 Mulatu Geleta
Mulatu Geleta Cecilia Gustafsson
Cecilia Gustafsson Jeffrey C. Glaubitz
Jeffrey C. Glaubitz Rodomiro Ortiz
Rodomiro Ortiz- 1Department of Plant Breeding, Swedish University of Agricultural Sciences, Alnarp, Sweden
- 2Institute of Biotechnology, Cornell University, Ithaca, NY, United States
Lepidium campestre has been targeted for domestication as future oilseed and catch crop. Three hundred eighty plants comprising genotypes of L. campestre, Lepidium heterophyllum, and their interspecific F2 mapping population were genotyped using genotyping by sequencing (GBS), and the generated polymorphic markers were used for the construction of high-density genetic linkage map. TASSEL-GBS, a reference genome-based pipeline, was used for this analysis using a draft L. campestre whole genome sequence. The analysis resulted in 120,438 biallelic single-nucleotide polymorphisms (SNPs) with minor allele frequency (MAF) above 0.01. The construction of genetic linkage map was conducted using MSTMap based on phased SNPs segregating in 1:2:1 ratio for the F2 individuals, followed by genetic mapping of segregating contig tag haplotypes as dominant markers against the linkage map. The final linkage map consisted of eight linkage groups (LGs) containing 2,330 SNP markers and spanned 881 Kosambi cM. Contigs (10,302) were genetically mapped to the eight LGs, which were assembled into pseudomolecules that covered a total of ∼120.6 Mbp. The final size of the pseudomolecules ranged from 9.4 Mbp (LG-4) to 20.4 Mpb (LG-7). The following major correspondence between the eight Lepidium LGs (LG-1 to LG-8) and the five Arabidopsis thaliana (At) chromosomes (Atx-1–Atx-5) was revealed through comparative genomics analysis: LG-1&2_Atx-1, LG-3_Atx-2&3, LG-4_Atx-2, LG-5_Atx-2&Atx-3, LG-6_Atx-4&5, LG-7_Atx-4, and LG-8_Atx-5. This analysis revealed that at least 66% of the sequences of the LGs showed high collinearity with At chromosomes. The sequence identity between the corresponding regions of the LGs and At chromosomes ranged from 80.6% (LG-6) to 86.4% (LG-8) with overall mean of 82.9%. The map positions on Lepidium LGs of the homologs of 24 genes that regulate various traits in A. thaliana were also identified. The eight LGs revealed in this study confirm the previously reported (1) haploid chromosome number of eight in L. campestre and L. heterophyllum and (2) chromosomal fusion, translocation, and inversion events during the evolution of n = 8 karyotype in ancestral species shared by Lepidium and Arabidopsis to n = 5 karyotype in A. thaliana. This study generated highly useful genomic tools and resources for Lepidium that can be used to accelerate its domestication.
Introduction
Today’s major crop species are the results of thousands of years of intentional and unintentional selection of traits that brought genetically determined changes in the ancestral wild plant species (Burger et al., 2008). Domestication of a crop species is generally a very slow and long-term process that leads to significant changes in major traits that are regarded as “domestication syndrome” traits, such as determinate growth habit, increased seed size, loss of seed dormancy, and reduced pod shattering (Harlan et al., 1973; Doebley et al., 2006; Weeden, 2007; Burger et al., 2008). However, progress in genomic research that include comparative genomics, gene identification, annotation of whole genome sequences (WGS), development of genome-wide molecular markers, and genome-wide association studies (GWAS) for various crops led to deep insight into the process of plant domestication and evolution (Geleta and Ortiz, 2016). The use of genomic tools and resources in combination with conventional plant breeding methods is becoming essential in the development of new crop cultivars in a relatively shorter time than before (Pérez-de-Castro et al., 2012). Genomic tools and resources, such as a variety of molecular markers, high-density genetic linkage, quantitative trait locus (QTL), and genome-wide association maps are becoming the cornerstone of plant breeding, as they facilitate marker-assisted and genomic selection (Collard and Mackill, 2008; Lorenz et al., 2012; Lopez-Cruz et al., 2015; Qu et al., 2017; Koech et al., 2019). Through the use of these tools and resources and the analyses of the genetics of “domestication syndrome” traits, a good insight into the evolutionary changes that have occurred during plant domestication have been gained and can be used to facilitate a rapid domestication of new plant species.
Lepidium L. is a large, undomesticated genus in the Brassicaceae family that comprises 231 species distributed around the world (Al-Shehbaz et al., 2006). Indigenous Lepidium species are found on all continents (Al-Shehbaz, 1986), and they often grow in habitats with less competition for resources and space, such as roadsides, railway sides, and disturbed areas. L. campestre (L.) R. Br. (field cress), an annual or biennial (Mulligan, 1961) diploid species with 2n = 2x = 16 chromosomes (Rice et al., 2015), has wide distribution in Europe including Nordic countries. Based on its various desirable characteristics including winter hardiness, promising potential for high seed yield, self-compatibility, synchronous seed maturity, and suitability as an undersown catch crop (Al-Shehbaz, 1986; Merker and Nilsson, 1995; Eriksson, 2009; Geleta et al., 2014), it has been considered for domestication as a future oilseed and catch crop in Sweden to contribute to increased global production of vegetable oil as well as diversification of agroecosystems. Although the oil content of L. campestre was initially reported to be ∼20% (Nilsson et al., 1998), further studies of a wide collection of European and North American L. campestre accessions showed that the oil content varies from 12 to 20% (Mulatu Geleta et al., SLU, unpublished data). Its seed oil is mainly composed of linolenic acid (C18:3; 34–39%), erucic acid (C22:1; 22–25%), oleic acid (C18:1; 12–16%), and linoleic acid (C18:2; 8–11%).
Research and breeding activities of varying intensity have been ongoing during the last 25 years at the Swedish University of Agricultural Sciences (SLU), contributing to the domestication of L. campestre (Merker and Nilsson, 1995; Andersson et al., 1999; Börjesdotter, 1999; Eriksson, 2009; Merker et al., 2010; Ivarson et al., 2013; Geleta et al., 2014; Ivarson et al., 2016; Gustafsson et al., 2018). Domestication of L. campestre should result in significant improvement in various major traits, such as oil content and quality, pod shatter resistance, and seed yield (Eriksson, 2009; Geleta et al., 2014). For use as edible oil, antinutritional compounds, such as glucosinolates and erucic acid, will have to be eliminated or drastically reduced through breeding (Andersson et al., 1999; Ivarson et al., 2016).
Given that perenniality is a favorable trait in catch crops, the overall goal of the domestication of L. campestre is to develop both biennial and perennial cultivars. However, developing perennial L. campestre requires its interspecific hybridization with perennial Lepidium species, such as L. heterophyllum Benth. and Lepidium hirtum (L.) Sm. (Mulligan, 1961; Mummenhoff et al., 1995). Both species are closely related to L. campestre (Mummenhoff et al., 2001; Lee et al., 2002) and share the same ploidy level and chromosome number (2n = 2x = 16) (Rice et al., 2015). The interspecific hybridization of L. campestre with these two species was successful, and perennial breeding lines derived from these hybrids have been developed (Mulatu Geleta et al., SLU, unpublished data). In addition to the perenniality trait, hybridization between these species led to a larger variation in various desirable traits as compared to the variation within either of the parental species, providing wider opportunities for further breeding. This study was conducted to develop genomic tools and resources for Lepidium in order to understand its genome as well as accelerate its domestication.
Materials and Methods
Plant Material
A total of 380 plants that comprises three genotypes of L. campestre (Par_1, Stu_7 and C92_2_3), two genotypes of L. heterophyllum (Par_2 and Hast_3), and a mapping population of 375 F2 plants derived from interspecific hybrid of Par_1 and Par_2 (parents) were used in this study. Par_1 and Stu_7 were genotypes collected from Arlid (unknown exact location) and Stuvsta (59°15′25″ N, 17°58′51″ E), Sweden, respectively. C92_2_3 was a genotype sampled from IPK (Germany) accession LEP-92 originally collected from Greece. Par_2 was a genotype that belongs to a US Department of Agriculture (USDA)-Agricultural Research Service (ARS) accession LH 597856 originally collected from Spain, whereas Hast_3 is a genotype collected from Hästveda, Sweden (56°17′21″ N, 13°56′09″ E). Genomic data from the mapping population and the two parents were used for genetic linkage mapping and various statistical analyses. The two L. campestre and one L. heterophyllum genotypes were included to estimate various genetic diversity parameters within and among the two Lepidium species.
DNA Extraction
Seeds from target samples were planted in a greenhouse at the Department of Plant Breeding, SLU, Alnarp, Sweden. Young leaf tissue was sampled in Eppendorf tubes from individual plants and immediately frozen in liquid nitrogen, and genomic DNA was extracted as described in Gustafsson et al. (2018). The quality and quantity of the extracted DNA was assessed using 1% agarose gel electrophoresis and a NanoDrop® ND-1000 Spectrophotometer (Saveen Warner, Sweden). The extracted DNA samples were then sent to the Genomic Diversity Facility, Cornell University, USA, for genotyping by sequencing (GBS).
GBS Optimization and Analysis
A number of restriction enzymes were tested to determine the best enzyme that produces fragment size distribution suitable for the construction of GBS library. At the end, ApeKI (G∗CWGC), a 4-base cutter enzyme was selected, as majority of fragments produced were < 500 bp and hence were appropriate for Illumina sequencing. The reference genome-based pipeline TASSEL-GBS (Glaubitz et al., 2014) was used for this GBS analysis, where a draft whole genome sequence of L. campestre assembled in-house was used as a reference genome. In this GBS analysis, a total of 7,591,461 tags were generated after merging, and these tags were aligned against the in-house assembled L. campestre genome. Of these tags (3,688,674 tags), 48.6% were uniquely aligned to the reference genome, whereas 8.8 and 42.6% were multiply aligned and unaligned, respectively. SNP calling after aligning the tags to the reference genome resulted in 165,892 SNPs. VCFtools version v0.1.12a (Danecek et al., 2011) was used to calculate heterozygosity as well as the depth and missingness of the resultant SNPs. Filtering out of SNPs with minor allele frequency (MAF) below 0.01 and missing data per site above 90% resulted in 126,859 SNPs, of which 120,438 were biallelic. PLINK version v1.07 was used to generate a multidimension scaling (MDS) plot based on the 120,438 genome-wide biallelic SNPs.
Linkage Map Construction With GBS SNPs
The 126,859 GBS SNP markers were processed using VCFtools version 0.1.15 (Danecek et al., 2011) in the following order: (1) only SNPs with MAF of at least 40% were retained; (2) genotypes supported by a read depth of less than seven were set to missing; (3) SNPs with more than 10% missing data were discarded; (4) SNPs deviating from 1:2:1 segregation with p < 0.01 were discarded; (5) the SNPs were thinned so that no two SNPs were <65 bases apart (i.e., only one SNP was retained per 64-base GBS tag locus); and (6) the genotypes were converted to a numerical format to facilitate further processing in R (R Core Team, 2016). The genotypes were then phased based upon the two parents, and the resultant 2,352 phased SNPs for the 375 F2 individuals were reformatted for input into MSTMap (Wu et al., 2008) with a custom R script. The parameters used for linkage map construction with MSTMap include Kosambi distance with cutoff P-value of 1e–12, number of map distance of 10, number of map size of 1, and missing threshold of 0.15.
The resulting linkage map of eight linkage groups was reformatted for loading into R with a custom awk command, and then, a custom R script was used to merge it with the genotypic data and count the number of crossovers per individual across the eight linkage groups. Nine outlier individuals with more than 66 crossovers were discarded from further analysis, and a new linkage map was constructed with MSTMap using the remaining 366 individuals (same parameters as above). Twenty-one outlier SNPs with more than 15 double crossovers were then identified using R and excluded, and a final linkage map was constructed with MSTMap for the remaining 2,331 SNPs (again with the same parameters). The R package R/qtl (Broman et al., 2003) was then used to correct genotyping errors and impute most of the missing genotypes [using the fill.geno() function with method = “maxmarginal,” map.function = “kosambi,” and min.prob = 0.8]. The resulting genotypes were visualized with a custom R script (Supplementary Figure S1) and output in R/qtl csvr format to facilitate conversion to hapmap format via a custom awk command.
