 Chenxin Li1,2
Chenxin Li1,2 Zhongshuai Sun
Zhongshuai Sun Ming Jiang
Ming Jiang Junmin Li
Junmin Li- 1College of Life Sciences and Medicine, Zhejiang Sci-Tech University, Hangzhou, China
- 2Zhejiang Provincial Key Laboratory of Plant Evolutionary Ecology and Conservation, Taizhou University, Taizhou, China
- 3School of Advanced Study, Taizhou University, Taizhou, China
Species identification is vital for protecting species diversity and selecting high-quality germplasm resources. Wild Fragaria spp. comprise rich and excellent germplasm resources; however, the variation and evolution of the whole chloroplast (cp) genomes in the genus Fragaria have been ignored. In the present study, 27 complete chloroplast genomes of 11 wild Fragaria species were sequenced using the Illumina platform. Then, the variation among complete cp genomes of Fragaria was analyzed, and phylogenetic relationships were reconstructed from those genome sequences. There was an overall high similarity of sequences, with some divergence. According to analysis with mVISTA, non-coding regions were more variable than coding regions. Inverted repeats (IRs) were observed to contract or expand to different degrees, which resulted in different sizes of cp genomes. Additionally, five variable loci, trnS-trnG, trnR-atpA, trnC-petN, rbcL-accD, and psbE-petL, were identified that could be used to develop DNA barcoding for identification of Fragaria species. Phylogenetic analyses based on the whole cp genomes supported clustering all species into two groups (A and B). Group A species were mainly distributed in western China, while group B contained several species from Europe and Americas. These results support allopolyploid origins of the octoploid species F. chiloensis and F. virginiana and the tetraploid species F. moupinensis and F. tibetica. The complete cp genomes of these Fragaria spp. provide valuable information for selecting high-quality Fragaria germplasm resources in the future.
Introduction
The genus Fragaria Linnaeus belongs to the family Rosaceae and is comprised of 25 species, including 13 diploids (2n), five tetraploids (4n), one pentaploid (5n), one hexaploid (6n), three octoploids (8n), and two decaploids (10n; Staudt, 1962, 1989, 2009; Davis et al., 2010; Hummer, 2012; Lei et al., 2017). Most Fragaria species are wild, except for F. × ananassa, which is a cultivated species and an economically important crop (Staudt, 1962; Potter et al., 2000; Cheng et al., 2017). China has been recognized as an important distribution center of wild strawberry resources in the world, as it has 14 wild Fragaria species including nine diploid species and five tetraploid species (Staudt, 1989, 2006; Lei et al., 2017). Compared with cultivated species, wild species have several advantages, including stronger resistance (Diamanti et al., 2012; Guo et al., 2018), unique fruit aromas (Urrutia et al., 2017), and richness in nutrients (Capocasa et al., 2008a,b; Tulipani et al., 2008). Therefore, elucidating the relationships among Fragaria spp. is vital to developing high-quality strawberry varieties.
As well as being an important plastid functioning in photosynthesis and carbon fixation (Neuhaus and Emes, 2000), the chloroplast (cp) contains a genome that can provide valuable information for taxonomic and phylogenic purposes, with the key advantages of its relatively small size, more conservative structure, and low nucleotide substitution rate (Palmer, 1985; Wolfe et al., 1987; Jansen et al., 2005; Wei et al., 2006; Wicke et al., 2011; Terakami et al., 2012; Liu et al., 2019). Furthermore, the cp genome is haploid and uniparentally inherited, so it is helpful for tracing source populations and conducting phylogenetic studies to resolve complex evolutionary relationships (Jansen et al., 2007; Parks et al., 2009; Ruhsam et al., 2015). To date, phylogenetic relationships based on complete cp genomes of many angiosperms have been fully studied in many genera (Hu et al., 2016; Zhang et al., 2016; Zhao et al., 2018). Njuguna et al. (2013) systematically studied the phylogenetic relationships of Fragaria based on cp genomes, but there were only 10 sequences assembled from total genomic data, and the coverage of PCR amplified sequences ranged from 60 to 90%, which may mean the assembled chloroplast sequences were incomplete. To the best of our knowledge, there is only one study that has focused on the molecular phylogenetic analysis of Fragaria genus based on whole complete chloroplast genome sequences (Sun et al., 2021), which revealed that 20 Fragaria species were clustered into northern group (eight species), southern group (11 species), and an oldest extant species (one species) based on whole cp genomes. However, this previous study focused on molecular clock analysis and ignored the variation and evolution of whole cp genomes of Fragaria, including, for example, the contraction and expansion of inverted repeats (IRs) regions (Plunkett and Downie, 2000; Kode et al., 2005; Ma et al., 2014; Lei et al., 2016).
In addition, although some Fragaria species can be identified by their morphological characteristics, some species that have similar morphological structures, such as F. mandshurica and F. orientalis (Staudt, 2003; Lei et al., 2017), are likely to be inaccurately identified if morphological indexes are not collected (Yang et al., 2020). Furthermore, Yang et al. (2020) found that most wild Fragaria in Yunnan were difficult to accurately identify without flowers and fruits of collected species. In total, owing to the stage of species development and subjective factors (such as differences in personal knowledge and experience), traditional morphology classification is often inconsistent and unreliable, which can also affect the results of species identification (Yang et al., 2020). Over the years, many molecular analyses have provided insights into the species taxonomy and identification. Currently, DNA barcoding such as rbcL, matK, psbA-trnH and ITS sequences (Potter et al., 2007; Fazekas et al., 2008; Chen et al., 2013; Xin et al., 2013; Tegally et al., 2019; Phi et al., 2020; Islam et al., 2021) has been used for species identification. In Fragaria, the study of DNA barcoding dates back to the phylogenetic analysis conducted by Potter et al. (2000) using ITS and trnL-trnF sequences, but the authors were unable to completely distinguish among species in this genus. Njuguna and Bassil (2011) analyzed psbA-trnH and ITS sequences and reported that the two DNA barcoding sequences are not suitable for species identification in Fragaria. Moreover, more and more studies demonstrated that the four universal barcoding sequences were problematic with low bootstrap support and inability to distinguish between species in land plants (Xin et al., 2013; Tegally et al., 2019; Phi et al., 2020; Islam et al., 2021). Therefore, it is urgent to excavate superior DNA barcoding for special land plants, including Fragaria species, utilizing complete cp genomes, which contain more important variation information for taxonomic and phylogenic purposes (Huang et al., 2014).
In the present study, we sequenced 27 complete cp genomes of 11 wild Fragaria species from different collection sites in China and downloaded the whole cp genome sequences of seven more Fragaria species. This research had the following objectives: (1) to describe the characteristics of Fragaria cp genomes; (2) to infer the phylogenetic relationships among Fragaria spp.; (3) to detect the variations among Fragaria cp genomes and infer the evolution of the whole cp genomes of Fragaria species; and (4) to provide candidate DNA barcodes for Fragaria species identification. These results provide new insights into the interspecies relationships and evolution of Fragaria spp. as well as basic reference material for the application of Fragaria germplasm resources.
Materials and Methods
Plant Material Collection and Genome Data Sources
Twenty-seven individuals belonging to 11 Fragaria species were included in the present study, as summarized in Table 1. All the sampled plants were cultured in the greenhouse facilities of Taizhou University under conditions of 70% relative humidity with temperatures of 25°C in the day and 20°C at night. The plants were identified by Professor Beifen Yang of Taizhou University. The specimens were stored in Zhejiang Provincial Key Laboratory of Plant Evolutionary Ecology and Conservation, Taizhou University, China.
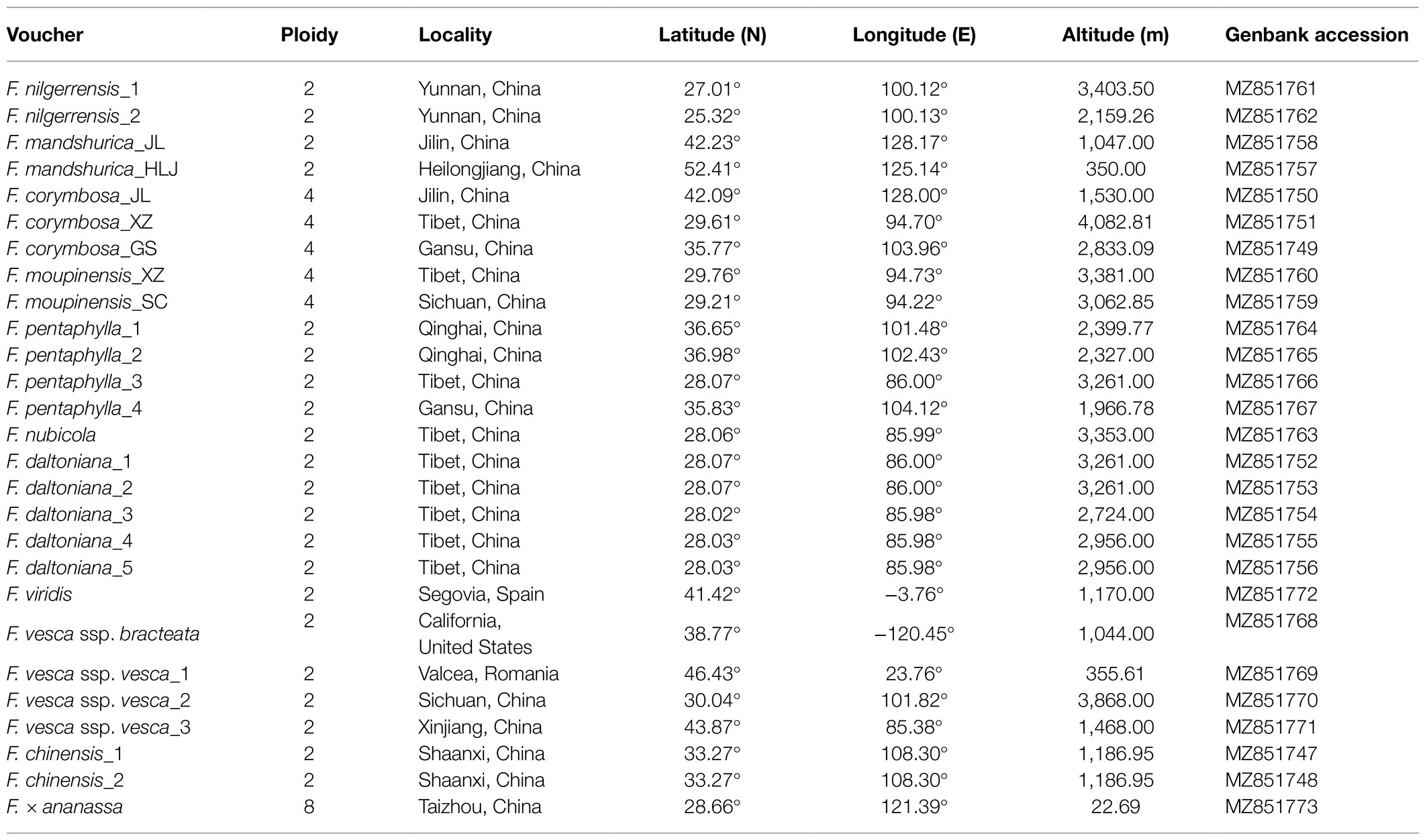
Table 1. Fragaria species collection information.
