new assembly.
FIGURE S2 | (A) Maximum likelihood tree, built using IQ-TREE v.1.5.2 (Nguyen et al., 2015), of amino acid sequences of Arabidopsis thaliana MADs-box and CmoCh08G001760 paralogs (in bold). Bootstrap values higher than 0.5 and lower than 0.95 are shown in the tree. Nodes with bootstrap values lower than 0.5 have been collapsed. (B) Conserved motifs found in all the C. moschata paralogs of gene CmoCH08G001760 by MEME Suite v.5.0.3. Red box represent the MAD motif.
TABLE S1 | List of SNP markers polymorphic in the F2 population derived from the cross PI 419083 × PI 604506. Their positions in the Cucurbita moschata genome are according to the Version 1 (http://cucurbitgenomics.org/). The positions in the genetic map is according to the genetic map constructed with the F2 plants in this study and used for QTL analysis.
TABLE S2 | SNPs used in all the QTLs validations. The chromosome and genetic position in each C. moschata × C. moschata and C. pepo × C. moschata linkage maps is indicated. Genomic position is shown in the last version of the C. moschata (v1.0), C. pepo (v4.1) and C. melo (v3.6.1) genomes available in Cucurbit Genomics Database (http://cucurbitgenomics.org). Flanking sequence of all markers is provided as well as the oligonucleotides used in the HRM genotyping assay. Markers that failed to show the expected polymorphism between parental sequences are marked as no data (−). Query cover, identity and E-value of Blast alignments with chromosome 11 of C. melo is also provide. Markers without significant similarity with chromosome 11 of C. melo, considering a minimum overlap between sequences of 70%, are shown as not applicable data (n/a). Striped and dotted lines are enclosing the interval position of the QTLs identified in the F2 (PI 419083 × PI 604506) and F2 (MU-CU-16 × PI 604506), respectively.
TABLE S3 | List of SNPs with their predicted effect on gene function on candidate region for C. moschata parents and for the six transcriptomes of C. moschata. Gene affected, SNP location, reference and alternative alleles, allele causing the predicted high impact, type of change and genotype for each accession is shown. Note that genotypes are coded with number of allele, where 0 means the reference allele and 1,2,3 refer to the alternative allele in the displayed order. Genes with related function to plant virus resistance are marked with ∗.
TABLE S4 | Genes annotated in the candidate region of chromosome 8 of C. moschata. Their paralog genes in chromosome 17 of C. moschata are shown, as well as BLAST alignment results with chromosome 17 of C. pepo. Those genes with none paralogs copies identified along the C. moschata genome are marked with ∗.
TABLE S5 | Statistical significance of synteny between homologous genes in candidate region for ToLCNDV resistance in chromosome 8 of C. moschata and chromosome 11 of C. melo genomes v1.0 and v3.6.1, respectively. Genes shown in bold are indicating the pick LOD in both QTLs analysis Cucurbita populations and C. melo, respectively. Dotted lines are defining the candidate region of C. melo were the major locus, responsible of the resistance to ToLCNDV, was identified.
TABLE S6 | Annotation results and fasta sequence of the Long Terminal Repeats (LTR) retrotransposon in C. pepo genome. The region was annotated according to the methodology for annotate repetitive elements described at Montero-Pau et al. (2018).
Footnotes
- ^ http://cucurbitgenomics.org
- ^ http://cucurbitgenomics.org
- ^ https://github.com/JoseBlanca/
- ^ https://www.dnazoo.org/assemblies/Cucurbita_moschata
- ^ https://www.dnazoo.org/assemblies/Cucurbita_pepo
- ^ http://cucurbitgenomics.org
- ^ https://www.dnazoo.org/assemblies/
References
Bio-Rad Laboratories, (2006). Real-Time PCR Applications Guide. Hercules, CA: Bio-Rad Laboratories, Inc, 42–43.
