Case report: Recombinant human epidermal growth factor gel plus kangfuxin solution in the treatment of aplasia cutis congenita in a case with Adams–Oliver syndrome
 Xiu-Fang Yang
Xiu-Fang Yang Shang-Wen Shi1
Shang-Wen Shi1 - 1Department of Neonatology, Zhongshan Hospital Affiliated to Sun Yat-Sen University, Zhongshan, China
- 2Molecular Inspection Center, Zhongshan Hospital Affiliated to Sun Yat-Sen University, Zhongshan, China
Background: Aplasia cutis congenita is a congenital disorder with the absence of skin, muscle and(or) bone. It usually affects the scalp. The presence of a large scalp defect can be potentially serious when complicated with hemorrhage and infection. Early healing of this condition is beneficial to improve the prognosis of infants.
Study case: A full-term newborn male was born with a round-shaped defect at the vertex of the scalp and skull (dimensions, 8 cm × 9 cm). The infant had a large deletion encompassing the 15.1 region of chromosome 15, including the DLL4 gene. Genetic testing was positive for Adams–Oliver syndrome (AOS). After two months of recombinant human epidermal growth factor gel combined with kangfuxin solution therapy, the skin defects of the scalp healed remarkably. The infant had regular follow-up appointments. At the age of 5 months, the defect became smaller, hairless, and showed good granulation tissue. At 2 years of age, the child's Gesell Developmental Schedules was 70.
Conclusion: Recombinant human epidermal growth factor gel combined with kangfuxin solution was a successful conservative treatment for an infant with a large scalp defect accompanied by AOS.
Introduction
Aplasia cutis congenita (ACC) is a defect that can affect the skin, dermis, and even subcutaneous tissue (including muscle and bone) in neonates (1, 2). A scalp defect is the most common side effect in infants with ACC, particularly in those with Adams–Oliver syndrome [AOS (MIM 100300)]. The clinical manifestations of AOS include congenital hypoplasia of parietal skin, tricuspid regurgitation, ventricular septal defect, skull defect, short finger syndrome, short and toe deformity, some patients with marble-like skin and nail dysplasia (3–6). The cause of Adams–Oliver syndrome is unknown, maybe sporadic or hereditary. A scalp defect, combined with bleeding and infection, can lead to adverse outcomes in children. Proper management strategies for scalp defects are thus extremely important for improving prognosis. Due to the lack of consensus or evidence-based guidelines for ACC management, treatment strategies are often individualized.
We report on a family in which the son has Adams–Oliver syndrome. The son carries with a 625.9 kb deletion including DLL4 (Delta Like Canonical Notch Ligand 4) gene (MIM 605185) which may cause AOS. The son with AOS has scalp defect and skull absence. The scalp defect of this case with AOS had been successfully treated by combining recombinant human epidermal growth factor gel with kangfuxin solution.
Patient, clinical signs, and outcomes
A full-term male newborn with a round-shaped defect at the vertex of the scalp and skull (dimensions, 8 cm × 9 cm) was was born in the Obstetrics Department of Zhongshan Hospital Affiliated with Sun Yat-Sen University (also known as Zhongshan City People's Hospital) on May 10, 2019 (Figure 1A). The infant weighed 2.5 kg (−2SD) (38 weeks gestational age). The height of the infant was 48.6 cm (−1SD). Th head circumference was 33.2 cm (–1SD). The infant's mother experienced premature rupture of amniotic membrane for 11 h prior to giving birth and had gestational diabetes mellitus. Her examination showed no placental abnormalities, and no herpes simplex, varicella-zoster, rubella, or cytomegalovirus infections. The mother denied having been exposed to teratogenic drugs, radiation, or chemicals during pregnancy. The pregnancy of his mother was G1P1. There was no history of hypertensive disorder complicating pregnancy. There were no family histories of aplasia cutis congenita and other congenital malformations.The Apgar score of the infant was 10 at 1, 5, and 10 min after birth. He had no congenital malformation of the digestive tract, nor skeletal or congenital heart malformations. Peripheral blood, white blood cell, and blood gas analyses, as well as lactic acid, serum C-reactive protein, blood ammonia, and hepatobiliary biochemistry test results, were all normal. The blood cultures revealed no bacterial growth.