The complete cp genome sequences of F. orientalis (NC_035501), Fragaria chiloensis (NC_019601), and F. virginiana (NC_019602) were downloaded from the National Center for Biotechnology Information (NCBI). Fragaria nipponica (KY769125) and F. iinumae (KC507759) were excluded for there is no supporting of publication reference. Fragaria × ananassa (KY358226) was also downloaded from NCBI to test the accuration of the sequencing and de novo assembly of cp genome.
Raw sequence data from three species, including F. gracilis (BOP214815), F. tibetica (BOP214818), and F. moschata (BOP214819; Sun et al., 2021), were downloaded from NCBI and de novo assembly was performed according to the following processes. The other seven species listed by Sun et al. (2021) were excluded for duplication of our own sequencing species.
Genome Sequencing and Assembly
Fresh and clean leaves of each sampled species were collected and frozen in liquid nitrogen immediately. The samples were used to extract the total DNA by the modified CTAB method (Doyle and Doyle, 1987). Then, paired-end sequencing (Insert size: 350bp) was performed using the Illumina Novaseq 6000 platform (Illumina, San Diego, CA, United States). Raw reads were filtered to obtain high-quality clean data. Then, de novo assembly was performed with the GetOrganelle software package (Jin et al., 2020).
Chloroplast Genome Annotation
Thirty cp genomes were annotated using the online GeSeq tool (Tyagi et al., 2020)1 with default parameters and F. chiloensis (NC_019601) was used as the reference to predict protein-coding genes (PCGs), transfer RNA (tRNA) genes, and ribosomal RNA (rRNA) genes. Then, the cp genome sequences were manually modified using Geneious Prime 2021.1.1 (Biomatters Ltd., Auckland, New Zealand). A circular diagram of the cp genomes was generated using the online OrganellarGenome DRAW tool (OGDRAW; Lohse et al., 2013; Bai et al., 2017).
Comparative Genome Analyses
The boundaries of the large single-copy region (LSC), short single-copy region (SSC), and IRs of 12 newly assembled complete cp genomes of Fragaria species (F. nilgerrensis_2, F. mandshurica_JL, F. corymbosa_GS, F. moupinensis_SC, F. pentaphylla_3, F. nubicola, F. daltoniana_1, F. viridis, F. vesca ssp. bracteata, F. vesca ssp. vesca_1, F. chinensis_1, F. × ananassa) in this study together with six complete cp genomes of F. gracilis (BOP214815), F. tibetica (BOP214818), F. moschata (BOP214819), F. orientalis (NC_035501), F. chiloensis (NC_019601), and F. virginiana (NC_019602) from NCBI were compared using IRscope software (Ali et al., 2018). The level of divergence among the 18 sequences was visualized with Shuffle-LAGAN mode (Cheng et al., 2017; Tyagi et al., 2020) in mVISTA software (Kawabe et al., 2018; Jeon and Kim, 2019) with the default settings, and F. gracilis was used as a reference sequence.
Hypervariable Site Identification
All 18 sequences were aligned using the Geneious prime 2021.1.1 plugin MAFFT v.7.450 (Katoh and Standley, 2013), and the alignment was manually adjusted. Then, we performed a sliding window analysis using DnaSP v6 software (Rozas et al., 2017; Jeon and Kim, 2019) to analyze nucleotide diversity (π) in order to detect hypervariable sites among Fragaria cp genomes. The window length was set to 600bp, and the step size was set to 200bp (Sun et al., 2021).
Phylogenetic Analysis
Thirty-four complete cp genome sequences were used to reconstruct the phylogenetic relationships among Fragaria spp. based on maximum likelihood (ML) using RAxML 8.2.10 (Stamatakis, 2014), with 1,000 bootstrap replicates employed for estimating node support. Potentilla fruticosa (NC_036423) and Drymocallis saviczii (NC_050966) were downloaded from Genbank and used as outgroups (Eriksson et al., 1998, 2003; Potter et al., 2000; Feng et al., 2017; Dimeglio et al., 2014). In total, 36 cp genome sequences were aligned using a Geneious prime 2021.1.1 plugin MAFFT v.7.450 (Katoh and Standley, 2013), and the alignment was manually adjusted when necessary. Modeltest 3.7 (Liu et al., 2013) was used to select the best-fit evolutionary model of Fragaria cp genome sequence evolution for reconstruction of the phylogenetic relationships of Fragaria.
Results
General Chloroplast Genome Characteristics
The total genome sequence lengths of Fragaria species ranged from 155,479bp (F. viridis) to 155,832bp (F. daltoniana_3). The cp genomes presented a typical quadripartite structure including a pair of IR regions with lengths of 51,872bp (F. vesca ssp. bracteata) to 51,948bp (F. corymbosa_JL), separated by a LSC region from 85,471bp (F. viridis) to 85,726bp (F. daltoniana_3) and a SSC region from 18,116bp (F. viridis) to 18,219bp (F. moupinensis_XZ). The GC contents ranged from 37.2 to 37.3%. Overall, the cp genome of Fragaria encodes a total of 130 genes, including 85 PCGs, 37 tRNA genes, and eight rRNA genes (Figure 1; Table 2). Among these genes, 15 contained a single intron (ndhA, ndhB, petB, petD, rpl2, rpl16, rpoC1, rps12, rps16, trnA-UGC, trnG-GCC, trnI-CAU, trnK-UUU, trnL-UAA, and trnV-UAC), while two harbored two introns (ycf3 and clpP). The trnK-UUU gene had the longest intron (2,492–2,496bp), which contained the matK gene, whereas trnL-UAA was smallest (420bp; Table 3).
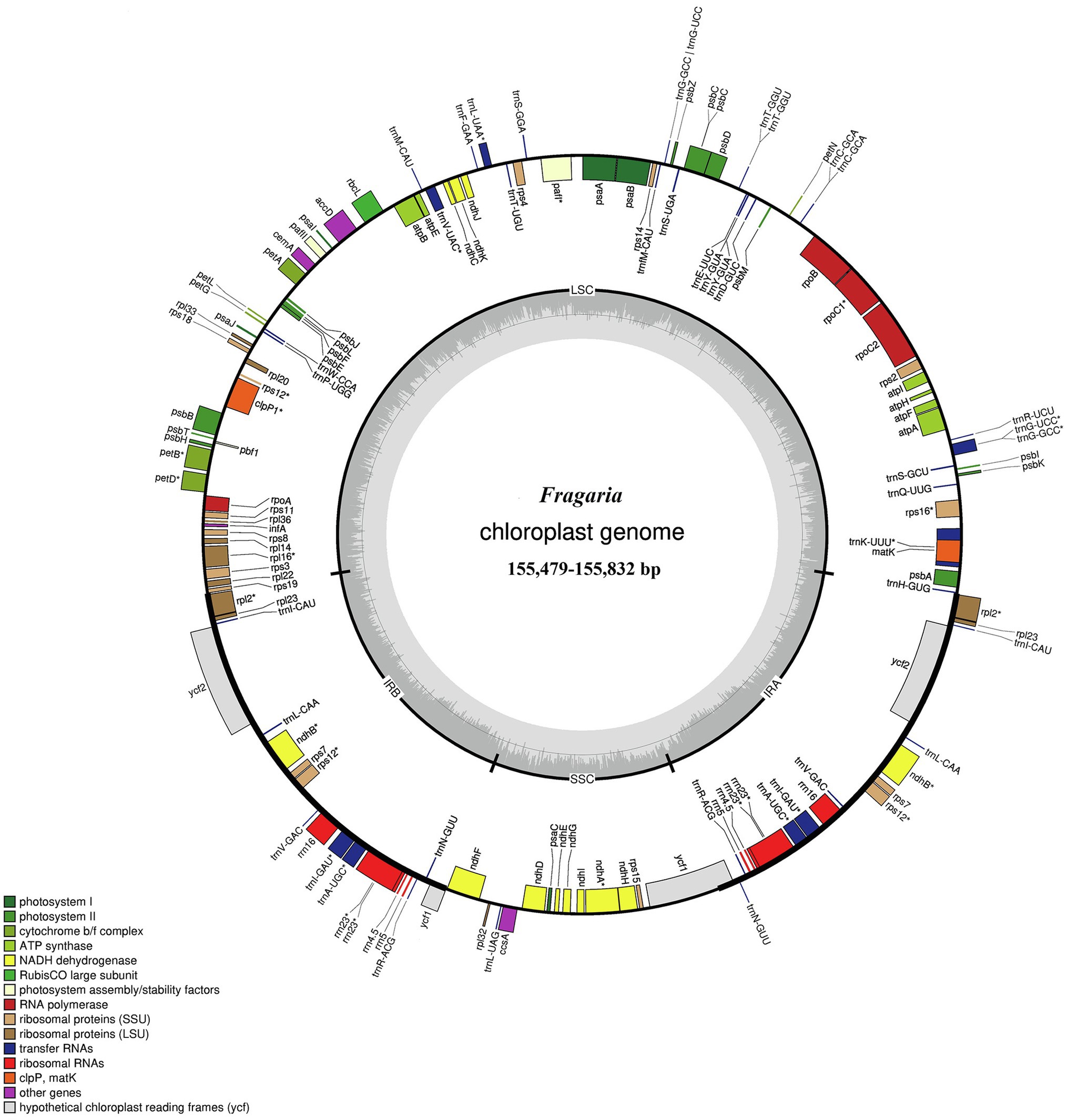
Figure 1. Gene map of the Fragaria chloroplast genome. Genes drawn inside the circle are transcribed clockwise, and those outside are transcribed counter clockwise. Color coding is used to show genes belonging to different functional groups. The inner circle indicates the range of the long single-copy region (LSC), short single-copy region (SSC), and inverted repeat regions (IRs) and also shows a GC content graph of the genome. In the GC content graph, the dark gray lines indicate GC content, while light gray lines indicate the AT content of each locus.