Capuozzo, C., Formisano, G., Iovieno, P., Andolfo, G., Tomassoli, L., Barbella, M. M., et al. (2017). Inheritance analysis and identification of SNP markers associated with ZYMV resistance in Cucurbita pepo. Mol. Breed. 37:99. doi: 10.1007/s11032-017-0698-5
Chang, H. H., Ku, H. M., Tsai, W. S., Chien, R. C., and Jan, F. J. (2010). Identification and characterization of a mechanical transmissible begomovirus causing leaf curl on oriental melon. Eur. J. Plant Pathol. 127, 219–228. doi: 10.1007/s10658-010-9586-0
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Källberg, M., et al. (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. doi: 10.1093/bioinformatics/btv710
Cingolani, P., Platts, A., Wang, le L, Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92. doi: 10.4161/fly.19695
Decker-Walters, D., and Walters, T. (2000). “Squash,” in The Cambridge World History of Food, eds K. Kiple and K. Ornelas (Cambridge, MA: Cambridge University Press), 335–351.
Dhillon, N. P. S., Monforte, A. J., Pitrat, M., Pandey, S., Singh, P. K., Reitsma, K. R., et al. (2012). Melon landraces of India: contributions and importance. Plant Breed. 35, 85–150. doi: 10.1002/9781118100509.ch3
Díaz, J. A., Nieto, C., Moriones, E., and Aranda, M. A. (2002). Spanish Melon necrotic spot virus isolate overcomes the resistance conferred by the recessive nsv gene of melon. Plant Dis. 86:694. doi: 10.1094/PDIS.2002.86.6.694C
Diaz-Pendón, J. A., Trunger, V., Nieto, C., García-Mas, J., Bendahmane, A., and Aranda, M. A. (2004). Advances in understanding recessive resistance to plant viruses. Mol. Plant Pathol. 5, 223–233. doi: 10.1111/j.1364-3703.2004.00223.x
EPPO (2017). European and Mediterranean Plant Protection Organization. Available at: https://www.eppo.int/QUARANTINE/Alert_List/viruses/ToLCNDV.htm (accessed on 28 August 2017).
EPPO (2019). European and Mediterranean Plant Protection Organization. Available at https://gd.eppo.int/taxon/TOLCND/distribution (accessed on 20 November 2019).
FAO (2017). FAOSTAT. Available at: http://www.fao.org/faostat/en/#data/QC (accessed 5 February 2020).
Gallois, J. L., Moury, B., and German-Retana, S. (2018). Role of the genetic background in resistance to plant viruses. Int. J. Mol. Sci. 19:2856. doi: 10.3390/ijms19102856
Garcia-Ruiz, H. (2018). Susceptibility genes to plant viruses. Viruses 10:484. doi: 10.3390/v10090484
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv [Preprint]. arXiv:1207.3907
Giner, A., Pascual, L., Bourgeois, M., Gyetvai, G., and Rios, P. (2017). A mutation in the melon vacuolar protein sorting 41 prevents systemic infection of Cucumber mosaic virus. Sci. Rep. 7:10471. doi: 10.1038/s41598-017-10783-3
Haider, M. S., Tahir, M., Latif, S., and Briddon, R. W. (2006). First report of tomato leaf curl New Delhi virus infecting Eclipta prostrata in Pakistan. Plant Pathol. 55:285. doi: 10.1111/j.1365-3059.2005.01278.x
Hashimoto, M., Neriya, Y., Yamaji, Y., and Namba, S. (2016). Recessive resistance to plant viruses: potential resistance genes beyond translation initiation factors. Front. Microbiol. 7:1695. doi: 10.3389/fmicb.2016.01695
Huang, T. S., Wei, T., Laliberté, J. F., and Wang, A. (2010). A host RNA helicase-like protein, AtRH8, interacts with the potyviral genome-linked protein, VPg, associates with the virus accumulation complex, and is essential for infection. Plant Physiol. 152, 255–266. doi: 10.1104/pp.109.147983
Huh, S. U., Choi, L. M., Lee, G. J., and Kim, Y. J. (2012). Capsicum annuum WRKY transcription factor d (CaWRKYd) regulates hypersensitive response and defense response upon tobacco mosaic virus infection. Plant Sci. 197, 50–58. doi: 10.1016/j.plantsci.2012.08.013
Hussain, M., Mansoor, S., Iram, S., Zafar, Y., and Briddon, R. W. (2004). First report of tomato leaf curl New Delhi virus affecting chilli pepper in Pakistan. Plant Pathol. 53:794. doi: 10.1111/j.1365-3059.2004.01073.x