Genetic Mapping of Segregating GBS Tag Sequences
Custom Tassel3 (Bradbury et al., 2007) code was used to filter the TagsOnPhysicalMap (TOPM) and TagsByTaxa (TBT) data structures produced in the GBS SNP calling pipeline (Glaubitz et al., 2014) so that they contained only tags with unique alignment positions with no sequence divergence from the contig-level reference assembly. The TBT was also filtered to retain only the F2 individuals present in the final linkage map and only tags that appeared to segregate in the F2 [present in at least 30 and no more than 256 (80%) of the 366 individuals]. Each tag was then genetically mapped as a dominant marker against the linkage map. Because GBS was performed at low sequencing depth, the absence of a tag in an F2 individual is not always informative; hence, only the progeny in which a given tag was observed were used to calculate the recombination rate between that tag and each SNP in the linkage map. For this subset of progeny, the recombination rate was calculated as: min[nHomPar_2/(nHomPar_1 + nHomPar_2), nHomPar_1/(nHomPar1 + nHomPar2)] where nHomPar_1 was the number of individuals with the tag that were homozygous for the parent_1 allele at the SNP in question, and nHomPar_2 was the number of individuals with the tag that were homozygous for the parent_2 allele. Heterozygous individuals at the SNP were excluded as non-informative. Tags were considered genetically mapped if the recombination rate was < 5%, and the sample size (nHomPar1 + nHomPar2) was at least 30 (two-tailed binomial P < 6e–8). The genetic mapping span (GeneticStart to GeneticEnd) for a tag was from the first to last SNP on the linkage group with the same minimum recombination rate, and the genetic mapping position (GeneticMean) was the mean of this span, where the SNP positions were numbered consecutively from 1 to the number of SNPs on the linkage group.
Genetic Mapping of Contig Tag Haplotypes and Assembly Into Pseudomolecules
In light of the low sequencing depth of GBS, the statistical power for mapping contigs was increased by combining all of the concordant genetically mapped tags for a contig into a single tag haplotype, with the presence of any given tag imputing the presence of the contig haplotype as a whole. For contigs with multiple genetically mapped tags, the consensus linkage group assignment was determined by a majority rule weighted by sample size. For those tags mapped to the same linkage group, the consensus genetic mapping position was determined as the sample size weighted average of the tag mapping positions (GeneticMean). Tags with genetic mapping positions on the same linkage group and within 50 SNPs of the consensus genetic mapping position were considered as agreeing with the consensus and were thus merged into a contig tag haplotype, by summing their tag counts across each taxon. The contig tag haplotypes were then genetically mapped in the same manner as the individual tags, except that a minimum recombination rate of 10% was used, and to prevent false positives from occasional sequencing errors, a contig tag haplotype was only considered present in a genotype if the count for that genotype was at least 5% of the mean of the non-zero counts across genotypes.
The genetically mapped contigs were ordered into pseudomolecules according to the following rules, using custom awk and bash commands: (1) contigs placed on the linkage map via segregating SNPs were kept in the same relative order, regardless of whether they were mapped by tag haplotype or not, in which (a) contigs with multiple mapped SNPs were quasi-oriented if the centimorgan position differed between the first and last SNP in the contig, with the first and last SNP defined by physical position in the contig, (b) a contig with multiple SNPs was not always contiguous in the map, as it was sometimes comingled with one or more additional contigs, and the order of comingled contigs was resolved by the position of their first SNP in the linkage map, and (c) contigs without genetic consensus (e.g., with an equal number of SNPs mapping to different linkage groups) were removed from the assembly; (2) contigs mapped only by tag haplotype were placed immediately after the contig containing their GeneticMean SNP, with the following sort order: GeneticStart, GeneticEnd, contig type (scaffold < contig), contig name; (3) ordered contigs on a linkage group/pseudomolecule were separated by 100 N’s; and (4) the linkage groups/pseudomolecules were named LG-1, LG-2, LG-3, LG-4, LG-5, LG-6, LG-7, and LG-8, by modifying the names of the linkage groups assigned by the MSTMap software.
Results
Multidimensional Scaling, Heterozygosity, and Inbreeding Coefficient
Ninety-five percent (120,438) of the 126,859 filtered SNPs were biallelic. Multidimensional scaling based on these genome-wide biallelic SNPs displayed the distribution of the 375 individuals of the mapping population, their parents, as well as the other three genotypes included in the study (Figure 1). In this analysis the two parents, Par_1 and Par_2, were positioned at the top right and bottom left corners of the plot, respectively, and the MDS clearly displayed the clustering of the three L. campestre genotypes (Par_1, Stu_7 and C92_2_3) and the two L. heterophyllum genotypes (Par_2 and Hast_3) at their respective corners (Figure 1). The two Swedish genotypes of L. campestre (Par_1 and Stu_7) were more closely related to each other than to L. campestre genotype originally collected from Greece (C92_2-3). The F2 individuals spread widely across the two dimensions with the highest concentration around the center (Figure 1). The distribution of these individuals shows that they represented the whole F2 population very well. Based on these data, it is possible to select individuals that are genetically more similar to L. campestre (the target species for domestication) for further breeding. For example, individuals such as Hy25_A24 and Hy25_A307 would be among the top candidates for further breeding or crossbreeding with L. campestre, if they have desirable traits such as perenniality.
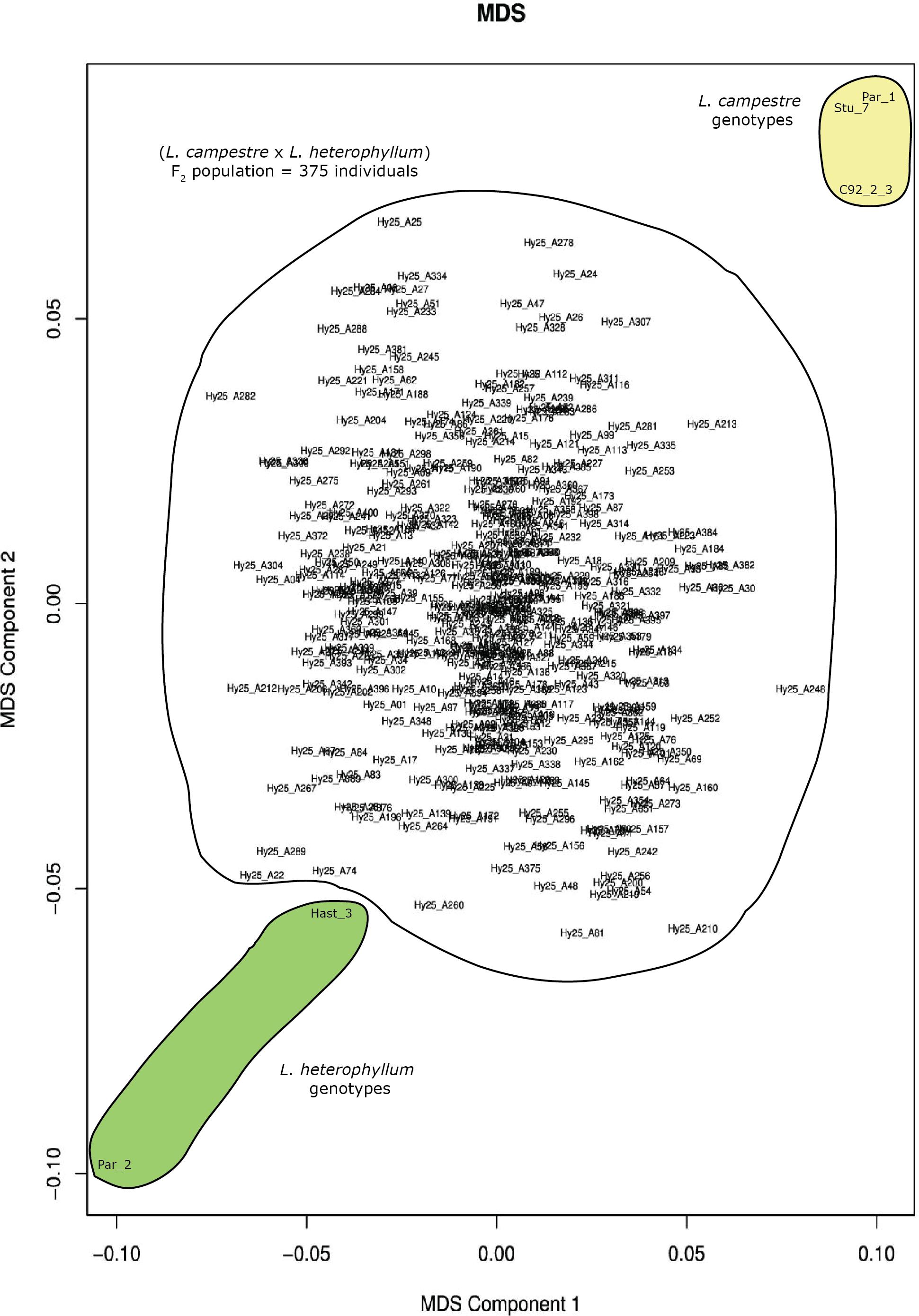
Figure 1. Multidimension scaling (MDS) plot generated for 380 individual plants comprising three L. campestre and two L. heterophyllum genotypes as well as 375 F2 mapping population based on 120,438 genome-wide biallelic SNPs.
Observed heterozygosity (Ho) and expected heterozygosity (He) as well as inbreeding coefficient (F) were calculated for the three L. campestre, two L. heterophyllum samples, as well as for of the F2 individuals across thousands of SNP loci (2,331–100,760 loci) (Table 1 and Figure 2). These parameters were calculated for each individual after removing loci with missing data. In the case of all filtered SNPs (126,859), 50,439–10,076 SNP loci remained per individual after removing the loci with missing data. Similarly, removing the loci with missing data for mapped SNPs resulted in 2,326–2331 loci per individual. For the three L. campestre genotypes, only 5.1–5.7% of the loci were heterozygous, and the mean heterozygosity was 5.4% (P_Ho = 0.054; Table 1). Similarly, heterozygous loci accounted for a mean of 5.5% in L. heterophyllum. Inbreeding coefficient (F) was 0.64 on average for both species. In the case of F2 population, the proportion of observed heterozygosity was 13.1 and 51.9% for all filtered and mapped SNPs, with corresponding inbreeding coefficient of 0.07 and −0.08, respectively (Table 1 and Figure 2).