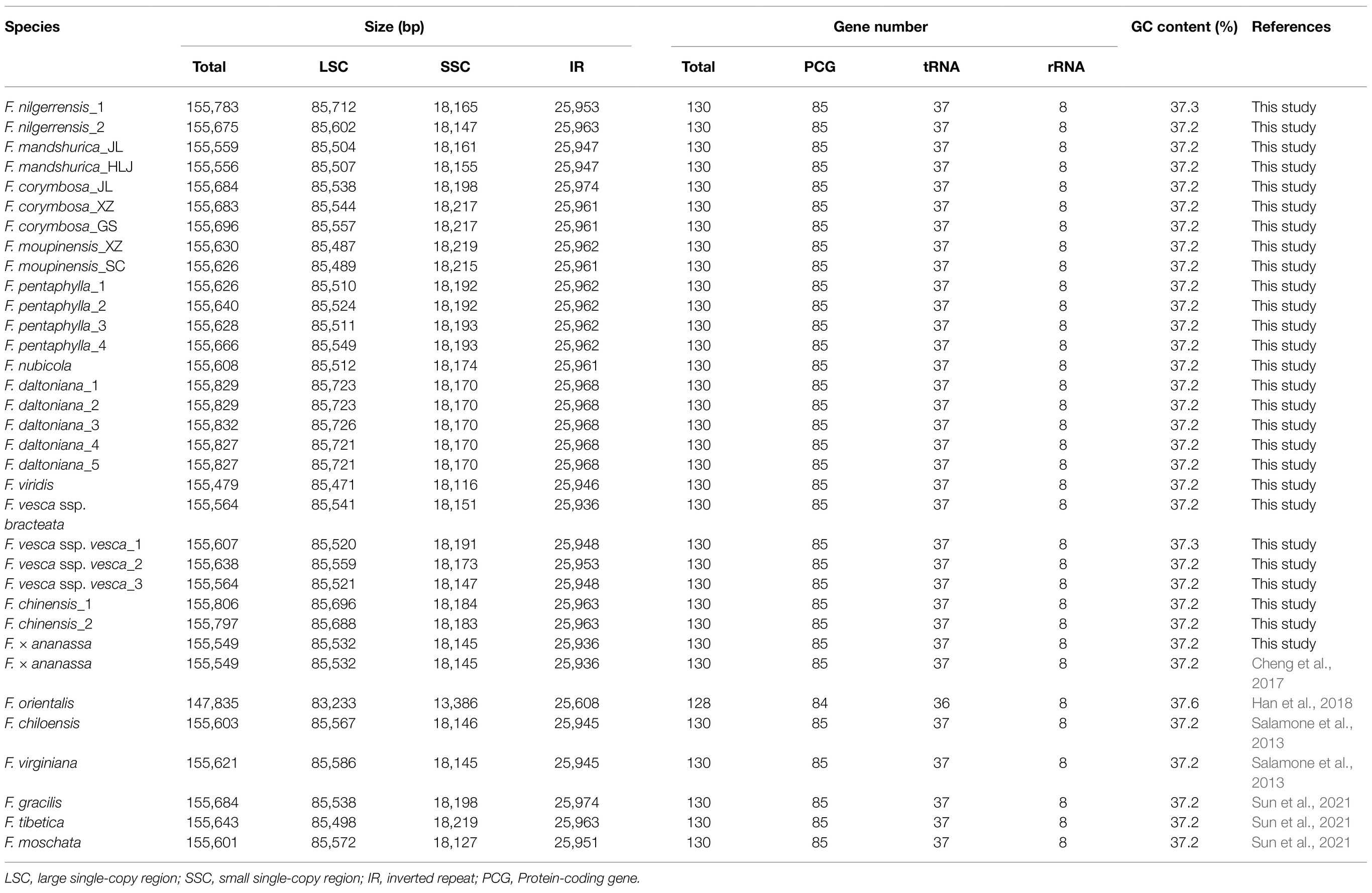
Table 2. Summary of 34 complete chloroplast genomes for Fragaria species.
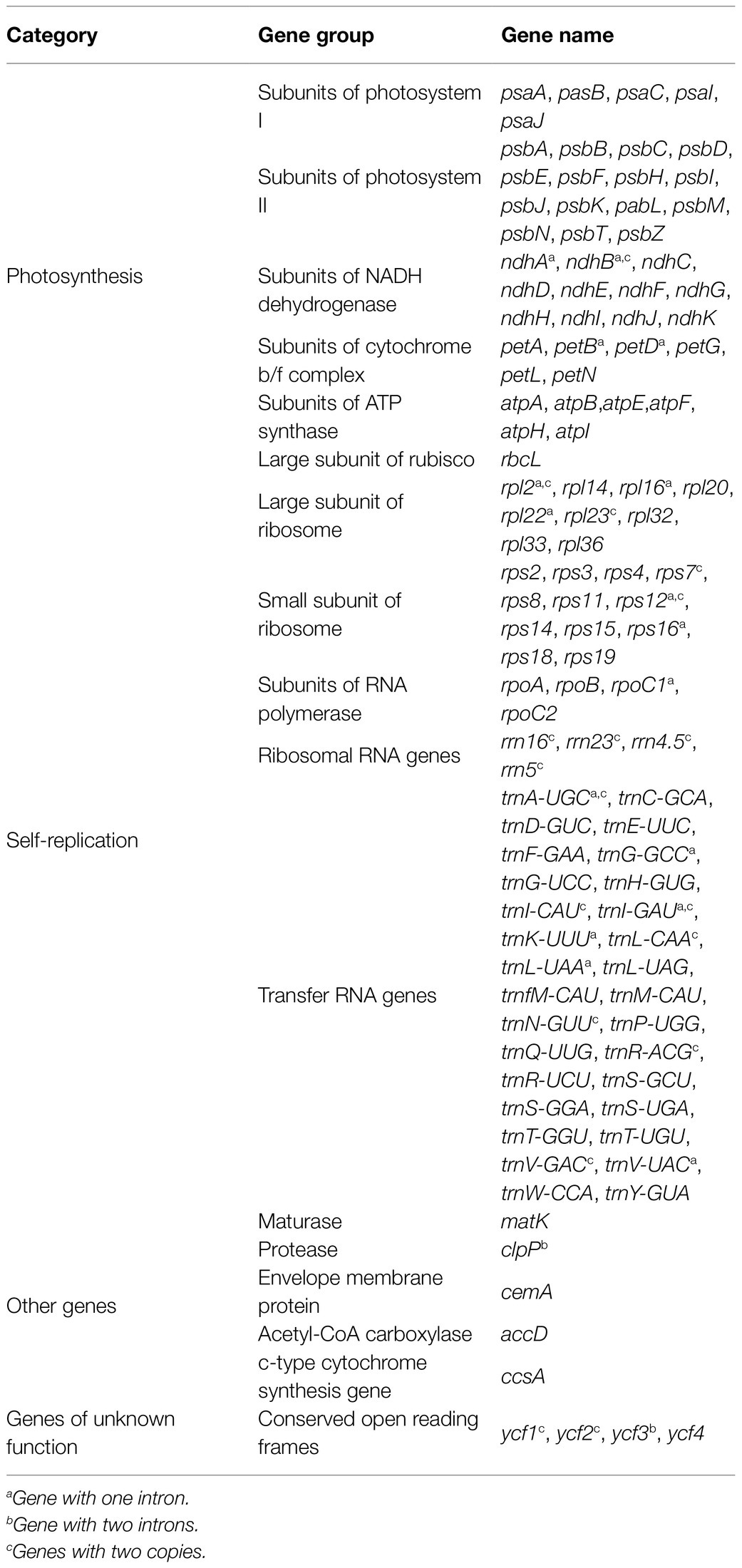
Table 3. Genes in the Fragaria chloroplast genome.