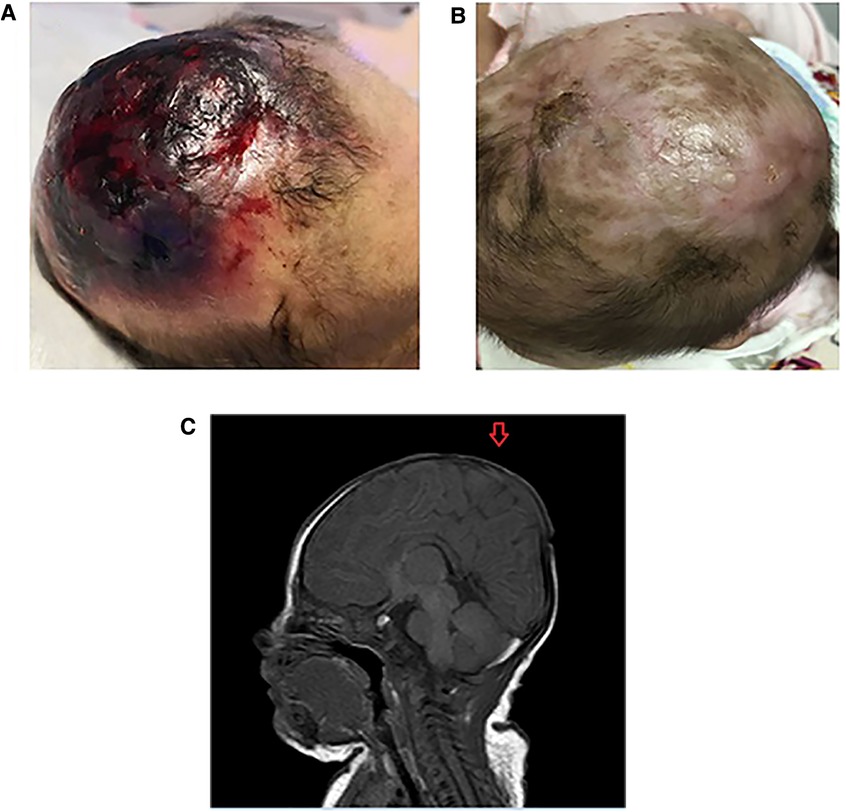
Figure 1. (A) The scalp defect of the patient with AOC at birth. (B) The scalp of the patient with AOC at 5 months old. (C) Cranial magnetic resonance imaging scan of the patient with aplasia cutis congenital (AOC). The scan shows a complete loss of continuity of the dermal, subcutaneous, and skull tissues (arrow).
The defect included scalp and skin layers and was confirmed by magnetic resonance imaging (MRI) examinations on May 13, 2019. The cranial MRI scan revealed complete loss of continuity of the dermal and subcutaneous tissues and skull (Figure 1B). Echocardiography, a chest x-ray, and abdominal ultrasonography revealed no apparent abnormalities. Peripheral blood chromosome karyotyping was 46, XY. The metaphases of the 20 cells were analyzed under the microscope. The list of organisms by chromosome count was normal. Chromosome structure was not broken, deletion, repeat, ectopic, inversion, ring and isobar chromosome aberrations. Genetic testing indicated a 625.9 kb deletion (Copy number variation 1) in the 15.1 (41043614-41669512) region of chromosome 15 of the infant, which involved in DLL4, KIF21B, DNAJC17, ZFYVE19, SPINT1, VPS18 gene, etc. The deletion of DLL4 gene was found to be related to the clinical manifestation of this infant through the GeneCards(the human gene database). The results of his parents showed that the copy number in this region was normal, which suggested that the heterozygous deletion of DLL4 gene was new in the infant. The defect of DLL4 gene is sporadic.The result of genetic qualitative dynamic mutation, verified by semi-quantitative polymerase chain reaction with a specific gene deletion, showed a large deletion of exon1, exon5, and exon11 of the DLL4 gene of the infant, which can cause AOS (Figure 2). The DLL4 gene is a protein coding gene. The deletion of DLL4 gene may cause Adams–Oliver syndrome type 6 (MIM 616589). The child was diagnosed with AOS, based on his clinical presentation and the genetic examination.
