Plasma Membrane Pores Drive Inflammatory Cell Death
 Benedikt Kolbrink
Benedikt Kolbrink Theresa Riebeling
Theresa Riebeling Ulrich Kunzendorf
Ulrich Kunzendorf  Stefan Krautwald
Stefan Krautwald- Department of Nephrology and Hypertension, University Hospital Schleswig-Holstein, Kiel, Germany
Necroptosis and pyroptosis are two forms of regulated cell death. They are executed by the proteins mixed-lineage kinase domain-like (MLKL) and gasdermin D (GSDMD), respectively. Once activated by numerous pathways, these proteins form membrane pores that allow the influx and efflux of various ions, proteins, and water, ultimately resulting in the death of the cell. These modalities of cell death are considered highly inflammatory because of the release of inflammatory cytokines and damage-associated molecular patterns, and are thereby not only deleterious for the dying cell itself, but also its environment or the entire organism. The relevance for these processes has been observed in various physiological and pathophysiological conditions, ranging from viral and bacterial infections over autoimmune and chronic inflammatory diseases to ischemic organ damage. In recent years, initial in vitro experiments have shed light on a range of connections between necroptosis and pyroptosis. Initial in vivo studies also indicate that, in many disease models, these two forms of cell death cannot be considered individually, as they demonstrate a complex interaction. In this article, we provide an overview of the currently known structure, pathways of activation, and functions of MLKL and GSDMD. With emerging evidence for an interconnection between necroptosis and pyroptosis in not only in vitro, but also in vivo models of disease, we highlight in particular the clinical relevance of the crosslinks between these two forms of inflammatory cell death and their implications for novel therapeutic strategies in a variety of diseases.
Introduction
In recent years, a variety of different forms of regulated cell death (RCD) has been discovered, and many of the underlying molecular mechanisms have been deciphered (Galluzzi et al., 2018; Tang et al., 2019). The permeabilization of the plasma membrane by pore-forming proteins plays a crucial role in only two distinct necrotic forms of RCD, namely necroptosis and pyroptosis. Both are considered inflammatory RCD, as the dying cells release miscellaneous damage-associated molecular patterns (DAMPs) and pro-inflammatory cytokines that induce profound inflammation of the surrounding tissue (Bergsbaken et al., 2009; Pasparakis and Vandenabeele, 2015). Apart from the obvious similarities, however, there are also subtle and less subtle differences, cross-links, and cooperation in the development of diseases between necroptosis and pyroptosis. Herein, we focus on the executing proteins mixed-lineage kinase domain-like (MLKL) and gasdermin D (GSDMD), and provide an overview of their known mechanisms of activation and membrane permeabilization and the clinical relevance of understanding their interconnections.
Necroptosis and MLKL
Mixed-lineage kinase domain-like is the most terminal known indispensable effector protein of necroptosis. It is a 54 kDa protein that consists of an N-terminal four-helix bundle (4HB) and the C-terminal pseudokinase domain (PKD), which are connected by a two-helix linker brace (Petrie et al., 2017). In necroptosis, MLKL is activated by receptor-interacting serine/threonine protein kinase 3 (RIPK3) via phosphorylation of the residues T357/S358 in humans or S345 in mice (Sun et al., 2012; Murphy et al., 2013; Rodriguez et al., 2016). To date, three different activation pathways of MLKL in the context of necroptosis have been described: the first and best characterized begins by death receptor signaling via TNFR1, TRAIL or Fas ligand, and is mediated by RIPK1. The second pathway follows the activation of toll-like receptor (TLR) 3 and 4 and is mediated via the adaptor protein TRIF (also known as TICAM1), whereas the third characterized pathway follows activation of the cytosolic RNA sensor ZBP1 (also known as DAI). Common to all these pathways is that, in the absence or impairment of caspase function, the RHIM domain-containing proteins RIPK1, TRIF, or ZBP1 bind RIPK3 via their respective RHIM domains to form the so-called necrosome, which is followed by RIPK3 autophosphorylation and consecutive activation of MLKL (Weinlich et al., 2017; Shan et al., 2018; Malireddi et al., 2019). Whether necroptosis is dependent on the generation of mitochondrial reactive oxygen species (ROS) has also been discussed, but more recent data have challenged this hypothesis. The evidence for and against this idea is described in detail (Marshall and Baines, 2014). Recently, it was shown that, at least for RIPK1-dependent necroptosis, ROS enhance the activation of RIPK1 by autophosphorylation in a positive feedback loop (Zhang et al., 2017). However, the exact mechanism of how MLKL is phosphorylated by RIPK3 and hence activated differs among species and has not been resolved. In human cells, RIPK3 needs to bind MLKL via its PKD to phosphorylate and activate it (Sun et al., 2012; Petrie et al., 2018), whereas in murine cells, MLKL is not recruited to RIPK3, and phosphorylation alone is sufficient for inducing necroptosis (Murphy et al., 2013; Hildebrand et al., 2014; Jacobsen et al., 2016). For different vertebrates, it was shown recently that varying conformations of the PKD in particular play an important role in species-specific activation of MLKL (Davies et al., 2020).
The structure of the membrane pore formed by MLKL has not been resolved to date. It is generally agreed that phosphorylation of the PKD leads to a conformational change that exposes the 4HB, which results in MLKL oligomerization in the cytoplasm, traffic to the cell periphery via Golgi-microtubule-actin-dependent mechanisms and subsequent membrane translocation by binding phosphatidylinositol phosphates (PIPs), but there is disagreement about whether MLKL forms trimers, tetramers, or hexamers (Cai et al., 2014; Chen et al., 2014; Dondelinger et al., 2014; Wang H. et al., 2014; Samson et al., 2020). Recently, we visualized necroptotic cell death in the presence of polyethylene glycols (PEGs) of different sizes and were able to estimate that the membrane pores executing necroptosis had inner diameters of around 4 nm (Ros et al., 2017). Once inserted into the plasma membrane, MLKL oligomers increase membrane permeability to cations, which leads to calcium and sodium influx and potassium efflux from the cell along the concentration gradients. This is followed by the influx of water into the cell along the osmotic gradient, and results in a unique morphology of the dying necroptotic cell, that is described as explosion-like (Chen et al., 2016; Xia et al., 2016).