Comparative Analyses
The detailed comparisons of LSC, SSC, and IR boundaries in 19 Fragaria complete cp genomes are shown in Figure 2, revealing differences at boundary regions. There were five genes around borders of these regions, including rps19, rpl2, ycf1, ndhF, and trnH, in which rpl2 and ycf1 had two copies. Overall, F. moschata, F. mandshurica_JL, F. viridis, F. vesca ssp. vesca_1, F. vesca ssp. bracteata, and F. × ananassa showed higher structural similarity, with rps19, rpl2, and trnH genes found at the same distances from the boundaries. The ycf1 gene in Fragaria species spanned 1,092bp from the SSC to IRb region, resulting in a non-functional ѱycf1 fragment gene with the same length in IRa. Additionally, ѱycf1 extended to the SSC region, with different distances ranging from 3bp (F. orientalis) to 37bp (F. daltoniana_1), with the cp genomes of F. mandshurica_JL, F. viridis, F. vesca ssp. vesca_1, F. vesca ssp. bracteata, F. × ananassa, F. chiloensis, and F. virginiana all showing the same gap of 13bp. Moreover, ѱycf1 and ndhF had more length polymorphisms at the IRa/SSC border (Figure 2).

Figure 2. Comparison of the LSC, SSC, and IR border regions among 18 Fragaria chloroplast genomes. Ѱ is used to indicate pseudogenes.
Annotation of the F. gracilis cp genome was used for plotting the total sequence identity of the 18 Fragaria cp genomes in mVISTA (Figure 3). Overall, there was a high similarity among Fragaria species, except F. chinensis_1, F. viridis, and F. orientalis. Furthermore, these results showed non-coding regions are more divergent than coding regions. In our research, the most divergent coding regions in the Fragaria cp genomes analyzed were rpoC2, psbJ, ycf1, and ndhA. In addition, the representative most divergent non-coding regions were rps16-trnQ, trnR-atpA, petN-psbM, trnT-trnL, ndhC-trnV, petA-psbJ, trnP-psaJ, rpl32-trnL, and rps15-ycf1.
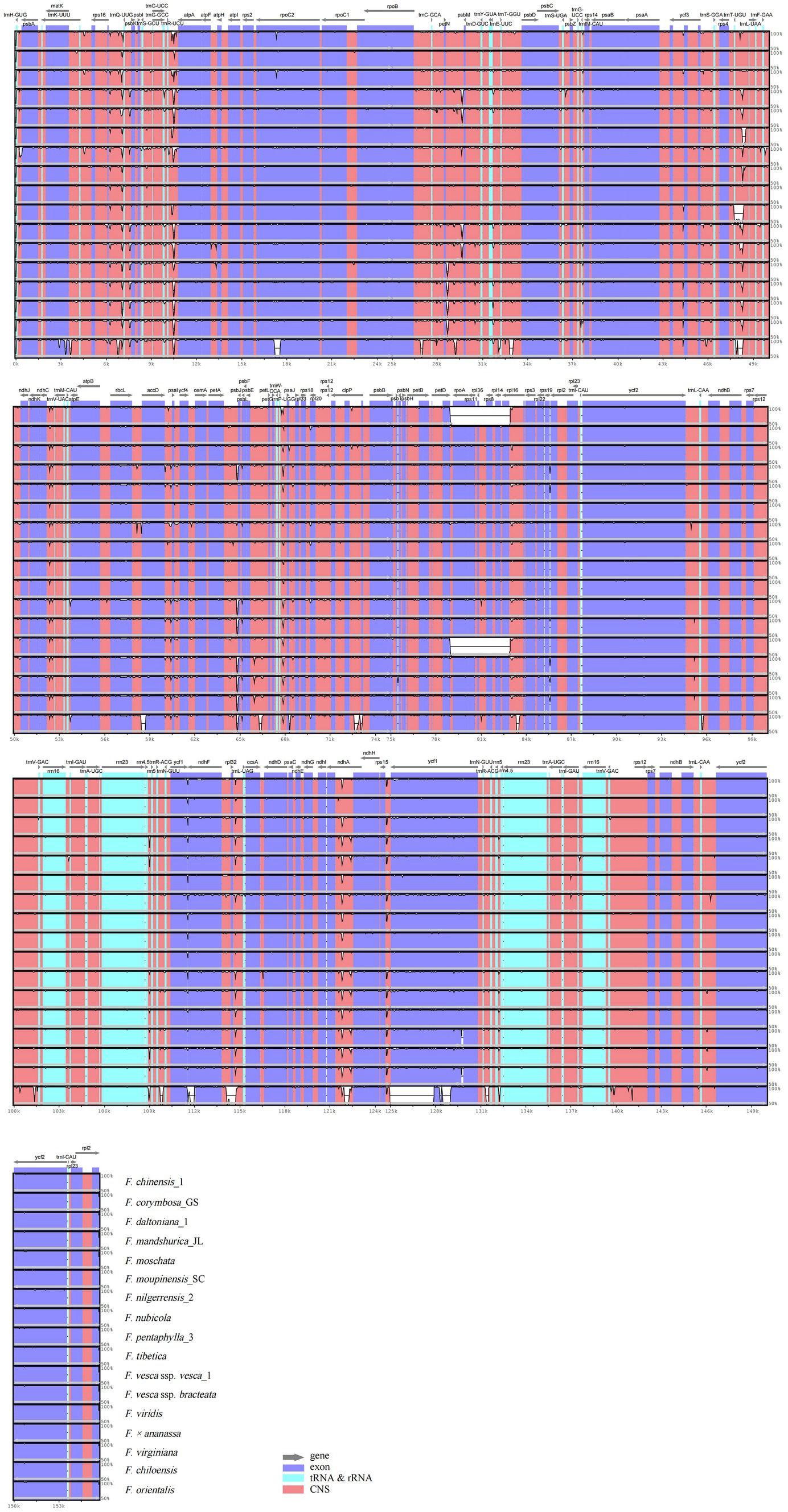
Figure 3. Visualized alignment of the Fragaria chloroplast genome sequences with annotations, using mVISTA. Each horizontal row shows the graph for the pairwise sequence identity with the Fragaria gracilis chloroplast genome sequence. The x-axis represents the base sequence of the alignment, and the y-axis represents the pairwise percent identity ranging from 50 to 100%. Gray arrows indicate the position and direction of each gene. Red indicates non-coding sequences (CNS); blue indicates the exons of protein-coding genes (exons); lime green indicates ribosomal RNA (rRNA) or transfer RNA (tRNA) sequences.
Nucleotide diversity (π) was calculated to evaluate the sequence variability level in Fragaria cp genomes using DnaSP 6.0 software (Figure 4). The values ranged from 0 to 0.01016, and the average value was 0.00167. Four more polymorphic regions (π>0.007) were identified, including trnS-trnG, trnR-atpA, trnC-petN, rbcL-accD, and psbE-petL, in which rbcL-accD had the highest π value, and all of these regions were located in the LSC region.
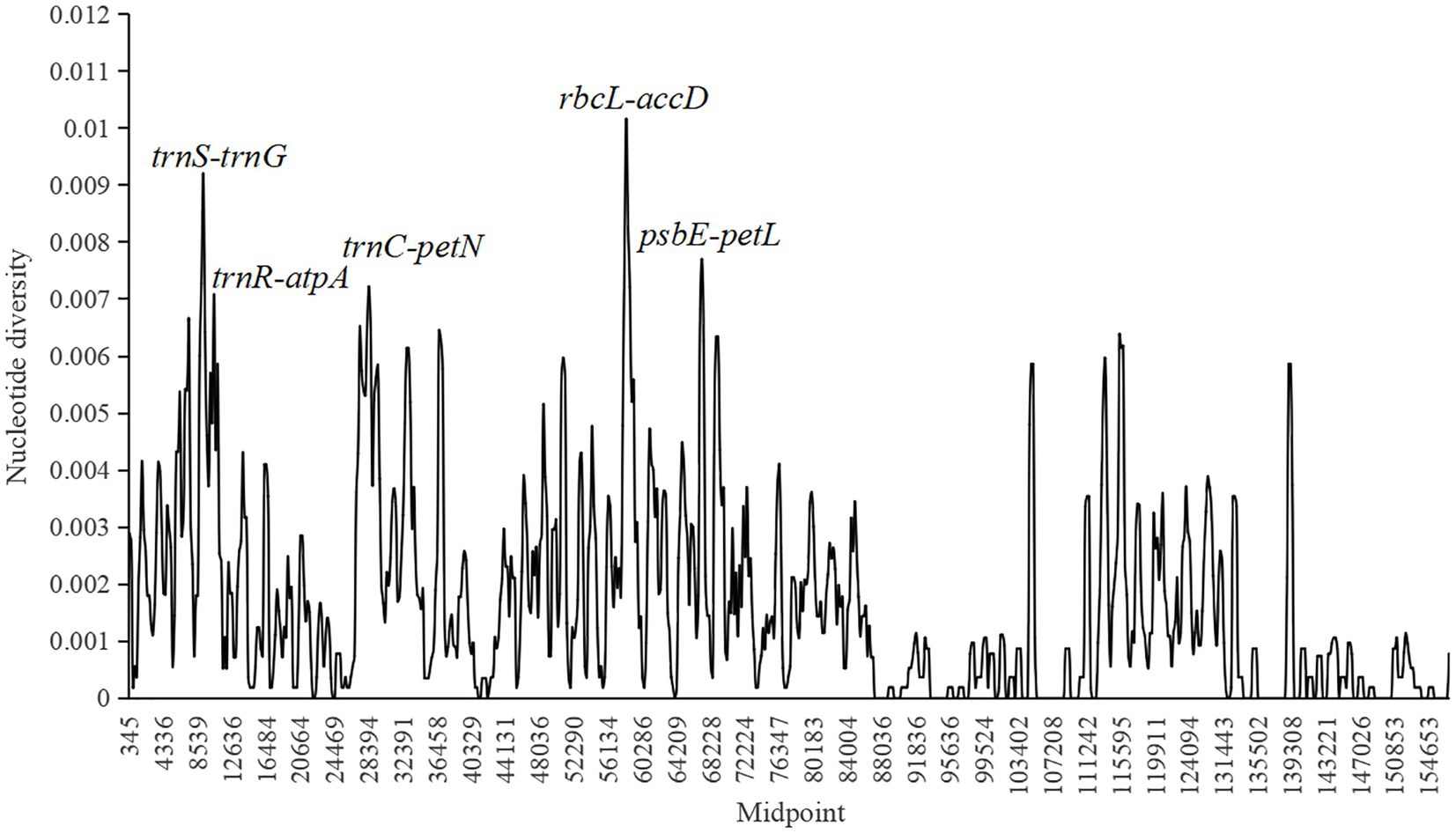
Figure 4. Sliding window analysis of 18 complete chloroplast (cp) genomes of Fragaria species (window length, 600bp; step size, 200bp; x-axis, position of the midpoint of a window; y-axis, nucleotide diversity of each window). Each highly polymorphic region of the Fragaria cp genomes is annotated on the graph.