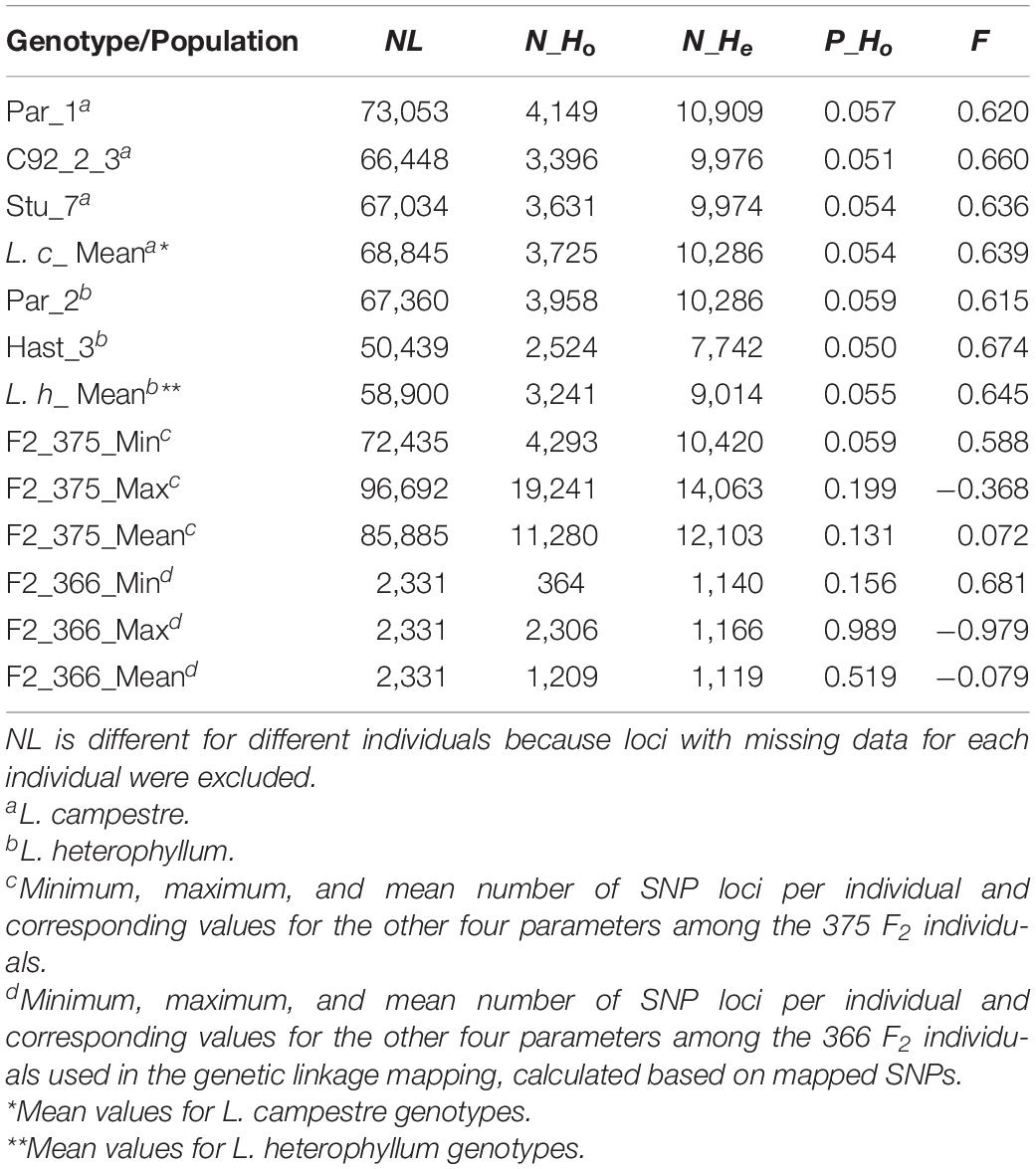
Table 1. Number of SNP loci (NL), number of observed heterozygosity (N_Ho), number of expected heterozygosity (N_He), proportion of observed heterozygosity (P_Ho), and inbreeding coefficient (F) for the three L. campestre, two L. heterophyllum genotypes, and the F2 mapping population.
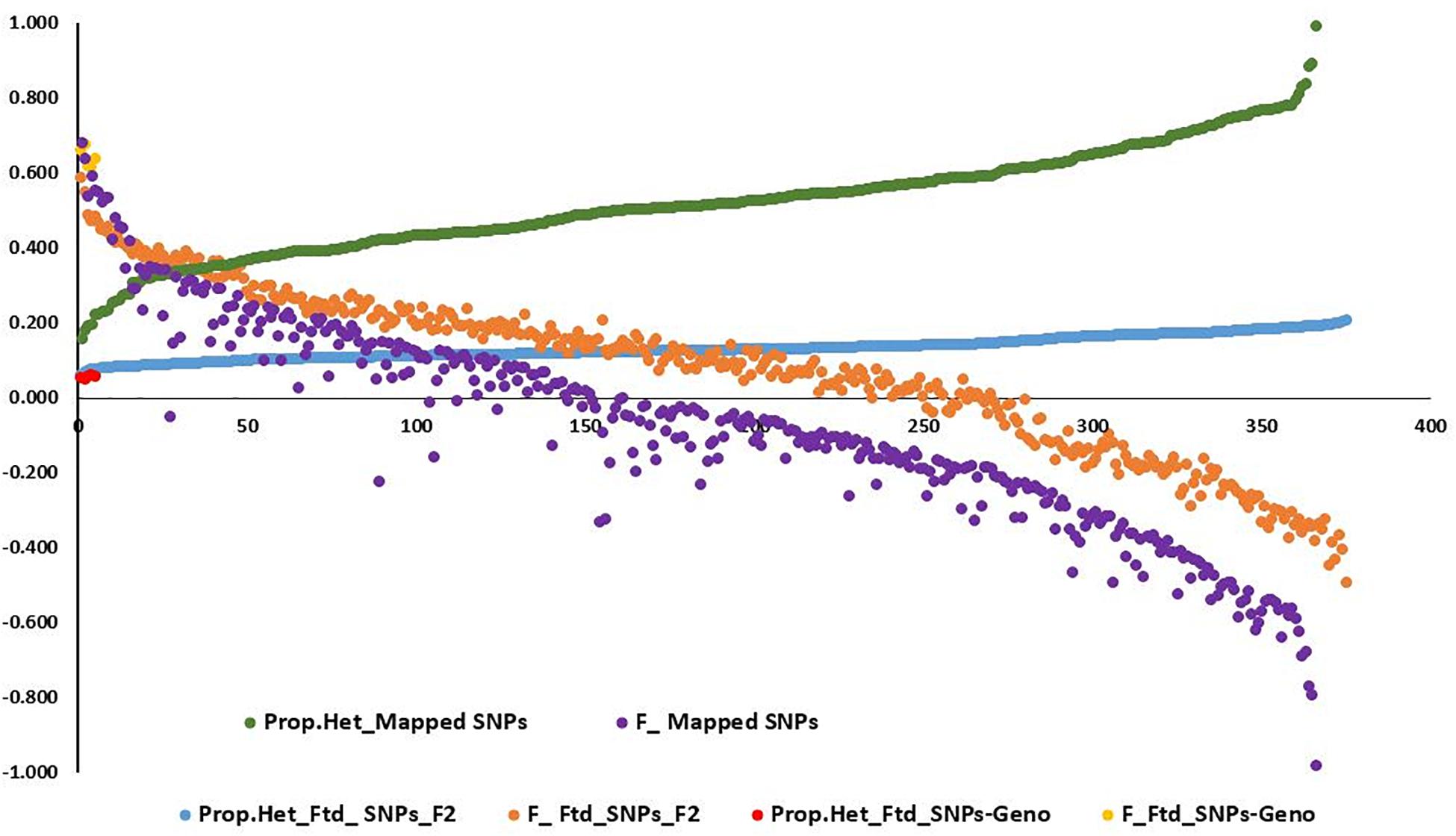
Figure 2. Graph showing the distribution of the proportion of heterozygosity at (1) mapped single-nucleotide polymorphism (SNP) loci for the 375 F2 population (green) and their corresponding inbreeding coefficient (purple), (2) filtered SNP loci for the 375 F2 population (blue) and their corresponding inbreeding coefficient (orange), and (3) filtered SNP loci for the five genotypes (three L. campestre and two L. heterophyllum) (red) and their corresponding inbreeding coefficient (yellow).
Linkage Map Construction With GBS SNPs
The final linkage map consisted of eight linkage groups (LGs) containing 2,331 SNP markers derived from 1,044 contigs, and spanned 881 Kosambi cM in total (Figure 3, see also Supplementary Figure S1). The number of mapped SNPs per contig varied from 1 to 66, and 305 (29.2%) of the 1,044 contigs contained more than one mapped SNPs (Supplementary Tables S1, S2). Of the 305 contigs possessing more than one mapped SNPs, only one was mapped to more than one linkage groups (scaffold140, with four SNPs on LG-2 and one SNP on LG-5). The SNP on LG-5 was excluded, and hence, the total number of SNPs shown on the linkage map is 2,330 (Figure 4 and Supplementary Tables S1, S2). The SNPs on each contig were mapped within 1 cM of each other for 81.0% of the contigs, within 5 cM for 96.4%, and within 10 cM for 99.7% (Figure 5 and Supplementary Tables S2). These results indicate that the assembly of paired end reads into contigs was highly accurate. The number of SNPs mapped to LG-1 to LG-8 were 238 (10.2%), 160 (6.9%), 354 (15.2%), 192 (8.2%), 436 (18.7%), 308 (13.2%), 197 (8.5%), and 445 (19.1%), respectively. These SNPs spanned 72.3, 50.5, 99.4, 105.8, 103.4, 98.0, 49.3, and 99.2 cM and hence have an average SNP density of 3.3, 3.2, 3.9, 3.3, 4.2, 3.1, 4.0, and 4.5 SNPs/cM for LG-1 to LG-8 in that order (Figure 4).
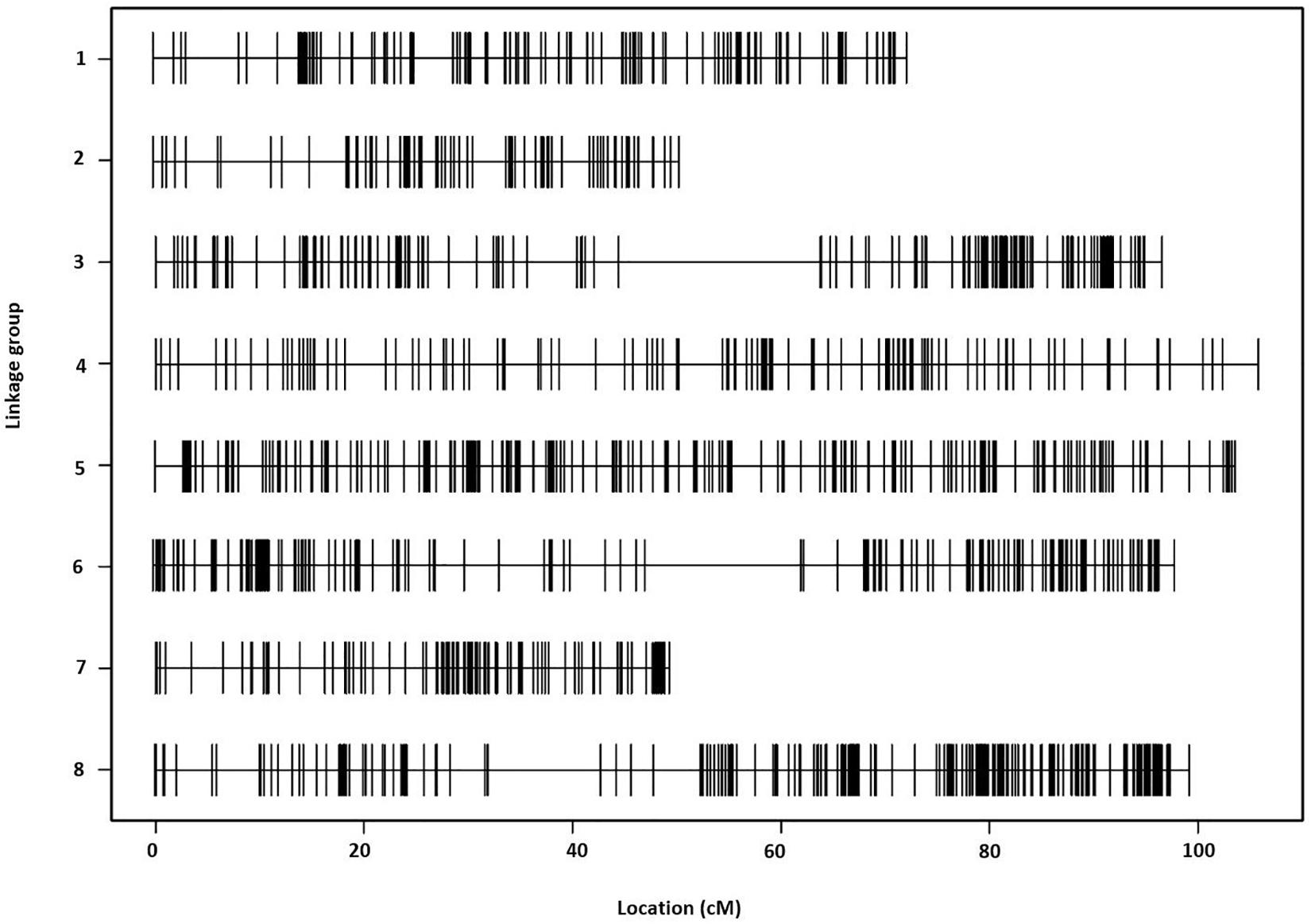
Figure 3. The Lepidium linkage map comprising eight linkage groups and showing the distribution of 2,330 single-nucleotide polymorphism (SNP) markers across a span of 881 Kosambi cM in total.