The relevance for necroptosis has been shown in the pathophysiology of various diseases, both infectious and non-infectious. From a historical perspective, the concept of necroptosis was originally discovered in that small molecules, the so-called necrostatins, could suppress necrotic cell death in certain settings (Degterev et al., 2005). The next step was the discovery of RIPK1 as the target molecule of necrostatins and the subsequent identification of the other proteins involved in necroptosis, which then enabled investigations in knockout animals and animal models of specific diseases (Degterev et al., 2008; Silke et al., 2015). In an infectious context, necroptosis is a double-edged sword. It is of particular importance in the defense against viral pathogens and intracellular bacteria, although the mechanisms vary and have not been definitively elucidated. For specific infections, necroptosis has positive effects on the removal of pathogens, as shown in murine models of herpes simplex virus, cytomegalovirus, vaccinia virus, and influenza A virus (Cho et al., 2009; Upton et al., 2012; Wang X. et al., 2014; Nogusa et al., 2016). Remarkably, the clearance of Listeria monocytogenes also relies on RIPK3 and MLKL, which in this case does not lead to the death of the host cell, but inhibits bacterial growth by direct binding of MLKL to the bacteria (Sai et al., 2019). However, there are adverse effects of necroptosis in infections. In mouse models of infection with Salmonella enterica or Staphylococcus aureus, triggering of necroptosis by the pathogens is a deleterious process for the host (Robinson et al., 2012; Kitur et al., 2015). Apart from infections, necroptosis also has a function in sterile pathologies. Genetic deletion or pharmacological inhibition of RIPK1/3 and MLKL improve the outcome in models of systemic inflammatory response syndrome (Zelic et al., 2018; Moerke et al., 2019), and ischemia-reperfusion injury of the kidney (Müller et al., 2017), brain (Naito et al., 2020), and heart (Luedde et al., 2014). The RIPK3-MLKL axis is also involved in crystallopathies induced by cytotoxic crystals (Mulay et al., 2016). Cancer metastasis also depends on necroptosis (Strilic et al., 2016). As shown recently, necroptosis also plays a role in inflammatory skin diseases. In a mouse model of psoriasis, it was shown that inhibition of the RIPK1-RIPK3-MLKL axis has a protective effect (Duan et al., 2020), and that, in the absence of RIPK1 in keratinocytes, ZBP1-induced necroptosis leads to massive inflammation of the skin (Devos et al., 2020). The fact that two very different stimuli can trigger necroptosis via different pathways underlines the complexity of this topic, especially with regard to possible therapeutic interventions. A function of the necroptosis signaling pathway has also been demonstrated in a broad range of other disease models, which is reviewed elsewhere in greater detail (Weinlich et al., 2017). Most recently, certain homozygous mutations in the Mlkl gene that lead to a lethal inflammatory phenotype in mice were described for the first time. For unknown reasons, this phenotype seems to be caused by hyperinflammation of the mediastinum and salivary glands. In heterozygous human individuals, however, these mutations are associated with increased incidence of the autoimmune disease chronic recurrent multifocal osteomyelitis (Hildebrand et al., 2020). Taken together, there is ever growing evidence for necroptosis as a promising target for therapeutic efforts in a multitude of highly relevant diseases, although the mechanisms require further clarification.
Pyroptosis and GSDMD
Pyroptosis is executed by the proteins of the gasdermin family. This chapter focuses on GSDMD, as it is so far the best characterized of these homologous pore-forming proteins and is conserved in humans and mice. GSDMD is a cytosolic 53 kDa protein expressed in immune cells and epithelia, and consists of a cytotoxic N-terminal domain (GSDMD-N), a linker region, and an inhibitory C-terminal domain (GSDMD-C) (Kuang et al., 2017). Although structurally similar at first glance, GSDMD is, in contrast to MLKL, not activated via phosphorylation but through proteolytic cleavage (Kayagaki et al., 2015; Shi et al., 2015). Two major pathways of GSDMD activation have been described. The so-called canonical pathway is dependent on the formation of multiprotein complexes called inflammasomes. Inflammasomes are formed by cytosolic pattern recognition receptors (PRRs) termed nucleotide-binding oligomerization domain (NOD) and leucine-rich repeat (LRR) receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors, and retinoic acid-inducible gene I (RIG I)-like receptors. One of the best studied of these is the NLR protein 3 (NLRP3) inflammasome (Broz and Dixit, 2016). Canonical activation of the NLRP3 inflammasome requires two steps: in the first priming step, DAMPs or pathogen-associated molecular patterns (PAMPs) activate various PRRs such as TLRs, which leads to upregulation in the synthesis of critical inflammasome components such as NLRP3 itself, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), procaspase-1, and the inactive precursor cytokines pro-interleukin (IL)-1β and pro-IL-18. A second step is needed for inflammasome activation. A critical element of this activation step are changes in intracellular potassium and chloride concentrations, the function of which we have gained better understanding in recent years. To assemble the NLRP3 inflammasome, cytosolic ASC oligomers or specks must first be formed, and this process is dependent on chloride efflux from the cell. This chloride efflux relies on the translocation of chloride intracellular channels (CLICs) to the plasma membrane and is an indispensable upstream event of NLRP3 inflammasome activation (Tang et al., 2017; Green et al., 2018). Besides the CLICs, chloride outflow from the volume-regulated anion channel (VRAC) is also described in this context, which can be inhibited by certain non-steroidal anti-inflammatory drugs (Daniels et al., 2016). In addition to chloride, potassium efflux is also necessary for inflammasome activation, and can be triggered by various mechanisms. Ionophores and bacterial toxins such as nigericin or streptolysin O can facilitate potassium efflux, making the NLRP3 inflammasome a sensor of deleterious microorganisms (Petrilli et al., 2007; Greaney et al., 2015). In addition, cell damage in the surrounding tissue, through which ATP enters the extracellular space, can be sensed by the ligand-gated purinergic P2X7 ion channel receptor (P2X7R). Thus, the cell membrane is permeabilized for cations, and potassium efflux becomes possible (Alves et al., 2014). The potassium efflux then leads to the association of NIMA-related kinase 7 (NEK7) with NLRP3 and subsequent inflammasome activation, which is followed by the cleavage of procaspase-1 into its active form and the processing and release of IL-1β and IL-18 (Munoz-Planillo et al., 2013; He et al., 2016). As with the execution of necroptosis, the influence of disturbances in the redox homeostasis of the cell and ROS generation from several sources on NLRP3 inflammasome activation has also been discussed, although the underlying mechanisms are not fully understood (reviewed in Abais et al., 2015). The further details of canonical inflammasome activation exceed the scope of this review, and are described excellently elsewhere (Schroder and Tschopp, 2010; Broz and Dixit, 2016). Besides the canonical pathway, there are other means of initiating pyroptosis. The non-canonical pathway is a “shortcut” for the activating GSDMD. Upon sensing intracellular bacterial lipopolysaccharide (LPS), the inflammatory caspases-4/5 (in humans) and caspase-11 (in mice) can directly cleave GSDMD and initiate pyroptosis (Shi et al., 2014). GSDMD can also be activated directly by caspase-8 when transforming growth factor β-activated kinase 1 (TAK1), a central regulator of cell death and survival in a variety of settings, is inhibited, as described in the case of Yersinia pseudotuberculosis infection (Orning et al., 2018; Sarhan et al., 2018). Apart from this, GSDMD activation by the proteases cathepsin G and neutrophil elastase has been reported in myeloid cells (Sollberger et al., 2018; Burgener et al., 2019).