Phylogenetic Analysis
The GTR+I+G model was selected as the best-fit evolutionary model by using Modeltest 3.7. The phylogenetic tree was constructed using 34 complete cp sequences of Fragaria species, with P. fruticosa and D. saviczii as outgroups. As shown in Figure 5, Fragaria species can be clustered into two groups, A and B, with 100% bootstrap support. Group A included two subgroups, A1 (F. chinensis and F. daltoniana) and A2. Subgroup A2 contained six Fragaria species (F. nubicola, F. pentaphylla, F. corymbosa, F. moupinensis, F. gracilis and F. tibetica) that are mainly distributed in western China. Group B was composed of the remaining species, which included F. nilgerrensis, F. mandshurica, F. viridis, F. orientalis, F. moschata, F. × ananassa, F. chiloensis, F. virginiana, F. vesca ssp. vesca and F. vesca ssp. bracteata. These latter species are from Europe and America, apart from F. nilgerrensis, which is mainly distributed in southeast Asia, F. mandshurica, which is distributed in northeast China, and F. orientalis, which is mainly found in northern Asia.
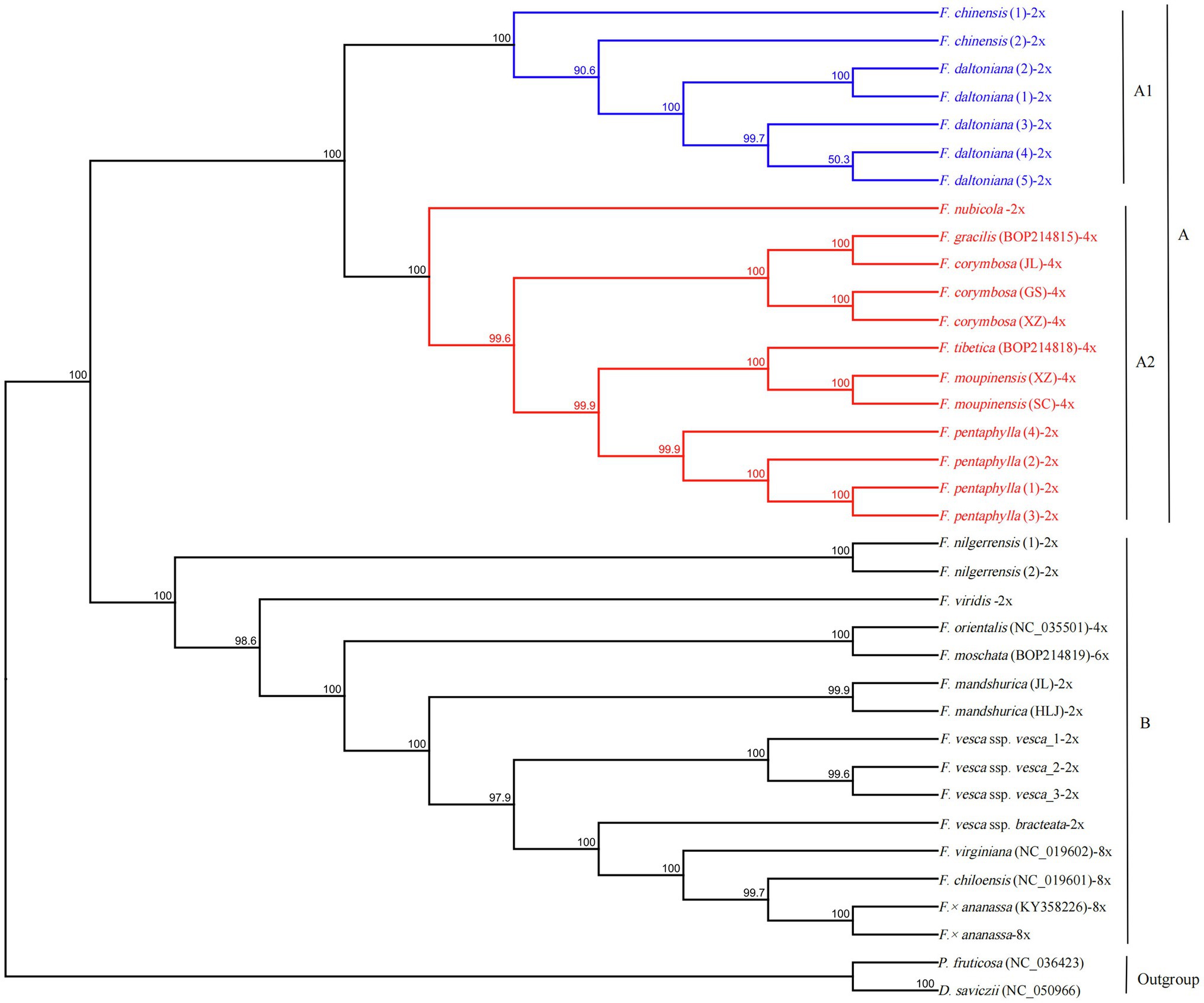
Figure 5. A maximum likelihood phylogenetic tree was reconstructed based on 34 Fragaria cp genomes. Potentilla fruticosa and Drymocallis saviczii were used as outgroups, and −2x, −4x, and −8x represent the different ploidies of Fragaria spp. Nodes marked with capital letters are discussed in the text.
Discussion
Variations and Evolution of Whole Cp Genomes of Fragaria spp.
In this study, 27 cp genomes of Fragaria species were sequenced and found to range in size from 155,479 to 155,832bp, which falls within the cp genome size range for angiosperms but tends to be smaller than the cp genomes of other Rosaceae species (Palmer, 1985; Cheng et al., 2017). LSC regions showed the most difference in size, ranging from 85,471 to 85,726bp. Additionally, the inferred structures and gene contents were in accordance with previous research (Sun et al., 2021).
Overall, Fragaria cp genomes were highly conservative, both in sequence and structure. Analysis with mVISTA showed that there is high similarity among Fragaria species apart from F. chinensis_1, F. viridis, and F. orientalis. We also observed that most variable regions were located in LSC, and non-coding regions were more variable than coding regions. This is a common phenomenon in the cp genomes of most angiosperms (Nazareno et al., 2015; Cheng et al., 2017; Asaf et al., 2018; Tyagi et al., 2020). Additionally, some of the most divergent regions of ycf1, rps16-trnQ, petN-psbM, and rpl32-trnL, as shown in Figure 5, were consistent with previous research (Cheng et al., 2017), indicating that these regions indeed evolve rapidly in Fragaria.
The size differences among cp genomes in angiosperms may be caused by both the contraction and expansion of IR regions (Raubeson et al., 2007; Zhao et al., 2015, 2018). To elucidate this mechanism in Fragaria, we compared IR/SC boundaries of Fragaria cp genomes. In general, the distribution of border genes is conserved, but the distances between genes and the borders do differ somewhat. The distances between genes and IR/SC borders of F. virginiana, F. orientalis, and F. × ananassa are in accordance with the prior report by Cheng et al. (2017). Fragaria mandshurica, F. viridis, F. moschata, F. × ananassa, F. vesca ssp. vesca and F. vesca ssp. bracteata showed the same gap between rps19, rpl2, ycf1, trnH, and IR/SC junctions, which may explain why these cp genomes are more conserved. Additionally, some species also exhibited the same distances between the rps19 and LSC/IRa border, including F. pentaphylla, F. moupinensis, and F. tibetica. These results also indicated a low level of molecular divergence in the genus Fragaria.
Additionally, rpl2 and rps19 differed in their distances from the LSC/IRa border, which may be owing to IR contraction and expansion. Compared with F. × ananassa, the IR regions of F. pentaphylla, F. daltoniana, F. nubicola, F. chinensis, F. corymbosa, F. moupinensis, F. orientalis, F. gracilis, F. tibetica, F. virginiana, and F. chiloensis expanded to different degrees. Therefore, IR regions of these species are longer than that of F. × ananassa. For the other species in the family Rosaceae, such as Malus (Terakami et al., 2012), Prinsepia (Wang et al., 2013), Pyrus (Li et al., 2018), and Prunus (Kim et al., 2019), rps19 crossed the LSC region and IR region. Additionally, rps19 extended to the IRa region, resulting in the presence of ѱrps19 having the same length within IRb region. Notably, we found that rps19 was located inside the LSC region. The contraction of rps19 inside the LSC region would result in the size of the Fragaria cp genomes being smaller than that of other species in Rosaceae.