Ishibashi, K., Miyashita, S., Katoh, E., and Ishikawa, M. (2012). Host membrane proteins involved in the replication of tobamovirus RNA. Curr. Opin. Virol. 2, 693–698. doi: 10.1016/j.coviro.2012.09.011
Islam, S., Munshi, A. D., Mandal, B., Behera, T. K., and Kumar, R. (2010). Genetics of resistance in Luffa cylindrical Roemagainst tomato leaf curl New Delhi virus. Euphytica 174, 83–89. doi: 10.1007/s10681-010-0138-7
Islam, S., Munshi, A. D., Verma, M., Arya, L., Mandal, B., Behera, T. K., et al. (2011). Screening of Luffa cylindrical Roem for resistance against tomato leaf curl New Delhi virus, inheritance of resistance, and identification of SRAP markers linked to the single dominant resistance gene. J. Hortic. Sci. Biotechnol. 86, 661–667. doi: 10.1080/14620316.2011.11512819
Ito, T., Sharma, P., Kittipakorn, K., and Ikegami, M. (2008). Complete nucleotide sequence of a new isolate of tomato leaf curl New Delhi virus infecting cucumber, bottle gourd and muskmelon in Thailand. Arch. Virol. 153, 611–613. doi: 10.1007/s00705-007-0029-y
Jamil, N., Rehman, A., Hamza, M., Hafeez, A., Ismail, H., Zubair, M., et al. (2017). First report of tomato leaf curl New Delhi virus, a bipartite begomovirus, infecting soybean (Glycine max). Plant Dis. 101, 845. doi: 10.1094/PDIS-09-16-1267-PDN
Janssen, D., Simon, A., Crespo, O., and Ruiz, L. (2017). Genetic population structure of Bemisia tabaci in Spain associated with Tomato leaf curl New Delhi virus. Plant Protect. Sci. 53, 25–31. doi: 10.17221/62/2016-PPS
Joehanes, R., and Nelson, J. C. (2008). QGene 4.0, an extensible Java QTL-analysis platform. Bioinformatics 24, 2788–2789. doi: 10.1093/bioinformatics/btn523
Juárez, M., Rabadán, M. P., Díaz Martínez, L., Tayahi, M., Grande-Pérez, A., and Gómez, P. (2019). Natural hosts and genetic diversity of the emerging tomato leaf curl New Delhi virus in spain. Front. Microbiol. 2019:140. doi: 10.3389/fmicb.2019.00140
Juárez, M., Tovar, R., Fiallo-Olivé, E., Aranda, M. A., Gosálvez, B., Castillo, P., et al. (2014). First detection of tomato leaf curl New Delhi virus infecting Zucchini in Spain. Plant Dis. 98, 857–858. doi: 10.1094/pdis-10-13-1050-PDN
Jyothsna, P., Haq, Q. M. I., Singh, P., Sumiya, K. V., Praveen, S., Rawat, R., et al. (2013). Infection of tomato leaf curl New Delhi virus (ToLCNDV), a bipartite begomovirus with betasatellites, results in enhanced level of helper virus components and antagonistic interaction between DNA B and betasatellites. Appl. Microbiol. Biotechnol. 97, 5457–5471. doi: 10.1007/s00253-012-4685-9
Kang, B. C., Yeam, I., and Jahn, M. M. (2005). Genetics of plant virus resistance. Annu. Rev. Phytopathol. 43, 581–621. doi: 10.1146/annurev.phyto.43.011205.141140
Kheireddine, A., Sifres, A., Sáez, C., Picó, B., and López, C. (2019). First report of tomato leaf curl New Delhi virus infecting cucurbit plants in Algeria. Plant Dis. 103:3291. doi: 10.1094/PDIS-05-19-1118-PDN
Kielbasa, S. M., Wan, R., Sato, K., Horton, P., and Frith, M. C. (2011). Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493. doi: 10.1101/gr.113985.110
Lander, E. S., Green, P., Abrahamson, J., Barlow, A., and Daly, M. J. (1987). MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 174–181. doi: 10.1016/0888-7543(87)90010-3
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. arXiv:1303.3997
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoecker, T. C. J. Jr., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lincoln, S., Daly, M., and Lander, E. (1992). Constructing genetic maps with MAPMAKER/EXP 3.0. Whitehead Institute Technical Report, 3rd Edn. Cambridge, MA: Whitehead Institute.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–DDCt method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
López, C., Ferriol, M., and Picó, M. B. (2015). Mechanical transmission of tomato leaf curl New Delhi virus to cucurbit germplasm: selection of tolerance sources in Cucumis melo. Euphytica 204, 679–691. doi: 10.1007/s10681-015-1371-x