Common to all these pathways initiating pyroptosis is the proteolytic cleavage of GSDMD in the linker region, which releases GSDMD-N from its autoinhibition by GSDMD-C. GSDMD-N forms oligomers that create membrane pores. Therefore, GSDMD-N translocates exclusively to the inner leaflet of cell membranes by binding PIPs, phosphatidic acid, and phosphatidylserine (Ding et al., 2016; Liu et al., 2016). The nanostructure of the gasdermin pore has been described for mouse gasdermin A3 via cryo-electron microscopy. It appears to be a 26–28-fold symmetric, anti-parallel β-barrel pore with an inner diameter of 18 nm (Ruan et al., 2018). The size of the human GSDMD-N pore is estimated to be 10–20 nm (Ding et al., 2016; Sborgi et al., 2016). GSDMD-N also has the ability to bind cardiolipin, and there are reports of gasdermin-mediated permeabilization of mitochondria contributing to pyroptotic cell death (de Vasconcelos et al., 2019; Rogers et al., 2019). The GSDMD-N pore is structurally and functionally different from the MLKL pore. Intracellular osmolality is unchanged during pyroptosis, as the GSDMD-N pore is unselective and allows free diffusion of ions and water as well as the release of processed cytokines, the first and foremost being IL-1β and IL-18 (Heilig et al., 2018). This difference also finds its expression in the varying morphology between necroptosis and pyroptosis. In pyroptosis, there is no swelling or explosive rupture of the cells, but a flattening of the cytoplasm, as cytoplasmic fluid leaks through the pores (Chen et al., 2016).
Analogous to necroptosis, most of the understanding of the (patho-)physiological role of pyroptosis has been generated through the utilization of animal models deficient for the inflammatory caspases, inflammasome components, IL-1 and IL-18, or GSDMD. Countering infections is doubtless one of the most crucial roles of pyroptosis. The clearance of viral, bacterial, fungal, and protozoan infections relies on it to differing extents, depending on the underlying microorganism and context (Man et al., 2017). Direct lysis of bacteria through its ability to bind cardiolipin in the bacterial cell wall has been described for GSDMD-N (Liu et al., 2016). Counterintuitively, and all the more surprising, is the fact that Gsdmd-deficient mice showed improved survival in a model of infectious peritonitis by Escherichia coli, as Gsdmd deficiency in this case prolonged neutrophil survival and therefore improved bacterial clearance (Kambara et al., 2018). These findings once again underscore the complexity of RCD in disease. Besides infectious diseases, there is also evidence that pyroptosis has a role in sterile diseases, and these largely overlap those in which necroptosis has also shown relevance, such as in ischemia-reperfusion injury of the heart (Qiu et al., 2017), brain (Yang F. et al., 2014), and kidney (Yang J.R. et al., 2014), crystallopathies (Ludwig-Portugall et al., 2016; Goldberg et al., 2017), Alzheimer’s disease (Han et al., 2020), and other neurodegenerative pathologies (McKenzie et al., 2020) and a multitude of other inflammatory diseases (Goldberg et al., 2017). Of particular note in this context is the recent discovery that ticagrelor – a substance usually administered in coronary heart disease due to its inhibitory properties on platelet aggregation – also inhibits the NLRP3 inflammasome and therefore pyroptosis by blocking the CLIC-dependent chloride efflux (Huang et al., 2020). This interesting double effect could explain part of the good efficacy after coronary interventions. However, much of this research has been conducted through genetic ablation or pharmacological inhibition of the NLRP3 inflammasome, and requires future testing of the models in specific Gsdmd knockout mice to clearly discriminate the exact contribution of pyroptosis itself from other inflammasome-driven effects.
Crosslinks Between Necroptosis and Pyroptosis
Although it was and still is necessary to consider the different forms of RCD in isolation to decipher their molecular pathways, there is emerging evidence for combined action of specific forms of RCD in particular diseases. We ourselves first demonstrated that ischemia-reperfusion injury of the kidney was not only mediated by necroptosis (Linkermann et al., 2012), but also by the combination of necroptosis and ferroptosis, an iron-dependent form of regulated cell death driven by phospholipid peroxidation (Müller et al., 2017), and was greatly ameliorated by combined targeting of necroptosis and the RCD cyclophilin D-mediated mitochondrial permeability transition (Linkermann et al., 2013). Other authors have reported the cooperation of apoptosis and necroptosis in the pathology of ischemic stroke (Naito et al., 2020).