Phylogenetic Analysis
The similar morphology of most Fragaria spp. and the geographical overlap in ranges may lead to taxonomic confusion among collected specimens (Johnson et al., 2014) and finally result in misjudgment of phylogenetic relationship. To explore the evolutionary relationships among Fragaria species, we constructed a phylogenetic tree of Fragaria species based on whole cp genomes with more than 99% bootstrap support across all nodes. Our results showed high consistency with previous results, especially for species clustering in group B (Njuguna et al., 2013; Sun et al., 2021). Additionally, our results include multiple individuals of each species from different collection sites, which increases the reliability of the phylogenetic analysis.
The phylogenetic analysis showed that F. × ananassa and two octoploids, F. chiloensis and F. virginiana, formed a group, which was in accordance with the inferred origin of F. × ananassa from a hybridization between F. virginiana and F. chiloensis (Staudt, 1962, 1989). In addition, the two octoploids were considered to share a common maternal ancestor that may be F. vesca or F. mandshurica (Harrison et al., 1997; Rousseau-Gueutina et al., 2009; Dimeglio et al., 2014). Fortunately, we observed that F. vesca ssp. bracteata is evolutionarily closely related to the two octoploids. Similar results were also presented by Sun et al. (2021). Additionally, Njuguna et al. (2013) and Edger et al. (2019) suggested that F. vesca ssp. bracteata was probably the last diploid progenitor to the octoploid species. Thus, the conclusion that F. vesca ssp. bracteata is an ancestor of octoploid species was strengthened. However, the other ancestors of the octoploid species remain uncertain.
The evolutionary relationships of F. nubicola, F. pentaphylla, F. chinensis, F. daltoniana, F. corymbosa, F. moupinensis, F. gracilis, and F. tibetica have never been clear (Rousseau-Gueutina et al., 2009; Sun et al., 2021). These species are mainly distributed in Western China (Lei et al., 2017) and, in our results, they were clustered into group A. In group A2, diploid F. pentaphylla was sister to the tetraploids F. moupinensis and F. tibetica with 99.9% bootstrap support (Figure 5), which was consistent with previous research (Njuguna et al., 2013; Sun et al., 2021). In addition, F. pentaphylla had been suggested to be the diploid ancestor to F. moupinensis (Rousseau-Gueutina et al., 2009; Kamneva et al., 2017). Lei et al. (2017) hypothesized that F. tibetica is a descendant of F. pentaphylla based on their runner branching and number of leaflets. Thus, it can be hypothesized that the tetraploid species F. moupinensis and F. tibetica may share the same female parent of F. pentaphylla, which is supported by their more similar morphological characteristics (Lei et al., 2017) and overlapping distribution in Southwestern China (Staudt, 1989; Johnson et al., 2014; Lei et al., 2017).
In our study, we revealed a sister relationship between F. corymbosa and F. gracilis, which was consistent with Rousseau-Gueutina et al. (2009). And F. corymbosa and F. gracilis have some similar morphological characteristics, such as runners are filiform and monopodial, petioles and peduncles have spreading hairs, fruits are red and tasteless, calyx is reflexed, etc. (Lei et al., 2017). So our results strengthened the point that F. corymbosa and F. gracilis may have the same ancestor (Rousseau-Gueutina et al., 2009). In addition, F. corymbosa and F. gracilis may be the descendent of F. chinensis (Staudt, 2009; Yang and Davis, 2017). Notably, in this study, all accessions of F. chinensis and F. daltoniana were clustered into group A1, in contrast with the findings of previous studies (Yang and Davis, 2017; Sun et al., 2021). Therefore, future research should explore the relationship among F. chinensis, F. corymbosa, and F. daltoniana. In the future, multiple molecular markers, including cp and nuclear sequence data from more samples from different geographical populations, should be combined with geographical distribution data to analyse ancestral state reconstruction to clarify their phylogenetic ancestor relationships among F. moupinensis and F. tibetica; F. chinensis, F. corymbosa, and F. daltoniana; F. vesca ssp. bracteata and the other octoploid species.
Chloroplast capture is an important process of plant evolution (Okuyama et al., 2005). Hybridization and repeated backcross, the cytoplasm of one species is replaced by the cytoplasm of another species through gene flow infiltration, so that the genetic components of the species not only have nuclear genome components inherited from parents, but also capture new chloroplast gene components (Fehrer et al., 2007). More and more studies have proved the phenomenon of organelle DNA introgression (Du et al., 2011), and the phenomenon of chloroplast introgression between plant species has also been observed in previous studies on hazelnut (Hu et al., 2020). In this study, the phylogeographical relationships among Fragaria species were not declared for the lack of geographical population collections. However, clear geographical patterns (Western China, Southwestern China, and Northeastern China) have been clearly inferred in phylogenic tree figure. Chloroplast capture could be another explanation why the chloroplast genome analysis does not appear to reflect the species phylogeographical relationship (Tsitrone et al., 2003). Further research should be conducted by combining multiple molecular tools (e.g., nuclear DNA sequences) together with more comprehensive sampling to clarify if chloroplast capture does occur in Fragaria genus.
Candidate Barcoding Sequences for Fragaria
Species identification based on morphology is affected by season, environment, and human factors, which may cause results to be unreliable (Yang et al., 2020). In recent years, DNA barcoding has been widely used to promote accurate species identification owing to its clear advantages (Tegally et al., 2019; Phi et al., 2020; Islam et al., 2021). The ideal DNA barcode would be a single locus that could be universally amplified and sequenced across a broad range of taxa and provide sufficient variation to reliably distinguish among closely related species (Song et al., 2017). Many introns, coding regions, and intergenic regions, such as trnL-trnF (Potter et al., 2000), accD-psaI (Wang et al., 2017), ycf1-ndhF (Amar, 2020), matK, and trnK (Hilu et al., 2008), have been used as barcodes for constructing phylogenetic relationships. In this study, with the threshold of nucleotide diversity as 0.007, the five intergenic regions, trnS-trnG, trnR-atpA, trnC-petN, rbcL-accD, and psbE-petL were found to be the most divergent and provide potential information for species identification and phylogenetic analyses of Fragaria. Among them, three intergenic regions, including trnS-atpA, rbcL-accD, and psbE-petL were also suggested in Sun et al. (2021). However, the threshold of nucleotide diversity used in Sun et al. (2021) was only 0.006, resulting in a low nucleotide diversity of the selected candidate barcoding sequences. In addition, the amplified fragments of the selected intergenic regions trnS-atpA more than 2,000bp in length would reduce the success of the sequencing. In our study, trnS-trnG and trnR-atpA are located in trnS-atpA regions and the length of these two regions are about 700bp, which will result in the high success of sequencing. Therefore, trnS-trnG and trnR-atpA are more suitable than trnS-atpA to be the potential candidate barcoding sequence.
Conclusion
This study provides 27 complete cp genome sequences of 11 wild Fragaria species. Comparative analysis of cp genomes of Fragaria species revealed that their genome structure is highly conserved. However, IR expansion or contraction was observed among different Fragaria cp genomes, resulting in cp genomes of different sizes. Five identified highly variable gene regions (trnS-trnG, trnR-atpA, trnC-petN, rbcL-accD, and psbE-petL) showed strong potential for species identification and phylogenetic relationship construction in the genus Fragaria. Phylogenetic analysis indicated that F. vesca ssp. bracteata may be one of the progenitor species of octoploids. Similarly, we hypothesize that F. pentaphylla is one of the progenitors of F. corymbosa and F. tibetica. The analysis of multiple molecular markers combined with morphological characters would be helpful for future research to test this hypothesis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository and accession number(s) can be found below: NCBI repository, accession numbers MZ851747 and MZ851773.
Author Contributions
JL designed the research. CL, CC, YT, ZS, and MJ performed the research and analyzed the data. CL wrote the first draft of the manuscript. All authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was financially supported by the Ten Thousand Talent Program of Zhejiang Province (No. 2019R52043) and the National Natural Science Foundation of China (No. 31261120580).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Beifen Yang of Taizhou University (Taizhou, China) for the help of identification of Fragaria species.