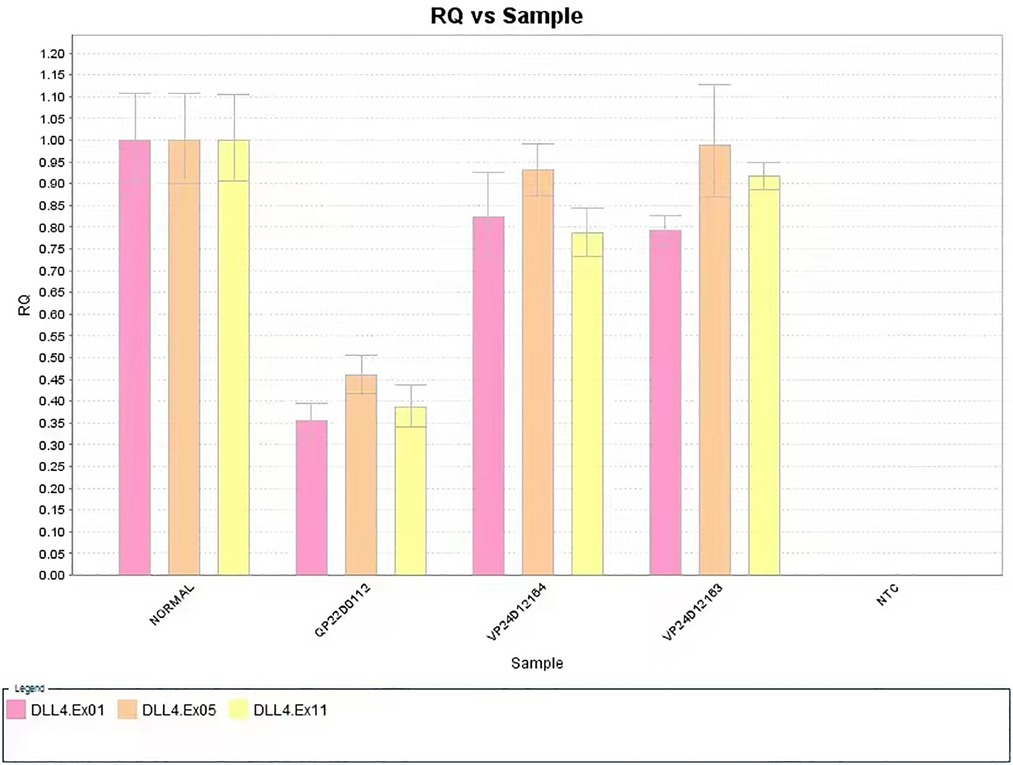
Figure 2. The large deletion of exon1, exon5, and exon11 of the DLL4 gene of the case study infant (sample no. QP22D0112, the patient; Sample no. VP24D12164, the patient's father; Sample no. VP24D12163, the patient's mother).
The lesion of scalp was treated with normal saline cleansing, recombinant human epidermal growth factor gel, and kangfuxin solution for two months. Following daily local treatment, the wound gradually healed, and the lesion of scalp exhibited good epithelialization and eschar formation. The child attended regular follow-up visits. Complete epithelialization was achieved and the defect of scalp became hairless with good granulation tissue at five months (Figure 1C). At 2 years old, the scalp defect became smaller and hairless (Figure 3), and Gesell Developmental Schedules was 70. For large defects involving the skull, surgical intervention maybe the treatment of most ACC patients. However, his parents refused to reexamine the cranial MRI and surgery.
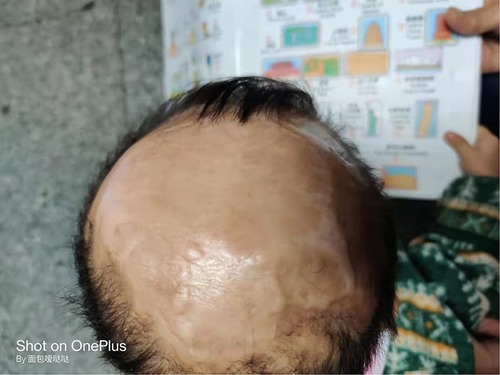
Figure 3. The scalp of the patient with aplasia cutis congenita at 2 years old.
Discussion
Adams–Oliver syndrome is a condition of scalp defects and/or limb deformities that were first reported by Addams and Oliver in 1945 (7). Its incidence is low and sporadic, but there is also a genetic tendency for developing the condition. To date, the etiology of AOS remains unknown. Vascular injury during embryogenesis and disruption of the blood supply to developmental structures, caused by placental thrombosis, has been identified as possible mechanisms of AOS (8, 9).