Recently, such an interaction has also become apparent between necroptosis and pyroptosis. Initial in vitro studies have shown RIPK3-dependent processing of IL-1 in the absence of the inhibitors of apoptosis proteins (IAPs), which play a critical role in several RCD pathways and human disease (Vince et al., 2012). The refined and complex mechanism of this process has been elucidated in further excellent research (Yabal et al., 2014; Lawlor et al., 2017). At the same time, a role for MLKL has been brought into play, and inflammasome activation dependent on the RIPK1-RIPK3-MLKL axis in dendritic cells has been reported (Kang et al., 2013) as well as MLKL-dependent and -independent RIPK3-mediated inflammasome activation (Kang et al., 2015; Lawlor et al., 2015). In 2017, it was shown that the activation of MLKL is sufficient for inflammasome assembly and the release of mature IL-1β even in absence of GSDMD, a prime example of how different forms of RCD can compensate for each other (Gutierrez et al., 2017). Only weeks later, a second group similarly reported a cell-intrinsic activation of the NLRP3 inflammasome by MLKL during the induction of necroptotic cell death (Conos et al., 2017). These two studies also clearly identified the underlying mechanism: MLKL-mediated potassium efflux from the cell is the indispensable activation step analogous to the canonical pathway of inflammasome activation (Figure 1).
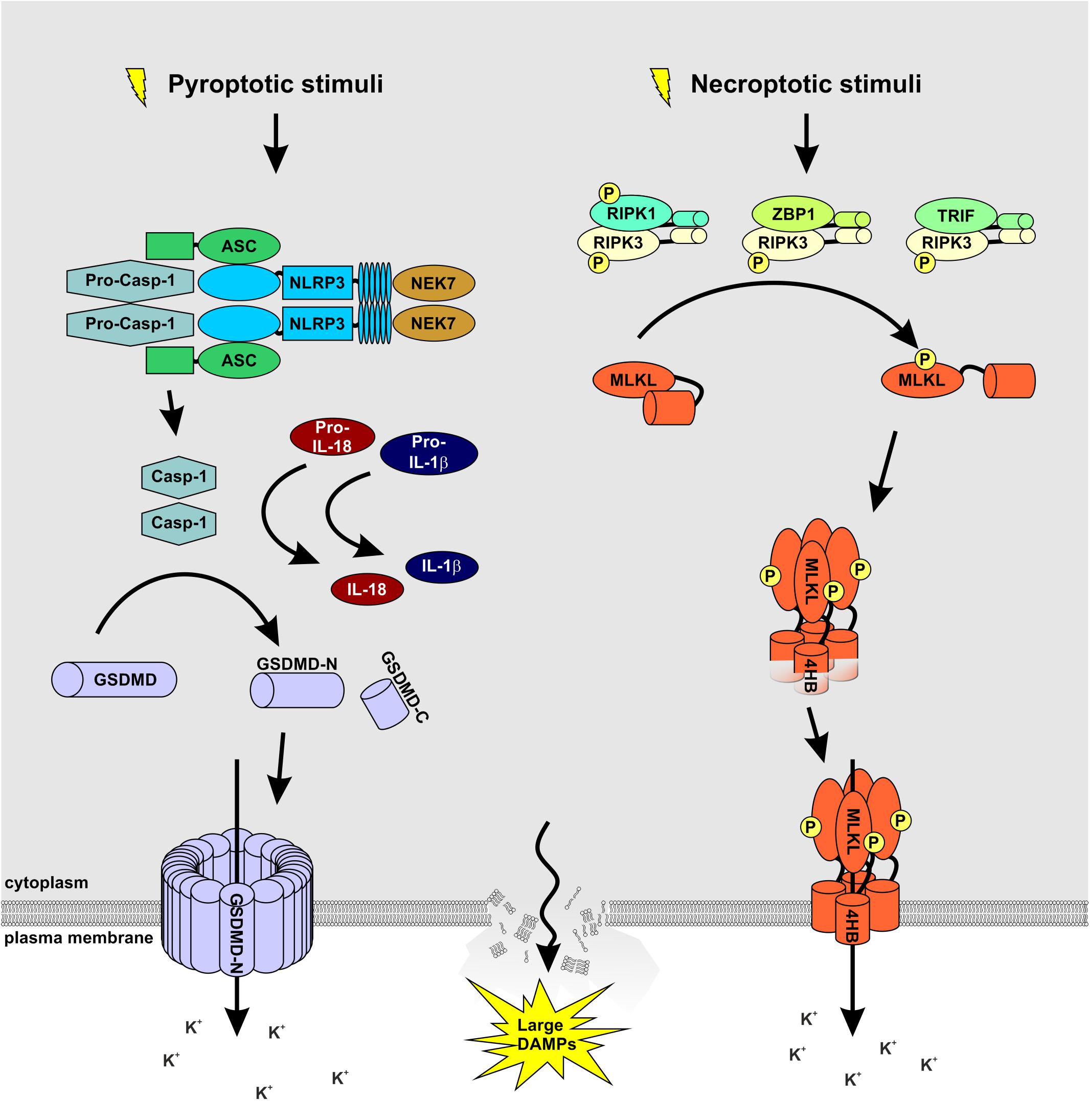
Figure 1. MLKL and GSDMD: partners in crime. A wide array of extracellular stimuli such as bacteria, viruses, and toxins serve as cellular stressors that activate NLRP3. The latter activates caspase-1 via the adaptor protein ASC. Caspase-1 processes and activates IL-1β and IL-18 and cleaves GSDMD to release the membrane pore-forming GSDMD-N domain. Necroptosis is induced by ligand binding to TNF family death domain receptors, PRRs, and virus sensors, which promote the assembly of RIPK3-MLKL complex. RIPK3-mediated phosphorylation of MLKL results in MLKL oligomerization followed by translocation to the plasma membrane to induce membrane rupture. Notably, both signaling pathways cause potassium efflux, accompanied by water influx, cell swelling, and osmotic lysis, propagating the pro-inflammatory responses triggered by NEK7 binding to NLRP3.