Footnotes
References
Ali, A., Jaakko, H., and Peter, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Amar, M. H. (2020). ycf1-ndhF genes, the most promising plastid genomic barcode, sheds light on phylogeny at low taxonomic levels in Prunus persica. J. Genet. Eng. Biotechnol. 18:42. doi: 10.1186/s43141-020-00057-3
Asaf, S., Khan, A. L., Khan, M. A., Shahzad, R., and Lee, I. J. (2018). Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinustaeda L.) with related species. PLoS One 13:e0192966. doi: 10.1371/journal.pone.0192966
Bai, L., Ye, Y., Chen, Q., and Tang, H. R. (2017). The complete chloroplast genome sequence of the white strawberry Fragaria pentaphylla. Conserv. Genet. Resour. 9, 659–661. doi: 10.1007/s12686-017-0713-5
Capocasa, F., Diamanti, J., Tulipani, S., and Battino, M. (2008a). Breeding strawberry (Fragaria × ananassa Duch) to increase fruit nutritional quality. Biofactors 34, 67–72. doi: 10.1002/biof.5520340107
Capocasa, F., Scalzo, J., Mezzetti, B., and Battino, M. (2008b). Combining quality and antioxidant attributes in the strawberry: the role of genotype. Food Chem. 111, 872–878. doi: 10.1016/j.foodchem.2008.04.068
Chen, X. C., Liao, B. S., Song, J. Y., Pang, X. H., Han, J. P., and Chen, S. L. (2013). A fast SNP identification and analysis of intraspecific variation in the medicinal Panax species based on DNA barcoding. Gene 530, 39–43. doi: 10.1016/j.gene.2013.07.097
Cheng, H., Li, J. F., Zhang, H., Cai, B. H., Gao, Z. H., Qiao, Y. S., et al. (2017). The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. PeerJ. 5:e3919. doi: 10.7717/peerj.3919
Davis, T. M., Shields, M. E., Reinhard, A. E., Reavey, P. A., Lin, J., Zhang, H., et al. (2010). Chloroplast DNA inheritance, ancestry, and sequencing in Fragaria. Acta Hortic. 859, 221–228. doi: 10.17660/ActaHortic.2010.859.25
Diamanti, J., Capocasa, F., Balducci, F., Battino, M., Hancock, J., and Mezzetti, B. (2012). Increasing strawberry fruit sensorial and nutritional quality using wild and cultivated germplasm. PLoS One 7:e46470. doi: 10.1371/journal.pone.0046470
Dimeglio, L. M., Staudt, G., Yu, H., and Davis, T. M. (2014). A phylogenetic analysis of the genus Fragaria (strawberry) using intron-containing sequence from the ADH-1 gene. PLoS One 9:e102237. doi: 10.1371/journal.pone.0102237
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15. doi: 10.1016/0031-9422(80)85004-7
Du, F. K., Peng, X. L., Liu, J. Q., Lascoux, M., Hu, F. S., and Petit, R. J. (2011). Direction and extent of organelle DNA introgression between two spruce species in the Qinghai-Tibetan plateau. New Phytol. 192, 1024–1033. doi: 10.1111/j.1469-8137.2011.03853.x
Edger, P. P., Poorten, T. J., VanBuren, R., Hardigan, M. A., Colle, M., Mckain, M. R., et al. (2019). Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547. doi: 10.1038/s41588-019-0356-4
Eriksson, T., Donoghue, M. J., and Hibbs, M. S. (1998). Phylogenetic analysis of Potentilla using DNA sequences of nuclear ribosomal internal transcribed spacers (ITS), and implications for the classification of Rosoideae (Rosaceae). Plant Syst. Evol. 211, 155–179. doi: 10.1007/BF00985357
Eriksson, T., Hibbs, M. S., Yoder, A. D., Delwiche, C. F., and Donoghue, M. J. (2003). The phylogeny of Rosoideae (Rosaceae) based on sequences of the internal transcribed spacers (ITS) of nuclear ribosomal DNA and the trnL/F region of chloroplast DNA. Int. J. Plant Sci. 164, 197–211. doi: 10.1086/346163
Fazekas, A. J., Burgess, K. S., Kesanakurti, P. R., Graham, S. W., Newmaster, S. G., Husband, B. C., et al. (2008). Multiple multilocus DNA barcodes from the plastid genome discriminate plant species equally well. PLoS One 3:e2802. doi: 10.1371/journal.pone.0002802
Fehrer, J., Gemeinholzer, B., Chrtek, J. J., and Braütigam, S. (2007). Incongruent plastid and nuclear DNA phylogenies reveal ancient intergeneric hybridization in Pilosella hawkweeds (Hieracium, Cichorieae, Asteraceae). Mol. Phylogenet. Evol. 42, 347–361. doi: 10.1016/j.ympev.2006.07.004
Feng, T., Moore, M. J., Yan, M. H., Sun, Y. X., Zhang, H. J., Meng, A. P., et al. (2017). Phylogenetic study of the tribe Potentilleae (Rosaceae), with further insight into the disintegration of Sibbaldia. J. Syst. Evol. 55, 177–191. doi: 10.1111/jse.12243
Guo, R. X., Xue, L., Luo, G. J., Zhang, T. C., and Lei, J. J. (2018). Investigation and taxonomy of wild Fragaria resources in Tibet, China. Genet. Resour. Crop Evol. 65, 405–415. doi: 10.1007/s10722-017-0541-1
Han, Y., Wu, H. B., and Liu, Y. (2018). The complete chloroplast genome sequence of Fragaria orientalis (Rosales: Rosaceae). Mitochondrial DNA B Resour. 3, 127–128. doi: 10.1080/23802359.2018.1424578
Harrison, R. E., Luby, J. J., and Furnier, G. R. (1997). Chloroplast DNA restriction fragment variation among strawberry (Fragaria spp.) taxa. J. Am. Soc. Hortic. Sci. 122, 63–68.
Hilu, K. W., Black, C., Diouf, D., and Burleigh, J. G. (2008). Phylogenetic signal in matK vs. trnK: a case study in early diverging eudicots (angiosperms). Mol. Phylogenet. Evol. 48, 1120–1130. doi: 10.1016/j.ympev.2008.05.021
Hu, G. L., Cheng, L. L., Huang, W. G., Cao, Q. C., Zhou, L., Jia, W. S., et al. (2020). Chloroplast genomes of seven species of Coryloideae (Betulaceae): structures and comparative analysis. Genome 63, 337–348. doi: 10.1139/gen-2019-0153
Hu, H., Hu, Q. J., Al-Shehbaz, I. A., Luo, X., Zeng, T. T., Guo, X. Y., et al. (2016). Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 7:1826. doi: 10.3389/fpls.2016.01826
Huang, H., Shi, C., Liu, Y., Mao, S. Y., and Gao, L. Z. (2014). Thirteen camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Hummer, K. E. (2012). A new species of Fragaria (Rosaceae) from Oregon. J. Bot. Res. Inst. Texas 6, 9–15.
Islam, S. U., Dar, T., Khuroo, A. A., Bhat, B. A., and Malik, A. H. (2021). DNA barcoding aids in identification of adulterants of trillium govanianum Wall. ex D. Don. J. Appl. Res. Med. Aroma 23:100305. doi: 10.1016/j.jarmap.2021.100305
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., and Boore, J. L. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U. S. A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jansen, R. K., Raubeson, L. A., Boore, J. L., Depamphilis, C. W., and Cui, L. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Jeon, J. H., and Kim, S. C. (2019). Comparative analysis of the complete chloroplast genome sequences of three closely related east-Asian wild roses (Rosa sect. Synstylae; Rosaceae). Genes 10:23. doi: 10.3390/genes10010023
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., de Pamphilis, C. W., Yi, T. S., et al. (2020). GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Johnson, A. L., Govindarajulu, R., and Ashman, T. (2014). Bioclimatic evaluation of geographical range in Fragaria (Rosaceae): consequences of variation in breeding system, ploidy and species age. Bot. J. Linn. Soc. 176, 99–114. doi: 10.1111/boj.12190
Kamneva, O. K., Syring, J., Liston, A., and Rosenberg, N. A. (2017). Evaluating allopolyploid origins in strawberries (Fragaria) using haplotypes generated from target capture sequencing. BMC Evol. Biol. 17:180. doi: 10.1186/s12862-017-1019-7
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kawabe, A., Nukii, H., and Furihata, H. Y. (2018). Exploring the history of chloroplast capture in Arabis using whole chloroplast genome sequencing. Int. J. Mol. Sci. 19:602. doi: 10.3390/ijms19020602
Kim, H. T., Kim, J. S., Lee, Y. M., Mun, J. H., and Kim, J. H. (2019). Molecular markers for phylogenetic applications derived from comparative plastome analysis of Prunus species. J. Syst. Evol. 57, 15–22. doi: 10.1111/jse.12453
Kode, V., Mudd, E. A., Iamtham, S., and Day, A. (2005). The tobacco plastid accD gene is essential and is required for leaf development. Plant J. 44, 237–244. doi: 10.1111/j.1365-313X.2005.02533.x
Lei, W. J., Ni, D. P., Wang, Y. J., Shao, J. J., Wang, X. C., Yang, D., et al. (2016). Intraspecific and heteroplasmic variations, gene losses and inversions in the chloroplast genome of Astragalus membranaceus. Sci. Rep. 6:21669. doi: 10.1038/srep21669
Lei, J. J., Xue, L., Guo, R. X., and Dai, H. P. (2017). The Fragaria species native to China and their geographical distribution. Acta Hortic. 1156, 37–46. doi: 10.17660/ActaHortic.2017.1156.5
Li, Y., Zhang, Z. R., Yang, J. B., and Lv, G. H. (2018). Complete chloroplast genome of seven Fritillaria species, variable DNA markers identification and phylogenetic relationships within the genus. PLoS One 13:e0194613. doi: 10.1371/journal.pone.0194613
Liu, X. L., Wen, J., Ni, Z. L., Johnson, G., Liang, Z. S., and Chang, Z. Y. (2013). Polyphyly of the Padus group of Prunus (Rosaceae) and the evolution of biogeographic disjunctions between eastern Asia and eastern North America. J. Plant Res. 126, 351–361. doi: 10.1007/s10265-012-0535-1