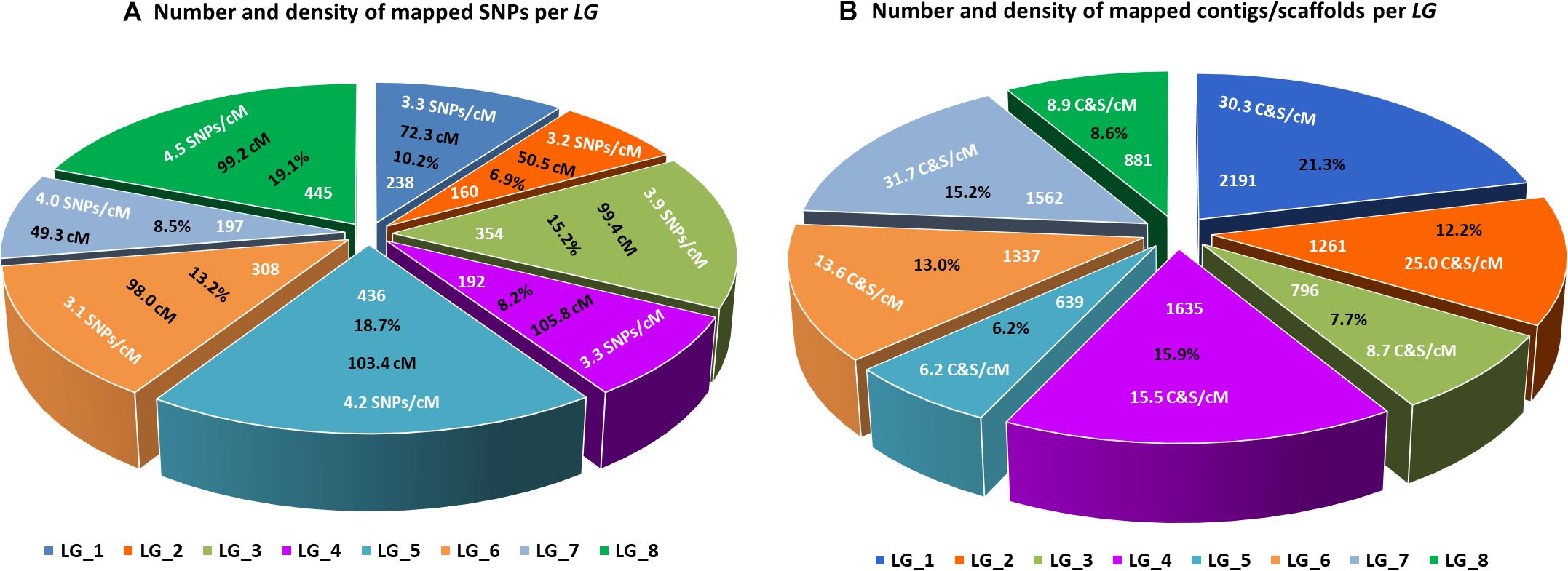
Figure 4. Lepidium linkage groups. (A) Size of each linkage group in centimorgans (cM), number and percentage of SNPs mapped to each linkage group, and average number of mapped SNPs per centimorgan. (B) Number and percentage of contigs/scaffolds mapped to each linkage group, and average number of mapped contigs/scaffolds per centimorgan.
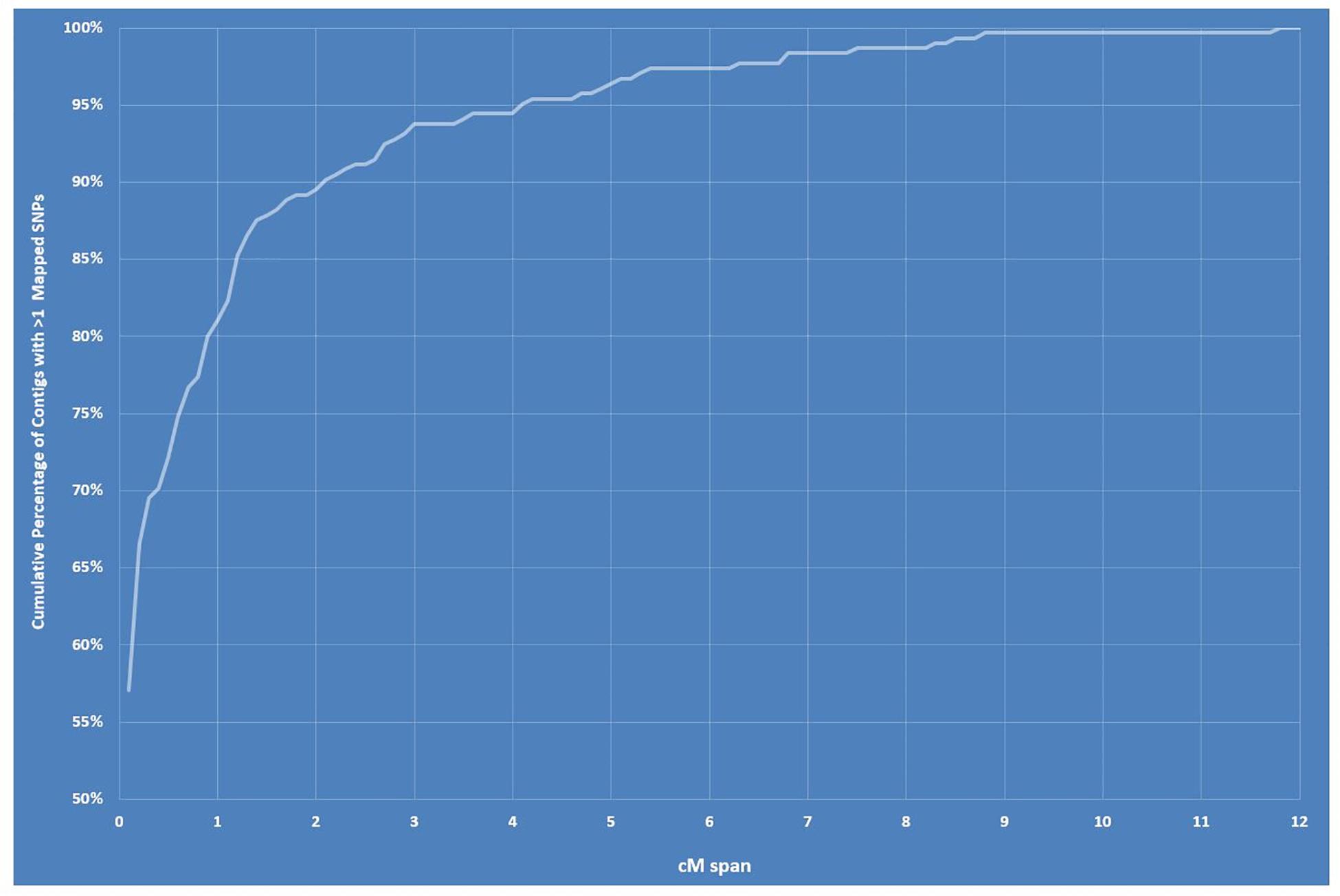
Figure 5. Graph showing cumulative percentage of contigs/scaffolds with > 1 mapped single-nucleotide polymorphisms (SNPs) within a centimorgan span between 0 and 12.
Genetic Mapping of Segregating GBS Tag Sequences and Contig Tag Haplotypes
In total, 34,342 segregating 64-base GBS tag sequences from 9,943 contigs were placed on the genetic map within a recombination fraction of 5% of one or more SNP markers (Supplementary Tables S3). Of these tags, 33,832 either agreed with the genetic consensus for their respective contig or were the sole representative tag, whereas 510 (1.5%) disagreed with the consensus and were excluded from further analysis, along with 12 contigs that did not display genetic consensus. The remaining 9,931 contigs with genetic consensus were all successfully mapped, as a segregating tag haplotype, to within a recombination fraction of 7.9% of one or more SNPs in the linkage map (Supplementary Tables S3). In total, 10,302 contigs were genetically mapped, with 371 mapped only by SNP, 673 mapped by both SNP and contig tag haplotype, and 9,258 mapped only by contig tag haplotype (Supplementary Tables S1). The sequences of the 10,302 mapped contigs have been deposited at DDBJ/ENA/GenBank as Whole Genome Shotgun project under the accession number WJSH00000000. These contigs were assembled into a pseudomolecule fasta file according to the ordering and orientation rules described in section Materials and Methods above. The pseudomolecules in the assembly covered 120,594,250 bases (∼120.6 Mbp) (Table 2), of which 116,577,053 (96.7%; ∼116.6 Mbp) were not N. The final size of the LGs ranged from 9.4 to 20.4 Mbp with LG-1 to LG-8 having a size of 18.9, 10.1, 13.1, 9.4, 15.2, 18.7, 20.4, and 14.9 Mbp (Table 2), and accounted for 15.6, 8.3, 10.9, 7.8, 12.6, 15.5, 16.9, and 12.3% of the 120.6 Mbp total assembled sequences, in that order. The G + C content of the LGs ranged from 34.9 (LG-4) to 35.9 (LG-3) with a mean of 35.5% (Table 2).
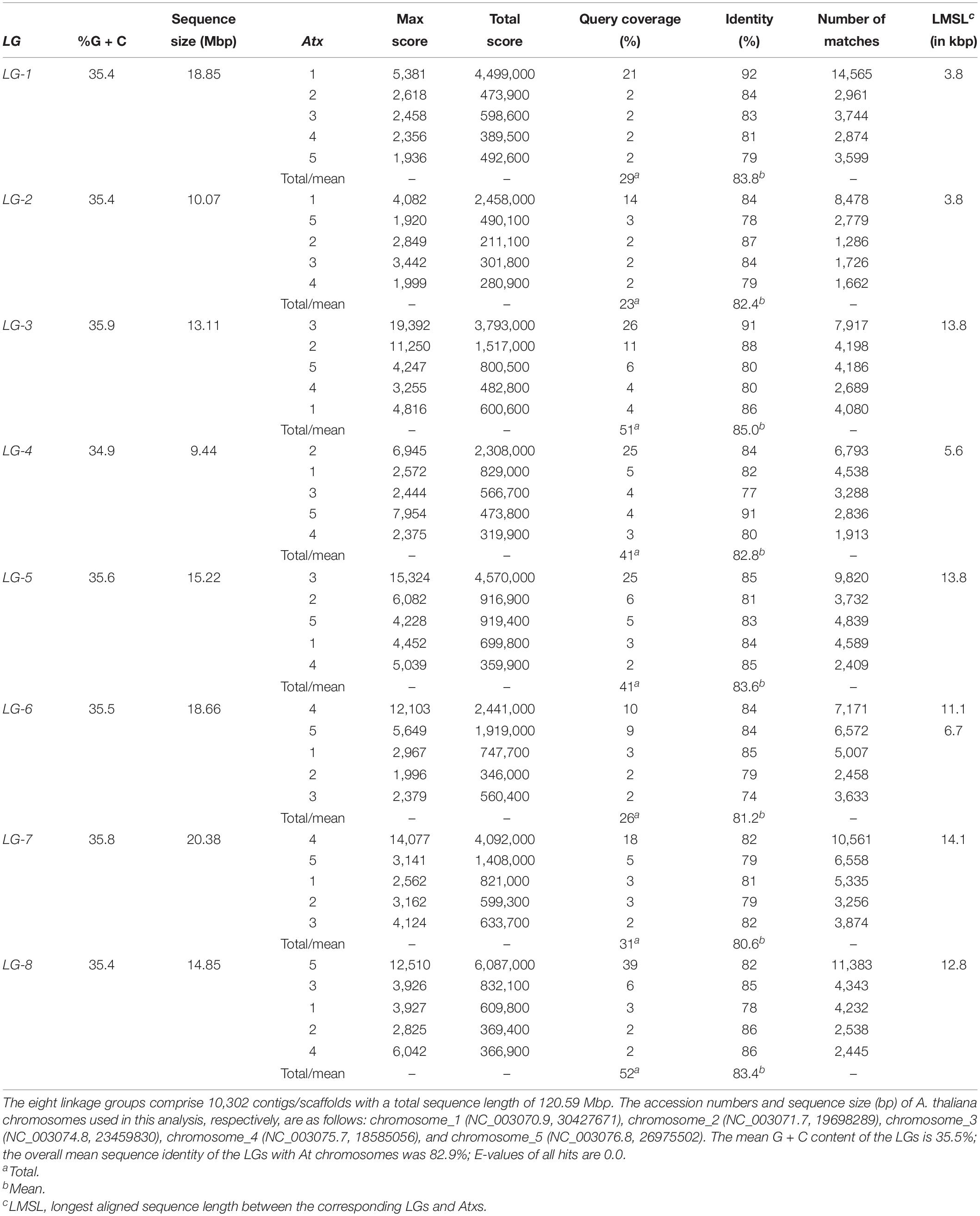
Table 2. The assembled size of the eight Lepidium linkage groups (LGs) in megabase pair and the homeology, sequence identity, and length of their sequences corresponding to A. thaliana chromosome (Atx) sequences.