The first strong indications of an in vivo connection have been reported in a mouse model of S. aureus pneumonia, in which the inhibition of MLKL-dependent necroptosis decreased caspase-1 activation and IL-1β processing (Kitur et al., 2015). Second, in a mouse model of oral infection with Salmonella enterica serovar Typhimurium, MLKL-dependent initiation of pyroptosis in the caecum was needed for effective clearance of the pathogen, whereas Mlkl-deficient mice died significantly faster than their wild-type counterparts (Yu et al., 2018). Especially interesting in this case is the fact that Yu and colleagues showed, via the generation of bone marrow chimeras, that MLKL and therefore pyroptosis in non-hematopoietic cells conferred the protective effect against infection. Up to that point, a connection between MLKL and inflammasome activation had, to our knowledge, only been reported in cells of myeloid heritage. Another noteworthy connection between necroptosis and pyroptosis was found in the effect of the small molecule necrosulfonamide (NSA). NSA was initially described in a groundbreaking work by Xiaodong Wang’s group (Sun et al., 2012). It covalently binds to human MLKL via the Cys86 residue, inhibiting its activation by RIPK3 and consequently necroptosis. Surprisingly, NSA is also capable of binding GSDMD via Cys191, inhibiting pyroptotic cell death not only in vitro in human and mouse cell lines but also in vivo in murine sepsis models (Rathkey et al., 2018). Most recently, a mouse model of combined Ripk3/Gsdmd and Mlkl/Gsdmd knockout was reported. The double knockout showed significantly better protection in a mouse model of polymicrobial sepsis, and by generating bone marrow chimeras, the relevance of hematopoietic as well as non-hematopoietic cells in the model could be documented (Chen et al., 2020). Latterly, a connection between the three different forms of RCD pyroptosis, apoptosis, and necroptosis (PAN) has been shown in vitro. Evidence that these three forms of cell death may be linked has been found in specific settings. In influenza A virus infection, ZBP1 is a central regulator of PAN (Kuriakose et al., 2016; Kesavardhana et al., 2017). Additionally, the genetic- or pathogen-induced loss of TAK1 can also trigger pyroptosis, apoptosis, and necroptosis (Malireddi et al., 2018; Orning et al., 2018; Podder et al., 2019). This threefold activation of the cell death pathways was eventually termed PANoptosis (Malireddi et al., 2019). The most recent research in this field shows that this process is under the control of a master protein complex termed the PANoptosome (Christgen et al., 2020). Despite all this excellent work, our mechanistic understanding of these processes in vivo remains very vague. Based on the available data, it can be assumed that necroptosis and pyroptosis in particular play a common role not only in sepsis, but in a variety of other diseases, and we eagerly await further research in this area.
Conclusion and Clinical Relevance
Over the last few years, it has become clear that the forms of inflammatory RCD have a critical position in the pathophysiology of almost all known diseases. Membrane pores and ion fluxes play a central role in these cell death processes and are already promising targets for therapeutic approaches. However, it is becoming equally clear that different cell death pathways have context-dependent positive or negative impacts in that they can compensate for each other and have synergistic or antagonistic effects. Therefore, their therapeutic applications in humans must be tested with great care, and it must be clarified beyond doubt whether and to what extent the different forms of RCD are advantageous or disadvantageous in the respective disease and how they are interconnected. A further complicating factor is that an attempt at therapeutic intervention in one pathway may unexpectedly have an impact on others. Besides testing combined gene knockouts in disease models, it would be of special interest to see whether a combined pharmacological approach of inhibiting necroptosis and pyroptosis would generate the same effect. Future efforts will have to focus on these interactions, as intervening in these pathways promises new and better treatment options for many previously poorly treatable diseases, including stroke, myocardial infarction, acute and chronic kidney failure, sepsis, systemic inflammatory response syndrome, and cancer.
Author Contributions
UK contributed valuable ideas. TR generated the figure. BK and SK wrote the manuscript with input from all authors.
Funding
Work in the Krautwald lab was supported by a grant by the Deutsche Forschungsgemeinschaft [DFG, German Research Foundation, Projektnummer 400339789 (KR 1690/6-1)] and by the Dr. Werner Jackstädt-Stiftung.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Maike Berger and Katja Bruch for their excellent technical assistance. We would like to apologize to the many authors whose work we could not cite due to space restrictions.
References
Abais, J. M., Xia, M., Zhang, Y., Boini, K. M., and Li, P. L. (2015). Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 22, 1111–1129. doi: 10.1089/ars.2014.5994
Alves, L. A., de Melo Reis, R. A., de Souza, C. A., de Freitas, M. S., Teixeira, P. C., Neto Moreira Ferreira, D., et al. (2014). The P2X7 receptor: shifting from a low- to a high-conductance channel - an enigmatic phenomenon? Biochim. Biophys. Acta 1838, 2578–2587. doi: 10.1016/j.bbamem.2014.05.015
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Broz, P., and Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Burgener, S. S., Leborgne, N. G. F., Snipas, S. J., Salvesen, G. S., Bird, P. I., and Benarafa, C. (2019). Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep. 27, 3646–3656.e5. doi: 10.1016/j.celrep.2019.05.065
Cai, Z., Jitkaew, S., Zhao, J., Chiang, H. C., Choksi, S., Liu, J., et al. (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell. Biol. 16, 55–65. doi: 10.1038/ncb2883
Chen, H., Li, Y., Wu, J., Li, G., Tao, X., Lai, K., et al. (2020). RIPK3 collaborates with GSDMD to drive tissue injury in lethal polymicrobial sepsis. Cell Death Differ. doi: 10.1038/s41418-020-0524-1 [Epub ahead of print].