Liu, E. X., Yang, C. Z., Liu, J. D., Jin, S. R., Harijati, N., Hu, Z. L., et al. (2019). Comparative analysis of complete chloroplast genome sequences of four major Amorphophallus species. Sci. Rep. 9:809. doi: 10.1038/s41598-018-37456-z
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Ma, J., Yang, B. X., Zhu, W., Sun, L. L., Tian, J. K., and Wang, X. M. (2014). The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 528, 120–131. doi: 10.1016/j.gene.2013.07.037
Nazareno, A. G., Carlsen, M., and Lohmann, L. G. (2015). Complete chloroplast genome of Tanaecium tetragonolobum: the first bignoniaceae plastome. PLoS One 10:e0129930. doi: 10.1371/journal.pone.0129930
Neuhaus, H. E., and Emes, M. J. (2000). Nonphotosynthetic metabolism in plastids. Annu. Rev. Plant Physiol. Plant Mol. Biol. 51, 111–140. doi: 10.1146/annurev.arplant.51.1.111
Njuguna, W., and Bassil, N. V. (2011). DNA barcoding: unsuccessful for species identification in Fragaria L. Acta Hortic. 918, 349–356. doi: 10.17660/ActaHortic.2011.918.45
Njuguna, W., Listonb, A., Cronn, R., Ashman, T. L., and Bassil, N. (2013). Insights into phylogeny, sex function and age of Fragaria based on whole chloroplast genome sequencing. Mol. Phylogenet. Evol. 66, 17–29. doi: 10.1016/j.ympev.2012.08.026
Okuyama, Y., Fujii, N., Wakabayashi, M., Kawakita, A., Ito, M., Watanabe, M., et al. (2005). Nonuniform concerted evolution and chloroplast capture: heterogeneity of observed introgression patterns in three molecular data partition phylogenies of Asian Mitella (Saxifragaceae). Mol. Biol. Evol. 22, 285–296. doi: 10.1093/molbev/msi016
Palmer, J. D. (1985). Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Parks, M., Cronn, R., and Liston, A. (2009). Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 7:84. doi: 10.1186/1741-7007-7-84
Phi, T. C. M., Chu, H. H., Le, N. T., and Nguyen, D. B. (2020). Phylogenetic relationship of Paramignya trimera and its relatives: an evidence for the wide sexual compatibility. Sci. Rep. 10:21662. doi: 10.1038/s41598-020-78448-2
Plunkett, G. M., and Downie, S. R. (2000). Expansion and contraction of the chloroplast inverted repeat in Apiaceae subfamily Apioideae. Syst. Bot. 25, 648–667. doi: 10.2307/2666726
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43. doi: 10.1007/s00606-007-0539-9
Potter, D., Luby, J. J., and Harrison, R. E. (2000). Phylogenetic relationships among species of Fragaria (Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences. Syst. Bot. 25, 337–348. doi: 10.2307/2666646
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Rousseau-Gueutina, M., Gastona, A., Aïnoucheb, A., Aïnoucheb, M. L., Olbricht, K., Staudt, G., et al. (2009). Tracking the evolutionary history of polyploidy in Fragaria L. (strawberry): new insights from phylogenetic analyses of low-copy nuclear genes. Mol. Phylogenet. Evol. 51, 515–530. doi: 10.1016/j.ympev.2008.12.024
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Ruhsam, M., Rai, H. S., Mathews, S., Ross, T. G., Graham, S. W., Raubeson, L. A., et al. (2015). Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Resour. 15, 1067–1078. doi: 10.1111/1755-0998.12375
Salamone, I., Govindarajulu, R., Falk, S., Parks, M., Liston, A., and Ashman, T. L. (2013). Bioclimatic, ecological, and phenotypic intermediacy and high genetic admixture in a natural hybrid of octoploid strawberries. Am. J. Bot. 100, 939–950. doi: 10.3732/ajb.1200624
Song, Y., Wang, S. J., Ding, Y. M., Xu, J., Li, M. F., Zhu, S. F., et al. (2017). Chloroplast genomic resource of Paris for species discrimination. Sci. Rep. 7:3427. doi: 10.1038/s41598-017-02083-7
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Staudt, G. (1962). Taxonomic studies in the genus Fragaria. Can. J. Bot. 40, 869–886. doi: 10.1139/b62-081
Staudt, G. (1989). The species of Fragaria, their taxonomy and geographical distribution. Acta Hortic. 265, 23–33. doi: 10.17660/ActaHortic.1989.265.1
Staudt, G. (2003). Notes on Asiatic Fragaria species: III. Fragaria orientalis Losinsk. and Fragaria mandshurica spec. nov. Bot. Jahrb. 124, 397–419. doi: 10.1127/0006-8152/2003/0124-0397
Staudt, G. (2006). Himalayan species of Fragaria (Rosaceae). Bot. Jahrb. 126, 483–508. doi: 10.1127/0006-8152/2006/0126-0483
Staudt, G. (2009). Strawberry biogeography, genetics and systematics. Acta Hortic. 842, 71–84. doi: 10.17660/ActaHortic.2009.842.1
Sun, J., Sun, R., Liu, H., Chang, L. L., Li, S. T., Zhao, M. Z., et al. (2021). Complete chloroplast genome sequencing of ten wild Fragaria species in China provides evidence for phylogenetic evolution of Fragaria. Genomics 113, 1170–1179. doi: 10.1016/j.ygeno.2021.01.027
Tegally, A., Jaufeerally-Fakim, Y., and Dulloo, M. E. (2019). Molecular characterisation of Solanum melongena L. and the crop wild relatives, S. violaceum Ortega and S. torvum Sw., using phylogenetic/DNA barcoding markers. Genet. Resour. Crop. Evol. 66, 1625–1634. doi: 10.1007/s10722-019-00827-0
Terakami, S., Matsumura, Y., Kurita, K., Kanamori, H., Katayose, Y., Yamamoto, T., et al. (2012). Complete sequence of the chloroplast genome from pear (Pyrus pyrifolia): genome structure and comparative analysis. Tree Genet. Genomes 8, 841–854. doi: 10.1007/s11295-012-0469-8
Tsitrone, A., Kirkpatrick, M., and Levin, D. A. (2003). A model for chloroplast capture. Evolution 57, 1776–1782. doi: 10.1111/j.0014-3820.2003.tb00585.x
Tulipani, S., Mezzetti, B., Capocasa, F., Bompadre, S., Beekwilder, J., de Voset, C. H. A., et al. (2008). Antioxidants, phenolic compounds, and nutritional quality of different strawberry genotypes. J. Agric. Food Chem. 56, 696–704. doi: 10.1021/jf0719959
Tyagi, S., Jung, J. A., Kim, J. S., and Won, S. Y. (2020). Comparative analysis of the complete chloroplast genome of mainland Aster spathulifolius and other Aster species. Plants 9:568. doi: 10.3390/plants90505680
Urrutia, M., Rambla, J. L., Alexiou, K. G., Granell, A., and Monfort, A. (2017). Genetic analysis of the wild strawberry (Fragaria vesca) volatile composition. Plant Physiol. Biochem. 121, 99–117. doi: 10.1016/j.plaphy.2017.10.015
Wang, S., Shi, C., and Gao, L. Z. (2013). Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into wvolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS One 8:e73946. doi: 10.1371/journal.pone.0073946
Wang, Y. H., Qu, X. J., Chen, S. Y., Li, D. Z., and Yi, T. S. (2017). Plastomes of Mimosoideae: structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet. Genomes 13:41. doi: 10.1007/s11295-017-1124-1
Wei, W., Zheng, Y. L., Chen, L., Wei, Y. M., Yan, Z. H., and Yang, R. W. (2006). PCR-RFLP analysis of cpDNA and mtDNA in the genus Houttuynia in some areas of China. Hereditas 142, 24–32. doi: 10.1111/j.1601-5223.2005.01704.x
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wolfe, K. H., Li, W., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Nati. Acad. Sci. U. S. A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Xin, T. Y., Yao, H., Gao, H. H., Zhou, X. Z., Ma, X. C., Xu, C. Q., et al. (2013). Super food Lycium barbarum (Solanaceae) traceability via an internal transcribed spacer 2 barcode. Food Res. Int. 54, 1699–1704. doi: 10.1016/j.foodres.2013.10.007
Yang, Y., and Davis, T. M. (2017). A new perspective on polyploid Fragaria (strawberry) genome composition based on large-scale, multi-locus phylogenetic analysis. Genome Biol. Evol. 9, 3433–3448. doi: 10.1093/gbe/evx214
Yang, J. Y., Wei, S. J., Su, D. B., Chen, S. Y., Luo, Z. W., Shen, X. M., et al. (2020). Molecular identification and evolutionary characteristics of Fragaria nilgerrensis in Yunnan based on nrDNA ITS and cpDNA psbA-trnH sequence analysis. J. South Agric. 51, 748–757.
Zhang, Y., Du, L., Liu, A., Chen, J. J., Wu, L., Hu, W. M., et al. (2016). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhao, M. L., Song, Y., Ni, J., Yao, X., Tan, Y. H., and Xu, Z. F. (2018). Comparative chloroplast genomics and phylogenetics of nine Lindera species (Lauraceae). Sci. Rep. 8:8844. doi: 10.1038/s41598-018-27090-0
Keywords: Fragaria, chloroplast genome, comparative analysis, wild species, phylogenetic
Citation: Li C, Cai C, Tao Y, Sun Z, Jiang M, Chen L and Li J (2021) Variation and Evolution of the Whole Chloroplast Genomes of Fragaria spp. (Rosaceae). Front. Plant Sci. 12:754209. doi: 10.3389/fpls.2021.754209
Edited by:
Wei Hu, Chinese Academy of Tropical Agricultural Sciences, ChinaReviewed by:
Xiwen Li, China Academy of Chinese Medical Sciences, ChinaYong Qi Zheng, Chinese Academy of Forestry, China
Guanglong Hu, Beijing Academy of Agricultural and Forestry Sciences, China
Copyright © 2021 Li, Cai, Tao, Sun, Jiang, Chen and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junmin Li, lijm@tzc.edu.cn