McCreight, J. D., Wiutemantel, W. M., Natwick, E. T., Sinclair, J. W., Crosby, K. M., and Gómez-Guillamón, M. L. (2017). Recessive resistance to CYSDV in melon TGR 1551. Acta Hortic. 1151, 101–108. doi: 10.17660/ActaHortic.2017.1151.17
Mnari-Hattab, M., Zammouri, S., Belkadhi, M. S., Bellon Doña, D., Ben Nahia, E., Hajlaoui, M. R., et al. (2015). First report of tomato leaf curl New Delhi virus infecting cucurbits in Tunisia. New Dis. Rep. 31:21. doi: 10.5197/j2044-05882015031021
Montero-Pau, J., Blanca, J., Bombarely, A., Ziarsolo, P., Esteras, C., Martí-Gómez, C., et al. (2018). De-novo assembly of zucchini genome reveals a whole genome duplication associated with the origin of the Cucurbita genus. Plant Biotechnol. J. 16, 1161–1171. doi: 10.1111/pbi.12860
Moriones, E., Praveen, S., and Chakraborty, S. (2017). Tomato leaf curl New Delhi virus: an emerging virus complex threatening vegetable and fiber crops. Viruses 9:264. doi: 10.3390/v9100264
Munger, H. M., and Provvidenti, R. (1987). Inheritance of resistance to zucchini yellow mosaic virus in Cucurbita moschata. Cucurbit Genet. Coop. Rep 10, 8–81.
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nicaise, V. (2014). Crop immunity against viruses: outcomes and future challenges. Front. Plant Sci. 5:660. doi: 10.3389/fpls.2014.00660
Nieto, C., Morales, M., Orjeda, G., Clepet, C., Monfort, A., Sturbois, B., et al. (2006). An eIF4E allele confers resistance to an uncapped and non-polyadenylated RNA virus in melon. Plant J. 48, 452–462. doi: 10.1111/j.1365-313X.2006.02885.x
Orfanidou, C. G., Malandraki, I., Beris, D., Kektsidou, O., Vassilakos, N., Varveri, C., et al. (2019). First report of tomato leaf curl New Delhi virus in zucchini crops in Greece. J. Plant Pathol. 101:799. doi: 10.1007/s42161-019-00265-y
Pachner, M., Paris, H. S., and Lelley, T. (2011). Genes for resistance to zucchini yellow mosaic in tropical pumpkin. J. Hered. 102, 330–335. doi: 10.1093/jhered/esr006
Pachner, M., Paris, H. S., Winkler, J., and Lelley, T. (2015). Phenotypic and marker-assisted pyramiding of genes for resistance to zucchini yellow mosaic virus in oilseed pumpkin (Cucurbita pepo). Plant Breed. 134, 121–128. doi: 10.1111/pbr.12219
Padidam, M., Beachy, R. N., and Fauquet, C. M. (1995). Tomato leaf curl geminivirus from India has a bipartite genome and coat protein is not essential for infectivity. J Gen. Virol. 76, 25–35. doi: 10.1099/0022-1317-76-1-25
Padmanabhan, C., Ma, Q., Shekasteband, R., Stewart, K. S., Hutton, S. F., Scott, J. W., et al. (2019). Comprehensive transcriptome analysis and functional characterization of PR-5 for its involvement in tomato Sw-7 resistance to tomato spotted wilt tospovirus. Sci. Rep. 9:7673. doi: 10.1038/s41598-019-44100-x
Panno, S., Iacono, G., Davino, M., Marchione, S., Zappardo, V., Bella, P., et al. (2016). First report of tomato leaf curl New Delhi virus affecting zucchini squash in an important horticultural area of southern Italy. New Dis. Rep. 33:6. doi: 10.5197/j2044-05882016033006
Paris, H. S., and Cohen, S. (2000). Oligogenic inheritance for resistance to zucchini yellow mosaic virus in Cucurbita pepo. Ann. Appl. Biol. 136, 209–214. doi: 10.1111/j.1744-7348.2000.tb00027.x
Paris, H. S., Cohen, S., Burger, Y., and Joseph, R. (1988). Single-gene resistance to zucchini yellow mosaic virus in Cucurbita moschata. Euphytica 37, 27–29. doi: 10.1007/BF00037219
Pratap, D., Kashikar, A. R., and Mukherjee, S. K. (2011). Molecular characterization and infectivity of a tomato leaf curl New Delhi virus variant associated with newly emerging yellow mosaic disease of eggplant in India. Virol. J. 8, 305. doi: 10.1186/1743-422X-8-305
Rai, N. K., Sahu, P. P., Gupta, S., Reddy, M. K., Ravishankar, K. V., Singh, M., et al. (2013). Identification and validation of an ISSR marker linked to Tomato leaf curl New Delhi virus resistant gene in a core set of tomato accessions. Vegetable Sci. 40, 1–6.