Chen, X., He, W. T., Hu, L., Li, J., Fang, Y., Wang, X., et al. (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26, 1007–1020. doi: 10.1038/cr.2016.100
Chen, X., Li, W., Ren, J., Huang, D., He, W. T., Song, Y., et al. (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24, 105–121. doi: 10.1038/cr.2013.171
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. doi: 10.1016/j.cell.2009.05.037
Christgen, S., Zheng, M., Kesavardhana, S., Karki, R., Malireddi, R. K. S., Banoth, B., et al. (2020). Identification of the PANoptosome: a molecular platform triggering pyroptosis, Apoptosis, and necroptosis (PANoptosis). Front. Cell. Infect. Microbiol. 10:237. doi: 10.3389/fcimb.2020.00237
Conos, S. A., Chen, K. W., De Nardo, D., Hara, H., Whitehead, L., Nunez, G., et al. (2017). Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc. Natl. Acad. Sci. U.S.A. 114, E961–E969. doi: 10.1073/pnas.1613305114
Daniels, M. J., Rivers-Auty, J., Schilling, T., Spencer, N. G., Watremez, W., Fasolino, V., et al. (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 7:12504. doi: 10.1038/ncomms12504
Davies, K. A., Fitzgibbon, C., Young, S. N., Garnish, S. E., Yeung, W., Coursier, D., et al. (2020). Distinct pseudokinase domain conformations underlie divergent activation mechanisms among vertebrate MLKL orthologues. Nat. Commun. 11:3060. doi: 10.1038/s41467-020-16823-3
de Vasconcelos, N. M., Van Opdenbosch, N., Van Gorp, H., Parthoens, E., and Lamkanfi, M. (2019). Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 26, 146–161. doi: 10.1038/s41418-018-0106-7
Degterev, A., Hitomi, J., Germscheid, M., Ch’en, I. L., Korkina, O., Teng, X., et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321. doi: 10.1038/nchembio.83
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119. doi: 10.1038/nchembio711
Devos, M., Tanghe, G., Gilbert, B., Dierick, E., Verheirstraeten, M., Nemegeer, J., et al. (2020). Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J. Exp. Med. 217:e20191913. doi: 10.1084/jem.20191913
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. doi: 10.1038/nature18590
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7, 971–981. doi: 10.1016/j.celrep.2014.04.026
Duan, X., Liu, X., Liu, N., Huang, Y., Jin, Z., Zhang, S., et al. (2020). Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis. 11:134. doi: 10.1038/s41419-020-2328-0
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 25, 486–541. doi: 10.1038/s41418-017-0012-4
Goldberg, E. L., Asher, J. L., Molony, R. D., Shaw, A. C., Zeiss, C. J., Wang, C., et al. (2017). beta-hydroxybutyrate deactivates neutrophil NLRP3 inflammasome to relieve gout flares. Cell Rep. 18, 2077–2087. doi: 10.1016/j.celrep.2017.02.004
Greaney, A. J., Leppla, S. H., and Moayeri, M. (2015). Bacterial exotoxins and the inflammasome. Front. Immunol. 6:570. doi: 10.3389/fimmu.2015.00570
Green, J. P., Yu, S., Martin-Sanchez, F., Pelegrin, P., Lopez-Castejon, G., Lawrence, C. B., et al. (2018). Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. U.S.A. 115, E9371–E9380. doi: 10.1073/pnas.1812744115
Gutierrez, K. D., Davis, M. A., Daniels, B. P., Olsen, T. M., Ralli-Jain, P., Tait, S. W., et al. (2017). MLKL activation triggers NLRP3-mediated processing and release of IL-1beta independently of Gasdermin-D. J. Immunol. 198, 2156–2164. doi: 10.4049/jimmunol.1601757
Han, C., Yang, Y., Guan, Q., Zhang, X., Shen, H., Sheng, Y., et al. (2020). New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J. Cell. Mol. Med. 24, 8078–8090. doi: 10.1111/jcmm.15439
He, Y., Zeng, M. Y., Yang, D., Motro, B., and Nunez, G. (2016). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. doi: 10.1038/nature16959
Heilig, R., Dick, M. S., Sborgi, L., Meunier, E., Hiller, S., and Broz, P. (2018). The gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 48, 584–592. doi: 10.1002/eji.201747404
Hildebrand, J. M., Kauppi, M., Majewski, I. J., Liu, Z., Cox, A. J., Miyake, S., et al. (2020). A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat. Commun. 11:3150. doi: 10.1038/s41467-020-16819-z
Hildebrand, J. M., Tanzer, M. C., Lucet, I. S., Young, S. N., Spall, S. K., Sharma, P., et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl. Acad. Sci. U.S.A. 111, 15072–15077. doi: 10.1073/pnas.1408987111
Huang, B., Qian, Y., Xie, S., Ye, X., Chen, H., Chen, Z., et al. (2020). Ticagrelor inhibits the NLRP3 inflammasome to protect against inflammatory disease independent of the P2Y12 signaling pathway. Cell. Mol. Immunol. doi: 10.1038/s41423-020-0444-5 [Epub ahead of print].