Gene mutations (ARHGAP31, DLL4, NOTCH1, RBPJ, DOCK6, and EOGT) are associated with AOS (10–12). The molecular mechanism of this syndrome, based on gene mutation, is guanosine-5′-triphosphate (GTP) being a primer for deoxyribonucleic acid replication, a 5′-guanosine monophosphate provider for transcription, and an energy carrier as part of the tricarboxylic acid cycle process (13). Guanosine-triphosphate-binding protein is a large family of hydrolases that can bind and hydrolyze GTP in a highly conserved G-domain (14). These proteins are important for signal transduction, protein biosynthesis, and protein and vesicle transport, respectively.
The results of proband gene detection indicated a large deletion in the 15.1 region of chromosome 15, including the DLL4 gene. The protein encoded by the DLL4 gene is part of the Notch signal transduction pathway (15). The DLL4 protein is essential for vascular development; patients with a DLL4 gene deletion may develop ACC and extremity defects, which may result from the abnormal development of small blood vessels, thrombosis, and vascular obstruction (16).
A diagnosis of AOS is based on the clinical presentation of the scalp and cranial bone defects, as well as limb anomalies that are visible following the birth of an infant. Other presentations include a cleft lip or palate, growth retardation, cardiovascular abnormalities, urinary system abnormalities, and intellectually disabled (4). The current case included scalp and skull defects, as well as intellectually disabled. The scalp defect was large, and if not treated correctly, may have affected the healing of the skin defect, prolonged the course of treatment, and increased the incidence of infection and septic shock (17).
Some infants with severe ACC require skin grafting to prevent bleeding and infection (18, 19). Kangfuxin solution (Periplaneta americana L. extract) is used for trauma wounds, ulcers, fistulas, burns, scalds, and bedsores (20, 21). The current authors combined recombinant human epidermal growth factor gel with kangfuxin solution in an external application to successfully treat a scalp defect wound.
In general, cutis aplasia with AOS is a potentially life-threatening disease. Surgery, combined with conservative treatment, is an important approach for treating large skin defects in this context. Based on the current case, we found that recombinant human epidermal growth factor gel combined with kangfuxin solution worked as a successful conservative treatment for a large scalp defect of an infant with AOS and did not cause any serious complications.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Zhongshan Hospital Affiliated to Sun Yat-Sen University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
Conception and design of the research: X-FY, S-WS; Acquisition of data: X-FY, S-WS; Analysis and interpretation of the data: S-WS, KC; Statistical analysis: KC; Obtaining financing: X-FY; Writing of the manuscript: X-FY, S-WS; Critical revision of the manuscript for intellectual content: X-FY, S-WS. All authors contributed to the article and approved the submitted version.
Acknowledgments
We would like to acknowledge the hard and dedicated work of all the staff that implemented the intervention and evaluation components of the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Alexandros B, Dimitrios G, Elias A, Evangelos D, Andreas M, Sotirios P, et al. Aplasia cutis congenita: two case reports and discussion of the literature. Surg Neurol Int. (2017) 8(1):273. doi: 10.4103/sni.sni_188_17
2. TaifourSuliman M, Quazi A. Aplasia cutis congenita of the trunk in a Saudi newborn. Br J Plast Surg. (2004) 57(6):582–4. doi: 10.1016/j.bjps.2003.12.026
3. Bilginer B, Önal MB, Bahadir S, Akalan N. Aplasia cutis congenita of the scalp, skull and dura associated with Adams–Oliver syndrome. Turk Neurosurg. (2008) 18(2):191–3. PMID: 18597236
4. Temtamy SA, Aglan MS, Ashour AM, Zaki MS. Adams–Oliver syndrome: further evidence of an autosomal recessive variant. Clin Dysmorphol. (2007) 16(3):141–9. doi: 10.1097/MCD.0b013e3280f9df22