Comparative Analysis of Lepidium Linkage Groups and Arabidopsis thaliana Chromosomes
Basic Local Alignment Search Tool (BLAST)1 was used to search Arabidopsis thaliana genome (taxid:3702) at the GenBank2 for comparative analysis of the eight Lepidium LGs with the five A. thaliana (At) chromosomes (Atx-1–Atx-5). All LGs had hits from multiple regions within the five At chromosomes, but to highly different extents (Table 2). This analysis revealed that the largest group of hits for LG-8 was from At chromosome 5 (Atx-5) and covered 39% of LG-8 sequences with mean sequence identity (mSI) of 82%, whereas the largest group of hits that covered 25% of LG-5 with mSI of 85% came from Atx-3. These are composed of 11,383 (LG-8) and 9,820 (LG-5) matching sequences on Atx-5 and Atx-3, respectively (Table 2). Similarly, the largest group of hits for LG-1 (mSI = 92%), LG-2 (mSI = 84%), LG-3 (mSI = 91%), LG-4 (mSI = 84%), LG-6 (mSI = 84%), and LG-7 (mSI = 82%) were from Atx-1, Atx-1, Atx-3, Atx-2, Atx-4, and Atx-4, respectively, and covered 21, 14, 26, 25, 10, and 18% of the corresponding LG sequences (Table 2). In the case of LG-3 and LG-6, 11 and 9% of the sequences matched sequences of Atx-2 and Atx-5, respectively. The sequence identity of the LGs with At chromosomes ranged from 80.6% (LG-7) to 85.0% (LG-3) with overall mean of 82.9%. Overall, 29, 28, 51, 41, 41, 26, 31, and 52% of LG-1 to LG-8 sequences, respectively, matched the sequences of At chromosomes (Table 2).
The contigs/scaffolds mapped to Lepidium LGs were blast searched against the At genome, and the position of corresponding sequences within At chromosome sequences were determined. Based on major sequence coverage and identity, the correspondence between the Lepidium LGs and At chromosomes were grouped into three groups (Figure 6A). Group 1 shows the correspondence of LG-1 and LG-2 with Atx-1. Group 2 contains LG-3, LG-4, and LG-5 as well as Atx-2 and Atx-3. LG-3 and LG-5 mainly correspond to Atx-3, although LG-3 also has a smaller portion that corresponds to Atx-2, whereas LG-4 corresponds to Atx-2. Group 3 contains LG-6, LG-7, and LG-8 as well as Atx-4 and Atx-5. LG-7 mainly correspond to Atx-4, whereas LG-8 corresponds to Atx-5. On the other hand, LG-6 has two major portions where one corresponds to Atx-4 and the other to Atx-5. Blast searching of the sequences of the LGs against At genome revealed that the largest matching sequences between LG-1_Atx-1, LG-2_Atx-1, LG-3_Atx3, LG-4_Atx-2, LG-5_Atx-3, LG-6_Atx-4, LG-6_Atx-5, LG-7_Atx-4, and LG-8_At-5 were 3.8, 3.7, 13.8, 6.5, 13.8, 11.1, 6.7, 14.1, and 12.8 kbp, respectively (Table 2).
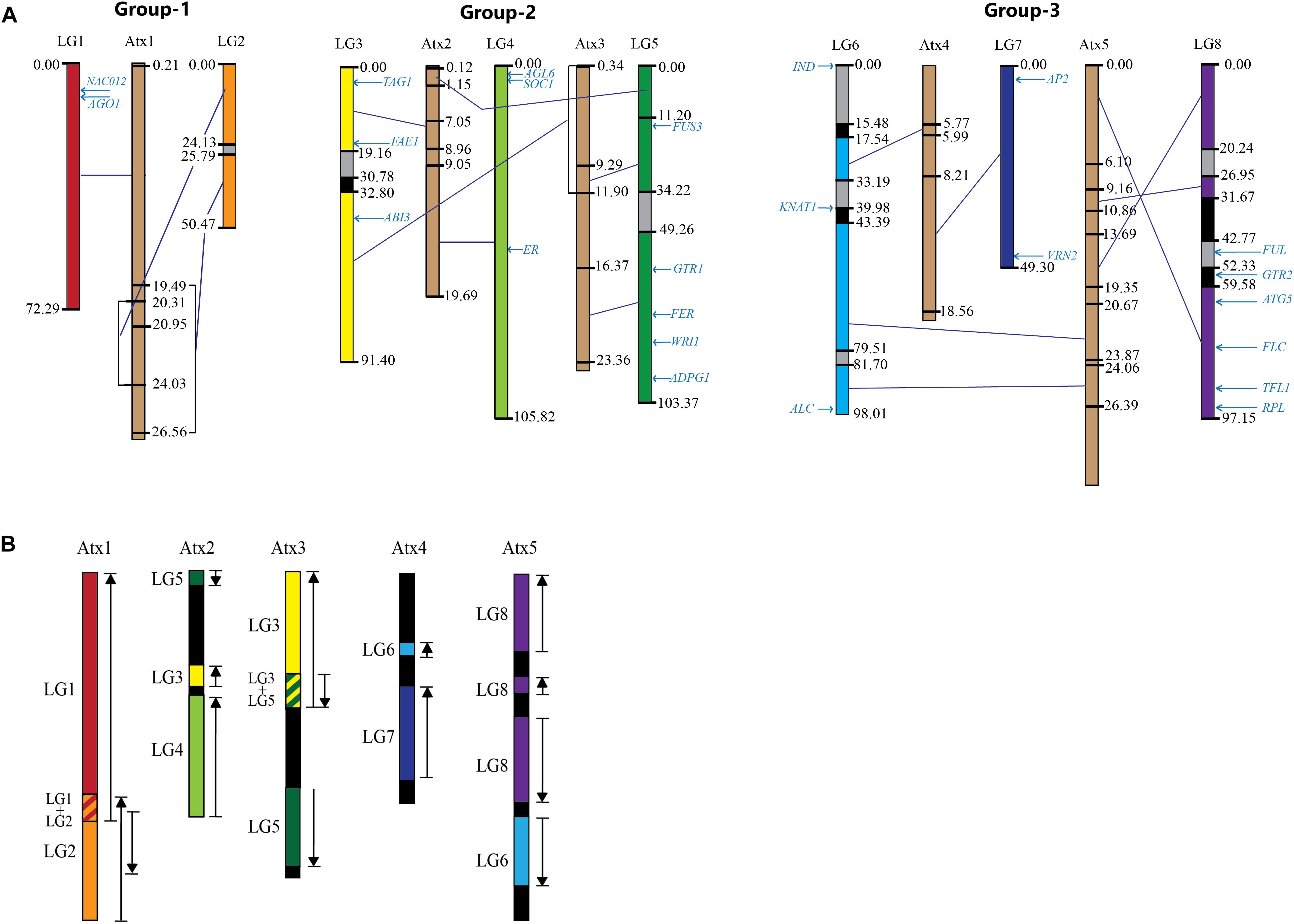
Figure 6. A comparison of the eight Lepidium linkage groups (LGs) and the five A. thaliana chromosomes (Atxs) forming three groups: (A) The LGs are shown in different colors whereas Atxs in brown; the numbers at the sides of the LGs and Atxs are map distance in centimorgan and sequence position in megabase pair, respectively, the text in light blue shows the map position of the homologs of genes known to regulate various traits in A. thaliana; gray and black regions within the LGs show regions lacking collinearity/matching sequences and mapped markers, respectively, deep blue lines connecting the LGs and Atxs show regions of high collinearity between them within the boundaries of the closest centimorgan (for LGs) and megabase pair (for Atxs); overlapping regions in collinearity are shown in square brackets. (B) The five Atxs displaying regions of collinearity with their corresponding LGs shown in different colors in (A); upward arrows show collinearity in the same direction, whereas downward arrows show collinearity in opposite direction as a result of inversion.
The comparison of the LGs with At chromosomes revealed three regions within the LG sequences as shown in Figure 6A: (1) regions shown in colors other than gray and black showed high collinearity with At chromosome sequences, (2) regions shown in gray are either did not show significant collinearity or did not match sequences of At chromosomes, and (3) regions shown in black do not have mapped contigs and hence could not be compared with At chromosomes. In the highly collinear regions, collinearity is either in the same or opposite direction as compared to the corresponding At chromosome sequences, as shown in black upward and downward arrows, respectively, in Figure 6B. At least 66% of the sequences of the LGs showed high collinearity with At chromosome sequences. However, significant portions of LG-3, LG-6, and LG-8 were either gray or black, whereas LG-2 and LG-5 have significant gray regions (Figure 6A). On the other hand, the whole regions of LG-1, LG-4, and LG-7 showed high collinearity with their corresponding At chromosome sequences. LG-2, LG-3, LG-5, LG-6, and LG-8 showed 92.7, 86.4, 85.2, 69.7, and 66.7% collinearity with their corresponding At chromosomes. On average, 87.6% of the LGs are collinear with the At chromosome sequences.
The map positions of the homologs of 24 At genes that are known to regulate various traits in Arabidopsis have been identified on Lepidium LGs (Table 3 and Figure 6A). LG-1 carries the homologs of NAC012 and AGO1 genes on Atx-1. The homologs of TAG1 (Atx-2), ABI3 (Atx-3), and FAE1 (Atx-4) were located on LG-3. LG-4 carries the homologs of AGL6, SOC1, and ER genes on Atx-2. Similarly, the homologs of five genes on Atx-3 (FUSCA3, GTR1, FER, WRI1, and ADPG1) were located on LG-5, whereas the homologs of IND and KNAT1 (Atx-4) were located on LG-6, which also contained the homolog of ALC gene (Atx-5). LG-7 carries the homologs of AP2 and VRN2 genes on Atx-4. The homologs of FUL, GTR2, ATG5, FLC, TFL1, and RPL genes that belong to Atx-5 were located on LG-8. None of the homologs of the 24 At genes were located on LG-2.