Jacobsen, A. V., Lowes, K. N., Tanzer, M. C., Lucet, I. S., Hildebrand, J. M., Petrie, E. J., et al. (2016). HSP90 activity is required for MLKL oligomerisation and membrane translocation and the induction of necroptotic cell death. Cell Death. Dis. 7:e2051. doi: 10.1038/cddis.2015.386
Kambara, H., Liu, F., Zhang, X., Liu, P., Bajrami, B., Teng, Y., et al. (2018). Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 22, 2924–2936. doi: 10.1016/j.celrep.2018.02.067
Kang, S., Fernandes-Alnemri, T., Rogers, C., Mayes, L., Wang, Y., Dillon, C., et al. (2015). Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 6:7515. doi: 10.1038/ncomms8515
Kang, T. B., Yang, S. H., Toth, B., Kovalenko, A., and Wallach, D. (2013). Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38, 27–40. doi: 10.1016/j.immuni.2012.09.015
Kayagaki, N., Stowe, I. B., Lee, B. L., O’Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kesavardhana, S., Kuriakose, T., Guy, C. S., Samir, P., Malireddi, R. K. S., Mishra, A., et al. (2017). ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J. Exp. Med. 214, 2217–2229. doi: 10.1084/jem.20170550
Kitur, K., Parker, D., Nieto, P., Ahn, D. S., Cohen, T. S., Chung, S., et al. (2015). Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 11:e1004820. doi: 10.1371/journal.ppat.1004820
Kuang, S., Zheng, J., Yang, H., Li, S., Duan, S., Shen, Y., et al. (2017). Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc. Natl. Acad. Sci. U.S.A. 114, 10642–10647. doi: 10.1073/pnas.1708194114
Kuriakose, T., Man, S. M., Subbarao Malireddi, R. K., Karki, R., Kesavardhana, S., Place, D. E., et al. (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1:aag2045. doi: 10.1126/sciimmunol.aag2045
Lawlor, K. E., Feltham, R., Yabal, M., Conos, S. A., Chen, K. W., Ziehe, S., et al. (2017). XIAP loss triggers RIPK3- and Caspase-8-driven IL-1beta activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 20, 668–682. doi: 10.1016/j.celrep.2017.06.073
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D’Cruz, A. A., et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6:6282. doi: 10.1038/ncomms7282
Linkermann, A., Bräsen, J. H., Darding, M., Jin, M. K., Sanz, A. B., Heller, J. O., et al. (2013). Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 110, 12024–12029. doi: 10.1073/pnas.1305538110
Linkermann, A., Bräsen, J. H., Himmerkus, N., Liu, S., Huber, T. B., Kunzendorf, U., et al. (2012). Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761. doi: 10.1038/ki.2011.450
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi: 10.1038/nature18629
Ludwig-Portugall, I., Bartok, E., Dhana, E., Evers, B. D., Primiano, M. J., Hall, J. P., et al. (2016). An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 90, 525–539. doi: 10.1016/j.kint.2016.03.035
Luedde, M., Lutz, M., Carter, N., Sosna, J., Jacoby, C., Vucur, M., et al. (2014). RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc. Res. 103, 206–216. doi: 10.1093/cvr/cvu146
Malireddi, R. K. S., Gurung, P., Mavuluri, J., Dasari, T. K., Klco, J. M., Chi, H., et al. (2018). TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J. Exp. Med. 215, 1023–1034. doi: 10.1084/jem.20171922
Malireddi, R. K. S., Kesavardhana, S., and Kanneganti, T.-D. (2019). ZBP1 and TAK1: master regulators of NLRP3 inflammasome/pyroptosis, Apoptosis, and necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9:406. doi: 10.3389/fcimb.2019.00406
Man, S. M., Karki, R., and Kanneganti, T. D. (2017). Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 277, 61–75. doi: 10.1111/imr.12534
Marshall, K. D., and Baines, C. P. (2014). Necroptosis: is there a role for mitochondria? Front. Physiol. 5:323. doi: 10.3389/fphys.2014.00323
McKenzie, B. A., Dixit, V. M., and Power, C. (2020). Fiery cell death: pyroptosis in the central nervous system. Trends Neurosci. 43, 55–73. doi: 10.1016/j.tins.2019.11.005
Moerke, C., Bleibaum, F., Kunzendorf, U., and Krautwald, S. (2019). Combined knockout of RIPK3 and MLKL reveals unexpected outcome in tissue injury and inflammation. Front. Cell. Dev. Biol. 7:19. doi: 10.3389/fcell.2019.00019
Mulay, S. R., Desai, J., Kumar, S. V., Eberhard, J. N., Thomasova, D., Romoli, S., et al. (2016). Cytotoxicity of crystals involves RIPK3-MLKL-mediated necroptosis. Nat. Commun. 7:10274. doi: 10.1038/ncomms10274
Müller, T., Dewitz, C., Schmitz, J., Schröder, A. S., Bräsen, J. H., Stockwell, B. R., et al. (2017). Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell. Mol. Life Sci. 74, 3631–3645. doi: 10.1007/s00018-017-2547-4
Munoz-Planillo, R., Kuffa, P., Martinez-Colon, G., Smith, B. L., Rajendiran, T. M., and Nunez, G. (2013). K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. doi: 10.1016/j.immuni.2013.05.016
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. doi: 10.1016/j.immuni.2013.06.018
Naito, M. G., Xu, D., Amin, P., Lee, J., Wang, H., Li, W., et al. (2020). Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. U.S.A. 117, 4959–4970. doi: 10.1073/pnas.1916427117
Nogusa, S., Thapa, R. J., Dillon, C. P., Liedmann, S., Oguin, T. H. III, Ingram, J. P., et al. (2016). RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated Apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24. doi: 10.1016/j.chom.2016.05.011
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. doi: 10.1126/science.aau2818
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature 517, 311–320. doi: 10.1038/nature14191
Petrie, E. J., Hildebrand, J. M., and Murphy, J. M. (2017). Insane in the membrane: a structural perspective of MLKL function in necroptosis. Immunol. Cell. Biol. 95, 152–159. doi: 10.1038/icb.2016.125
Petrie, E. J., Sandow, J. J., Jacobsen, A. V., Smith, B. J., Griffin, M. D. W., Lucet, I. S., et al. (2018). Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9:2422. doi: 10.1038/s41467-018-04714-7
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., and Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 14, 1583–1589. doi: 10.1038/sj.cdd.4402195