5. Narang T, Kanwar AJ, Dogra S. Adams–Oliver syndrome: a sporadic occurrence with minimal disease expression. Pediatr Dermatol. (2008) 25(1):115–6. doi: 10.1111/j.1525-1470.2007.00598.x
6. Dehdashtian A, Dehdashtian M. Adams–Oliver syndrome: a case with full expression. Pediatr Rep. (2016) 8(2):6517. doi: 10.4081/pr.2016.6517
7. Adams FH, Oliver CP. Hereditary deformities in man: due to arrested development. J Hered. (1945) 36:3–7. doi: 10.1093/oxfordjournals.jhered.a105415
8. Hoyme HE, Jones KL, Van Allen MI, Saunders BS, Benirschke K. Vascular pathogenesis of transverse limb reduction defects. J Pediatr. (1982) 101(5):839–43. doi: 10.1016/S0022-3476(82)80343-0
9. Fryns JP, Legius E, Demaerel P, van den Berghe H. Congenital scalp defect, distal limb reduction anomalies, right spastic hemiplegia and hypoplasia of the left arteri cerebri media: further evidence that interruption of early embryonic blood supply may result in Adams–Oliver (plus) syndrome. Clin Genet. (1996) 50(6):505–9. doi: 10.1111/j.1399-0004.1996.tb02723.x
10. Hassiem F, Cavanna AE. Multiple tics in a patient with Adams–Oliver syndrome. J Neuropsychiatry Clin Neurosci. (2015) 27(1):e80. doi: 10.1176/appi.neuropsych.13120377
11. Nagasaka M, Taniguchi-Ikeda M, Inagaki H, Ouchi Y, Kurokawa D, Yamana K, et al. Novel missense mutation in DLL4 in a Japanese sporadic case of Adams–Oliver syndrome. J Hum Genet. (2017) 62(9):851–5. doi: 10.1038/jhg.2017.48
12. Hassed S, Li S, Mulvihill J, Aston C, Palmer S. Adams–Oliver syndrome review of the literature: refining the diagnostic phenotype. Am J Med Genet A. (2017) 173(3):790–800. doi: 10.1002/ajmg.a.37889
13. Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, et al. Gain-of-function mutations of ARHGAP31, a Cdc42 /Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet. (2011) 88(5):574–85. doi: 10.1016/j.ajhg.2011.04.013
14. Southgate L, Sukalo M, Karountzos ASV, Taylor EJ, Collinson CS, Ruddy D, et al. Haploinsufficiency of the NOTCH1 receptor as a cause of Adams–Oliver syndrome with variable cardiac anomalies. Circ Cardiovasc Genet. (2015) 8(4):572–81. doi: 10.1161/CIRCGENETICS.115.001086
15. Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong ZY, Li H, et al. Mutations in NOTCH1 cause Adams–Oliver syndrome. Am J Hum Genet. (2014) 95(3):275–84. doi: 10.1016/j.ajhg.2014.07.011
16. Gridley T. Notch signaling in vascular development and physiology. Development. (2007) 134(15):2709–18. doi: 10.1242/dev.004184
17. Kadry I, Koratum T, Obaid L. Approach in management of aplasia cutis congenita in a newborn with Adams–Oliver syndrome. Emirates Med J. (2020) 2(1):42–4. doi: 10.2174/0250688201999200518121808
18. Khashab ME, Rhee ST, Pierce SD, et al. Management of large scalp and skull defects in a severe case of Adams–Oliver syndrome. J Neurosurg Pediatr. (2009) 4(6):523–7. doi: 10.3171/2009.7.PEDS09220
19. Khashab ME, Rhee ST, Pierce SD, Khashab YE, Nejat F, Fried A. Surgical treatment of aplasia cutis in the Adams–Oliver syndrome. J Craniofac Surg. (2001) 12(6):569–72. doi: 10.1097/00001665-200111000-00014
20. Zeng A, Gu G, Deng L. Effect of kangfuxin solution fumigation bath on postoperative patients with hemorrhoid PPH and influence on the postoperative complications. Evid Based Complementary Altern Med. (2019) 6(8):1–8. doi: 10.1155/2021/6473754
Keywords: neonates, aplasia cutis congenita, DLL4 gene, Adams–Oliver syndrome, case report
Citation: Yang X, Shi S and Chen K (2023) Case report: Recombinant human epidermal growth factor gel plus kangfuxin solution in the treatment of aplasia cutis congenita in a case with Adams–Oliver syndrome. Front. Surg. 9:1072021. doi: 10.3389/fsurg.2022.1072021
Received: 17 October 2022; Accepted: 5 December 2022;
Published: 11 January 2023.
Edited by:
Antonino Morabito, University of Florence, ItalyReviewed by:
Francisco Cammarata-Scalisi, Regional Hospital of Antofagasta, ChileJiangyuan Zhou, Sichuan University, China
Teresa Oranges, Meyer Children's Hospital, Italy
© 2023 Yang, Shi and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiu-Fang Yang yangxuifanfyxf9kc@126.com
Specialty Section: This article was submitted to Pediatric Surgery, a section of the journal Frontiers in Surgery