Romay, G., Pitrat, M., Lecoq, H., Wipf-Scheibel, C., Millot, P., Girardot, G., et al. (2019). Resistance against melon chlorotic mosaic virus and tomato leaf curl New Delhi virus in melon. Plant Dis. 103, 2913–2919. doi: 10.1094/PDIS-02-19-0298-RE
Rosen, R., Kanakala, S., Kliot, A., Pakkianathan, B. C., Farich, B. A., Santana-Magal, N., et al. (2015). Persistent, circulative transmission of begomoviruses by whitefly vectors. Curr. Opin. Virol. 15, 1–8. doi: 10.1016/j.coviro.2015.06.008
Roy, A., Spoorthi, P., Panwar, G., Bag, M. K., Prasad, T. V., Kumar, G., et al. (2013). Molecular evidence for occurrence of tomato leaf curl New Delhi virus in ash gourd (Benincasa hispida) germplasm showing a severe yellow stunt disease in India. Indian J. Virol. 24, 74–77. doi: 10.1007/s13337-012-0115-y
Sáez, C., Esteras, C., Martínez, C., Ferriol, M., Narinder, P. S. D., López, C., et al. (2017). Resistance to tomato Leaf Curl New Delhi Virus in melon is controlled by a major QTL located in chromosome 11. Plant Cell Rep. 36, 1571–1584. doi: 10.1007/s00299-017-2175-3
Sáez, C., Martínez, C., Ferriol, M., Manzano, S., Velasco, L., Jamilena, M., et al. (2016). Resistance to tomato leaf curl New Delhi virus in Cucurbita spp. Ann. Appl. Biol. 169, 91–105. doi: 10.1111/aab.12283
Sifres, A., Sáez, C., Ferriol, M., Selmani, E., Riado, J., Picó, B., et al. (2018). First Report of tomato leaf curl New Delhi virus infecting Zucchini in morocco. Plant Dis. 102:1045. doi: 10.1094/PDIS-10-17-1600-PDN
Singh, A. K., Mishra, K. K., Chattopadhyay, B., and Chakraborty, S. (2009). Biological and molecular characterization of a begomovirus associated with yellow mosaic vein mosaic disease of pumpkin from Northern India. Virus Genes 39, 359–370. doi: 10.1007/s11262-009-0396-4
Sohrab, S. S., Karim, S., Varma, A., Abuzenadah, A. M., Chaudhary, A. G., Damanhouri, G. A., et al. (2013). Characterization of tomato leaf curl New Delhi virus infecting cucurbits: evidence for sap transmission in a host specific manner. Afr. J. Biotechnol. 12, 5000–5009. doi: 10.5897/AJB2013.12012
Sohrab, S. S., Mandal, B., Pant, R. P., and Varma, A. (2003). First report of association of tomato leaf curl virus-New Delhi with yellow mosaic disease of Luffa cylindrica in India. Plant Dis. 87:1148. doi: 10.1094/PDIS.2003.87.9.1148A
Srivastava, A., Kumar, S., Jaidi, M., Raj, S. K., and Shukla, S. K. (2016). First report of tomato leaf curl New Delhi virus on opium poppy (Papaver somniferum) in India. Plant Dis. 100:232. doi: 10.1094/PDIS-08-15-0883-PDN
Srivastava, K. M., Hallan, V., Raizada, R. K., Chandra, G., Singh, B. P., and Sane, P. V. (1995). Molecular cloning of Indian tomato leaf curl virus genome following a simple method of concentrating the supercoiled replicative form of viral DNA. J. Virol. Methods 51, 297–304. doi: 10.1016/0166-0934(94)00122-w
Sun, H., Wu, S., Zhang, G., Jiao, C., Guo, S., Ren, Y., et al. (2017). Karyotype stability and unbiased fractionation in the paleo-allotetraploid cucurbita genomes. Mol. Plant. 10, 1293–1306. doi: 10.1016/j.molp.2017.09.003