Podder, B., Gutta, C., Rozanc, J., Gerlach, E., Feoktistova, M., Panayotova-Dimitrova, D., et al. (2019). TAK1 suppresses RIPK1-dependent cell death and is associated with disease progression in melanoma. Cell Death Differ. 26, 2520–2534. doi: 10.1038/s41418-019-0315-8
Qiu, Z., Lei, S., Zhao, B., Wu, Y., Su, W., Liu, M., et al. (2017). NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid. Med. Cell. Longev. 2017:9743280. doi: 10.1155/2017/9743280
Rathkey, J. K., Zhao, J., Liu, Z., Chen, Y., Yang, J., Kondolf, H. C., et al. (2018). Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 3:eaat2738. doi: 10.1126/sciimmunol.aat2738
Robinson, N., McComb, S., Mulligan, R., Dudani, R., Krishnan, L., and Sad, S. (2012). Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar typhimurium. Nat. Immunol. 13, 954–962. doi: 10.1038/ni.2397
Rodriguez, D. A., Weinlich, R., Brown, S., Guy, C., Fitzgerald, P., Dillon, C. P., et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88. doi: 10.1038/cdd.2015.70
Rogers, C., Erkes, D. A., Nardone, A., Aplin, A. E., Fernandes-Alnemri, T., and Alnemri, E. S. (2019). Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10:1689. doi: 10.1038/s41467-019-09397-2
Ros, U., Pena-Blanco, A., Hanggi, K., Kunzendorf, U., Krautwald, S., Wong, W. W., et al. (2017). Necroptosis execution is mediated by plasma membrane nanopores independent of calcium. Cell Rep. 19, 175–187. doi: 10.1016/j.celrep.2017.03.024
Ruan, J., Xia, S., Liu, X., Lieberman, J., and Wu, H. (2018). Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. doi: 10.1038/s41586-018-0058-6
Sai, K., Parsons, C., House, J. S., Kathariou, S., and Ninomiya-Tsuji, J. (2019). Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J. Cell. Biol. 218, 1994–2005. doi: 10.1083/jcb.201810014
Samson, A. L., Zhang, Y., Geoghegan, N. D., Gavin, X. J., Davies, K. A., Mlodzianoski, M. J., et al. (2020). MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 11:3151. doi: 10.1038/s41467-020-16887-1
Sarhan, J., Liu, B. C., Muendlein, H. I., Li, P., Nilson, R., Tang, A. Y., et al. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U.S.A. 115, E10888–E10897. doi: 10.1073/pnas.1809548115
Sborgi, L., Ruhl, S., Mulvihill, E., Pipercevic, J., Heilig, R., Stahlberg, H., et al. (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35, 1766–1778. doi: 10.15252/embj.201694696
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Shan, B., Pan, H., Najafov, A., and Yuan, J. (2018). Necroptosis in development and diseases. Genes Dev. 32, 327–340. doi: 10.1101/gad.312561.118
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. doi: 10.1038/nature13683
Silke, J., Rickard, J. A., and Gerlic, M. (2015). The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 16, 689–697. doi: 10.1038/ni.3206
Sollberger, G., Choidas, A., Burn, G. L., Habenberger, P., Di Lucrezia, R., Kordes, S., et al. (2018). Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
Strilic, B., Yang, L., Albarran-Juarez, J., Wachsmuth, L., Han, K., Müller, U. C., et al. (2016). Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 536, 215–218. doi: 10.1038/nature19076
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi: 10.1016/j.cell.2011.11.031
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. doi: 10.1038/s41422-019-0164-5
Tang, T., Lang, X., Xu, C., Wang, X., Gong, T., Yang, Y., et al. (2017). CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat. Commun. 8:202. doi: 10.1038/s41467-017-00227-x
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. doi: 10.1016/j.chom.2012.01.016
Vince, J. E., Wong, W. W., Gentle, I., Lawlor, K. E., Allam, R., O’Reilly, L., et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36, 215–227. doi: 10.1016/j.immuni.2012.01.012
Wang, H., Sun, L., Su, L., Rizo, J., Liu, L., Wang, L. F., et al. (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. doi: 10.1016/j.molcel.2014.03.003
Wang, X., Li, Y., Liu, S., Yu, X., Li, L., Shi, C., et al. (2014). Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443. doi: 10.1073/pnas.1412767111
Weinlich, R., Oberst, A., Beere, H. M., and Green, D. R. (2017). Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136. doi: 10.1038/nrm.2016.149
Xia, B., Fang, S., Chen, X., Hu, H., Chen, P., Wang, H., et al. (2016). MLKL forms cation channels. Cell Res. 26, 517–528. doi: 10.1038/cr.2016.26
Yabal, M., Müller, N., Adler, H., Knies, N., Gross, C. J., Damgaard, R. B., et al. (2014). XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7, 1796–1808. doi: 10.1016/j.celrep.2014.05.008
Yang, F., Wang, Z., Wei, X., Han, H., Meng, X., Zhang, Y., et al. (2014). NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J. Cereb. Blood Flow Metab. 34, 660–667. doi: 10.1038/jcbfm.2013.242
Yang, J. R., Yao, F. H., Zhang, J. G., Ji, Z. Y., Li, K. L., Zhan, J., et al. (2014). Ischemia-reperfusion induces renal tubule pyroptosis via the CHOP-caspase-11 pathway. Am. J. Physiol. Renal Physiol. 306, F75–F84. doi: 10.1152/ajprenal.00117.2013
Yu, S. X., Chen, W., Liu, Z. Z., Zhou, F. H., Yan, S. Q., Hu, G. Q., et al. (2018). Non-hematopoietic MLKL protects against salmonella mucosal infection by enhancing inflammasome activation. Front. Immunol. 9:119. doi: 10.3389/fimmu.2018.00119
Zelic, M., Roderick, J. E., O’Donnell, J. A., Lehman, J., Lim, S. E., Janardhan, H. P., et al. (2018). RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clin. Invest. 128, 2064–2075. doi: 10.1172/JCI96147
Keywords: necroptosis, pyroptosis, MLKL, GSDMD, regulated cell death, necroinflammation
Citation: Kolbrink B, Riebeling T, Kunzendorf U and Krautwald S (2020) Plasma Membrane Pores Drive Inflammatory Cell Death. Front. Cell Dev. Biol. 8:817. doi: 10.3389/fcell.2020.00817
Received: 30 June 2020; Accepted: 03 August 2020;
Published: 21 August 2020.
Edited by:
Yasunobu Okada, National Institute for Physiological Sciences (NIPS), JapanReviewed by:
Elodie Lafont, INSERM U1242 Laboratoire COSS, FranceCarsten Culmsee, University of Marburg, Germany
Copyright © 2020 Kolbrink, Riebeling, Kunzendorf and Krautwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Krautwald, krautwald@nephro.uni-kiel.de