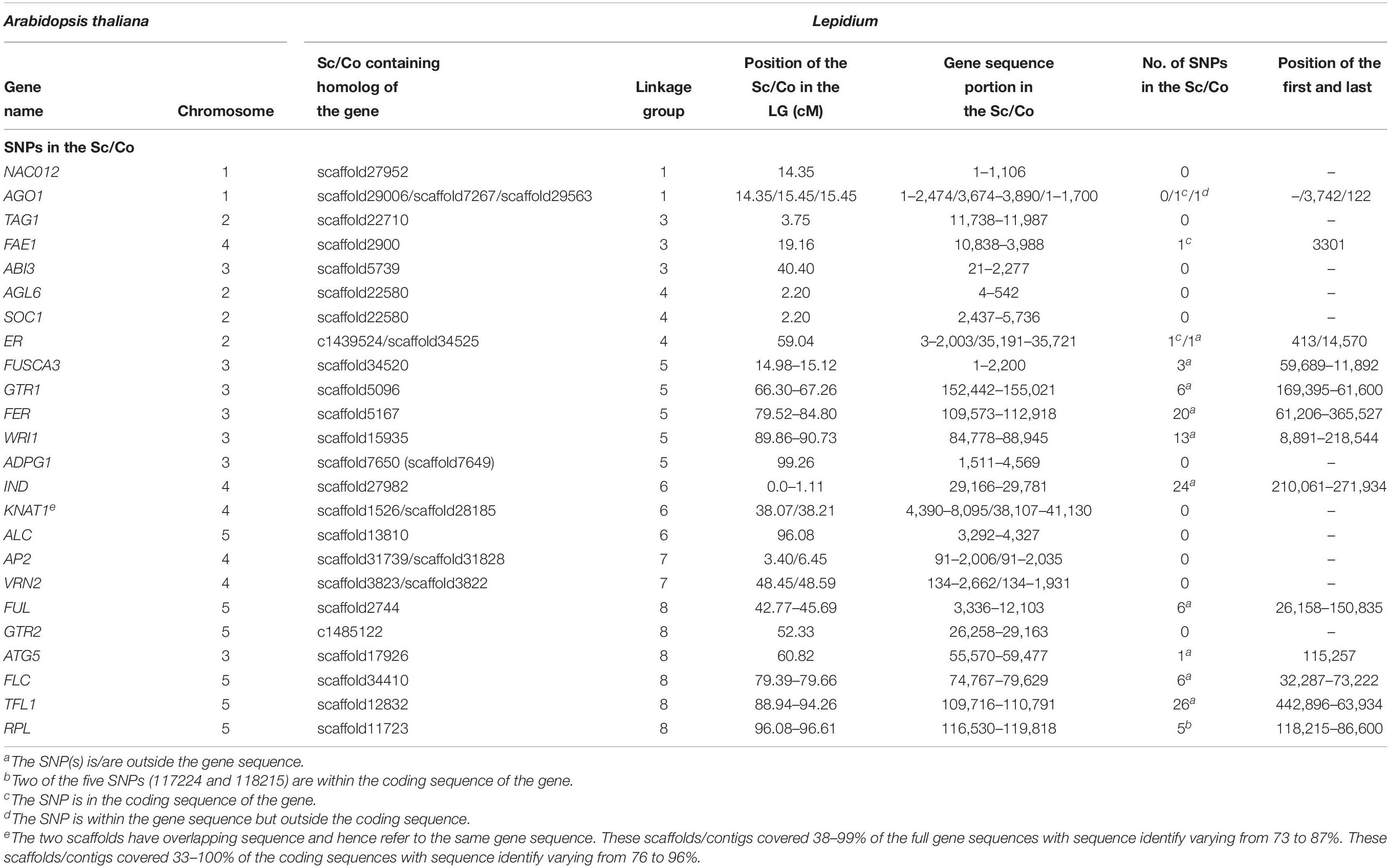
Table 3. List of mapped scaffolds/contigs (Sc/Co) containing sequences of homologs of Arabidopsis genes regulating various traits.
Discussion
Advanced next generation sequencing technologies allow identification of thousands of polymorphic markers that have various applications including the determination of genetic diversity and development of high-density genetic linkage map in plant species. The present study revealed an average observed heterozygosity (Ho) of <6% in both L. campestre and L. heterophyllum signifying that both species are predominantly inbreeders. In the F2 mapping population, only 13.1% of the filtered SNPs were heterozygous, on average, which is significantly lower than the 50% heterozygosity expected for the whole F2 population derived from the crosses of the two parental plants. Although random F2 seeds were planted, the seedlings of some of them were extremely weak or unhealthy at very young age, and consequently, leaf tissue was not sampled from such plants for DNA extraction. Given the obtained result of low proportion of heterozygous SNPs in the mapping population, it is likely that most of the unfit seedlings had higher proportion of heterozygosity across their genome. This finding may suggest that plants with higher proportion of heterozygosity across their genome perform poorly, the case that can be generally regarded as heterozygote disadvantage. This is in line with theoretical prediction that homozygosity is fixed easily in strongly selfing plant species if rearrangements reduce fitness (Charlesworth, 1992). The 51.9% heterozygosity for the 2,331 mapped SNPs is in line with the expected 50%, and this has been obtained through discarding SNPs deviating from 1:2:1 segregation with P < 0.01 to make it suitable for the linkage mapping.
Construction of linkage map is an important step in the identification of genes and molecular markers for its application in plant breeding. In this study, we used the GBS method (Baird et al., 2008; Elshire et al., 2011) to simultaneously discover new SNP markers and genotype individual samples from two Lepidium species (L. campestre and L. heterophyllum) as well as an F2 mapping population of interspecific hybrid between genotypes of the two species. SNP markers discovered through GBS has previously been successfully used for the construction of high-density genetic linkage mapping in various plant species including barley and wheat (Poland et al., 2012), Aethionema arabicum (Nguyen et al., 2019), Avena species (Latta et al., 2019), and grapevine (Tello et al., 2019). The eight linkage groups constructed in this study correspond to the eight haploid chromosome number previously reported for both L. campestre and L. heterophyllum (Rice et al., 2015).
The Brassicaceae family has been described as having four major evolutionary lineages (Bailey et al., 2006; Couvreur et al., 2010). The ancestral karyotype of lineages I and II is composed of eight chromosomes, which later evolved into the ancestral Camelineae karyotype of eight chromosomes and the Calepineae karyotype of seven chromosomes (Murat et al., 2015). Lepidium and Arabidopsis belong to lineage 1 and evolved from ancestral Camelineae karyotype of eight chromosomes, which makes it suitable to transfer genomic information from Arabidopsis (a model species) to Lepidium (species of agronomic interest). Hence, the sequences of A. thaliana chromosomes were used as a tool for comparative analyses of Lepidium and Arabidopsis genomes in this study.
The LGs of L. campestre are named from LG-1 to LG-8 in a way that they match previously reported chromosome nomenclature of Brassicaeae species with a haploid chromosome number of eight (n = 8), such as Arabidposis lyrata (Boivin et al., 2004; Kuittinen et al., 2004; Yogeeswaran et al., 2005; Hu et al., 2011; Murat et al., 2015). LG-1 to LG-8 match chromosomes 1–8 of A. lyrata in that order. Studies on different Brassicaceae species have revealed chromosomal events through which a karyotype of n = 8 in an ancestral species shared by Lepidium and Arabidopsis have evolved to a karyotype of n = 5 in A. thaliana (Boivin et al., 2004; Kuittinen et al., 2004; Yogeeswaran et al., 2005; Koch and Kiefer, 2005; Lysak et al., 2006; Hu et al., 2011; Murat et al., 2015). The comparative genomic analysis between A. lyrata (n = 8) and A. thaliana (n = 5) revealed that more than 50% the A. lyrata genome is absent in A. thaliana, whereas ∼25% the A. thaliana genome is missing in A. lyrata (Hu et al., 2011), which is the result of accumulated chromosomal and point mutations since the two species separated roughly 5–6 million years ago (mya) (Koch and Kiefer, 2005). However, the overall sequence identity between their homologous sequences is >80% (Hu et al., 2011), which is comparable with the overall sequence identity of 82.9% between homologous sequences of L. campestre and A. thaliana.
The degree to which genes and genomic regions are maintained on corresponding chromosomes (remain syntenic) and in corresponding orders (remain collinear) over a period of time varies among eukaryotic genomes (Coghlan et al., 2005). Correlating arrangements of genomic regions of a model species with a related species allows inference of shared ancestry of genes as well as utilization of known genetic information of the model species to study less-well-understood systems (Tang et al., 2008). About 10 major rearrangements have been reported between A. thaliana and A. lyrata, including two reciprocal translocations and three chromosomal fusions (Kuittinen et al., 2004; Yogeeswaran et al., 2005; Lysak et al., 2006) that resulted in the formation of a karyotype of five chromosomes in A. thaliana from the ancestral state of eight chromosomes that still exist in other Brassicaceae species, such as A. lyrata. The fusions and translocations reported based on the karyotypes of the two Arabidopsis species are also evident in this study through comparison of the L. campestre and A. thaliana genomes. As a result of the fusion of ancestral chromosomes, LG-1 and LG-2 matched Atx-1, LG-3, and LG-4 matched Atx-2, and LG-8 and LG-6 matched Atx-5. In all three pairs, the first LGs matched the upper part of their corresponding At chromosomes. LG-5 corresponds to Atx-3, whereas LG-7 corresponds to Atx-4.
The inversion within the lower arm of Atx-1 that corresponds to ancestral chromosome-2 (Lysak et al., 2006) was also evident in this study (inversion of corresponding region of LG-2). Following the fusion of ancestral chromosomes 3 and 4, unequal reciprocal translocation occurred between the fused chromosome (at region corresponding to chromosome 3) and the upper part of ancestral chromosome 5 (Lysak et al., 2006; Hu et al., 2011), which is in line with the present study (Figures 6A,B). However, unlike the case between A. lyrata and A. thaliana, the comparison of Lepidium and Arabidopsis genomes revealed inversion of the translocated block to the fused chromosome, suggesting the occurrence of further major chromosomal rearrangements after the Arabidopsis and Lepidium lineages were separated. Following the fusion of ancestral chromosomes 6 and 8, unequal reciprocal translocation occurred between the fused chromosome (at region corresponding to chromosome 6) and the upper part of ancestral chromosome 7 (Lysak et al., 2006; Hu et al., 2011), which is in agreement with the present study (Figures 6A,B). Among the two inversions previously reported (Lysak et al., 2006; Hu et al., 2011), the inversion within Atx-5 was evident but not the one within Atx-4.
Parkin et al. (2005) identified 21 shared syntenic blocks between A. thaliana and Brassica napus genomes representing collinear regions maintained since the divergence of their lineages approximately 20 mya. The genomes of A. thaliana and A. lyrata are ∼90% syntenic that predominately showed highly conserved collinear arrangements, although multiple inversions also exist between the genomes (Hu et al., 2011). Similarly, the present study showed an average of 87.6% synteny between the sequences of Lepidium LGs and A. thaliana chromosomes, and inversion of small segments are common throughout the Lepidium LGs. Except in LG-1, LG-4, and LG-7, the other LGs have significant regions that are unalignable with A. thaliana chromosomes similar to the case between A. thaliana and A. lyrata that have unalignable regions throughout the genome. The latter case is mainly due to deletions throughout A. thaliana genome, suggesting that deletions are favored over insertions and hence smaller genome (Hu et al., 2011).
Through comparative analysis of genomic sequences, the linkage map positions of the homologs of 24 genes that are known to regulate various traits in A. thaliana have been located on Lepidium LGs (Table 3 and Figure 6A). These genes regulate traits that are targeted for improvement within the domestication project of L. campestre. The homologs of the NAC DOMAIN CONTAINING PROTEIN 12 (NAC012) and ARGONAUTE 1(AGO1) were mapped to LG-1. NAC012 contributes to the regulation of pod shattering in A. thaliana through controlling the development of secondary walls in siliques (Rajani and Sundaresan, 2001; Liljegren et al., 2004). The sequence identity between the NAC012 partial coding sequences of these two species was 92% (Gustafsson et al., 2018). AGO1 is involved in the determination of inflorescence architecture in Arabidopsis through suppressing the TERMINAL FLOWER 1 (TFL1) (Ferrándiz-Nohales et al., 2014).
The homologs of TRIACYLGLYCEROL BIOSYNTHESIS DEFECT 1 (TAG1), ABA-insensitive 3 (ABI3), and FATTY ACID ELONGATION 1 (FAE1) were mapped to LG-3. TAG1 is one of the genes involved in the biosynthesis of fatty acids, and it regulates oil production (Routaboul et al., 1999; Jako et al., 2001); FAE1 controls seed oil composition (James et al., 1995), and ABI3 regulates seed dormancy (Ooms et al., 1993) in Arabidopsis. The sequence identity of TAG1 and FAE1 between the partial coding sequences of these two species was 93 and 88%, respectively (Gustafsson et al., 2018). The homologs of A. thaliana AGAMOUS LIKE 6 (AGL6), SUPRESSOR OF OVEREXPRESSION OF CO1 (SOC1), and ERECTA (ER) were mapped to LG-4. AGL6 and SOC1 are flowering time genes that regulate flowering in Arabidopsis. The sequence identity of AGL6 and SOC1 between the partial coding sequences of these two species was 98 and 94%, respectively (Gustafsson et al., 2018). ER regulates traits such as internode length and angles of pods (Douglas et al., 2002; Venglat et al., 2002), which have direct effect on seed yield through the determination of the number of pods on each inflorescence.