Sundararaj, D., Jesse, M., Gunasekaran, D., Riyaz, M., Thangavelu, R., Kathiravan, K., et al. (2019). First report of tomato leaf curl New Delhi virus infecting Crossandra infundibuliformis (L.) in India. Plant Dis. 104. doi: 10.1094/PDIS-08-19-1764-PDN
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3–new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Usharani, K. S., Surendranath, B., Paul-Khurana, S. M., Garg, I. D., and Malathi, V. G. (2004). Potato leaf curl – a new disease of potato in northern India caused by a strain of tomato leaf curl New Delhi virus. Plant Pathol. 53:235. doi: 10.1111/j.0032-0862.2004.00959.x
Van Ooijen, J. W. (2009). MapQTL® 6 Software for the Mapping of Quantitative Trait Loci in Experimental Population of Diploid Species. Wageningen: Kayzma BV.
Vossen, R. H., Aten, E., Roos, A., and den Dunnen, J. T. (2009). High-resolution melting analysis (HRMA) - More than just sequence variant screening. Hum. Mutat. 30, 860–866. doi: 10.1002/humu.21019
Wessel-Beaver, L. (2005). Cultivar and germplasm release. Release of ‘Soler’ tropical pumpkin. J. Agric. Univ. 89, 263–266.
Yang, L., Koo, D., Li, D., Zhang, T., Jiang, J., Luan, F., et al. (2014). Next-generation sequencing, FISH mapping and synteny-based modeling reveal mechanisms of decreasing dysploidy in Cucumis. Plant J. 77, 16–30. doi: 10.1111/tpj.12355
Yang, X., Yang, X., Caro, M., Hutton, S., Scott, J., Guo, Y., et al. (2014). Fine mapping of the tomato yellow leaf curl virus resistance gene Ty-2 on chromosome 11 of tomato. Mol. Breed. 34, 749–760. doi: 10.1007/s11032-014-0072-9
Zaidi, S. S., Shafiq, M., Amin, I., Scheffler, B. E., Scheffler, J. A., Briddon, R. W., et al. (2016). Frequent occurrence of tomato leaf curl New Delhi virus in cotton leaf curl disease affected cotton in Pakistan. PLoS One 1:e0155520. doi: 10.1371/journal.pone.0155520
Zaidi, S. S., Shakir, S., Malik, H. J., Farooq, M., Amin, I., and Mansoor, S. (2017). First Report of tomato leaf curl New Delhi virus on Calotropis procera, a weed as potential reservoir begomovirus host in Pakistan. Plant Dis. 101, 1071–1072. doi: 10.1094/PDIS-10-16-1539-PDN
Keywords: ToLCNDV, resistance, Cucurbita, zucchini, QTL, synteny
Citation: Sáez C, Martínez C, Montero-Pau J, Esteras C, Sifres A, Blanca J, Ferriol M, López C and Picó B (2020) A Major QTL Located in Chromosome 8 of Cucurbita moschata Is Responsible for Resistance to Tomato Leaf Curl New Delhi Virus. Front. Plant Sci. 11:207. doi: 10.3389/fpls.2020.00207
Received: 16 December 2019; Accepted: 11 February 2020;
Published: 20 March 2020.
Edited by:
Amnon Levi, Agricultural Research Service, United States Department of Agriculture, United StatesReviewed by:
Umesh K. Reddy, West Virginia State University, United StatesYounghoon Park, Pusan National University, South Korea
Geoffrey Meru, University of Florida, United States
Kyle Edward LaPlant, Cornell University, United States
Copyright © 2020 Sáez, Martínez, Montero-Pau, Esteras, Sifres, Blanca, Ferriol, López and Picó. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Belén Picó, mpicosi@btc.upv.es