The homologs of FUSCA 3 (FUS3), GLUCOSINOLATE TRANSPORTER-1 (GTR1), FERONIA (FER), WRINKLED (WRI1), and ARABIDOPSIS DEHISCENCE ZONE POLYGALACTURONASE 1 (ADPG1) were mapped to LG-5. FUS3 regulates seed dormancy (Luerssen et al., 1998), whereas GTR1 regulates glucosinolates transport (Andersen and Halkier, 2014; Saito et al., 2015) in Arabidopsis. FER is a host-plant resistance gene (Kessler et al., 2010), WRI1 regulates oil production (Focks and Benning, 1998), and ADPG1 is one of the genes regulating fruit dehiscence (Ogawa et al., 2009) in Arabidopsis. A. thaliana and L. campestre showed sequence identity of 93, 88, and 91% for coding region partial sequences of FER, WRI1, and ADPG1, respectively (Gustafsson et al., 2018). The homologs of INDEHISCENT (IND), KNOTTED-LIKE FROM ARABIDOPSIS THALIANA-1 (KNAT1), and ALCATRAZ (ALC) were mapped to LG-6. IND and ALC are valve identity genes responsible for the establishment of the valve margin in the seed-containing pod and thereby regulate pod shattering in Arabidopsis (Rajani and Sundaresan, 2001; Liljegren et al., 2004). KNAT1 is a gene that determines pod density through regulating traits such as internode length and angles of pods (Douglas et al., 2002; Venglat et al., 2002). The alignment of A. thaliana and L. campestre partial coding sequences showed 85 and 83% sequence identity for IND and ALC genes, respectively (Gustafsson et al., 2018).
The homologs of APETALA2 (AP2) and VERNALIZATION 2 (VRN2) were mapped to LG-7. AP2 is a transcription factor gene involved in the regulation of flowering and seed development (Okamuro et al., 1997) as well as controlling seed yield (Jofuku et al., 2005), whereas VRN2 regulates vernalization responses (Gendall et al., 2001) in Arabidopsis. AP2 and VRN2 showed 89 and 90% sequence identity, respectively, between A. thaliana and L. campestre in their partial coding sequences (Gustafsson et al., 2018). The homologs of FRUITFULL (FUL), GLUCOSINOLATE TRANSPORTER-2 (GTR2), AUTOPHAGY RELATED 5 (ATG5), FLOWERING LOCUS C (FLC), TERMINAL FLOWER 1 (TFL1), and REPLUMLESS (RPL) were mapped to LG-8. FUL controls the development of the wall of seedpods (valve) (Gu et al., 1998; Ferrándiz et al., 2000) and thereby regulates pod shattering. GTR2 regulates glucosinolates transport (Andersen and Halkier, 2014), whereas ATG5 is a gene involved in plant defense (Yoshimoto et al., 2009). FLC is a MADS-box gene that plays a key role in regulating plant developmental responses to temperature as well as flowering (Michaels and Amasino, 1999; Sheldon et al., 2000). TFL1 is a gene involved in the determination of inflorescence architecture in Arabidopsis (Ferrándiz-Nohales et al., 2014), whereas RPL is one of several genes responsible for the establishment of the valve margin in the seed-containing pod and thereby involved in the regulation of pod shattering (Roeder et al., 2003). According to Gustafsson et al. (2018), the partial coding sequences of FUL, GTR2, ATG5, FLC, and RPL showed 92, 88, 81, 90, and 89% sequence identity, in that order, between A. thaliana and L. campestre.
In summary, the eight LGs revealed in this study confirm the previously reported (1) haploid chromosome number of eight in L. campestre and L. heterophyllum; (2) chromosomal fusion, translocation, and inversion events during the evolution of n = 8 karyotype in ancestral species shared by Lepidium and Arabidopsis to n = 5 karyotype in A. thaliana. The construction of high-density genetic linkage map bearing thousands of polymorphic markers and the identification of homologs of various desirable genes in the present study are significant steps toward the application of marker-aided and genomic selection in L. campestre for its accelerated domestication.
Data Availability Statement
The datasets generated for this study can be found in the DDBJ/ENA/GenBank under the accession WJSH00000000. The version described in this paper is version WJSH01000000.
Author Contributions
RO and MG secured the funding. CG and MG developed the mapping population and extracted DNA and conducted comparative genomics analysis. JG generated the GBS data and constructed genetic linkage map. MG and JG wrote the manuscript. All authors conceived and designed the study, contributed to data analysis, revised the manuscript, and read and approved the final version of the manuscript for publication.
Funding
This work was financed by grants from the Swedish Foundation for Strategic Research (SSF), the Swedish Foundation for Strategic Environmental Research (MISTRA), and Swedish University of Agricultural Sciences (SLU).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank PlantLink for bioinformatics support. We would also like to acknowledge Professor Sten Stymne for his major role in the development of the SSF project.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00448/full#supplementary-material
FIGURE S1 | A framework genetic map comprising 2331 SNP markers on eight linkage groups: The rows are F2 individuals and the columns (each only a few pixels wide) are markers; the vertical black lines separate the linkage groups; blue is heterozygous, green is homozygous for parent-1, yellow is homozygous for parent-2, and pink is missing data.
TABLE S1 | (A) list of the 2330 mapped SNPs on 1044 contigs/scaffolds, (B) final ordering of the 10302 genetically mapped contigs/scaffolds, (C) List of contigs/scaffolds with mapped SNPs for each linkage group arranged according to the number of mapped SNPs, (D) summary of data provided in (A), (B) and (C).
TABLE S2 | (A) List of contigs/scaffolds mapped by SNPs, and (B) cumulative percentage of contigs with >1 mapped SNPs.
TABLE S3 | (A) List of the 34342 genetically mapped tags from contigs, and (B) list of the 9931 mapped contig tag haplotypes.
Footnotes
References
Al-Shehbaz, I. A. (1986). The genera of Lepidieae (Cruciferae: Brassicaceae) in the southeastern United States. J. Arnold Arbor. 67, 265–311. doi: 10.5962/bhl.part.27392
Al-Shehbaz, I. A., Beilstein, M. A., and Kellogg, E. A. (2006). Systematics and phylogeny of the Brassicaceae (Cruciferae): an overview. Plant Syst. Evol. 259, 89–120. doi: 10.1007/s00606-006-0415-z
Andersen, T. G., and Halkier, B. A. (2014). Upon bolting the GTR1 and GTR2 transporters mediate transport of glucosinolates to the inflorescence rather than roots. Plant Signal. Behav. 9:e27740. doi: 10.4161/psb.27740
Andersson, A. A., Merker, A., Nilsson, P., Sørensen, H., and Åman, P. (1999). Chemical composition of the potential new oilseed crops Barbarea vulgaris, Barbarea verna and Lepidium campestre. J. Sci. Food Agric. 79, 179–186. doi: 10.1002/(sici)1097-0010(199902)79:2<179::aid-jsfa163>3.0.co;2-n
Bailey, C. D., Koch, M. A., Mayer, M., Mummenhoff, K., O’Kane, S. L.Jr., Warwick, S. I., et al. (2006). Toward a global phylogeny of the Brassicaceae. Mol. Biol. Evol. 23, 2142–2160. doi: 10.1093/molbev/msl087
Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., et al. (2008). Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3:e3376. doi: 10.1371/journal.pone.0003376
Boivin, K., Acarkan, A., Mbulu, R.-S., Clarenz, O., and Schmidt, R. (2004). The Arabidopsis genome sequence as a tool for genome analysis in Brassicaceae. A comparison of the Arabidopsis and Capsella rubella Genomes. Plant Physiol. 135, 735–744. doi: 10.1104/pp.104.040030
Börjesdotter, D. (1999). Potential Oil Crops. Cultivation of Barbarea verna, Barbarea vulgaris and Lepidium campestre. Uppsala: Swedish University of Agricultural Sciences.
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Broman, K. W., Wu, H., Sen, Ś, and Churchill, G. A. (2003). R/qtl: QTL mapping in experimental crosses. Bioinformatics 19, 889–890. doi: 10.1093/bioinformatics/btg112
Burger, J. C., Chapman, M. A., and Burke, J. M. (2008). Molecular insights into the evolution of crop plants. Am. J. Bot. 95, 113–122. doi: 10.3732/ajb.95.2.113
Charlesworth, B. (1992). Evolutionary rates in partially self-fertilizing species. Am. Nat. 140, 126–148. doi: 10.1086/285406
Coghlan, A., Eichler, E. E., Oliver, S. G., Paterson, A. H., and Stein, L. (2005). Chromosome evolution in eukaryotes: a multi-kingdom perspective. Trends Genet. 21, 673–682. doi: 10.1016/j.tig.2005.09.009
Collard, B. C. Y., and Mackill, D. J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. B 363, 557–572. doi: 10.1098/rstb.2007.2170
Couvreur, T. L. P., Franzke, A., Al-Shehbaz, I. A., Bakker, F. T., Koch, M. A., and Mummenhoff, K. (2010). Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 27, 55–71. doi: 10.1093/molbev/msp202
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Doebley, J. F., Gaut, B. S., and Smith, B. D. (2006). The molecular genetics of crop domestication. Cell 127, 1309–1321. doi: 10.1016/j.cell.2006.12.006
Douglas, S. J., Chuck, G., Dengler, R. E., Pelecanda, L., and Riggs, C. D. (2002). KNAT1 and ERECTA regulate inflorescence architecture in Arabidopsis. Plant Cell 14, 547–558. doi: 10.1105/tpc.010391
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple Genotyping- by-Sequencing (GBS) approach for high diversity species. PLoS One 6:e19379. doi: 10.1371/journal.pone.0019379
Eriksson, D. (2009). Towards the domestication of Lepidium Campestre as an undersown oilseed crop. Acta Univ. Agric. Suec. Agrar. 2009:65.
Ferrándiz, C., Liljegren, S. J., and Yanofsky, M. F. (2000). Negative regulation of the SHATTERPROOF genes by FRUITFULL during Arabidopsis fruit development. Science 289, 436–438. doi: 10.1126/science.289.5478.436
Ferrándiz-Nohales, P., Domenech, M. J., de Alba, A. E. M., Micol, J. L., Ponce, M. R., and Madueno, F. (2014). AGO1 controls Arabidopsis inflorescence architecture possibly by regulating TFL1 expression. Ann. Bot. 114, 1471–1481. doi: 10.1093/aob/mcu132
Focks, N., and Benning, C. (1998). wrinkled1: a novel, low-seed-oil mutant of Arabidopsis with a deficiency in the seed-specific regulation of carbohydrate metabolism. Plant Physiol. 118, 91–101. doi: 10.1104/pp.118.1.91
Geleta, M., and Ortiz, R. (2016). Molecular and genomic tools provide insights on crop domestication and evolution. Adv. Agron. 135, 181–223.
Geleta, M., Zhu, L.-H., Stymne, S., Lehrman, A., and Hansson, S. O. (2014). “Domestication of Lepidium campestre as part of Mistra Biotech, a research programme focused on agro-biotechnology for sustainable food. Paper presented at the perennial crops for food security,” in Proceedings of the FAO Expert Workshop, Rome, Italy.
Gendall, A. R., Levy, Y. Y., Wilson, A., and Dean, C. (2001). The VERNALIZATION 2 gene mediates the epigenetic regulation of vernalization in Arabidopsis. Cell 107, 525–535. doi: 10.1016/s0092-8674(01)00573-6
Glaubitz, J. C., Casstevens, T. M., Lu, F., Harriman, J., Elshire, R. J., Sun, Q., et al. (2014). TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9:e90346. doi: 10.1371/journal.pone.0090346
Gu, Q., Ferrandiz, C., Yanofsky, M. F., and Martienssen, R. (1998). The FRUITFULL MADS-box gene mediates cell differentiation during Arabidopsis fruit development. Development 125, 1509–1517.
Gustafsson, C., Willforss, J., Lopes-Pinto, F., Ortiz, R., and Geleta, M. (2018). Identification of genes regulating traits targeted for domestication of field cress (Lepidium campestre) as a biennial and perennial oilseed crop. BMC Genetics 19:36. doi: 10.1186/s12863-018-0624-9
Harlan, J. R., DeWet, M. J. J., and Price, E. G. (1973). Comparative evolution of cereals. Evol. Int. J. Org. Evol. 27, 311–325. doi: 10.1111/j.1558-5646.1973.tb00676.x
Hu, T. T., Pattyn, P., Bakker, E. G., Cao, J., Cheng, J. F., Clark, R. M., et al. (2011). The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–481. doi: 10.1038/ng.807
Ivarson, E., Ahlman, A., Lager, I., and Zhu, L.-H. (2016). Significant increase of oleic acid level in the wild species Lepidium campestre through direct gene silencing. Plant Cell Rep. 35, 2055–2063. doi: 10.1007/s00299-016-2016-9
Ivarson, E., Ahlman, A., Li, X., and Zhu, L.-H. (2013). Development of an efficient regeneration and transformation method for the new potential oilseed crop Lepidium campestre. BMC Plant Boil. 13:1. doi: 10.1186/1471-2229-13-115
Jako, C., Kumar, A., Wei, Y., Zou, J., Barton, D. L., Giblin, E. M., et al. (2001). Seed-specific over-expression of an Arabidopsis cDNA encoding a diacylglycerol acyltransferase enhances seed oil content and seed weight. Plant Physiol. 126, 861–874. doi: 10.1104/pp.126.2.861
James, D. W. Jr., Lim, E., Keller, J., Plooy, I., Ralston, E., and Dooner, H. K. (1995). Directed tagging of the Arabidopsis FATTY ACID ELONGATION1 FAE1; gene with the maize transposon activator. Plant Cell 7, 309–319. doi: 10.1105/tpc.7.3.309
Jofuku, K. D., Omidyar, P. K., Gee, Z., and Okamuro, J. K. (2005). Control of seed mass and seed yield by the floral homeotic gene APETALA2. Proc. Nat. Acad. Sci. U.S.A. 102, 3117–3122. doi: 10.1073/pnas.0409893102
Kessler, S. A., Shimosato-Asano, H., Keinath, N. F., Wuest, S. E., Ingram, G., Panstruga, R., et al. (2010). Conserved molecular components for pollen tube reception and fungal invasion. Science 330, 968–971. doi: 10.1126/science.1195211
Koch, M., and Kiefer, M. (2005). Genome evolution among cruciferous plants: a lecture from the comparison of the genetic maps of three diploid species—Capsella rubella, Arabidopsis lyrata subsp. petraea, and A. thaliana. Am. J. Bot. 92, 761–767. doi: 10.3732/ajb.92.4.761
Koech, R. K., Mose, R., Kamunya, S. M., and Apostolides, Z. (2019). Combined linkage and association mapping of putative QTLs controlling black tea quality and drought tolerance traits. Euphytica 215:162.
Kuittinen, H., de Haan, A. A., Vogl, C., Oikarinen, S., Leppala, J., Koch, M., et al. (2004). Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics 168, 1575–1584. doi: 10.1534/genetics.103.022343
Latta, R. G., Bekele, W. A., Wight, C. P., and Tinker, N. A. (2019). Comparative linkage mapping of diploid, tetraploid, and hexaploid Avena species suggests extensive chromosome rearrangement in ancestral diploids. Sci. Rep. 9:12298. doi: 10.1038/s41598-019-48639-7
Lee, J.-Y., Mummenhoff, K., and Bowman, J. L. (2002). Allopolyploidization and evolution of species with reduced floral structures in Lepidium L. (Brassicaceae). ıProc. Natl. Acad. Sci. U.S.A. 99, 16835–16840. doi: 10.1073/pnas.242415399
Liljegren, S. J., Roeder, A. H., Kempin, S. A., Gremski, K., Ostergaard, L., Guimil, S., et al. (2004). Control of fruit patterning in Arabidopsis by INDEHISCENT. Cell 116, 843–853. doi: 10.1016/s0092-8674(04)00217-x
Lopez-Cruz, M., Crossa, J., Bonnett, D., Dreisigacker, S., Poland, J., Jean-Luc Jannink, J.-L., et al. (2015). Increased prediction accuracy in wheat breeding trials using a marker x environment interaction genomic selection model. G3 5, 569–582. doi: 10.1534/g3.114.016097
Lorenz, A. J., Smith, K. P., and Jannink, J.-L. (2012). Potential and optimization of genomic selection for Fusarium head blight resistance in six-row barley. Crop Sci. 52, 1609–1621. doi: 10.2135/cropsci2011.09.0503
Luerssen, H., Kirik, V., Herrmann, P., and Miséra, S. (1998). FUSCA3 encodes a protein with a conserved VP1/AB13-like B3 domain which is of functional importance for the regulation of seed maturation in Arabidopsis thaliana. Plant J. 15, 755–764. doi: 10.1046/j.1365-313x.1998.00259.x
Lysak, M. A., Berr, A., Pecinka, A., Schmidt, R., McBreen, K., and Schubert, I. (2006). Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl. Acad. Sci. U.S.A. 103, 5224–5229. doi: 10.1073/pnas.0510791103
Merker, A., Eriksson, D., and Bertholdsson, N.-O. (2010). Barley yield increases with undersown Lepidium campestre. Acta Agric. Scand. B S. P. 60, 269–273. doi: 10.1080/09064710902903747
Merker, A., and Nilsson, P. (1995). Some oil crop properties in wild Barbarea and Lepidium species. Swed. J. Agric. Res. 25, 173–178.
Michaels, S. D., and Amasino, R. M. (1999). FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11, 949–956. doi: 10.1105/tpc.11.5.949
Mulligan, G. A. (1961). The genus Lepidium in canada. Madroño 16, 77–90. doi: 10.1094/PDIS-93-1-0108B
Mummenhoff, K., Brüggemann, H., and Bowman, J. L. (2001). Chloroplast DNA phylogeny and biogeography of Lepidium (Brassicaceae). Am. J. Bot. 88, 2051–2063. doi: 10.2307/3558431
Mummenhoff, K., Kuhnt, E., Koch, M., and Zunk, K. (1995). Systematic implications of chloroplast DNA variation in Lepidium sections Cardamon, Lepiocardamon and Lepia (Brassicaceae). Plant Syst. Evol. 196, 75–88. doi: 10.1007/bf00985336
Murat, F., Louis, A., Maumus, F., Armero, A., Cooke, R., Quesneville, H., et al. (2015). Understanding Brassicaceae evolution through ancestral genome reconstruction. Genome Biol. 16:262. doi: 10.1186/s13059-015-0814-y
Nguyen, T.-P., Mühlich, C., Mohammadin, S., van den Bergh, E., Platts, A. E., Haas, F. B., et al. (2019). Genome improvement and genetic map construction for Aethionema arabicum, the first divergent branch in the Brassicaceae family. G3 9, 3521–3530. doi: 10.1534/g3.119.400657
Nilsson, P., Johansson, S. Å., and Merker, A. (1998). Variation in seed oil composition of species from the genera Barbarea and Lepidium. Acta Agric. Scand. B S. P 48, 159–164.
Ogawa, M., Kay, P., Wilson, S., and Swain, S. M. (2009). Arabidopsis Dehiscence Zone Polygalacturonase1 ADPG1, ADPG2, and QUARTET2 are polygalacturonases required for cell separation during reproductive development in Arabidopsis. Plant Cell 21, 216–233. doi: 10.1105/tpc.108.063768
Okamuro, J. K., Caster, B., Villarroe, R., Van Montagu, M., and Jofuku, K. D. (1997). The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 94, 7076–7081. doi: 10.1073/pnas.94.13.7076
Ooms, J. J. J., Le’on-Kloosterziel, K. M., Bartels, D., Koornneef, M., and Karssen, C. M. (1993). Acquisition of desiccation tolerance and longevity in seeds of Arabidopsis thaliana (a comparative study using abscisic acid-insensitive abi3 mutants). Plant Physiol. 102, 1185–1191. doi: 10.1104/pp.102.4.1185
Parkin, I. A. P., Gulden, S. M., Sharpe, A. G., Lukens, L., Trick, M., Osborn, T. C., et al. (2005). Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics 171, 765–781.
Pérez-de-Castro, A. M., Vilanova, S., Cañizares, J., Pascual, L., Blanca, J. M., Díez, M. J., et al. (2012). Application of genomic tools in plant breeding. Curr. Genomics 13, 179–195. doi: 10.2174/138920212800543084
Poland, J. A., Brown, P. J., Sorrells, M. E., and Jannink, J.-L. (2012). Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7:e32253. doi: 10.1371/journal.pone.0032253
Qu, C., Jia, L., Fu, F., Zhao, H., Lu, K., Wei, L., et al. (2017). Genome-wide association mapping and identification of candidate genes for fatty acid composition in Brassica napus L. using SNP markers. BMC Genomics 18:232. doi: 10.1186/s12864-017-3607-8
R Core Team (2016). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rajani, S., and Sundaresan, V. (2001). The Arabidopsis myc/bHLH gene ALCATRAZ enables cell separation in fruit dehiscence. Curr. Biol. 11, 1914–1922.
Rice, A., Glick, L., Abadi, S., Einhorn, M., Kopelman, N. M., Salman-Minkov, A., et al. (2015). The chromosome counts database (CCDB) – a community resource of plant chromosome numbers. New Phytol. 206, 19–26.
Roeder, A. H., Ferrandiz, C., and Yanofsky, M. F. (2003). The role of the REPLUMLESS homeodomain protein in patterning the Arabidopsis fruit. Curr. Biol. 13, 1630–1635.
Routaboul, J. M., Benning, C., Bechtold, N., Caboche, M., and Lepiniec, L. (1999). The TAG1 locus of Arabidopsis encodes for a diacylglycerol acyltransferase. Plant Physiol. Biochem. 37, 831–840.
Saito, H., Oikawa, T., Hamamoto, S., Ishimaru, Y., Kanamori-Sato, M., Sasaki-Sekimoto, Y., et al. (2015). The jasmonate-responsive GTR1 transporter is required for gibberellin-mediated stamen development in Arabidopsis. Nat. Commun. 6:6095. doi: 10.1038/ncomms7095
Sheldon, C. C., Rouse, D. T., Finnegan, E. J., Peacock, W. J., and Dennis, E. S. (2000). The molecular basis of vernalization: the central role of FLOWERING LOCUS C FLC. Proc. Natl. Acad. Sci. U.S.A. 97, 3753–3758.
Tang, H., Bowers, J. E., Wang, X., Ming, R., Alam, M., and Paterson, A. H. (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. doi: 10.1126/science.1153917
Tello, J., Roux, C., Chouiki, H., Laucou, V., Sarah, G., Weber, A., et al. (2019). A novel high-density grapevine (Vitis vinifera L.) integrated linkage map using GBS in a half-diallel population. Theor. Appl. Genet. 132, 2237–2252. doi: 10.1007/s00122-019-03351-y
Venglat, S. P., Dumonceaux, T., Rozwadowski, K., Parnell, L., Babic, V., and Keller, W. (2002). The homeoboxgene BREVIPEDICELLUS is a key regulator of inflorescence architecture in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 99, 4730–4735.
Weeden, N. F. (2007). Genetic changes accompanying the domestication of Pisum sativum: is there a common genetic basis to the ‘Domestication Syndrome’ for legumes? Ann. Bot. 100, 1017–1025.
Wu, Y., Bhat, P. R., Close, T. J., and Lonardi, S. (2008). Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genetics 4:e1000212. doi: 10.1371/journal.pgen.1000212
Yogeeswaran, K., Frary, A., York, T. L., Amenta, A., Lesser, A. H., Nasrallah, J. B., et al. (2005). Comparative genome analyses of Arabidopsis spp.: inferring chromosomal rearrangement events in the evolutionary history of A. thaliana. Genome Res. 15, 505–515.
Yoshimoto, K., Jikumaru, Y., Kamiya, Y., Kusano, M., Consonni, C., Panstruga, R., et al. (2009). Autophagy negatively regulates cell death by controlling NPR1-dependent salicylic acid signaling during senescence and the innate immune response in Arabidopsis. Plant Cell 21, 2914–2927. doi: 10.1105/tpc.109.068635
Keywords: contig tag haplotype, field cress, genetic linkage mapping, genotyping by sequencing, Lepidium, linkage group, single-nucleotide polymorphism
Citation: Geleta M, Gustafsson C, Glaubitz JC and Ortiz R (2020) High-Density Genetic Linkage Mapping of Lepidium Based on Genotyping-by-Sequencing SNPs and Segregating Contig Tag Haplotypes. Front. Plant Sci. 11:448. doi: 10.3389/fpls.2020.00448
Received: 27 November 2019; Accepted: 26 March 2020;
Published: 30 April 2020.
Edited by:
Jose Luis Gonzalez Hernandez, South Dakota State University, United StatesReviewed by:
Leonardo Velasco, Institute for Sustainable Agriculture, Spanish National Research Council, SpainSanghyeob Lee, Sejong University, South Korea
Copyright © 2020 Geleta, Gustafsson, Glaubitz and Ortiz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mulatu Geleta, Mulatu.Geleta.Dida@slu.se